
Figure 8.
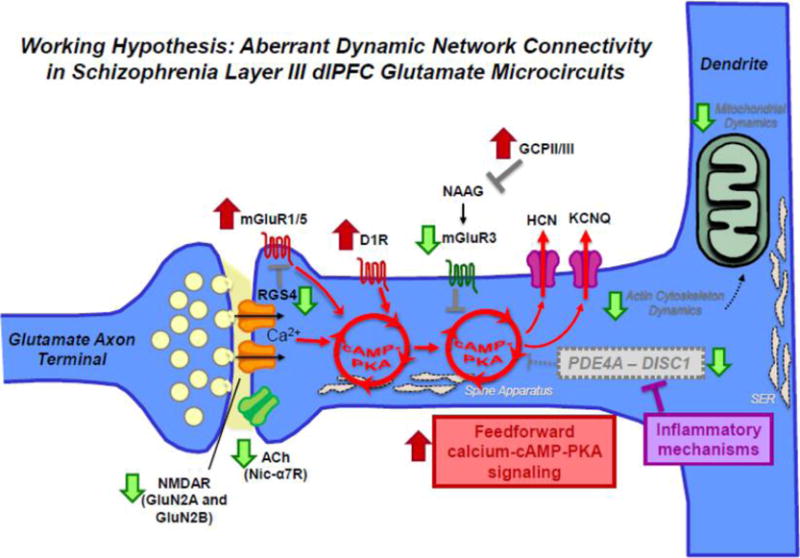
Aberrant DNC mechanisms in schizophrenia dlPFC layer III. Schematic summarizing changes in DNC mechanisms in schizophrenia dlPFC layer III due to genetic predispositions and neuropathological changes from postmortem studies. For example, both genetic and postmortem studies have revealed a decrement in acetylcholine nic-α7R and glutamate NMDAR signaling. Translocations in the Disc1 gene have been implicated in mental illness and in particular schizophrenia. DISC1 serves as a scaffold for the family of phosphodiesterases (e.g., PDE4A) that hydrolyze cAMP signaling, which have also been genetically linked to schizophrenia. ImmunoEM studies of monkey dlPFC layer III spines show that DISC1 anchors PDE4A near the spine apparatus, adjacent to HCN channels. In vitro studies show that PDE4 activity is inhibited by MK2 signaling during inflammation. If similar actions occur in human PFC in response to inflammation, e.g. in utero and/or during adolescence at the onset of illness, dysregulated cAMP–calcium signaling may lead to weaker connections and loss of spines. Schizophrenia is also associated with elevation in mGluR1α which drives internal calcium release. The activity of mGluR1α is inhibited by regulator of G protein signaling 4 (RGS4), positioned perisynaptically to gate signaling. Recent layer III pyramidal cell-type specific studies have also revealed downregulation of RGS4 mRNA in schizophrenia, which is predicted to result in dysregulated feedforward cAMP–calcium signaling. Finally, neuroimaging studies have suggested elevation in D1R in drug naïve patients which would further exaggerate a vicious cycle. These perturbations are thought to converge of impairments in actin cytoskeleton and mitochondria in dlPFC layer III pyramidal cells in schizophrenia, leading to enhanced vulnerability and weakening of network connectivity. In sum, various studies in schizophrenia suggest alterations that increase the generation of cAMP–calcium signaling and decrease mechanisms that moderate cAMP–calcium signaling.