

Abstract
The endoplasmic reticulum (ER) is a critical organelle for protein synthesis, folding and modification, and lipid synthesis and calcium storage. Dysregulation of ER functions leads to the accumulation of misfolded- or unfolded-protein in the ER lumen, and this triggers the unfolded protein response (UPR), which restores ER homeostasis. The UPR is characterized by three distinct downstream signaling pathways that promote cell survival or apoptosis depending on the stressor, the intensity and duration of ER stress, and the cell type. Mammalian cells express the UPR transducers IRE1, PERK, and ATF6, which control transcriptional and translational responses to ER stress. Direct links between ER stress and immune responses are also evident, but the mechanisms by which UPR signaling cascades are coordinated with immunity remain unclear. This review discusses recent investigations of the roles of ER stress in immune responses that lead to differentiation, maturation, and cytokine expression in immune cells. Further understanding of how ER stress contributes to the pathogenesis of immune disorders will facilitate the development of novel therapies that target UPR pathways.
Keywords: ER stress, immune response, inositol requiring enzyme 1 (IRE1), unfolded protein response (UPR), X-box binding protein 1 (XBP1)
INTRODUCTION
Secreted or transmembrane proteins are synthesized, folded, and modified in the endoplasmic reticulum (ER), which also plays crucial roles in lipid biosynthesis, calcium storage, and detoxification (Braakman and Bulleid, 2011; Chen et al., 2010; Marisa et al., 2018)(Fig. 1). Upon entry into the ER lumen, newly synthesized polypeptides are folded and modified to ensure precise conformations and function. Impairments of this process lead to ER stress due to the accumulation of misfolded- or unfolded-proteins in the ER (Byrd and Brewer, 2012). ER homeostasis is sensitive to multiple physiological and pathological processes, and perturbations have been associated with excessive protein synthesis, abnormal ER calcium content, lipid overload, hypoxia, oxidative stress, iron imbalance, nutrient deprivation, cancer, and infection (Kaufman et al., 2002; Marciniak and Ron, 2006; Martins et al., 2016).
Fig. 1. ER stress and the unfolded protein response (UPR).
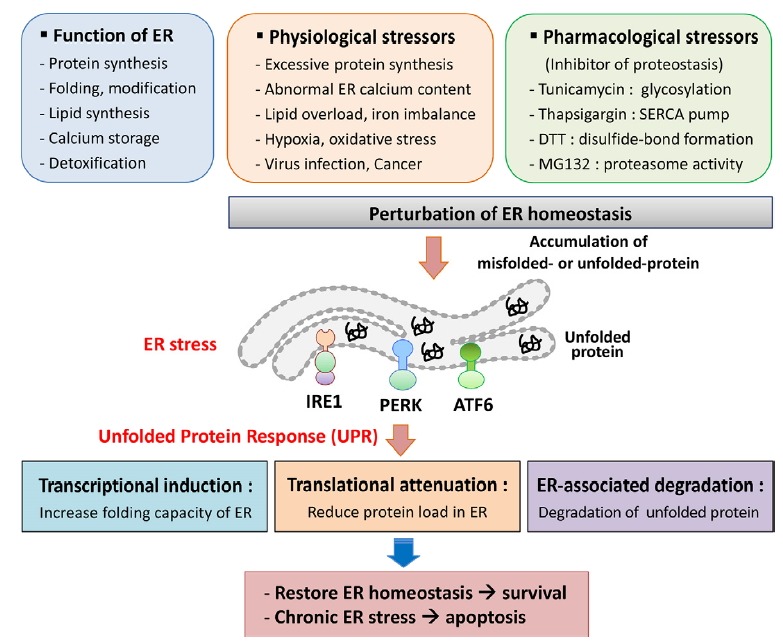
The endoplasmic reticulum (ER) is an essential organelle for protein synthesis, folding, and modification. The ER also plays important roles in lipid biosynthesis, calcium storage, and detoxification. ER homeostasis is disturbed by physiological and pharmacological stressors, and the resulting accumulations of misfolded proteins cause ER stress. ER stress triggers the UPR, which is an adaptive cellular response that is mediated by the three mammalian UPR transducers IRE1, PERK, and ATF6. Under conditions of ER stress, these proteins have various signal-mediated transcriptional effects that ameliorate ER stress. Proteins that are induced by IRE1, PERK, and ATF6 are involved in protein folding and ER expansion, and some attenuate protein translation. In addition, the UPR induces ERAD, which mediates degradation of unfolded proteins by proteasome. Under overwhelming conditions of ER stress, the UPR initiates apoptosis. IRE1, inositol-requiring enzyme 1; PERK, protein kinase R-like ER kinase; ATF6, activating transcription factor 6; ERAD, ER-associated degradation.
Protein quality control is monitored by a complex system that modulates ER function to resolve ER stress and dysfunction. This protective mechanism is known as the unfolded protein response (UPR) and is an adaptive cellular response that elicits specific intracellular signaling pathways to protect against ER stress (Hotamisligil, 2010)(Fig. 1). The UPR restores ER homeostasis by attenuating protein synthesis or by inducing the expression of various genes encoding molecular chaperones and protein processing enzymes that promote protein folding and post-translational modification. Proteins that remain improperly folded are degraded by the proteasome through a pathway known as ER-associated degradation (ERAD)(Adachi et al., 2008). The UPR also enhances ER capacity by promoting expansions of the ER membrane through increased expression of genes involved in lipid metabolism (Lee et al., 2008; Volmer and Ron, 2015; Walter and Ron, 2011). Finally, when these mechanisms fail to resolve chronic or overwhelming ER stress, the UPR stimulates apoptosis (Hetz, 2012).
Accumulating evidence suggests that ER stress is a major contributor to the pathogenesis of several diseases, including obesity, diabetes, cancer, neurodegenerative disorders, and inflammatory and autoimmune diseases, and can increase the severity of these (Hetz et al., 2013; Hotamisligil, 2010; Marciniak and Ron, 2006; Oyadomari et al., 2008; Özcan et al., 2004; Wang and Kaufman, 2012). Whereas sustained ER stress has been associated with the expression of pro-inflammatory cytokines and potential induction of inflammatory responses, causal relationships between immunity and ER stress remain an incipient area of interest (Smith, 2018). Herein, I discuss the underlying mechanisms by which ER stress affects immune responses, with a focus on differentiation and cytokine expression in immune cells and the etiology of immune disorders.
GENERAL MECHANISMS OF THE UPR
ER stress is recognized by the ER-resident proteins inositol-requiring enzyme 1 (IRE1), protein kinase R (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6) (Bettigole and Glimcher, 2015)(Fig. 2). These transmembrane proteins are major transducers of the UPR in mammals and mediate multiple distinct signals from the ER to the cytosol. As a UPR initiating response to perturbations of protein folding, the ER chaperone Grp78 dissociates from the ER luminal domain and binds to the unfolded protein to prevent aggregation (Bertolotti et al., 2000; Ron and Walter, 2007; Shen et al., 2002). Various pharmacological agents have been shown to disturb ER functions. Among these, tunicamycin (Tun) prevents N-linked glycosylation, thapsigargin (Tg) depletes Ca2+ from the ER by inhibiting the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pump, dithiothreitol (DTT) interferes with the formation of protein disulfide bonds, and MG132 inhibits the proteasome (Oslowski and Urano, 2011)(Fig. 1).
Fig. 2. Signaling pathways of the UPR.
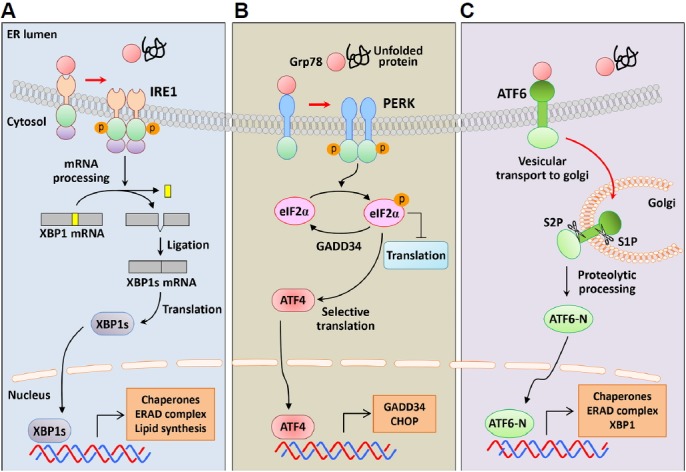
ER-resident proteins IRE1, PERK, and ATF6 sense ER stress and deliver distinct signals from the ER to the cytosol. Under normal conditions, Grp78 binds to the ER luminal domains of sensor proteins and inhibits their activation. However, Grp78 dissociates from the sensors in response to ER stress and binds to unfolded proteins, leading to activation of the sensors. (A) IRE1 pathway; IRE1 has Ser/Thr kinase and RNase domain in the cytoplasmic region, and ER stress induces IRE1 oligomerization and autophosphorylation of the kinase domain. The RNase domain of activated IRE1 performs unconventional splicing and cleaves 26 intronic nucleotides from XBP1 mRNA in mammalian cells. This splicing induces a translational frame-shift, and the truncated XBP1 mRNA encodes XBP1s, which contains a new carboxyl terminus. As a transcription factor, XBP1s activates UPR-related genes including ER chaperones, ERAD components, and lipid-biosynthetic enzymes. (B) PERK pathway; PERK is a protein Ser/Thr kinase that undergoes oligomerization and autophosphorylation of the kinase domain under conditions of ER stress. Activated PERK phosphorylates eIF2α at serine 51, resulting in general inhibition of protein translation. However, phosphorylated eIF2α selectively increases the translation of ATF4, which upregulates CHOP and GADD34 mRNA. As a negative feedback mechanism, GADD34 promotes dephosphorylation of eIF2α to restore protein synthesis following elimination of ER stress. However, failure to alleviate ER stress leads to CHOP-mediated apoptosis. (C) ATF6 pathway; ATF6 has a bZIP domain in the cytosol and translocates from the ER to the Golgi apparatus under ER stress. ATF6 is then cleaved by the proteases S1P and S2P to produce the aminoterminus of ATF6 (ATF6-N), which then migrates to the nucleus and upregulates target genes encoding ER chaperones, ERAD components, and XBP1. RNase, endoribonuclease; XBP1, X-box binding protein 1; eIF2α, α-subunit of eukaryotic translation initiation factor 2; ATF4, activating transcription factor 4; CHOP, C/EBP homologous protein; GADD34, growth arrest and DNA damage-inducible protein 34; S1P, site-1 protease.
The proteins IRE1 and PERK are central type-I transmembrane proteins of the UPR. IRE1 has Ser/Thr kinase and endoribonuclease (RNase) domains in the cytoplasmic region, and comparisons of yeast IRE1 with the mammalian homologs IRE1α and IRE1β reveal a high level of conservation (Fig. 2A). Whereas IRE1α is expressed in most cells and tissues, IRE1β expression is primarily restricted to epithelial cells of the gastrointestinal tract (Kaufman et al., 2002). The ER-luminal domain of the yeast IRE1 senses misfolded proteins following dissociation of Grp78 and via direct interactions with unfolded proteins, but does not bind to unfolded proteins in mammals (Oikawa et al., 2009). Subsequent IRE1 oligomerization induces autophosphorylation in the kinase domain, and the resulting RNase activity of IRE1 leads to unconventional splicing of yeast HAC1 or mammalian X-box binding protein 1 (XBP1) mRNA (Yoshida et al., 2001). XBP1 mRNA encodes the short unstable protein “unspliced form XBP1” (267-amino acids), and IRE1-mediated excision of 26 intronic nucleotides produces the spliced form that encodes “XBP1s” (371-amino acids). Due to the resulting translational frame-shift, this protein contains a novel carboxyl terminus (Calfon et al., 2002; Lee et al., 2003). Specifically, XBP1s contains a basic leucine zipper (bZIP) domain in the C-terminus and acts as a transcription factor for UPR-related genes, including the ER chaperones ERdj4, HEDJ, Grp58, and p58IPK, the ERAD components EDEM, Herp, and Hrd1, the protein disulfide isomerase PDI-P5, and the ER translocon Sec61α (Byrd and Brewer, 2012; Lee et al., 2003; Yamamoto et al., 2008; Yoshida et al., 2001). XBP1s binds to the UPR element (UPRE) containing the consensus sequence TGACGTGG/A and regulates the transcription of target genes in a cell type- and condition-specific manner (Yamamoto et al., 2004). These genes play distinct roles in various physiological processes, including secretion, lipid metabolism, glucose homeostasis, and inflammation. (Acosta-Alvear et al., 2007; Byrd and Brewer, 2012).
PERK is a protein Ser/Thr kinase with a stress-sensing domain in the ER lumen (Bertolotti et al., 2000) (Fig. 2B). Similar to IRE1, Grp78 dissociates from the luminal domain of PERK under ER stress, which activates oligomerization of PERK and autophosphorylation in the cytosolic kinase domain (Ron and Walter, 2007). PERK then phosphorylates the α-subunit of eukaryotic translation initiation factor 2 (eIF2α) at Serine 51, leading to general inhibition of protein translation. Although eIF2α phosphorylation decreases global protein synthesis and reduces protein uptake into the ER, it induces the translation of activating transcription factor 4 (ATF4), which controls specific transcriptional programs of the UPR (Walter and Ron, 2011). The 5′-untranslated region (5′-UTR) of ATF4 has two short upstream open reading frames (uORF) that prevent ATF4 translation in unstressed cells. However, eIF2α phosphorylation induces ribosome bypass of inhibitory uORF, resulting in increased translation from the ATF4 ORF (Lu et al., 2004). ATF4 activates the transcription of target genes encoding C/EBP homologous protein (CHOP) and growth arrest and DNA damage-inducible protein 34 (GADD34) (Fawcett et al., 1999; Ma and Hendershot, 2003; Ma et al., 2002). As a negative feedback mechanism, GADD34 then associates with protein phosphatase 1 (PP1) and promotes dephosphorylation of eIF2α to restore protein synthesis upon stress relief (Harding et al., 2009; Novoa et al., 2003). When ER stress remains unmitigated, CHOP induces apoptosis via the genes encoding death receptor 5 (DR5) and Bim (Puthalakath et al., 2007; Raffaella and Cristina, 2015; Yamaguchi and Wang, 2004; Zinszner et al., 1998). DR5 induces extrinsic apoptosis pathways by recruiting the adaptor protein FADD, which activates caspase-8 (Raffaella and Cristina, 2015). Concomitantly, the pro-apoptotic protein Bim activates Bax and Bak and induces permeabilization of the mitochondrial membrane to release cytochrome c (Urra et al., 2013).
ATF6 is a type-II ER transmembrane protein that contains a bZIP domain in the cytosol and a stress-sensing domain in ER lumen (Adachi et al., 2008)(Fig. 2C). Under conditions of protein homeostasis, ATF6 is maintained in the ER through interactions with Grp78, but is translocated from the ER to the Golgi apparatus under conditions of ER stress, where it is sequentially cleaved at two sites by site-1 (S1P) and site-2 (S2P) proteases (Ye et al., 2000). The released aminoterminal of ATF6 (ATF6-N) then migrates to the nucleus and binds to the ER stress response element (ERSE) containing the consensus sequence CCAAT-N9-CCACG to activate genes encoding ER chaperones, ERAD components, and XBP1 (Chen et al., 2010; Yamamoto et al., 2004; Yoshida et al., 2001).
CROSSTALK BETWEEN ER STRESS AND IMMUNE RESPONSES
ER stress profoundly affects innate and adaptive immune responses and is associated with autoimmune and inflammatory disorders such as diabetes, atherosclerosis, myositis, and inflammatory bowel disease (Heazlewood et al., 2008; Kaser et al., 2008; Vattemi et al., 2004; Zhang and Kaufman, 2008). Many recent studies show immune roles of the UPR via direct crosstalk between ER stress-induced signaling pathways and immune responses (Bettigole and Glimcher, 2015; Cao et al., 2016; Garg et al., 2012; Janssens et al., 2014; Zhang and Kaufman, 2008).
Increased expression of pro-inflammatory cytokines during ER stress suggests roles of the UPR in inflammatory disorders (Smith, 2018; Wheeler et al., 2008) (Fig. 3A). In agreement, several studies suggest that ER stress induces inflammatory responses by activating UPR transcription factors such as XBP1s, ATF6, and cAMP response element-binding protein H (CREBH) (Kaser et al., 2008; Shkoda et al., 2007; Zhang et al., 2006). A missense mutation in the Muc2 gene, which encodes the major constituent of intestinal mucus mucin, led to the accumulation of the MUC2 precursor in mouse goblet cells, thereby increasing ER stress and Grp78 and XBP1s expression (Heazlewood et al., 2008). These mice were susceptible to intestinal inflammation and had increased levels of inflammatory cytokines IL-1β, TNF-α, and IFN-γ in the colon. UPR activation was also demonstrated during chronic inflammation, with increased Grp78 expression in intestinal epithelial cells of IL-10-deficient mice and in patients suffering inflammatory bowel disease (Shkoda et al., 2007). In this study, treatments with the anti-inflammatory cytokine IL-10 reduced Grp78 expression in MODE-K epithelial cells, reflecting suppressed recruitment of ATF6 to the Grp78 promoter following inhibition of nuclear translocation of ATF6. In contrast, XBP1-deletion in mouse intestinal epithelial cells led to ER stress and increased pro-inflammatory responses to flagellin (Kaser et al., 2008). These mice also had impaired antimicrobial peptide secretory activities of Paneth cells, and thereby developed spontaneous enteritis. In another study, hepatocyte-specific transcription factor CREBH was cleaved by S1P and S2P in response to ER stress, and the aminoterminal fragment of CREBH upregulated acute phase proteins such as serum amyloid P-component (SAP) and C-reactive protein (CRP), contributing to the inflammatory response (Zhang et al., 2006).
Fig. 3. Crosstalk between ER stress and inflammatory responses.

ER stress-induced signaling pathways play important roles in inflammatory responses. (A) Increased inflammation due to ER stress; ER stress has been implicated in the pathogenesis of inflammatory and autoimmune diseases, such as obesity, diabetes, atherosclerosis, myositis, and inflammatory bowel disease. ER stress induces inflammatory responses by activating UPR transcription factors, such as XBP1s, ATF6, and CREBH. These transcription factors upregulate the pro-inflammatory cytokines IL-1β, TNF-α, and IFN-γ, and the acute phase proteins SAP and CRP. (B) Activation of inflammatory signaling by the UPR; ER stress induces interactions between UPR components and inflammatory signaling cascades. Activated IRE1 interacts with IKK via the adaptor protein TRAF2 and induces NF-κB by initiating proteasomal degradation of IκBα. Phosphorylation of eIF2α inhibits the translation of IκBα and lowers expression levels, thereby activating NF-κB. IRE1 also activates JNK by binding TFAF2 at its cytoplasmic region. Activation of NF-κB and AP-1 by ER stress upregulates inflammatory genes, and is considered a mechanism for inducing inflammatory responses. (C) Activation of the UPR by inflammation; pro-inflammatory cytokines, such as IL-1β, IL-6, and TNF-α, induce ER stress and activate the UPR. LPS also increases the expression of inflammatory cytokines and ER stress-related genes. After inflammatory activation, the UPR causes excessive inflammation and induces apoptosis, thus exacerbating disease conditions. CREBH, cAMP responsive element binding protein H; SAP, serum amyloid P-component; CRP, C-reactive protein; IKK, IκB kinase; TRAF2, TNF-α receptor associated factor 2; IκB, inhibitor of κB; JNK, c-Jun N-terminal kinase; LPS, lipopolysaccharide.
Accumulating evidence suggests that ER stress activates inflammatory signaling cascades through the interaction between UPR components and canonical cytokine-regulatory transcription factors (Smith et al., 2008)(Fig. 3B). ER stress reportedly activates NF-κB signaling, and this was considered a mechanism for inducing inflammatory responses (Deng et al., 2004; Hu et al., 2006). ER stress induced the formation of a complex between IRE1α and IκB kinase (IKK) through the adaptor protein TNF-α receptor associated factor 2 (TRAF2), thus promoting TNF-α production by enhancing IκBα degradation and NF-κB activation (Hu et al., 2006). In addition, NF-κB was activated by eIF2α phosphorylation, which induces translational suppression of IκBα (Deng et al., 2004). Following binding of TRAF2 to IRE1 cytoplasmic regions, IRE1α and IRE1β activated c-Jun N-terminal kinase (JNK) (Urano et al., 2000). These data were also confirmed in XBP1-deficient small intestines, with IRE1α hyperactivation and increased phosphorylation of JNK (Kaser et al., 2008). Although these studies collectively indicate immunological roles of the UPR, further studies are required to characterize and identify the related regulatory mechanisms and signaling molecules.
Several studies show that inflammation augments ER stress responses (Fig. 3C). In particular, injections of pro-inflammatory cytokines such as IL-1β and IL-6, induced ER stress and activated the UPR in the livers of mice (Zhang et al., 2006). The inflammatory cytokine TNF-α also increased eIF2α phosphorylation and XBP1s expression by elevating reactive oxygen species (ROS) levels in L929 murine fibrosarcoma cells (Xue et al., 2005). The immunostimulant lipopolysaccharide (LPS) increases the production of inflammatory cytokines, and intraperitoneal injections of LPS into mice activated the transcription of ER stress-related genes, including Xbp1s, Atf4, and Chop (Endo et al., 2005). Among encoded proteins, CHOP plays a key role in the apoptosis of lung cells. LPS injection also increased protein levels of XBP1s, Grp78, CHOP, and ER degradation-enhancing α-mannosidase-like protein (EDEM) in the livers of mice (Zhang et al., 2006). However, the mechanisms by which LPS activates the UPR pathway have not been precisely demonstrated and are likely key factors in the link between inflammatory responses and ER stress.
ROLES OF THE UPR IN IMMUNE CELLS
Because the UPR is required for ER expansion and protein secretion, it is essential for development, differentiation, and the precise function of highly secretory cells, including pancreatic β cells, exocrine acinar cells, hepatocytes, and salivary gland epithelial cells (Bettigole and Glimcher, 2015; Harding et al., 2001; Lee et al., 2005; Lipson et al., 2006; Marisa et al., 2018; Reimold et al., 2000; Rutkowski and Hegde, 2010)(Fig. 4A). The UPR also has crucial roles in the development of immune cells, such as plasma cells, dendritic cells (DCs), and eosinophils (Bettigole et al., 2015; Iwakoshi et al., 2007; Lee et al., 2003; Reimold et al., 2001)(Fig. 4B). Many recent studies of various immune cell types show roles of ER stress in immune processes, including differentiation, immune activation, and cytokine expression.
Fig. 4. Roles of the UPR in immune cells.
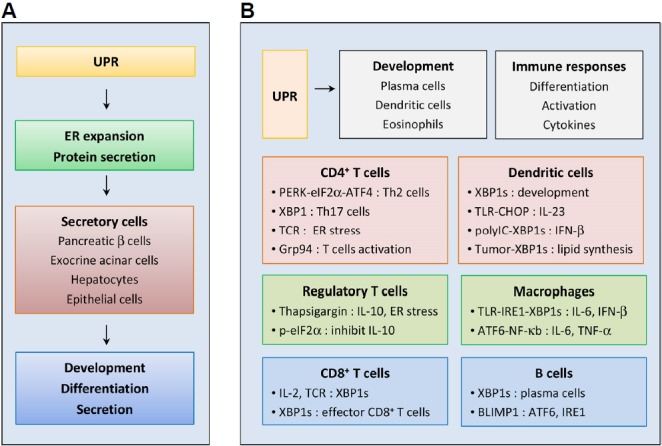
(A) Roles of the UPR in secretory cells; the UPR regulates ER expansion and protein secretion, and is therefore essential for development, differentiation, and specialized functions of highly secretory cells, such as pancreatic β cells, exocrine acinar cells, hepatocytes, and salivary gland epithelial cells. (B) Roles of the UPR in immune cells; the UPR plays crucial roles in the development of immune cells, such as plasma cells, DCs, and eosinophils. ER stress also regulates immune cell differentiation, activation, and cytokine expression. In CD4+ T cells, the UPR regulates differentiation to Th2 cells, Th17 cells, and Tregs, and has an important function in TCR-mediated activation. The IRE1–XBP1 pathway is important for differentiation of CD8+ T cells into effector cells. The UPR is also activated during B cell differentiation, and XBP1s is essential for antibody secretion from plasma cells. ER stress strongly influences innate and adaptive immune responses by modulating the production of pro-inflammatory cytokines in B cells, macrophages, and DCs. In macrophages, XBP1s and ATF6 function as positive regulators of inflammatory cytokine expression following TLR stimulation. XBP1s also regulates developmental and inflammatory responses of DCs. However, in DCs of tumor microenvironments, XBP1s induces abnormal lipid accumulation and inhibits anti-tumor immunity. TCR, T cell receptor; Th2, T helper type 2; Tregs, regulatory T cells; TLR, Toll-like receptor.
CD4+ T cells
Depending on microenvironments, naive CD4+ T cells differentiate into one of several lineages of helper T (Th) cells, including T helper type 1 (Th1), Th2, Th9, Th17, and regulatory T cells (Tregs) (Stadhouders et al., 2018). Effector T cell subsets secrete distinguishing cytokines and coordinately regulate different aspects of immune responses. Potentially, the UPR plays fundamental roles in T cell differentiation upon antigen recognition. This hypothesis is supported by observations of activated ER stress responses during differentiation of Th2 cells, and the PERK-eIF2α axis has been implicated in this process (Scheu et al., 2006). Specifically, primed Th2 cells showed increased phosphorylation of eIF2α and upregulation of ATF4 and its target genes Gadd34, Herp, Chop, and Grp78. Moreover, restimulation of Th2 cells resulted in rapid dephosphorylation of eIF2α, thereby activating translation and production of IL-4. XBP1 is important for the differentiation of Th17 cells, which are associated with various inflammatory and autoimmune diseases (Brucklacher-Waldert et al., 2017; Stadhouders et al., 2018). In this study, cellular stress, such as low oxygen or glucose levels and ionic pressure, enhanced the generation of Th17 cells by increasing cytoplasmic calcium levels and XBP1 activity (Brucklacher-Waldert et al., 2017). In contrast, inhibition of cellular stress and conditional deletion of XBP1 in lymphocytes suppressed Th17 cell-dependent autoimmunity in a mouse model of experimental autoimmune encephalomyelitis (EAE). Moreover, adoptive transfer of Th17 cells generated by ER stress inducer increased the disease severity of EAE.
A recent study showed that ER stress responses are important for CD4+ T cell activation (Thaxton et al., 2017). In their study, T cell receptor (TCR) ligation-mediated ER stress increased Grp94 expression in CD4+ T cells, whereas deletion of Grp94 resulted in a critical activation defect. In addition, Grp94-deficient T cells showed impaired Ca2+ mobilization upon TCR engagement and failed to accomplish activation-induced glycolysis. ER molecular chaperone Grp94, upregulated by ER stress, has diverse immunological activities (Ansa-Addo et al., 2016).
Regulatory T cells (Tregs)
The effects of ER stress on differentiation have been investigated in Tregs that maintain tolerance to self-antigens and prevent autoimmune disease (Franco et al., 2010). Specifically, T cells from patients with Kawasaki disease produced high levels of the anti-inflammatory cytokines IL-10 and TGF-β, and Foxp3, which is a critical transcription factor for Tregs (Franco et al., 2010). Treatments of these cells with thapsigargin induced the expression of the ER stress-response genes Grp78, Chop, and Gadd34, and the Il-10. Conversely, inhibition of eIF2α dephosphorylation by the agent salubrinal limited Il-10 transcription, suggesting that eIF2α phosphorylation suppresses the differentiation of IL-10-producing Tregs.
CD8+ T cells
In the presence of pathogens, CD8+ cytotoxic T cells secrete various cytokines and cytotoxic molecules to eliminate infected cells. In a study of CD8+ T cells by Kamimura and Bevan, the IRE1–XBP1 pathway was activated by acute infection and was required for differentiation into effector T cells (Kamimura and Bevan, 2008). These investigators also showed that viral or bacterial infection induces IRE1-mediated splicing and increases XBP1s mRNA expression in Ag-specific CD8+ T cells (Kamimura and Bevan, 2008). Finally, splicing of XBP1 mRNA was enriched in terminally differentiated effector cells expressing killer cell lectin-like receptor G1 (KLRG1), whereas XBP1-deficiencies in CD8+ T cells decreased the proportion of KLRG1high effector cells.
B cells
The physiological importance of the UPR was first described in B cells that were differentiating into plasma cells. In these studies, the UPR was activated during differentiation of B cells, and XBP1s was deemed essential for antibody production from plasma cells (Gass et al., 2002; Reimold et al., 2001; Shaffer et al., 2004; Todd et al., 2009). Accordingly, XBP1s induced ER expansion in plasma cells, therefore accommodating the synthesis of large amounts of immunoglobulin (Shaffer et al., 2004). Whereas XBP1-deficient B cells developed normally and formed germinal centers upon activation, they failed to secrete immunoglobulins (Reimold et al., 2001; Taubenheim et al., 2012). As a transcription factor, Blimp-1 necessary for plasma cell differentiation, and Blimp-1-deficient B cells failed to activate transcription of most plasma cell-related genes, including Xbp1 (Shaffer et al., 2004). In contrast, Blimp-1 was normally induced in XBP1-deficient B cells, indicating that XBP1 acts downstream of Blimp-1. Moreover, XBP1-deficiency prevented upregulation of genes encoding secretory pathway components. A recent study suggested that Blimp-1 also plays crucial roles in the UPR in plasma cells, reflecting direct transcriptional regulation of Atf6 and Ern1 (encoding IRE1) (Tellier et al., 2016).
ER stress also upregulated the production of pro-inflammatory cytokines in B cells, and expression of Ag with the ER-retention motif KDEL in B cells induced the ER stress-responsive genes Gadd34, Grp78, and Chop (Wheeler et al., 2008). Thapsigargin treatment induced transcription of those UPR genes and pro-inflammatory cytokine genes, IL-23p19, IL-6, TNF-α, and IL-2 in B cells. It follows that these B cells induced the production of IFN-γ and TNF-α in CD4+ T cells.
Macrophages
Macrophages are crucial effector cells of the innate immune system that phagocytose pathogens and produce various inflammatory mediators (Hirayama et al., 2018). XBP1s is a positive regulator of toll-like receptor (TLR) responses in macrophages, and TLR2 and TLR4 activated the IRE1α–XBP1s axis, but not the PERK or ATF6α pathways (Martinon et al., 2010). Moreover, XBP1s was required to produce pro-inflammatory cytokines, such as IL-6, TNF, and INF-β. TLR-mediated splicing of XBP1 mRNA was dependent on signaling via TIRAP, MyD88, TRIF, TRAF6, and NADPH oxidase 2 (NOX2) as a TRAF-bound enzyme complex. Under condition of XBP1-deficiency, IL-6 production was reduced in macrophages, and the bacterial burden was increased in mice infected with an intracellular pathogen. Conversely, overexpression of XPB1s or thapsigargin treatments increased LPS-stimulated IFN-β production in RAW267.4 cells (Smith et al., 2008).
Prolonged ischemia induces the expression of pro-inflammatory cytokines in liver Kupffer cells, primarily through ER stress-activated ATF6 (Rao et al., 2014). Accordingly, knockdown of ATF6 in macrophages suppressed the expression of TNF-α and IL-6 by limiting NF-κB activity. In agreement, alleviation of ER stress using the chemical chaperone 4-phenylbutyrate (4-PBA) or ATF6 siRNA led to decreased inflammatory responses in Kupffer cells against ischemia-reperfusion.
Dendritic cells (DCs)
DCs are professional antigen-presenting cells, and developmental and survival roles of XBP1s have been demonstrated in these cells (Iwakoshi et al., 2007). In agreement, XBP1s expression was greater in plasmacytoid DCs than in immature conventional DCs, and XBP1-deficient chimeric mice had decreased numbers of conventional and plasmacytoid DCs. Under conditions of ER stress, treatment with TLR agonists significantly enhanced IL-23p19 expression in DCs (Goodall et al., 2010). The gene Il-23 is a target of CHOP whereby both ER stress and TLR stimulation enhanced binding of CHOP to its promoter. Consequently, knockdown of CHOP reduced the expression of IL-23 in U937 cells and in Chlamydia trachomatis-infected myeloid cells. ER stress also significantly increased the levels of inflammatory cytokines and IFN-β in DCs stimulated by polyIC (Fanlei et al., 2011). Whereas silencing of XBP1 inhibited polyIC-induced IFN-β expression, overexpression of XBP1s synergistically augmented inflammatory responses in polyIC-stimulated DCs. In addition, XBP1s-induced IFN-β effectively inhibited vesicular stomatitis virus infections.
A recent study associated tumor microenvironments with ER stress in DCs, and showed that XBP1s is important for the suppression of anti-tumor immunity (Cubillos-Ruiz et al., 2015). Specifically, XBP1s promoted the triglyceride biosynthetic program in DCs, which led to abnormal lipid accumulation and impaired antigen presentation. Conversely, DC-specific deletion of XBP1 restored immunostimulatory activities and increased anti-tumor responses.
ROLES OF REGULATED IRE1-DEPENDENT DECAY (RIDD) IN IMMUNE RESPONSES
Several recent studies show that IRE1 recognizes unspliced XBP1 mRNA and various other mRNAs as substrates to be degraded, and the associated process is called regulated IRE1-dependent decay (RIDD) (Coelho and Domingos, 2014; Hollien and Weissman, 2006; Hollien et al., 2009)(Fig. 5). Because IRE1 mediates the degradation of mRNAs encoding ER-targeted proteins, RIDD was originally thought to reduce ER protein loads during ER stress (Hollien and Weissman, 2006). In mammalian cells, RIDD targets have specific motifs with the consensus sequence CUGCAG and secondary structures that are similar to the stem-loop of XBP1 mRNA (Moore and Hollien, 2015; Oikawa et al., 2010; So et al., 2015)(Fig. 5A). Emerging evidence indicates that RIDD targets various mRNAs and therefore has diverse physiological functions, particularly in drug and lipid metabolism, and ER homeostasis in goblet cells and pancreatic β-cells (Han et al., 2009; Hur et al., 2012; Iqbal et al., 2008; Lipson et al., 2008; So et al., 2012; Tsuru et al., 2013)(Fig. 5B).
Fig. 5. Regulated IRE1-dependent decay (RIDD).
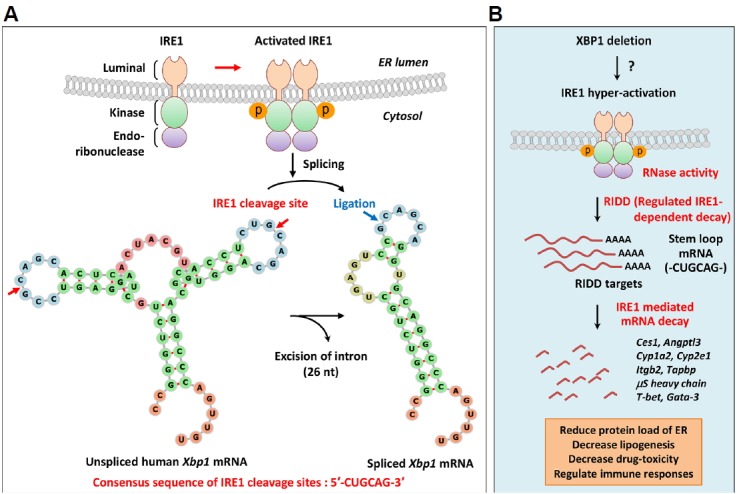
(A) Unconventional splicing of XBP1 mRNA by IRE1; ER stress results in activation of the RNase domain of IRE1 by inducing IRE1 oligomerization and autophosphorylation of kinase domains. The RNase domain of activated IRE1 recognizes two stem-loop structures on XBP1 mRNA and splices motifs with the consensus sequence CUGCAG. The 26-nucleotides intron is cleaved from XBP1 mRNA, and the exons are joined by tRNA ligase to generate spliced XBP1 mRNA encoding XBP1s. (B) Regulated IRE1-dependent decay (RIDD); in addition to XBP1 mRNA, IRE1 recognizes ER-bound mRNAs and promotes their degradation to reduce the protein folding load of ER. This process is known as regulated IRE1-dependent decay (RIDD). IRE1 recognition motifs of RIDD substrates carry the CUGCAG consensus sequence and a secondary structure that is similar to the stem-loop of XBP1 mRNA. XBP1 deficiency induces IRE1 hyperactivation in various cells by as yet unknown mechanisms. RIDD targets various mRNAs to achieve diverse physiological functions, such as reducing plasma lipid levels (Ces1, Angptl3), protecting against drug toxicity (Cyp1a2, Cyp2e1), maintaining ER homeostasis in goblet cells (MUC2), and causing dysfunction of pancreatic β-cells (Ins1, Ins2, PC1, PC2, CPE). RIDD also regulates immune processes, such as antigen presentation by CD8α+ DCs (Itgb2, Tapbp), synthesis of secretory IgM in B cells (μS heavy chain), and cytokine production from iNKT cells (T-bet, Gata-3). Ces1, carboxylesterase 1; Angptl3, angiopoietin-like protein 3; Cyp1a2, cytochrome P450 1A2; Cyp2e1, cytochrome P450 2E1; MUC2, mucin 2; Ins1, insulin 1; Ins2, insulin 2; PC1, prohormone convertase 1; CPE, carboxypeptidase E; Itgb2, integrin subunit beta 2; Tapbp, TAP binding protein; μS, secretory μ chain; iNKT, invariant natural killer T.
RIDD has also been implicated in the regulation of immune responses, with critical functions in the cross-presentation of cell-derived antigens via MHC class I between CD8+ T cells and CD8α+ conventional DCs (cDCs) (Osorio et al., 2014). Deletion of XBP1 in CD8α+ cDCs resulted in hyperactivation of IRE1α, and the RNase activity of IRE1α limited the expression of CD18 integrins and components of the MHC class I machinery by degrading Itgb2, which encodes a common β2 integrin chain, and Tapbp, which encodes the TAP-binding protein tapasin. RIDD also differentially regulates the survival of mucosal cDCs (Tavernier et al., 2017). In this study, XBP1 deletion induced apoptosis of cDCs in lung but not intestine tissues. Under these conditions, inhibition of RIDD reduced the population of intestinal cDCs, suggesting a protective role of RIDD on the survival of intestinal cDCs in the absence of XBP1. IRE1α was also shown to be active in XBP1-deficient B cells, where it cleaved mRNA encoding secretory μ (μS) heavy chains of immunoglobulin (Sandrine et al., 2013). These observations indicate the mechanisms by which RIDD decreases the synthesis of secretory IgM and IgM responses following T-dependent and independent vaccinations. A recent study demonstrated that XBP1-deficiency in B cells induced IRE1α phosphorylation at S729 (Tang et al., 2018). In accordance, S729A mutation of this protein increased the expression of μS mRNA in B cells and the production of IgM in immunized mice, indicating a critical role for S729 phosphorylation on RIDD. IRE1α also degraded T-bet and Gata-3 mRNA in invariant natural killer T (iNKT) cells, and this RIDD activity was promoted by palmitic acid, leading to the inhibition of IL-4 and IFN-γ production in iNKT cells and suppression of arthritis (Ko et al., 2017).
Several miRNAs have been identified as RIDD targets, and these reportedly increase the expression of other mRNAs during ER stress (Upton et al., 2012). IRE1α improved the stability of thioredoxin-interacting protein (TXNIP) mRNA by inducing cleavage of miR17, which destabilizes TXNIP (Lerner et al., 2012). Consequent augmentation of TXNIP likely activates the NLRP3 inflammasome, thus inducing caspase-1 cleavage and IL-1β secretion, and promoting inflammation and programmed cell death.
CONCLUSION
The ER is an essential organelle for protein homeostasis, and perturbations of ER homeostasis by physiological or pathological stressors cause accumulations of misfolded proteins in the ER. ER stress-induced UPR restores ER homeostasis through dynamic intracellular signaling pathways but gains its relationship with various diseases by regulating cell-specific functions. Much remains unknown regarding the impacts of ER stress on immune responses, although recent studies suggest that ER stress has specific immune regulatory activities in specialized cells and tissues. These studies collectively suggest that the UPR is a fundamental mediator of the pathogenesis of inflammatory disorders. In line with this suggestion, ER stress is known to induce inflammatory responses through UPR signaling, and immune cells are extensively dependent on the signaling branches of the UPR to perform their specific functions. Improved understanding of the molecular mechanisms underlying ER stress-regulated immune responses may lead to the discovery of new therapeutic targets for immune disorders. However, investigations of the UPR branches and their associated molecules in specific immune cells and under specific immune disorders remain challenging.
ACKNOWLEDGMENTS
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government, the Ministry of Science and ICT (MSIT) (No. NRF-2017R1C1B5018313).
REFERENCES
- Acosta-Alvear D., Zhou Y., Blais A., Tsikitis M., Lents N.H., Arias C., Lennon C.J., Kluger Y., Dynlacht B.D. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell. 2007;27:53–66. doi: 10.1016/j.molcel.2007.06.011. [DOI] [PubMed] [Google Scholar]
- Adachi Y., Yamamoto K., Okada T., Yoshida H., Harada A., Mori K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct. 2008;33:75–89. doi: 10.1247/csf.07044. [DOI] [PubMed] [Google Scholar]
- Ansa-Addo E.A., Thaxton J., Hong F., Wu B.X., Zhang Y., Fugle C.W., Metelli A., Riesenberg B., Williams K., Gewirth D.T., et al. Clients and oncogenic roles of molecular chaperone gp96/grp94. Curr Top Med Chem. 2016;16:2765–2778. doi: 10.2174/1568026616666160413141613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotti A., Zhang Y., Hendershot L.M., Harding H.P., Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Bettigole S.E., Glimcher L.H. Endoplasmic reticulum stress in immunity. Annu Rev Immunol. 2015;33:107–138. doi: 10.1146/annurev-immunol-032414-112116. [DOI] [PubMed] [Google Scholar]
- Bettigole S.E., Lis R., Adoro S., Lee A.-H., Spencer L.A., Weller P.F., Glimcher L.H. The transcription factor XBP1 is selectively required for eosinophil differentiation. Nat Immunol. 2015;16:829–837. doi: 10.1038/ni.3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braakman I., Bulleid N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- Brucklacher-Waldert V., Ferreira C., Stebegg M., Fesneau O., Innocentin S., Marie J.C., Veldhoen M. Cellular stress in the context of an inflammatory environment supports TGF-β-independent T helper-17 differentiation. Cell Rep. 2017;19:2357–2370. doi: 10.1016/j.celrep.2017.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd A.E., Brewer J.W. Intricately regulated: a cellular toolbox for fine-tuning XBP1 expression and activity. Cells. 2012;1:738–753. doi: 10.3390/cells1040738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfon M., Zeng H., Urano F., Till J.H., Hubbard S.R., Harding H.P., Clark S.G., Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Cao S.S., Luo K.L., Shi L. Endoplasmic reticulum stress interacts with inflammation in human diseases. J Cell Physiol. 2016;231:288–294. doi: 10.1002/jcp.25098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Karnovsky A., Sans M.D., Andrews P.C., Williams J.A. Molecular characterization of the endoplasmic reticulum: insights from proteomic studies. Proteomics. 2010;10:4040–4052. doi: 10.1002/pmic.201000234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho D.S., Domingos P.M. Physiological roles of regulated Ire1 dependent decay. Front Genet. 2014;5:76. doi: 10.3389/fgene.2014.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubillos-Ruiz J.R., Silberman P.C., Rutkowski M.R., Chopra S., Perales-Puchalt A., Song M., Zhang S., Bettigole S.E., Gupta D., Holcomb K., et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161:1527–1538. doi: 10.1016/j.cell.2015.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J., Lu P.D., Zhang Y., Scheuner D., Kaufman R.J., Sonenberg N., Harding H.P., Ron D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol. 2004;24:10161–10168. doi: 10.1128/MCB.24.23.10161-10168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo M., Oyadomari S., Suga M., Mori M., Gotoh T. The ER stress pathway involving CHOP is activated in the lungs of LPS-treated mice. J Biochem. 2005;138:501–507. doi: 10.1093/jb/mvi143. [DOI] [PubMed] [Google Scholar]
- Fanlei H., Xiaofei Y., Hongxia W., Daming Z., Chunqing G., Huanfa Y., Boaz T., R.S.J., Xiaoyan Q., Xiang-Yang W. ER stress and its regulator X-box-binding protein-1 enhance polyIC-induced innate immune response in dendritic cells. Eur J Immunol. 2011;41:1086–1097. doi: 10.1002/eji.201040831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett T.W., Martindale J.L., Guyton K.Z., Hai T., Holbrook N.J. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem J. 1999;339:135–141. [PMC free article] [PubMed] [Google Scholar]
- Franco A., Almanza G., Burns J.C., Wheeler M., Zanetti M. Endoplasmic reticulum stress drives a regulatory phenotype in human T-cell clones. Cell Immunol. 2010;266:1–6. doi: 10.1016/j.cellimm.2010.09.006. [DOI] [PubMed] [Google Scholar]
- Garg A.D., Kaczmarek A., Krysko O., Vandenabeele P., Krysko D.V., Agostinis P. ER stress-induced inflammation: does it aid or impede disease progression? Trends Mol Med. 2012;18:589–598. doi: 10.1016/j.molmed.2012.06.010. [DOI] [PubMed] [Google Scholar]
- Gass J.N., Gifford N.M., Brewer J.W. Activation of an unfolded protein response during differentiation of antibody-secreting B cells. J Biol Chem. 2002;277:49047–49054. doi: 10.1074/jbc.M205011200. [DOI] [PubMed] [Google Scholar]
- Goodall J.C., Wu C., Zhang Y., McNeill L., Ellis L., Saudek V., Gaston J.S.H. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc Natl Acad Sci USA. 2010;107:17698–17703. doi: 10.1073/pnas.1011736107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D., Lerner A.G., Vande Walle L., Upton J.-P., Xu W., Hagen A., Backes B.J., Oakes S.A., Papa F.R. IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding H.P., Zeng H., Zhang Y., Jungries R., Chung P., Plesken H., Sabatini D.D., Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- Harding H.P., Zhang Y., Scheuner D., Chen J.-J., Kaufman R.J., Ron D. Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2α) dephosphorylation in mammalian development. Proc Natl Acad Sci USA. 2009;106:1832–1837. doi: 10.1073/pnas.0809632106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heazlewood C.K., Cook M.C., Eri R., Price G.R., Tauro S.B., Taupin D., Thornton D.J., Png C.W., Crockford T.L., Cornall R.J., et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLOS Med. 2008;5:e54. doi: 10.1371/journal.pmed.0050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- Hetz C., Chevet E., Harding H.P. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 2013;12:703–719. doi: 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]
- Hirayama D., Iida T., Nakase H. The phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. Int J Mol Sci. 2018;19:92. doi: 10.3390/ijms19010092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J., Weissman J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- Hollien J., Lin J.H., Li H., Stevens N., Walter P., Weissman J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil G.S. Endoplasmic reticulum stress and atherosclerosis. Nat Med. 2010;16:396–399. doi: 10.1038/nm0410-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P., Han Z., Couvillon A.D., Kaufman R.J., Exton J.H. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1α-mediated NF-κB activation and down-regulation of TRAF2 expression. Mol Cell Biol. 2006;26:3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur K.Y., So J.-S., Ruda V., Frank-Kamenetsky M., Fitzgerald K., Koteliansky V., Iwawaki T., Glimcher L.H., Lee A.-H. IRE1α activation protects mice against acetaminophen-induced hepatotoxicity. J Exp Med. 2012;209:307–318. doi: 10.1084/jem.20111298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal J., Dai K., Seimon T., Jungreis R., Oyadomari M., Kuriakose G., Ron D., Tabas I., Hussain M.M. IRE1β inhibits chylomicron production by selectively degrading MTP mRNA. Cell Metab. 2008;7:445–455. doi: 10.1016/j.cmet.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakoshi N.N., Pypaert M., Glimcher L.H. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J Exp Med. 2007;204:2267–2275. doi: 10.1084/jem.20070525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens S., Pulendran B., Lambrecht B.N. Emerging functions of the unfolded protein response in immunity. Nat Immunol. 2014;15:910–919. doi: 10.1038/ni.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura D., Bevan M.J. Endoplasmic reticulum stress regulator XBP-1 contributes to effector CD8+ T cell differentiation during acute infection. J Immunol. 2008;181:5433–5441. doi: 10.4049/jimmunol.181.8.5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser A., Lee A.-H., Franke A., Glickman J.N., Zeissig S., Tilg H., Nieuwenhuis E.E.S., Higgins D.E., Schreiber S., Glimcher L.H., et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman R.J., Scheuner D., Schröder M., Shen X., Lee K., Liu C.Y., Arnold S.M. The unfolded protein response in nutrient sensing and differentiation. Nat Rev Mol Cell Biol. 2002;3:411–421. doi: 10.1038/nrm829. [DOI] [PubMed] [Google Scholar]
- Ko J.S., Koh J.M., So J.-S., Jeon Y.K., Kim H.Y., Chung D.H. Palmitate inhibits arthritis by inducing t-bet and gata-3 mRNA degradation in iNKT cells via IRE1α-dependent decay. Sci Rep. 2017;7:14940. doi: 10.1038/s41598-017-14780-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.-H., Iwakoshi N.N., Glimcher L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A., Chu G.C., Iwakoshi N.N., Glimcher L.H. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005;24:4368–4380. doi: 10.1038/sj.emboj.7600903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.-H., Scapa E.F., Cohen D.E., Glimcher L.H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320:1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner A.G., Upton J.-P., Praveen P.V.K., Ghosh R., Nakagawa Y., Igbaria A., Shen S., Nguyen V., Backes B.J., Heiman M., et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death during endoplasmic reticulum stress. Cell Metab. 2012;16:250–264. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson K.L., Fonseca S.G., Ishigaki S., Nguyen L.X., Foss E., Bortell R., Rossini A.A., Urano F. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab. 2006;4:245–254. doi: 10.1016/j.cmet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Lipson K.L., Ghosh R., Urano F. The role of IRE1α in the degradation of insulin mRNA in pancreatic β-cells. PLoS One. 2008;3:e1648. doi: 10.1371/journal.pone.0001648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P.D., Harding H.P., Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol. 2004;167:27–33. doi: 10.1083/jcb.200408003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y., Hendershot L.M. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J Biol Chem. 2003;278:34864–34873. doi: 10.1074/jbc.M301107200. [DOI] [PubMed] [Google Scholar]
- Ma Y., Brewer J.W., Alan Diehl J., Hendershot L.M. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318:1351–1365. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- Marciniak S.J., Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006;86:1133–1149. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- Marisa R., Andreia M., J.A.R., Evelina G., Philippe P. At the crossway of ER-stress and proinflammatory responses. FEBS J. 2018 doi: 10.1111/febs.14391. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Martinon F., Chen X., Lee A.-H., Glimcher L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins A.S., Alves I., Helguero L., Domingues M.R., Neves B.M. The unfolded protein response in homeostasis and modulation of mammalian immune cells. Int Rev Immunol. 2016;35:457–476. doi: 10.3109/08830185.2015.1110151. [DOI] [PubMed] [Google Scholar]
- Moore K., Hollien J. Ire1-mediated decay in mammalian cells relies on mRNA sequence, structure, and translational status. Mol Biol Cell. 2015;26:2873–2884. doi: 10.1091/mbc.E15-02-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa I., Zhang Y., Zeng H., Jungreis R., Harding H.P., Ron D. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 2003;22:1180–1187. doi: 10.1093/emboj/cdg112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa D., Kimata Y., Kohno K., Iwawaki T. Activation of mammalian IRE1α upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp Cell Res. 2009;315:2496–2504. doi: 10.1016/j.yexcr.2009.06.009. [DOI] [PubMed] [Google Scholar]
- Oikawa D., Tokuda M., Hosoda A., Iwawaki T. Identification of a consensus element recognized and cleaved by IRE1α. Nucleic Acids Res. 2010;38:6265–6273. doi: 10.1093/nar/gkq452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oslowski C.M., Urano F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011;490:71–92. doi: 10.1016/B978-0-12-385114-7.00004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio F., Tavernier S.J., Hoffmann E., Saeys Y., Martens L., Vetters J., Delrue I., De Rycke R., Parthoens E., Pouliot P., et al. The unfolded-protein-response sensor IRE-1α regulates the function of CD8α+ dendritic cells. Nat Immunol. 2014;15:248–257. doi: 10.1038/ni.2808. [DOI] [PubMed] [Google Scholar]
- Oyadomari S., Harding H.P., Zhang Y., Oyadomari M., Ron D. Dephosphorylation of translation initiation factor 2α enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008;7:520–532. doi: 10.1016/j.cmet.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özcan U., Cao Q., Yilmaz E., Lee A.-H., Iwakoshi N.N.Özdelen ETuncman G., Görgün C., Glimcher L.H., Hotamisligil G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Puthalakath H., O’Reilly L.A., Gunn P., Lee L., Kelly P.N., Huntington N.D., Hughes P.D., Michalak E.M., McKimm-Breschkin J., Motoyama N., et al. ER Stress Triggers Apoptosis by Activating BH3-Only Protein Bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- Raffaella I., Cristina M. Cell death induced by endoplasmic reticulum stress. FEBS J. 2015;283:2640–2652. doi: 10.1111/febs.13598. [DOI] [PubMed] [Google Scholar]
- Rao J., Yue S., Fu Y., Zhu J., Wang X., Busuttil R.W., Kupiec-Weglinski J.W., Lu L., Zhai Y. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia reperfusion injury. Am J Transplant. 2014;14:1552–1561. doi: 10.1111/ajt.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimold A.M., Etkin A., Clauss I., Perkins A., Friend D.S., Zhang J., Horton H.F., Scott A., Orkin S.H., Byrne M.C., et al. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000;14:152–157. [PMC free article] [PubMed] [Google Scholar]
- Reimold A.M., Iwakoshi N.N., Manis J., Vallabhajosyula P., Szomolanyi-Tsuda E., Gravallese E.M., Friend D., Grusby M.J., Alt F., Glimcher L.H. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- Ron D., Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rutkowski D.T., Hegde R.S. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J Cell Biol. 2010;189:783–794. doi: 10.1083/jcb.201003138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandrine B., Rivka H., Takao I., Jae-Seon S., Ann-Hwee L., Boaz T. Regulated IRE1-dependent decay participates in curtailing immunoglobulin secretion from plasma cells. Eur J Immunol. 2013;44:867–876. doi: 10.1002/eji.201343953. [DOI] [PubMed] [Google Scholar]
- Scheu S., Stetson D.B., Reinhardt R.L., Leber J.H., Mohrs M., Locksley R.M. Activation of the integrated stress response during T helper cell differentiation. Nat Immunol. 2006;7:644–651. doi: 10.1038/ni1338. [DOI] [PubMed] [Google Scholar]
- Shaffer A.L., Shapiro-Shelef M., Iwakoshi N.N., Lee A.-H., Qian S.-B., Zhao H., Yu X., Yang L., Tan B.K., Rosenwald A., et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Shen J., Chen X., Hendershot L., Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of golgi localization signals. Dev Cell. 2002;3:99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- Shkoda A., Ruiz P.A., Daniel H., Kim S.C., Rogler G., Sartor R.B., Haller D. Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: impact on chronic inflammation. Gastroenterology. 2007;132:190–207. doi: 10.1053/j.gastro.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Smith J.A. Regulation of cytokine production by the unfolded protein response; implications for infection and autoimmunity. Front Immunol. 2018;9:422. doi: 10.3389/fimmu.2018.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.A., Turner M.J., DeLay M.L., Klenk E.I., Sowders D.P., Colbert R.A. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-β induction via X-box binding protein 1. Eur J Immunol. 2008;38:1194–1203. doi: 10.1002/eji.200737882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So J.-S., Hur K.Y., Tarrio M., Ruda V., Frank-Kamenetsky M., Fitzgerald K., Koteliansky V., Lichtman A.H., Iwawaki T., Glimcher L.H., et al. Silencing of lipid metabolism genes through IRE1α-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012;16:487–499. doi: 10.1016/j.cmet.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So J.-S., Cho S., Min S.-H., Kimball S.R., Lee A.-H. IRE1α-dependent decay of CReP/Ppp1r15b mRNA increases eukaryotic initiation factor 2α phosphorylation and suppresses protein synthesis. Mol Cell Biol. 2015;35:2761–2770. doi: 10.1128/MCB.00215-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadhouders R., Lubberts E., Hendriks R.W. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J Autoimmun. 2018;87:1–15. doi: 10.1016/j.jaut.2017.12.007. [DOI] [PubMed] [Google Scholar]
- Tang C.-H.A., Chang S., Paton A.W., Paton J.C., Gabrilovich D.I., Ploegh H.L., Del Valle J.R., Hu C.-C.A. Phosphorylation of IRE1 at S729 regulates RIDD in B cells and antibody production after immunization. J Cell Biol. 2018;217:1739–1755. doi: 10.1083/jcb.201709137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenheim N., Tarlinton D.M., Crawford S., Corcoran L.M., Hodgkin P.D., Nutt S.L. High rate of antibody secretion is not integral to plasma cell differentiation as revealed by XBP-1 deficiency. J Immunol. 2012;189:3328–3338. doi: 10.4049/jimmunol.1201042. [DOI] [PubMed] [Google Scholar]
- Tavernier S.J., Osorio F., Vandersarren L., Vetters J., Vanlangenakker N., Van Isterdael G., Vergote K., De Rycke R., Parthoens E., van de Laar L., et al. Regulated IRE1-dependent mRNA decay sets the threshold for dendritic cell survival. Nat Cell Biol. 2017;19:698–710. doi: 10.1038/ncb3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellier J., Shi W., Minnich M., Liao Y., Crawford S., Smyth G.K., Kallies A., Busslinger M., Nutt S.L. Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat Immunol. 2016;17:323–330. doi: 10.1038/ni.3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaxton J.E., Wallace C., Riesenberg B., Zhang Y., Paulos C.M., Beeson C.C., Liu B., Li Z. Modulation of endoplasmic reticulum stress controls CD4+ T-cell activation and antitumor function. Cancer Immunol Res. 2017;5:666–675. doi: 10.1158/2326-6066.CIR-17-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd D.J., McHeyzer-Williams L.J., Kowal C., Lee A.-H., Volpe B.T., Diamond B., McHeyzer-Williams M.G., Glimcher L.H. XBP1 governs late events in plasma cell differentiation and is not required for antigen-specific memory B cell development. J Exp Med. 2009;206:2151–2159. doi: 10.1084/jem.20090738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuru A., Fujimoto N., Takahashi S., Saito M., Nakamura D., Iwano M., Iwawaki T., Kadokura H., Ron D., Kohno K. Negative feedback by IRE1β optimizes mucin production in goblet cells. Proc Natl Acad Sci USA. 2013;110:2864. doi: 10.1073/pnas.1212484110. LP-2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton J.-P., Wang L., Han D., Wang E.S., Huskey N.E., Lim L., Truitt M., McManus M.T., Ruggero D., Goga A., et al. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic caspase-2. Science. 2012;338:818–822. doi: 10.1126/science.1226191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano F., Wang X., Bertolotti A., Zhang Y., Chung P., Harding H.P., Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- Urra H., Dufey E., Lisbona F., Rojas-Rivera D., Hetz C. When ER stress reaches a dead end. Biochim Biophys Acta Mol Cell Res. 2013;1833:3507–3517. doi: 10.1016/j.bbamcr.2013.07.024. [DOI] [PubMed] [Google Scholar]
- Vattemi G., Engel W.K., McFerrin J., Askanas V. Endoplasmic reticulum stress and unfolded protein response in inclusion body myositis muscle. Am J Pathol. 2004;164:1–7. doi: 10.1016/S0002-9440(10)63089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volmer R., Ron D. Lipid-dependent regulation of the unfolded protein response. Curr Opin Cell Biol. 2015;33:67–73. doi: 10.1016/j.ceb.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P., Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang S., Kaufman R.J. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler M.C., Rizzi M., Sasik R., Almanza G., Hardiman G., Zanetti M. KDEL-retained antigen in B lymphocytes induces a proinflammatory response: a possible role for endoplasmic reticulum stress in adaptive T cell immunity. J Immunol. 2008;181:256–264. doi: 10.4049/jimmunol.181.1.256. [DOI] [PubMed] [Google Scholar]
- Xue X., Piao J.-H., Nakajima A., Sakon-Komazawa S., Kojima Y., Mori K., Yagita H., Okumura K., Harding H., Nakano H. Tumor necrosis factor α (TNFα) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFα. J Biol Chem. 2005;280:33917–33925. doi: 10.1074/jbc.M505818200. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H., Wang H.-G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem. 2004;279:45495–45502. doi: 10.1074/jbc.M406933200. [DOI] [PubMed] [Google Scholar]
- Yamamoto K., Yoshida H., Kokame K., Kaufman R.J., Mori K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J Biochem. 2004;136:343–350. doi: 10.1093/jb/mvh122. [DOI] [PubMed] [Google Scholar]
- Yamamoto K., Suzuki N., Wada T., Okada T., Yoshida H., Kaufman R.J., Mori K. Human HRD1 promoter carries a functional unfolded protein response element to which XBP1 but not ATF6 directly binds. J Biochem. 2008;144:477–486. doi: 10.1093/jb/mvn091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J., Rawson R.B., Komuro R., Chen X., Davé U.P., Prywes R., Brown M.S., Goldstein J.L. ER Stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Matsui T., Yamamoto A., Okada T., Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Zhang K., Kaufman R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Shen X., Wu J., Sakaki K., Saunders T., Rutkowski D.T., Back S.H., Kaufman R.J. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- Zinszner H., Kuroda M., Wang X., Batchvarova N., Lightfoot R.T., Remotti H., Stevens J.L., Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]