

Abstract
BACKGROUND AND PURPOSE:
Ischemic brain injury induces both functional and structural disarray affecting the blood–brain barrier (BBB) which in return aggravates stroke outcomes. Complement system and its bioactive proteins are important molecular responders to ischemia. C5a protein along with its receptor C5a receptor 1 is a key component of this system with potent pro-inflammatory and chemoattractant properties. The purpose of this study is to investigate the role of C5a protein and its receptor which are believed to participate in the inflammatory response that follows ischemic insult.
MATERIALS AND METHODS:
To mimic an ischemic in vivo event in which C5a may contact brain endothelial cells after injury, we studied oxygen-glucose deprivation (OGD) followed by reperfusion in brain microvascular endothelial cells (b.End. 3) by only added C5a at the time of reperfusion. Cell death and viability were estimated by trypan blue and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assays, respectively. Tight junction protein zonula occluden (ZO-1) levels were analyzed by Western blot analysis, and nitric oxide (NO) was assessed using the Griess reagent.
RESULTS:
Brain-derived endothelial cell was susceptible to OGD-induced injury in a duration-dependent manner as was the presence of ZO-1 protein. However, the addition of C5a protein had no notable effects even when used at high concentrations up to 100 nM. While OGD led to reduction in ZO-1 protein levels, no change was seen following the addition of C5a. Finally, OGD led unexpectedly to small decreases in NO generation, but this was again unaltered by C5a.
CONCLUSIONS:
Our study suggests that complement system protein C5a may not have a direct role in the disruption of BBB, following brain ischemia. This is in contrary with previous literature that suggests a possible role of this protein in the inflammatory response to ischemia.
Keywords: Blood–brain barrier, C5a complement protein, in vitro model, ischemia-like injury, zonula occluden-1
Introduction
Despite remarkable advancements in elucidating the hallmarks of ischemic brain injury, stroke remains a leading cause of morbidity and mortality worldwide due to the lack of effective therapies.[1] Among the various pathobiological processes associated with ischemia, there is emerging evidence that highlights the possible role of neurovascular unit in worsening the damage following stroke. This unit includes endothelial cells which are a central player in controlling blood–brain barrier (BBB) permeability and maintenance.[2,3] The integrity of BBB is susceptible to a wide range of processes that are also believed to exacerbate stroke severity and outcomes.[4,5,6,7] Systemic inflammation is an important example of such processes and is in part controlled by the complement system. As a host defense mechanism that functions to eliminate foreign pathogens and opsonize necrotic cells, the complement system is considered a vital component of our innate immunity. It consists of more than sixty plasma and membrane bound proteins, receptors, and regulators that collectively work together to confer immunity. Currently, four biochemical pathways have been discovered that activate the complement system: classical complement pathway, alternative complement pathway, lectin pathway, and recently discovered extrinsic protease pathway.[8,9,10,11,12,13,14] First three pathways are initiated by the binding of complement factor C1q to antigen–antibody complexes, foreign surface, or by the binding of mannan-binding lectin to specifically arranged mannose residues. This eventually leads to the formation of C3 and C5 convertase enzymes, which cleave their respective inactive complement factors, C3 and C5, into their active fragments: C3a, C3b and C5a, C5b, respectively. In addition, the fourth pathway is initiated through the direct cleavage of C5 by a series of proteolytic enzymes such as kallikrein and thrombin, resulting in the release of bioactive C5a fragment.[14,15]
C5a is a fluid-phase inflammatory mediator that consists of 74 amino acids. It acts as an anaphylatoxin with potent pro-inflammatory and chemoattractant properties. C5a both recruits immune cells to the site of injury through chemotaxis and activates them to induce an inflammatory response. It exerts its major effects through binding to the membrane-bound G-protein coupled first-identified C5a receptor 1 (C5AR1).[16] Thus, depending on the pathologic condition and cell type, C5a signaling can lead to various outcomes, including phagocytosis.
Unlike peripheral tissues, the central nervous system (CNS) is immunologically separated from systemic complement system due to the restriction of cell penetration by the BBB.[17] However, CNS can internally express all complement factors, including C5, and cell-derived proteases necessary to generate C5a.[18] Furthermore, primary and secondary cellular receptors including C5a, C5AR1, and C5a-like receptor 2 have been reported to be on all CNS cells suggesting a functional role for C5a in the brain.[19]
On the other hand, pathological complement system activation and the generation of C5a could also have damaging effects exacerbating CNS diseases.[19] For example, following ischemia, the complement system activates a set of bioactive molecules, including C5a.[20] C5a generation and the activation of C5AR1 are gaining increasing attention as potential players in the neuroinflammatory response, following an ischemic insult.[20,21,22,23,24] Thus, its regulation and control of this system are salient as a potential target for future stroke therapies. However, all prior investigations have been limited to investigating only one strategy: C5AR1 blocking.[23,24,25] The goal of the present study is to further elucidate the direct role of C5a in an in vitro BBB model of ischemia-reperfusion injury. To this end, we looked at oxygen-glucose deprivation (OGD) followed by reperfusion in murine brain microvascular endothelial cells (b. End. 3) subjected to OGD with and without C5a protein.
Materials and Methods
Chemicals and reagents
Reagents of analytical grade and MilliQ water were used. Dulbecco's Modified Eagle Medium (DMEM) and the immortalized b. End. 3 were purchased from American Type Culture Collection (Manassas, VA, USA). Fetal bovine serum (FBS) was purchased from Hyclone Laboratories (Logan, UT, USA). Trypsin/ethylenediaminetetraacetic acid solution, polyclonal rabbit anti-zonula occluden (ZO-1) antibody, and other cell culture reagents were purchased from Invitrogen Inc. (Camarillo, CA, USA) and the UCSF Cell Culture Facility (UCSF, San Francisco, CA, USA). Mouse recombinant C5a was purchased from ProSpec (East Brunswick, NJ, USA). C5a was dissolved in sterile 18 M-cm H2O (at stock concentration 100 μg/ml), and aliquots were stored for later use at − 20°C. C5a reconstitution was performed according to the manufacturer recommendations. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and Greiss reagent were purchased from Sigma (St. Louis, MO, USA). Balanced salt solution containing 116 mM NaCl, 1.8 mM CaCl2, 0.8 mM MgSO4, 5.4 mM KCl, 1 mM NaH2 PO4, 14.7 mM NaHCO3, 10 mM N-(2-hydroxyethyl) piperazine-N-ethane sulfonic acid (HEPES), and 10 mg/l phenol red at pH 7.4 (BSS0, glucose free) and 5.5 mM glucose (BSS5.5, with glucose) were used in the experiments with several OGD conditions. Culture flasks and plates were from BD Biosciences Labware. Protease inhibitor cocktail complete mini was obtained from Roche diagnostics.
Cell culture, treatment, and ischemia-like injury model
The immortalized mouse b.END.3 was used in this study. Cells were cultured in adjusted DMEM supplemented with 4.5 g/ml glucose, 10% FBS defined, and antibiotics.
All cells were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air. All experiments were performed in approximately 80% confluent monolayers with a cell density of nearly 0.5 × 106, verified before use. Cells were treated by recombinant mouse C5a protein (10–100 nM). Cultures were washed three times in C5a containing media and then were returned to the incubator. Control cultures were washed only in plating media.
For ischemia-like injury model, OGD followed by reperfusion was carried out. Cell cultures were deprived of glucose and oxygen by transferring into an anaerobic chamber (O2 <0.2%, COY Lab Products, MI, USA) and changing the culture medium to oxygen-, glucose-free balanced salt solution (BSS0) for 2, 4, and 6 h. After incubation at OGD conditions, the cultures were returned to the normoxia by adding 5.5 mM glucose to the culture medium (reperfusion step) and transferring into the incubator. Control cultures were washed only in BSS5.5 but were maintained in the normoxic environment. Recombinant mouse C5a protein was added on set up before the reperfusion step with a tested beforehand intermediate concentration not causing toxicity to primary cells. The total observation period for OGD experiments with following reperfusion was 24 h. The cultures that were not exposed to OGD were considered to be kept at normoxia (controls).
Cell death and viability assessment
Cell death and viability were assessed by trypan blue staining and MTT functional assay, respectively. Five random high power fields were selected, and total cell counts and trypan blue positive (dead) cell counts were made.
The MTT assay measures mitochondrial respiration, an index of cell viability. At the indicated times, appreciate volumes of 5 mg/ml MTT solution in PBS were added to each well for 4 h at 37°C. After removal of the medium, dimethyl sulfoxide was added to each well to dissolve the formazan crystals. The absorbance at 570 nm was determined using a microplate reader. All experiments were performed in triplicates. The percentage of absorbance in each sample versus the control was calculated to assess the cell viability.
Nitric oxide assay
Nitric oxide (NO) levels in culture media, BSS0 and BSS5.5, were estimated for 2 and 4 h of OGD with 5 and 24 h of reperfusion period using Greiss reagent (1% sulfanilamide and 0.1% N-1-naphthy-ethylenediamine dihydrochloride in 5% phosphoric acid) with nitrite as the standard according to protocol recommendation.[26,27] The absorbance at 570 nm was then determined using a microplate reader and a standard curve was generated using NaNO2. Total cellular protein was measured by the bicinchoninic acid (BCA)-based method (Pierce, USA).
Western blot analysis of zonula occludens-1 protein expression
Cells were harvested by scraping and washing in ice-cold PBS and lysed with ice-cold cell lysis buffers (50 mM Tris, pH 8.0; 150 mM NaCl, 1% triton X-100, and protease inhibitor cocktail complete mini), followed by sonication and centrifugation for 5 min. BCA protein assay (Pierce, Rockford, IL, USA) was used for the determination of protein concentration in the supernatant. The supernatant was used for further steps. Equal amounts of protein were loaded on 7.5% polyacrylamide gels, and SDS-PAGE (Bio-Rad Laboratories, Inc., USA) was performed according the previously described protocol.[27,28,29] After separation, proteins were electrophoretically transferred to polyvinyl difluoride membranes (Immuno-Blot PVDF membrane, Bio-Rad, USA) and blocked with 4% fat-free milk in PBS (pH 7.4) overnight. The membranes then were incubated with a 1:1000 dilution of primary rabbit polyclonal anti-mouse ZO-1 antibody for 1 h at room temperature and then incubated with a 1:10,000 dilution of secondary antibody conjugated with horseradish peroxidase for 1 h at room temperature. After the blots were incubated with secondary antibodies, bands were visualized using the enhanced Amersham ECL Western Blotting Detection Kit (GE Healthcare Life Sciences, US). B-actin was used as a housekeeping control. All experiments were performed in triplicates and analyzed by an investigator blinded to experimental conditions.
Data analysis
Statistical analysis was performed using one-way ANOVA (GraphPad 5 Software, Graphpad Software Inc., La Jolla, CA, USA). All data were expressed as mean ± standard deviation. Significance was assumed for P < 0.05. All experiments were carried out in a randomized fashion, with each experiment performed three times from three different culture sets. Investigators analyzing the data did so in a blinded fashion.
Results
To investigate the direct role of C5a in an ischemia-like injury model of BBB, we first estimated the influence of OGD duration on b. End. 3 cells. This allowed us to determine the duration of OGD necessary to cause <40% and >80% cell death. Cell death was estimated by trypan blue staining and viability was measured by MTT assay. Results showed that OGD for 2 h did not affect cell death or viability. However, there was ~40% and ~80% cell death [Figure 1a] and ~20% and 5% cell viability [Figure 1b] when OGD was continued for 4 and 6 h, respectively, and assessed 24 h later.
Figure 1.
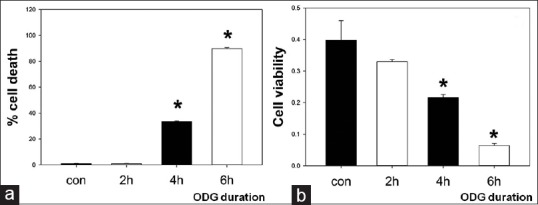
Duration--dependent endothelial cell death due to oxygen-glucose deprivation. Cultures of murine brain-derived endothelial cells (b.End. 3) were exposed to varying durations of oxygen-glucose deprivation followed by 24 h reperfusion. Cell death was estimated by trypan blue (a), whereas cell viability was estimated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (b). oxygen-glucose deprivation durations of 4 and 6 h led to dose-dependent cell death (~40% after 4 h, ~90% after 6 h) and cell viability (~20% after 4 h, 5% after 6 h), whereas 2 h oxygen-glucose deprivation had little effect on either. *P < 0.01 versus controls. OGD: Oxygen-glucose deprivation, Con: Controls
Before evaluating any effect of C5a on in vitro model of BBB, we examined cytotoxicity of C5a on b. End. 3 cells under normoxia conditions and found that C5a, even at concentrations up to 100 nM, did not affect cell viability (data are not presented).
To model a potential in vivo scenario where C5a may contact brain endothelial cells during stroke, we tested C5a effect on cell viability with dosage range of 10–100 nM by adding it only at the onset of reperfusion. Interestingly, the addition of C5a did not affect cell death [Figure 2]. In particular, there were no significant differences (P > 0.05) in the mean values of cell death (trypan blue) or cell viability (optical density, MTT assay) after OGD exposure of 4 h with (32.63 ± 1.96 and 0.25 ± 0.026) and without (33.35 ± 3.59 and 0.22 ± 0.028) C5a added at the time of reperfusion (return of cultures to normoxia with the addition of 5.5 mM glucose to the culture media).
Figure 2.
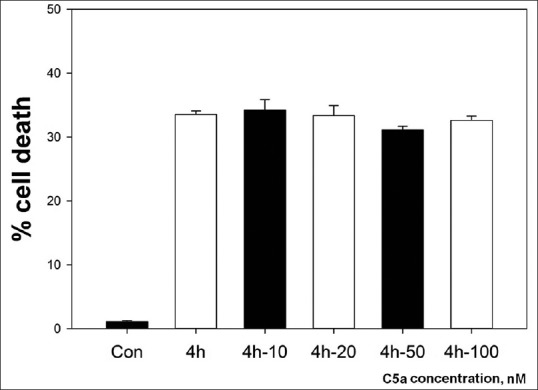
C5a does not affect cell death in endothelial cells after oxygen-glucose deprivation. b.End. 3 cells were exposed to 4 h oxygen-glucose deprivation with and without added C5a. Cell death was assessed 24 h later by trypan blue staining and compared to uninjured controls. C5a concentrations of 10–100 nM (added C5a denoted in the X-axis labels) failed to affect the extent of oxygen-glucose deprivation-induced cell death. OGD: Oxygen-glucose deprivation, Con: Controls
NO levels were estimated using the Griess reagent. b. End. 3 subjected to 4 h of OGD was somewhat lower than baseline levels although this did not reach statistical significance [Figure 3]. The addition of C5a protein at the time of reperfusion did not affect NO generation compared to OGD alone.
Figure 3.
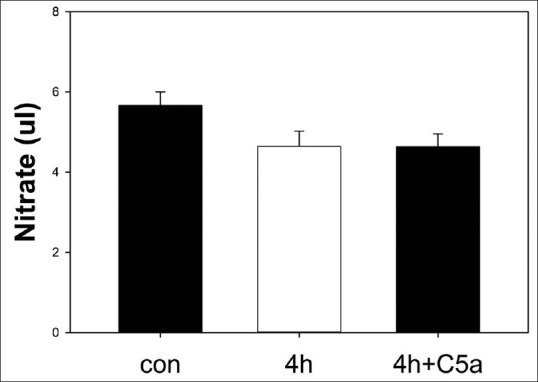
Nitric oxide generation in the media of cell cultures subjected to oxygen-glucose deprivation. b.End. 3 cells were exposed to 4 h oxygen-glucose deprivation followed by 24 h reperfusion. Estimates of nitric oxide accumulation (nitrate) from culture media were made using the Griess reagent. Nonsignificant decreases in nitric oxide accumulation were detected in the media of oxygen-glucose deprivation exposed cultures (4 h) compared to uninjured controls, but this was not affected by the addition of 100 nM C5a (4 h + C5a). OGD: Oxygen-glucose deprivation, NO: Nitric oxide, Con: Controls
Western blots of ZO-1 protein were used to estimate BBB integrity. ZO-1 is a major tight junction (TJ) protein, and TJs are thought to form the basis of the BBB by forming a scaffold between the transmembrane proteins and the actin cytoskeleton.[30] Four hours OGD led to marked reduction in ZO-1 levels, whereas 2 h OGD did not [Figure 4a]. This was the rationale behind using 2 h OGD to test our hypothesis that the addition of C5a might reduce ZO-1 levels. However, C5a addition failed to show any impact on ZO-1 levels [Figure 4b and c].
Figure 4.
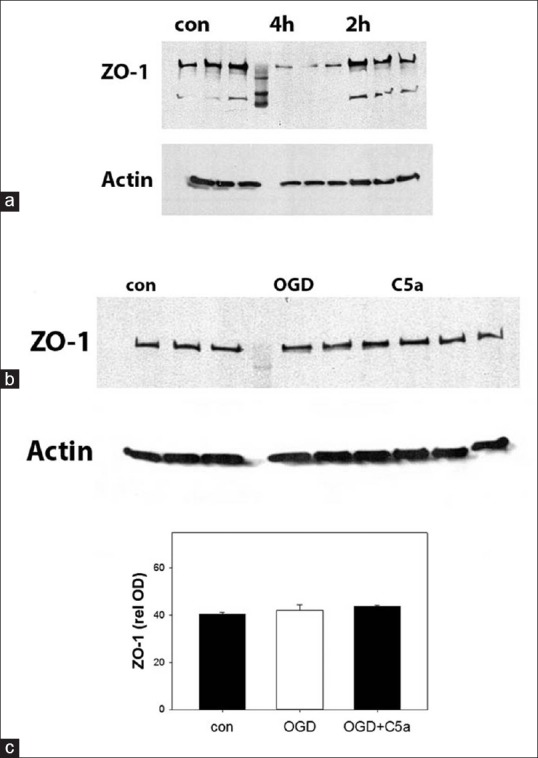
C5a does not alter zonula occludens-1 levels in endothelial cells exposed to oxygen-glucose deprivation. Representative Western blots of zonula occludens-1 protein are shown. b.End. 3 cells exposed to 2 h oxygen-glucose deprivation failed to show any changes in zonula occludens-1 protein, whereas 4 h oxygen-glucose deprivation led to marked decreases (a). Blot shows three replicates of each condition plus a maker lane (center). A Western blot of actin protein is shown as a housekeeping control. Endothelial cell cultures were then exposed to 2 h oxygen-glucose deprivation, and C5a was added to some oxygen-glucose deprivation exposed cultures at a concentration of 100 nM (C5a). A representative Western blot shows that the addition of C5a failed to change zonula occludens-1 expression compared to uninjured controls (b). Quantification of Western blots from 3 samples collected from 3 separate experiments (9 observations total) is shown in the graph (c). Relative optical densities from the blots are shown normalized to the OD of the actin proteins. OGD: Oxygen-glucose deprivation, ZO: Zonula occludens, ODs: Optical densities, Con: Controls
Discussion
BBB consists of a network of cerebral endothelial cells and associated TJs. Those junctions are formed by transmembrane molecules (claudin, occludin, and junctional adhesion molecules) that form structures with several actin cytoskeleton and cytoplasmic proteins, ZO-1, ZO-2, and ZO-3.[4,31] As was shown in previous research,[32] brain microvascular endothelial cell line, b. End. 3, demonstrates similar functional and structural characteristics of BBB. Those include formation of functional barriers, expression of the TJ proteins including ZO-1 as well as junctional adhesion molecules.[32] It has been shown that, like primary endothelial cells, b. End. 3 cells express C5AR1 on their membranes.[33]
The role of the complement system and its various components in ischemic brain injury has been the subject of much debate. Immunohistochemical analysis of human brains identified the presence of multiple components of the complement system in ischemic brain including the classic pathway products (C1q, C3, C4d) and other components such as MASP-2 of lectin pathway, factor B of alternative pathway, and C9.[34] Furthermore, the activation of complement alternative pathway, as well as the protective effect of factor B deficiency and CR2-fH treatment in pathogenesis of murine ischemic stroke, was demonstrated.[35] A In addition, elevated levels of C3a and C5a anaphylatoxins were observed in blood samples from human patients with ischemic stroke.[36] This was further proven in an in vitro study that looked at various types of ischemic insults in different neuronal cells and showed that hypothermia-induced neuroprotection is enhanced by blocking C5AR1.[22] Two other studies with C5 deficient mice subjected to middle cerebral artery occlusion (MCAO) revealed that lack of C5 led to protective effects from ischemic injury, reduced lesion size, and improved neurological scores.[21,37] Similar results were obtained in the experiments with C5AR1 (−/−) mice subjected to MCAO.[25] However, it was also shown that those beneficial effects were concentration- and time-dependent.[24,37] On the other hand, Mocco et al. reported that C5 deficiency is not protective after focal cerebral ischemia.[36] All these studies implicated significance of blocking C5AR1 in terms of neuroprotection.
Recently, Jacob et al. using serum from lupus mouse models as a source of C5a and b. End. 3 cells demonstrated that C5a/C5AR1 signaling alters BBB integrity in an NF-κB-dependent manner.[33] Using treatment with a C5AR1 antagonist and silencing the C5AR1 gene with siRNA, it has been shown that C5aR is responsible for vascular endothelial cell injury in cultured mouse dermal microvascular endothelial cells.[32] This controversy and lack of studies on C5a direct role in BBB disruption following ischemia necessitates further investigation and here where the root of our paper lies.
Interestingly, our study reveals no effect of C5a protein, even when used in high doses, on b. End. 3 cells viability and ZO-1 protein levels in ischemia-like model. One possible explanation for this discrepancy with previous research may be that C5aR has multiple ligands capable initiating downstream signaling cascades. It is known that C5aR also binds to C3a and ribosomal protein S19, but with lower binding affinity compared to C5a.[38,39] It also seems that C5 and C5a target neurons during ischemia/reperfusion rather than directly affecting the BBB. An in vitro study of OGD demonstrated the possibility of the proteolytic cleavage of C5 protein generated by neurons, leading to increased apoptotic cell death (in a C5a-dependent manner) and upregulation in C5a/C5AR1 signaling.[22] Thus, our inability to detect any effect of added C5a might be explained in part by the lack of critical co-factors that have yet to be identified. In addition, there are minor issues in the experimental design that might limit the ability of detect C5a effect on BBB cells. First, there are no actual data on the C5a levels in the brains during ischemia/reperfusion, which forced us to use a wide concentration range to test the effects of the protein on the cell viability or BBB integrity in a model of ischemia-like injury. Next, it is known that pH is changed in the brain during ischemia due to accumulation of lactate and pyruvate. However, pH drop is not implemented in the current model of BBB injury, and protocol modifications are needed in future to account for these changes. However, the ODG model is considered to be adequate to study the BBB changes and is widely used in research.
Conclusion
Overall, we conclude that C5a alone has no direct effects on endothelial cell viability or BBB integrity in an ischemic-like environment. However, further studies are necessary to shed more light on this controversial question. The application of ischemia exposed sera to cells in the presence and absence of C5a blockade or studying models that include other BBB elements such as astrocytes could be the subject of future investigation.
Financial support and sponsorship
This study was funded by grants from the National Institutes of Health (NS40516), Department of Defense and the Veteran's Merit Award (I01 BX000589) to MY, and a Fulbright Fellowship (to AK). Grants to MY was administered by the Northern California Institute for Research and Education and supported by resources of the Veterans Affairs Medical Center, San Francisco, California.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Mackay J, Mensah G. The Atlas of Heart Disease and Stroke. World Health Organization; 2005. [Last accessed on 2017 Jan 16]. Available from: http://www.who.int/cardiovascular_diseases/resources/atlas/en/ [Google Scholar]
- 2.Bazzoni G, Martinez-Estrada OM, Orsenigo F, Cordenonsi M, Citi S, Dejana E. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J Biol Chem. 2000;275:20520–6. doi: 10.1074/jbc.M905251199. [DOI] [PubMed] [Google Scholar]
- 3.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Kvietys PR, Granger DN. Role of reactive oxygen and nitrogen species in the vascular responses to inflammation. Free Radic Biol Med. 2012;52:556–92. doi: 10.1016/j.freeradbiomed.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg GA. Neurological diseases in relation to the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32:1139–51. doi: 10.1038/jcbfm.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184:53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denes A, Thornton P, Rothwell NJ, Allan SM. Inflammation and brain injury: Acute cerebral ischaemia, peripheral and central inflammation. Brain Behav Immun. 2010;24:708–23. doi: 10.1016/j.bbi.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 8.Nauta AJ, Roos A, Daha MR. A regulatory role for complement in innate immunity and autoimmunity. Int Arch Allergy Immunol. 2004;134:310–23. doi: 10.1159/000079261. [DOI] [PubMed] [Google Scholar]
- 9.Cole DS, Morgan BP. Beyond lysis: How complement influences cell fate. Clin Sci (Lond) 2003;104:455–66. doi: 10.1042/CS20020362. [DOI] [PubMed] [Google Scholar]
- 10.Villiers CL, Villiers MB, Marche PN. Role of the complement C3 protein in the control of the specific immune response. Ann Biol Clin (Paris) 1999;57:127–35. [PubMed] [Google Scholar]
- 11.Sakamoto M, Fujisawa Y, Nishioka K. Physiologic role of the complement system in host defense, disease, and malnutrition. Nutrition. 1998;14:391–8. doi: 10.1016/s0899-9007(97)00473-5. [DOI] [PubMed] [Google Scholar]
- 12.Brodsky RA. Complement in health and disease. Hematol Oncol Clin North Am. 2015;29:xi. doi: 10.1016/j.hoc.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alsenz J, Avila D, Huemer HP, Esparza I, Becherer JD, Kinoshita T, et al. Phylogeny of the third component of complement, C3: Analysis of the conservation of human CR1, CR2, H, and B binding sites, concanavalin A binding sites, and thiolester bond in the C3 from different species. Dev Comp Immunol. 1992;16:63–76. doi: 10.1016/0145-305x(92)90052-e. [DOI] [PubMed] [Google Scholar]
- 14.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, et al. Generation of C5a in the absence of C3: A new complement activation pathway. Nat Med. 2006;12:682–7. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 15.Lee H, Whitfeld PL, Mackay CR. Receptors for complement C5a. The importance of C5aR and the enigmatic role of C5L2. Immunol Cell Biol. 2008;86:153–60. doi: 10.1038/sj.icb.7100166. [DOI] [PubMed] [Google Scholar]
- 16.Gerard NP, Gerard C. The chemotactic receptor for human C5a anaphylatoxin. Nature. 1991;349:614–7. doi: 10.1038/349614a0. [DOI] [PubMed] [Google Scholar]
- 17.Kleine TO, Benes L. Immune surveillance of the human central nervous system (CNS): Different migration pathways of immune cells through the blood-brain barrier and blood-cerebrospinal fluid barrier in healthy persons. Cytometry A. 2006;69:147–51. doi: 10.1002/cyto.a.20225. [DOI] [PubMed] [Google Scholar]
- 18.Huber-Lang M, Younkin EM, Sarma JV, Riedemann N, McGuire SR, Lu KT, et al. Generation of C5a by phagocytic cells. Am J Pathol. 2002;161:1849–59. doi: 10.1016/S0002-9440(10)64461-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woodruff TM, Ager RR, Tenner AJ, Noakes PG, Taylor SM. The role of the complement system and the activation fragment C5a in the central nervous system. Neuromolecular Med. 2010;12:179–92. doi: 10.1007/s12017-009-8085-y. [DOI] [PubMed] [Google Scholar]
- 20.Ducruet AF, Zacharia BE, Hickman ZL, Grobelny BT, Yeh ML, Sosunov SA, et al. The complement cascade as a therapeutic target in intracerebral hemorrhage. Exp Neurol. 2009;219:398–403. doi: 10.1016/j.expneurol.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Costa C, Zhao L, Shen Y, Su X, Hao L, Colgan SP, et al. Role of complement component C5 in cerebral ischemia/reperfusion injury. Brain Res. 2006;1100:142–51. doi: 10.1016/j.brainres.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 22.Thundyil J, Pavlovski D, Hsieh YH, Gelderblom M, Magnus T, Fairlie DP, et al. C5a receptor (CD88) inhibition improves hypothermia-induced neuroprotection in an in vitro ischemic model. Neuromolecular Med. 2012;14:30–9. doi: 10.1007/s12017-012-8167-0. [DOI] [PubMed] [Google Scholar]
- 23.Arumugam TV, Woodruff TM, Lathia JD, Selvaraj PK, Mattson MP, Taylor SM. Neuroprotection in stroke by complement inhibition and immunoglobulin therapy. Neuroscience. 2009;158:1074–89. doi: 10.1016/j.neuroscience.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim GH, Mocco J, Hahn DK, Kellner CP, Komotar RJ, Ducruet AF, et al. Protective effect of C5a receptor inhibition after murine reperfused stroke. Neurosurgery. 2008;63:122–5. doi: 10.1227/01.NEU.0000335079.70222.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pavlovski D, Thundyil J, Monk PN, Wetsel RA, Taylor SM, Woodruff TM. Generation of complement component C5a by ischemic neurons promotes neuronal apoptosis. FASEB J. 2012;26:3680–90. doi: 10.1096/fj.11-202382. [DOI] [PubMed] [Google Scholar]
- 26.Han HS, Qiao Y, Karabiyikoglu M, Giffard RG, Yenari MA. Influence of mild hypothermia on inducible nitric oxide synthase expression and reactive nitrogen production in experimental stroke and inflammation. J Neurosci. 2002;22:3921–8. doi: 10.1523/JNEUROSCI.22-10-03921.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kacimi R, Giffard RG, Yenari MA. Endotoxin-activated microglia injure brain derived endothelial cells via NF-κB, JAK-STAT and JNK stress kinase pathways. J Inflamm (Lond) 2011;8:7. doi: 10.1186/1476-9255-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kacimi R, Chentoufi J, Honbo N, Long CS, Karliner JS. Hypoxia differentially regulates stress proteins in cultured cardiomyocytes: Role of the p38 stress-activated kinase signaling cascade, and relation to cytoprotection. Cardiovasc Res. 2000;46:139–50. doi: 10.1016/s0008-6363(00)00007-9. [DOI] [PubMed] [Google Scholar]
- 29.Kacimi R, Gerdes AM. Alterations in G protein and MAP kinase signaling pathways during cardiac remodeling in hypertension and heart failure. Hypertension. 2003;41:968–77. doi: 10.1161/01.HYP.0000062465.60601.CC. [DOI] [PubMed] [Google Scholar]
- 30.Luissint AC, Artus C, Glacial F, Ganeshamoorthy K, Couraud PO. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation. Fluids Barriers CNS. 2012;9:23. doi: 10.1186/2045-8118-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liebner S, Kniesel U, Kalbacher H, Wolburg H. Correlation of tight junction morphology with the expression of tight junction proteins in blood-brain barrier endothelial cells. Eur J Cell Biol. 2000;79:707–17. doi: 10.1078/0171-9335-00101. [DOI] [PubMed] [Google Scholar]
- 32.Li G, Simon MJ, Cancel LM, Shi ZD, Ji X, Tarbell JM, et al. Permeability of endothelial and astrocyte cocultures: In vitro blood-brain barrier models for drug delivery studies. Ann Biomed Eng. 2010;38:2499–511. doi: 10.1007/s10439-010-0023-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacob A, Hack B, Chen P, Quigg RJ, Alexander JJ. C5a/CD88 signaling alters blood-brain barrier integrity in lupus through nuclear factor-κB. J Neurochem. 2011;119:1041–51. doi: 10.1111/j.1471-4159.2011.07490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pedersen ED, Løberg EM, Vege E, Daha MR, Maehlen J, Mollnes TE. In situ deposition of complement in human acute brain ischaemia. Scand J Immunol. 2009;69:555–62. doi: 10.1111/j.1365-3083.2009.02253.x. [DOI] [PubMed] [Google Scholar]
- 35.Elvington A, Atkinson C, Zhu H, Yu J, Takahashi K, Stahl GL, et al. The alternative complement pathway propagates inflammation and injury in murine ischemic stroke. J Immunol. 2012;189:4640–7. doi: 10.4049/jimmunol.1201904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mocco J, Mack WJ, Ducruet AF, Sosunov SA, Sughrue ME, Hassid BG, et al. Complement component C3 mediates inflammatory injury following focal cerebral ischemia. Circ Res. 2006;99:209–17. doi: 10.1161/01.RES.0000232544.90675.42. [DOI] [PubMed] [Google Scholar]
- 37.Arumugam TV, Tang SC, Lathia JD, Cheng A, Mughal MR, Chigurupati S, et al. Intravenous immunoglobulin (IVIG) protects the brain against experimental stroke by preventing complement-mediated neuronal cell death. Proc Natl Acad Sci U S A. 2007;104:14104–9. doi: 10.1073/pnas.0700506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Filip AM, Klug J, Cayli S, Fröhlich S, Henke T, Lacher P, et al. Ribosomal protein S19 interacts with macrophage migration inhibitory factor and attenuates its pro-inflammatory function. J Biol Chem. 2009;284:7977–85. doi: 10.1074/jbc.M808620200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishiura H, Nonaka H, Revollo IS, Semba U, Li Y, Ota Y, et al. Pro- and anti-apoptotic dual functions of the C5a receptor: Involvement of regulator of G protein signaling 3 and extracellular signal-regulated kinase. Lab Invest. 2009;89:676–94. doi: 10.1038/labinvest.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]