

Abstract
We report here numerous novel genes and multiple new signatures which robustly predict prostate cancer (PC) recurrence. We extracted 696 differentially expressed genes relative to a reported PC signature from the TCGA dataset (n = 492) and built a 15‐gene signature (SigMuc1NW) using Elastic‐net with 10‐fold cross‐validation through analyzing their expressions at 1.5 standard deviation/SD below and 2 SD above a population mean. SigMuc1NW predicts biochemical recurrence (BCR) following surgery with 56.4% sensitivity, 72.6% specificity, and 63.24 median months disease free (MMDF) (P = 1.12e‐12). The prediction accuracy is improved with the use of SigMuc1NW's cutpoint (P = 3e‐15) and is further enhanced (sensitivity 67%, specificity 75.7%, MMDF 45.2, P = 0) when all 15 genes were analyzed through their cutpoints instead of their SDs. These genes individually associate with BCR using either SD or cutpoint as the cutoff points. Eight of 15 genes are individual risk factors after adjusting for age at diagnosis, Gleason score, surgical margin, and tumor stage. Eleven of 15 genes are novel to PC. SigMuc1NW discriminates BCR with time‐dependent AUC (tAUC) values of 76.6% at 11.5 months (76.6%–11.5 m), 73.8%‐22.3 m, 78.5%‐32.1 m, and 76.4%–48.4 m. SigMuc1NW is correlated with adverse features of PC, high Gleason scores (odds ratio/OR 1.48, P < 2e‐16), and advanced tumor stages (OR 1.33, P = 4.37e‐13). SigMuc1NW remains an independent risk factor of BCR (HR 2.44, 95% CI 1.53–3.87, P = 1.62e‐4) after adjusting for age at diagnosis, Gleason score, surgical margin, and tumor stage. In an independent PC (MSKCC) cohort (n = 140), these 15 genes were altered in PC vs normal tissue, metastatic PCs vs primary PCs, and recurrent PCs vs nonrecurrent PCs. Importantly, a 10‐gene subsignature SigMuc1NW1 predicts BCR in MSKCC (P = 3.11e‐15) and TCGA (P = 3.13e‐12); SigMuc1NW1 discriminates BCR at 18.4 m with tAUC as 82.5%. Collectively, our analyses support SigMuc1NW as a novel and robust signature in predicting BCR of PC.
Keywords: biomarkers, MUC1, prostate cancer, prostate cancer recurrence
Abbreviations
- ADT
androgen deprivation therapy
- BCR
biochemical recurrence
- CRPC
castration‐resistant prostate cancer
- DEGs
differentially expressed genes
- DFS
disease‐free survival
- GS
Gleason score
- MMDF
median months disease free
- OS
overall survival
- PC
prostate cancer
- RP
radical prostatectomy
1. Introduction
Prostate cancer (PC) is the most common malignancy in men in the developed countries (Ferlay et al., 2015). The disease progresses with a large degree of disparity. While a large proportion of the low grade [Gleason score 6/WHO grade (group) I or ISUP (the International Society of Urological pathology) grade 1] tumors are not life‐threatening, approximately 30% of patients after radical prostatectomy (RP) will experience disease recurrence with a rise in serum prostate‐specific antigen (PSA) (Zaorsky et al., 2013); this biochemical recurrence (BCR) indicates significantly increased risk for PC metastasis and castration‐resistant prostate cancer (CRPC) (Semenas et al., 2012). Metastasis is the leading cause of PC death. The standard treatment for metastatic PC is androgen deprivation therapy (ADT), which offers palliative care as resistance in the form of CRPC always occurs. In this regard, intervention at the point of BCR will be more effective than at time when PC has advanced to later stages. Thus, effectively assessing PCs with increased risk of BCR is highly desirable.
Recent developments have yielded three commercially available mRNA expression‐based multigene panels, Oncotype DX (Genomic Prostate Score/GPS), Prolaris (cell cycle progression/CCP), and Decipher (Genomic Classifier/GC). Both the 17‐gene Oncotype DX and the 31‐gene Prolaris improve risk stratification of patients with high risk of PC recurrence at time of diagnosis (Albala et al., 2016; Cuzick et al., 2011; Klein et al., 2014; Knezevic et al., 2013; Oderda et al., 2017) and after radical prostatectomy (RP) (Cooperberg et al., 2013; Cullen et al., 2015). The 22‐gene Decipher predicts metastasis following RP (Erho et al., 2013; Karnes et al., 2013; Klein et al., 2016). While these and other biomarkers assist decision making and thus improve patient management, their clinical application requires further validation (Lamy et al., 2017; Martin, 2016; McGrath et al., 2016; Patel and Gnanapragasam, 2016; Ross et al., 2016; Zhuang and Johnson, 2016). There is a clear need to improve our ability to stratify PCs with high risk of recurrence following RP. The challenge in accurately predicting PC recurrence is in part attributable to a complex network of pathways that drive the disease development.
The Mucin 1 (MUC1) network plays a role in BCR after RP (Eminaga et al., 2016; Lin et al., 2017). MUC1 is a tumor‐associated antigen that has been intensively investigated (Apostolopoulos et al., 2015; Kufe, 2009; Nath and Mukherjee, 2014). MUC1 is a glycoprotein that is expressed on the apical surface of most epithelial tissues (de Paula Peres et al., 2015; Wurz et al., 2014); its glycosylation is altered in over 70% of cancers (Kufe, 2009; de Paula Peres et al., 2015). In PC, MUC1 expression is upregulated and aberrantly glycosylated (Arai et al., 2005; Cozzi et al., 2005; Rabiau et al., 2009). These abnormalities are associated with angiogenesis (Papadopoulos et al., 2001) and adverse clinical features (Eminaga et al., 2016). MUC1 upregulation weakly correlates with shortening in disease‐free survival (DFS) and overall survival (OS) (Eminaga et al., 2016) and associates with adverse histopathology following RP (Durrani et al., 2015). A 3‐protein panel (AZGP1, MUC1, and p53) is related to poor prognosis in men with local PC (Severi et al., 2014). Increases in MUC1 mRNA expression were detected in metastatic PC. Genomic alterations in a 25‐gene MUC1 network were marginally associated with PC recurrence (Wong et al., 2016). Among these 25 genes, genomic alterations in nine genes substantially enhanced the association (Lin et al., 2017).
To further explore the biomarker value of the MUC1 network, we examined the transcriptome of the 9‐gene MUC1 genomic signature using the TCGA Provisional dataset within cBioPortal, and established 696 differentially expressed genes (DEGs). From these DEGs, a 15‐gene panel and multiple subpanels were constructed. These signatures robustly associate with reductions in DFS following RP in two independent PC datasets (n = 492 and n = 140). Cutpoints have been derived, which not only enhance the power of these signatures in the stratification of men with higher risk of BCR but also provide a guideline for the subsequent validation and clinical application. Taken together, we have constructed a set of novel and robust signatures to assess PC recurrence following RP.
2. Materials and methods
2.1. cBioPortal
The cBioPortal (Cerami et al., 2012; Gao et al., 2013) (http://www.cbioportal.org/index.do) database contains the most well‐organized and comprehensive data on cancer genetics for various cancer types. The TCGA Provisional datasets for individual cancer types cover genetic abnormalities, transcriptomes determined by either cDNA microarray or RNA sequencing, and the detailed clinical characteristics including disease outcomes (recurrence and mortality). The TCGA Provisional PC dataset has 492 patients with localized PC.
2.2. Establishing of multigene panel signatures
The largest TCGA Provisional dataset within the cBioPortal database (Cerami et al., 2012; Gao et al., 2013) (http://www.cbioportal.org/index.do), which includes 492 patients with follow‐up data, was used to derive 696 DEGs that are associated with the 9‐gene signature of the MUC1 genomic network (Lin et al., 2017). These DEGs were defined at q < 0.001. Follow‐up period, recurrence, and other clinical data were also extracted. Elastic‐net logistic regression within the glmnet package in R was used to select variables with major impacts on BCR with 10‐fold cross‐validation; the mixing parameter of Elastic‐net α was used at: 0.2 and 0.8. When α = 0, Elastic‐net operates as Ridge regression which does not perform covariate selection but shrink the coefficients of correlated predictors toward one another. When α = 1, it runs as Lasso which tends to select one covariate among a group of related covariates; this will make a signature less robust. To enhance selection of highly related variables as a group while maintaining the number of covariates to minimum, we used a range of α value: 0.2 and 0.8. With this system, a 15‐gene panel was selected.
2.3. Assignment of signature scores to patients/tumors
Individual component genes have been examined to predict BCR using univariate Cox proportional hazards (PH) regression; the Cox coefficients for individual component genes were obtained. The PH assumption was also determined. This analysis was performed using the R ‘survival’ package. The signature scores for individual patients were given using Sum (coef1 + coef2 + … … + coefn), where coef1 … coefn are the coefs of individual genes.
2.4. Cutpoint estimation
Cutpoint of signature in separation of recurrent tumor from those without BCR was estimated using Maximally Selected Rank Statistics (the Maxstat package) in R. We also retrieved the RNA expression data for each component gene from the TCGA dataset; the cutpoints to discriminate PCs with BCR from those without BCR for each RNA expression data were also derived.
2.5. Regression analyses
Logistic regression was performed using R. Cox proportional hazards (Cox PH) regression analyses were carried out using the R survival package. The PH assumption was examined.
2.6. Pathway enrichment analysis
The GAGE (Luo et al., 2009) and Reactome (Yu and He, 2016) packages in R were used to analyze gene sets and pathways that were enriched in DEGs using the KEGG (Kyoto Encyclopedia of Genes and Genomes) and GO (gene ontology) databases.
2.7. Statistical analysis
Fisher's exact test was performed using the GraphPad Prism 5 software. Kaplan–Meier surviving curves and log‐rank test were carried out using the R survival package, and tools provided by cBioPortal. Univariate and multivariate Cox regression analyses were run using the R survival package. Time‐dependent receive operating characteristic (tROC) analysis was performed using the R timeROC package. A value of P < 0.05 is considered statistically significant.
3. Results
3.1. Identification of DEGs which are associated with the 9‐gene MUC1 genomic signature
Biochemical recurrence (BCR) after surgical resection occurs in 30–40% of patients (Punnen et al., 2014); approximately 40% of these patients will develop metastatic disease (Briganti et al., 2015; Den et al., 2014). Improving our ability in predicting BCR risk is clearly critical in preventing metastatic progression. We have recently constructed a 9‐gene genomic signature from the MUC1 genomic network (Lin et al., 2017); the signature effectively predicts BCR using the TCGA Provisional dataset: sensitivity 34.8%, specificity 83.6%, and median months disease free (MMDF) 73.36 months (P = 5.57e‐5) (Lin et al., 2017). BCR is a complex process driven by multiple pathway alterations. In this regard, we reasoned that the transcriptome associated with the 9‐gene genomic signature may yield a better signature. To investigate this possibility, we analyzed the 9‐gene signature‐associated transcriptome using the TCGA Provisional dataset within the cBioPortal database following the strategy outlined in Fig. 1A. Among 492 patients/tumors, 100 were positive for the signature (Fig. 1A). A comparison to the mean expression of individual genes between these 100 PCs and other 392 PCs revealed a total of 696 differentially expressed genes (DEGs), which were defined at q < 0.001 (Table S1). These DEGs contained 416 downregulations and 280 upregulations (Fig. 1A; Table S1). Geneset enrichment analysis of these DEGs using the KEGG (kegg) kegg.set.hs dataset and Gaga package in R revealed the upregulation of the genesets regulating cell cycle, oocyte meiosis, progesterone‐mediated oocyte maturation (Table S2A), and downregulation of the genesets mediating focal adhesion and others (Table S2B). With the Gene Ontology (go) go.sets.hs dataset, the upregulated genesets include those regulating multiple aspects of cell cycle progression, DNA metabolism, and other processes related to cell proliferation (Table S2C). Downregulated genesets contain those that mediate cell adhesion, extracellular processes, and other events (Table S2D). Pathway enrichment analysis of the 696 DEGs using the Reactome package in R identified pathways regulating G1, M, DNA replication, and chromatid segregation (Table S2E). Collectively, the above analyses reveal an association of the 696 DEGs with PC cell proliferation, implying their potential in predicting PC progression.
Figure 1.
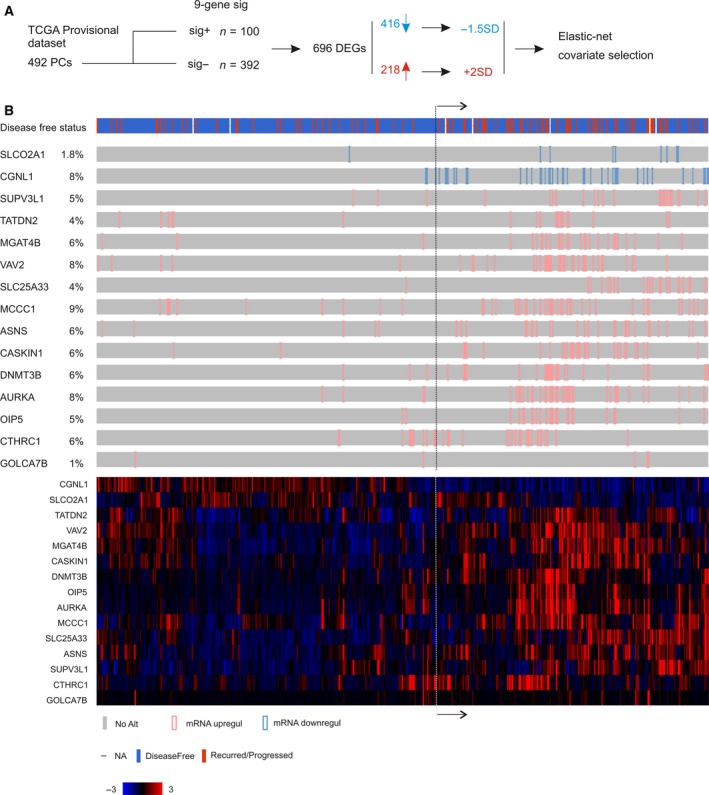
Construction of a 15‐gene signature. (A) Strategy used to produce the signature. The TCGA Provisional dataset within cBioPortal has 492 prostate cancers with gene expression profiled by RNA sequencing. The cohort was first divided into two populations: one (n = 100) positive for a 9‐gene signature derived from a MUC1 genomic network (Lin et al., 2017) and another (n = 392) negative for the signature. From these two populations, 696 differentially expressed genes (DEGs) were identified based on the mean mRNA expression and q < 0.001. These DEGs consist of 461 downregulated genes and 218 upregulated genes. For the downregulated genes, we have assigned tumors with gene expression at 1.5 SD (standard deviation) lower than a reference population mean (−1.5 SD); for those upregulated genes, we have located PCs with these gene expression at 2 SD above the population mean. We then performed model‐building using regularization‐coupled covariate selection of these 696 DEGs for their impact on BCR using the Elastic‐net penalty in the R glmnet package (Fig S1 for a typical selection), which resulted in a 15‐gene signature (SigMuc1NW). (B) PCs of the TCGA cohort with −1.5 SD (SLCP2A1 and CGNL1) and 2 SD expression are shown using OncoPrint (top gray illustration) and clustered (bottom color image). The disease‐free status is also included. The illustration was generated using tools provided by cBioPortal.
3.2. Construction of a 15‐gene signature SigMuc1NW to predict BCR following radical prostatectomy (RP)
We then analyzed the contributions of these 696 DEGs to BCR using the TCGA Provisional cohort, in which the primary treatment was RP (cBioPortal). While the classic system to construct a signature is to randomly divide a dataset into a training set and testing set (Lin et al., 2017), we chose to use the system of cross‐validation. This system is selected due to our large number of DEGs to be assessed for their impact on BCR and the availability of the powerful machine learning programs in the glmnet R package. Based on the heterogeneity of PCs, we reasoned that these DEGs may affect BCR when their expression is beyond a threshold level. For the downregulated DEGs, we separated PCs with their expression lower than 1.5 SD (standard deviation) of a reference population mean from those without this level of downregulation. For the upregulated DEGs, we grouped PCs with DEG expressions above 2 SD from the reference population mean (Fig. 1A). A reference population was either tumors within the dataset that are diploid for the gene of interest or the intact tumor population (http://www.cbioportal.org/faq.jsp). The justifications of using the levels of −1.5 SD downregulation and 2 SD upregulation here were based on our publication (Ojo et al., 2017) and to maintain a sufficient number of DEGs available for variable selection as a value below −1.5 SD or above 2 SD significantly reduced the number of qualified DEGs (data not shown).
Using this re‐organized dataset containing the downregulations, upregulations, follow‐up period, and recurrence status for each patient, we then performed covariate selection with regularization using Elastic‐net logistic regression within the R glmnet package (Fig. 1A). To balance the selection of highly correlated covariates and minimization of the number of covariates, we ran Elastic‐net with the mixing parameter α set at 0.2 or 0.8. A 10‐fold cross‐validation was used in all selection settings. As expected, more covariates were selected at α = 0.2 (n = 17) than α = 0.8 (n = 5) (Fig. S1). We also performed covariate selection with a different setting (s = 0.5) which resulted in more covariates than the setting of α = 0.2. We then removed those DEGs with coefficient < 0.01 in the s = 0.5 setting and < 0.001 in the α = 0.2 setting. This resulted in a panel of 15 genes (SigMuc1NW; NW referring to network), including all 5 genes selected at α = 0.8, 14 genes selected from α = 0.2 (including all 5 genes selected at α = 0.8), and 15 DEGs from s = 0.5 (including all 14 genes selected at α = 0.2) (Table 1).
Table 1.
The component genes of SigMuc1NW
| Gene | Locus | Name | Role in PC/other tumorigenesis | References |
|---|---|---|---|---|
| SLCO2A1a | 3q22.1‐q22.2 | Solute carrier organic anion transporter family member 2A1 | Unknown/inactivation of it facilitates color cancer formation | Guda et al., 2014; |
| CGNL1a | 15q21.3 | Cingulin like 1 | Unknown/unknown | NA |
| SUPV3L1b | 10q22.1 | Suv3 like RNA helicase | Unknown/unknown | NA |
| TATDN2b | 3p25.3 | TatD DNase domain containing 2 | Unknown/unknown | NA |
| MGAT4Bb | 5q35.3 | Mannosyl (alpha‐1,3‐)‐glycoprotein β‐1,4‐N‐acetylglucosaminyltransferase, isozyme B | Unknown/upregulation in murine hepatocellular carcinoma | Blomme et al., 2013; |
| VAV2b | 9q34.2 | Vav guanine nucleotide exchange factor 2 | An androgen receptor (AR) coactivator; enhancing AR signaling in PC/ | Magani et al., 2017; |
| SLC25A33b | 1p36.22 | Solute carrier family 25 member 33 | Unknown/a mitochondrial UTP carrier; contributing to IGF‐induced cell growth | Lyons et al., 2017; |
| MCCC1b | 3q27.1 | Methylcrotonyl‐CoA carboxylase 1 | Unknown/gain of function was reported in oral squamous cell carcinoma | Ribeiro et al., 2014; |
| ASNSb | 7q21.3 | Asparagine synthetase | Contributing to CRPC/ | Sircar et al., 2012; |
| CASKIN1b | 16p13.3 | CASK interacting protein 1 | Unknown/unknown | NA |
| DNMT3Bb | 20q11.21 | DNA methyltransferase 3 beta | Likely facilitating CRPC/ | Gravina et al., 2011; |
| AURKAb | 20q13.2 | Aurora kinase A | Contributing to CRPC/ | Mosquera et al., 2013; |
| OIP5b | 15q15.1 | Opa interacting protein 5 | Unknown/a cancer testis antigen detected in colorectal cancer | Tarnowski et al., 2016; |
| CTHRC1b | 8q22.3 | Collagen triple helix repeat containing 1 | Unknown/promoting tumorigenesis in multiple cancer types | Ke et al., 2014; |
| GOLGA7Bb | 10q24.2 | Golgin A7 family member B | Unknown/unknown | NA |
−1.5 SD downregulated genes.
2 SD upregulated genes.
NA: not available.
Among the 15 genes, SLCO2A1 and CGNL1 are downregulated and the rest are upregulated (Table 1). Five genes CGNL1, SUPV3L1, TATDN2, CASKIN1, and GOLGA7B are of unknown functions in either prostate cancer tumorigenesis or tumorigenesis in general (Table 1). Six genes (SLCO2A1, MGAT4B, SLC25A33, MCCC1, OIP5, and CTHRC1) have been shown to affect the tumorigenesis of other cancer types but not PC (Blomme et al., 2013; Chen et al., 2013; Guda et al., 2014; Ke et al., 2014; Lyons et al., 2017; Ribeiro et al., 2014; Tarnowski et al., 2016) (Table 1). OIP5 (Opa interacting protein 5) is a cancer testis antigen and has been reported in other cancer types as a type of tumor‐associated antigen (TAA) (Tarnowski et al., 2016); its detection in PC here suggests OIP5 being a TAA for PC. The remaining four genes VAV2 (VAV guanine nucleotide exchange factor 2), ASNS (asparagine synthesis), DNMT3B (DNA methyltransferase 3 beta), and AURKA (Aurora kinase A) not only all promote PC pathogenesis but also play a role in the development of CRPC (Gravina et al., 2011; Magani et al., 2017; Mosquera et al., 2013; Sircar et al., 2012). VAV2 is a coactivator of androgen receptor (AR) and sustains AR signaling under androgen deprivation therapy (ADT) (Magani et al., 2017); it also promotes angiogenesis and metastasis (Barrio‐Real and Kazanietz, 2012). AURKA plays a critical role in mitosis (Dominguez‐Brauer et al., 2015; Plotnikova et al., 2015) and promotes the development of neuroendocrine PC under ADT (Beltran et al., 2011; Mosquera et al., 2013). DNMT3B may regulate epigenetic events to facilitate CRPC development (Hoffmann et al., 2007). Collectively, evidence supports an association of SigMuc1NW with PC recurrence.
In line with this possibility, univariate Cox proportional hazards (PH) analysis revealed that all component genes at the defined level expression (−1.5 SD downregulation and 2 SD upregulation) significantly predict BCR (Table 2). Except for TATDN2 and OIP5, the PH assumption of the Cox model was confirmed. The prediction for some genes (MGAT4B, ASNS, DNMT3B, and OIP5) is robust (Table 2), particularly considering the prediction being individual gene‐based.
Table 2.
Association of the component genes of SigMuc1NW with PC recurrencea
| Genes | Coefb | HRc | 95% CId | P‐value |
|---|---|---|---|---|
| SLCO2A1e | 1.5813 | 4.861 | 1.763–13.4 | 0.00225** |
| CGNL1e | 0.9902 | 2.692 | 1.546–4.686 | 0.000464*** |
| SUPV3L1f | 0.8437 | 2.325 | 1.168–4.629 | 0.0163* |
| TATDN2f | 1.3132 | 3.718 | 1.855–7.45 | 0.000213*** |
| MGAT4Bf | 1.5178 | 4.562 | 2.245–9.272 | 2.73e‐5*** |
| VAV2f | 1.1027 | 3.012 | 1.671–5.429 | 0.000244*** |
| SLC25A33f | 1.096 | 2.992 | 1.55–5.777 | 0.00109** |
| MCCC1f | 0.8336 | 2.302 | 1.322–4.007 | 0.00321** |
| ASNSf | 1.3456 | 3.84 | 2.064–7.145 | 2.15e‐5*** |
| CASKIN1f | 1.0286 | 2.797 | 1.55–5.047 | 0.000636*** |
| DNMT3Bf | 1.2919 | 3.64 | 1.928–6.87 | 6.73e‐5*** |
| AURKAf | 1.0966 | 2.994 | 1.692–5.298 | 0.000166*** |
| OIP5f | 1.365 | 3.914 | 2.022–7.576 | 5.13e‐5*** |
| CTHRC1f | 0.7981 | 2.221 | 1.15–4.289 | 0.0174* |
| GOLGA7Bf | 2.0406 | 7.695 | 2.388–24.79 | 0.00063*** |
Univariate Cox analysis was performed using the TCGA Provisional cohort (n = 492).
Cox coefficient.
Hazard ratio.
Confidence interval.
Gene expression was < −1.5 SD of the reference population mean.
Gene expression was at > 2 SD of the reference population mean.
*P < 0.05; **P < 0.01; ***P < 0.001.
In support of our selection of related genes, changes in the 15 genes show an overlapping profile (Fig. 1B, up panel) and their expression can be clustered (Fig. 1B, bottom panel). The downregulation/upregulation‐based alterations and gene expression‐derived cluster are well matched (Fig. 1B), providing a validation for our covariate selection. Importantly, patients with these changes are at risk of developing recurrent PC; that is, these patients are enriched with recurrent tumors (Fig. 1B, see the ‘Disease‐free status’ illustration). Tumors positive to SigMuc1NW are also robustly associated with reductions in disease‐free survival (DFS) (Fig. 2A, P = 1.12e‐12). The association has a sensitivity of 56.4% and specificity of 72.6%, which are significantly improved from the initially reported 9‐gene signature (sensitivity of 34.8%, specificity of 83.6%, P = 5.57e‐5) (Lin et al., 2017). Considering the TCGA cohort had 10 total mortality, it is intriguing that 8 of these 10 deaths occurred in patients with SigMuc1NW‐positive PC (Fig. 2B, P = 0.00212), which are consistent with VAV2, ASNS, DNMT3B, and AURKA being factors promoting CRPC development (Gravina et al., 2011; Magani et al., 2017; Mosquera et al., 2013; Sircar et al., 2012). As expected, SigMuc1NW displays an overlapping pattern with the 9‐gene genomic signature used to select DEGs (Fig. S2). Inclusion of SigMuc1NW substantially enhanced the association of the 9‐gene signature with BCR (Fig. S3A,C) and significantly correlates with a reduction in overall survival (OS) (Fig. S3B).
Figure 2.
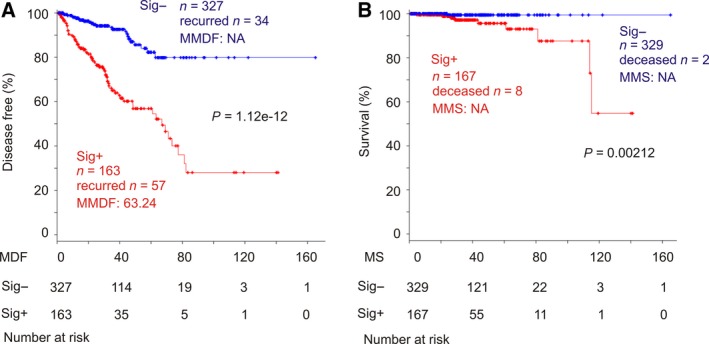
SigMuc1NW is associated with reductions in disease‐free survival (DFS) and overall survival (OS) in patients with PC. The TCGA Provisional cohort was used in these analyses. (A) The effect of SigMuc1NW on DFS. MDF: months disease free; MS: months survival; MMDF: median months disease free; NA: not available as MMDF being not reached. Numbers of patient at risk at the start of the indicated follow‐up period were included. (B) The impact of SigMuc1NW on OS. MMS: median months survival. Kaplan–Meier and log‐rank test were performed using the R survival Package.
3.3. SigMuc1NW effectively discriminates recurrent PCs from those without BCR
To examine the effectiveness of SigMuc1NW in separation of recurrent PC from those without BCR, we have assigned the alterations of the 15 genes with their Cox efficient (Table 2). The cumulative scores of SigMuc1NW for individual patients were then calculated as ∑(f i)n (f i: Cox coefficient of genei, n = 15) (Table S3). The sensitivity and specificity of the scores derived from SigMuc1NW in discrimination of BCR was analyzed using time‐dependent ROC (tROC). The scores discriminate recurrent PC with tAUC (area under curve) ranging from 74.9% at 11.5 and 32.1 months to 69.7% at 48.4 months (Fig. 3A), revealing SigMuc1NW being particularly effective in predicting earlier BCR. To further investigate this application, we determined the cutpoint of the SigMuc1NW scores in the separation of recurrent from nonrecurrent PC using Maximally Selected Rank Statistics using the Maxstat package in R (Fig. S4) and converted the scores into binary code; scores ≤ 1.7833 (cutpoint, Fig. S4) were assigned ‘0’ and scores > 1.7833 were assigned ‘1’. PCs with scores above the cutpoint have a dynamically faster profile of BCR than those with scores not above the cutpoint (Fig. 3B). Intriguingly, the cutpoint‐positive tumors even developed BCR in a shorter time frame (Fig. 3B; MMDF 33.1, 95% CI 30.9–73.4) compared to SigMuc1NW‐positive PCs (Fig. 2A; MMDF 63.2, 95% CI 40–77.3). The cutpoint thus not only will facilitate clinical examination of SigMuc1NW but also enhances its predictive power. Additionally, both mean and quartile 3 (Q3) scores can stratify patients with high risk of BCR with comparable effectiveness as SigMuc1NW (comparing Fig. 3C,D to Fig. 2A). Both mean and Q3 scores cover 48 and 46 recurrent PCs, respectively (Fig. 3C,D) which are more than the 41 recurrent PCs marked by the cutpoint (Fig. 3A). Thus, the mean (0.918), Q3 (1.019), and cutpoint (1.7883) scores can also be used to predict BCR following RP with a range of BCR risk. We further demonstrated SigMuc1NW (1.62e‐4), cutpoint (P = 2.05e‐5) (Table 3), Mean (P = 1.19e‐4), and Q3 (P = 1.67e‐4) (data not shown) being independent risk factors for PC recurrence after adjusting for age at diagnosis, RP Gleason scores, surgical margin, and TMN tumor stage. When the World Health Organization (WHO) PC grading system [WHO grade (group) I‐V] or its equivalent ISUP (the International Society of Urological Pathology) grade (Egevad et al., 2016; Gordetsky and Epstein, 2016) (Table S3 for details) instead of Gleason grade was used, SigMuc1NW (P = 2.05e‐4), cutpoint (P = 1.91e‐5), Mean (P = 1.37e‐4), and Q3 (P = 1.86e‐4) remain an independent risk factor for BCR. The demographics of the TCGA dataset with respect to the clinical characteristics used in the above multivariate Cox analyses are included (Table S4).
Figure 3.
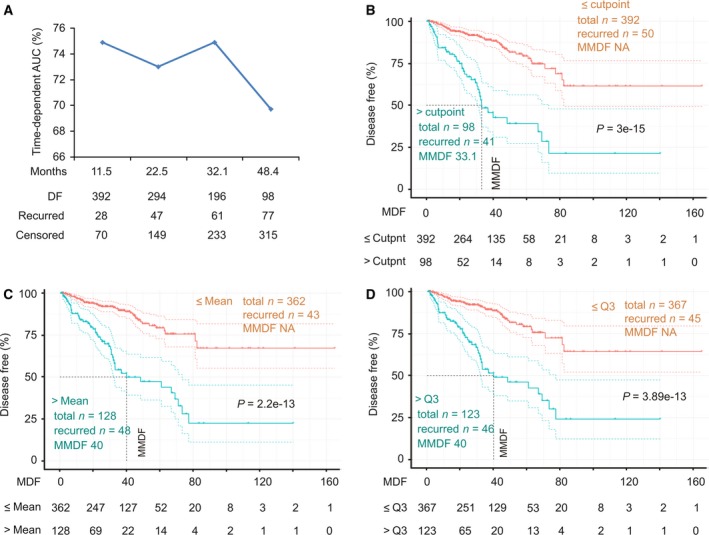
SigMuc1NW scores effectively stratify PCs with a high risk of recurrence. (A) All tumors within the TCGA Provisional cohort were scored for SigMuc1NW (see Results for details). The scores were analyzed for discrimination of tumors with high risk of recurrence using tROC. AUC at the indicated period of time (tAUC) along with the status of disease recurrence are indicated. DF: disease free. (B) The cutpoint of SigMuc1NW scores for effectively separating PCs with high risk of recurrence from low risk PCs was estimated (Fig S4 for details), followed by assigning binary codes to tumors based on the cutpoint (see Results for details). The effects of cutpoint on DFS of the patients in the TCGA cohort were then determined. (C, D) The effects of Mean and Q3 scores of SigMuc1NW on BCR in PC patients in the TCGA Provisional cohort. Kaplan–Meier and log‐rank test were performed using the R survival Package. The vertical dot line shows MMDF. The color dot curves are for 95% CI.
Table 3.
Univariate and multivariate Cox analysis of SigMuc1NW for PC recurrence
| Factors | Univariate Cox analysis | Multivariate Cox analysis | Multivariate Cox analysis | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HR | 95% CI | P‐value | HR | 95% CI | P‐value | HR | 95% CI | P‐value | |
| Siga | 4.16 | 2.74–6.36 | 5.54e‐11* | 2.44 | 1.53–3.87 | 1.62e‐4* | NA | NA | NA |
| Cutpointb | 4.6 | 3.03–6.97 | 6.44e‐13* | NA | NA | NA | 2.67 | 1.70–4.20 | 2.05e‐5* |
| Agec | 1.03 | 0.99–1.06 | 0.0981 | 0.999 | 0.97–1.03 | 0.9711 | 1.001 | 0.97–1.03 | 0.9756 |
| GSd | 2.19 | 1.76–2.72 | 1.49e‐12* | 1.62 | 1.25‐2.11 | 2.71e‐4* | 1.62 | 1.25–2.10 | 2.86e–4* |
| SMargine | 2.25 | 1.48–3.41 | 0.000137* | 1.25 | 0.79–1.98 | 0.3306 | 1.28 | 0.81–2.02 | 0.2976 |
| TumStgef | 3.68 | 2.08–6.51 | 8.19e‐6* | 1.82 | 0.97–3.40 | 0.0614 | 1.82 | 0.96–3.45 | 0.0668 |
SigMuc1NW.
SigMuc1NW‐derived cutpoint.
Age at diagnosis.
Radical prostatectomy Gleason score.
Surgical margin.
Tumor stages (0 for ≤ T2; 1 for T3 and T4).
HR, hazard ratio; CI, confidence interval; NA, not available.
*P < 0.05.
3.4. Enhancing the predictive efficiency of SigMuc1NW
To further demonstrate SigMuc1NW being effective and robust, we analyzed the signature using the actual gene expression data instead of using SD (standard deviation)‐based distribution. For this purpose, the RNA sequencing data for all 15 SigMuc1NW genes were retrieved from the TCGA dataset and estimated for cutpoints in separating recurrent PCs (Table 4). All tumors were given a binary code for all 15 genes as described above with exception for both downregulated genes SLCO2A1 and CGNL1 in which tumors with expression less than the cutpoint were assigned ‘1’. Univariate Cox PH analysis was carried out with the PH assumption confirmed for all genes. All 15 genes, as defined by their cutpoint, significantly predict BCR (Fig. 4). Additionally, SLCO2A1, SUPV3L1, TATDN2, MGAT4B, VAV2, SLC25A33, ASNS, and OIP5 remain as independent risk factors of BCR after adjusting for age at diagnosis, RP Gleason scores, surgical margin, and TMN tumor stage (Table 5). These observations are appealing considering their single gene‐based nature, and that 8/15 component genes of SigMuc1NW possesses independent predicting value to BCR, which further supports SigMuc1NW as a signature for BCR.
Table 4.
SigMuc1NWa component genes defined at their cutpoints associate with BCR
| Genes | Cutpointb | P‐value | Coefc | P‐value |
|---|---|---|---|---|
| SLCO2A1 | 497.3292 | 0.09128 | 0.7967 | 0.00499** |
| CGNL1 | 3066.229 | 0.004126** | 0.7966 | 0.000372*** |
| SUPV3L1 | 545.8928 | 0.007953** | 0.7992 | 0.000187*** |
| TATDN2 | 1756.057 | 0.002471** | 0.8731d | 8.48e‐5*** |
| MGAT4B | 1818.718 | 6.389e‐5*** | 1.0331 | 2.61e‐6*** |
| VAV2 | 1489.06 | 0.000547*** | 0.9402 | 9.94e‐6*** |
| SLC25A33 | 297.5508 | 0.2522 | 0.8503 | 0.0218* |
| MCCC1 | 1233.159 | 0.001077** | 1.0179 | 1.2e‐5*** |
| ASNS | 1041.086 | 0.01123* | 1.0544 | 0.000109*** |
| CASKIN1 | 106.4046 | 0.02646* | 0.7006 | 0.00125** |
| DNMT3B | 61.4086 | 0.008576** | 0.9082 | 0.000175*** |
| AURKA | 81.1249 | 3.807e‐5*** | 1.0223 | 1.12e‐6*** |
| OIP5 | 16.4317 | 4.237e‐7*** | 1.242d | 2.64e‐8*** |
| CTHRC1 | 180.8622 | 0.01389* | 0.7608 | 0.000537*** |
| GOLGA7B | 23.2022 | 0.01249* | 0.7623 | 0.000581*** |
RNA sequencing data of SigMuc1NW's component genes were retrieved from the TCGA Provisional dataset (cBioPortal).
Cutpoint was estimated using Maximally Selected Rank Statistics in R.
Coefficient to BCR was determined using univariate Cox proportion hazard analysis.
PH assumption was at P < 0.05.
*P < 0.05; **P < 0.01; ***P < 0.001.
Figure 4.
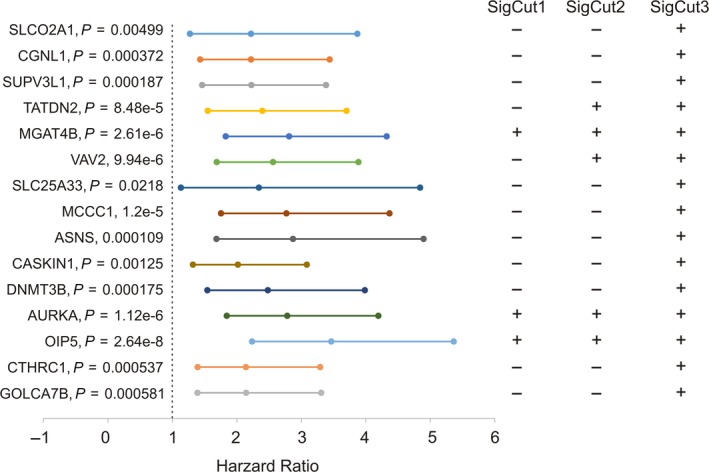
All 15 component genes are significantly associated with PC recurrence and the formulation of three subsignatures. The mRNA expression data for the 15 genes were retrieved from the TCGA Provisional dataset (cBioPortal). Individual cutpoints were derived, and binary codes were assigned to all tumors. The hazard ratio (HR) of PC recurrence for all individual genes was determined using the univariate Cox proportional hazards (PH) mode. The PH assumption was evaluated and confirmed. These analyses were carried out using the R survival package. Individual HR, the 95% CI, and P‐value are included. The inclusion of component genes in SigCut1, SigCut2, and SigCut3 were shown, which was based on the P‐values.
Table 5.
Univariate and multivariate Cox analysis of SigMuc1NW component genes defined at cutpoint for PC recurrence
| Factors | Univariate Cox analysis | Multivariate Cox analysis | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P‐value | HR | 95% CI | P‐value | |
| Agea | 1.03 | 0.99–1.06 | 0.0981 | NSe | ||
| GSb | 2.19 | 1.76–2.72 | 1.49e‐12* | 1.71–1.89f | (1.32–1.46)–(2.20‐2.41)f | 4.48e‐7*–1.4e‐5*,f |
| SMarginc | 2.25 | 1.48–3.41 | 0.000137* | NSe | ||
| TumStged | 3.68 | 2.08–6.51 | 8.19e‐6* | 1.62–2.07 | (0.85–1.08)‐(3.08–3.96)f | 0.0272*,h–0.139f,g |
| SLCO2A1 | 2.22 | 1.27–3.87 | 0.00499* | 1.82 | 1.04–3.19 | 0.0369* |
| SUPV3L1 | 2.22 | 1.46–3.38 | 1.87e‐4* | 2.08 | 1.36–3.19 | 7.98e‐4* |
| TATDN2 | 2.39 | 1.55–3.70 | 8.48e‐5* | 2.15 | 1.37–3.37 | 8.35e‐4* |
| MGAT4B | 2.81 | 1.83–4.32 | 2.61e‐6* | 1.77 | 1.23–2.78 | 0.0128* |
| VAV2 | 2.56 | 1.69–3.89 | 9.94e‐6* | 1.93 | 1.26–2.95 | 0.0024* |
| SLC25A33 | 2.34 | 1.13–4.84 | 0.0218* | 2.25 | 1.08–4.67 | 0.0297* |
| ASNS | 2.87 | 1.68–4.90 | 1.09e‐4* | 1.91 | 1.09–3.36 | 0.0239* |
| OIP5 | 3.46 | 2.24–5.36 | 2.64e‐8* | 1.94 | 1.20–3.12 | 0.00638* |
Age at diagnosis.
Radical prostatectomy Gleason score.
Surgical margin.
Tumor stages (0 for ≤ T2; 1 for T3 and T4).
Not significant.
Range of HR, 95% CI, and P‐values resulted from multivariate Cox analysis with the individual genes.
The P‐values for SLCO2A1 (P = 0.0749), MGAT4B (P = 0.0891), ASNS (P = 0.0917), and OIP5 (P = 0.139).
hThe P‐values for SUPV3L1 (P = 0.0431*), TATDN2 (P = 0.0272*), VAV2 (P = 0.0364*), and SLC25A33 (P = 0.0334*).
HR, hazard ratio; CI, confidence interval.
Using the Cox coefficients (Table 4), all cutpoint‐positive events were converted to the respective coefficient values (Table S5). Based on the robustness defined by P‐values (Fig. 4), we formulated three subsignatures SigCut1, SigCut2, and SigCut3 (Fig. 4). All tumors were then scored for SigCut1, SigCut2, and SigCut3 using ∑(f i)n (f i: Cox coefficient of genei, n = 3, 6, or 15). All three subsignatures discriminate recurrent PC effectively with tAUC > 70% (Fig. 5A). The respective cutpoints were determined: 1.0331/P = 6.166e‐8 for SigCut1, 4.0135/P = 1.005e‐11 for SigCut2, and 5.4067/P = 7.97e‐15 for SigCut3. The respective binary code for individual subsignature was then assigned to all tumors, which was used to perform survival analysis. All three subsignatures dramatically associate with reductions in DFS with SigCut2 and SigCut3 being more robust (Fig. 5B–D). Nonetheless, they predict BCR with a range of effectiveness in terms of the number of recurrent tumors included, the duration of MMDF, and sensitivity/specificity: 71.4%/63.9% for SigCut1, 41.8%/87.5% for SigCut2, and 67.7%/75.7% for SigCut3 (Fig. 5B–D). These three subsignatures can thus be used together to predict recurrent PCs; this will significantly enhance their predictive power.
Figure 5.
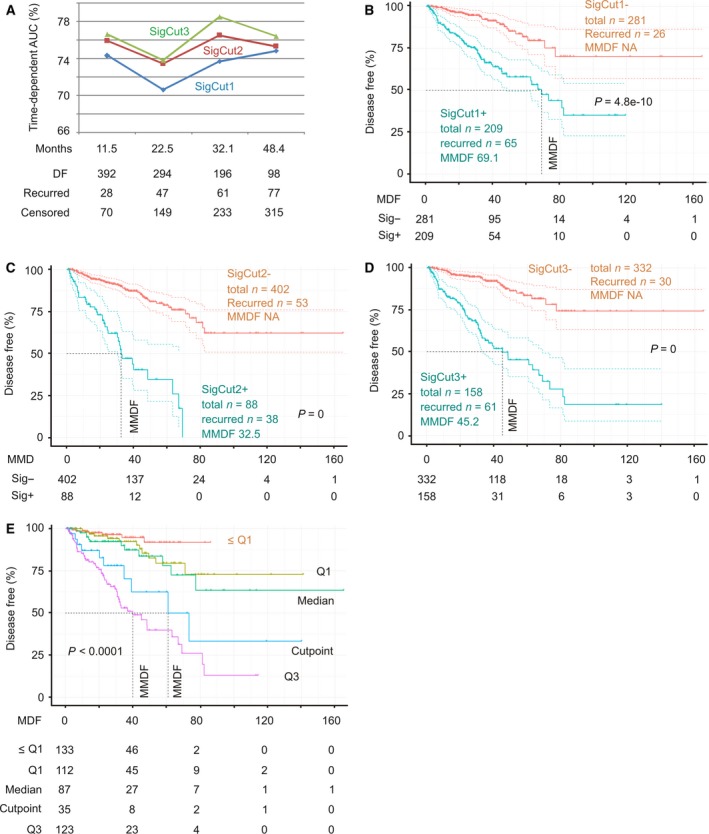
Analyses of SigCut1, SigCut2, and SigCut3 for their association with reductions in DFS. The TCGA Provisional dataset was used here. (A) All tumors were scored for SigCut1, SigCut2, and SigCut3 using the respective Cox coefficient. Time‐dependent AUCs for individual signature at the current follow‐up period and the corresponding recurrent status are shown. (B‐D) The associations of SigCut1, SigCut2, and SigCut3 with BCR. (E) The Q1, Median, Cutpoint, and Q3 scores of SigCut3 were analyzed for the stratification of PC with high risk of recurrence. The number of risk individuals at the indicated follow‐up period is included. The multiple Kaplan–Meier curves and log‐rank test were performed using the R survival package.
The Q1 (1.647), Median (3.589), and Q3 (6.386) scores all effectively stratify PC with high risk of BCR with a range of effectiveness in terms of sensitivity/specificity/MMDF (median month disease free)/P‐value being 93.4%/31.8%/81.2/6.76e‐6 for Q1, 80.2%/56.9%/66.9/6.73e‐11 for Median, and 56%/82%/40/0 for Q3 (Fig. S5). When Q1, Median, Q3, and cutpoint of SigCut3 are used together, it offers an impressive system to stratify recurrent and nonrecurrent PCs with only a few recurrent cases in tumors with score < Q1 (Fig. 5E).
Furthermore, in comparison with SD‐defined SigMuc1NW (Fig. 2A), SigCut3 is clearly more effective (Fig. 5D). After adjusting for age at diagnosis, RP Gleason scores, surgical margin, and TMN tumor stage, SigCut1 (P = 0.00308), SigCut2 (P = 1.55e‐5), and SigCut3 (P = 2.97e‐6) independently predict BCR, respectively. All three signatures are associated with adverse features of PC: high tumor stages (T3 and T4) at odds ratio/95% CI of 1.78/1.51–2.12 (P = 2.39e‐11) for SigCut1, 1.55/1.37–1.77 (P = 1.33e‐11) for SigCut2, and 1.33/1.23–1.44 (P = 8.47e‐13) as well as for Gleason scores (8–10) at the respective odds ratio/95% CI of 2.19/1.86–2.6 (P < 2e‐16), 1.84/1.62–2.1 (P < 2e‐16), and 1.48/1.37–1.61 (P < 2e‐16). Taken together, these observations validate the efficacy of SigMuc1NW.
3.5. Validation of SigMuc1NW
We have made an effort to determine the individual component gene expression in PCs. The MKSCC (Cancer Cell 2010) (Taylor et al., 2010) dataset within cBioPortal has 216 PCs/patients with mRNA expression profiled using microarray; the expression data were organized for comparison between normal prostate tissues and PC (cBioPortal). Importantly, all primary PCs have been treated and the follow‐up information is available; this cohort thus supports survival analysis. To further validate SigMuc1NW constructed using RNA sequencing data from the TCGA Provisional dataset, mRNA expression data for all 15 component genes along with all clinical information were extracted from the MKSCC dataset. Tissues can be grouped into normal prostate (n = 29), primary PCs (n = 149), recurred PCs (n = 36), and metastatic PCs (n = 9) (cBioPortal). Using this setting, we demonstrated significant reductions of CGNL1 in primary PCs over normal prostate tissues, in metastatic PCs compared to localized PCs, and in recurrent PCs compared to nonrecurrent PCs among the two downregulated genes (SLCO2A1 and CGNL1) of SigMuc1NW (Fig. 6A–C). Significantly higher levels for most upregulated genes identified in SigMuc1NW were shown in the above comparisons (Fig. 6A–C), supporting the authenticity of SigMuc1NW.
Figure 6.
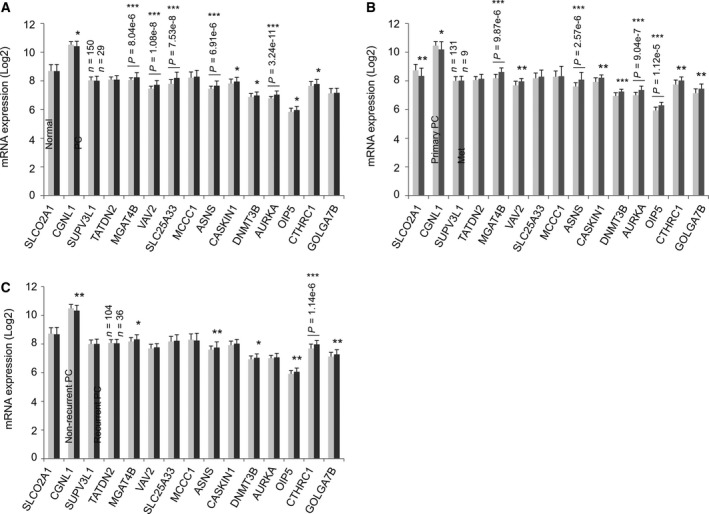
Alterations in the expression of the component genes in an independent PC population. Gene expression data determined by microarray were extracted from the MSKCC dataset (Robinson et al., 2015) within cBioPortal. The mRNA levels in normal and PC tissues (A), in primary PC and metastatic PC (B), and in nonrecurrent and recurrent PC (C) were determined. The number of cases used in the comparisons is indicated. Means ± SD are graphed. Statistical analyses were performed using Student's t‐test (2‐tailed). *P < 0.05, **P < 0.01, and ***P < 0.001.
Following our system described above, cutpoints for all 15 genes were estimated, binary codes were assigned, and association of individual genes with BCR was determined using Cox PH regression (Table 6). Except MCCC1 being reversely associated with DFS and four genes without a significant correlation with DFS, other 10 genes significantly or robustly (CGNL1 and CTHRC1) predict BCR risk (Table 6). We then formulated a subsignature with these 10 genes (SigMuc1NW1). As described above, all tumors were scored for SigMuc1NW1 using their coefficients (Table 6). Analysis with tROC shows tAUC values being from 76.6% to 82.5% (Fig. 7A). SigMuc1NW1 thus effectively discriminates recurred PCs from nonrecurrent tumors across all follow‐up period from 18.4 months to 65 months (Fig. 7A); this efficiency matches that of SigMuc1NW in the discrimination of recurrent PCs in the TCGA cohort (Fig. 5A). Additionally, using the binary code derived from Q1 (0), Median (1.805), Q3 (3.727), and cutpoint (6.2136) scores of SigMuc1NW1, all these classifications significantly stratify recurrent PCs (Fig. 7B–E). The respective sensitivity/specificity/PPV (positive predictive value) are 36.1%/98.1%/86.7% for cutpoint, 97.2%/35.6%/34.3% for Q1, 75%/59.6%/39.1% for Median, and 52.8%/84.6%/54.3% for Q3 (Fig. 7B–E). The PPV for cutpoint is robust (86.7%). Collectively, through combination of Q1, Median, Q3, and cutpoint, PC recurrence could be effectively predicted for patients in the MSKCC cohort. The similar situation was also demonstrated in the TCGA cohort using SigMuc1NW. In a reverse validation effort, we demonstrated that SigMuc1NW1 is also robustly associated with BCR in the TCGA cohort and significantly correlates with a reduction in OS in the TCGA dataset (Fig. 8A,B). Taken together, we provide a thorough validation of SigMuc1NW and SigMuc1NW1.
Table 6.
Cutpoint and Cox coefficients of SigMuc1NW component genes in the MSKCC cohorta
| Genes | Cutpointb | P‐value | Coefc | HR | 95% CI | P‐value |
|---|---|---|---|---|---|---|
| SLCO2A1 | 8.155098 | 0.7073 | 0.6364 | 1.89 | 0.7835–4.558 | 0.157 |
| CGNL1 | 10.02132 | 0.004758** | 1.4679 | 4.34 | 2.084–9.038 | 8.8e‐5*** |
| SUPV3L1 | 7.655546 | 0.7029 | −0.6931 | 0.5 | 0.2277–1.098 | 0.0841 |
| TATDN2 | 7.755133 | 0.969 | −0.5149 | 0.5976 | 0.2476–1.442 | 0.252 |
| MGAT4B | 8.536576 | 0.01469* | 1.3245 | 3.76 | 1.833–7.712 | 0.000302*** |
| VAV2 | 7.801308 | 0.2076 | 0.8258 | 2.284 | 1.184–4.405 | 0.0138* |
| SLC25A33 | 8.653056 | 1 | 0.4752 | 1.608 | 0.6248–4.14 | 0.325 |
| MCCC1 | 7.789343 | 0.2982 | −1.0768 | 0.3407 | 0.1467–0.7911 | 0.0122* |
| ASNS | 7.946625 | 0.01918* | 1.1815 | 3.259 | 1.567–6.78 | 0.00157** |
| CASKIN1 | 8.142854 | 0.04935* | 1.0985 | 3 | 1.529–5.886 | 0.0014** |
| DNMT3B | 7.199673 | 0.06077 | 1.0373 | 2.822 | 1.385–5.749 | 0.00428** |
| AURKA | 7.215284 | 0.03781* | 1.0552 | 2.873 | 1.435–5.75 | 0.00288** |
| OIP5 | 6.026397 | 0.05557 | 0.9789 | 2.662 | 1.374–5.156 | 0.00372** |
| CTHRC1 | 7.827664 | 0.0001814*** | 1.631 | 5.109 | 2.4–10.88 | 2.33e‐5*** |
| GOLGA7B | 7.534541 | 0.1695 | 1.1095 | 3.033 | 1.371–6.71 | 0.00617** |
Microarray data of SigMuc1NW's component genes were retrieved from the MSKCC dataset (cBioPortal).
Cutpoint was estimated using Maximally Selected Rank Statistics in R.
Coefficient to BCR was determined using univariate Cox proportion hazard analysis.
*P < 0.05; **P < 0.01; ***P < 0.001.
Figure 7.
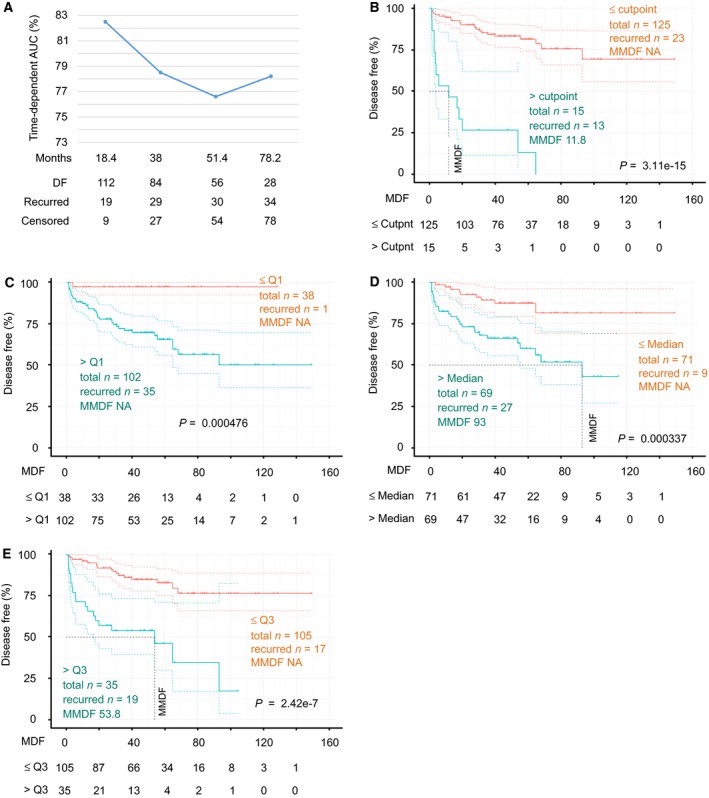
SigMuc1NW1 robustly predicts PC recurrent in an independent PC dataset. The follow‐up data along with mRNA expression data for all 15 genes were retrieved from the MSKCC dataset (Robinson et al., 2015). SigMuc1NW1 was formed using 10 genes (see Results for details). Time‐dependent AUCs were derived (A). The stratification of PC with increased risk of recurrence was analyzed using the cutpoint (B), Q1 (C), Median (D), and Q3 (E) scores of SigCut1NW1. Numbers of risk individuals at the current follow‐up period are also included.
Figure 8.
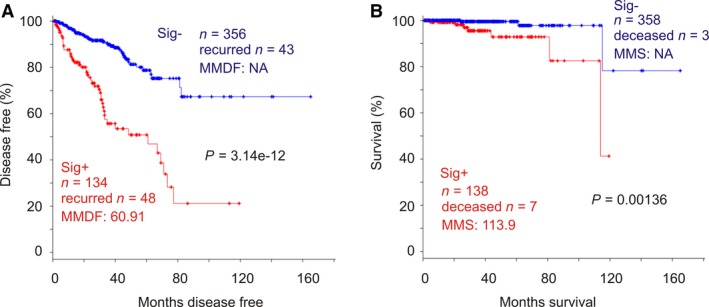
SigMuc1NW1 significantly correlates with reductions in DFS and OS in PC patients. The analyses were performed using the TCGA Provisional dataset. SigMuc1NW1 gene expression was based on the SD levels. Kaplan–Meier curve and log‐rank test were performed using tools provided by cBioPortal.
Finally, we made an attempt to compare the performance of SigMuc1NW to Prolaris (cell cycle progression/CPC) (Cuzick et al., 2011) in predicting BCR. The basis for this comparison was the similarities between SigMuc1NW to CPC: (a) like CPC, SigMuc1NW affects cell cycle progression (Table S2A and S2C; also see Discussion), and (b) similar to CPC, SigMuc1NW predicts BCR. As the CPC component genes promote cell cycle progression, we analyzed their effects on BCR using the 2 SD expression level. In the TCGA Provisional cohort, CPC is not correlated with a reduction in OS but significantly associated with BCR (Fig. S6). However, the predictive accuracy is lower than SigMuc1NW (comparing Fig. 2 and Fig. S6). Considering Prolaris being a real‐time PCR‐based signature and SigMuc1NW being derived from RNA seq, this comparison may not fully realize Prolaris effectiveness in predicting BCR. Nonetheless, it suggests that SigMuc1NW (Fig. 2A, MMDF 63.24, P = 1.12e‐12) offers comparable efficacy to Prolaris (Fig. S6, MMDF 66.89, P = 1.34e‐4) in assessing PC recurrence.
4. Discussion
Progression to biochemical recurrence is a major turning point in PC development; from there, a large proportion of PC will metastasize (Shipley et al., 2017), leading to ultimate death. The current treatments to metastatic PC are essentially palliative. It is thus highly desirable to effectively stratify PCs with higher risk of BCR following RP, allowing early intervention prior to metastatic progression.
MUC1 drives tumor progression in multiple tumor types (Kufe, 2009; de Paula Peres et al., 2015; Wurz et al., 2014) through activating important oncogenic proteins including EGFR, β‐catenin, NF‐κB, PKM2, and other pathways (Kufe, 2009; Singh and Hollingsworth, 2006; Wong et al., 2015). In line with its functions, a 9‐gene genomic signature was recently constructed from the MUC1 genomic network, which predicts BCR with a relatively good effectiveness (Lin et al., 2017). Using a novel system, we report here a robust improvement of this 9‐gene genomic signature in predicting BCR by systemically exploring its associated transcriptome. To our best knowledge, this is the first thorough analysis not on a single gene‐associated but rather on a multigene signature‐associated transcriptome consisting of 696 genes (Table S1). Because of the complex nature of cancer progression, in this case the progression to BCR, we chose not to focus on a specific aspect or pathway of tumorigenesis and instead performed a systemic examination of these 696 genes for their predictive power in BCR.
This novel and comprehensive analytic approach has resulted in a new 15‐gene panel. In the panel, 73.3% (11/15) of genes have not been reported to associate with PC. These 11 new PC genes include MGAT4B and OIP5. The former may play a role in the alteration of protein glycosylation, which is well known for being an important aspect of tumorigenesis (Munkley et al., 2016). Abnormalities in MUC1 glycosylation have been well demonstrated in tumorigenesis (Kufe, 2009; de Paula Peres et al., 2015). Thus, the inclusion of MGAT4B in the 15‐gene panel is in accordance with the panel being derived from a 9‐gene MUC1 genomic signature (Lin et al., 2017). The presence of OIP5 in SigMuc1NW suggests the protein as a tumor‐associated antigen (TAA) in PC. TAAs have been extensively investigated in cancer diagnosis and therapy (Scheid et al., 2016). In this regard, the OIP5's potential in PC diagnosis and therapy should be pursued.
As the construction of SigMuc1NW was not aimed on specific pathways, the gene panel covers multiple pathways. In addition to the potential effects on protein glycosylation though MGAT4B, the panel contains proteins with RNA helicase activity (SUPV3L1, Table 1) and DNA methyltransferase activity (DNMT3B, Table 1). These activities are important in gene expression and epigenetic alterations, which are well known to facilitate caner progression. SigMuc1NW also have a component of cell proliferation. AURKA is emerging as an important regulator of mitosis and a critical player in tumorigenesis. As such, AURKA is a hotly pursued in cancer therapy (Dominguez‐Brauer et al., 2015; Plotnikova et al., 2015). Additionally, OIP5 is also known as Mis18β which has recently been shown to play an important role in chromatid separation during mitosis (Nardi et al., 2016; Stellfox et al., 2016), adding another appealing feature for its inclusion in SigMuc1NW. Intriguingly, among the 15 genes, only four are known to function in PC and all four genes facilitate CRPC development, which is in accordance with the detection of SigMuc1NW elevation in mCRPCs (Table 6). As alterations in gene expression and the epigenetic patterns are involved in CRPC, the 15‐gene panel may also predict CRPC development, which will be examined in the future.
Inclusion of genes functioning in multiple pathways is likely a major attributor for the robust nature of the signature. SigMuc1NW and a set of its subsignatures all effectively stratify PC with increased risk of BCR with P‐value being the lowest (0) and are able to discriminate recurrent PC with tAUC >75%. Through combination of the subsignatures, sensitivity, specificity, and PPV can be achieved at high levels, 97.2%/, 98.1%, and 86.7% (Fig. 7B–E). Collectively, these evidences strongly indicate that the signatures constructed in this study will have important clinical applications in predicting PC recurrence.
This possible clinical application is supported by that the 15‐gene panel is likely not overfitted. (a) The overfitting issue is largely taken care of by modeling the 696 DEGs with covariate selection coupled with regularization (Elastic‐net penalty in R) with 10‐fold cross‐validation. (b) The component genes were directly examined using a different system: maximally selected rank statistics‐derived cutpoint; importantly, this system clearly improved the effectiveness of the SD‐based signature. (c) The signatures were robust in two independent PC cohorts (TCGA Provisional and MSKCC). (d) RNA was profiled through RNA sequencing (TCGA) and microarray analysis (MSKCC). (e) The 15‐gene panel is robustly associated with adverse feature of PC: Gleason scores and tumor stages. These associations likely resulted in the reduced HR of Gleason scores and tumor stages when they were analyzed with SigMuc1NW in multivariate Cox analysis (Table 3).
Between two commercially available multigene panels, Oncotype DX (12 genes plus 5 reference genes) and Prolaris (31 genes), there are no overlapping genes (Cuzick et al., 2011; Knezevic et al., 2013). This suggests the coexistence of different genesets with predictive values toward PC recurrence, which might be attributable to the complex mechanisms involved in disease progression. In this regard, our newly established SigMuc1NW, which contains a different set of genes from Oncotype DX and Prolaris, will enrich our ability to assess the risk of PC recurrence. While our research comprehensively supports that the signatures constructed here will have attractive clinical applications, realization of this potential requires further investigation.
5. Conclusions
We have formulated a novel strategy to derive differentially expressed genes (DEGs) relative to a reported PC signature from the most comprehensive and large PC genomic dataset (the TCGA dataset) and to systemically analyze these DEGs (n = 696) for pathways affected and impacts on PC recurrence. In this effort, a novel multigene set (n = 15 genes, SigMuc1NW) has been constructed. SigMuc1NW robustly predicts PC recurrence and is an independent risk factor of PC recurrence after adjusting for age at diagnosis, Gleason score, surgical margin, and tumor stage. Among these 15 component genes include 5 candidate oncogenic genes and 6 novel PC genes; within these 11 novel genes affecting PC recurrence, 6 genes (SLCO2A1, SUPV3L1, TATDN2, MGAT4B, SLC25A33, and OIP5) individually predict PC recurrence after adjusting for the above clinical factors. Collectively, we have identified novel genes affecting oncogenesis in general and PC pathogenesis in particular as well as constructed a novel and robust multigene set predicting PC recurrence using our system reported here. This system will have applications in exploration of publically available datasets for factors affecting cancer progression.
Author contributions
YJ, WM, and DT performed literature search and initial analyses. YG, XL, LH, and HZ contributed to the analysis. FW, XW, HY, PM, and DT designed the research. PM and DT supervised the project. YJ, WM, YG, PM, and DT prepared the manuscript. All authors edited the manuscript and approved the final manuscript for submission.
Supporting information
Fig. S1. Covariate selection from 696 DEGs using Elastic‐net penalty.
Fig. S2. Overlapping between the 9‐gene genomic signature which we have previously reported (Lin et al., 2017) and the current signature (SigMuc1NW). Graph was produced using the TCGA Provisional dataset (n = 492, cBioPortal).
Fig. S3. The combined signature is significantly associated with reductions in DFS and OS in PC patients.
Fig. S4. Cutpoint estimation.
Fig. S5. SigMuc1NW scores effectively stratify PCs with elevated risk of recurrence following RP.
Fig. S6. CPC geneset is associated with a reduction in DFS but not OS in PC patients.
Table S1. Differentially expression genes (DEGs) of a 9‐gene signature identified in the TCGA Provisional dataset.
Table S2. (A) Upregulation of gene sets among the 696 DEGs associated with the 9‐gene genomic signature within the kegg.sets.hs dataset. (B) Downregulation of gene sets among the 696 DEGs within the kegg.sets.hs dataset. (C) Upregulation of gene sets among the 696 DEGs within the GO.sets.hs dataset. (D) Downregulation of gene sets among the 696 DEGs within the GO.sets.hs dataset. (E) Pathways affected by the 696 DEGs associated with the 9‐gene genomic signature.
Table S3. Scores of the component genes and some clinical characteristics of patients with prostate cancer in the TCGA Provisional dataset within cBioPortal.
Table S4. Demographics of the TCGA patient population. The clinical characteristics were extracted from the TCGA Provisional dataset within cBioPortal along with the indicated clinical data.
Table S5. Cutpoints of individual gene expression determined by RNA sequencing.
Acknowledgements
The results shown here are in part based upon data generated by the TCGA Research Network (http://cancergenome.nih.gov/). FW is supported by grants from the Natural Science Foundation of Guangdong Province (Grant No. 2014A030313749), Foundation from Science and Technology Innovation Committee of Shenzhen (Grant No. JCYJ20150403094227974), and Research Foundation of Guangdong medical science and Technology Research (Grant No. 2016118111251132). DT is supported by an Award from Teresa Cascioli Charitable Foundation Research Award in Women's Health and grants from Canadian Cancer Society (grant #: 319412) and Cancer Research Society.
Contributor Information
Pierre Major, Email: majorp@hhsc.ca.
Damu Tang, Email: damut@mcmaster.ca.
References
- Albala D, Kemeter MJ, Febbo PG, Lu R, John V, Stoy D, Denes B, McCall M, Shindel AW and Dubeck F (2016) Health economic impact and prospective clinical utility of oncotype DX(R) genomic prostate score. Rev Urol 18, 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolopoulos V, Stojanovska L and Gargosky SE (2015) MUC1 (CD227): a multi‐tasked molecule. Cell Mol Life Sci 72, 4475–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Fujita K, Fujime M and Irimura T (2005) Expression of sialylated MUC1 in prostate cancer: relationship to clinical stage and prognosis. Int J Urol 12, 654–661. [DOI] [PubMed] [Google Scholar]
- Barrio‐Real L and Kazanietz MG (2012) Rho GEFs and cancer: linking gene expression and metastatic dissemination. Sci Signal 5, pe43. [DOI] [PubMed] [Google Scholar]
- Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, Wang Y, Sheikh KL, Terry S, Tagawa ST et al (2011) Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov 1, 487–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomme B, Heindryckx F, Stassen JM, Geerts A, Colle I and Van Vlierberghe H (2013) Serum protein N‐glycan alterations of diethylnitrosamine‐induced hepatocellular carcinoma mice and their evolution after inhibition of the placental growth factor. Mol Cell Biochem 372, 199–210. [DOI] [PubMed] [Google Scholar]
- Briganti A, Karnes RJ, Gandaglia G, Spahn M, Gontero P, Tosco L, Kneitz B, Chun FK, Zaffuto E, Sun M et al (2015) Natural history of surgically treated high‐risk prostate cancer. Urol Oncol 33, 163. e167‐113. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YL, Wang TH, Hsu HC, Yuan RH and Jeng YM (2013) Overexpression of CTHRC1 in hepatocellular carcinoma promotes tumor invasion and predicts poor prognosis. PLoS One 8, e70324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooperberg MR, Simko JP, Cowan JE, Reid JE, Djalilvand A, Bhatnagar S, Gutin A, Lanchbury JS, Swanson GP, Stone S et al (2013) Validation of a cell‐cycle progression gene panel to improve risk stratification in a contemporary prostatectomy cohort. J Clin Oncol 31, 1428–1434. [DOI] [PubMed] [Google Scholar]
- Cozzi PJ, Wang J, Delprado W, Perkins AC, Allen BJ, Russell PJ and Li Y (2005) MUC1, MUC2, MUC4, MUC5AC and MUC6 expression in the progression of prostate cancer. Clin Exp Metas 22, 565–573. [DOI] [PubMed] [Google Scholar]
- Cullen J, Rosner IL, Brand TC, Zhang N, Tsiatis AC, Moncur J, Ali A, Chen Y, Knezevic D, Maddala T et al (2015) A Biopsy‐based 17‐gene genomic prostate score predicts recurrence after radical prostatectomy and adverse surgical pathology in a racially diverse population of men with clinically low‐ and intermediate‐risk prostate cancer. Eur Urol 68, 123–131. [DOI] [PubMed] [Google Scholar]
- Cuzick J, Swanson GP, Fisher G, Brothman AR, Berney DM, Reid JE, Mesher D, Speights VO, Stankiewicz E, Foster CS et al (2011) Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. Lancet Oncol 12, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Den RB, Feng FY, Showalter TN, Mishra MV, Trabulsi EJ, Lallas CD, Gomella LG, Kelly WK, Birbe RC, McCue PA et al (2014) Genomic prostate cancer classifier predicts biochemical failure and metastases in patients after postoperative radiation therapy. Int J Radiat Oncol Biol Phys 89, 1038–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez‐Brauer C, Thu KL, Mason JM, Blaser H, Bray MR and Mak TW (2015) Targeting mitosis in cancer: emerging strategies. Mol Cell 60, 524–536. [DOI] [PubMed] [Google Scholar]
- Durrani N, Waldron M, Chae C, Harewood L, Frydenberg M, Pedersen J and Mills J (2015) Assessing expression of MUC1 and ZAG protein biomarkers in prostate biopsies improves prediction of adverse pathology following radical prostatectomy. Open Prostate Cancer J 8, 1–9. [Google Scholar]
- Egevad L, Delahunt B, Srigley JR and Samaratunga H (2016) International Society of Urological Pathology (ISUP) grading of prostate cancer ‐ An ISUP consensus on contemporary grading. APMIS: acta pathologica, microbiologica, et immunologica. Scandinavica 124, 433–435. [DOI] [PubMed] [Google Scholar]
- Eminaga O, Wei W, Hawley SJ, Auman H, Newcomb LF, Simko J, Hurtado‐Coll A, Troyer DA, Carroll PR, Gleave ME et al (2016) MUC1 expression by immunohistochemistry is associated with adverse pathologic features in prostate cancer: a multi‐institutional study. PLoS One 11, e0165236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erho N, Crisan A, Vergara IA, Mitra AP, Ghadessi M, Buerki C, Bergstralh EJ, Kollmeyer T, Fink S, Haddad Z et al (2013) Discovery and validation of a prostate cancer genomic classifier that predicts early metastasis following radical prostatectomy. PLoS One 8, e66855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136, E359–E386. [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordetsky J and Epstein J (2016) Grading of prostatic adenocarcinoma: current state and prognostic implications. Diagn Pathol 11, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravina GL, Marampon F, Piccolella M, Motta M, Ventura L, Pomante R, Popov VM, Zani BM, Pestell RG, Tombolini V et al (2011) Hormonal therapy promotes hormone‐resistant phenotype by increasing DNMT activity and expression in prostate cancer models. Endocrinology 152, 4550–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guda K, Fink SP, Milne GL, Molyneaux N, Ravi L, Lewis SM, Dannenberg AJ, Montgomery CG, Zhang S, Willis J et al (2014) Inactivating mutation in the prostaglandin transporter gene, SLCO2A1, associated with familial digital clubbing, colon neoplasia, and NSAID resistance. Cancer Prev Res 7, 805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann MJ, Engers R, Florl AR, Otte AP, Muller M and Schulz WA (2007) Expression changes in EZH2, but not in BMI‐1, SIRT1, DNMT1 or DNMT3B are associated with DNA methylation changes in prostate cancer. Cancer Biol Ther 6, 1403–1412. [DOI] [PubMed] [Google Scholar]
- Karnes RJ, Bergstralh EJ, Davicioni E, Ghadessi M, Buerki C, Mitra AP, Crisan A, Erho N, Vergara IA, Lam LL et al (2013) Validation of a genomic classifier that predicts metastasis following radical prostatectomy in an at risk patient population. J Urol 190, 2047–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Z, He W, Lai Y, Guo X, Chen S, Li S, Wang Y and Wang L (2014) Overexpression of collagen triple helix repeat containing 1 (CTHRC1) is associated with tumour aggressiveness and poor prognosis in human non‐small cell lung cancer. Oncotarget 5, 9410–9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein EA, Cooperberg MR, Magi‐Galluzzi C, Simko JP, Falzarano SM, Maddala T, Chan JM, Li J, Cowan JE, Tsiatis AC et al (2014) A 17‐gene assay to predict prostate cancer aggressiveness in the context of Gleason grade heterogeneity, tumor multifocality, and biopsy undersampling. Eur Urol 66, 550–560. [DOI] [PubMed] [Google Scholar]
- Klein EA, Haddad Z, Yousefi K, Lam LL, Wang Q, Choeurng V, Palmer‐Aronsten B, Buerki C, Davicioni E, Li J et al (2016) Decipher genomic classifier measured on prostate biopsy predicts metastasis risk. Urology 90, 148–152. [DOI] [PubMed] [Google Scholar]
- Knezevic D, Goddard AD, Natraj N, Cherbavaz DB, Clark‐Langone KM, Snable J, Watson D, Falzarano SM, Magi‐Galluzzi C, Klein EA et al (2013) Analytical validation of the Oncotype DX prostate cancer assay ‐ a clinical RT‐PCR assay optimized for prostate needle biopsies. BMC Genom 14, 690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufe DW (2009) Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer 9, 874–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamy PJ, Allory Y, Gauchez AS, Asselain B, Beuzeboc P, de Cremoux P, Fontugne J, Georges A, Hennequin C, Lehmann‐Che J et al (2017) Prognostic biomarkers used for localised prostate cancer management: a systematic review. Eur Urol Focus pii: S2405‐4569(17)30065‐2. 10.1016/j.euf.2017.02.017. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Lin X, Gu Y, Kapoor A, Wei F, Aziz T, Ojo D, Jiang Y, Bonert M, Shayegan B, Yang H et al (2017) Overexpression of MUC1 and genomic alterations in its network associate with prostate cancer progression. Neoplasia 19, 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Friedman MS, Shedden K, Hankenson KD and Woolf PJ (2009) GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons A, Coleman M, Riis S, Favre C, O'Flanagan CH, Zhdanov AV, Papkovsky DB, Hursting SD and O'Connor R (2017) Insulin‐like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J Biol Chem 292, 16983–16998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magani F, Peacock SO, Rice MA, Martinez MJ, Greene AM, Magani PS, Lyles R, Weitz JR and Burnstein KL (2017) Targeting AR variant‐coactivator interactions to exploit prostate cancer vulnerabilities. Mol Cancer Res 15, 1469–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin NE (2016) New developments in prostate cancer biomarkers. Curr Opin Oncol 28, 248–252. [DOI] [PubMed] [Google Scholar]
- McGrath S, Christidis D, Perera M, Hong SK, Manning T, Vela I and Lawrentschuk N (2016) Prostate cancer biomarkers: are we hitting the mark? Prostate Int 4, 130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosquera JM, Beltran H, Park K, MacDonald TY, Robinson BD, Tagawa ST, Perner S, Bismar TA, Erbersdobler A, Dhir R et al (2013) Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment‐related neuroendocrine prostate cancer. Neoplasia 15, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munkley J, Vodak D, Livermore KE, James K, Wilson BT, Knight B, McCullagh P, McGrath J, Crundwell M, Harries LW et al (2016) Glycosylation is an androgen‐regulated process essential for prostate cancer cell viability. EBioMedicine 8, 103–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardi IK, Zasadzinska E, Stellfox ME, Knippler CM and Foltz DR (2016) Licensing of centromeric chromatin assembly through the Mis18alpha‐Mis18beta heterotetramer. Mol Cell 61, 774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath S and Mukherjee P (2014) MUC1: a multifaceted oncoprotein with a key role in cancer progression. Trends Mol Med 20, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oderda M, Cozzi G, Daniele L, Sapino A, Munegato S, Renne G, De Cobelli O and Gontero P (2017) Cell‐cycle progression‐score might improve the current risk assessment in newly diagnosed prostate cancer patients. Urology 102, 73–78. [DOI] [PubMed] [Google Scholar]
- Ojo D, Seliman M and Tang D (2017) Signatures derived from increase in SHARPIN gene copy number are associated with poor prognosis in patients with breast cancer. BBA Clin 8, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos I, Sivridis E, Giatromanolaki A and Koukourakis MI (2001) Tumor angiogenesis is associated with MUC1 overexpression and loss of prostate‐specific antigen expression in prostate cancer. Clin Cancer Res 7, 1533–1538. [PubMed] [Google Scholar]
- Patel KM and Gnanapragasam VJ (2016) Novel concepts for risk stratification in prostate cancer. J Clin Urol 9, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Paula Peres L, da Luz FA, Dos Anjos Pultz B, Brigido PC, de Araujo RA, Goulart LR and Silva MJ (2015) Peptide vaccines in breast cancer: the immunological basis for clinical response. Biotechnol Adv 33, 1868–1877. [DOI] [PubMed] [Google Scholar]
- Plotnikova OV, Seo S, Cottle DL, Conduit S, Hakim S, Dyson JM, Mitchell CA and Smyth IM (2015) INPP5E interacts with AURKA, linking phosphoinositide signaling to primary cilium stability. J Cell Sci 128, 364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punnen S, Freedland SJ, Presti JC Jr, Aronson WJ, Terris MK, Kane CJ, Amling CL, Carroll PR and Cooperberg MR (2014) Multi‐institutional validation of the CAPRA‐S score to predict disease recurrence and mortality after radical prostatectomy. Eur Urol 65, 1171–1177. [DOI] [PubMed] [Google Scholar]
- Rabiau N, Dechelotte P, Guy L, Satih S, Bosviel R, Fontana L, Kemeny JL, Boiteux JP, Bignon YJ and Bernard‐Gallon D (2009) Immunohistochemical staining of mucin 1 in prostate tissues. In Vivo 23, 203–207. [PubMed] [Google Scholar]
- Ribeiro IP, Marques F, Caramelo F, Ferrao J, Prazeres H, Juliao MJ, Rifi W, Savola S, de Melo JB, Baptista IP et al (2014) Genetic imbalances detected by multiplex ligation‐dependent probe amplification in a cohort of patients with oral squamous cell carcinoma‐the first step towards clinical personalized medicine. Tumour Biol 35, 4687–4695. [DOI] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G et al (2015) Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross AE, D'Amico AV and Freedland SJ (2016) Which, when and why? Rational use of tissue‐based molecular testing in localized prostate cancer. Prostate Cancer Prostatic Dis 19, 1–6. [DOI] [PubMed] [Google Scholar]
- Scheid E, Major P, Bergeron A, Finn OJ, Salter RD, Eady R, Yassine‐Diab B, Favre D, Peretz Y, Landry C et al (2016) Tn‐MUC1 DC vaccination of rhesus macaques and a phase I/II trial in patients with nonmetastatic castrate‐resistant prostate cancer. Cancer Immunol Res 4, 881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenas J, Allegrucci C, Boorjian SA, Mongan NP and Persson JL (2012) Overcoming drug resistance and treating advanced prostate cancer. Curr Drug Targets 13, 1308–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severi G, FitzGerald LM, Muller DC, Pedersen J, Longano A, Southey MC, Hopper JL, English DR, Giles GG and Mills J (2014) A three‐protein biomarker panel assessed in diagnostic tissue predicts death from prostate cancer for men with localized disease. Cancer Med 3, 1266–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipley WU, Seiferheld W, Lukka HR, Major PP, Heney NM, Grignon DJ, Sartor O, Patel MP, Bahary JP, Zietman AL et al (2017) Radiation with or without antiandrogen therapy in recurrent prostate cancer. N Engl J Med 376, 417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh PK and Hollingsworth MA (2006) Cell surface‐associated mucins in signal transduction. Trends Cell Biol 16, 467–476. [DOI] [PubMed] [Google Scholar]
- Sircar K, Huang H, Hu L, Cogdell D, Dhillon J, Tzelepi V, Efstathiou E, Koumakpayi IH, Saad F, Luo D et al (2012) Integrative molecular profiling reveals asparagine synthetase is a target in castration‐resistant prostate cancer. Am J Pathol 180, 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellfox ME, Nardi IK, Knippler CM and Foltz DR (2016) Differential binding partners of the Mis18alpha/beta YIPPEE domains regulate Mis18 complex recruitment to centromeres. Cell Rep 15, 2127–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarnowski M, Czerewaty M, Deskur A, Safranow K, Marlicz W, Urasinska E, Ratajczak MZ and Starzynska T (2016) Expression of cancer testis antigens in colorectal cancer: new prognostic and therapeutic implications. Dis Markers 2016, 1987505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B et al (2010) Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong N, Major P, Kapoor A, Wei F, Yan J, Aziz T, Zheng M, Jayasekera D, Cutz JC, Chow MJ et al (2016) Amplification of MUC1 in prostate cancer metastasis and CRPC development. Oncotarget 7, 83115–83133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong N, Ojo D, Yan J and Tang D (2015) PKM2 contributes to cancer metabolism. Cancer Lett 356, 184–191. [DOI] [PubMed] [Google Scholar]
- Wurz GT, Kao CJ, Wolf M and DeGregorio MW (2014) Tecemotide: an antigen‐specific cancer immunotherapy. Hum Vaccin Immunother 10, 3383–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G and He QY (2016) ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol BioSyst 12, 477–479. [DOI] [PubMed] [Google Scholar]
- Zaorsky NG, Raj GV, Trabulsi EJ, Lin J and Den RB (2013) The dilemma of a rising prostate‐specific antigen level after local therapy: what are our options? Semin Oncol 40, 322–336. [DOI] [PubMed] [Google Scholar]
- Zhuang L and Johnson MT (2016) How precisely can prostate cancer be managed? Int Neurourol J 20, S120–S130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Covariate selection from 696 DEGs using Elastic‐net penalty.
Fig. S2. Overlapping between the 9‐gene genomic signature which we have previously reported (Lin et al., 2017) and the current signature (SigMuc1NW). Graph was produced using the TCGA Provisional dataset (n = 492, cBioPortal).
Fig. S3. The combined signature is significantly associated with reductions in DFS and OS in PC patients.
Fig. S4. Cutpoint estimation.
Fig. S5. SigMuc1NW scores effectively stratify PCs with elevated risk of recurrence following RP.
Fig. S6. CPC geneset is associated with a reduction in DFS but not OS in PC patients.
Table S1. Differentially expression genes (DEGs) of a 9‐gene signature identified in the TCGA Provisional dataset.
Table S2. (A) Upregulation of gene sets among the 696 DEGs associated with the 9‐gene genomic signature within the kegg.sets.hs dataset. (B) Downregulation of gene sets among the 696 DEGs within the kegg.sets.hs dataset. (C) Upregulation of gene sets among the 696 DEGs within the GO.sets.hs dataset. (D) Downregulation of gene sets among the 696 DEGs within the GO.sets.hs dataset. (E) Pathways affected by the 696 DEGs associated with the 9‐gene genomic signature.
Table S3. Scores of the component genes and some clinical characteristics of patients with prostate cancer in the TCGA Provisional dataset within cBioPortal.
Table S4. Demographics of the TCGA patient population. The clinical characteristics were extracted from the TCGA Provisional dataset within cBioPortal along with the indicated clinical data.
Table S5. Cutpoints of individual gene expression determined by RNA sequencing.