

Abstract
Akt is a pro‐survival kinase frequently activated in human cancers and is associated with more aggressive tumors that resist therapy. Here, we connect Akt pathway activation to reduced sensitivity to chemotherapy via Akt phosphorylation of Bax at residue S184, one of the pro‐apoptotic Bcl‐2 family proteins required for cells to undergo apoptosis. We show that phosphorylation by Akt converts the pro‐apoptotic protein Bax into an anti‐apoptotic protein. Mechanistically, we show that phosphorylation (i) enables Bax binding to pro‐apoptotic BH3 proteins in solution, and (ii) prevents Bax inserting into mitochondria. Together, these alterations promote resistance to apoptotic stimuli by sequestering pro‐apoptotic activator BH3 proteins. Bax phosphorylation correlates with cellular resistance to BH3 mimetics in primary ovarian cancer cells. Further, analysis of the TCGA database reveals that 98% of cancer patients with increased BAX levels also have an upregulated Akt pathway, compared to 47% of patients with unchanged or decreased BAX levels. These results suggest that in patients, increased phosphorylated anti‐apoptotic Bax promotes resistance of cancer cells to inherent and drug‐induced apoptosis.
Keywords: Akt, Bax, Bcl‐2 family proteins, cancer, drug resistance
Subject Categories: Autophagy & Cell Death, Cancer, Signal Transduction
Introduction
Activation of the mitochondrial apoptosis pathway in cancer cells serves as an important route of cell death following treatment with chemotherapeutic agents. Alteration of this pathway can cause resistance to therapy in cancer cells. A decisive step for commitment to apoptosis is mitochondrial outer membrane permeabilization (MOMP) by the pro‐apoptotic Bax and/or Bak proteins that release intermembrane space proteins including cytochrome c into cytosol 1, 2. Membrane permeabilization by Bax and Bak is provoked by activator proteins including the BH3 proteins Bim and Bid. Pro‐survival Bcl‐2 proteins (Bcl‐2, Bcl‐XL, Mcl‐1, Bfl‐1, and Bcl‐W) inhibit MOMP by sequestering either activator BH3 proteins or Bax and Bak 3, 4. Other so‐called sensitizer BH3 proteins, including Bad, Noxa, and Bik, cannot activate Bax or Bak, but rather exert a pro‐death function by competing for the BH3 binding sites of pro‐survival proteins 2, 5. Differences in the affinities of the interactions, expression levels, and post‐translational modifications of these proteins together determine the fate of the cell.
Measurement of MOMP upon incubating BH3 domain‐derived peptides with mitochondria and identifying differential response patterns was successfully translated into an assay called BH3 profiling 6, 7. By interpreting the pattern of mitochondrial sensitivity to BH3 peptides of different affinities for anti‐apoptotic proteins, BH3 profiling can be used to identify dependence on individual anti‐apoptotic Bcl‐2 proteins for survival and sensitivity to inhibitors. Certain BH3 domain peptides, including those from Bid and Bim, interact with all known anti‐apoptotic proteins. Mitochondrial sensitivity to these peptides can be interpreted as a measure of how close a cell is to the threshold of apoptosis, or how “primed” a cell is for death 6, 8. The degree of priming predicts how sensitive the cell will be to toxic insults, and correlates with clinical response to chemotherapy 9.
In cancer, particularly in breast cancer, upregulation of the Akt pathway is strongly associated with poor prognosis and resistance to therapy 10. PTEN (phosphatase and tensin homolog deleted on chromosome 10) functions as a lipid phosphatase to restrain Akt pathway activation by diminishing the phosphatidylinositol‐3,4,5‐biphosphate (PIP3) cellular pool through hydrolysis of 3‐phosphate on PIP3 to generate phosphatidylinositol‐4,5‐biphosphate (PIP2). PI3Ks phosphorylate PIP2 to regenerate PIP3 which promotes Akt recruitment to plasma membrane through binding its pleckstrin‐homology (PH) domain. Following recruitment to the plasma membrane by PIP3, Akt is phosphorylated by PDK1 at T308 and by mTORC2 at S473 which leads to its activation 11. Hence, inactivation or loss of PTEN results in increased accumulation of PIP3 and constitutively active Akt signaling which promotes cell growth and survival. The Akt pathway regulates fundamental processes in cells, including survival, cell cycle progression, and metabolism. Upregulation of the Akt signaling pathway is commonly detected in a wide spectrum of human cancers. Several mechanisms including genomic amplification of Akt or growth factor receptors, PTEN deletion or mutations, or activating mutations in pathway genes can activate Akt in cancer cells.
Importantly, Akt blocks pro‐death signaling upstream of MOMP 12. However, it is still unclear how pro‐survival Akt signaling makes the critical connection to the Bcl‐2 family that controls the mitochondrial apoptosis pathway. Some suggest an indirect effect, for instance, via transcriptional control of pro‐apoptotic Bcl‐2 family proteins via the FOXO family of transcriptional regulators 11. Akt could also play a more direct role since it can phosphorylate the pro‐apoptotic BH3 protein Bad. However, Bad is dispensable for apoptosis induced by several mechanisms 13, 14, suggesting that a more central Bcl‐2 family protein such as Bax might also be controlled by AKT 15, 16. However, reports on the function of phosphorylated Bax are inconsistent—one suggests that S184 phosphorylation activates Bax 17, while others suggest that S184‐phosphorylated Bax is inhibited 15, 16, 18, 19.
Here, we show that Akt directly phosphorylates Bax and can localize to mitochondria. Unexpectedly, phosphorylation switches the function of Bax from pro‐ to anti‐apoptotic, thereby impeding mitochondrial priming for apoptosis. Mechanistically, we show (i) that phosphorylation of Bax blocks its insertion into membranes upstream of the oligomerization essential for its pro‐apoptotic membrane permeabilization function, and (ii) that after phosphorylation, Bax acts as a dominant negative by binding to and sequestering activator BH3 proteins thereby inhibiting BH3‐protein‐mediated apoptosis. Consistent with a role for this switch in Bax function in human disease, we find that in cancer patients with elevated Bax, Bax expression is positively associated with an increase in expression of genes in the Akt pathway. Our data suggest that in cancers with upregulated Akt pathway signaling, one means of inhibiting apoptosis is to select for increased levels of Bax which is then phosphorylated switching on its anti‐apoptotic function. Given the established role of Bax as a critical effector of MOMP downstream of many pro‐apoptotic signals, phosphorylation of Bax is likely a key mediator of the anti‐apoptotic effect exerted by upregulated Akt in cancer.
Results
A post‐translational modification alters mitochondrial priming in ABT‐737‐resistant cell lines
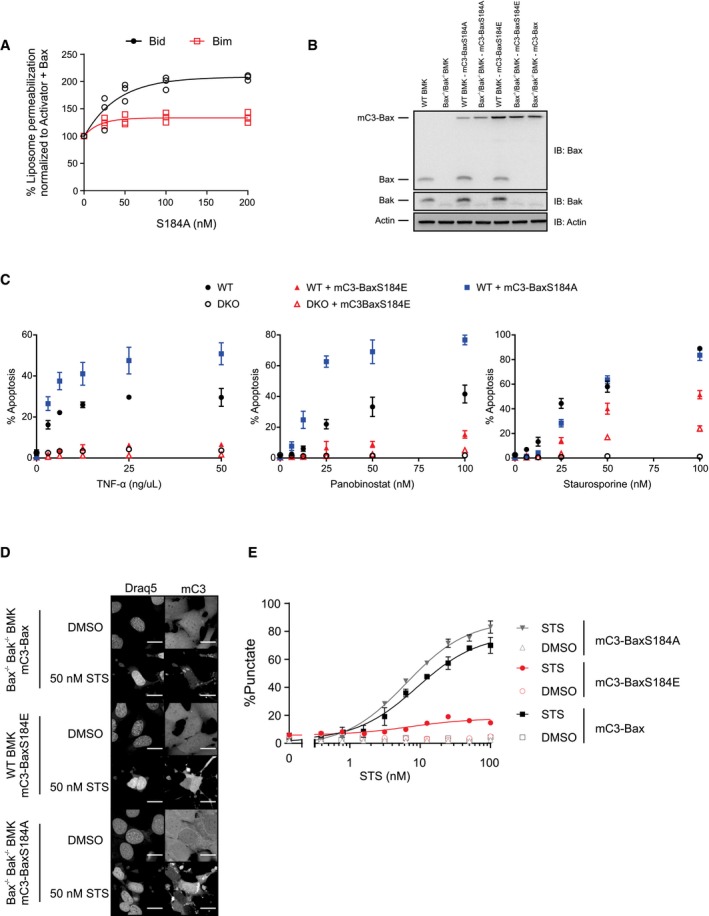
BH3 profiling with Bad can predict cellular sensitivity to Bcl‐2 antagonism by ABT‐737 6, 8, 20. To extend this observation, we tested this correlation in a new cancer cell line panel. As shown in Fig 1A, we found that mitochondria from MDA‐MB‐435, MDA‐MB‐468, SF539, and 768‐O cells were at least as sensitive to Bad as MCF‐7, T47‐D, or ZR‐75‐1 cell lines. Surprisingly, mitochondrial sensitivity was not predictive of cellular responses as only MDA‐MB‐435 cells were sensitive to ABT‐737 (EC50, 32 nM), and other cells were relatively insensitive to ABT‐737 with EC50 values at micromolar levels (Fig 1B).
Figure 1. A post‐translational modification alters priming of mitochondria from cancer cell lines.
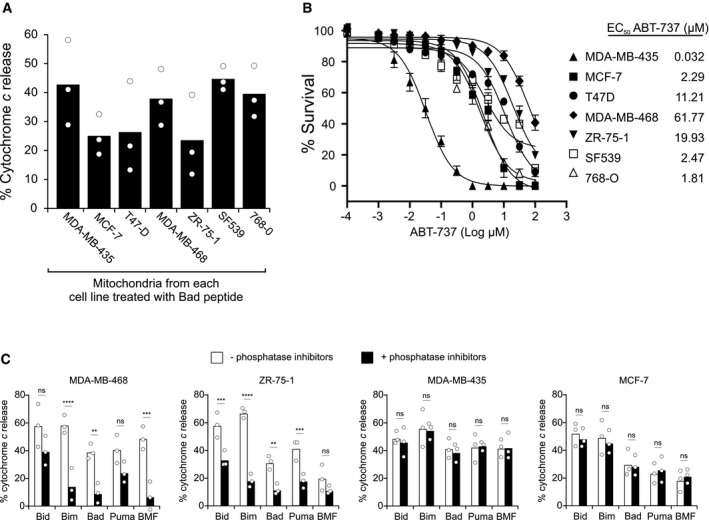
- Mitochondria isolated from the indicated cell lines were incubated with Bad peptide and MOMP measured as % cytochrome c release determined by ELISA. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment.
- Cells were treated with indicated concentrations of ABT‐737 for 48 h. ABT‐737 EC50 values of cancer cell lines were determined by using MTT assay (mean ± SEM, n = 4 experimental replicates).
- Mitochondria isolated from the indicated cell lines were incubated with the indicated BH3 peptides in the presence or absence of phosphatase inhibitor cocktail PhosSTOP in all buffers. MOMP was measured as % cytochrome c release determined by ELISA. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment. One‐way ANOVA was used followed by post hoc t‐tests with Bonferroni correction for multiple comparisons (ns = not significant). MDA‐MB‐468 (Bim ****P < 0.0001; Bad **P = 0.0059; BMF ***P = 0.0002). ZR‐75‐1 (Bid ***P = 0.0003; Bim ****P < 0.0001; Bad **P = 0.0041; Puma ***P = 0.0007).
One possible explanation for the deviation from sensitivity predicted by BH3 profiling is that post‐translational modifications (PTMs) are present in cells that are not present in our in vitro system. Since the primary post‐translational modification of Bcl‐2 family proteins is phosphorylation 21, 22, we tested whether including phosphatase inhibitors in mitochondrial isolation buffer and during BH3 profiling in experimental buffer would modulate the priming profile (Fig 1C). We found that phosphatase inhibitors significantly reduced sensitivity to BH3 peptides in ABT‐737‐resistant cells (MDA‐MB‐468 and ZR‐75‐1) but had no significant effect on BH3 peptide response in ABT‐737‐sensitive cells (MDA‐MB‐435 and MCF‐7). Taken together, our results suggest that a kinase is unpriming mitochondria selectively in the ABT‐737‐resistant cell lines. For this reason, we used a phosphatase inhibitor cocktail in all subsequent BH3 profiling experiments.
Bax is phosphorylated in ABT‐737‐resistant cell lines
Resistance to the BH3 mimetic ABT‐199 is reportedly overcome by inhibition of the Akt pathway 23. Furthermore, since Bax can be phosphorylated by Akt at residue S184 and inhibited 15, 16, we examined the phosphorylation status of Bax in ABT‐737‐resistant cells. Immunoprecipitating Bax and blotting for phosphoserine suggest Bax is phosphorylated, while blotting with a phospho‐Bax S184 antibody demonstrated that residue S184 is phosphorylated in ABT‐737‐resistant MDA‐MB‐468 cells, but not in ABT‐737‐sensitive MDA‐MB‐435 cells (Figs 2A and EV1A).
Figure 2. Bax is phosphorylated in ABT‐737‐resistant cell lines.
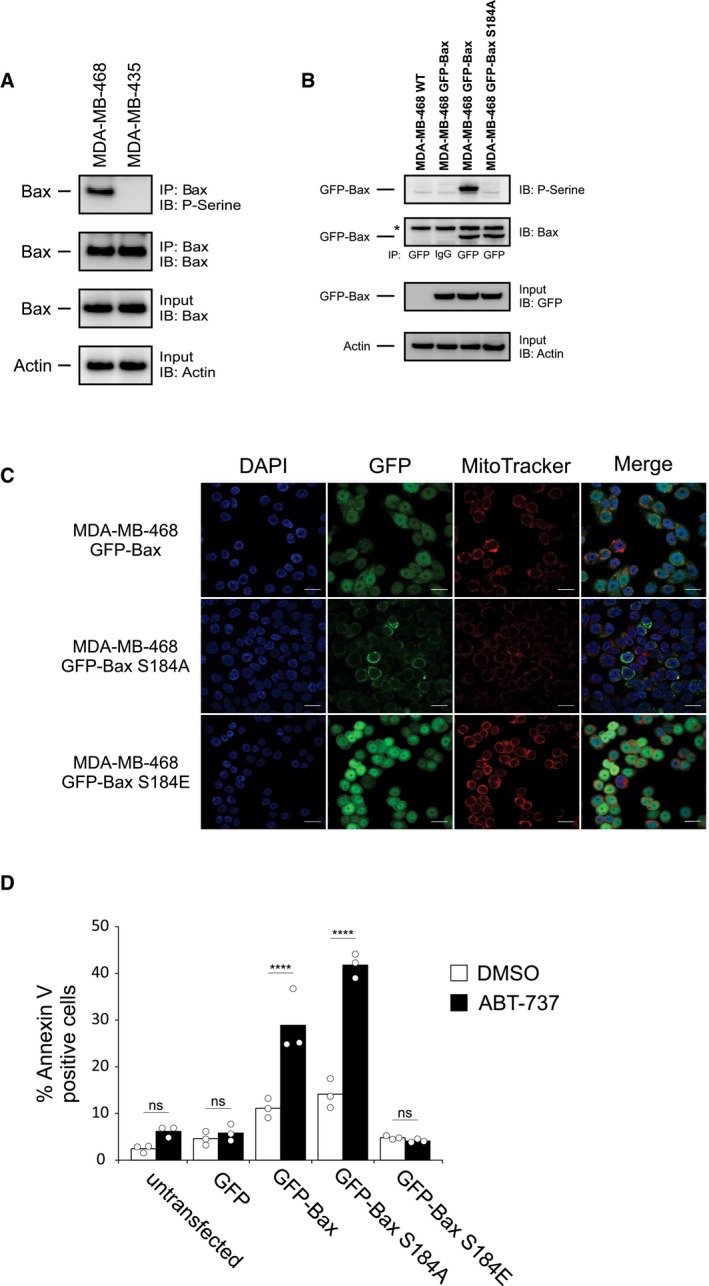
- Top two panels: Phosphorylation of Bax was evaluated using lysates from MDA‐MB‐435 and MDA‐MB‐468 cells by Western blotting with the indicated antibodies (IB) after immunoprecipitation with the indicated antibodies (IP). Lower two panels: 5–10% of the total lysates (input) were probed for Bax (Bax ∆21 antibody) and actin as expression and loading controls, respectively, by Western blotting with the indicated antibodies (IB).
- Phosphorylation of Bax was evaluated using lysates from GFP‐Bax‐ and GFP‐Bax S184A‐expressing MDA‐MB‐468 cells by blotting with the antibodies indicated at the right after precipitation with the antibodies indicated below the panels. Lysates from untransfected cells (MDA‐MB‐468 WT) and IgG IP are used as negative controls. Lower panels: 5–10% of the total lysates were probed for GFP and actin as expression and loading controls, respectively. Asterisk (*) indicates a cross‐reacting band. Bax delta‐21 antibody was used for IB of Bax.
- MDA‐MB‐468 cells were transfected with plasmids encoding GFP‐Bax, GFP‐Bax S184A, or GFP‐Bax S184E, and the localization of Bax was evaluated by confocal microscopy (upper panel). Cells were co‐stained with MitoTracker Red CMXRos (mitochondria) and DAPI (nucleus). Whole‐image Pearson's correlation coefficients between MitoTracker and GFP for the single images shown were 0.644, 0.607, and 0.206 for GFP‐Bax, GFP‐Bax S184A, and GFP‐Bax S184E, respectively (see Appendix Fig S1). Scale bars: 20 μm.
- MDA‐MB‐468 cells that were either untransfected or expressing GFP, GFP‐Bax, GFP‐Bax S184A, or GFP‐Bax S184E were treated with ABT‐737 (100 nM, 48 h), and apoptosis was evaluated by Annexin V staining followed by fluorescence‐activated cell sorting (FACS). Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment. One‐way ANOVA was used followed by post hoc t‐tests with Bonferroni correction (****P < 0.001, ns = not significant).
Figure EV1. Related to Fig 2: Bax is phosphorylated on residue S184.
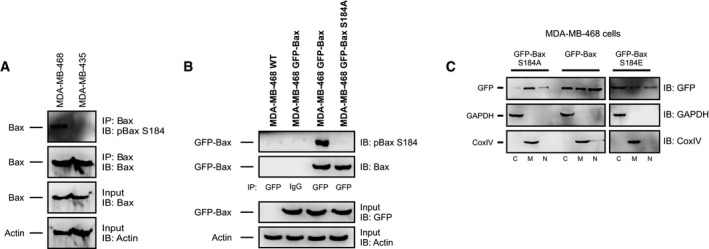
- Top two panels: Phosphorylation of Bax S184 was evaluated using lysates from MDA‐MB‐435 and MDA‐MB‐468 cells by Western blotting with the indicated antibodies (IB) after immunoprecipitation with the indicated antibodies (IP). Lower two panels: 5–10% of the total lysates (input) were probed for Bax and actin as expression and loading controls, respectively, by Western blotting with the indicated antibodies (IB).
- Top two panels: Phosphorylation of Bax S184 was evaluated using lysates from GFP‐Bax‐ and GFP‐Bax S184A‐expressing MDA‐MB‐468 cells by blotting with the antibodies indicated at the right after precipitation with the antibodies indicate below the panels. Lysates from untransfected cells (MDA‐MB‐468 WT) and IgG IP are used as negative controls. Lower panels: 5–10% of the total lysates were probed for GFP and actin as expression and loading controls, respectively.
- Localization of GFP‐Bax constructs in MDA‐MB‐468 cells. MDA‐MB‐468 cells transiently expressing GFP‐Bax S184A, GFP‐Bax, or GFP‐Bax S184E were lysed and separated into cytosolic (C), mitochondrial (M), or nuclear fractions (N) and then immunoblotted for GFP. GAPDH and CoxIV were immunoblotted for cytosolic and mitochondrial marker proteins, respectively.
Bax can be phosphorylated at multiple residues 21. To further confirm the specific phosphorylation of Bax on residue S184 in the ABT‐737‐resistant MDA‐MB‐468 cells, we created a mutant Bax (S184A) that would prevent Bax phosphorylation at residue S184. In ABT‐737‐resistant MDA‐MB‐468 cells, GFP immunoprecipitation revealed that GFP‐Bax was phosphorylated at residue S184, whereas GFP‐Bax S184A was not (Figs 2B and EV1B). These data support S184 as a primary phosphorylation site on Bax in the ABT‐737‐resistant cells and corroborate other studies that also observed Bax S184 phosphorylation by Akt 15, 16, 19, 24.
While Bax is localized both in the cytosol and at mitochondria of many cells, migration to the mitochondrion is essential for Bax pro‐apoptotic function. We evaluated the subcellular localization of GFP‐tagged WT Bax and the S184A (phosphorylation‐resistant) and S184E (phosphomimetic) mutants in ABT‐737‐resistant MDA‐MB‐468 cells. GFP‐Bax was localized diffusely throughout the cell and also co‐localized with mitochondria, while GFP‐Bax S184A showed punctate, mostly mitochondrial localization (Fig 2C). In contrast, GFP‐Bax S184E showed a diffuse cytoplasmic and nuclear pattern without mitochondrial localization (Fig 2C). These data were confirmed by subcellular fractionation, where GFP‐Bax S184A is predominantly mitochondrial, GFP‐Bax S184E is cytosolic and nuclear, and GFP‐Bax levels are similar in all fractions (Fig EV1C).
In transient transfection experiments, both GFP‐Bax and GFP‐Bax S184A were highly toxic to MDA‐MB‐468 cells. Spontaneous cell death was also increased in cells stably expressing these constructs (Fig 2D), whereas there was minimal basal cell death in GFP‐Bax S184E‐expressing cells. Consistent with these results, GFP‐Bax‐ and GFP‐Bax S184A‐expressing MDA‐MB‐468 cells were significantly more sensitive to ABT‐737 treatment. By contrast, cells expressing GFP‐Bax S184E remained resistant to ABT‐737 (Fig 2D).
Akt phosphorylates Bax in ABT‐737‐resistant cells
Since Bax can be phosphorylated at residue S184 by Akt, this pathway may mediate the resistance to ABT‐737 we observed in cancer cells (Fig 1B). To test this hypothesis, we examined the effect of small molecule inhibitors of the Akt pathway on mitochondrial priming. As predicted, treatment of ABT‐737‐resistant MDA‐MB‐468 cells (Fig 3A, top left panel) or ZR‐75‐1 cells (Fig 3A, bottom left panel) with Akt pathway inhibitors significantly enhanced priming in the majority of cases as shown by increased response to BH3 peptides (Appendix Table S1). As expected, there was no significant difference in the priming of ABT‐737‐sensitive MDA‐MB‐435 cells (Fig 3A, top right panel) or MCF‐7 cells (Fig 3A, bottom right panel) when treated with Akt pathway inhibitors (Appendix Table S1). Furthermore, Bax phosphorylation at S184 is substantially reduced when the ABT‐737‐resistant cell lines, MDA‐MB‐468 and ZR‐75‐1, are treated with AKT pathway inhibitors (Figs 3B and EV2A), directly linking Akt activation to Bax S184 phosphorylation.
Figure 3. AKT phosphorylates and inhibits Bax, preventing mitochondrial cytochrome c release in response to BH3 peptides and proteins.
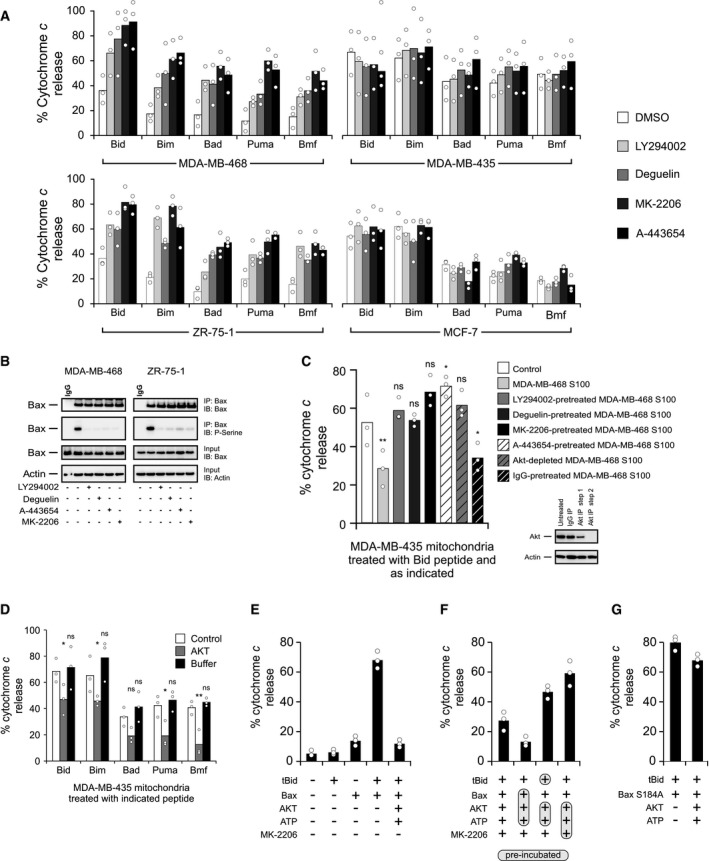
- BH3‐peptide‐resistant (left panels; MDA‐MB‐468 and ZR‐75‐1) and BH3‐peptide‐sensitive (right panels; MDA‐MB‐435 and MCF‐7) cells were treated with direct Akt inhibitors (MK‐2206 [1 μM], A‐443654 [0.4 μM]) or Akt pathway inhibitors (LY294002 [25 μM], deguelin [10 nM]) for 6 h, and BH3 profiles were analyzed. The phosphatase inhibitor cocktail PhosSTOP was used in all buffers for these experiments. Cytochrome c release was determined by ELISA. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment. Two‐way ANOVA was conducted on the influence of two independent variables (BH3 peptide, drug) on cytochrome c release of isolated mitochondria for each cell line followed by post hoc t‐tests with Bonferroni correction for multiple comparisons. Each drug treatment was statistically compared to DMSO control within each peptide treatment group. The percentage of P‐values that were significant (DMSO control to each drug) was 65% for MDA‐MB‐468, 0% for MDA‐MB‐435, 95% for ZR‐75‐1, and 0% for MDA‐MB‐435. See Appendix Table S1 for P‐values of each comparison.
- Top two panels: MDA‐MB‐468 and ZR‐75‐1 cells were treated with 1 μM MK‐2206, 0.4 μM A‐443654, 25 μM LY294002, or 10 nM deguelin where indicated. Phosphorylation of Bax was evaluated using lysates from the treated cells by Western blotting with the indicated antibodies (IB) after immunoprecipitation with the indicated antibodies (IP). Lower two panels: 5%–10% of the total lysates (input) were probed for Bax and actin as expression and loading controls, respectively, by Western blotting with the indicated antibodies (IB). Bax ∆21 antibody was used for both IP and IB of Bax.
- S100 fractions from untreated cells (MDA‐MB‐468) or from cells treated with direct Akt inhibitors (MK‐2206 [1 μM], A‐443654 [0.4 μM]) or Akt pathway inhibitors (LY294002 [25 μM], deguelin [10 nM]) were isolated. Akt was immunodepleted by sequential immunoprecipitation of untreated S100 fractions, and the efficiency of immunodepletion was tested by immunoblot analysis. IgG was used as a negative control for immunodepletion experiments (inset). Indicated S100 fractions were incubated with MDA‐MB‐435 mitochondrial preparations, and the resulting extent of priming was assessed by measuring the cytochrome c released from MDA‐MB‐435 mitochondria by Bid BH3 peptide (treatments with Bim, Bad, Puma, and Bmf are located in Fig EV2B). Cytochrome c release was determined by ELISA. The phosphatase inhibitor cocktail PhosSTOP was used in all buffers for these experiments. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment. Two‐way ANOVA was conducted on the influence of two independent variables (BH3 peptide, conditions) on cytochrome c release of isolated mitochondria followed by post hoc t‐tests with Bonferroni correction for multiple comparisons. Each condition was statistically compared to control (white bar) within each peptide treatment group. Shown here are P‐values for treatment with Bid BH3 peptide (ns = not significant). MDA‐MB‐468 S100 (**P = 0.0031), A443654‐pre‐treated MDA‐MB‐468 S100 (*P = 0.0324), and IgG‐pre‐treated MDA‐MB‐468 S100 (*P = 0.0432). See Appendix Table S2 for P‐values.
- MDA‐MB‐435 mitochondria were incubated with recombinant active Akt in kinase assay buffer containing ATP, and alteration of priming in MDA‐MB‐435 mitochondria was detected by using BH3 profiling. Cytochrome c release was determined by ELISA. The phosphatase inhibitor cocktail PhosSTOP was used in all buffers for these experiments. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment. Two‐way ANOVA was conducted on the influence of two independent variables (BH3 peptide, treatment) on cytochrome c release. BH3 peptides included Bid, Bim, Bad, Puma, and Bmf. Treatment included Akt or buffer alone. Each drug treatment was statistically compared to control (white bar) within each peptide group using t‐tests with Bonferroni correction for multiple comparisons (ns = not significant). Bid, Akt (*P = 0.0283); Bim, Akt (*P = 0.0482); Puma, Akt (*P = 0.0170); Bmf, Akt (**P = 0.0035).
- tBid (5 nM) and Bax (50 nM), with or without active Akt plus ATP, were incubated with mitochondria isolated from Bax−/− Bak−/− DKO MEFs for 1 h, and mitochondrial cytochrome c release was determined by ELISA. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment.
- tBid, Bax, Akt, ATP, and the Akt inhibitor MK‐2206, as indicated, were incubated with mitochondria isolated from Bax−/− Bak−/− DKO MEFs for 1 h, and mitochondrial cytochrome c release was determined by ELISA. The shaded and circled reagents were pre‐incubated before the addition of the other indicated reagents. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment.
- tBid (5 nM) and Bax S184A (50 nM), with or without active Akt plus ATP, were incubated with mitochondria isolated from Bax−/− Bak−/− DKO MEFs for 1 h, and mitochondrial cytochrome c release was determined by ELISA. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment.
Figure EV2. Related to Fig 3: Akt pathway inhibitors change the primed state of mitochondria, the localization of Akt, and the phosphorylation status of Bax S184.
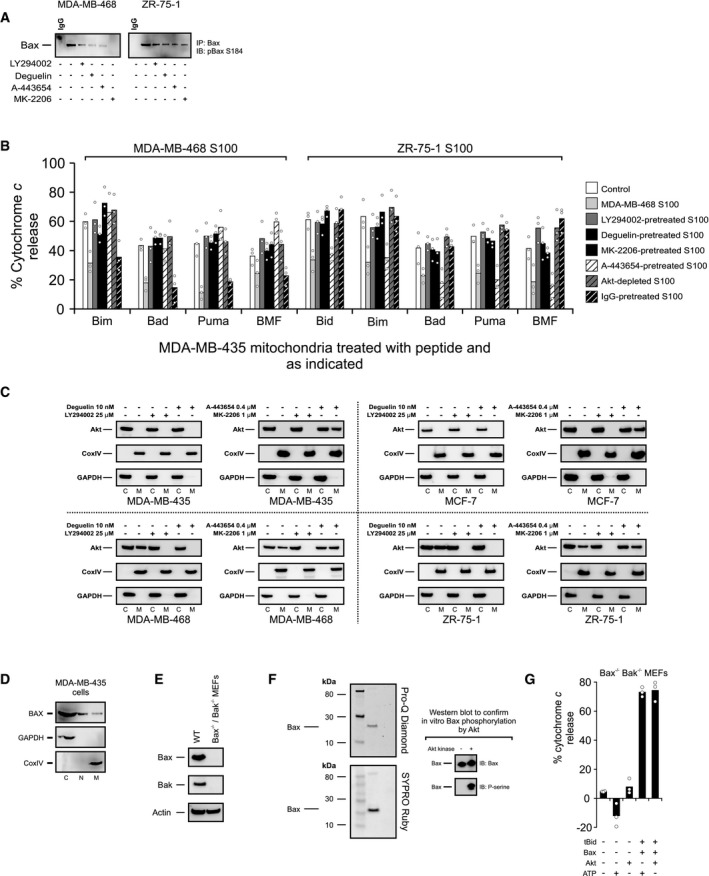
- MDA‐MB‐468 and ZR‐75‐1 cells were treated with 1 μM MK‐2206, 0.4 μM A‐443654, 25 μM LY294002, or 10 nM deguelin where indicated. Phosphorylation of Bax S184 was evaluated using lysates from the treated cells by Western blotting with the indicated antibodies (IB) after immunoprecipitation with the indicated antibodies (IP).
- S100 fractions from untreated cells (MDA‐MB‐468) or from cells treated with direct Akt inhibitors (MK‐2206 [1 μM], A‐443654 [0.4 μM]) or Akt pathway inhibitors (LY294002 [25 μM], deguelin [10 nM]) were isolated. Akt was immunodepleted by sequential immunoprecipitation in untreated S100 fractions, and the efficiency of immunodepletion was tested by immunoblot analysis. IgG was used as a negative control for immunodepletion experiments. The indicated S100 fractions were incubated with MDA‐MB‐435 mitochondrial preparations, and change in priming was assessed by using BH3 profiling. Responses to various BH3 peptides are shown. The phosphatase inhibitor cocktail PhosSTOP was used in all buffers. Cytochrome c release was determined by ELISA. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment. Two‐way ANOVA was conducted on the influence of two independent variables (BH3 peptide, treatment) on cytochrome c release of isolated mitochondria from ABT‐737‐sensitive MDA‐MB‐435 cells that were treated with the S100 fraction isolated from ABT‐737‐resistant MDA‐MB‐468 or ZR‐75‐1 cells. Each treatment was statistically compared to control within each peptide group using t‐tests with Bonferroni correction for multiple comparisons. See Appendix Table S2 for P‐values.
- ABT‐737‐resistant MDA‐MB‐468 and ZR‐75‐1 and ABT‐737‐sensitive MDA‐MB‐435 and MCF‐7 cells were treated with direct Akt inhibitors (MK‐2206 [1 μM], A‐443654 [0.4 μM]) or Akt pathway inhibitors (LY294002 [25 μM], deguelin [10 nM]), and mitochondrial and cytosolic fractions were immunoblotted for Akt. CoxIV was probed as a mitochondrial marker protein, and GAPDH was used as a cytosolic marker protein (M, mitochondrial fraction; C, cytosolic fraction).
- MDA‐MB‐435 cells were lysed and separated into cytosolic (C), mitochondrial (M), or nuclear fractions (N) and then immunoblotted for Bax. GAPDH and CoxIV were immunoblotted for cytosolic and mitochondrial marker proteins, respectively.
- Lack of expression of Bax and Bak in Bax−/− Bak−/− DKO MEFs was detected by immunoblotting with Bax and Bak antibodies. Actin was probed as a loading control. Bax ∆21 antibody was used for IB of Bax.
- Following kinase reaction with recombinant Akt, Bax was separated by SDS–PAGE. The resulting gels were stained with Pro‐Q Diamond (stains phosphoproteins) or SYPRO Ruby (stains total protein) and imaged. Additionally, phosphorylation of recombinant Bax following kinase reaction by Akt was evaluated by immunoblotting with phosphoserine antibody. Bax was blotted as a loading control.
- Akt or ATP did not alter cytochrome c release triggered by tBid/Bax treatment when used alone. Mitochondria from Bax−/− Bak−/− DKO MEFs were directly exposed to Akt or ATP or after incubated Akt or ATP with tBid and Bax as designated. Cytochrome c release was determined by ELISA. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment.
To test whether Akt directly alters mitochondrial priming, cytosolic (S100) fractions from ABT‐737‐resistant MDA‐MB‐468 cells (Figs 3C and EV2B) or ZR‐75‐1 cells (Fig EV2B) were isolated and pre‐incubated with mitochondria isolated from ABT‐737‐sensitive MDA‐MB‐435 cells before BH3 profiling experiments. This pre‐incubation led to decreased priming of MDA‐MB‐435 mitochondria, with decreased cytochrome c release in response to Bid and other BH3 peptides (Figs 3C and EV2B). Pre‐treatment of ABT‐737‐resistant cells with Akt pathway inhibitors or immunodepletion of Akt from the S100 fraction abrogated inhibition of priming by the S100 fraction (Figs 3C and EV2B). In general across all peptides, when compared to control, treatment of MDA‐MB‐435 mitochondria with the S100 fraction or IgG‐pre‐treated S100 fraction from either MDA‐MB‐468 or ZR‐75‐1 cells was associated with a significant decrease in priming, whereas most other treatments resulted in non‐significant changes in the level of priming (Appendix Table S2). Incubation of isolated MDA‐MB‐435 mitochondria with active recombinant Akt unprimes mitochondria resulting in a significant decrease in cytochrome c release mediated by BH3 peptides (Fig 3D). Thus, only when Akt was present and active, BH3‐peptide‐mediated cytochrome c release was inhibited. Additionally, Akt is localized to both the cytosol and mitochondria in ABT‐737‐resistant MDA‐MB‐468 and ZR‐75‐1 cells, but mostly in the cytosol of ABT‐737‐sensitive MDA‐MB 435 and MCF‐7 cells (Fig EV2C). Treatment with the inhibitors of Akt signaling, deguelin, LY294002, and MK‐2206, prevented Akt from localizing to mitochondria of MDA‐MB‐468 or ZR‐75‐1 cells but did not alter Akt localization in MDA‐MB 435 or MCF‐7 cells (Fig EV2C). In contrast, A‐443654 promoted Akt localization to mitochondria; however, this drug is known to promote paradoxical Akt phosphorylation, indicative of active Akt, with concomitant Akt kinase inhibition (Fig EV2C) 25, 26. A fraction of endogenous Bax was localized to the mitochondria in MDA‐MB‐435 cells where Bax was primarily cytosolic, suggesting that Akt can act on Bax in these experiments (Fig EV2D). Taken together, our results indicate a potent effect of Akt to prevent priming of mitochondria, likely through the phosphorylation of Bax.
To test Akt‐mediated Bax inhibition in a well‐defined model system, we reconstituted Bax‐regulated MOMP using mitochondria from Bax−/− Bak−/− DKO MEFs (Bax and Bak knockout was confirmed by Western blots; Fig EV2E). Incubation of Bax and Akt results in phosphorylation of Bax (Fig EV2F). Consistent with these results, combining tBid and Bax treatments triggered cytochrome c release from mitochondria that was substantially inhibited when tBid and Bax were incubated with active Akt (Fig 3E). Further, excluding ATP from the reaction eliminated the protective effect of Akt, while adding ATP alone to tBid/Bax did not have any impact on mitochondrial permeabilization (Fig EV2G). Adding the Akt inhibitor MK‐2206 to a kinase assay reaction composed of Akt, ATP, tBid, and Bax attenuated the inhibitory effect of Akt on cytochrome c release, confirming that Akt kinase activity is required for inhibition of tBid/Bax‐mediated mitochondrial permeabilization (Fig 3F). Pre‐incubating Bax, but not tBid, with active Akt plus ATP before exposing them to mitochondria decreased cytochrome c release considerably, whereas pre‐incubation of Akt, ATP, and MK‐2206 prevented Akt‐mediated inhibition of tBid/Bax‐mediated cytochrome c release (Fig 3F). Incubating Akt with Bax S184A had no effect on tBid/Bax‐mediated cytochrome c release, showing that Bax S184 phosphorylation by Akt, rather than phosphorylation of an unknown protein by Akt at the mitochondria, is responsible for inhibition of MOMP (Fig 3G).
Phosphomimetic Bax can bind cBid, but does not insert helices 5 and 9 into the membrane inhibiting pore formation
We examined how Bax phosphorylation at S184 affects Bax‐mediated membrane permeabilization using a phosphomimetic mutant of Bax (S184E). This mutant was used to ensure that all of the Bax functioned as a phosphorylated protein because in vitro Akt phosphorylation of recombinant Bax was incomplete (data not shown). We also assessed whether a phosphorylation‐resistant mutant of Bax (S184A) alters the activation mechanism of Bax. When transitioning from a soluble monomer to a membrane‐embedded oligomer, Bax undergoes sequential activation steps (Fig 4A) that can be examined using fluorescence spectroscopy with purified proteins 27, 28. The sequence begins with recruitment of Bax to the membrane by interaction with an activator BH3 protein on the membrane surface. Bax then inserts helices 5, 6, and 9 into the membrane followed by oligomerization and membrane permeabilization (Fig 4A).
Figure 4. Phosphomimetic Bax can bind cBid, but cannot insert helices 5 and 9 into the membrane abrogating pore formation.
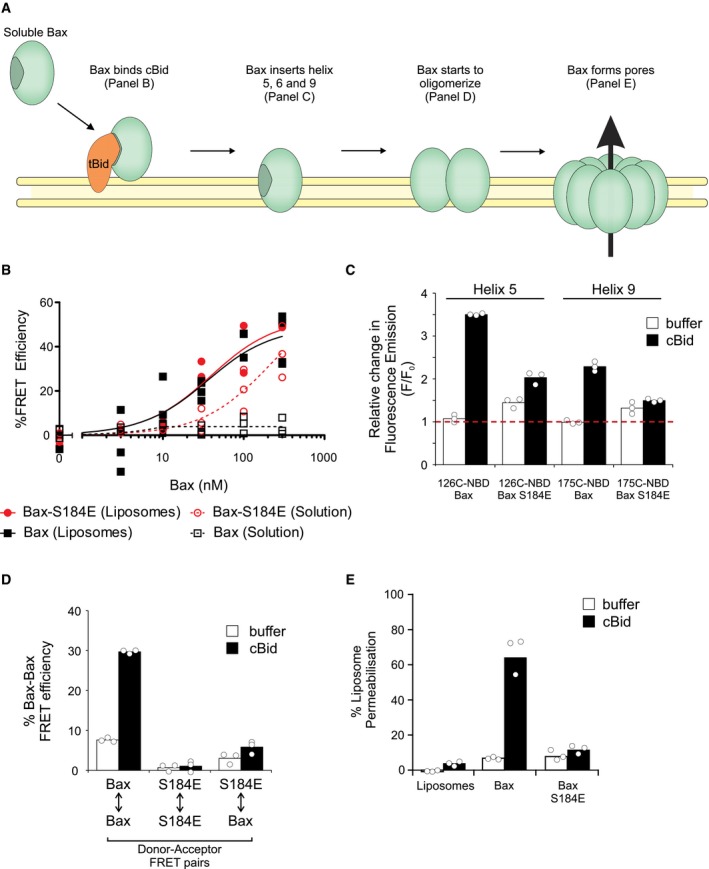
- Schematic of the activation mechanism of Bax. Caspase‐8 cleaved Bid (cBid) spontaneously targets to and embeds within the lipid bilayer. Here, the amino‐terminal portion of cBid dissociates and the carboxy‐terminal portion becomes tBid (truncated Bid) 56. Soluble Bax interacts with tBid at the membrane (tested in panel B). This interaction activates Bax and allows Bax to insert alpha‐helices 5, 6, and 9 into the bilayer (tested in panel C). After insertion of Bax within the lipid bilayer, Bax oligomerizes and permeabilizes the membrane (tested in panels D and E, respectively).
- The apparent affinity between cBid and Bax (step 1) was measured with liposomes and in solution using FRET. Samples containing liposomes were incubated with 20 nM cBid 126C‐DAC, and Bax‐NBD was added as indicated. FRET efficiency was calculated at 2‐h endpoint. Symbols indicate the mean of at least two technical replicates for each independent experiment (n = 3). These data were fit to an equation that describes the equilibrium binding between two proteins (lines; see Materials and Methods).
- Insertion of alpha‐helices 5 and 9 of Bax into the liposome bilayer (step 2) was measured using the environment‐sensitive dye NBD covalently attached to cysteine residues at the indicated locations in Bax. The relative change of NBD emission (F/F 0) was calculated at a 1‐h endpoint with 20 nM cBid (cBid) or buffer as a control (buffer) in the presence of liposomes. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment. The red dotted line indicates no change in fluorescence (F/F 0 = 1).
- Bax oligomerization (step 3) was measured by FRET between Bax monomers. Samples containing liposomes were incubated with 10 nM DAC (donor)‐labeled WT or S184E Bax and 100 nM of NBD (acceptor)‐labeled WT or S184E Bax in the absence (buffer) or presence of 20 nM cBid. FRET efficiency was calculated at 2‐h endpoint. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment.
- The liposome permeabilization activity of Bax was measured using an ANTS/DPX release assay. ANTS/DPX liposomes were incubated with 100 nM of the indicated Bax in the absence (buffer) or presence of 20 nM cBid. Liposomes indicates a control reaction without added Bax. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment.
To assess how phosphorylation at S184 affects the first step, the interaction between cBid and Bax was measured using fluorescence resonance energy transfer (FRET) for the proteins labeled with the dyes DAC and NBD as the donor and acceptor 28. As expected, WT Bax and Bax S184A bound to cBid only in the presence of membranes (Figs 4B and EV3A). Unexpectedly, Bax S184E bound to both cBid (Fig 4B) and Bim (Fig EV3B). Furthermore, WT Bax and Bax S184E bound cBid with similar affinities (30 and 33 nM, respectively). Unlike WT Bax, Bax S184E also bound cBid in solution.
Figure EV3. Related to Fig 4: Bax S184A is a functional Bax mutant that spontaneously inserts into membranes.
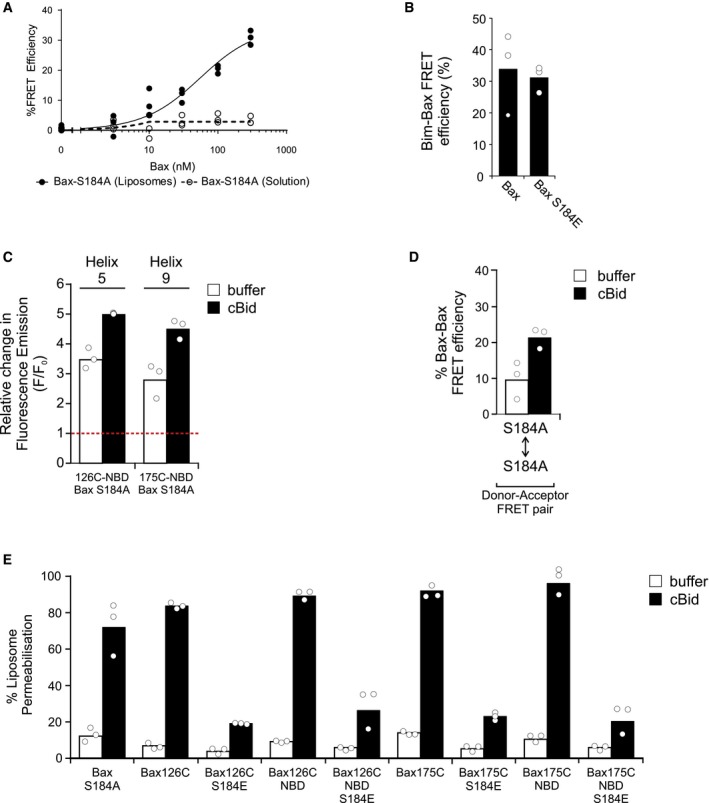
- The apparent affinity between cBid and Bax S184A was measured with liposomes and in solution using FRET. Samples containing liposomes were incubated with 20 nM cBid 126C‐DAC, and Bax‐126C‐NBD S184A was added as indicated. FRET efficiency was calculated at 2‐h endpoint. Symbols indicate the mean of at least two technical replicates for each independent experiment (n = 3). These data were fit to an equation that describes the equilibrium binding between two proteins (lines; see Materials and Methods).
- The interaction between Bim and Bax was measured by FRET. Samples containing liposomes were incubated with 20 nM BIM 41C‐DAC and 100 nM of Bax175C‐NBD or Bax 175C‐NBD S184E. FRET efficiency was calculated at 2‐h endpoint. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment.
- Insertion of alpha‐helices 5 and 9 of Bax S184A into the liposome bilayer was measured using the environment‐sensitive dye NBD covalently attached to cysteine residues at the indicated locations in Bax S184A. The relative change of NBD emission (F/F 0) was calculated at a 1‐h endpoint with 20 nM cBid (cBid) or buffer as a control (buffer) in the presence of liposomes. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment. The red dotted line indicates no change in fluorescence (F/F 0 = 1).
- Bax S184A oligomerization was measured by FRET between Bax monomers. Samples containing liposomes were incubated with 10 nM DAC (donor)‐labeled Bax S184A and 100 nM of NBD (acceptor)‐labeled Bax S184A in the absence (buffer) or presence of 20 nM cBid. FRET efficiency was calculated at 2‐h endpoint. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment.
- The liposome permeabilization activity of Bax S184A or single cysteine mutants of Bax and Bax S184E (labeled and unlabeled) was measured using an ANTS/DPX release assay. ANTS/DPX liposomes were incubated with 100 nM of the indicated Bax in the absence (buffer) or presence of 20 nM cBid. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment.
As Bax S184E binds cBid and Bim, we determined whether it was impaired for insertion into the lipid bilayer. To examine the insertion of specific residues of Bax into membranes, the residue to be probed was exchanged for a cysteine in an otherwise cysteine‐less Bax and the mutant protein was labeled with an iodoacetamide–NBD dye. Positioning the environment‐sensitive NBD dye on cysteines located at either residue 126 in helix 5 or residue 175 in helix 9 enabled detection of the transition of the labeled region of the protein into the lipid bilayer by monitoring the resulting increase in NBD fluorescence. Upon activation of Bax by cBid, residues 126 and 175 of Bax become protected from chemical labeling in cell lysates, suggesting that in cells, they insert into the mitochondrial outer membrane (MOM) 29. Thus, we interpret the increase in NBD emission of Bax labeled at residues 126C and 175C as the insertion of these residues of Bax into membranes. In response to the addition of cBid to NBD‐labeled Bax and liposomes, the fluorescence of NBD at either location increased, confirming that these residues of helices 5 and 9 insert into membranes (Fig 4C). In contrast, NBD‐labeled phosphomimetic Bax did not insert either residue into liposome membranes since the fluorescence emission from NBD probes at 126C and 175C did not increase substantially. Consistent with previous reports and the localization data above, residues 126 and 175 of the control protein Bax S184A spontaneously inserted into the membrane in the absence of cBid (Fig EV3C). These data indicate that insertion of Bax into membranes is inhibited by phosphorylation of Bax S184.
Because phosphomimetic Bax bound to cBid in solution, it is possible that cBid triggers phosphomimetic Bax oligomerization in the absence of Bax helix 5 and 9 insertion into membranes. To measure Bax oligomerization by FRET, Bax molecules labeled with either the donor or acceptor dye were incubated with liposomes. As expected, after incubation with cBid, FRET was detected between Bax proteins (Fig 4D) and Bax S184A (Fig EV3D). However, FRET was not detected for Bax S184E or between Bax and Bax S184E, supporting that phosphorylation prevents the upstream step Bax insertion into membranes, therefore also inhibiting Bax oligomerization (Fig 4D).
To corroborate our results suggesting that Bax S184E does not insert into membranes, we assessed membrane permeabilization by Bax S184E compared to WT Bax. To test this final step in Bax activation, we used a dye release assay in which permeabilization of liposomes releases both the entrapped dye ANTS and its quencher DPX. Release from liposomes dilutes both species resulting in a dramatic increase in the ANTS fluorescence. Compared to the wild‐type protein, the phosphomimetic S184E mutation greatly attenuated ANTS release from liposomes in response to cBid (Fig 4E). This assay was also used in control experiments to confirm that neither the S184A point mutation, labeling with the fluorescent dyes, nor single cysteine mutations (required for NBD or DAC labeling) altered the membrane permeabilizing function of Bax (Fig EV3E).
Phosphorylation of Bax at residue 184 converts Bax into an anti‐apoptotic protein protecting cells from death stimuli in the cytoplasm
Anti‐apoptotic proteins have two modes of inhibition of MOMP 30. Mode 1 inhibition occurs when anti‐apoptotic proteins sequester BH3 proteins, thus preventing BH3‐mediated activation of Bax. Mode 2 inhibition occurs when anti‐apoptotic proteins bind to active Bax and Bak, thus inhibiting their oligomerization. Both modes result in inhibition of MOMP and subsequent cell death. Bax S184E is blocked at insertion into membranes but can bind BH3 activator proteins, suggesting that phosphomimetic Bax could inhibit MOMP by sequestering BH3 proteins. Consistent with this hypothesis, phosphomimetic Bax reduced liposome permeabilization by WT Bax and cBid or Bim in a dose‐dependent manner as shown by a decrease in ANTS and DPX release (Fig 5A), whereas incubation with Bax S184A had the opposite effect (Fig EV4A). Thus, in contrast to previous studies suggesting phosphorylation of Bax abrogates function, Bax S184E functions to impair liposome permeabilization by sequestering BH3 proteins.
Figure 5. Bax S184E protects from apoptotic stimuli and is primarily localized to the cytosol.
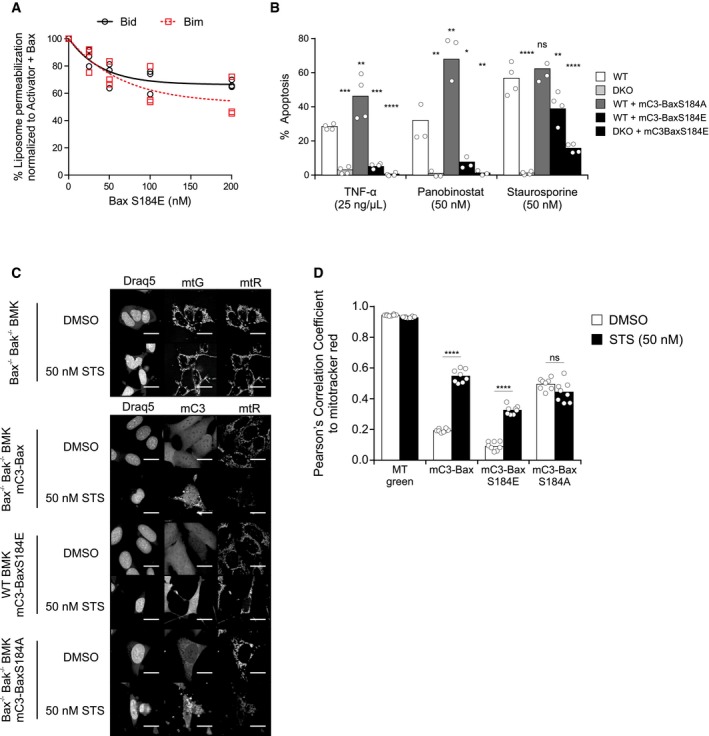
-
AInhibition of liposome permeabilization by phosphomimetic Bax. Liposome permeabilization measured as an increase in fluorescence due to release of ANTS/DPX from liposomes. Fluorescence was normalized using samples not containing Bax S184E. Samples were incubated with 50 nM WT Bax, 20 nM cBid (black circles), or Bim (red squares) and the indicated concentration of Bax S184E. Symbols indicate the mean of at least two technical replicates for each independent experiment (n = 3). These data were fit to a simple exponential decay (lines).
-
BWT BMK or Bax−/− Bak−/− DKO BMK cells that are either untransfected or stably expressing the indicated mCerulean3 (mC3) Bax constructs were treated with the indicated cell death stimuli for 24 h. Cells treated with TNF‐alpha were also treated with 5 μg/ml cycloheximide. Cells stained with DRAQ5, Annexin V conjugated to Alexa 488, and TMRE were imaged on an Opera Phenix automated confocal microscope. Intensity of Annexin V and TMRE staining was measured for > 500 cells per independent experiment. A threshold was used to automatically determine Annexin V‐positive and TMRE‐negative cells. Percent apoptosis was calculated by taking the total number of Annexin V–Alexa 488‐positive and TMRE‐negative cells and dividing by the total number of cells. Bars indicate the mean of four independent experiments (n = 4). Symbols indicate the mean of at least two technical replicates (≈250–500 cells per condition) for each independent experiment. One‐way ANOVA was used, and cell line was statistically compared to WT (white bar) for each drug treatment using t‐tests with Bonferroni correction for multiple comparisons (ns = not significant). TNF‐alpha: DKO (***P = 0.0001), WT+mC3‐BaxS184A (**P = 0.0031), WT+mC3‐BaxS184E (***P = 0.0002), DKO+mC3‐BaxS184E (****P < 0.0001); panobinostat: DKO (**P = 0.0024), WT+mC3‐BaxS184A (**P = 0.0010), WT+mC3‐BaxS184E (*P = 0.0145), DKO+mC3‐BaxS184E (**P = 0.0030); STS: DKO (****P < 0.0001), WT+mC3‐BaxS184E (**P = 0.0057), DKO+mC3‐BaxS184E (****P < 0.0001)
-
C, DThe indicated cell lines (left) were treated with 50 nM STS and 10 μM of caspase inhibitor (Q‐VD‐OPH) for 24 h, stained with DRAQ5 and MitoTracker Red (mtR). Untransfected Bax−/− Bak−/− DKO BMK cells (top panel) were stained with MitoTracker Green (mtG) as a positive control for co‐localization with MitoTracker Red. Cells were imaged using an Opera Phenix automated confocal microscope (C, representative images; and D, co‐localization analysis). (C) Representative images (63× water‐immersion objective) of cells treated with caspase inhibitor (10 μM Q‐VD‐OPH) and either DMSO or 50 nM STS are shown. Images shown are cropped, highlighting single cells to aid in visual discrimination of localization. Scale bars: 20 μm. (D) Cells were imaged as above, and the Pearson's correlation coefficient between the MitoTracker Red channel and the indicated channel below was calculated for each cell. Symbols indicate the mean Pearson's correlation coefficient per cell for each of eight technical replicates (> 50 cells per technical replicate). Bars indicated the mean Pearson's correlation coefficient per cell across two independent experiments (n = 2). The data were heteroscedastic; therefore, a one‐way Welch's ANOVA was used followed by post hoc Welch's t‐tests with Bonferroni correction. Comparisons were made between DMSO control and STS within each group: mC3‐Bax (****P < 0.0001); mC3‐Bax S184E (****P < 0.0001); mC3‐BaxS184A (ns = not significant).
Figure EV4. Related to Fig 5: mCerulean3‐Bax‐S184E protects cells from apoptosis but Bax S184A does not.
-
ALiposome permeabilization was measured as an increase in fluorescence due to release of ANTS/DPX from liposomes. Fluorescence was normalized using samples not containing Bax S184A. Samples were incubated with 50 nM WT Bax, 20 nM cBid (black circles) or Bim (red squares), and the indicated concentration of Bax S184A. Symbols indicate the mean of at least two technical replicates for each independent experiment (n = 3). These data were fit to a simple exponential decay (lines).
-
BWestern blot of cell lysates of the indicated cell lines (top) either untransfected or stably expressing mC3‐Bax. Constructs were analyzed by gel electrophoresis followed by Western blot using the antibodies indicated on the right. Bax (#2772; Cell Signaling) was used for IB of Bax.
-
CWT BMK or Bax−/− Bak−/− DKO BMK cells that are either untransfected or stably expressing the indicated mCerulean3 (mC3) Bax constructs were treated with increasing concentrations of the indicated cell death stimuli for 24 h. Cells treated with TNF‐alpha were also treated with 5 μg/ml cycloheximide. Cells stained with DRAQ5, Annexin V conjugated to Alexa 488, and TMRE were imaged on an Opera Phenix automated confocal microscope. Intensity of Annexin V and TMRE staining was measured for > 500 cells per independent experiment. A threshold was used to automatically determine Annexin V‐positive and TMRE‐negative cells. Percent apoptosis was calculated by taking the total number of Annexin V–Alexa 488‐positive and TMRE‐negative cells and dividing by the total number of cells. Each point represents the mean % apoptosis calculated from four independent experiments (n = 4) each with two technical replicates. Shown is mean ± SEM.
-
D, EThe indicated cell lines (left) were treated with increasing doses of STS (0–100 nM) and 10 μM of caspase inhibitor (Q‐VD‐OPH) for 20 h, stained with DRAQ5 (DNA), and imaged using an Opera Phenix automated confocal microscope (D, representative images; and E, image analysis). (D) Representative images (63× water‐immersion objective) of cells treated with caspase inhibitor (10 μM Q‐VD‐OPH) and either DMSO or 50 nM STS are shown. Images shown are cropped, highlighting single cells to aid in visual discrimination of localization. Scale bars: 20 μm. (E) Cells were imaged as representative images in EV4D above except with a 40× water‐immersion lens. The percent of cells with a punctate mCerulean3 localization pattern was quantified based on automated assessment of intensity fluctuations for cells expressing the indicated mC3‐Bax construct in the absence (DMSO control, open symbols) or presence of increasing concentrations of STS (closed symbols). Symbols indicate the mean of at least two technical replicates (≈400–1,600 cells per condition) for each independent experiment (n = 3). These data were fit to a simple log(dose) vs. response model with a hill slope of 1 (lines). Shown is mean ± SEM.
If our in vitro results translate to cells, phosphorylation of Bax may be sufficient to protect cells from apoptotic stimuli. To test this, Bax S184E or the control Bax S184A was stably expressed in cells with a fluorescence protein tag mCerulean3 (mC3) to enable monitoring expression of the mutant in WT or Bax−/−/Bak−/− DKO BMK. Expression of each construct and confirmation of Bax and Bak expression or lack thereof in WT and Bax−/−/Bak−/− DKO BMK cells was confirmed by Western blot (Fig EV4B). Stable Bax‐S184E expression in WT BMK cells, which have endogenous expression of Bax and Bak, had a significant protective effect against cell death induced by the pro‐death cytokine TNF‐α and cycloheximide, the pan‐kinase inhibitor staurosporine, and the HDAC inhibitor panobinostat (Figs 5B and EV4C). In BMK cells, apoptosis was dependent on endogenous WT Bax and Bak since Bax−/− Bak−/− double‐knockout (DKO) BMK cells did not die from exposure to low concentrations of these agents. The control cell lines stably expressing Bax S184A had a marked increase in apoptosis in response to each drug, whereas Bax−/−/Bak−/− DKO BMK cells stably expressing Bax S184E were refractory to apoptosis. These data suggest that sequestration of BH3 proteins by Bax S184E likely inhibits both Bax and Bak‐induced MOMP. That some STS‐treated BMK cells and DKO BMK cells expressing Bax S184E died suggests that this drug may also activate Bax independently of BH3 protein binding. In these experiments, exogenous Bax S184E protein levels were similar to those of endogenous Bax (Fig EV4B). Moreover, expression of Bax S184E in WT BMK cells did not alter the levels of endogenous Bax and resulted in a small increase in Bak. Thus, the decrease in apoptosis due to expression of Bax S184E was not due to decreased endogenous Bax or Bak compared to WT cells, nor was the decrease in apoptosis due an artifact of transfection since Bax S184A expression in the WT BMK cells resulted in enhanced apoptosis.
Bax S184E binds activator BH3 proteins in solution (Fig 4B) and has a mainly cytoplasmic localization in MDA‐MB‐468 cells (Figs 2C and EV1C), suggesting that phosphomimetic Bax sequesters activator BH3 proteins in the cytosol thereby preventing apoptosis. To test whether Bax S184E remains in the cytoplasm under cell death conditions, we compared the co‐localization of mCerulean3‐Bax proteins to that of MitoTracker Red (Fig 5C and D) and the localization pattern of mCerulean3‐Bax proteins in the presence and absence of STS (Fig EV4D and E). In addition to STS, cells were treated with a caspase inhibitor in order to delay the later stages of apoptosis for optimal imaging of mCerulean3‐fused WT Bax, Bax S184E, or Bax S184A. MitoTracker Green, a positive control for co‐localization, had a Pearson's correlation coefficient close to 1, indicating a near‐perfect correlation between two dyes that stain mitochondria which did not significantly change in response to STS.
Both WT Bax and Bax S184E had a significantly increased co‐localization with MitoTracker Red in response to STS, whereas there was no significant change in Bax S184A mitochondrial localization in response to STS (Fig 5D). Additionally, in response to apoptotic stimuli, Bax localizes to mitochondria resulting in a punctate pattern of mCerulean3 fluorescence (Fig EV4D). This is most clearly seen when the proportion of cells with a punctate localization pattern for mCerulean3 was quantified (Fig EV4E). The localization pattern of mC3‐fused WT Bax and Bax S184A transitions from a diffuse to punctate pattern in response to STS in a dose‐dependent manner, whereas the majority of mC3‐fused Bax S184E remains diffuse under the same conditions (Fig EV4E and D). These data indicate that in response to STS, Bax S184A remains localized to the mitochondria but both Bax and Bax S184E transition from the cytoplasm to the mitochondria where compared to Bax a smaller fraction of Bax S184E localizes to the mitochondria under apoptotic conditions. Taken together, the results strongly suggest that phosphorylation of Bax at position 184 generates a protein that competes with WT Bax for binding to activator BH3 proteins in a manner analogous to mode 1, but not mode 2, inhibition of apoptosis by multidomain anti‐apoptotic proteins since phosphorylated Bax only sequesters BH3 proteins but not other Bax molecules 30, 31. Furthermore, we predict that sequestration of activator BH3 proteins by phosphorylated Bax occurs primarily in the cytosol allowing interception of activator BH3 proteins diffusing to the MOM before they activate WT Bax and Bak 28. However, it appears that Bax S184E can also sequester BH3 proteins at the mitochondria since Bax S184E also localizes to mitochondria in response to apoptotic stimuli.
In human cancer, pBax S184 status is correlated with cellular response to ABT‐737 and overexpression of Bax is countered by Akt pathway activation
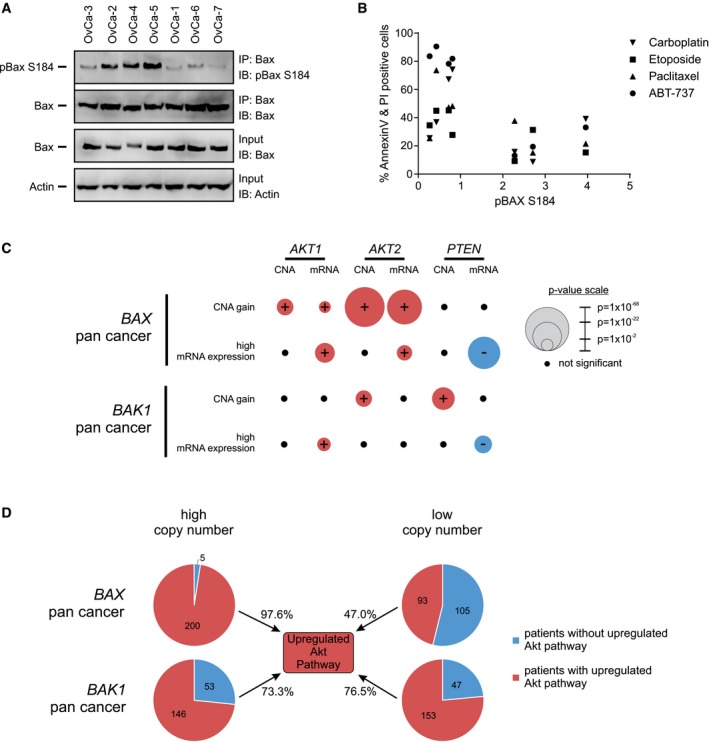
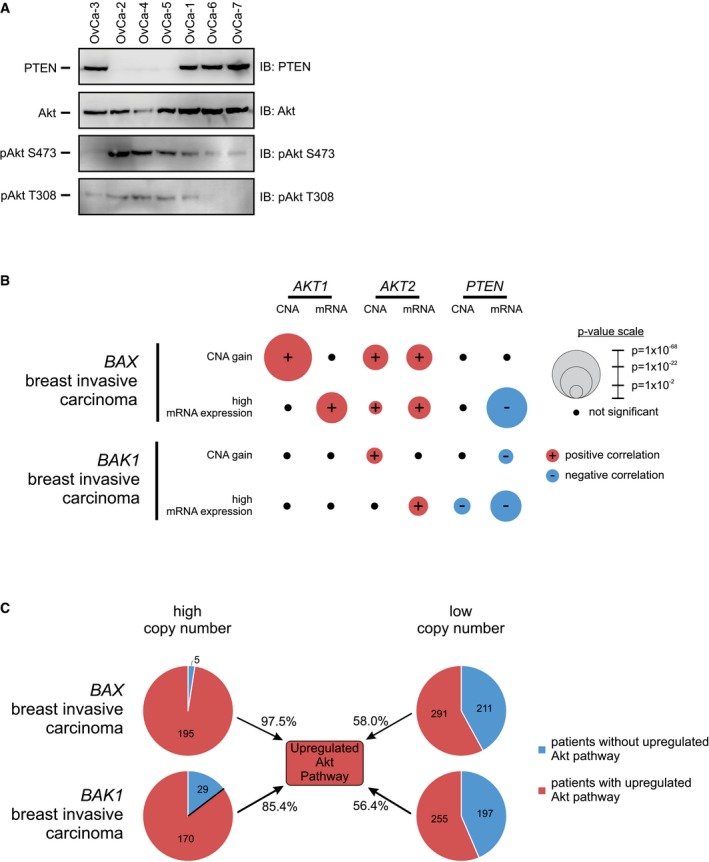
The Akt pathway is frequently upregulated in cancers 32, and we have shown that Akt phosphorylates Bax at position S184 converting Bax into an anti‐apoptotic protein. Although Bax is generally considered an anti‐oncogene and reduced expression is associated with poor prognosis 33, 34, we hypothesized that certain cancers may be protected from apoptosis due to an increase in the amount of phosphorylated Bax. To address this hypothesis directly, we assayed whether ovarian cancer cells isolated from patients had any correlation between pBax S184 levels and response to chemotherapy agents (Fig 6A and B). Bax S184 phosphorylation levels were significantly correlated (P < 0.05) with resistance to ABT‐737 treatment; however, correlations between Bax S184 phosphorylation and responses to other chemotherapeutic drugs tested were not statistically significant (Fig 6B). This may be due to the relatively poor induction of apoptosis by these drugs and/or because of killing by both apoptotic and non‐apoptotic mechanisms. As expected, pBax S184 levels were positively correlated with activated Akt (pAkt S473 and pS308) and negatively correlated with PTEN status (Fig EV5A).
Figure 6. S184‐phosphorylated Bax is correlated with ABT‐737 resistance in primary ovarian cancer cells, while overexpression of Bax is countered by Akt pathway activation in human cancers.
- S184‐phosphorylated Bax is correlated with ABT‐737 resistance in primary ovarian cancer cells. Phosphorylation of Bax S184 was evaluated using lysates from the indicated primary ovarian cancer cells by Western blotting with the indicated antibodies (IB) after immunoprecipitation with the indicated antibodies (IP) (left panel). 5–10% of the total lysates (input) were probed for Bax (Bax ∆21 antibody) and actin as expression and loading controls, respectively, by Western blotting with the indicated antibodies (IB).
- To evaluate apoptotic response in primary ovarian cancer cells, cells from each OvCa cell line indicated in Fig 6A were treated with either carboplatin (100 μM), etoposide (50 μM), paclitaxel (0.1 μM), and ABT‐737 (0.5 μM) for 48 h, and apoptotic cells were evaluated by Annexin V/PI staining and flow cytometry (right panel). Apoptotic response to each drug was plotted against the pBaxS184/Bax ratio (calculated from Fig 6A) for each primary ovarian cancer sample. Correlation between pBaxS184 and response to drug treatment was evaluated two‐tailed Spearman correlation. Only ABT‐737 resistance was significantly (P < 0.05) correlated with pBax S184 levels. Symbols indicate the mean of two experimental repeats (n = 2), each having two technical repeats for each of the seven OvCa cell lines treated with the indicated drug.
- TCGA database of all human cancers was probed for significant associations between the row heading (e.g., BAX CNA gain) and copy number (CNA) gain/loss (red/blue) or mRNA up/downregulation (red/blue) of the indicated genes (AKT1, AKT2, and PTEN). Association strength is proportional to −log(P‐value), reflected in the radii of the circles. Black circles indicates non‐significant association (P‐value cutoff = 0.02). The data used to generate this figure are located in Appendix Table S3.
- Each circle represents the total number of patients, across all cancers in the TCGA database, with either high BAX or BAK1 (higher than the mean, left) or low BAX or BAK1 (lower than the mean, right) copy number. Within each circle, the proportion of patients with upregulated Akt pathway (red) is compared to the proportion of patients without upregulated Akt pathway (blue). Upregulated Akt pathway is defined as an increase in AKT1 or AKT2 or PIK3CA or decrease in PTEN copy number.
Figure EV5. Related to Fig 6: Overexpression of Bax is countered by Akt pathway activation in breast invasive carcinoma.
- PTEN, Akt, pAkt S473, and pAkt T308 levels were evaluated using lysates from the indicated primary ovarian cancer cells by Western blotting with the indicated antibodies (IB) after immunoprecipitation with the indicated antibodies (IP).
- TCGA database of invasive breast carcinoma was probed for significant associations between the row heading (e.g., BAX CNA gain) and copy number (CNA) gain/loss (red/blue) or mRNA up/downregulation (red/blue) of the indicated genes (AKT1, AKT2, and PTEN). Association strength is proportional to −log(P‐value), reflected in the radii of the circles. Black circles indicates non‐significant association (P‐value cutoff = 0.02). The data used to generate this figure are located in Appendix Table S3.
- Each circle represents the total number of patients with invasive breast carcinoma from the TCGA database, with either high BAX or BAK1 (higher than the mean, left) or low BAX or BAK1 (lower than the mean, right) copy number. Within each circle, the proportion of patients with upregulated Akt pathway (red) is compared to the proportion of patients without upregulated Akt pathway (blue). Upregulated Akt pathway is defined as an increase in AKT1 or AKT2 or PIK3CA or decrease in PTEN copy number.
As an alternative albeit indirect approach to assess our hypothesis across all cancers, we examined publically available patient data for gene copy number and mRNA expression data. If, as our results suggest, Akt is a major enzyme responsible for phosphorylating Bax a prediction would be that in cancers overexpressing Bax the activity and/or amount of Akt would be increased. Therefore, gene copy number alterations (CNA) and mRNA expression data were examined to determine whether high Bax expression correlates with increased expression/activation of the Akt pathway genes (AKT1, AKT2, PIK3CA, PIK3R1) and the Akt pathway inhibitor gene PTEN.
An initial examination of all available CNA data (16,557 samples) from cBioPortal revealed that BAX duplications were significantly associated with Akt pathway duplications (P < 10−10) as 879/925 (95%) of patients with a BAX duplication also had at a duplication of at least one Akt pathway gene. In contrast, only 41% of all samples in the database had Akt pathway gene duplications. In addition, 183/925 (20%) of patients with BAX duplications had duplications of three or more Akt pathways genes. Furthermore, there were deletions in the Akt negative regulator gene PTEN in 11% of patient samples with BAX duplications compared to PTEN deletions in 17% of all patient samples suggesting a slight but not statistically significant negative association. However, among the 46 patients with BAX duplications, but no Akt pathway gene duplications, 13 (28%) had PTEN deletions (P = 0.045). This suggests cancers that have BAX amplifications tend to have upregulated Akt either by Akt pathway amplifications or PTEN deletions.
Amplification of the Akt pathway may permit, but not directly result in, the overexpression of Bax. However, due to the anti‐oncogenic nature of Bax, we hypothesized that overexpression of Bax in cancer requires its inhibition and that upregulation of the Akt pathway could be a major mechanism. Thus, we examined associations of both BAX and BAK1 CNA gain and mRNA overexpression in relation to Akt pathway genes from the TCGA database (Figs 6C and EV5B; Appendix Table S3). BAK1 was used as a control for BAX specificity, since Bax and Bak are functionally redundant, if there is a specific link between Bax and Akt, there may not be one with Bak. Associations were examined in the pan‐cancer (Fig 6C, Appendix Table S3) and in breast invasive carcinoma (Fig EV5B, Appendix Table S3) samples as an example from a single cancer type. The latter was chosen since five of the seven cell lines studied here are breast cancer cell lines and it is a relatively data rich set. In breast invasive carcinoma, we observe similar trends to that of the pan‐cancer dataset. However, fewer of the associations are statistically significant, a likely consequence of the smaller size of this dataset.
As expected from the results obtained with data from cBioPortal, BAX gain in the pan‐cancer dataset was associated with gain of AKT1, AKT2, and PIK3R1 and increased expression of AKT1 and AKT2 mRNA (Fig 6C, Appendix Table S3). Furthermore, high BAX mRNA expression is associated with an increase in AKT1 and AKT2 and a decrease in PTEN mRNA (Fig 6C, Appendix Table S3). Akt directly targets Bax and thus the increased Akt is consistent with the hypothesis that in cancers with high levels of Bax the protein is phosphorylated and anti‐apoptotic. However, contrary to our hypothesis, there was a significant association between either BAX gain or high BAX mRNA expression and decreased levels PIK3CA and PIK3R1 mRNA (Appendix Table S3). As PI3K signaling is upstream of Akt and regulates many pathways, there could be a number of explanations for this observation. One possibility is that low levels of PI3K activity due to lower expression of PI3K genes could be countered by the decreased expression of PTEN that we observe when BAX (CNA and mRNA) levels are high. As a control, the same analysis was repeated for BAK1 (CNA and mRNA levels). We found that, compared to BAX, there were more non‐significant associations between BAK1 and Akt pathway genes and of the significant associations found the P‐values were much higher (Fig 6C, Appendix Table S3). The most significant association for BAK1 was an increase in copy number for PTEN that was not seen for BAX. Similar results were found in the breast invasive carcinoma dataset (Fig EV5B, Appendix Table S3). These data suggest that overall the BAX and AKT positive associations and the BAX and PTEN negative associations are stronger compared to that of BAK1 and AKT or PTEN.
Due to the positive associations between increased BAX levels and Akt pathway genes, we hypothesize a higher probability of Akt pathway upregulation in patients with high BAX levels compared to that with low BAX levels (Fig 6D). Probing the TCGA database, 97.6% (200/205) of patients showing increase in BAX copy number also show upregulation of the Akt pathway (defined here as an increases in AKT1 OR AKT2 OR PIK3CA OR decrease in PTEN copy number). This positive association is highly significant when compared to the whole population (P = 7e‐40). On the other hand, only 58% (4,868/8,364) of patients where Bax is neither increased nor decreased and 47% (93/198) of patients showing a decrease in BAX copy number also show upregulation of the Akt pathway. There was very little difference in the upregulation of the Akt pathway in cancer patients with high or low BAK1 levels (Fig 6D). When looking at breast invasive carcinoma, the association of BAX levels and Akt pathway activation was similar to the pan‐cancer dataset (Fig EV5C). Thus, 97.5% (195/200) of breast cancer patients showing increase in BAX copy number also show upregulation of the Akt pathway compared to 50% (128/255) of patients where BAX is neither increased or decreased and 58% (291/502) of breast cancer patients with low BAX (P = 2e‐36). The difference in Akt pathway activation between low and high BAK1 copy number in breast cancer patients is smaller than that found with BAX (Fig EV5C). Overall, these results suggest that BAX copy number is strongly associated with Akt pathway upregulation compared to BAK1, both in pan‐cancer and breast cancer patients. Taken together, the bioinformatics data suggest that in tumors with elevated BAX the protein may be phosphorylated and therefore anti‐apoptotic due to upregulation of the Akt pathway.
Discussion
Residue S184 on Bax was reported to control Bax activation based on a series of point mutations at S184 that were found to alter Bax pro‐apoptotic function 35. Akt has also been shown to phosphorylate Bax at S184 thereby inhibiting activation and mitochondrial localization in response to various perturbations in several normal tissues, including neutrophils and endothelial cells 15, 19, 36. Activation of Akt and phosphorylation of Bax at S184 are induced by nicotine in lung cancer cell lines where it protects cells from cisplatin 16. However, a recent study also reported that either co‐expression of Bax and Akt or expression of S184D phosphomimetic Bax results in the activation of Bax 17. Here, we have confirmed that Akt phosphorylates Bax at position S184 (Fig 3) and that both Akt phosphorylated and phosphomimetic Bax are defective in mediating membrane permeabilization in multiple in vitro systems using isolated mitochondria (Fig 3), liposomes (Fig 4), mammalian cells (Fig 5), and primary patient samples (Fig 6). Although we cannot rule out or account for the localization or post‐translational modifications of other Bcl‐2 family proteins in our experimental assays, our data strongly suggest that the Akt target responsible for unpriming mitochondria is Bax and not another mitochondrial protein, since incubation of mitochondria with Akt and Bax, but not the phosphorylation‐resistant mutant Bax S184A, prevents cytochrome c release (Fig 3E–G).
Although previous studies correlated Bax inactivation and S184 phosphorylation, the mechanism remained unclear 15, 18. Previous results were interpreted as suggesting that S184 phosphorylation of Bax abrogated all steps in the Bax activation pathway (Fig 4A) 15, 16, 19. By assaying each step in the Bax activation pathway individually, we identified that insertion of Bax into membranes is inhibited by this post‐translational modification. Subsequent steps including Bax homo‐oligomerization and pore formation were also prevented suggesting the temporal order identified previously 28 results from each step being dependent on successful completion of the one before it. Since BH3 protein binding by Bax occurs at the membrane but before insertion of Bax into membranes, it is logical that some Bax S184E bound to activator BH3 proteins at membranes. This is further supported by subcellular fractionation (Fig EV1C) experiments under normal conditions and co‐localization assays (Figs 5D and EV4E) under apoptotic conditions showing some Bax S184E at the mitochondria, albeit less than that of WT Bax or Bax S184A. BH3 protein binding in solution was unexpected because when Bax is in solution the BH3 binding site is normally occluded by helix 9 and as a result, binding was not detected for Bax and BH3 proteins in the absence of membranes 28. We speculate that the addition of a negative charge within the hydrophobic groove by helix 9 by phosphorylation or mutation reduces the affinity of the helix 9 interaction and “opens” the BH3 binding site on Bax allowing BH3 activator binding in solution. Conversely, since Bax S184A spontaneously targets membranes but does not interact with cBid in solution, we speculate that helix 9 of the S184A mutant occludes the hydrophobic groove in solution and only once a membrane is present can helix 9 disengage allowing BH3 binding.
A phosphorylated Bax that does not permeabilize membranes yet binds activator BH3 proteins inhibits MOMP by sequestering the BH3 protein similar to mode 1 inhibition of BH3 proteins by anti‐apoptotic Bcl‐2 family proteins 30, 31. As phosphomimetic Bax competitively sequesters activator BH3 proteins, but does not bind WT Bax (Fig 4), it is expected to be less effective at inhibiting apoptosis than anti‐apoptotic Bcl‐2 proteins. Thus, phosphorylation of Bax by Akt converts Bax from pro‐ to anti‐apoptotic, resulting in a diminished sensitivity of mitochondria to apoptotic signaling by BH3 proteins. As a consequence, cancer cell lines and primary patient cells are resistant to drugs that activate apoptosis, including the Bcl‐2/Bcl‐XL/Bcl‐w antagonist ABT‐737 (Figs 1B and 6A). Consistent with this conclusion, expression of Bax S184E in cells with endogenous Bax and Bak protected cells from apoptotic stimuli while having a predominately cytosolic localization suggesting that phosphorylated Bax likely sequesters activator BH3 proteins in the cytoplasm (Fig 5B–D). However, some Bax S184E did target to mitochondria under apoptotic conditions; thus, Bax S184E and S184‐phosphorylated Bax may inhibit BH3 proteins at the mitochondria as well.
In primary patient samples, the amount of S184‐phosphorylated Bax directly correlates with resistance to BH3 mimetics (Fig 6A and B). A predicted consequence of inhibition of apoptosis by phosphorylated Bax is that Bax phosphorylation in cells by upregulated Akt likely provides a selective advantage for cancer cells that have increased levels of Bax. As a tumor suppressor, Bax expression is usually decreased in cancer cells. Indeed, overexpression of Bax sensitizes cancer cells to apoptosis induced by chemotherapy or radiation 37, 38, 39, 40, 41 and can be correlated with improved clinical outcomes in ovarian cancer and gastric cancer 42, 43. However, in other cancers, high Bax expression is associated with poorer outcomes and an increased risk of relapse 44, 45, 46. Our analysis clearly demonstrates that when Bax copy number is increased, 97.6% of the patients also show upregulation of the Akt pathway (Fig 6D), suggesting the latter observation may be more prevalent. Phosphorylation of Bax by Akt provides an explanation for these observations as S184‐phosphorylated Bax would allow cells to evade apoptosis by competing with WT Bax and Bak for binding to BH3 proteins (Fig 7). Furthermore, phosphorylated Bax may have a binding advantage for BH3 proteins since phosphomimetic Bax binds to activator BH3 proteins in solution and at membranes, whereas WT Bax and Bak only bind membrane bound activator BH3 proteins. Thus, increased expression of Bax in cells with upregulated Akt represents a novel mechanism for cancer cells to prevent apoptosis and acquire drug resistance.
Figure 7. Phosphorylated Bax protects cells from apoptosis.
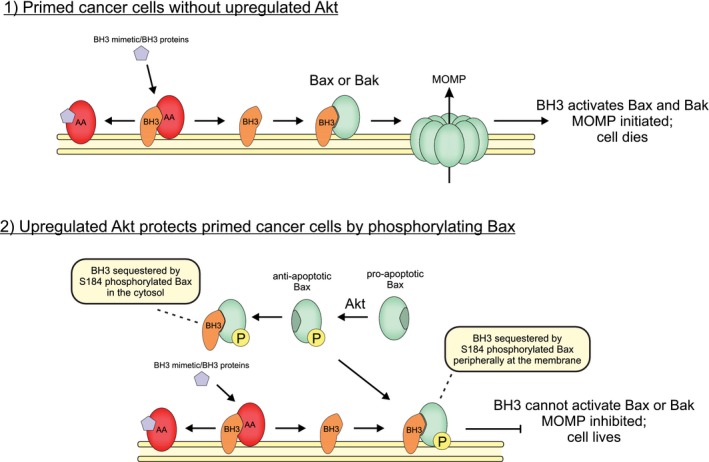
Cartoon showing how phosphorylation by Akt switches Bax activity from pro‐ to anti‐apoptotic. (1) In cells without upregulated Akt signaling, cells are sensitive to pro‐apoptotic signaling and BH3 mimetics (purple pentagon), and mitochondria are primed for death due to anti‐apoptotic proteins (AA, red) like Bcl‐XL, Bcl‐2, and Mcl‐1, binding activator BH3 proteins (BH3, orange) like tBid and Bim, at the mitochondrial outer membrane. Pro‐apoptotic signaling or addition of a BH3 mimetic such as ABT‐737 displaces activator BH3 proteins from anti‐apoptotic proteins, whereupon tBid or Bim activates Bax (green) leading to MOMP and cell death. (2) In cells with upregulated Akt signaling, cells are resistant to pro‐apoptotic signaling and BH3 mimetics (purple pentagon) because Bax is phosphorylated (P, yellow) at position S184 by Akt. Phosphorylation prevents Bax inserting into the mitochondrial outer membrane while allowing activator BH3 binding. Cells become resistant to BH3 mimetics and pro‐apoptotic BH3 proteins, because activator BH3 proteins released from anti‐apoptotic proteins are sequestered by phosphorylated Bax mainly in the cytosol but also peripherally at mitochondria via binding of BH3 proteins inserted into the bilayer. Sequestration by Bax prevents BH3 proteins from activating non‐phosphorylated Bax and Bak.
Our findings are of clinical importance because activation of the Akt pathway occurs often in cancer cells as a result of loss of PTEN, aberrant receptor tyrosine kinase activity or PIK3CA or AKT1 activating mutations. This heterogeneity has made it difficult to establish predictive biomarkers. An important role for Bax phosphorylation in the anti‐apoptotic effects of Akt as elucidated here suggests that phosphorylation of Bax at S184 is a potential predictive, and perhaps pharmacodynamic, biomarker for pharmacologic inhibitors of the Akt pathway in cancer clinical trials. Intriguingly, Choudhary et al 23 showed that non‐Hodgkin lymphoma cell lines with acquired resistance to ABT‐199 had substantial Akt activation. These cells became sensitive to ABT‐199 only after treatment with dual AKT/mTOR pathway inhibitors. Although this group did not look at the phosphorylation status of Bax, we would predict that acquired resistance to ABT‐199 in these cells was due in part to the anti‐apoptotic effects of phosphorylated Bax. Considering the growing interest in BH3 mimetics as anti‐cancer agents, and the ongoing development of Akt pathway inhibitors, we suggest that examination of possible combinatorial therapies for cancers with upregulated Akt is warranted.
Materials and Methods
Antibodies, reagents, and plasmids
Antibodies
Akt (#9272), Actin (#4967), Bax (#2772), Bak (#3814), and GAPDH (#2118) were from Cell Signaling. Cytochrome c (7H8.2C12) was from BD Biosciences. Phosphoserine (4A9; #525284) was purchased from EMD Millipore. β‐Actin (#Y00005‐002) was from Immune Biosolutions (used in Fig EV4B). GFP (19F7) was from Memorial Sloan‐Kettering Monoclonal Antibody Facility. Bax ∆21 antibody (#sc‐6236) was from Santa Cruz. pBax S184 antibody (ab111391) was from Abcam.
Reagents
ABT‐737, negative control enantiomer, and A‐443654 were obtained from Abbott Laboratories. MK‐2206 and LY294002 were purchased from Selleck Chemicals. Deguelin was obtained from Tocris Bioscience, Minneapolis. Recombinant Human Active Akt1 was obtained from R&D Systems (1775‐KS) and Sigma (A8729).
Plasmids
GFP‐Bax (hBax C3‐EGFP, Addgene plasmid 19741), GFP‐Bax S184A (hBax S184A C3‐EGFP, Addgene plasmid 19742), and GFP‐Bax S184E (hBax S184E C3‐EGFP, Addgene plasmid 19743) were previously described 35. GFP (PCDNA3‐EGFP, Addgene plasmid 13031) was used as a control for transfections. Single cysteine Bax 126C (pMAC1731) and single cysteine cBid 126C (pMAC2118) were previously described 28. The cDNA for single cysteine Bax 175C was transferred from the corresponding retroviral vector 29 into the IMPACT expression system (New England Biolabs, Ipswich, MA, USA) generating pMAC 1739. Bax S184E and S184A mutation was introduced into the Bax WT, Bax 126C, and Bax 175C plasmids via QuikChange™ mutagenesis (Stratagene).
The cDNA from a plasmid encoding mCerulean3 protein 47 was put in place of the EGFP coding region in pEGFP‐C1 (Clontech). The plasmid encoding mCerulean3 was a gift from Mark Rizzo (University of Maryland School of Medicine), and the pEGFP‐C1 plasmid was a gift from Ray Truant (McMaster University). To generate a plasmid containing mCerulean3‐Bax S184E, the cDNA of Bax‐α was amplified by PCR and inserted after the mCerulean3 coding region. The entire coding region of mCerulean3‐Bax was cut out from this plasmid and inserted into the MCS1 of pVitro2‐Blasti‐MCS (Invivogen plasmid pvitro2‐bmcs) that was shortened to remove one of the two gene expression elements (CMV Enhancer, hFerL promoter, chEF1 5'UTR, and MCS2). Site‐directed mutagenesis was used on this construct as above to generate mCerulean3‐BaxS184E and mCerulean3‐BaxS184A plasmids.
Cell culture
MDA‐MB‐435, MCF‐7, T47‐D, MDA‐MB‐468, ZR‐75‐1, and SF539 AND 768‐O cells were grown in DMEM/F12 (Invitrogen), and WT MEFs and Bax−/− Bak−/− DKO MEFs were grown in DMEM (Invitrogen) supplemented with 2 mM l‐glutamine, 10% heat‐inactivated fetal bovine serum (Sigma), 100 IU/ml penicillin, and 100 mg/ml streptomycin (Invitrogen) in a humidified incubator at 37°C and 5% CO2. MDA‐MB‐468 cells were stably transfected with vectors containing GFP, GFP‐Bax, GFP‐Bax S184A, and GFP‐Bax S184E plasmids using FuGENE 6 (Roche), and the clonal selection was carried out in the presence of G418 (1.2–1.6 mg/ml, Sigma). Selected clones were maintained in growth medium containing 0.16 mg/ml G418.
WT baby mouse kidney (BMK) cells and their Bax−/− Bak−/− DKO derivative were gifts from Eileen White (Rutgers University). The WT and Bax−/− Bak−/− DKO BMK cells were cultured in DMEM supplemented with 10% FBS and 1% MEM NEAA. WT BMK cells were stably transfected with a vector encoding mCerulean3‐Bax‐S184E using FuGENE HD (Roche), and clonal selection was carried out in the presence of 5 μg/ml blasticidin (BLA477; Bioshop). WT BMK cells stably transfected with a plasmid encoding mCerulean3‐Bax‐S184E were subjected to two rounds of fluorescence‐activated cell sorting to isolate cells stably expressing mCerulean3‐BaxS184E.
Immunoblotting, immunoprecipitation, and coimmunoprecipitation
Total cell lysates were prepared in 1% CHAPS buffer [5 mM MgCl2, 137 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% CHAPS, 20 mM Tris–HCl (pH 7.5), protease inhibitors (Complete, Roche), and phosphatase inhibitors (PhosSTOP; Roche)] as reported previously 48. For coimmunoprecipitations, proteins (800 μg–10 mg) were immunoprecipitated with indicated antibodies at 4°C for 2 h or O/N, and coimmunoprecipitates were captured by Dynabeads Protein G at 4°C for 2 h or O/N. Beads were recovered using DynaMag Spin Magnet and washed thrice in 1% CHAPS buffer. For Bax immunoprecipitations for phosphorylation analysis, proteins were isolated in a modified RIPA buffer [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1% NP‐40, 0.5% Na‐deoxycholate, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, protease inhibitors (Complete; Roche), and phosphatase inhibitors (PhosSTOP; Roche)] and immunoprecipitated with Bax ∆21 antibody (#sc‐6236; Santa Cruz). Immunoprecipitates, total cell extracts, and subfractionation lysates were separated on NuPAGE 10% Bis‐Tris gels. Following SDS–PAGE, proteins were transferred onto PVDF membranes (Immobilon) and then blocked with 5% dried milk in PBS–Tween‐20. Membranes were incubated with a 1:1,000 dilution of primary antibodies (as indicated and described above) and 1:5,000 dilution of secondary antibodies (GE Healthcare Life Sciences), in a buffer containing 10% milk diluent blocking concentrate (KPL), and detected with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific). For phosphorylation blots, 4% BSA in TBS–Tween was used for blocking and antibody preparation and TBS–Tween was used for membrane washing steps. Blots were imaged with LAS4000 image analyzer (Fujifilm) on chemiluminescence mode.
For blots in Fig EV4B, cells were harvested and washed in ice‐cold PBS, pelleted, and resuspended in 1 ml cell lysis buffer [1% Triton X‐100, 150 nM NaCl, 10 mM Tris (pH 8.0)], heated for 5 min at 98°C, and pelleted. Whole‐cell lysate protein concentration was quantified by Bradford (Bio‐Rad Protein Assay) and standardized to 1 mg/ml. Whole‐cell lysate proteins (15 μg per lane) were separated by SDS–PAGE, and Bax, Bak, and Actin were detected by immunoblotting. Primary antibodies were used at 1:1,000 dilution for Bax (Rabbit‐α‐Bax, #2772; Cell Signaling), Bak (Rabbit‐α‐Bak, #3814, Cell Signaling), and β‐actin (Chicken‐α‐β‐actin, #Y00005‐002, Immune Biosolutions). Secondary antibodies were used at a 1:20,000 dilution from Jackson ImmunoResearch Inc. (for Bax and Bak; Donkey‐α‐Rabbit, #711‐035‐152) and Immune Biosolutions (for β‐actin; Aurora Alpaca‐α‐Chicken, #Y00008‐HRP). Immunoblots were developed using chemiluminescence, and intensities were recorded using a CCD camera (DNR).
Peptide synthesis
Peptides were synthesized by Tufts University Core Facility and purified by HPLC. Peptide identities were confirmed by mass spectrometry, and stock solutions were prepared in DMSO. tBid generated from His‐tagged human Bid cleaved with recombinant caspase‐8 was a generous gift from Claudio Hetz (University of Chile, Santiago, Chile).
Phosphoprotein gel staining
Protein samples after kinase reactions were separated on 10% Bis‐Tris NuPAGE gels and stained with Pro‐Q Diamond phosphoprotein gel stain and SYPRO Ruby total protein gel stain (Invitrogen) according to manufacturer's protocol. Gels were imaged with LAS4000 image analyzer (Fujifilm), using the same exposure settings for different samples on green and blue fluorescence epi‐illumination mode, and the quantitation of bands was performed using ImageJ software.
Protein purification and labeling, and liposome preparation
WT and single cysteine Bax mutants, 126C and 175C with or without the S184E or S184A mutation, were expressed, purified, and labeled (when indicated) with DAC or NBD, as previously described 27. Single cysteine His‐Bid 126C was expressed, purified, and labeled with DAC as previously described 27. Liposomes were prepared as previously published 27. Briefly, a 1 mg mitochondria‐like lipid film [0.46 mg of egg phosphatidylcholine (PC), 0.25 mg of egg phosphatidylethanolamine (PE), 0.11 mg of bovine liver phosphatidylinositol (PI), 0.10 mg 18:1 dioleoyl phosphatidylserine (DOPS), and 0.08 mg tetraoleoyl cardiolipin (TOCL)] was hydrated 1 with ml of assay buffer (10 mM HEPES pH 7.2, 200 mM KCl, 1 mM MgCl2; all lipids were acquired from Avanti Polar Lipids). Hydrated lipid films underwent 10 freeze‐thaw cycles and were extruded 11 times through a 100‐nm‐pore‐size nucleopore track‐etched polycarbonate membrane (Whatman).
Subcellular fractionation
Subcellular fractionation was performed as reported before 49. Briefly, cells were harvested and washed in ice‐cold PBS and then resuspended in an isotonic buffer [250 mM sucrose, 20 mM HEPES (pH 7.5), 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, and protease inhibitors (Complete, Roche)] on ice for 20 min. Phosphatase inhibitors (PhosSTOP; Roche) were added as indicated. After incubation, cells were homogenized with Dounce homogenizer and centrifuged at 800 g for 10 min at 4°C. The remaining supernatant was centrifuged at 8,000 g for 20 min at 4°C to obtain mitochondria‐enriched HM pellet and cytosolic fractions. To obtain S‐100 fractions, the remaining supernatant (cytosolic fraction) was further centrifuged at 100,000 g for 1 h at 4°C. The resulting pellet constituted LM and the supernatant was saved as S‐100. Mitochondria‐enriched pellets were lysed in 1% CHAPS buffer for immunoblot analysis.
Nuclear protein isolation
Cells were washed with ice‐cold phosphate‐buffered saline (PBS) and harvested by centrifugation. Cells were lysed by incubation for 10 min in cold hypotonic buffer [10 mM HEPES/KOH (pH 7.9), 10 mM KCl, 2 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 0.5 mM PMSF, protease inhibitors, and Nonidet P‐40 (0.2%)]. After centrifugation at 15,000 g for 30 s, supernatants containing cytoplasmic proteins were removed. Nuclear protein isolation was carried out by incubation of the pellet for 20 min on ice in a cold saline buffer [20 mM HEPES/KOH (pH 7.9), 1.5 mM MgCl2, 0.2 mM EDTA, 650 mM NaCl, glycerol (25%, v/v), 1 mM DTT, 0.5 mM PMSF, and protease inhibitors]. After centrifugation at 15,000 g for 20 min at 4°C, supernatants were stored as nuclear proteins. GAPDH was used as cytosolic marker, and COXIV was used as mitochondrial marker for immunoblots.
BH3 profiling
BH3 profiling assays and peptide sequences were previously reported 6, 20. Briefly, 0.5 mg of protein/ml mitochondria was suspended in mitochondria buffer (125 mM KCl, 10 mM Tris–MOPS, pH 7.4, 5 mM glutamate, 2.5 mM malate, 1 mM KPO4, and 10 μM EGTA–Tris, pH 7.4) and exposed to BH3 peptides (100 μM) for 40 min at room temperature. Phosphatase inhibitors (PhosSTOP) were added as indicated in the figures to the mitochondria buffer. Cytochrome c release was calculated by dividing the amount of cytochrome c detected in the supernatant fraction by the total amount of cytochrome c in the supernatant and pellet fractions. The amount of cytochrome c in each fraction was determined by ELISA (#SCTC0; R&D Systems) and expressed as % of untreated control. For BH3 experiments using mitochondria treated with S100 fractions, S100 fractions were immunodepleted for Akt by sequential immunoprecipitations using anti‐Akt antibody (Cell Signaling, #9272) and Dynabeads twice. IgG immunoprecipitations were done in parallel as a negative control. Mitochondria were incubated with immunodepleted S100 fractions for 1 h at 30°C, washed twice with mitochondria buffer containing PhosSTOP, and incubated with BH3 peptides for 40 min at room temperature. Cytochrome c release was determined by ELISA, as above. For BH3 experiments using mitochondria treated with recombinant active Akt, mitochondria (0.1 mg/ml) were incubated with recombinant active Akt (1 μg) and ATP (400 μM) for 1 h in kinase assay buffer [25 mM Tris–HCl (pH 7.5), 5 mM beta‐glycerophosphate, 2 mM dithiothreitol (DTT), 0.1 mM Na3VO4, 10 mM MgCl2] for 2 h at 30°C, washed twice with mitochondria buffer containing PhosSTOP, and treated with BH3 peptides for 40 min at room temperature. Cytochrome c release was determined by ELISA, as above.
Mitochondrial permeabilization assay
Mitochondria were purified from Bax−/− Bak−/− DKO MEFs and resuspended in mitochondria buffer (0.5 mg of protein/ml mitochondria) and incubated with tBid and Bax for 1 h at room temperature. tBid and Bax were pre‐incubated with recombinant active Akt (400 ng) and ATP (200 μM) in kinase assay buffer for 2 h at 30°C as indicated some experiments. Pre‐incubation with 120 nM MK‐2206 for 1 h was used to block Akt kinase activity as indicated. Cytochrome c release was determined by ELISA, as above.
Apoptosis and cell viability assays
Cell proliferation kit (MTT; Roche) and alamar blue (Invitrogen) were used to determine cell viability and survival of cells as described by the manufacturer. For Fig 2D, apoptosis was evaluated by Annexin V–FITC and Annexin V–Cy3 (BioVision) staining using a FACSCanto (BD Biosciences) flow cytometer and WinMDI 2.9 software (Scripps Institute).
Confocal microscopy
For Fig 2C, GFP‐tagged protein‐expressing cells were grown in Lab‐Tek II chamber slides (Thermo Fisher Scientific) and co‐stained with MitoTracker Red CMXRos (Molecular Probes, Invitrogen) before fixation in 4% PFA. Monolayer cells fixed with 4% paraformaldehyde were mounted with Vectashield mounting medium with DAPI (Vector Laboratories). Cells were imaged by using spinning disk confocal microscope (Yokogawa) using an Andor iXon EM‐CCD camera with a 40× oil objective. Three channels were captured: (i) DAPI (excitation laser: 405 nm; emission filter: 455/50 nm bandpass); (ii) Alexa 488 (excitation laser: 488 nm; emission filter: 525/36 nm bandpass); and (iii) MitoTracker Red (excitation laser: 561 nm; emission filter: 605/52 nm bandpass). Images were pseudo‐colored and analyzed by using ImageJ software. Co‐localization analysis for GFP‐Bax image was performed using CoLocalizer Pro software for Mac (3.0.2). Pearson's correlation coefficient was used to evaluate the co‐localization of GFP‐Bax and MitoTracker channels.
Live cell imaging and analysis
WT BMK cells, WT BMK cells with a stable expression of mCerulean3‐Bax‐S184E or Bax mCerulean3‐Bax‐S184A, Bax−/− Bak−/− DKO BMK cells, or Bax−/− Bak−/− DKO BMK cells with a stable expression of mCerulean3‐Bax, mCerulean3‐Bax‐S184E, or mCerulean3‐Bax‐S184A were seeded at ~2,000 cells/well in a 384‐well imaging plate (PerkinElmer Cell Carrier Ultra). Twenty hours later, cells were incubated with indicated concentrations of TNF‐α (S0419041; Invitrogen), staurosporine (S5921; Sigma‐Aldrich), panobinostat (A10518; Adooq Bioscience), or DMSO (as a negative control) for 20 h. Where indicated, the cells were also treated with 10 μM caspase inhibitor Q‐OVD‐OPH (03OPH10903; MP Biomedicals) for the 20‐h incubation.
For Figs 5B and EV4C: 30 min prior to imaging, cells were co‐stained with 5 μM DRAQ5 (Biostatus), a 1/1,000 dilution of TMRE (Life technologies) and 1/1,000 dilution of Annexin V conjugated to Alexa 488 which was prepared in our laboratory and used as described previously 50. DRAQ5 staining was used to identify all cells, TMRE, a dye that is only fluorescent at mitochondria when there is a membrane potential, was used to measure loss of mitochondria membrane potential, and Annexin V–Alexa 488 staining was used to identify apoptotic cells. Cells were imaged using an Opera Phenix automated confocal microscope (PerkinElmer) with a 20× air objective. Four channels were captured: (i) DRAQ5 (excitation laser: 640 nm; emission filter: 650–760 nm); (ii) Annexin V–Alexa 488 (excitation laser: 488 nm; emission filter: 500–550); (iii) TMRE (excitation laser: 561 nm; emission filter: 570–630 nm); and (iv) mCerulean3 (excitation laser: 425 nm; emission filter: 435–550 nm). Image acquisition, calculation of intensity features for each channel, and image analysis were performed using Harmony software (PerkinElmer). The DRAQ5 channel was used identify the nucleus and cytoplasm of each cell. TMRE‐negative cells were assigned based on a per‐cell cytoplasmic TMRE intensity less than a threshold of 2 standard deviations above the per‐cell cytoplasmic TMRE intensity in 100 nM STS‐treated WT BMK cells (our cell death positive control). Annexin V–Alexa 488‐positive cells were assigned based on a per‐cell Alexa 488 intensity greater than a threshold of 2 standard deviations above the per‐cell Annexin V–Alexa 488 intensity of untreated DMSO controls. The percentage of apoptosis was calculated by taking the total number of TMRE‐negative and Annexin V–Alexa 488‐positive cells dividing by the total number of cells imaged under each condition. Two technical replicates were performed for each of four independent experiments (n = 4). Each technical replicate was in a separate well, and a minimum of five different fields of view (~50 cells per field) were acquired.
For Fig 5C and D: After 20‐h treatment with caspase inhibitor and either STS or DMSO, cells were co‐stained with 5 μM DRAQ5 (to visualize the nucleus) and 1/1,000 dilution of MitoTracker Red CMXRos 30 min before imaging. Where indicated, the cells were also stained with a 1/1,000 dilution of MitoTracker Green. Cells were imaged on the Opera Phenix automated confocal microscope (PerkinElmer) with water‐immersion objectives at 63× (for representative images, Fig 5C) and 40× (for image analysis, Fig 5D). Three channels were captured: (i) DRAQ5 (excitation laser: 640 nm; emission filter: 650–760 nm); (ii) MitoTracker Red (excitation laser: 561 nm; emission filter: 570–630); and (iii) mCerulean3/MitoTracker Green (excitation laser: 425 nm; emission filter: 435–550). Image acquisition, calculation of intensity features for each channel, and image analysis were performed using Harmony software (PerkinElmer). The DRAQ5 channel was used to identify the nucleus and the cytoplasm of each cell. The average per‐cell Pearson's correlation coefficient between the MitoTracker Red and either MitoTracker Green (positive control for co‐localization to mitochondria) or mCerulean3 in the cytoplasm was calculated using built in co‐localization analysis in Harmony software. Four technical replicates were performed for each of two independent experiments (n = 2). Each technical replicate was in a separate well, and a minimum of 15 different fields of view (≈50 cells per field) were acquired.
For Fig EV4D and E: After 20‐h treatment with caspase inhibitor and either STS or DMSO, cells were stained with 5 μM DRAQ5 (to visualize the nucleus) 30 min before imaging on the Opera Phenix automated confocal microscope (PerkinElmer) with water‐immersion objectives at 63× (for representative images, Fig EV4D) and 40× (for image analysis, Fig EV4E). Two channels were captured: (i) DRAQ5 (excitation laser: 640 nm; emission filter: 650–760 nm); and (ii) mCerulean3 (excitation laser: 425 nm; emission filter: 435–550). Image acquisition, calculation of intensity features for each channel, and image analysis were performed using Harmony software (PerkinElmer). The DRAQ5 channel was used to identify the nucleus and the cytoplasm of each cell. Both the average and standard deviation of the mCerulean3 intensity in the cytoplasm were calculated for each cell. Diffuse and punctate localization patterns have a lower and higher, respectively, per‐cell standard deviation of fluorescence intensity. To identify cells with a punctate localization pattern, the standard deviation in mCerulean3 fluorescence was calculated for all imaged cells. A cell was assigned a punctate localization pattern if the standard deviation of mCerulean3 fluorescence was more than two standard deviations higher than the average standard deviation of mCerulean3 intensity in DMSO‐treated control cells. The percentage of punctate cells was calculated by taking the total number of cells identified as punctate and dividing by the total number of cells identified. Two technical replicates were performed for each of three independent experiments (n = 3). Each technical replicate was in a separate well, and a minimum of 15 different fields of view (≈50 cells per field) were acquired.
Bax membrane permeabilization: ANTS/DPX release assay
For membrane permeabilization assays, a mitochondria‐like lipid film was hydrated with 1 ml of assay buffer supplemented with 12.5 mM of the fluorophore 8‐aminonaphthalene‐1,3,6‐trisulfonic acid (ANTS) and 45 mM of the quencher p‐xylene‐bis‐pyridinium bromide (DPX; Molecular Probes). Liposomes were prepared as above and after extrusion, and unencapsulated ANTS and DPX were removed by passing the liposomes over a 10‐ml CL2B gel‐filtration column (GE Healthcare Life Sciences). ANTS/DPX release assay was performed as previously described 28, 31. Briefly, 8 μl of ANTS/DPX encapsulated liposomes was added to a reaction of 100 μl final volume in assay buffer. Fluorescence of ANTS (λex = 355 nm, λem = 520 nm) was measured over the course of 2 h at 37°C. Percent ANTS release was calculated as % release = [(F final − F 0)/(F 100 − F 0)]*100, where F final is the fluorescence at a 2‐h endpoint, F 0 is the fluorescence in the absence of proteins, and F 100 is the fluorescence of liposomes permeabilized with 0.2% v/v Triton X‐100.
Fluorescence experiments: measuring protein–protein interactions via FRET and Bax insertion into membranes
NBD emission assays and FRET experiments were performed as previously described 28. In a black 96‐well half‐area non‐binding surface assay plate (Corning #3881), 10 μl of 1 mg/ml mitochondria‐like liposomes was added to a reaction of 100 μl final volume in assay buffer. Fluorescence emission of 100 nM Bax 175C‐NBD and Bax 175C‐NBD S184E (λex = 475 nm, λem = 530 nm) incubated with or without 20 nM recombinant cBid was recorded using a Tecan Infinite M1000 plate reader (Tecan) at 2‐h endpoint 37°C. Relative change in NBD emission was expressed as F/F 0 = (F final − F bckg)/(F initial − F bckg), where F bckg is the fluorescence signal of liposomes and buffer, F initial is the fluorescence signal once NBD‐labeled protein was added to the well, and F final is the fluorescence signal at 1‐h endpoint. cBid–Bax FRET was similarly measured; however, the emission of cBid 126C‐DAC donor (λex = 380 nm, λem = 460 nm) was recorded. FRET efficiency was calculated as % FRET efficiency = 1 − [(F DA − F bckg)/(F D − F bckg)] × 100, where F DA is the fluorescence emission of 20 nM cBid 126C‐DAC donor in the presence of 100 nM NBD‐labeled Bax acceptor at 2‐h endpoint, F D is the fluorescence emission of 20 nM cBid 126C‐DAC donor alone in the presence of unlabeled Bax 126C at 2‐h endpoint, and F bckg is the fluorescence signal of liposomes and assay buffer alone. Bax–Bax FRET was measured as cBid–Bax FRET; however, measurements were recorded in a PTI QM‐40 fluorometer (Photon Technology International), using a 1.8‐ml quartz cuvette containing 100 μl lipids in a final reaction volume of 1 ml in assay buffer at 37°C. FRET efficiency was calculated as above; however, F DA is the fluorescence emission of 10 nM DAC‐labeled Bax donor in the presence of 100 nM NBD‐labeled Bax acceptor at 2‐h endpoint, and F D is the fluorescence emission of 10 nM DAC‐labeled Bax donor in the presence of 100 nM unlabeled Bax acceptor.
For Fig 4B, the dissociation constant (KD) for the interaction between cBid and Bax was extracted from the titration curve by fitting the data to Equation (1) which models the FRET that occurs between two interacting proteins. Here, we assume that the binding between cBid (A) and Bax (B) is in equilibrium () 28.
| (1) |
where F fret([B T]) is a function describing the FRET efficiency at any concentration of B. F max is the maximum FRET efficiency given that all of A is bound to B. [A T] and [B T] are the total concentrations of protein A and B, respectively, and KD is described by:
| (2) |
In this case, [A], [B], and [AB] are the equilibrium concentrations of cBid, Bax, and a cBid:Bax heterodimer, respectively, and [A T] and [B T] are the total concentrations of cBid and Bax, respectively.
Ovarian cancer cells
Primary ovarian cancer cells were isolated as described before 51. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki, and it was approved by the ethics committee of Baskent University School of Medicine (KA15/230). Informed consent was obtained from all participants.
Briefly, malignant ascites samples (20–50 ml) were collected from ovarian cancer patients, and cells were pelleted, resuspended in PBS, and separated on a discontinuous gradient consisting of Lymphoprep (Axis‐Shield, Norway), Lymphoprep/Krebs HEPES Ring solution 3:1, and Lymphoprep/Krebs HEPES Ring solution 1:2 (Krebs HEPES Ring solution: 137 mM NaCl, 5.4 mM KCl, 0.34 mM Na2HPO4, 0.35 mM KH2PO4, 8 mM MgSO4, 1 mM HEPES pH 7.4). Cells were then centrifuged at 1,500 × g for 20 min, and tumor cells were collected at the interphase between the top and middle layers, washed, and plated. Cells were grown at 37°C in 5% CO2 in RPMI‐1640 medium supplemented with 10% FCS and 1% Pen/Strep. All patients were chemotherapy naïve, FIGO stage IIIC, with a median age of diagnosis at 61.2 years. Tumors were identified as serous epithelial ovarian carcinoma.
To evaluate apoptotic response in ovarian cancer cells, cells were treated with carboplatin (100 μM), etoposide (50 μM), paclitaxel (100 nM), and ABT‐737 (0.5 μM) for 48 h, and apoptotic cells were evaluated by Annexin V/PI staining and flow cytometry. pBAXS184/BAX ratio was determined by using Image Studio Digits V3.1 software. Correlation of pBAXS184 level and response to drug treatment in ovarian cancer cells was evaluated two‐tailed Spearman correlation by using GraphPad 5.0 software.
Statistical analysis of in vitro data
GraphPad Prism 7.0 software (GraphPad Software, San Diego, CA, USA) was used for data analysis and to determine the significance of statistical differences between data. All data were assumed to have a Gaussian distribution. Homoscedasticity (equal variance) of the data was tested using the Brown–Forsythe test. If the data were homoscedastic (passes the Brown–Forsythe test), then either a one‐way or two‐way ANOVA (where indicated in figure legends) was used followed by followed by post hoc unpaired t‐tests (two‐tailed with α = 0.05) with Bonferroni correction for multiple comparisons. Only in the case of Fig 5D, did the data fail the Brown–Forsythe test and the data had unequal variances. In this case, Welch's ANOVA followed post hoc unpaired Welch's t‐tests (two‐tailed with α = 0.05) with Bonferroni correction for multiple comparisons were used as these statistical tests do not assume homoscedastic data. All P‐values are multiplicity adjusted P‐values calculated in GraphPad Prism and are either listed in the figure legends or in Appendix Tables S1 and S2. Symbols in figures indicate P‐values: ns (P > 0.05), *(P ≤ 0.05), **(P ≤ 0.01), ***(P ≤ 0.001), and ****(P < 0.0001).
Bioinformatics analysis and statistics
CNA analysis
CNA data were obtained from cBioPortal 52, 53. Duplications and deletions were defined as log2CNA > 0.5 or < −0.5, respectively. Association with the Akt pathway was evaluated using a Cochran–Armitage test for trend on the number of Akt pathway genes (AKT1, AKT2, AKT3, PIK3CA, PIK3R1) that were duplicated in patients carrying BAX or BAK duplications. Association with PTEN was evaluated using a Fisher's exact test.
TCGA pan‐cancer gene‐level analysis
This analysis covered expression (n = 9,755), copy number (n = 10,843), and mutation data (n = 6,901) across 42 cancers. Data were obtained from the Cancer Browser 54. This database uses the software GISTIC2 55 to normalize copy number gain or copy number loss, which were defined in the present work as a GISTIC2 call ≥ 1 or ≤ 1, respectively. Gene expression association was evaluated by ranking expression values of the reference gene and applying a two‐tailed unpaired Student's t‐test to evaluate differential gene expression between 1st and 10th deciles across genes. Bonferroni correction was applied to determine the associations that were significant (P‐value cutoff of 0.02 was chosen arbitrarily). High and low Bax copy numbers (Figs 6D and EV5C) were defined as the n patients with the top highest/lowest Bax GISTIC2 call. Upregulated Akt pathway was defined as an increase in AKT1 or AKT2 or PIK3CA or decrease in PTEN copy number.
Author contributions
Conceptualization: JK, OK, BL, AL, and DWA; methodology: JK, OK, GCB, TSA, BL, AL, and DWA; formal analysis: GCB and TSA; investigation: JK, OK, GCB, and TSA; resources: AL and DWA; writing of the original draft: JK; review and editing of the manuscript: JK, OK, TSA., BL, AL, and DWA; supervision: BL, AL, and DWA; and funding acquisition: JK, OK, AL, and DWA.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
O.K. and A.L. would like to gratefully acknowledge financial support from the NIH via R01 CA129974 and P01 CA139980 and from the Terri Brodeur Breast Cancer Foundation. A.L. was a Leukemia and Lymphoma Society Scholar. The work was also supported by Grant FRN12517 from the Canadian Institute of Health Research (CIHR) and a Tier 1 Canada Research Chair to D.W.A. J.K. was a recipient of a doctoral scholarship from the Canadian Breast Cancer Foundation, Ontario Division.
EMBO Reports (2018) 19: e45235
Contributor Information
Anthony Letai, Email: anthony_letai@dfci.harvard.edu.
David W Andrews, Email: david.andrews@sri.utoronto.ca.
References
- 1. Brunelle JK, Letai A (2009) Control of mitochondrial apoptosis by the Bcl‐2 family. J Cell Sci 122: 437–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shamas‐Din A, Kale J, Leber B, Andrews DW (2013) Mechanisms of action of Bcl‐2 family proteins. Cold Spring Harb Perspect Biol 5: a008714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cheng EHYA, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ (2001) BCL‐2, BCL‐X‐L sequester BH3 domain‐only molecules preventing BAX‐ and BAK‐mediated mitochondrial apoptosis. Mol Cell 8: 705–711 [DOI] [PubMed] [Google Scholar]
- 4. Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P et al (2007) Apoptosis initiated when BH3 ligands engage multiple Bcl‐2 homologs, not Bax or Bak. Science 315: 856–859 [DOI] [PubMed] [Google Scholar]
- 5. Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183–192 [DOI] [PubMed] [Google Scholar]
- 6. Certo M, Moore VD, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A (2006) Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL‐2 family members. Cancer Cell 9: 351–365 [DOI] [PubMed] [Google Scholar]
- 7. Ryan JA, Brunelle JK, Letai A (2010) Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4(+) CD8(+) thymocytes. Proc Natl Acad Sci USA 107: 12895–12900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A (2007) Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT‐737. J Clin Invest 117: 112–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chonghaile TN, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore VD, Deng J, Anderson KC, Richardson P, Tai YT et al (2011) Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 334: 1129–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Salmena L, Carracedo A, Pandolfi PP (2008) Tenets of PTEN tumor suppression. Cell 133: 403–414 [DOI] [PubMed] [Google Scholar]
- 11. Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129: 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kennedy SG, Kandel ES, Cross TK, Hay N (1999) Akt protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol 19: 5800–5810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang SW, Denny TA, Steinbrecher UP, Duronio V (2005) Phosphorylation of Bad is not essential for PKB‐mediated survival signaling in hemopoietic cells. Apoptosis 10: 341–348 [DOI] [PubMed] [Google Scholar]
- 14. Ranger AM, Zha J, Harada H, Datta SR, Danial NN, Gilmore AP, Kutok JL, Le Beau MM, Greenberg ME, Korsmeyer SJ (2003) Bad‐deficient mice develop diffuse large B cell lymphoma. Proc Natl Acad Sci USA 100: 9324–9329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gardai SJ, Hildeman DA, Frankel SK, Whitlock BB, Frasch SC, Borregaard N, Marrack P, Bratton DL, Henson PM (2004) Phosphorylation of Bax Ser(184) by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem 279: 21085–21095 [DOI] [PubMed] [Google Scholar]
- 16. Xin MG, Deng XM (2005) Nicotine inactivation of the proapoptotic function of Bax through phosphorylation. J Biol Chem 280: 10781–10789 [DOI] [PubMed] [Google Scholar]
- 17. Simonyan L, Renault TT, da Costa Novais MJ, Sousa MJ, Corte‐Real M, Camougrand N, Gonzalez C, Manon S (2016) Regulation of Bax/mitochondria interaction by AKT. FEBS Lett 590: 13–21 [DOI] [PubMed] [Google Scholar]
- 18. Quast SA, Berger A, Eberle J (2013) ROS‐dependent phosphorylation of Bax by wortmannin sensitizes melanoma cells for TRAIL‐induced apoptosis. Cell Death Dis 4: e839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang QH, Sun SY, Khuri F, Curran WJ, Deng XM (2010) Mono‐ or double‐site phosphorylation distinctly regulates the proapoptotic function of Bax. PLoS One 5: e13393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A (2007) BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT‐737 and conventional chemotherapeutic agents. Cancer Cell 12: 171–185 [DOI] [PubMed] [Google Scholar]
- 21. Kale J, Osterlund EJ, Andrews DW (2018) BCL‐2 family proteins: changing partners in the dance towards death. Cell Death Differ 25: 65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kutuk O, Letai A (2008) Regulation of Bcl‐2 family proteins by posttranslational modifications. Curr Mol Med 8: 102–118 [DOI] [PubMed] [Google Scholar]
- 23. Choudhary GS, Al‐Harbi S, Mazumder S, Hill BT, Smith MR, Bodo J, Hsi ED, Almasan A (2015) MCL‐1 and BCL‐xL‐dependent resistance to the BCL‐2 inhibitor ABT‐199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis 6: e1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamaguchi H, Wang HG (2001) The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene 20: 7779–7786 [DOI] [PubMed] [Google Scholar]
- 25. Han EK, Leverson JD, McGonigal T, Shah OJ, Woods KW, Hunter T, Giranda VL, Luo Y (2007) Akt inhibitor A‐443654 induces rapid Akt Ser‐473 phosphorylation independent of mTORC1 inhibition. Oncogene 26: 5655–5661 [DOI] [PubMed] [Google Scholar]
- 26. Luo Y, Shoemaker AR, Liu X, Woods KW, Thomas SA, de Jong R, Han EK, Li T, Stoll VS, Powlas JA et al (2005) Potent and selective inhibitors of Akt kinases slow the progress of tumors in vivo . Mol Cancer Ther 4: 977–986 [DOI] [PubMed] [Google Scholar]
- 27. Kale J, Chi X, Leber B, Andrews D (2014) Examining the molecular mechanism of bcl‐2 family proteins at membranes by fluorescence spectroscopy. Methods Enzymol 544: 1–23 [DOI] [PubMed] [Google Scholar]
- 28. Lovell JF, Billen LP, Bindner S, Shamas‐Din A, Fradin C, Leber B, Andrews DW (2008) Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135: 1074–1084 [DOI] [PubMed] [Google Scholar]
- 29. Annis MG, Soucie EL, Dlugosz PJ, Cruz‐Aguado JA, Penn LZ, Leber B, Andrews DW (2005) Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J 24: 2096–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Llambi F, Moldoveanu T, Tait SW, Bouchier‐Hayes L, Temirov J, McCormick LL, Dillon CP, Green DR (2011) A unified model of mammalian BCL‐2 protein family interactions at the mitochondria. Mol Cell 44: 517–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Billen LP, Kokoski CL, Lovell JF, Leber B, Andrews DW (2008) Bcl‐XL inhibits membrane permeabilization by competing with Bax. PLoS Biol 6: e147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bellacosa A, Kumar CC, Di Cristofano A, Testa JR (2005) Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res 94: 29–86 [DOI] [PubMed] [Google Scholar]
- 33. Schuyer M, van der Burg ME, Henzen‐Logmans SC, Fieret JH, Klijn JG, Look MP, Foekens JA, Stoter G, Berns EM (2001) Reduced expression of BAX is associated with poor prognosis in patients with epithelial ovarian cancer: a multifactorial analysis of TP53, p21, BAX and BCL‐2. Br J Cancer 85: 1359–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Krajewski S, Blomqvist C, Franssila K, Krajewska M, Wasenius VM, Niskanen E, Nordling S, Reed JC (1995) Reduced expression of proapoptotic gene BAX is associated with poor response rates to combination chemotherapy and shorter survival in women with metastatic breast adenocarcinoma. Can Res 55: 4471–4478 [PubMed] [Google Scholar]
- 35. Nechushtan A, Smith CL, Hsu YT, Youle RJ (1999) Conformation of the Bax C‐terminus regulates subcellular location and cell death. EMBO J 18: 2330–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kolliputi N, Waxman AB (2009) IL‐6 cytoprotection in hyperoxic acute lung injury occurs via PI3K/Akt‐mediated Bax phosphorylation. Am J Physiol Lung Cell Mol Physiol 297: L6–L16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sakakura C, Sweeney EA, Shirahama T, Igarashi Y, Hakomori S, Tsujimoto H, Imanishi T, Ogaki M, Ohyama T, Yamazaki J et al (1997) Overexpression of bax sensitizes breast cancer MCF‐7 cells to cisplatin and etoposide. Surg Today 27: 676–679 [DOI] [PubMed] [Google Scholar]
- 38. Sakakura C, Sweeney EA, Shirahama T, Igarashi Y, Hakomori S, Nakatani H, Tsujimoto H, Imanishi T, Ohgaki M, Ohyama T et al (1996) Overexpression of bax sensitizes human breast cancer MCF‐7 cells to radiation‐induced apoptosis. Int J Cancer 67: 101–105 [DOI] [PubMed] [Google Scholar]
- 39. Lin PH, Pan Z, Zheng L, Li N, Danielpour D, Ma JJ (2005) Overexpression of Bax sensitizes prostate cancer cells to TGF‐beta induced apoptosis. Cell Res 15: 160–166 [DOI] [PubMed] [Google Scholar]
- 40. Bargou RC, Wagener C, Bommert K, Mapara MY, Daniel PT, Arnold W, Dietel M, Guski H, Feller A, Royer HD et al (1996) Overexpression of the death‐promoting gene bax‐alpha which is downregulated in breast cancer restores sensitivity to different apoptotic stimuli and reduces tumor growth in SCID mice. J Clin Investig 97: 2651–2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xu ZW, Friess H, Buchler MW, Solioz M (2002) Overexpression of Bax sensitizes human pancreatic cancer cells to apoptosis induced by chemotherapeutic agents. Cancer Chemother Pharmacol 49: 504–510 [DOI] [PubMed] [Google Scholar]
- 42. Tai YT, Lee S, Niloff E, Weisman C, Strobel T, Cannistra SA (1998) BAX protein expression and clinical outcome in epithelial ovarian cancer. J Clin Oncol 16: 2583–2590 [DOI] [PubMed] [Google Scholar]
- 43. Pietrantonio F, Biondani P, de Braud F, Pellegrinelli A, Bianchini G, Perrone F, Formisano B, Di Bartolomeo M (2012) Bax expression is predictive of favorable clinical outcome in chemonaive advanced gastric cancer patients treated with capecitabine, oxaliplatin, and irinotecan regimen. Transl Oncol 5: 155–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kohler T, Schill C, Deininger MW, Krahl R, Borchert S, Hasenclever D, Leiblein S, Wagner O, Niederwieser D (2002) High Bad and Bax mRNA expression correlate with negative outcome in acute myeloid leukemia (AML). Leukemia 16: 22–29 [DOI] [PubMed] [Google Scholar]
- 45. Kaparou M, Choumerianou D, Perdikogianni C, Martimianaki G, Kalmanti M, Stiakaki E (2013) Enhanced levels of the apoptotic BAX/BCL‐2 ratio in children with acute lymphoblastic leukemia and high‐risk features. Genet Mol Biol 36: 7–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hogarth LA, Hall AG (1999) Increased BAX expression is associated with an increased risk of relapse in childhood acute lymphocytic leukemia. Blood 93: 2671–2678 [PubMed] [Google Scholar]
- 47. Markwardt ML, Kremers GJ, Kraft CA, Ray K, Cranfill PJ, Wilson KA, Day RN, Wachter RM, Davidson MW, Rizzo MA (2011) An improved cerulean fluorescent protein with enhanced brightness and reduced reversible photoswitching. PLoS One 6: e17896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Leu JI, Dumont P, Hafey M, Murphy ME, George DL (2004) Mitochondrial p53 activates Bak and causes disruption of a Bak‐Mcl1 complex. Nat Cell Biol 6: 443–450 [DOI] [PubMed] [Google Scholar]
- 49. Ruiz‐Vela A, Opferman JT, Cheng EH, Korsmeyer SJ (2005) Proapoptotic BAX and BAK control multiple initiator caspases. EMBO Rep 6: 379–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Logue SE, Elgendy M, Martin SJ (2009) Expression, purification and use of recombinant annexin V for the detection of apoptotic cells. Nat Protoc 4: 1383–1395 [DOI] [PubMed] [Google Scholar]
- 51. Wintzell M, Hjerpe E, Avall Lundqvist E, Shoshan M (2012) Protein markers of cancer‐associated fibroblasts and tumor‐initiating cells reveal subpopulations in freshly isolated ovarian cancer ascites. BMC Cancer 12: 359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cline MS, Craft B, Swatloski T, Goldman M, Ma S, Haussler D, Zhu J (2013) Exploring TCGA pan‐cancer data at the UCSC cancer genomics browser. Sci Rep 3: 2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G (2011) GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy‐number alteration in human cancers. Genome Biol 12: R41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shamas‐Din A, Bindner S, Zhu W, Zaltsman Y, Campbell C, Gross A, Leber B, Andrews DW, Fradin C (2013) tBid undergoes multiple conformational changes at the membrane required for Bax activation. J Biol Chem 288: 22111–22127 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File