

Abstract
Stem cell maturation is a fundamental, yet poorly understood aspect of human development. We devised a DNA methylation signature deeply reminiscent of embryonic stem cells (a fetal cell origin signature, FCO) to interrogate the evolving character of multiple human tissues. The cell fraction displaying this FCO signature was highly dependent upon developmental stage (fetal versus adult), and in leukocytes, it described a dynamic transition during the first 5 yr of life. Significant individual variation in the FCO signature of leukocytes was evident at birth, in childhood, and throughout adult life. The genes characterizing the signature included transcription factors and proteins intimately involved in embryonic development. We defined and applied a DNA methylation signature common among human fetal hematopoietic progenitor cells and have shown that this signature traces the lineage of cells and informs the study of stem cell heterogeneity in humans under homeostatic conditions.
Studies of hematopoiesis have laid the foundation for advances in stem cell biology; however, the sources and diversity of hematopoietic stem cells (HSCs) remain controversial (Orkin and Zon 2008). Heterogeneity within HSC populations is well established (Muller-Sieburg et al. 2012) with hematopoiesis in fetal and early life representing dynamic periods of stem cell transition and maturation (Dykstra and de Haan 2008; Copley and Eaves 2013; Herzenberg 2015). In mice, potential regulators of HSC maturation include Polycomb repressor complex 2 proteins (PRC2s) (Mochizuki-Kashio et al. 2011; Xie et al. 2014; Oshima et al. 2016), SOX17 (He et al. 2011), ARID3A (Ratliff et al. 2014), and let-7b miRNA (Copley et al. 2013; Rowe et al. 2016). Direct tracking of stem cell lineage and diversity has been achieved in experimental animal models by enumerating chromosomal translocations, retroviral insertions, and molecular barcodes in repopulating cells during hematopoietic reconstitution (Eaves 2015). Most recently, lineage tracing studies using genetically labeled HSCs, which permits stem cell tracking without engraftment, have produced contrasting data on the relative contributions of HSCs and progenitors in steady-state hematopoiesis (Sun et al. 2014b; Busch et al. 2015; McKenna et al. 2016; Sawai et al. 2016; Säwén et al. 2016). At the same time, because genetic lineage tracing is not feasible in humans, effective strategies for identifying and defining markers capable of capturing both progenitor and stem cell lineages in human populations remain to be developed.
Naturally occurring epigenetic marks such as DNA methylation provide a promising alternative for assessing progenitor and stem cell diversity in vivo (Ji et al. 2010; Beerman et al. 2013; Farlik et al. 2016). Following fertilization, DNA methylation is erased and reestablished in concert with lineage commitment and cellular differentiation (Lee et al. 2014). Because lineage-specific marks of DNA methylation have been successfully employed to detect the relative abundance of individual cell types in blood mixtures (Houseman et al. 2012; Accomando et al. 2014; Koestler et al. 2016; Salas et al. 2018) and because a significant proportion of progenitor and stem cell methylation events are mitotically stable throughout differentiation, it is possible that a common set of unchanging DNA methylation markers can trace a common cell ontogeny (Kim et al. 2010).
Here, we describe a novel analytical pipeline that involves generating a library of stable CpG loci that are markers of the cell of origin for studying peripheral blood leukocytes. The pipeline is based upon the observation that a subset of CpG-specific methylation marks are inherited in progeny cells irrespective of lineage differentiation. These candidate marker loci, reflecting the progenitors from which they are derived, are identified and selected as an initial step in the pipeline. In a second filtering process, we select a subset of these candidate loci that optimize the discrimination of fetal and adult differentiated leukocytes. This second step provides CpG marker loci that are different among fetal and adult progenitors; these loci form what we refer to as a fetal cell origin (FCO) signature. Finally, we employed the FCO signature in conjunction with our established algorithm for cell mixture deconvolution (Houseman et al. 2012) for estimating the proportion of cells in a mixture of cell types that are of fetal cell origins.
Results
In this study, we used several genome-scale DNA methylation data sets from newborn and adult leukocyte populations to identify a common set of CpG loci among fetal leukocyte subtypes (the FCO signature) and applied it to trace the proportion of cells with the progenitor phenotype in several tissue types across the lifecourse (Supplemental Table S1). We hypothesized that invariant methylation marks with high potential to be indicative of a FCO would be differentially methylated in newborns compared with adults and shared across six major blood cell lineages (granulocytes [Gran], monocytes [Mono], B lymphocytes [Bcell], CD4+ T lymphocytes [CD4T], CD8+ T lymphocytes [CD8T], and natural killer lymphocytes [NK]). The analytic pipeline for identification of candidate FCO CpGs from libraries of Illumina HumanMethylation450 array data is shown in Supplemental Figure S1. We initially compared genome-scale DNA methylation profiles of each of the six major blood cell lineages separately between umbilical cord blood (UCB) and adult whole peripheral blood (AWB) DNA samples. Across the separate models fit to each blood cell type, we identified 1255 CpG sites (false discovery rate [FDR] < 0.05) with shared, significant differential methylation between newborns and adults. Then we filtered this lineage invariant subset of CpG loci to arrive at CpGs exhibiting both a consistent direction of differential methylation across all lineage groups and an absolute change in methylation >10% between newborns and adults, resulting in n = 1218 CpGs associated with 518 genes (Supplemental File S1). We further reduced the list of candidate FCO CpG loci (Supplemental Fig. S2A) to minimize potential cell-type–specific contribution by selecting CpGs with minimal residual cell-specific effects, resulting in 27 CpGs (Supplemental Fig. S2B). We accomplished this by using a principal component (PC) regression analysis in which the standardized and rotated scores of the first four PCs captured most of the variation in DNA methylation across the 1218 candidate CpGs. The first PC explained 79.4% of the variance and was significantly associated with both methylation age (P = 4.62 × 10−62) and UCB versus adult peripheral blood (P = 9.56 × 10−123). Some residual variability, 13.4%, was significantly associated with cell type in the second to fourth PCs (Supplemental Fig. S2A, lower heatmap). Once filtered to 27 CpGs, 84.6% of the variance was explained by the first PC, which was significantly associated with both methylation age (P = 1.89 × 10−63) and UCB versus adult peripheral blood (P = 3.81 × 10−110). However, cell type was no longer significantly associated with any of the first four PCs (94.1% of the total variance) (Supplemental Fig. S2B, lower heatmap). The library of 27 CpGs we identified represents a phenotypic block of differentially methylated regions (DMRs), with a FCO phenotype here defined as the FCO signature. The name FCO signature summarizes the idea of a common invariant biomarker of a cell that originated during the prenatal period, which is present across different cell lineage subtypes but also is reduced or lost during lineage commitment of progenitor cells in the adult.
Next, we used the FCO library in conjunction with the constrained projection quadratic programming approach of Houseman and coworkers (Houseman et al. 2012; Accomando et al. 2014; Koestler et al. 2016) to estimate the proportion of cells exhibiting the FCO signature in a manner agnostic to variation in underlying proportions of cell types in any given sample, as well as independent of a sample's DNA methylation age (Hannum et al. 2013; Horvath 2013). The proportion of cells with the FCO signature was estimated for each sample in the discovery data set of newborn and adult leukocytes. As expected, UCB samples were predicted to harbor a very high proportion of cells of fetal origin (mean = 85.4%), significantly higher than adult leukocytes (mean = 0.6%, P = 2.11 × 10−191) (Fig. 1A). To replicate our findings, we applied the same estimation approach to an independent data set that included leukocyte-specific methylation measurements collected from newborn and adult sources. In the replication data set, we observed similar differences in proportions of cells with the stem cell lineage signature between cord blood and adults (P = 8.35 × 10−81) (Fig. 1B), where the proportion of cells exhibiting the FCO signature was again higher in the cord blood samples compared to the adult samples (89.9% vs. 2.0% for UCB and AWB samples, respectively). Together, these results suggest that the FCO signature captures a population of lineage invariant, developmentally sensitive cells.
Figure 1.

Discovery (A) and replication (B) of the deconvolution algorithm using lineage-invariant, developmentally sensitive CpG loci in newborn and adult peripheral blood leukocytes. Estimated mean percentage (standard deviation [SD]) FCO methylation fractions are 85.4% (6.0) for umbilical cord blood (UCB) and 0.6% (1.7) for peripheral adult blood in A; P = 2.11 × 10−191. In the replication (B), estimated FCO methylation fractions are 89.9% (3.8) for UCB and 2.0% (3.5) for peripheral adult blood; P = 8.35 × 10−81.
Once we obtained concordant results in the validation data, we assessed the classification performance of the 27 CpGs in the FCO signature compared to randomly selected sets of CpGs. We included five independent data sets (Supplemental Table S1, AUROC data sets) consisting of n = 123 UCB and n = 34 AWB samples. As a previous publication (Morin et al. 2017) had interrogated the potential of maternal blood contamination using these data sets, we evaluated if any of the samples showed evident maternal blood contamination. Using a combination of the 10 CpGs reported by Morin et al. (2017) and the calculation of DNA methylation age, we found one cord blood sample in the paired maternal–newborn GSE54399 data set (Montoya-Williams et al. 2017) was mostly maternal blood (DNA methylation age 44.5 yr corresponding to the paired 45 yr in the maternal sample and an adult hypermethylated pattern using the 10 markers of Morin et al. 2017). After removing this sample, we applied our FCO signature to these data and assessed how well it classified fetal from adult tissues by computing the area under the receiver operating characteristic curve (AUROC). The AUROC for our 27 CpG FCO signature was estimated to be 0.996 based on a combined analysis of the five data sets described above. To gauge whether the AUROC was statistically significant, and thus, that our 27 CpG FCO signature represents a statistically significant subset, we conducted an analysis in which we generated the empirical null distribution of the AUROC by (1) randomly selecting subsets of CpGs of size 27, followed by (2) calculation of the AUROC for the randomly selected subset. We repeated steps (1–2 above) 10,000 times and computed the probability of observing an AUROC as large or larger than what we computed based on our 27 CpG FCO signature. The P from our randomization-based test was P = 0.0193, meaning that there was only a 1.9% chance of observing an AUROC as large or larger than what was observed based on our FCO signature. In addition, we used this same data set to evaluate how stable the estimations would be if we excluded some of the 27 markers using a leave-one-out combination, leave-two-out combination, until five probes combination were removed. Although the estimates are stable in the absence of several of the probes, the potential error increases per probe removed (average RMSE: 10 when removing one probe, 15 when removing two, 19 when removing three, 22 with four and 25 with five) (see Supplemental Methods S1). To further demonstrate the validity and reliability of our signature, we generated reference synthetic cell mixtures by mixing cord blood and adult peripheral blood DNA methylation signatures in silico (Supplemental Table S1, synthetic mixtures data sets), varying the fraction of fetal cord blood across mixtures. Application of our algorithm to the reference synthetic cell mixtures showed a high concordance correlation coefficient (CCC) between the estimated fraction of cells carrying the FCO signature and the known mixture proportions (Supplemental Fig. S3, concordance correlation coefficient, CCC = 0.97).
To explore the ontogeny of the FCO signature, we next deconvoluted methylation array data from embryonic stem cell lines, induced pluripotent cells (iPCSs), fetal CD34+ stem/progenitor cells, and bone marrow adult CD34+ stem/progenitor cells. The results indicated concordance of the leukocyte-derived FCO signature with embryonic and pluripotent methylomes (Fig. 2; Supplemental Table S2). However, we were intrigued by the fact that among the embryonic stem cells (ESCs) and iPCs, there was a wide range of the estimated FCO signature. Using information on the number of passages (subcultures) per sample (mean = 27.2 passages, SD: 16.8), we modeled the estimated FCO fraction against the number of cell culture passages using a linear regression model. For every additional passage, we observed a reduction of 0.14%, on average, in the estimated FCO signature (P = 0.01) after adjusting for each sample's estimated DNA methylation age (Supplemental Fig. S4). This trend was observed in both ESCs and iPCs; however, when stratifying by cell type, the magnitude of the reduction was higher for ESCs (a mean reduction of 0.18% per passage), and it was attenuated in the induced pluripotent stem cells (iPSCs; a reduction of 0.07% per passage). The P of interaction for cell passage and cell type was not statistically significant (P = 0.11).
Figure 2.
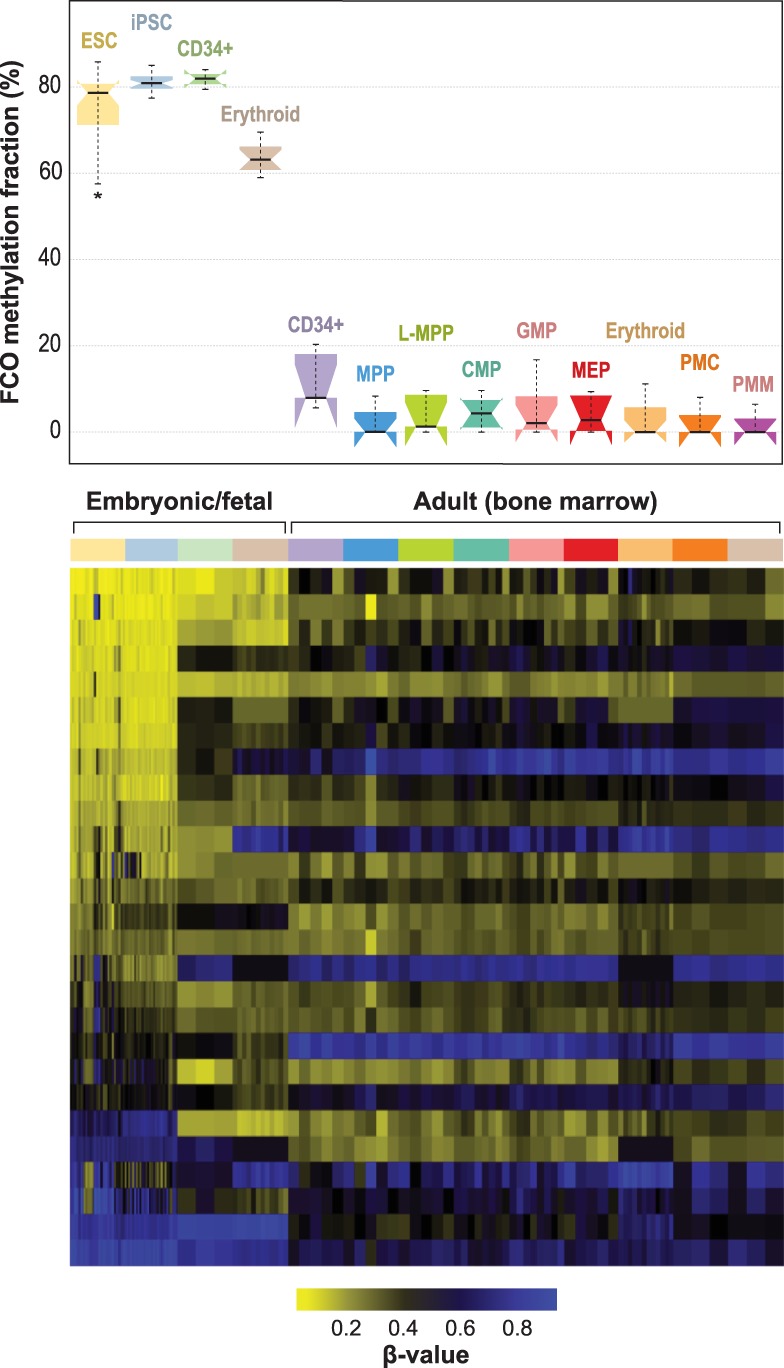
Developmentally sensitive methylation signature deconvolution in pluripotent, fetal progenitors, and adult CD34+ stem/progenitor cells. Mean (SD) estimated FCO methylation fractions for embryonic/fetal cells are 75.9% (8.5) and 4.4% (5.1) for adult progenitors (bone marrow); P = 1.81 × 10−86. In the boxplots: (1) The box shows the interquartile range (IQR), (2) the whiskers show the inner fences (1.5 × IQR out of the box), (3) the bolded line shows the median of the data, and the notches-horns display the 95% confidence interval of the median. (ESC) Embryonic stem cells; (iPSC) induced pluripotent stem cells; (CD34+ fetal) fresh cord blood cells expressing CD34+; (erythroid fetal) fetal liver CD34+ cells, differentiated ex vivo to express transferrin receptor and glycophorin; (CD34+ adult) bone marrow expressing CD34+ CD38− CD90+ CD45RA−; (MPP) multipotent progenitors; (L-MPP) lymphoid primed multipotent progenitors; (CMP) common myeloid progenitors; (GMP) granulocyte/macrophage progenitors; (MEP) megakaryocyte-erythroid progenitors; (erythroid adult) adult bone marrow CD34+ cells, differentiated ex vivo to express transferrin receptor and glycophorin; (PMC) promyelocyte/myelocyte; (PMN) metamyelocyte/band-myelocyte.
A potential caveat of our approach for deriving the FCO signature is the use of lineage committed neonatal cord and adult peripheral blood cells rather than the use of undifferentiated fetal and adult progenitor cells. One reason for this is the fact that considerable heterogeneity exists in isolating undifferentiated cells, making it problematic to generate a true “gold standard.” As an approximation and to estimate the relative variability and sources of uncertainty of our FCO signature, we applied a similar pipeline and filter criteria to a small data set of fetal and adult pluripotent cells. In this sensitivity analysis, we compared the DNA methylation between 19 undifferentiated ESCs and five adult hematopoietic stem cells (CD34+ CD38− CD90+ CD45RA−) as proxies of common pluripotent cells at the embryonic and adult ages, respectively. We observed 113 differentially methylated sites (FDR < 0.05) that overlapped with the original 1255 candidate list (9% overlap) generated from differentiated cells. Of those 113 differentially methylated sites, five out of the 27 CpGs (19%) in the FCO signature were represented. However, when we applied the same filtering process to those CpGs to remove lineage-specific effects (see Methods), only two CpGs out of the 113 CpGs were retained. When we explored the 113 overlapping CpGs using our discovery data set, we observed cell population stratification. The second PC variance increased from 6.0% using the 27 CpGs (Supplemental Fig. S2B) to 9.8% using the 113 CpGs, and in contrast to our approach as applied to differentiated blood cells, these 113 CpGs discriminated myeloid and lymphoid subpopulations in both the fetal and adult cells of the discovery data set. The distribution and the variance explained resembled the distribution observed using the 1218 CpGs from the candidate list (Supplemental Fig. S2A). This finding suggests a highly heterogeneous ESC population in this small sensitivity analysis, which is also consistent with the observed variance in FCO fraction of ESCs explained by cell culture passage number. However, these results also suggest that our FCO signature shares some CpG loci in common with those derived from a pipeline that starts with ESCs and adult progenitors.
Next, we reasoned that if part of the FCO signature were an indicator of ESC lineage, it would also be detectable among nonhematopoietic fetal tissues. Figure 3A shows the high FCO fraction in diverse fetal tissues (3–26 wk of gestational age) and in sharp contrast, the minimal representation of the FCO signature in adult tissues. The FCO signature demonstrated higher variability in fetal/embryonic brain and muscle, showing a dramatic drop of the signature with later gestational age (Fig. 3B) compared to other tissues including the liver (a hematopoietic tissue in the fetus).
Figure 3.
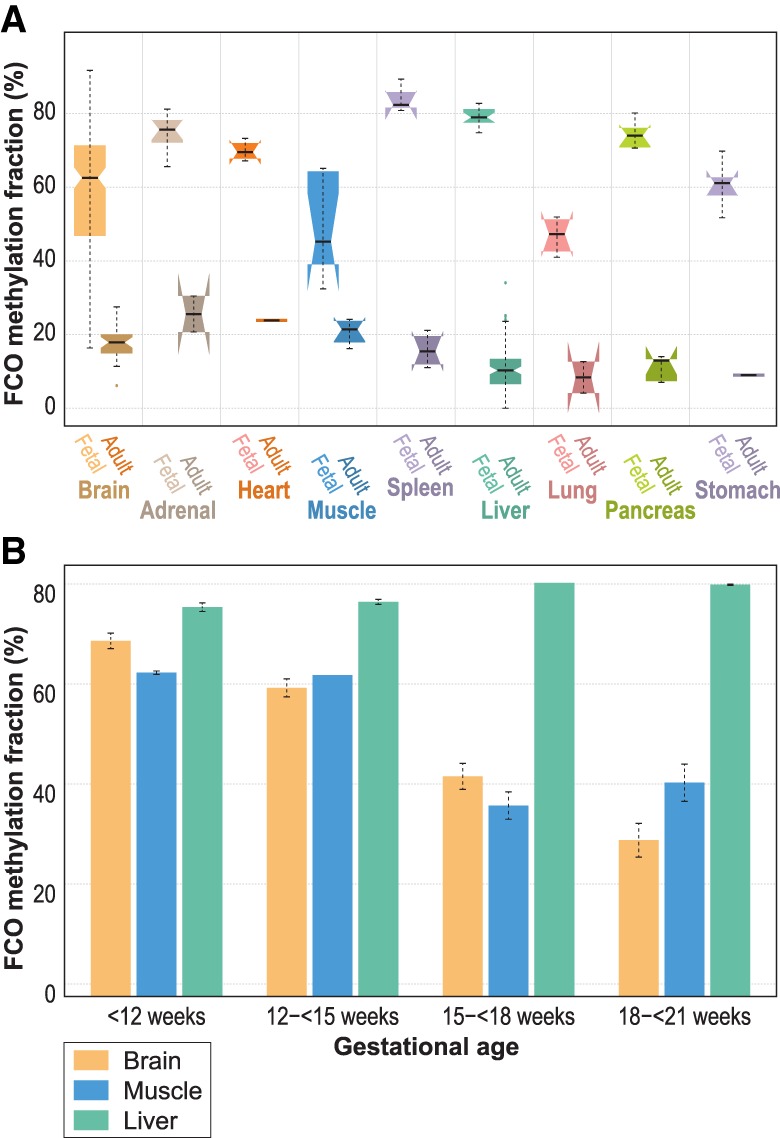
FCO methylation signature deconvolution in fetal/embryonic and adult tissues. Panel A compares the estimated FCO methylation fraction between fetal/embryonic and adult tissues. In the boxplots: (1) The box shows the interquartile range (IQR), (2) the whiskers show the inner fences (1.5 × IQR out of the box), (3) the bolded line shows the median of the data, and the notches-horns display the 95% confidence interval of the median. Panel B compares the estimated mean FCO methylation signature in three fetal/embryonic tissues in four gestational periods: Brain and muscle showed a marked reduction of the signature after the 15th week of gestational age. In contrast, fetal/embryonic liver showed a persistently high level of the FCO signature.
We also sought to explore the potential biologic functions of the FCO signature. To include sufficient genes in this analysis, we returned to the filtered lineage invariant FCO candidate CpG list (n = 1218 CpGs, associated with 518 genes) and applied a test of enrichment using information from the MSigDB curated databases v. 6.0 (Liberzon et al. 2011) and the Progenitor Cell Biology Consortium database (Salomonis et al. 2016). We used three different approaches to test for enrichment using the curated molecular signatures database (MSigDB): (1) ToppGene (Chen et al. 2009), (2) GREAT (McLean et al. 2010), and (3) missMethyl (Phipson et al. 2016). ToppGene and missMethyl used the 518 genes associated with the CpG site; in contrast, GREAT used 1238 genes within 1 Mb of the CpG site (cis-regulatory genes). In total, 18, 20, and 27 pathways were statistically significant after FDR correction, respectively. Of those, we found a significant statistical association in nine pathways using the three approaches, and in six pathways overlapping the ToppGene and missMethyl approaches (Supplemental Table S3). Among the nine overlapping the three approaches, there was a statistically significant association with pathways related to epigenetic marks in embryonic stem cells and progenitor cells. When restricting to the FCO signature CpGs, there was an interesting pattern in the chromatin features of 11 out of the 27 sites that changed from a poised promoter to a repressed state in umbilical vein endothelial cells (Supplemental Table S4). In addition, among the candidate stem cell gene list were 13 homeobox transcription factors as well as 14 others that play key roles in embryonic development (e.g., FOXD2, FOXE3, FOXI2, FOXL2, ARID3A, NFIX, PRDM16, SOX18) (Supplemental Table S5). Most notable were genes previously implicated in fetal to adult transitions in hematopoiesis. ARID3A plays a critical role in lineage commitment in early hematopoiesis (Ratliff et al. 2014). Among our targets was SOX18, a paralog of SOX17, the latter being shown to maintain fetal characteristics of HSCs in mice (He et al. 2011). PRC2 targets were overrepresented in FCO signature loci (Supplemental Tables S3, S4). EZH2, one of three PRC2 components, is indispensable for fetal liver hematopoiesis but largely dispensable for adult bone marrow hematopoiesis (Mochizuki-Kashio et al. 2011; Xie et al. 2014; Oshima et al. 2016). Among the larger set of loci used to derive the FCO signature, there are five DMRs within the MIRLET7BHG locus (Fig. 4). The LIN28A-LIN28B/let-7 axis is a highly evolutionarily conserved developmental regulator and has emerged as a prominent feature of the fetal to adult switch in murine hematopoiesis (Copley et al. 2013; Rowe et al. 2016). The DMR region we identified encompasses exon and intron 1 of the MIRLET7BHG. Methylation in this region displayed an inverse relationship within fetal and adult cells for CpG boundary probes that colocate with active histone marks, DNase I hypersensitivity, and transcription factor binding sites (Fig. 4). In addition, a middle region, which is devoid of regulatory motifs, displayed contrasting methylation features in each type of cell. The entire 15.5-kb region forms a bipartite methylation pattern with hypomethylated loci in adult cells demarcated by hypermethylation, whereas in embryonic cells, the bipartite region is bounded by hypermethylated loci demarcated by hypomethylation. In addition, overrepresentation of genes expressed in ESCs to embryoid body differentiation were among the FCO methylation gene loci (Supplemental Table S6). Taken together, we have developed a deconvolution algorithm based on DNA methylation that indicates the fraction of differentiated cells with FCOs that could represent a proxy for ESC origin.
Figure 4.

Candidate CpGs in the FCO methylation signature in the MIRLET7BHG locus on Chromosome 22. Box plots compare the DNA methylation levels (as β-values) at each CpG site for ESC (in yellow), UCB (in orange), adult progenitors (in green), and adult whole blood (in magenta). In the boxplots: (1) The box shows the interquartile range (IQR), (2) the whiskers show the inner fences (1.5 × IQR out of the box), (3) the bolded line shows the median of the data, and the notches-horns display the 95% confidence interval of the median. We rearranged the scale of the boxplots to approximate the different genomic context measured by the probes. Above the boxplots, tracks from the UCSC Genome Browser show the epigenomic features of normal adult CD14+ monocytes including activating histone marks, DNase I hypersensitivity clusters, and transcription factor binding sites. (ORegAnno) Open Regulatory Annotation Database (Lesurf et al. 2016). Differences in DNA methylation between fetal cells (ESC and UCB) and adult cells (adult progenitors and adult whole blood) were statistically significant at P < 2.0 × 10−16 after Bonferroni correction for all five CpG sites. Differences in DNA methylation between ESC and adult progenitors were significant for four out of five CpGs (P < 5.9 × 10−4) after Bonferroni correction (cg03684807 was not significant; P = 0.26).
The perinatal and early childhood periods are times of dramatic transition in erythropoiesis and leukocyte function. Therefore, we hypothesized that this time of life would be marked by variations in embryonic to adult-driven stem cell hematopoiesis. To test this idea, we examined the relative proportion of cells with the FCO signature in blood leukocytes from birth through old age (Fig. 5A). Dramatic and rapid decreases in the FCO cell fraction occurred over the first 5 yr of life (Fig. 5A,B; Supplemental Table S7). A reduction in the proportion of cells with the FCO signature of ∼60% was observed at 1.5 yr, and by age 5, the fraction was reduced by 80%. Most adults (>18 yr) demonstrated nondetectable levels of cells with the FCO signature. However, ∼10% of adults (18–65 yr), were observed to have a relatively high fraction of leukocytes with the FCO signature (range = 10%–25%). The FCO fraction among adults with detectable FCO levels (more than 0%) showed a poor linear correlation (r = −0.12) with age. However, when restricting to those with FCO levels ≥3%, this correlation between FCO and age was no longer significant (r = −0.12, P > 0.05). Of further note, there was no overlap in the loci comprising the FCO signature with the previously described CpGs used to calculate DNA methylation age (Lowe et al. 2016). Although age associated in the early postnatal period, the FCO signature loci did not overlap with Horvath's age-related epigenetic clock and/or other epigenetic clocks (Lowe et al. 2016). In addition, none of the CpG loci identified during HSC aging in mice (Sun et al. 2014a) overlap with our FCO signature. Our results indicate a distinction between aging and developmentally timed maturation events signaling variations in the fetal origin cell compartment (Rossi et al. 2008).
Figure 5.
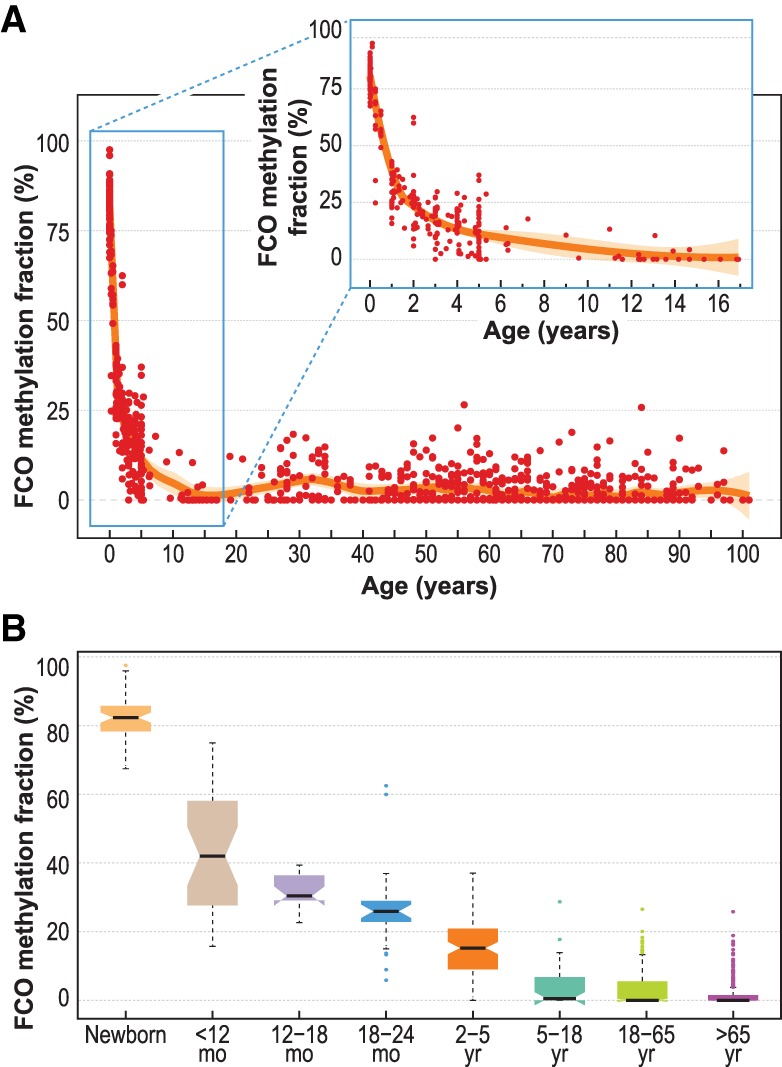
FCO methylation signature deconvolution in blood leukocytes sampled at birth through childhood and adult ages. Panel A shows the loess smoothing curve across different ages ranging from newborn to 101 yr. In the top subplot of the panel is an enlarged depiction of the marked decrease of the fraction of cells showing the FCO signature during the first 18 yr of life. Panel B summarizes the reduction of the FCO signature at different age intervals. In the boxplots: (1) The box shows the interquartile range (IQR), (2) the whiskers show the inner fences (1.5 × IQR out of the box), (3) the bolded line shows the median of the data, and the notches-horns display the 95% confidence interval of the median.
Discussion
This work represents a conceptual departure from previous studies that have focused on DMRs that mark fate determination during terminal differentiation. Most of the characteristic DMRs of stem/progenitor cells are what we would term unstable to differentiation as they undergo transitions within the progeny as cells differentiate (Ji et al. 2010; Beerman et al. 2013; Farlik et al. 2016). In contrast, a smaller set of DMRs retain their status throughout the differentiation sequence and thus form a memory trace of cell origin. By restricting our initial CpG selection to lineage invariant loci, we filtered out unstable loci (loci with additional sources of variability unrelated to the stem cell/progenitor origin). By subsetting invariant loci according to their differential methylation in newborn versus adult leukocytes, we obtained an “orthogonal” set of developmentally sensitive loci. The potential advantage of DNA methylation as a tracking strategy compared with previous methods (e.g., retroviral insertion, molecular barcodes) is that it is a natural feature of stem cells. DNA methylation–based methods can be applied to human cells without manipulation, in fresh or archival specimens (such as those of ongoing birth cohorts), and provide a window into in vivo cell ontogeny dynamics. An example of the utility of this approach is evident in our study of newborns, infants, and children that revealed a dramatic shift in hematopoietic ontogeny from birth to age 5 with evidence of wide individual variability. There is a great deal of interest in how the timing of early life developmental events shape life-long health outcomes (Gluckman et al. 2008). The FCO may be an easily applied developmental marker of early immunologic maturation in such studies.
The loci represented in the FCO signature are themselves potential candidates with regulatory function in stem cell maturation. A notable example is our finding of DMRs in the Chromosome 22 region containing a cluster of let-7 microRNAs. Extensive research has shown that expression of let-7 microRNAs play essential roles in the differentiation of ESCs (Lee et al. 2016). The maintenance of the pluripotent state requires suppression of let-7. The DMR region we identified encompasses exon and intron 1 of MIRLET7BHG. Methylation in this region displayed a bipartite pattern and described an inverse relationship within fetal and adult cells wherein regulatory regions were hypermethylated in the fetal cells. This novel pattern was unexpected as hypermethylation in MIRLET7BHG has only been reported in infant leukemic cells (Nishi et al. 2013), wherein methylation silenced MIRLET7BHG expression. In contrast, the primary physiologic mechanism for let-7 regulation has been thought to involve post-transcriptional interference with microRNA biogenesis promoted through the actions of the LIN28A and LIN28B proteins (Lee et al. 2016). LIN28A/LIN28B proteins are essential for normal development and contribute to the pluripotent state by preventing the maturation of let-7 pre-RNA (Piskounova et al. 2008, 2011). In turn, let-7 feeds back and dampens the expression of LIN28A/LIN28B, thus forming a reciprocal negative feedback loop, and acts as a bimodal switch (Rybak et al. 2008; Melton et al. 2010). Recent studies have identified novel DNA binding properties of Lin28 in mouse ESCs that may also modulate DNA methylation levels (Zeng et al. 2016). Our data are consistent with a DNA methylation mediated suppression of MIRLET7BHG in stem cells and its reversal via demethylation during the developmental switch, leading to ESC differentiation. However, more work testing the expression of MIRLET7BHG in differentiated fetal leukocytes is required to establish the functional importance of our observations and is beyond the scope of the current study.
The selection of the candidates for the FCO signature took advantage of isolated subtypes of adult and newborn blood cells instead of using ESCs or hematopoietic progenitors. Our rationale for this approach is based on the requirement in the discovery step of making comparisons between homogeneous populations present in both newborns and adults and the fact that such data do not currently exist for the respective fetal and adult HSCs. Although we implemented an analysis using ESCs and adult HSCs, we foresaw that the dynamic state within ESC subpopulations cannot correctly discriminate stochastic noise due to stem cell dynamics from the potential variation due to early cell commitment or coexistent cell states as observed in mouse models (Singer et al. 2014). We acknowledge that starting with differentiated cells as we did introduces some cell subpopulation heterogeneity (e.g., lymphocyte subpopulations) that cannot be controlled in our models. Nonetheless, using UCB and AWB sorted blood samples allowed a clear contrast between the more general immune cell lineages in vivo. We believe that under very controlled experimental conditions, this same approach would have yielded a similar or an improved signature using ESCs and a selected adult cell counterpart. Our sensitivity analysis using ESCs and adult CD34+ cells suggested that at least 19% of the FCO signature was shared when using this approach. Our results, however, also suggest a practical problem: When using ESCs, the ex vivo conditions may generate heterogeneous populations of ESCs, making them poor gold standards for comparison. In the absence of better standards, the proposed FCO signature provides a good proxy of the common fetal cell compartment. It is possible that the reduced FCO estimated fractions in higher passaged embryonic cells points to in vitro conditions leading to instability in the fetal epigenome and may constitute a quality control issue during the ex vivo manipulation of stem cells. The FCO fraction may provide one indicator of epigenome stability that could be useful in evaluating fetal cells expanded in vitro. An ongoing concern in adoptive cell transfer therapies is the paucity of informative markers reflecting epigenomic stability of expanded cell populations, as for example, in the expansion of UCB-derived T-regulatory cells (Seay et al. 2017).
We think our observations have additional implications and potential applications for future research. In clinical and epidemiological studies, the currently used cell correction methods (Teschendorff et al. 2017; Titus et al. 2017) could benefit from the additional information on cell heterogeneity provided by the FCO signature. As an adjunct to current cell correction methods, the FCO can reduce variability in methylation signals due to cell composition and increase the specificity of EWAS analyses in identifying non-cell-type causal factors. Large-scale population studies must also account for the now well-documented effects of age on a subset of DNA methylation loci, the so-called Horvath clock CpG loci (Horvath 2013), which we show here to be distinct from those forming the FCO signature. Aging in humans is well known to alter hematopoiesis, and recent studies in mice illustrate how it manifests in HSCs at multiple layers of the epigenome, including DNA methylation (Sun et al. 2014a). However, we did not see any obvious parallels of age-related HSC methylation with the FCO signature. None of the HSC age loci described in mice overlap with the FCO target loci. The phenomenon of clonal hematopoiesis of indeterminate potential (CHIP) is another age-related hematopoietic variation of great potential clinical import (Jaiswal et al. 2014, 2017). It is known that CHIP occurs in ∼10% of otherwise healthy persons of advanced age, which is similar to our FCO observations (Supplemental Table S7). However, in our study of 784 different adult samples (>18 yr), we found no significant correlation of the FCO with the age of blood donors. In the absence of an age-related explanation for increased FCO fractions in some adults, we are led to ask if there is a heretofore unrecognized cell component in adult blood with a distinct fetal cell ontogeny. In this regard, the FCO may provide a tool to help resolve a long-debated controversy about the occurrence of a B1 subtype of B-lymphocytes in humans (Descatoire et al. 2011; Griffin et al. 2011; Hardy and Hayakawa 2015). In mice, B1 cells are well described as long-lived self-renewing fetal-derived B cells that produce natural antibodies in the absence of apparent antigenic stimulation and that localize in pleural and peritoneal cavities in adults (Kantor and Herzenberg 1993; Ghosn and Yang 2015; Hardy and Hayakawa 2015). Furthermore, an important role has been established for Let-7 microRNA in mouse B1 cell development (Yuan et al. 2012), and our studies have linked differential methylation of MIRLET7BHG with our human fetal signature. To explore the hypothesis that the blood FCO signal can arise from a unique B-cell population will require isolation of candidate B1-cell populations and simultaneous measurement of the FCO fraction. Human resident macrophages are another potential fetal-derived cell type in adult tissues (Hoeffel and Ginhoux 2015, 2018); the FCO signature could provide a means to explore epigenetic features of the ontogeny of these cells as well.
While these results point to the potential for DNA methylation to mark developmentally regulated changes in the stem cell compartment, we recognize some limitations. The current work does not indicate whether cell intrinsic or extrinsic factors are involved in the early developmental transitions in hematopoiesis. We also have not yet examined the distribution of the FCO signature over time among different myeloid and lymphoid lineages or applied the approach to clonal single cells. As currently applied, our deconvolution approach does not capture the potential diversity of progenitors beyond what we consider a core set of methylation events that are shared by embryonic stem cells. It is conceivable that several or many adult stem cells give rise to leukocytes during adult life; our data would only indicate that most of these do not share the features of ESCs. Finally, it is of great interest that nonhematopoietic tissues also demonstrated a marked developmental age variation in the FCO signature fraction in fetal tissues. There was evidence of heterogeneity in the FCO signature fraction in brain and muscle according to fetal gestational age. This observation, which is consistent with previous studies in fetal brain (Jaffe et al. 2016), suggests that the transition observed postnatally in hematopoietic cells occurs prenatally in a tissue-dependent fashion. This observation begs the question whether the kinetics of stem cell maturation are unique to each tissue. Therefore, the FCO signature may be a tool that is useful to explore stem cell heterogeneity more broadly in human development. In conclusion, we defined and applied a DNA methylation signature common among human fetal hematopoietic progenitor cells and have shown that this signature traces the lineage of cells and informs the study of stem cell heterogeneity in humans under homeostatic conditions.
Methods
Discovery data sets
For the discovery of CpG markers, we used three publicly available data sets containing purified cell types (granulocytes: Gran, CD14+ monocytes: Mono, CD19+ B lymphocytes: Bcell, CD4+ T lymphocytes: CD4T, CD8+ T lymphocytes: CD8T, and CD56+ natural killer lymphocytes: NK cells) from peripheral blood in adults and cord blood in newborns (for details, see Supplemental Table S1, discovery data sets). In brief, discovery data sets contained whole blood and purified cell subtypes from several subjects: (1) GSE35069 (Reinius et al. 2012) contained purified cells from six adult subjects; (2) FlowSorted.CordBlood.450K (Bakulski et al. 2016) contained samples from 17 newborns; and (3) FlowSorted.CordBloodNorway.450K (Gervin et al. 2016) contained samples from 11 newborns.
Biomarker discovery: creation of a lineage-invariant and developmentally sensitive DNA methylation signature (the FCO signature)
We hypothesized that embryonic and adult hematopoietic stem cells contain CpG loci that are unique to each of these types of stem cells but that are invariant with respect to the lineage specification of their progeny. Thus, a selection strategy was undertaken in two steps: Using our discovery data sets, first we identified lineage invariant CpG sites within isolated leukocyte populations from UCB (fetal cells) and in adult whole blood (AWB), and second, among these CpG loci, we identified the subset that provided optimal discrimination between all subtypes of UCB and adult leukocytes (Fig. 1).
The aforementioned three data sets were pooled and included purified Gran, Mono, Bcell, CD4T, CD8T, and NK cells only. Data sets were harmonized to include sex, DNA methylation age (Horvath 2013; Lowe et al. 2016), and a subject indicator. Horvath's DNA methylation age was calculated using the agep function in the wateRmelon R-package (Pidsley et al. 2013). For newborns, we estimated the Knight's DNA methylation gestational age (Knight et al. 2016). The pooled data set was normalized using Funnorm (Fortin et al. 2014). Once normalized, we identified CpG loci exhibiting differential patterns of methylation between newborns and adults using two similar but distinct approaches. In the first approach, a series of linear models adjusted for sex- and sample-specific estimated DNA methylation age were fit independently to each of the J CpGs and to each cell type separately (Equation 1).
In Equation 1, represents the methylation β-value among subject i (i = 1,2,…,N), CpG j (j = 1,2,…,J), and cell type k (k = 1,2,…,K). For each of the J × K models that were fit, we tested the hypothesis that the mean methylation β-value is equivalent between fetal and adult tissues (e.g., ) and retained CpG loci exhibiting a statistically significant difference (FDR < 0.05). In the second approach, a series of linear mixed effect models adjusted for sex, sample-specific estimated DNA methylation age, and cell type (to obtain invariant loci across cell types), and including a subject-specific random intercept, were used to identify differentially methylated CpG loci between adult versus fetal tissues (Equation 2).
For each of the J fitted models, we tested the hypothesis that the mean methylation β-value is equivalent between fetal and adult tissues (e.g., H0 : β1j = 0) and retained CpG loci exhibiting a statistically significant differences (FDR < 0.05) for further analysis. While our strategy for identifying developmentally variant loci involved fitting a series of linear regression and linear mixed effects models, treating the methylation β-values as the response, we note the existence of alternative models (Du et al. 2010; Saadati and Benner 2014) that could be used as a substitute or in addition to the models considered here.
We next compared the results of the seven models (e.g., six linear models, one fit to each cell type, along with the linear mixed effects model) to identify CpG loci exhibiting statistically significant (FDR < 0.05) differences between fetal and adult tissues across all seven models (1255 CpG loci). Of those, CpG loci exhibiting inconsistent patterns of differential methylation fetal and adult tissues across any of two the seven models were filtered out. This resulted in a set of loci that exhibited consistent patterns of differential methylation across all cell types. Among those, we prioritized loci that showed absolute differences in methylation between fetal versus adult tissues greater than 0.1 across all cell types (1218 CpGs). The filtered candidate CpG list was then subject to a test for enrichment to identify biological pathways enriched with the associated genes using the MSigDB v6.0 curated database 2 using three different approaches: (1) ToppGene, which uses a classical hypergeometric distribution test (Chen et al. 2009); (2) GREAT v3.0.0 (Genomic Regions Enrichment of Annotations Tool) (McLean et al. 2010), which interrogates potential cis-regulatory regions (5000 bp upstream and 1000 bp downstream, as well as an extended region 1 Mbp of the CpG site) that are not captured using the genes associated to the CpG site; and (3) the R-package missMethyl to account for the potential microarray bias (Phipson et al. 2016). To mitigate the potential for bias, we restricted the background to consider only those genes interrogated in the Illumina HumanMethylation 450K array. We selected the pathways that overlap among the three approaches. In addition, we used ToppGene to test for enrichment of loci on the Progenitor Cell Biology Consortium database (Chen et al. 2009; Salomonis et al. 2016).
The next step involved reducing the candidate CpGs to a short instrumental list that provided optimal discrimination between adult and fetal tissues but minimal residual cell-specific effects. For this step, a confirmatory PC analysis was used to quantitatively compare differences in the components of the candidate list. The first PC should account for differences between adult and fetal, whereas subsequent PCs should account for inter-subject variability, residual cell type confounding, and other sources of technical noise. Indeed, in our analysis we observed that the first PC associated strongly with origin of the cell type (i.e., fetal versus adult), whereas the second PC indicated a small, but noticeable cell-specific effect (Supplemental Fig. S2). To identify loci with residual cell-specific effects, we computed the geometric angle between the x-axis (direction of the first PC) and the vector formed by loadings for PC1 (x) and PC2 (y) for each CpG. The geometric angle calculation uses x and y as the legs of the triangle, and then using the inverse trigonometric function arctangent (atan), the geometric angle is obtained as degrees = atan(x/y) × (180/π) with a known distribution between −90 and +90. CpGs with angles close to 0° represent those predominantly influencing PC1 (i.e., fetal versus adult differences), whereas angles away from 0° are indicative of contribution to PC2 (i.e., cell-specific effects). To minimize cell-specific signal among CpGs, we selected only those CpGs whose angle was close to 0° to form our FCO signature. Using the derived FCO signature, we proceeded to deconvolute the fetal versus adult cell fraction using constrained projection quadratic programming (CP/QP) proposed by Houseman (Houseman et al. 2012), substituting the default reference library with the library identified based on the above analysis (Supplemental File S1). For analyses using GEO data sets, no additional normalization steps were employed to the already preprocessed β-values. β-value distributions were, however, inspected for irregularities, and where relevant, k nearest neighbors was performed for missing value imputation.
Replication
We used purified Gran, Mono, Bcell, CD4T, CD8T, and NK from three replication data sets: (1) GSE68456 (de Goede et al. 2015) included samples from cord blood of 12 newborns; (2) GSE30870 (Heyn et al. 2012) contains purified CD4T of one adult and one newborn; and (3) GSE59065 (Tserel et al. 2015) included 99 CD4T and 100 CD8T samples.
AUROC, stability of the FCO estimations, and synthetic mixture statistical validation
We used five independent data sets to evaluate the classification AUROC of the FCO signature and the stability of the FCO estimations (Supplemental Methods S1): GSE80310 (Knight et al. 2016), GSE74738 (Hanna et al. 2016), GSE54399 (Montoya-Williams et al. 2017), GSE79056 (Knight et al. 2016), and GSE62924 (Rojas et al. 2015). To simulate synthetic mixtures, we used two additional DNA methylation data sets: GSE66459, a fetal UCB (n = 22) data set (Fernando et al. 2015), and GSE43976, restricting to those samples of adult peripheral blood (n = 52) data set (see Supplemental Methods S2; Marabita et al. 2013).
Embryonic stem cells (ESC), induced pluripotent stem cells (iPSC), and hematopoietic cell progenitors
To explore the ontogeny of the stem cell methylation signature, we examined several databases of arrayed hematopoietic progenitors: (1) GSE31848 (Nazor et al. 2012), undifferentiated ESC (n = 19) and iPSC (n = 29); (2) GSE40799 (Weidner et al. 2013), three fresh CD34+ stem/progenitor cells from fresh UCB; (3) GSE56491 (Lessard et al. 2015), 12 CD34+ cells from fetal liver and 12 from adult bone marrow, which were differentiated ex vivo to erythroid cells; (4) GSE50797 (Rönnerblad et al. 2014), three adult bone marrow samples were used to isolate two different CD34+ myeloid progenitors (common myeloid progenitors [CMP] and granulocyte/macrophage progenitors [GMP]) and two different CD34− immature myeloid progenitors (promyelocyte/myelocyte [PMC] and metamyelocyte/band-myelocyte [PMN]); and finally, (5) GSE63409 (Jung et al. 2015), five adult bone marrow samples including six different isolated CD34+ progenitors (CD34+adult stem cells, multipotent progenitors [MPP], lymphoid primed multipotent progenitors [L-MPP], CMP, GMP, megakaryocyte-erythroid progenitors [MEP]) (see Supplemental Table S1.
Fetal/embryonic and adult somatic tissue
We applied the FCO algorithm to data from nonhematopoietic tissues to explore the specificity of the DNA methylation signature among tissues derived from diverse embryonic layers and progenitors. For this purpose, we included six additional data sets restricted to those organs with at least one adult (necropsies) and one fetal (abortuses) sample (see Supplemental Table S1): (1) GSE61279 (Bonder et al. 2014), liver samples (fetuses n = 14, adults n = 96); (2) GSE31848 (Nazor et al. 2012), different organ biopsies (fetal n = 28, adults n = 13); (3) GSE56515 (Slieker et al. 2015), different organ biopsies (fetal n = 26); (4) GSE48472 (Slieker et al. 2013), different organ biopsies (adults n = 18); (5) GSE58885 (Spiers et al. 2015), brain samples (fetal/embryonic n = 179); and (6) GSE41826 (Guintivano et al. 2013), frontal brain neurons (adult n = 29).
Functional annotation of selection regions
We explored the regulatory features of candidate FCO loci using ENCODE (Rosenbloom et al. 2013; Sloan et al. 2016) and annotated the functional features of the 27 candidates using the human ESCs and human umbilical vein endothelial cell feature available therein.
Age-dependent changes in the FCO methylation signature in human populations
The following step took advantage of several data sets with subjects of different ages. Five data sets were selected for this purpose: (1) GSE83334 (Urdinguio et al. 2016), 15 paired samples (cord blood and 5-yr-old whole blood cells [WBC]); (2) GSE62219 (Acevedo et al. 2015), WBC samples from 10 children; (3) GSE36054 (Alisch et al. 2012), 176 WBC of children; and (4) GSE40279 (Hannum et al. 2013), 656 adult WBC samples. Finally, WBC and peripheral blood mononuclear cells samples available from the discovery and replication data sets were pooled (see Supplemental Table S1).
Sensitivity analyses
As per the method of Morin et al. (2017), we evaluated whether any of the UCB samples used in this manuscript showed any evidence of maternal blood contamination. We used the 10 CpGs described in the manuscript to cluster the samples. UCB samples showing evident hypermethylation and with inconsistent DNA methylation age (>3.6 yr margin of error reported by Horvath 2013) were excluded from the analyses (for details, see Supplemental Methods S3).
Supplementary Material
Acknowledgments
Work was supported by the National Institutes of Health with grants R01CA207360, R01CA52689, and P50CA097257 to J.K.W., R01CA207110 to K.T.K., and R01DE022772 and R01CA216265 to B.C.C. Support to J.K.W. was also provided by the Loglio Collective and the Robert Magnin Newman Endowed Chair in Neuro-oncology. D.C.K. was supported by the Kansas IDeA Network of Biomedical Research Excellence (K-INBRE) Bioinformatics Core, supported in part by the National Institute of General Medical Science award P20GM103418.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.233213.117.
Freely available online through the Genome Research Open Access option.
References
- Accomando WP, Wiencke JK, Houseman EA, Nelson HH, Kelsey KT. 2014. Quantitative reconstruction of leukocyte subsets using DNA methylation. Genome Biol 15: R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acevedo N, Reinius LE, Vitezic M, Fortino V, Söderhäll C, Honkanen H, Veijola R, Simell O, Toppari J, Ilonen J, et al. 2015. Age-associated DNA methylation changes in immune genes, histone modifiers and chromatin remodeling factors within 5 years after birth in human blood leukocytes. Clin Epigenetics 7: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alisch RS, Barwick BG, Chopra P, Myrick LK, Satten GA, Conneely KN, Warren ST. 2012. Age-associated DNA methylation in pediatric populations. Genome Res 22: 623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakulski KM, Feinberg JI, Andrews SV, Yang J, Brown S, L. McKenney S, Witter F, Walston J, Feinberg AP, Fallin MD. 2016. DNA methylation of cord blood cell types: applications for mixed cell birth studies. Epigenetics 11: 354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerman I, Bock C, Garrison BS, Smith ZD, Gu H, Meissner A, Rossi DJ. 2013. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 12: 413–425. [DOI] [PubMed] [Google Scholar]
- Bonder MJ, Kasela S, Kals M, Tamm R, Lokk K, Barragan I, Buurman WA, Deelen P, Greve J, Ivanov M, et al. 2014. Genetic and epigenetic regulation of gene expression in fetal and adult human livers. BMC Genomics 15: 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch K, Klapproth K, Barile M, Flossdorf M, Holland-Letz T, Schlenner SM, Reth M, Höfer T, Rodewald H-R. 2015. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 518: 542–546. [DOI] [PubMed] [Google Scholar]
- Chen J, Bardes EE, Aronow BJ, Jegga AG. 2009. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 37: 305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copley MR, Eaves CJ. 2013. Developmental changes in hematopoietic stem cell properties. Exp Mol Med 45: e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copley MR, Babovic S, Benz C, Knapp DJHF, Beer PA, Kent DG, Wohrer S, Treloar DQ, Day C, Rowe K, et al. 2013. The Lin28b-let-7-Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat Cell Biol 15: 916–925. [DOI] [PubMed] [Google Scholar]
- de Goede OM, Razzaghian HR, Price EM, Jones MJ, Kobor MS, Robinson WP, Lavoie PM. 2015. Nucleated red blood cells impact DNA methylation and expression analyses of cord blood hematopoietic cells. Clin Epigenetics 7: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descatoire M, Weill J-C, Reynaud C-A, Weller S. 2011. A human equivalent of mouse B-1 cells? J Exp Med 208: 2563–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P, Zhang X, Huang C-C, Jafari N, Kibbe WA, Hou L, Lin SM. 2010. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 11: 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykstra B, de Haan G. 2008. Hematopoietic stem cell aging and self-renewal. Cell Tissue Res 331: 91–101. [DOI] [PubMed] [Google Scholar]
- Eaves CJ. 2015. Hematopoietic stem cells: concepts, definitions, and the new reality. Blood 125: 2605–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlik M, Halbritter F, Müller F, Choudry FA, Ebert P, Klughammer J, Farrow S, Santoro A, Ciaurro V, Mathur A, et al. 2016. DNA methylation dynamics of human hematopoietic stem cell differentiation. Cell Stem Cell 19: 808–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando F, Keijser R, Henneman P, van der Kevie-Kersemaekers A-MF, Mannens MM, van der Post JA, Afink GB, Ris-Stalpers C. 2015. The idiopathic preterm delivery methylation profile in umbilical cord blood DNA. BMC Genomics 16: 736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin J, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, Greenwood C, Hansen KD. 2014. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol 15: 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervin K, Page CM, Aass HCD, Jansen MA, Fjeldstad HE, Andreassen BK, Duijts L, van Meurs JB, van Zelm MC, Jaddoe VW, et al. 2016. Cell type specific DNA methylation in cord blood: a 450K-reference data set and cell count-based validation of estimated cell type composition. Epigenetics 11: 690– 698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosn EEB, Yang Y. 2015. Hematopoietic stem cell-independent B-1a lineage. Ann NY Acad Sci 1362: 23–38. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Cooper C, Thornburg KL. 2008. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med 359: 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin DO, Holodick NE, Rothstein TL. 2011. Human B1 cells are CD3–: a reply to “A human equivalent of mouse B-1 cells?” and “The nature of circulating CD27+CD43+ B cells”. J Exp Med 208: 2566–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guintivano J, Aryee MJ, Kaminsky ZA. 2013. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics 8: 290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna CW, Peñaherrera MS, Saadeh H, Andrews S, McFadden DE, Kelsey G, Robinson WP. 2016. Pervasive polymorphic imprinted methylation in the human placenta. Genome Res 26: 756–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, et al. 2013. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 49: 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy RR, Hayakawa K. 2015. Perspectives on fetal derived CD5+ B1 B cells. Eur J Immunol 45: 2978–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Kim I, Lim MS, Morrison SJ. 2011. Sox17 expression confers self-renewal potential and fetal stem cell characteristics upon adult hematopoietic progenitors. Genes Dev 25: 1613–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzenberg LA. 2015. Layered evolution in the immune system: a view from history. Ann N Y Acad Sci 1362: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn H, Li N, Ferreira HHJ, Moran S, Pisano DG, Gomez A, Diez J. 2012. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci 109: 10522–10527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffel G, Ginhoux F. 2015. Ontogeny of tissue-resident macrophages. Front Immunol 6: 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffel G, Ginhoux F. 2018. Fetal monocytes and the origins of tissue-resident macrophages. Cell Immunol 10.1016/j.cellimm.2018.01.001. [DOI] [PubMed] [Google Scholar]
- Horvath S. 2013. DNA methylation age of human tissues and cell types. Genome Biol 14: R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. 2012. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AE, Gao Y, Deep-Soboslay A, Tao R, Hyde TM, Weinberger DR, Kleinman JE. 2016. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci 19: 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. 2014. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371: 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Natarajan P, Ebert BL. 2017. Clonal hematopoiesis and atherosclerosis. N Engl J Med 377: 1400–1402. [DOI] [PubMed] [Google Scholar]
- Ji H, Ehrlich LIR, Seita J, Murakami P, Doi A, Lindau P, Lee H, Aryee MJ, Irizarry RA, Kim K, et al. 2010. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 467: 338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung N, Dai B, Gentles AJ, Majeti R, Feinberg AP. 2015. An LSC epigenetic signature is largely mutation independent and implicates the HOXA cluster in AML pathogenesis. Nat Commun 6: 8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor AB, Herzenberg LA. 1993. Origin of murine B cell lineages. Annu Rev Immunol 11: 501–538. [DOI] [PubMed] [Google Scholar]
- Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LIR, et al. 2010. Epigenetic memory in induced pluripotent stem cells. Nature 467: 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight AK, Craig JM, Theda C, Bækvad-Hansen M, Bybjerg-Grauholm J, Hansen CS, Hollegaard MV, Hougaard DM, Mortensen PB, Weinsheimer SM, et al. 2016. An epigenetic clock for gestational age at birth based on blood methylation data. Genome Biol 17: 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koestler DC, Jones MJ, Usset J, Christensen BC, Butler RA, Kobor MS, Wiencke JK, Kelsey KT. 2016. Improving cell mixture deconvolution by identifying optimal DNA methylation libraries (IDOL). BMC Bioinformatics 17: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Hore TA, Reik W. 2014. Reprogramming the methylome: erasing memory and creating diversity. Cell Stem Cell 14: 710–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Han S, Kwon CS, Lee D. 2016. Biogenesis and regulation of the let-7 miRNAs and their functional implications. Protein Cell 7: 100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard S, Beaudoin M, Benkirane K, Lettre G. 2015. Comparison of DNA methylation profiles in human fetal and adult red blood cell progenitors. Genome Med 7: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesurf R, Cotto KC, Wang G, Griffith M, Kasaian K, Jones SJ, Montgomery SB, Griffith OL; Open Regulatory Annotation Consortium. 2016. ORegAnno 3.0: a community-driven resource for curated regulatory annotation. Nucleic Acids Res 44: D126–D132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. 2011. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27: 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe D, Horvath S, Raj K. 2016. Epigenetic clock analyses of cellular senescence and ageing. Oncotarget 7: 8524–8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marabita F, Almgren M, Lindholm ME, Ruhrmann S, Fagerström-Billai F, Jagodic M, Sundberg CJ, Ekström TJ, Teschendorff AE, Tegnér J, et al. 2013. An evaluation of analysis pipelines for DNA methylation profiling using the Illumina HumanMethylation450 BeadChip platform. Epigenetics 8: 333–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Findlay GM, Gagnon JA, Horwitz MS, Schier AF, Shendure J. 2016. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 353: aaf7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melton C, Judson RL, Blelloch R. 2010. Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature 463: 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki-Kashio M, Mishima Y, Miyagi S, Negishi M, Saraya A, Konuma T, Shinga J, Koseki H, Iwama A. 2011. Dependency on the polycomb gene Ezh2 distinguishes fetal from adult hematopoietic stem cells. Blood 118: 6553–6561. [DOI] [PubMed] [Google Scholar]
- Montoya-Williams D, Quinlan J, Clukay C, Rodney NC, Kertes DA, Mulligan CJ. 2017. Associations between maternal prenatal stress, methylation changes in IGF1 and IGF2, and birth weight. J Dev Orig Health Dis 9: 215–222. [DOI] [PubMed] [Google Scholar]
- Morin AM, Gatev E, McEwen LM, MacIsaac JL, Lin DTS, Koen N, Czamara D, Räikkönen K, Zar HJ, Koenen K, et al. 2017. Maternal blood contamination of collected cord blood can be identified using DNA methylation at three CpGs. Clin Epigenetics 9: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Sieburg CE, Sieburg HB, Bernitz JM, Cattarossi G. 2012. Stem cell heterogeneity: implications for aging and regenerative medicine. Blood 119: 3900–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazor KL, Altun G, Lynch C, Tran H, Harness JV, Slavin I, Garitaonandia I, Müller FJ, Wang YC, Boscolo FS, et al. 2012. Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell 10: 620–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi M, Eguchi-Ishimae M, Wu Z, Gao W, Iwabuki H, Kawakami S, Tauchi H, Inukai T, Sugita K, Hamasaki Y, et al. 2013. Suppression of the let-7b microRNA pathway by DNA hypermethylation in infant acute lymphoblastic leukemia with MLL gene rearrangements. Leukemia 27: 389–397. [DOI] [PubMed] [Google Scholar]
- Orkin SH, Zon LI. 2008. Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132: 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima M, Hasegawa N, Mochizuki-Kashio M, Muto T, Miyagi S, Koide S, Yabata S, Wendt GR, Saraya A, Wang C, et al. 2016. Ezh2 regulates the Lin28/let-7 pathway to restrict activation of fetal gene signature in adult hematopoietic stem cells. Exp Hematol 44: 282–296.e3. [DOI] [PubMed] [Google Scholar]
- Phipson B, Maksimovic J, Oshlack A. 2016. missMethyl: an R package for analyzing data from Illumina's HumanMethylation450 platform. Bioinformatics 32: 286–288. [DOI] [PubMed] [Google Scholar]
- Pidsley R, Y Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC. 2013. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics 14: 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskounova E, Viswanathan SR, Janas M, LaPierre RJ, Daley GQ, Sliz P, Gregory RI. 2008. Determinants of microRNA processing inhibition by the developmentally regulated RNA-binding protein Lin28. J Biol Chem 283: 21310–21314. [DOI] [PubMed] [Google Scholar]
- Piskounova E, Polytarchou C, Thornton JE, LaPierre RJ, Pothoulakis C, Hagan JP, Iliopoulos D, Gregory RI. 2011. Lin28A and Lin28B inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell 147: 1066–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratliff ML, Templeton TD, Ward JM, Webb CF. 2014. The bright side of hematopoiesis: regulatory roles of ARID3a/bright in human and mouse hematopoiesis. Front Immunol 5: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén SE, Greco D, Söderhäll C, Scheynius A, Kere J. 2012. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One 7: e41361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas D, Rager JE, Smeester L, Bailey KA, Drobná Z, Rubio-Andrade M, Stýblo M, García-Vargas G, Fry RC. 2015. Prenatal arsenic exposure and the epigenome: identifying sites of 5-methylcytosine alterations that predict functional changes in gene expression in newborn cord blood and subsequent birth outcomes. Toxicol Sci 143: 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rönnerblad M, Andersson R, Olofsson T, Douagi I, Karimi M, Lehmann S, Hoof I, de Hoon M, Itoh M, Nagao-Sato S, et al. 2014. Analysis of the DNA methylome and transcriptome in granulopoiesis reveals timed changes and dynamic enhancer methylation. Blood 123: e79–e89. [DOI] [PubMed] [Google Scholar]
- Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, et al. 2013. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res 41: D56–D63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, Jamieson CHM, Weissman IL. 2008. Stems cells and the pathways to aging and cancer. Cell 132: 681–696. [DOI] [PubMed] [Google Scholar]
- Rowe RG, Wang LD, Coma S, Han A, Mathieu R, Pearson DS, Ross S, Sousa P, Nguyen PT, Rodriguez A, et al. 2016. Developmental regulation of myeloerythroid progenitor function by the Lin28b-let-7-Hmga2 axis. J Exp Med 213: 1497–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak A, Fuchs H, Smirnova L, Brandt C, Pohl EE, Nitsch R, Wulczyn FG. 2008. A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nat Cell Biol 10: 987–993. [DOI] [PubMed] [Google Scholar]
- Saadati M, Benner A. 2014. Statistical challenges of high-dimensional methylation data. Stat Med 33: 5347–5357. [DOI] [PubMed] [Google Scholar]
- Salas LA, Koestler DC, Butler RA, Hansen HM, Wiencke JK, Kelsey KT, Christensen BC. 2018. An optimized library for reference-based deconvolution of whole-blood biospecimens assayed using the Illumina HumanMethylationEPIC BeadArray. Genome Biol 19: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomonis N, Dexheimer PJ, Omberg L, Schroll R, Bush S, Huo J, Schriml L, Ho Sui S, Keddache M, Mayhew C, et al. 2016. Integrated genomic analysis of diverse induced pluripotent stem cells from the Progenitor Cell Biology Consortium. Stem Cell Reports 7: 110–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai CM, Babovic S, Upadhaya S, Knapp DJHF, Lavin Y, Lau CM, Goloborodko A, Feng J, Fujisaki J, Ding L, et al. 2016. Hematopoietic stem cells are the major source of multilineage hematopoiesis in adult animals. Immunity 45: 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Säwén P, Lang S, Mandal P, Rossi DJ, Soneji S, Bryder D. 2016. Mitotic history reveals distinct stem cell populations and their contributions to hematopoiesis. Cell Rep 14: 2809–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seay HR, Putnam AL, Cserny J, Posgai AL, Rosenau EH, Wingard JR, Girard KF, Kraus M, Lares AP, Brown HL, et al. 2017. Expansion of human tregs from cryopreserved umbilical cord blood for GMP-compliant autologous adoptive cell transfer therapy. Mol Ther Methods Clin Dev 4: 178–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer ZS, Yong J, Tischler J, Hackett JA, Altinok A, Surani MA, Cai L, Elowitz MB. 2014. Dynamic heterogeneity and DNA methylation in embryonic stem cells. Mol Cell 55: 319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slieker RC, Bos SD, Goeman JJ, Bovée JV, Talens RP, van der Breggen R, Suchiman HED, Lameijer E-W, Putter H, van den Akker EB, et al. 2013. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slieker RC, Roost MS, van Iperen L, Suchiman HED, Tobi EW, Carlotti F, de Koning EJP, Slagboom PE, Heijmans BT, Chuva de Sousa Lopes SM. 2015. DNA methylation landscapes of human fetal development. PLoS Genet 11: e1005583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan CA, Chan ET, Davidson JM, Malladi VS, Strattan JS, Hitz BC, Gabdank I, Narayanan AK, Ho M, Lee BT, et al. 2016. ENCODE data at the ENCODE portal. Nucleic Acids Res 44: D726–D732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiers H, Hannon E, Schalkwyk LC, Smith R, Wong CCY, O'Donovan MC, Bray NJ, Mill J. 2015. Methylomic trajectories across human fetal brain development. Genome Res 25: 338–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, Wang H, Le T, Faull KF, Chen R, et al. 2014a. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 14: 673–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho Y-J, Klein A, Hofmann O, Camargo FD. 2014b. Clonal dynamics of native haematopoiesis. Nature 514: 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Breeze CE, Zheng SC, Beck S. 2017. A comparison of reference-based algorithms for correcting cell-type heterogeneity in Epigenome-Wide Association Studies. BMC Bioinformatics 18: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus AJ, Gallimore RM, Salas LA, Christensen BC. 2017. Cell-type deconvolution from DNA methylation: a review of recent applications. Hum Mol Genet 26: R216–R224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tserel L, Kolde R, Limbach M, Tretyakov K, Kasela S, Kisand K, Saare M, Vilo J, Metspalu A, Milani L, et al. 2015. Age-related profiling of DNA methylation in CD8+ T cells reveals changes in immune response and transcriptional regulator genes. Sci Rep 5: 13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urdinguio RG, Torró MI, Bayón GF, Álvarez-Pitti J, Fernández AF, Redon P, Fraga MF, Lurbe E. 2016. Longitudinal study of DNA methylation during the first 5 years of life. J Transl Med 14: 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner CI, Walenda T, Lin Q, Wölfler MM, Denecke B, Costa IG, Zenke M, Wagner W. 2013. Hematopoietic stem and progenitor cells acquire distinct DNA-hypermethylation during in vitro culture. Sci Rep 3: 3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, Orkin SH. 2014. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell 14: 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Nguyen CK, Liu X, Kanellopoulou C, Muljo SA. 2012. Lin28b reprograms adult bone marrow hematopoietic progenitors to mediate fetal-like lymphopoiesis. Science 335: 1195–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Yao B, Shin J, Lin L, Kim N, Song Q, Liu S, Su Y, Guo JU, Huang L, et al. 2016. Lin28A binds active promoters and recruits Tet1 to regulate gene expression. Mol Cell 61: 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.