

Abstract
In this study, sequencing of the 16S rRNA gene targeting the V4-V6 regions was conducted to assess the cecal microbial alterations in response to dietary supplementation with a yeast derived mannan rich fraction (MRF) in standard commercial broiler production settings across four separate broiler trials. The resulting data was analysed to identify consistent changes in the bacterial community structure of the broiler cecum in response to MRF supplementation. Subsequently, the datasets from each individual trial were pooled and analysed for differences between control and MRF supplemented diets at day 35 posthatch. The results from this analysis showed that Phylum Firmicutes was decreased and Phylum Bacteroidetes was increased across all four trials at day 35 posthatch when compared to the control. An extension of the random forest bioinformatics approach to discover a highly relevant set of microbial operational taxonomic units (OTUs) which are indicative of MRF supplementation in the broiler cecum was then used. This approach has enabled the identification of a novel set of yeast-mannan sensitive bacterial OTUs in the cecal microbiome. This information will be helpful in developing potential future nutritional strategies and will be favourable to the poultry industry.
Introduction
The health and nutritional state of the broiler is largely interlinked with the gastrointestinal (GI) microbiome. The GI microbiome contains complex populations of microbes that affect the physiology and health of the host1. It has both direct and indirect effects on gut morphology, the pathogenesis of disease, immune response, nutrient digestion and uptake2. Microbial interactions influence the intestinal environment and as a consequence affect the development and responses of the host immune system against pathogenic and non-pathogenic bacteria3.
Prebiotics have been used as gut microbial modifiers to enhance the host’s natural defence by modulating the GI microbiome4. Prebiotics are defined as ‘selectively fermented ingredients that allow specific changes in both the composition and/or activity in the GI microbiome which in turn confer benefits upon host well being and health’5. As prebiotics, mannan rich fractions (MRFs) are derived from the yeast cell wall of Saccharomyces cerevisiae, specifically the mannoprotein portion of the outer layer. MRF’s prebiotic effect on the gut microbiome benefit the host to allow better feed digestion and absorption, which in turn aid the bird in performing more closely to genetic potential6–8.
Recent studies in many animal species have shown that the GI microbiome mediates key physiological processes and in doing so can be both detrimental and beneficial to the host9,10. However, one consistent observation when studying the GI microbiome is the significant inter-individual variation arising from both genetic and environmental influences11,12. This variation makes it difficult to identify key microbial species that affect the host health or that may be affected as a result of a nutritional intervention. In addition, the complexity of the microbial community and the ability to characterise and culture these same microbes adds difficulty to identifying microbial species indicative of health status. For example, the phyla Firmicutes and Bacteroidetes dominate the broiler cecum microbiome, which is estimated to contain between 500 and 1,000 species with a large percentage of these species yet to be identified13. Early GI microbiome studies focused on culture-dependent techniques. However, the advent of molecular techniques like high-throughput sequencing has revolutionised our ability to characterise the microbiome. Within most complex communities a small number of phylotypes dominate the population structure hiding the appearance of distinct but lowly abundant microbial taxa14. Therefore in order to compare the microbial populations of these complex communities and the influence of supplements, it is necessary to compare thousands of PCR amplified sequences to detect differences in the rare taxa15. Advances in bioinformatic techniques are now making it possible to identify differences in these distinct but low abundant microbial taxa and implicate them with a physiological outcome16,17.
These recent advances in biological data acquisition and sequencing technologies are enabling identification of thousands, sometimes millions, of features for a sample18. A common desire is to combine these data with important metadata and identify the features relevant to understanding a phenotype of interest. The focus of this research was the selection and ranking of features (OTUs) that are relevant to a phenotype of interest (MRF supplemented microbiome) from a number of metagenomic sequencing datasets. A key component underlying our approach is the use of novel analytical methods that are specialised for this type of scenario. An alternative approach from the domain of machine learning called Random Forest (RF) has been proposed for prediction, feature selection, and feature ranking in fields related to computational biomedicine19. An Extended Conditional Inference Forest (ECIF) approach, developed by our team to remove variables less relevant than random predictors and used successfully on multiple applications, is a novel method for better feature selection and ranking accuracy, which are critical tasks for addressing the challenges outlined above. It was based on the Boruta Package20. This method has been used by the team successfully for other research efforts and was applied in this analysis21,22.
In this study we analysed four broiler experiments to identify consistent changes in the bacterial community structure in the cecum in response to dietary supplementation with an MRF supplement. Two of these trials had previously been analysed by 454-sequencing and found that dietary MRF significantly altered the bacterial community composition (BCC) in a commercial production setting6. Specifically MRF supplementation altered the BCC from 7 days through to 35 days supplementation across both trials. Phylum Bacteroidetes appeared to be replacing Phylum Firmicutes as a result of supplementation, with the most noticeable effects after 35 days. PICRUSt was also used to identify differences between the functional potential of the bacterial communities as a result of MRF supplementation which indicated that alterations of the bacterial communities as a result of MRF are likely to alter the functional capability of the cecal microbiome.
Using more complex bioinformatic techniques, the extended random forests approach, we then pooled this published dataset with two additional broiler trial datasets, also sequenced using 454-technology, to identify a highly relevant set of microbial OTUs indicative of MRF supplementation. The description and understanding of nutritional strategies is central to further developing their use in poultry and for the appropriate manipulation of diets to improve poultry performance and health. This analytical approach has enabled us to discover a novel set of candidate OTUs for the classification of an MRF sensitive cecal microbiome. This work has helped understand how the GI microbiota may influence the phenotypic effects usually associated with MRF (Actigen™) supplementation in broilers. This information will be helpful in developing potential future nutritional strategies and will be favourable to the poultry industry23.
Methods
Ethics Statement
Animals were selected from commercial production units and were raised under animal welfare guidelines as set forth by the European Union. Birds were euthanized in accordance with humane killing protocols as set forth in European Union Council Regulation (EC) 1099/2009.
Experimental Design, Sample Collection and Preservation
A total of four separate broiler trials were conducted at two different commercial production sites in Ireland (3 trials) and England (1 trial) (between late August and October). On arrival from the hatchery, 10,000 birds were randomly assigned to commercial production units where they received either a control standard commercial corn-soy diet or a standard diet plus MRF (Actigen™, Alltech Inc, Nicholasville, Kentucky) at the manufacturers recommended inclusion rates (800 g t−1 starter ration and 400 g t−1 grower ration). The birds were reared as per typical commercial production conditions receiving feed and water ad libitum. All other conditions were kept uniform among sheds. At days 7, 21 and 35 post-hatch the intact cecal pouches of 12 randomly selected birds per shed were euthanized and cecal contents were placed into sterile tubes containing sterilised 20% (w/v) maltodextrin which acts as a lyoprotectant. The tubes were frozen on dry ice, transported within 8 hours, lyophilised and stored at −80 °C. Animals were euthanized by cervical dislocation in accordance with humane killing protocols as set forth in EU Regulation 1099/2009.
Two of these trials (2 from Ireland) have been analysed previously for alterations in bacterial community structure as a result of yeast-mannan supplementation and the findings published6. For the purpose of this study, the two trials which had not been analysed previously (herein called Trial A (Ireland) and Trial B (England)) were now analysed separately for alterations in bacterial community structure as a result of yeast-mannan supplementation. Following this analysis, sequences from the day 35 timepoint for all four trials were then pooled to assess for the overall broad scale alterations in BCC. Finally, this pooled sequence dataset of four trials was then analysed using the extended random forests approach to enable the identification of bacterial OTUs which were consistently altered as a result of yeast-mannan supplementation.
Nucleic acid extraction and PCR amplification
Total DNA was extracted in triplicate from each cecal sample using a modified cetyltrimethylammonium bromide (CTAB) extraction method as previously described6. This method uses the ionic detergent CTAB to disrupt cell membranes and a chloroform-isoamyl alcohol mixture that separates contaminants into the organic phase and nucleic acids into the aqueous phase. Resulting DNA was purified using a High Pure PCR product purification kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions and was eluted in a final volume of 50 μL.
The V4-V6 region from the bacterial 16S rRNA operon was amplified from cecal DNA using a universal primer set, 16S-0515F (5′-TGYCAGCMGCCGCGGTA-3′) and 16S-1061R (5′-TCACGRCACGAGCTGACG-3′) tailed on each end with the Roche multiplex identifiers (MIDs). This barcode-based primer approach allowed sequencing of multiple samples in a single sequencing run without the need for physical partitioning. PCR conditions and reagents were similar to those described previously, and a standard concentration of 50 ng of cecal DNA was used in each reaction7. PCR products were purified using a High Pure PCR product purification kit (Roche, Basel, Switzerland).
Sequencing, OTU picking and phylogenetic diversity analysis
Sequencing of the V4-V6 16S rRNA PCR products from the two previously unpublished trials was carried out as described by6. Sequencing was carried out on a Roche 454 GS-FLX titanium platform with average read lengths of approximately 530 bp generated. Following sequencing all barcodes were sorted, removed and reads were quality assessed. The MIRA v. 3.2 assembler was used to assemble the resulting forward and reverse sequences, typically with overlaps over 90% of their length, into contigs and singletons, with a 98% sequence similarity requirement. The assembler has a 454 specific error model and is able to correct for base call errors. Combining forward and reverse sequences, it recovers the high quality V4-V6 amplicon consensus sequence, even for low abundant taxa. For taxonomic assignments, BlastN analysis was used on the assembled reads against an in-house curated version of the RDP database (Michigan State University) release 10.29, containing only non-redundant sequence entries with sufficiently detailed phylogenetic assignments24,25. The best 25 blast hits per contig or singleton were assigned to OTUs from the NCBI taxonomy with MEGAN v.4. MEGAN considers both blast score and taxonomic level of the blast hits in order to pick the appropriate OTU26. OTU counts were corrected for sequence numbers per contig, so as to obtain final OTU tables26. The Generalized Unifrac approach (R Bioconductor v. 2) was used to estimate pairwise distances between samples and establish beta diversity after alpha rarefaction27. PCA analysis was performed to establish two-dimensional projections of samples, reflecting time point and control/treatment status.
Construction of datasets for Random Forest classification
The challenge of finding relevant OTUs was achieved through a pipeline of binary classification and feature selection28. The primary algorithm used was the Extended Random Forest (ERF). The ERF algorithm builds a random forest decision model for a classification problem and through the usage of permutation tests, identifies which OTUs have the most predictive value. The ERF approach has the benefit of capturing variable importance even when there is an order of magnitude more variables than samples, as well as considering variable importance in the presence of multivariate interactions with other variables29.
To understand what OTUs were significant in differentiating MRF supplemented from control groups, a binary classification was established between the two groups and the ERF algorithm used. This step was followed by stability analysis and feature permutation to identify a minimal optimal set of highly relevant OTUs. The original feature set was extended with a permuted set of features, called shadow features. For each original feature, a shadow feature was created by randomly permuting the values from all observations. The measure for feature relevance was the likelihood that a feature has a higher mean variable importance metric (VIM) than its shadow feature. This was estimated by the p-value of a one-sided t-test between each feature and the shadow with the highest mean VIM.
Phylogenetic tree construction
OTUs that were considered to be highly relevant by ERF were used to construct phylogenetic trees using tools in the Ribosome Database Project25.
Results
A recent study by6 identified that MRF supplementation consistently altered the BCC across two concurrent trials6. The goal of this research was to gain new insights into the specific and reproducible effects of MRF supplementation on the cecal BCC. We sought to merge the existing knowledge of these two published trials with two additional broiler trials to identify the dominant changes in the principal taxonomic categories and also to assess if we could identify a consistent set of MRF sensitive bacterial OTUs in the broiler cecum using an extended RFM of classification.
Dietary supplementation with MRF alters bacterial community composition of the broiler cecum
The influence of MRF supplementation on the BCC of the broiler ceca from two previously unanalysed trials, one based in Ireland (trial A) and one based in England (trial B), was analysed by 454 sequencing. Approximately 500,000 high quality reads were obtained from 144 samples collected from the cecal contents of 12 birds on days 7, 21 and 35 posthatch for each of the control and supplemented groups. The sequences were analysed as described by6 and taxonomically assigned at the 3% distance level6. Alterations in the BCC identified in each trial and at each time point were visualised using GUniFrac from the R package. The permutational multivariate analysis of variance (PMAoV) was used to test for significance. Results indicated that cecal BCC was significantly altered as a result of dietary MRF in both trials. MRF supplementation significantly altered BCC at days 7, 21 and 35 posthatch in trial A (Fig. 1a–c) whilst significant alterations at days 7 and 21 but not at 35 days posthatch were observed for trial B (Fig. 2a–c) (P < 0.05).
Figure 1.
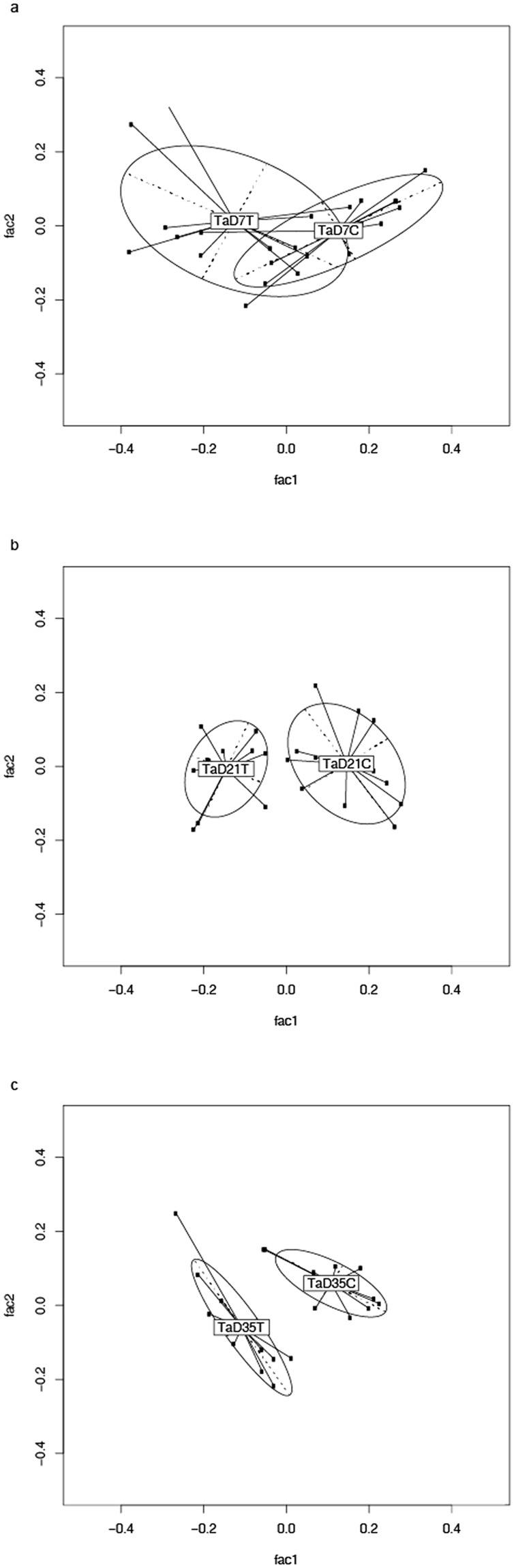
(a–c) PCA analysis was performed on pairwise distance estimates obtained from Megan 4 OTU picking followed by Generalized Unifrac analysis to assess for differences between control and MRF supplemented groups at each time point from Trial A. (a = 7 days posthatch, b = 21 days posthatch, c = 35 days posthatch, n = 12 for each group). T = trial, D = day, C = control, T = MRF supplemented.
Figure 2.
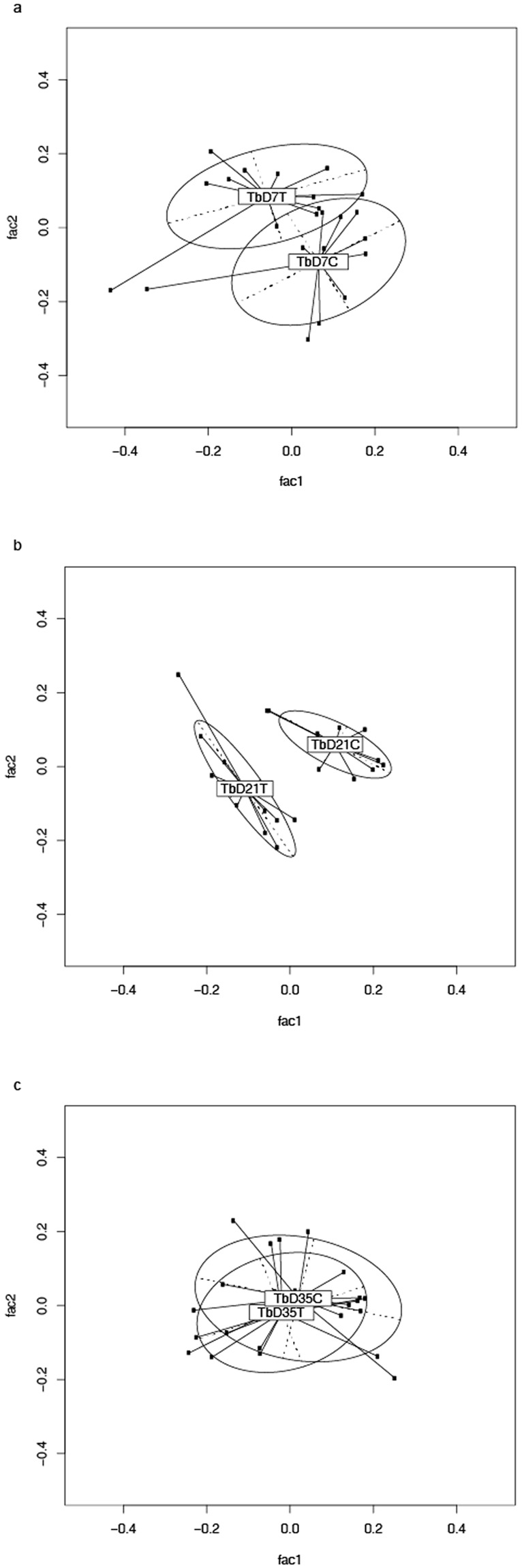
(a–c) PCA analysis was performed on pairwise distance estimates obtained from Megan 4 OTU picking followed by Generalized Unifrac analysis to assess for differences between control and MRF supplemented groups at each time point from Trial B. (a = 7 days posthatch, b = 21 days posthatch, c = 35 days posthatch, n = 12 for each group). T = trial, D = day, C = control, T = MRF supplemented.
Dietary MRF consistently alters bacterial Phyla
Following confirmation that MRF supplementation significantly altered BCC in trials A and B it was decided to pool these datasets with the previously published sequences6 in order to assess trends in alterations of the cecal microbiome at the taxonomic level. This involved combining and rebinning all sequence datasets to assess overall effects on BCC. Analysis was limited to day 35 posthatch given the results from previous experiments which showed the effect of MRF supplementation was strongest at this timepoint. BlastN analysis against the RDP database was used for taxonomic assignments. Eleven bacterial phyla were identified from this combined data set. Phlya identified included Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, Cyanobacteria Tenericutes, Euryarchaeota, Deferribacteres, Verrucomicrobia, Lentisphaerae, and Thermotogae in order of relative abundance. The top six most abundant phyla identified accounted for approximately 98% of all sequences in both the control (Fig. 3a) and MRF-supplemented groups (Fig. 3b). Comparison of the microbiota composition of the combined datasets revealed stark alterations in composition as high as at the phylum level. The relative abundance of Phylum Bacteroidetes increased in the MRF-supplemented birds by an average of 24%; Phylum Firmicutes was decreased on average by 17%, and the Phyla Actinobacteria, Proteobacteria, Cyanobacteria and Tenericutes were all decreased by as a result of MRF supplementation relative to the controls. At a family level, the main increases were observed in the Coriobacteriaceae, Bacteroidaceae, Prevotellaceae, Barnsiellaceae, and Streptococaceae whilst decreases in families Bifidobacteriaceae, Rikenellaceae, Porphyromonadaceae, Lactobacillaceae, Ruminococcaceae, Veillonellaceae, Erysipelotrichaceae, Alcaligenaceae, Campylobacteraceae, Helicobacteraceae, and Enterobacteraceae were observed in the MRF supplemented group compared to the control.
Figure 3.

Bacterial phyla distributions (percentage relative abundance) at day 35 posthatch in control (a) and MRF supplemented (b) groups for the four broiler trials combined (n = 48 for each group).
Identification of yeast-mannan sensitive bacterial OTUs in the cecal microbiome
Results from 454 sequencing showed that the BCC was altered in four broiler trials and two locations. In addition, at 35 days posthatch these alterations were mainly due to increases in phylum Bacteroidetes and decreases in phylum Firmicutes. To identify bacterial OTUs that were indicative of differentiating MRF supplemented and control BCC we employed a supervised machine learning approach; the conditional extended random forest approach. This method also applies an importance score to each OTU identified, indicating how important the selected OTUs are at contributing to differences. In our analysis we considered an OTU to be highly predictive if it’s importance score was at least 0.001. This method identified 47 individual bacterial OTUs across all four trials as being important at defining differences in control and MRF supplemented cecal microbiome with a likelihood of 100% (Fig. 4). Many other OTUs were identified as important (S1 Table) for defining differences in control and MRF supplemented microbiome composition with likelihoods of >97%. It must be noted that the cut-off point at 100% is an arbitrary figure as those ranking higher than the shadow feature are still relevant. We then assessed whether OTUs identified as relevant in distinguishing BCC were either over- or under-represented in MRF supplemented groups. We found 23 OTUs were decreased and 24 OTUs were increased in MRF supplemented birds relative to the control. OTUs were then assessed phylogenetically to identify specific trends related to MRF supplementation (Fig. 5a–c). The 47 OTUs identified were members of 4 bacterial phyla; Bacteroidetes (13), Firmicutes (27), Proteobacteria (6) and Actinobacteria (1).
Figure 4.

Extended conditional inference forest variable importance matrix. OTUs and phylogenetic associations identified as important at defining differences in control and MRF supplemented cecal microbiome.
Figure 5.

(a–c) Phylogenetic trees of OTUs from all 4 datasets identified as significant when comparing Control and MRF supplemented BCC. The orange circles represent OTUs that were more abundant in the MRF supplemented BCC, and blue circles represent OTUs that were less abundant in the MRF supplemented BCC. The size of the circles is directly proportional to the magnitude of the abundance. All of these OTUs were considered to be significant features by ERF. Panel a shows the order Bacteroidales OTUs with Coprococcus catus as the outgroup. Panel b shows the orders Clostridiales, Bifidobacteriales, and Bdellovibrionales with Lactobacillus salivarius as the outgroup. Panel c shows the orders Burkholderiales, Aeromonadales, Selenomonadales, Erysipelotrichales, Lactobacillales, and Kiloniellales with Coprobacillus cateniformis as the outgroup.
OTUs of the phylum Bacteroidetes showed a general trend that identified all but one of 13 OTUs increased in response to MRF supplementation (Fig. 5a). The majority of these OTUs (8 of 13) clustered within the family Bacteroidaceae, and were related to genus Bacteroides. Of the remaining Bacteroidetes-related OTUs which were increased in response to MRF supplementation, three were phylogenetically clustered within family Rikenellaceae, genus Alistipes and one OTU clustered with family Porphyomonadaceae, genus Barnsiella. One OTU relating closely to Parabacteroides spp, was shown to be decreased within this phylum (family Porphyomonadaceae).
Within the phylum Firmicutes, 27 OTUs were identified as important at distinguishing between MRF supplemented and control diets. The majority of the OTUs (18) were decreased by dietary MRF while nine were increased (Fig. 5 Panel b,c). OTUs identified were phylogenetically clustered into three main families; Lachnospiraceae, Ruminococcaceae and Veillonellaceae, while two OTUs clustered into family Erysipelotrichaceae and a single OTU each clustered into families Lactobacillaceae and Clostridiaceae. Within family Lachnospiraceae, a diverse family within the phylum Firmicutes, OTUs were differentially affected in response to MRF, with six OTUs decreased and five increased relative to the control (Fig. 5b). Two OTUs clustered strongly with known bacterial genera closely related to Coprococcus catus and Anaerostipes butyraticus with both increasing in birds given dietary MRF.
The Ruminococcaceae another diverse family within the phylum Firmicutes and together with the Lachnospiraceae make up two of the most abundant families from the order Clostridiales within the intestine30. OTUs identified within this family showed a discernible pattern with 6 of 7 OTUs decreased in response to dietary MRF and clustered strongly with the genus Faecalibacterium (Fig. 5b).
Within family Veillonellaceae five OTUs were identified as important in response to MRF supplementation with two decreasing and three increasing (Fig. 5c). Two OTUs that increased clustered tightly with identifiable bacterial genera Megasphaera elsdenii and Megamonas hypermegale.
A decrease in one OTU of the family Lactobacillaceae, whose closest match was Lactobacillus salivarius and a decrease in two OTUs of family Erysipelotrichaceae which matched closely to Coprobacillus spp, and Clostridium ramosum was observed in response to MRF supplementation. A decrease in one OTU of the family Clostridiaceae, whose closest match was Guggenheimella bovis was observed in response to MRF supplementation (Fig. 5b,c).
There were six OTUs of the phylum Proteobacteria identified as important at distinguishing dietary supplemented microbiota composition. Three of these OTUs increased while three decreased, however, there was no clear pattern in response to MRF supplementation (Fig. 5b,c). The OTUs clustered within the bacterial families Succinovibrionaceae, Alcaligenaceae, Rhodospirillaceae and one poorly defined OTU.
One OTU of the phylum Actinobacteria was identified as important at distinguishing between supplemented groups (Fig. 5b). This OTU was decreased in response to MRF supplementation and clustered within family Bifidobacteriaceae with the closest match to genus Bifidobacterium spp.
Discussion
In studies of the GI microbiome’s effect on health and productivity of the broiler much of the research to date has focused on optimising microbial community composition to aid the bird’s natural defences against pathogens such as Salmonella spp. and pathogenic Clostridia2,31–34. These infections cause poor flock health, reduced performance and increased mortalities resulting in economic losses to producers3,35. In addition, many of the studies into the effects of prebiotics on the GI microbiome and on host health have focused on specific beneficial bacterial populations such as Lactobacillus spp., or Bifidobacterium spp., which are known to affect intestinal health36,37. However, while the effects of prebiotics on poultry health have been well reviewed in both of these regards, there has been less research concerning their impact on the microbiome as a whole, on host health and performance. In recent years, some studies have shown that MRF supplementation in poultry alters the overall bacterial community structure4,38. The results demonstrated in Figs 1 and 2 have substantiated these studies and further confirmed that community structure changes as a result of MRF supplementation regardless of location, age and production environment6–8. The authors interpretation of these results suggests MRFs can be effective for manipulation of the cecal microbiome in the starter, grower and finisher diets as alterations were observed from as early as 7 days through 35 days of supplementation6–8. Understanding whether changes in BCC in response to diet are consistent and if they might contribute to host health is important as there is mounting evidence that the GI microbiome influences animal performance39. A previous broiler study showed there is a correlation between shifts in cecal microbiome composition and the efficiency of feed nutrient conversion40. As such it may prove beneficial to spread the scope of future studies beyond focusing on the ‘traditional’ bacterial genera to better understand nutritional effects on other gut microbes that could contribute to broiler performance2,39. If optimization of microbial balance in the cecal environment leads to good performance, it will be of great importance to establish or develop strategies to provide a competitive advantage for representatives of taxa identified as beneficial. It is with this goal in mind that we sought to taxonomically identify what bacteria were contributing to this shift in composition as a result of MRF supplementation across all four studies.
The sequencing datasets of all 4 studies were pooled and taxonomic analysis showed notable shifts at the phylum level between control and MRF supplemented microbiomes. A review by41 found that phylum Firmicutes dominated the broiler cecal microbiome accounting for an average of 70% of all sequences with Bacteroidetes (12%) and Proteobacteria (9%) accounting for the second and third most abundant phyla. These relative abundances are similar to that observed in the control groups of the four broiler trials discussed in this study. However, in the cecal microbiome of the MRF supplemented birds across the four trials assessed in this present study, a large shift in the bacterial taxonomic composition to a Bacteroidetes prominent profile was observed. Changes in bacterial profile corresponding to an increase in bacterial complexity have generally been associated with improved host health and microbiome stability with decreased bacterial complexity associated with higher incidence of disease42–45. In particular, increases in the phylum Bacteroidetes within the gut microbiome have been associated with many favourable outcomes. For example, intestinal microbial health in humans has been restored through microbiota transplantation from a healthy to a diseased individual resulting in a recovery of the microbial balance in the gut46. The recipient changed from a Firmicutes and Bacteroidetes deficient configuration to a community dominated by Bacteroides spp, resulting in the disappearance of disease like symptoms in the patient46. If a similar microbial balance in the poultry microbiome could lead to reduced incidence of enteric disease, thereby reducing the need for antibiotic use, this would be beneficial to both producers and consumers2,47. A study by48 examined the BCC changes in high feed conversion ratio (FCR) and low-FCR broilers and found that those birds with a lower FCR tended to be associated with higher levels of Bacteroidetes12,48. Enriched Bacteroidetes and depleted Firmicutes phyla were also noted in differences in intestinal microbiota between European and African children with the latter having higher levels of Bacteroidetes49. This has been hypothesised to be linked with differences in diet with increased Bacteroidetes allowing improvement of energy uptake from a typical fibre-rich African diet. Increased fibre digestion as a result of an increase in Bacteroidetes related bacteria could be beneficial to poultry farmers as more energy could be extracted from the diet resulting in improved feed efficiency50. A more varied taxonomic composition may also influence the stability of the microbiota due to an increased functional capacity that accompanies a more complex microbiome and allowing for quicker adaptation to change51. Previously, shifts in Firmicutes-dominant to Bacteroidetes-dominant bacterial profiles have been predicted to alter the functional potential of the microbiome by enriching pathways predicated to be associated with energy metabolism and carbon fixation in broilers6. There is much evidence in the literature to suggest that MRF supplementation is linked with improved broiler health and performance52,53 however, whether a link exists between this consistent phylogenetic shift as a result of MRF supplementation and host health and performance is yet to be elucidated.
Having observed this consistent higher level taxonomic shift between control and MRF supplemented birds we decided to seek methods of analysis which would allow identification of a specific set of bacterial OTUs which distinguished the MRF supplemented microbiome from the control. An extension of the random forest approach was therefore chosen to identify bacterial OTUs consistent with MRF supplementation from the sequencing datasets. This bioinformatic method was developed to analyse high dimensional omics datasets to allow identification of biomarkers. Using this classifier we have identified a consistent set of bacterial OTUs that are sensitive to MRF supplementation across all four trials. OTUs were identified from 4 main bacterial phyla and represent candidate biomarkers of MRF supplementation, with many having noted benefits in the literature for improved broiler health and performance. For example OTU candidates identified from phylum Bacteroidetes were mostly associated with family Bacteroidaceae and related mainly to Bacteroides spp. All eight of these OTUs increased in response to MRF supplementation. Bacteroides spp., are common bacteria in the intestine, involved in many important metabolic activities, including polysaccharide degradation, carbohydrate fermentation, and prevention of pathogen colonization54,55. Bacteroides spp., along with Alistipes spp., of the family Rikenellaceae are some of the main short chain fatty acid producers in the intestine56. An increase in short chain fatty acid production could be beneficial for gut health57,58. Barnesiella spp., have been shown to inhibit colonization by vancomycin-resistant Enterococci in mice59. The single Bacteroidetes-related OTU that decreased in response to dietary MRF clustered with the genus Parabacteroides of the family Porphyromonadaceae, a common bacterium identified in the poultry cecum for which the function is unknown41. In general, the results shown here suggest that phylum Bacteroidetes are positively influenced by MRF supplementation in the broiler cecum.
The influence of MRF supplementation on candidate OTUs identified from the phylum Firmicutes is not as clear as that of the Bacteroides phylum. Within this very diverse phylum OTUs were clustered into three main bacterial families; Lachnospiraceae, Ruminococcaceae and Veillonellaceae. Within the family lachnospiraceae it was difficult to define a clear trend with regard to MRF supplementation as 6 OTUs decreased and 5 OTUs increased. Two OTUs clustered strongly with the known genera Coprococcus catus and Anaerostipes butyraticus. Increases in OTUs related to these species may have a positive influence on broiler production by providing energy to the host in the form of short chain fatty acids. Coprococcus catus produces butyrate from fructose and produces propionate from lactate. Anaerostipes butyraticus is a known butyrate producer which is important for gut health60. These OTUs may be part of a bacterial network responsible for biosynthesis of the major microbial metabolites resulting from carbohydrate fermentation.
Candidate OTUs identified from the family Ruminococcaceae appeared to show a more specific trend as a result of dietary MRF inclusion with 6/7 OTUs decreased. These OTUs tended to cluster strongly with the genus Faecalibacterium spp. which are frequently found in the mammalian gut environment and have commonly been associated with gut health30. In studies of the poultry microbiome, correlations have been found between the co-occurrence of Campylobacter spp. and Faecalibacterium spp.61–63. These authors have shown that Faecalibacterium spp., presence typically indicates increased Campylobacter spp. presence. This might suggest a positive role for decreased Faecalibacterium-related OTUs in the broiler cecum, as a reduction in the presence of Campylobacter spp. in the broiler intestine is of significant interest to poultry producers. Further in-depth studies would be required to confirm these associations.
The response of candidate OTUs from family Veillonellaceae to MRF supplementation was also variable with two OTUs decreased and 3 OTUs increased. Of possible interest are two OTUs which clustered strongly with Megasphaera esldenii and Megamonas spp. Megasphaera esldenii has been investigated as a probiotic for ruminants as it may provide benefits for energy balance and animal productivity owing to its capability of producing various volatile fatty acids in the rumen64. Megamonas spp. have been associated with Campylobacter jejuni suppression in broilers, increasing bacterial OTUs which suppress this zoonotic pathogen would be beneficial to both poultry producers and human health65.
A decrease in one OTU of the family Lactobacillaceae, whose closest match was Lactobacillus salivarius was observed in response to MRF supplementation. Lactobacillus spp. are noted as probiotic bacteria and typically an increase in this OTU would be desirable66. However, although there are some strains of Lactobacillus known to improve performance some are retailed as weight loss probiotics and others reported to reduce obesity67. In a study by68 they found that some Lactobacillus spp, correlated with a negative effect on performance in poultry. This highlights that specific OTUs need to be identified and verified as having either beneficial or negative effects on health and performance in each candidate host and that not all strains or species of a particular genus can have the same effect67. A decrease in one OTU of the family Clostridiaceae, whose closest match was Guggenheimella bovis was observed in response to MRF supplementation. This bacterium has been associated with digital dermatitis in cows leading to production losses for farmers69. This genus of bacteria is not frequently identified in the poultry GI tract. Two OTUs related to family Erysipelotrichaeae were also shown to decrease in response to MRF supplementation relating to Clostridium ramosum and Coprobacillus cateniformis. Members of the family Erysipelotrichaeae have been implicated in weight gain in obese humans and also identified as potential probiotic candidates in poultry due to their association with improved FCR70,71. However, this is a diverse family with varying functional capabilities making it difficult to suggest a potential positive or negative role for its reduction in terms of MRF supplementation.
There were six OTUs of the phylum Proteobacteria identified as important at distinguishing dietary supplemented microbiota composition. Some of these OTUs increased while others decreased however, there was no clear pattern in response to MRF supplementation (Fig. 5b,c). The OTUs clustered within the bacterial families Succinovibrionaceae, Alcaligenaceae, Rhodospirillaceae and one poorly defined OTU. Very little is known with regard to the possible relationship of these OTUs to poultry intestinal health. This further highlights the need to expand our scope of investigation beyond the ‘traditional’ microbiome. One OTU related to Bifidobacterium spp., of the phylum Actinobacteria was also shown to decrease. Similar to Lactobacillus spp., Bifidobacterium spp., are also commonly used as probiotics in poultry but not all species of Bifidobacterium are beneficial to improving broiler performance72,73. Further investigations of this specific OTU and its possible implications on host health would be required to understand the possible functional impact of its reduction as a result of MRF supplementation.
Conclusions
Based on this large scale survey it is clear that MRF supplementation has a consistent and reproducible effect at altering BCC most notably by causing a large shift in the relative abundance of the Firmicutes and Bacteroidetes bacterial phyla. These higher level bacterial community changes have been implicated with improved host health in many species and in this case may be indicative of an MRF supplemented phenotype. More specifically, consistent differences were noted at the OTU level, mainly on OTUs related to Ruminococcaceae and Bacteroidaceae. These results were revealed using the random forest approach and would not have been detected by conventional bacterial community analysis methods due to the inherent problems with inferring associations using relative abundances74. The identification of diet sensitive bacterial OTUs opens possibilities to further investigate if changes in specific OTUs are implicated in specific phenotypic outcomes associated with diet. The 16S rRNA gene sequences generated in this study have established the basis for developing quantitative assays for the enumeration and identification of specific yeast-mannan sensitive OTUs.
Electronic supplementary material
Acknowledgements
The authors would like to thank Dennis Roche for his assistance with sample collection, preparation and DNA extraction and Brian Fay for his assistance with DNA extraction.
Author Contributions
A.C. and R.M. conceived and designed the experiment. A.C. performed experiments, analysed data and prepared manuscript. N.R., M.W., L.A., C.B. and B.W. performed extended bioinformatic analysis, produced data images and aided in manuscript preparation.
Data Availability
Sequencing data are accessible in the European Nucleotide Archive (Short Read Archive) under accession numbers ERP009698 and ERP016138.
Competing Interests
Dr. Corrigan and Dr. Murphy are employed by Alltech Biotechnology which funded the work within this manuscript. Alltech Biotechnology produce Actigen™. N. Russell, M. Welge, L. Auvil, C. Bushell, B.A., White. are all employees of the University of Illinois and received payment for their work.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-31438-x.
References
- 1.Yeoman CJ, White BA. Gastrointestinal Tract Microbiota and Probiotics in ProductionAnimals. Annu. Rev. Anim. Biosci. 2014;2:469–486. doi: 10.1146/annurev-animal-022513-114149. [DOI] [PubMed] [Google Scholar]
- 2.Rinttilä T, Apajalahti J. Intestinal microbiota and metabolites—Implications for broiler chicken health andperformance1. J. Appl. Poult. Res. 2013;22:647–658. doi: 10.3382/japr.2013-00742. [DOI] [Google Scholar]
- 3.Yegani M, Korver DR. Factors affecting intestinal health in poultry. Poult. Sci. 2008;87:2052–2063. doi: 10.3382/ps.2008-00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang Y, Iji PA, Choct M. Dietary modulation of gut microflora in broiler chickens: a review of the role of six kinds of alternatives to in-feed antibiotics. World’s Poult. Sci. J. 2009;65:97–114. doi: 10.1017/S0043933909000087. [DOI] [Google Scholar]
- 5.Rastall RA, Gibson GR. Recent developments in prebiotics to selectively impact beneficial microbes and promote intestinal health. Curr. Opin. Biotechnol. 2015;32:42–46. doi: 10.1016/j.copbio.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Corrigan A, de Leeuw M, Penaud-Frézet S, Dimova D, Murphy RA. Phylogenetic and Functional Alterations in Bacterial Community Compositions in Broiler Ceca as a Result of Mannan Oligosaccharide Supplementation. Appl. Environ. Microbiol. 2015;81:3460–3470. doi: 10.1128/AEM.04194-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corrigan A, Horgan K, Clipson N, Murphy RA. Effect of Dietary Supplementation with a Saccharomyces cerevisiae Mannan Oligosaccharide on the Bacterial Community Structure of Broiler Cecal Contents. Appl. Environ. Microbiol. 2011;77:6653–6662. doi: 10.1128/AEM.05028-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corrigan A, Horgan K, Clipson N, Murphy RA. Effect of dietary prebiotic (mannan oligosaccharide) supplementation on the caecal bacterial community structure of turkeys. Microb. Ecol. 2012;64:826–836. doi: 10.1007/s00248-012-0046-6. [DOI] [PubMed] [Google Scholar]
- 9.Sekirov I, Russell SL, Antunes LCM, Finlay BB. Gut Microbiota in Health and Disease. Physiol. Rev. 2010;90:859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- 10.Marchesi JR, et al. The gut microbiota and host health: a new clinical frontier. Gut. 2016;65:330–339. doi: 10.1136/gutjnl-2015-309990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salonen A, et al. Impact of diet and individual variation on intestinal microbiota composition and fermentation products in obese men. ISME J. 2014;8:2218–2230. doi: 10.1038/ismej.2014.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stanley D, Geier MS, Hughes RJ, Denman SE, Moore RJ. Highly Variable Microbiota Development in the Chicken Gastrointestinal Tract. PLoS ONE. 2013;8:e84290. doi: 10.1371/journal.pone.0084290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deusch S, Tilocca B, Camarinha-Silva A, Seifert J. News in livestock research — use of Omics-technologies to study the microbiota in the gastrointestinal tract of farm animals. Comput. Struct. Biotechnol. J. 2015;13:55–63. doi: 10.1016/j.csbj.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gagic D, Maclean PH, Li D, Attwood GT, Moon CD. Improving the genetic representation of rare taxa within complex microbial communities using DNA normalization methods. Mol. Ecol. Resour. 2015;15:464–476. doi: 10.1111/1755-0998.12321. [DOI] [PubMed] [Google Scholar]
- 15.Zhou J, et al. High-Throughput Metagenomic Technologies for Complex Microbial Community Analysis: Open and Closed Formats. mBio. 2015;6:e02288–02214. doi: 10.1128/mBio.02288-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beck D, Foster JA. Machine Learning Techniques Accurately Classify Microbial Communities by Bacterial Vaginosis Characteristics. PLoS ONE. 2014;9:e87830. doi: 10.1371/journal.pone.0087830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ross EM, Moate PJ, Marett LC, Cocks BG, Hayes BJ. Metagenomic Predictions: From Microbiome to Complex Health and Environmental Phenotypes in Humans and Cattle. PLoS ONE. 2013;8:e73056. doi: 10.1371/journal.pone.0073056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pengyi Y, Yee Hwa Y, Bing BZ, Albert YZ. A Review of Ensemble Methods in Bioinformatics. Curr. Bioinform. 2010;5:296–308. doi: 10.2174/157489310794072508. [DOI] [Google Scholar]
- 19.Brieman L. Random Forests. Mach. Learn. 2001;45:5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- 20.Kursa MB, Rudnicki WR. Feature Selection with the Boruta Package. J. Stat. Softw. 2010;36:1–13. doi: 10.18637/jss.v036.i11. [DOI] [Google Scholar]
- 21.Braundmeier-Fleming, A. et al. Stool-based biomarkers of interstitial cystitis/bladder pain syndrome. Sci. Rep. (6) 26083, (2016). [DOI] [PMC free article] [PubMed]
- 22.Hagler, M. et al. Identification of Novel microRNA profiles in patients with Myxomatous Mitral Vlave Disease. American Heart Association, Orlando, FL (2015).
- 23.Corrigan, A. & Murphy, R. In 67th Annual Meeting of the European Association for Animal Production. (ed EAAP scientific committee) 323 (Wageningen Academic Publishers).
- 24.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 25.Cole JR, et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42:633–642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitra S, Stark M, Huson DH. Analysis of 16S rRNA environmental sequences using MEGAN. BMC genomics. 2011;12(Suppl 3):S17. doi: 10.1186/1471-2164-12-S3-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen J, et al. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics. 2012;28:2106–2113. doi: 10.1093/bioinformatics/bts342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen X, Ishwaran H. Random Forests for Genomic Data Analysis. Genomics. 2012;99:323–329. doi: 10.1016/j.ygeno.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winham SJ, et al. SNP interaction detection with Random Forests in high-dimensional genetic data. BMC Bioinformatics. 2012;13:1–13. doi: 10.1186/1471-2105-13-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biddle A, Stewart L, Blanchard J, Leschine S. Untangling the Genetic Basis of Fibrolytic Specialization by Lachnospiraceae and Ruminococcaceae in Diverse Gut Communities. Diversity. 2013;5:627–640. doi: 10.3390/d5030627. [DOI] [Google Scholar]
- 31.McDevitt RM, Brooker JD, Acamovic T, Sparks NHC. Necrotic enteritis; a continuing challenge for the poultry industry. World’s Poult. Sci. J. 2006;62:221–247. doi: 10.1079/WPS200593. [DOI] [Google Scholar]
- 32.Santos EG, et al. Protective effect of mannan oligosaccharides against early colonization by Salmonella Enteritidis in chicks is improved by higher dietary threonine levels. J. Appl. Microbiol. 2013;114:1158–1165. doi: 10.1111/jam.12108. [DOI] [PubMed] [Google Scholar]
- 33.Baurhoo B, Ferket P, Ashwell CM, de Oliviera J, Zhao X. Cell Walls of Saccharomyces cerevisiae Differentially Modulated Innate Immunity and Glucose Metabolism during Late Systemic Inflammation. PLoS ONE. 2012;7:e30323. doi: 10.1371/journal.pone.0030323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yitbarek A, et al. Innate immune response to yeast-derived carbohydrates in broiler chickens fed organic diets and challenged with Clostridium perfringens. Poult. Sci. 2012;91:1105–1112. doi: 10.3382/ps.2011-02109. [DOI] [PubMed] [Google Scholar]
- 35.Roberts T, et al. New issues and science in broiler chicken intestinal health: intestinal microbial composition, shifts, and impacts. World’s Poult. Sci. J. 2015;71:259–270. doi: 10.1017/S0043933915000276. [DOI] [Google Scholar]
- 36.Mika, A. et al. Dietary prebiotics increase Bifidobacterium spp. and Lactobacillus spp. in the gut and promote stress resistance. Brain Behav. Immun. 40(Supplement), e45, (2014).
- 37.Mookiah S, Sieo CC, Ramasamy K, Abdullah N, Ho YW. Effects of dietary prebiotics, probiotic and synbiotics on performance, caecal bacterial populations and caecal fermentation concentrations of broiler chickens. J. Sci. Food Agr. 2014;94:341–348. doi: 10.1002/jsfa.6365. [DOI] [PubMed] [Google Scholar]
- 38.Sugiharto S. Role of nutraceuticals in gut health and growth performance of poultry. Journal of the Saudi Society of Agricultural Sciences. 2016;15:99–111. doi: 10.1016/j.jssas.2014.06.001. [DOI] [Google Scholar]
- 39.Roberts T, et al. New issues and science in broiler chicken intestinal health: Emerging technology and alternative interventions. J. Appl. Poult. Res. 2015;24:257–266. doi: 10.3382/japr/pfv023. [DOI] [Google Scholar]
- 40.Torok VA, et al. Identification and characterization of potential performance-related gut microbiotas in broiler chickens across various feeding trials. Appl. Environ. Microbiol. 2011;77:5868–5878. doi: 10.1128/AEM.00165-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei S, Morrison M, Yu Z. Bacterial census of poultry intestinal microbiome. Poult. Sci. 2013;92:671–683. doi: 10.3382/ps.2012-02822. [DOI] [PubMed] [Google Scholar]
- 42.Turnbaugh PJ, et al. The human microbiome project: exploring the microbial part of ourselves in a changing world. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flores GE, et al. Temporal variability is a personalized feature of the human microbiome. Genome Biol. 2014;15:531. doi: 10.1186/s13059-014-0531-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lloyd-Price J, Abu-Ali G, Huttenhower C. The healthy human microbiome. Genome Med. 2016;8:51. doi: 10.1186/s13073-016-0307-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stecher B, et al. Like Will to Like: Abundances of Closely Related Species Can Predict Susceptibility to Intestinal Colonization by Pathogenic and Commensal Bacteria. PLOS Pathog. 2010;6:e1000711. doi: 10.1371/journal.ppat.1000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J. Clin. Gastroenterol. 2010;44:354–360. doi: 10.1097/MCG.0b013e3181c87e02. [DOI] [PubMed] [Google Scholar]
- 47.Mead G, et al. Scientific and technical factors affecting the setting of Salmonella criteria for raw poultry: a global perspective. J. Food Prot. 2010;73:1566–1590. doi: 10.4315/0362-028X-73.8.1566. [DOI] [PubMed] [Google Scholar]
- 48.Stanley D, et al. Intestinal microbiota associated with differential feed conversion efficiency in chickens. Appl. Microbiol. Biotechnol. 2012;96:1361–1369. doi: 10.1007/s00253-011-3847-5. [DOI] [PubMed] [Google Scholar]
- 49.De Filippo C, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mateos GG, Jiménez-Moreno E, Serrano MP, Lázaro RP. Poultry response to high levels of dietary fiber sources varying in physical and chemicalcharacteristics1. J. Appl. Poult. Res. 2012;21:156–174. doi: 10.3382/japr.2011-00477. [DOI] [Google Scholar]
- 51.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spring P, Wenk C, Connolly A, Kiers A. A review of 733 published trials on Bio-Mos®, a mannan oligosaccharide, and Actigen®, a second generation mannose rich fraction, on farm and companion animals. J. Appl. Anim. Nutr. 2015;3:e8. doi: 10.1017/jan.2015.6. [DOI] [Google Scholar]
- 53.Hooge DM, Connolly A. Meta-Analysis Summary of Broiler Chicken Trials with Dietary Actigen® (2009–2011) Clin. Microbiol. Rev. 2011;10:819–824. [Google Scholar]
- 54.Sergeant MJ, et al. Extensive Microbial and Functional Diversity within the Chicken Cecal Microbiome. PLoS ONE. 2014;9:e91941. doi: 10.1371/journal.pone.0091941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wexler HM. Bacteroides: the Good, the Bad, and the Nitty-Gritty. Clin. Microbiol. Rev. 2007;20:593–621. doi: 10.1128/CMR.00008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mohd Shaufi MA, Sieo CC, Chong CW, Gan HM, Ho YW. Deciphering chicken gut microbial dynamics based on high-throughput 16S rRNA metagenomics analyses. Gut Pathog. 2015;7:1–12. doi: 10.1186/s13099-015-0051-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.den Besten G, et al. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013;54:2325–2340. doi: 10.1194/jlr.R036012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khan SH, Iqbal J. Recent advances in the role of organic acids in poultry nutrition. J. Appl. Anim. Res. 2016;44:359–369. doi: 10.1080/09712119.2015.1079527. [DOI] [Google Scholar]
- 59.Ubeda C, et al. Intestinal Microbiota Containing Barnesiella Species Cures Vancomycin-Resistant Enterococcus faecium Colonization. Infect. Immun. 2013;81:965–973. doi: 10.1128/IAI.01197-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eeckhaut V, et al. Anaerostipes butyraticus sp. nov., an anaerobic, butyrate-producing bacterium from Clostridium cluster XIVa isolated from broiler chicken caecal content, and emended description of the genus Anaerostipes. Int. J. Syst. Evol. Microbiol. 2010;60:1108–1112. doi: 10.1099/ijs.0.015289-0. [DOI] [PubMed] [Google Scholar]
- 61.Oakley BB, et al. The poultry-associated microbiome: network analysis and farm-to-fork characterizations. PLoS One. 2013;8:e57190. doi: 10.1371/journal.pone.0057190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaakoush N, et al. The interplay between Campylobacter and Helicobacter species and other gastrointestinal microbiota of commercial broiler chickens. Gut Pathog. 2014;6:18. doi: 10.1186/1757-4749-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thibodeau A, et al. Chicken Caecal Microbiome Modifications Induced by Campylobacter jejuni Colonization and by a Non-Antibiotic Feed Additive. PLoS ONE. 2015;10:e0131978. doi: 10.1371/journal.pone.0131978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marx H, et al. Genome Sequence of the Ruminal Bacterium Megasphaera elsdenii. J. Bacteriol. 2011;193:5578–5579. doi: 10.1128/JB.05861-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scupham AJ, Jones JA, Rettedal EA, Weber TE. Antibiotic Manipulation of Intestinal Microbiota To Identify Microbes Associated with Campylobacter jejuni Exclusion in Poultry. Appl. Environ. Microbiol. 2010;76:8026–8032. doi: 10.1128/AEM.00678-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ehrmann MA, Kurzak P, Bauer J, Vogel RF. Characterization of lactobacilli towards their use as probiotic adjuncts in poultry. J. Appl. Microbiol. 2002;92:966–975. doi: 10.1046/j.1365-2672.2002.01608.x. [DOI] [PubMed] [Google Scholar]
- 67.Fåk F, Bäckhed F. Lactobacillus reuteri Prevents Diet-Induced Obesity, but not Atherosclerosis, in a Strain Dependent Fashion in Apoe−/− Mice. PLoS ONE. 2012;7:e46837. doi: 10.1371/journal.pone.0046837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Torok V, Allison G, Percy N, Ophel-Keller K, Hughes R. Influence of antimicrobial feed additives on broiler commensal posthatch gut microbiota development and performance. Appl. Environ. Microbiol. 2011;77:3380–3390. doi: 10.1128/AEM.02300-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schlafer S, et al. Involvement of Guggenheimella bovis in digital dermatitis lesions of dairy cows. Vet. Microbiol. 2008;128:118–125. doi: 10.1016/j.vetmic.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 70.Stanley D, Hughes RJ, Geier MS, Moore RJ. Bacteria within the Gastrointestinal Tract Microbiota Correlated with Improved Growth and Feed Conversion: Challenges Presented for the Identification of Performance Enhancing Probiotic Bacteria. Front. Microbiol. 2016;7:187. doi: 10.3389/fmicb.2016.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang H, et al. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. USA. 2009;106:2365–2370. doi: 10.1073/pnas.0812600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jung SJ, Houde R, Baurhoo B, Zhao X, Lee BH. Effects of galacto-oligosaccharides and a Bifidobacteria lactis-based probiotic strain on the growth performance and fecal microflora of broiler chickens. Poult. Sci. 2008;87:1694–1699. doi: 10.3382/ps.2007-00489. [DOI] [PubMed] [Google Scholar]
- 73.Stanley D, Hughes RJ, Moore RJ. Microbiota of the chicken gastrointestinal tract: influence on health, productivity and disease. Appl. Microbiol. Biotechnol. 2014;98:4301–4310. doi: 10.1007/s00253-014-5646-2. [DOI] [PubMed] [Google Scholar]
- 74.Friedman J, Alm EJ. Inferring Correlation Networks from Genomic Survey Data. PLoS Comput. Biol. 2012;8:e1002687. doi: 10.1371/journal.pcbi.1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data are accessible in the European Nucleotide Archive (Short Read Archive) under accession numbers ERP009698 and ERP016138.