
Figure EV2. Related to Fig 3: Akt pathway inhibitors change the primed state of mitochondria, the localization of Akt, and the phosphorylation status of Bax S184.
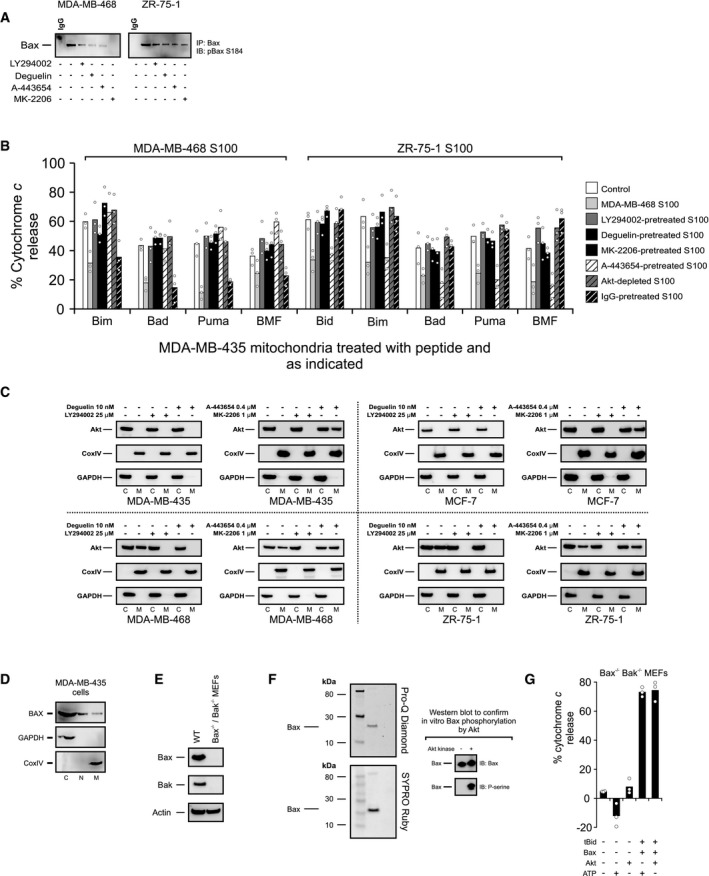
- MDA‐MB‐468 and ZR‐75‐1 cells were treated with 1 μM MK‐2206, 0.4 μM A‐443654, 25 μM LY294002, or 10 nM deguelin where indicated. Phosphorylation of Bax S184 was evaluated using lysates from the treated cells by Western blotting with the indicated antibodies (IB) after immunoprecipitation with the indicated antibodies (IP).
- S100 fractions from untreated cells (MDA‐MB‐468) or from cells treated with direct Akt inhibitors (MK‐2206 [1 μM], A‐443654 [0.4 μM]) or Akt pathway inhibitors (LY294002 [25 μM], deguelin [10 nM]) were isolated. Akt was immunodepleted by sequential immunoprecipitation in untreated S100 fractions, and the efficiency of immunodepletion was tested by immunoblot analysis. IgG was used as a negative control for immunodepletion experiments. The indicated S100 fractions were incubated with MDA‐MB‐435 mitochondrial preparations, and change in priming was assessed by using BH3 profiling. Responses to various BH3 peptides are shown. The phosphatase inhibitor cocktail PhosSTOP was used in all buffers. Cytochrome c release was determined by ELISA. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment. Two‐way ANOVA was conducted on the influence of two independent variables (BH3 peptide, treatment) on cytochrome c release of isolated mitochondria from ABT‐737‐sensitive MDA‐MB‐435 cells that were treated with the S100 fraction isolated from ABT‐737‐resistant MDA‐MB‐468 or ZR‐75‐1 cells. Each treatment was statistically compared to control within each peptide group using t‐tests with Bonferroni correction for multiple comparisons. See Appendix Table S2 for P‐values.
- ABT‐737‐resistant MDA‐MB‐468 and ZR‐75‐1 and ABT‐737‐sensitive MDA‐MB‐435 and MCF‐7 cells were treated with direct Akt inhibitors (MK‐2206 [1 μM], A‐443654 [0.4 μM]) or Akt pathway inhibitors (LY294002 [25 μM], deguelin [10 nM]), and mitochondrial and cytosolic fractions were immunoblotted for Akt. CoxIV was probed as a mitochondrial marker protein, and GAPDH was used as a cytosolic marker protein (M, mitochondrial fraction; C, cytosolic fraction).
- MDA‐MB‐435 cells were lysed and separated into cytosolic (C), mitochondrial (M), or nuclear fractions (N) and then immunoblotted for Bax. GAPDH and CoxIV were immunoblotted for cytosolic and mitochondrial marker proteins, respectively.
- Lack of expression of Bax and Bak in Bax−/− Bak−/− DKO MEFs was detected by immunoblotting with Bax and Bak antibodies. Actin was probed as a loading control. Bax ∆21 antibody was used for IB of Bax.
- Following kinase reaction with recombinant Akt, Bax was separated by SDS–PAGE. The resulting gels were stained with Pro‐Q Diamond (stains phosphoproteins) or SYPRO Ruby (stains total protein) and imaged. Additionally, phosphorylation of recombinant Bax following kinase reaction by Akt was evaluated by immunoblotting with phosphoserine antibody. Bax was blotted as a loading control.
- Akt or ATP did not alter cytochrome c release triggered by tBid/Bax treatment when used alone. Mitochondria from Bax−/− Bak−/− DKO MEFs were directly exposed to Akt or ATP or after incubated Akt or ATP with tBid and Bax as designated. Cytochrome c release was determined by ELISA. Bars indicate the mean of three independent experiments (n = 3). Symbols indicate the mean of at least two technical replicates for each independent experiment.