

Abstract
There is currently a demand for new highly efficient and specific drugs to treat osteoporosis, a chronic bone disease affecting millions of people worldwide. We have developed a combinatorial strategy for engineering bispecific inhibitors that simultaneously target the unique combination of c-FMS and αvβ3 integrin, which act in concert to facilitate bone resorption by osteoclasts. Using functional fluorescence-activated cell sorting (FACS)-based screening assays of random mutagenesis macrophage colony-stimulating factor (M-CSF) libraries against c-FMS and αvβ3 integrin, we engineered dual-specific M-CSF mutants with high affinity to both receptors. These bispecific mutants act as functional antagonists of c-FMS and αvβ3 integrin activation and hence of osteoclast differentiation in vitro and osteoclast activity in vivo. This study thus introduces a versatile platform for the creation of new-generation therapeutics with high efficacy and specificity for osteoporosis and other bone diseases. It also provides new tools for studying molecular mechanisms and the cell signaling pathways that mediate osteoclast differentiation and function.
Author summary
Many bone diseases—including osteoporosis, in which the bones become brittle and fragile from loss of tissue—are characterized by excessive and uncontrolled bone resorption by bone-destroying cells known as osteoclasts. Therefore, controlled and specific inhibition of osteoclast activity is a desired outcome in treatments for bone diseases. Osteoclast differentiation and function are coordinated by cell surface receptors, including c-FMS and αvβ3 integrin, which cooperate with one another to drive signals that are essential for osteoclast functions. Here, we describe the engineering, characterization, and testing of novel proteins that can target and inhibit both c-FMS and αvβ3 integrin at the same time, thereby providing a way of controlling osteoclast function. The study represents the first example of engineering a natural ligand, which acts as a signaling molecule, as a scaffold for binding not only its target protein but also a second target. We show that these engineered proteins inhibit osteoclast activity in a mouse model of osteoporosis. Our study describes potential inhibitors that target all the known functions resulting from c-FMS/integrin αvβ3 crosstalk and paves the way to create novel targeting proteins that could be used to treat osteoporosis. It also expands our understanding of the role of the c-FMS/αvβ3 integrin pathway in the regulation of osteoclast differentiation and function.
Introduction
Osteoporosis, a chronic skeletal disorder common in both women and men beyond the age of 50 [1], is the underlying cause of more than 8.9 million fractures annually worldwide, with the consequent high burden of social and economic costs [2]. The drugs initially used for the management of osteoporosis in women were based on estrogens, but long-term administration of these drugs is associated with increased risk of breast cancer, cardiovascular disease, and dementia [3–5]. To date, the most commonly prescribed drugs for osteoporosis are bisphosphonates, but these drugs, too, have nonspecific adverse side effects, such as gastrointestinal toxicity, renal toxicity, hypercalcemia, osteonecrosis of the jaw, and more [6]. A different therapeutic direction was recently opened with the Food and Drug Administration (FDA) approval of the receptor for activation of the nuclear factor–kappa B (NF-кB) ligand (RANKL) antibody, denosumab, but the clinical promise of this drug has been offset by reports of adverse side effects, such as hypocalcemia [7] and atypical hip fractures [8]. Thus, there is a pressing need for new efficient and highly specific drugs for the management of osteoporosis.
Central to the pathogenesis of osteoporosis is excessive bone resorption by osteoclasts [9]. These cells differentiate from cells of the monocyte/macrophage lineage upon stimulation of two essential factors, monocyte/macrophage colony-stimulating factor (M-CSF) and RANKL [10]. The importance of M-CSF and its receptor c-FMS in osteoclast function has been clearly illustrated in a study showing that both M-CSF–deficient and c-FMS–deficient mice suffer from retarded skeletal growth and osteopetrosis [11]. Another factor that is essential for osteoclast functioning is αvβ3 integrin, as indicated, for example, in studies showing increased bone mass in integrin β3 knockout mice due to a functional defect in their osteoclasts [12–15]. The interaction of αvβ3 integrin with the bone matrix induces a cytoskeleton organization that polarizes the osteoclast's resorptive machinery to the bone/cell interface, where it creates an isolated compartment consisting of an actin ring surrounding a ruffled border essential for resorption by the matured osteoclasts.
Importantly, in addition to the distinctive roles of c-FMS and αvβ3 integrin in osteoclast activity, these two factors also play a cooperative role in bone resorption. M-CSF signaling regulates bone resorption by crosstalk through its receptor c-FMS, with the signaling being activated by αvβ3 integrin. This signaling regulates cytoskeleton rearrangement, as both M-CSF and αvβ3 integrin stimulate the same c-Src–initiated signaling complex essential for regulation of the osteoclast cytoskeleton [16–18]. In addition, c-FMS alters the conformation of αvβ3 integrin from a low-affinity to a high-affinity state [19], and it activates the Rho GTPase family member Rac (which is essential for osteoclast cytoskeleton organization) in an αvβ3-integrin–dependent manner [16, 20]. Moreover, it has been demonstrated that β3-integrin–deficient mice have elevated concentrations of serum M-CSF and that impaired expression of differentiation markers of β3-integrin–deficient osteoclasts can be rescued by an increased concentration of M-CSF, suggesting a compensation mechanism between the actions of these receptors [16]. In addition, c-FMS and αvβ3 integrin colocalize to the osteoclast cytoskeleton, suggesting that the crosstalk between these receptors takes place at the same cellular sites. Indeed, it has been shown that an endogenic complex of c-FMS and αvβ3 integrin coprecipitates from differentiated osteoclasts [21]. Thus, the functional and spatial coupling of c-FMS and αvβ3 integrin in osteoclast differentiation and function, together with the osteoclast-exclusive presentation and crosstalk of these two receptors, suggests that an antagonist that could bind simultaneously to these two target receptors would serve as a highly specific and effective antiresorptive drug [19, 22].
We thus aimed to exploit both the osteoclast's need for functional c-FMS and αvβ3 integrin and the specific expression of these two factors as a platform for developing a new generation of bispecific c-FMS/αvβ3 integrin antagonists for application in osteoporosis therapy and as tools to study osteoclast biology. In our rational design, two focused libraries were constructed, in each of which an Arginine-Glycine-Aspartic acid (RGD) sequence (a motif crucial for αvβ3 integrin ligand binding) flanked by random amino acids was introduced into the dimerization interface of the M-CSF ligand with the aim to change its activity from agonistic to antagonistic. This modification blocks the interactions between two M-CSF monomers and hence their subsequent homodimerization and activation of the M-CSF/c-FMS signal transduction cascade (Fig 1). Our combinatorial strategy to further develop this unique antagonist involved the screening of the abovementioned focused libraries against αvβ3 integrin. Thereafter, functional FACS-based screening assays were applied to quantitatively select for M-CSF mutants with the highest expression, stabilities and affinities for both αvβ3 integrin and c-FMS. With this approach, we generated a number of unique dual-specific M-CSF mutants capable of binding to and antagonizing the biological activity of both c-FMS and αvβ3 integrin in vitro and in vivo.
Fig 1. Schematic representation of the different M-CSF constructs.

(A) WT M-CSF (designated M-CSFWT) constituting a disulfide-bond–linked homodimer. (B) Monospecific M-CSF that can bind αvβ3 integrin (designated M-CSFαvβ3) via an RGD motif but not c-FMS because of mutations in positions 9 and 15. (C) Monospecific M-CSF that can bind only c-FMS (designated M-CSFc-FMS) because of two point mutations that change the RGD motif to RDG, thereby preventing its binding to αvβ3 integrin. (D) Libraries (designated M-CSFRGD) created by changing two loops in the M-CSF dimerization site to an RGD motif with three random amino acids on each side, thereby enabling binding to αvβ3 integrin. M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid; WT, wild type.
Results
Testing the compatibility of the M-CSF/c-FMS system with the yeast surface display method
The M-CSF glycoprotein is secreted as a homodimer that, upon binding to two c-FMS receptors, induces c-FMS autophosphorylation followed by activation of downstream signaling pathways [23]. Our strategy for converting M-CSF from an agonist into an antagonist of the c-FMS receptor relied on impairing the ability of M-CSF to dimerize. To this end, we exploited the critical role of the cysteine in position 31 in the covalent dimerization of wild-type M-CSF (M-CSFWT) [24], namely, we generated a mutant M-CSF with impaired covalent dimerization capability (designated M-CSFC31S) by replacing C31 with a serine residue (S1A Fig). To determine whether the M-CSFC31S/c-FMS system was indeed compatible with the yeast surface display (YSD) method, we transformed the M-CSFC31S gene into Saccharomyces cerevisiae and determined M-CSFC31S protein expression (S2A and S2B Fig) and binding to soluble c-FMS (S2C Fig). To evaluate the binding affinity of M-CSFC31S for c-FMS in the YSD system, we measured its apparent binding to different concentrations of soluble c-FMS (kD,app = 20 nM; S2D Fig). The observed KD,app did indeed resemble the KD of 13.6 nM known from the literature for the M-CSF homodimer [25], thereby validating the compatibility of YSD with the M-CSFC31S/c-FMS system.
Engineering and selection of M-CSF harboring RGD motif proteins with high affinity for c-FMS and αvβ3 integrin
To construct a dual-specific M-CSF monomer that can bind both c-FMS and αvβ3 integrin, two libraries were created by replacing one of two loops on the M-CSFC31S scaffold with RGD, flanked by three random amino acids on each side, namely, by XXXRGDXXX, where X represents any random amino acid (designated M-CSFRGD) (S1B and S1C Fig). To identify and isolate M-CSF variants with high affinity for both c-FMS and αvβ3 integrin, we generated a YSD construct (S3 Fig) that allowed us to separate high-affinity clones from low-affinity clones by using flow cytometry. After confirming that M-CSFRGD libraries were well expressed on the surface of the yeast cell wall and that these libraries did indeed bind 100 nM soluble c-FMS and 500 nM αvβ3 integrin (S4 Fig), we merged the two libraries and regarded them as a single library that consisted of 1.1 × 107 variants. For affinity maturation, the M-CSFRGD library was sorted in a stepwise manner against αvβ3 integrin and c-FMS by using FACS (Fig 2 and S5 Fig). To identify the specific variants that would bind both c-FMS and αvβ3 integrin, we tested individual clones for c-FMS and αvβ3 integrin binding and identified three unique clones with high affinity for both receptors (S6 Fig). These M-CSFRGD variants were designated 4.22, 4.24 (both from sort four), and 5.6 (from sort five) (S1F Fig). Selective binding of these three variants to αvβ3 integrin and not to other integrins was confirmed by evaluating their binding to αvβ3, α4β7, αiibβ3 and α5β1 integrins (250 nM) (S6 Fig). Since all three variants showed high specificity for αvβ3 integrin, they were regarded as good candidates for osteoclast-specific drug development, since osteoclasts express high levels of αvβ3 integrin as early as 48 h of differentiation [26].
Fig 2. FACS dot plot of M-CSFRGD affinity maturation process.

Yeast-displayed mutant pools were tested for binding to (A) 200 nM c-FMS, (B) 500 nM αvβ3 integrin, (C) 250 nM αvβ3 integrin, (D) 100 nM αvβ3 integrin, (E) 20 nM αvβ3 integrin, and (F) 50 nM c-FMS. High target binders were sorted as indicated in each figure with black square- or polygon-shaped gates. M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid.
Production, purification, and characterization of bispecific M-CSFRGD variants and monospecific M-CSF controls
We produced the bispecific M-CSFRGD protein variants and their monospecific controls, M-CSFc-FMS (S1E Fig) and M-CSFαvβ3 (S1D Fig), with the aim to characterize these proteins in their soluble forms. The proteins were expressed in Pichia pastoris, glycans were removed, and the nonglycosylated proteins were purified by using affinity and size exclusion chromatography, giving the expected size of approximately 21 kDa (S7A and S7E Fig). Circular dichroism (CD) spectra of the purified proteins showed similar curves, corresponding to a protein that consists mostly of α-helix motifs, which means that the RGD loops did not cause a significant change in the protein secondary structure (S7C Fig). The CD spectra were also exploited to determine the melting temperatures of the proteins, giving 73 °C for variant 4.22, 77.5 °C for variant 4.24, 85 °C for variant 5.6, and 68.7 °C for M-CSFc-FMS; the melting temperature for M-CSFαvβ3 could not be determined (S7D Fig). The exact molecular weights of the purified variants and monospecific controls, determined by mass spectrometry, were as follows: 21.7 kDa for both variant 4.22 (S7B Fig) and variant 4.24, 22 kDa for variant 5.6, 21.8 kDa for M-CSFc-FMS, and 21.7 kDa for M-CSFαvβ3.
To ensure that our purified proteins did not dimerize and would thus be able to inhibit c-FMS dimerization and downstream signaling, we incubated the proteins with the cross-linker reagent BS3 (bis[sulfosuccinimidyl]suberate) to enhance noncovalent intermolecular interactions and to enable visualization of these interactions. As expected, the three bispecific M-CSFRGD proteins as well as the monospecific M-CSFαvβ3 and M-CSFc-FMS proteins did not dimerize even at a very high BS3 concentration of 2500 μM (S8 Fig). In contrast, M-CSFWT did dimerize at the very low BS3 concentration of 25 μM. Since M-CSFRGD and M-CSFc-FMS do not dimerize in solution, they are expected to bind c-FMS and prevent its dimerization and activation.
Purified bispecific proteins bind c-FMS and αvβ3 integrin with different affinities but do not bind other RGD binding integrins
An optimal competitor for native protein receptors should exhibit a higher binding affinity (lower KD) than the binding affinities of their natural ligands. The KD of M-CSFWT to its c-FMS receptor lies in the low nanomolar range (13.6 nM) [25]. The binding affinity of αvβ3 integrin to its natural ligand, vitronectin, is 40.7 nM [27]. The binding affinities of M-CSFRGD, M-CSFαvβ3, and M-CSFc-FMS to c-FMS and αvβ3 integrin were obtained from the equilibrium-binding phase of the sensorgrams measured by surface plasmon resonance (SPR) (S9 Fig). The affinities of the protein variants to c-FMS were lower than those of M-CSFWT as a result of the RGD loop substitution, resulting in KD values of 152 nM for 4.22, 219 nM for 4.24, 96 nM for 5.6, and 195 nM for M-CSFc-FMS (Fig 3 and S1 Table). As expected, M-CSFαvβ3 did not bind to c-FMS. The binding affinities of the M-CSFRGD proteins to αvβ3 integrin were 199 nM for 4.22, 108 nM for 4.24, and 245 nM for 5.6, and the KD of M-CSFαvβ3 was 231 nM. As expected, M-CSFc-FMS did not bind to αvβ3 integrin. Thus, consistent with the YSD results, the three M-CSFRGD proteins demonstrated different affinities for αvβ3 integrin and c-FMS: 4.24 had a low affinity for αvβ3 integrin and a higher affinity for c-FMS, 5.6 exhibited high affinity for c-FMS and lower affinity for αvβ3 integrin, and 4.22 had intermediate affinity levels for the two receptors. This heterogeneity in binding affinities underlines the plasticity of ligand-based bispecific antagonists, which can provide the most sensitive balance of efficacy and specificity. This binding heterogeneity may also enable us to predict the contribution of each of the two targets, c-FMS and αvβ3 integrin, to osteoclast activation and differentiation (see following sections).
Fig 3. SPR binding sensorgrams of the different purified M-CSF proteins.

Binding of M-CSFαvβ3 (A and F), M-CSFc-FMS (B and G), 4.22 (C and H), 4.24 (D and I), and 5.6 (E and J) to c-FMS (A-E) and αvβ3 integrin (F-J) at concentrations of 12.5 nM, 25 nM, 50 nM, 100 nM, and 200 nM. Source data can be found in S1 Data. M-CSF, macrophage colony-stimulating factor; RUs, response units; SPR, surface plasmon resonance.
To determine the specificity of the three M-CSFRGD variants for αvβ3 integrin vis-à-vis the other integrins produced in the human body, SPR was used to evaluate the binding of M-CSFRGD variants (1 μM) to four different RGD-binding integrins (α3β1, α4β7, α5β1, and αvβ3). Consistent with our previous results, αvβ3 integrin bound the soluble M-CSFRGD proteins with high affinity (S10A Fig). Integrins α3β1, α4β7, and α5β1 did not bind to the three M-CSFRGD proteins (S10B–S10D Fig).
Computational docking model of αvβ3 integrin and M-CSFRGD/c-FMS complex predicts the interaction interface
To better understand the interactions between M-CSFRGD variant 4.22 and αvβ3 integrin, a molecular docking procedure was performed with PatchDock [28] both for the M-CSFC31S (S11 Fig) and 4.22 with the binding domains of αvβ3 integrin. For further refinement, clustering was performed on the docking solutions, and the most abundant structure was used for the analysis of the model. The dimerization loop 1 of M-CSF is located on the opposite side of the molecule to the c-FMS binding domain. Thus, monomeric M-CSFRGD is capable of simultaneous binding to both c-FMS and αvβ3 integrin (Fig 4), since there is no steric clash between the native receptor c-FMS and αvβ3 integrin. In spatial terms, the ligand could thus serve as a bispecific agent for both receptors.
Fig 4. Docking model of the M-CSFRGD variant 4.22/c-FMS-αvβ3 integrin complex.

M-CSFRGD variant 4.22 is shown in pink, c-FMS in cyan, αv in yellow, β3 in green, and QTSRGDSPS mutant residues in red. M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid.
R28 of RGD is the only residue contacting the αv subunit. The rest of the loop is located closer to β3. Almost all the mutant residues lie on the β3 surface, making contact with integrin residues but still exposed to the surroundings (S12 Fig). This configuration creates a total interaction surface of 1,483 Å2 between M-CSFRGD variant 4.22 and αvβ3 integrin. The differences in RGD orientation in comparison to the crystal structure of αvβ3 integrin in a complex with cyclic RGD (cRGD) [29] are shown in S13 Fig. The D side of the ligand is buried deeper in the β3 subunit in our model than in the crystal. The location of G in the middle is similar in both structures, while R points in a different direction and thus has a very limited contact with αv.
M-CSFRGD variants bind to both c-FMS and αvβ3 integrin expressed on cells
Assays for direct cell binding were performed on breast cancer cell line MD Anderson metastatic breast 231 (MDA-MB-231) and murine bone-marrow–derived monocytes (BMMs) for the following reasons. MDA-MB-231 cells express human αvβ3 integrin and c-FMS on the cell membrane in a protein expression pattern that imitates the osteoclast state just before reaching full differentiation and maturation (S14C and S14D Fig). This cell line showed binding to 1 μM, 2.5 μM, and 7.5 μM of each of the M-CSFRGD variants 4.22 and 5.6 in a dose-dependent manner (Fig 5A), with variant 5.6 showing similar binding to 4.22. Murine BMMs express high levels of c-FMS and low levels of αvβ3 integrin (S14A and S14B Fig), a state that imitates an early osteoclast differentiation stage. For these cells, variants 4.22 and 5.6 showed similar dose-dependent binding. To show that M-CSFRGD variants do indeed bind to c-FMS and αvβ3 integrin expressed on cells, we used specific competitors to these receptors, namely, cRGD, which will compete with αvβ3 integrin binding, and M-CSFWT, which will compete with c-FMS binding. M-CSFRGD variant 5.6 showed reduced binding to MDA-MB-231 in the presence of each competitor versus in the absence of inhibitors. Strikingly, the binding to the cells was significantly reduced in the presence of both inhibitors, thereby emphasizing that the ability to bind both receptors contributes to the total binding capability of the bispecific proteins. Moreover, the fact that the binding of variant 5.6 to the cells was stronger than that of M-CSFc-FMS and M-CSFαvβ3 confirmed the advantage of the bispecific protein over the monospecific proteins (Fig 5C).
Fig 5. Cell binding, c-FMS, and Akt activation assays.

Purified M-CSFRGD variants 4.22 and 5.6 were tested for binding to (A) MDA-MB-231 breast cancer cell line and (B) murine BMMs at different protein concentrations (1 μM, 2.5 μM, and 7.5 μM). The cellular expression levels of c-FMS and αvβ3 integrin are indicated as superscripts. (C) Cell competition binding assay for variant 5.6 in the presence and absence of the two competitors, namely 10 μM cRGD and 5 μM M-CSFWT. Binding of M-CSFc-FMS and M-CSFαvβ3 to the cells is shown for comparison. (D) Tyrosine phosphorylation of c-FMS and (F) serine phosphorylation of Akt in murine BMMs. Different gel runs are separated by a black line. (E) Relative c-FMS and (G) Akt phosphorylation levels of BMMs following incubation with M-CSFc-FMS, M-CSFαvβ3, and M-CSFRGD variants 4.22, 4.24, and 5.6 in the presence of recombinant M-CSF as a competitor. Chemiluminescence read-outs were quantified by densitometry. Data are means ± SEM of triplicates. *p < 0.05, **p < 0.01, ***p < 0.001. The aspect ratios of the membranes in panels D and F were changed. Source data and analysis can be found in S2 Data. BMM, bone-marrow–derived monocyte; cRGD, cyclic RGD; M-CSF, macrophage colony-stimulating factor; MDA-MB-231, MD Anderson metastatic breast 231; RGD, Arginine-Glycine-Aspartic acid.
M-CSFRGD variants inhibit induction of Akt and c-FMS phosphorylation
Upon binding to c-FMS, M-CSFWT induces c-FMS phosphorylation, which is essential for downstream signaling. To test whether binding of M-CSFRGD inhibits the activation of c-FMS, we monitored its phosphorylation in the presence and absence of recombinant M-CSF in murine BMMs. As expected, the variants did not activate c-FMS (S15A Fig), and the inhibition of c-FMS phosphorylation correlated with the affinity of the different M-CSFRGD proteins for c-FMS, with variant 5.6 (which binds c-FMS with the highest affinity) being the most efficient inhibitor and variant 4.24 (which has the lowest c-FMS binding affinity) the least efficient (Fig 5D and 5E). These data show the correlation between inhibition of c-FMS by the M-CSFRGD proteins and the binding affinity of the variant proteins to c-FMS. It is well known that human M-CSFWT can bind mouse c-FMS, but the binding affinity is much lower than that for mouse M-CSF to mouse c-FMS [30]. Nonetheless, the M-CSFRGD variants significantly inhibited c-FMS phosphorylation and successfully competed with murine M-CSF in cells expressing murine c-FMS even though the M-CSFRGD variants were of human origin. This finding suggests that the M-CSFRGD variants should be highly effective as antagonists even in mouse cell models. The same experiment was performed for Akt activation with and without recombinant M-CSF. Akt activation has previously been shown to prolong cell survival in response to c-FMS and αvβ3 integrin activation [31]. As expected, our protein variants did not spontaneously activate Akt phosphorylation (S15B Fig), and they significantly inhibited M-CSF–induced Akt phosphorylation (Fig 5F). The M-CSFRGD variants, M-CSFc-FMS, and M-CSFαvβ3 inhibited Akt phosphorylation, with the normalized phosphorylation levels being lower than the basal level of the negative control (Fig 5G).
M-CSFRGD inhibits actin belt formation in mature osteoclasts
For a mature osteoclast to resorb bone properly, it must form a unique structure termed the "sealing zone"; this structure consists of an actin ring made from a dense actin mesh connected by adhesion complexes called podosomes. The formation of the actin ring involves several stages: at the beginning, podosomes are formed throughout the cell in a scattered manner; then groups of podosomes form small actin structures, known as belts, and upon osteoclast maturation, the belts move to the cell periphery, cluster, and form an actin ring. Podosomes contain high levels of αvβ3 integrin, which is essential for the formation on bones of proper actin rings with condensed podosome networks [32, 33]. As opposed to cells grown on bone, osteoclasts cultured on glass produce actin belts but are unable to form stable actin rings. However, the preceding stages of the podosome network are similar for cells grown on bone and on glass and are known to be regulated by αvβ3 integrin [34]. To test whether the inhibition of αvβ3 integrin by bispecific M-CSFRGD variants affects the organization of podosome networks, mouse BMMs cultured on glass were allowed to differentiate into mature osteoclasts to the point that multinucleated cells were formed and exhibited an actin belt structure. At this point, the inhibitors were added, and the cells were incubated for an additional 24 h before they were fixed and stained. Cells incubated only with M-CSF and RANKL without any inhibitor (i.e., positive control) formed a dense continuous actin belt structure. Addition of the monospecific M-CSFc-FMS did not drastically inhibit actin belt formation and abundance, and osteoclasts presenting dense continuous actin belts were observed (Fig 6A and 6B); nevertheless, scattered podosomes presenting immature belts were more abundant than in the positive controls (Fig 6, lower panel). Addition of either cRGD peptide, a well-established αvβ3 integrin inhibitor [35], or the monospecific M-CSFαvβ3 decreased actin belts’ distribution (Fig 6B). The actin belts in cells incubated with these inhibitors had wider and fainter actin stain, probably as a failure of the podosomes to form condensed structures as higher distribution of scattered podosomes was observed in these cells. The bispecific M-CSFRGD variant 5.6, which exhibited the lowest affinity to αvβ3 integrin, showed the same phenotype as cRGD and M-CSFαvβ3. Strikingly, the bispecific M-CSFRGD variants 4.22 and 4.24 had the highest impact on actin belt formation and density. Dense actin structures and even scattered podosomes were hard to detect, and instead, the actin stain in osteoclasts cultured in the presence of these inhibitors had a diffusible appearance throughout the cell (Fig 6, S16 Fig).
Fig 6. Actin belt formation in mature osteoclasts incubated with M-CSFRGD variants.

(A) Murine BMMs were allowed to differentiate into osteoclasts in the presence of M-CSF and RANKL for 72 h. Then, the cells were incubated for an additional 24 h without (positive control) or with inhibitors (5 μM), followed by fixation and staining for F-actin and nuclei. The cells formed solid actin belts (white arrowheads), actin belts with "scattered" podosomes (white arrows), or "amorphous" actin stain distribution (barbed arrowheads). (B) The actin belts’ formation was quantified by normalizing the numbers of solid actin belt to the number of osteoclasts. Pictures are representatives of 35 images acquired from five different wells per sample. Data are means ± SEM of triplicates. *p < 0.05, **p < 0.01, ***p < 0.001. Source data can be found in S3 Data. BMM, bone-marrow–derived monocyte; cRGD, cyclic RGD; M-CSF, macrophage colony-stimulating factor; RANKL, receptor activator of the nuclear factor–kappa-B ligand; RGD, Arginine-Glycine-Aspartic Acid.
M-CSFRGD variants inhibit osteoclast differentiation in vitro
To assess the ability of the three bispecific M-CSFRGD variants and their monospecific controls, M-CSFc-FMS and M-CSFαvβ3, to inhibit osteoclast differentiation, we monitored the effects of elevated concentrations of these proteins in cultures of differentiating BMMs or human peripheral blood CD14+ monocytes. The proteins were added together with recombinant RANKL and human (for CD14+ cells) or murine (for BMMs) M-CSF to initiate osteoclast differentiation. We note that as osteoclasts differentiate, the number of nuclei and the surface area increase as a result of cell fusion and cytoskeletal rearrangements, respectively. Strikingly, in cultures of differentiating BMMs and CD14+ cells, all three M-CSFRGD variants inhibited the nuclei number, surface area, total osteoclast number, and tartrate-resistant acid phosphatase (TRAP) absorbance, with inhibition being significant at a concentration as low as 50 nM (Fig 7 and Fig 8). To further validate the ability of the M-CSFRGD variants to inhibit osteoclast differentiation, the mRNA expression levels of osteoclast-associated receptor (OSCAR) and NFATc1—two well-known osteoclast differentiation markers exhibiting increased expression during osteoclast differentiation—were evaluated [36, 37]. The three M-CSFRGD variants significantly inhibited OSCAR and NFATc1 expression while M-CSFαvβ3 had no inhibitory effect (Fig 7F and 7G). In general, the ability of the M-CSF monospecific controls to inhibit osteoclast differentiation was inferior to that of the bispecific M-CSFRGD variants, particularly in the human CD14+ cell cultures. Since M-CSF signaling is known to regulate osteoclast survival, inhibition of M-CSF is expected to lead to a decrease in osteoclast survival. Indeed, a smaller number of cells was observed in osteoclasts cultured in the presence of the M-CSFRGD variants. In addition, an assay for cell death, based on propidium iodide (PI) permeability, in differentiating osteoclast cultures showed that the three M-CSFRGD inhibitors and the M-CSFc-FMS significantly increased cell death versus the small molecule GW2580 (a known inhibitor for c-FMS phosphorylation) used in the control assay. We note that M-CSFαvβ3 was unable to induce cell death, probably because of its inability to bind c-FMS (Fig 7H). To rule out nonspecific toxic effects of the M-CSFRGD variants on cell proliferation and survival, we added these inhibitors to murine bone-marrow–derived mesenchymal stromal cells (BMSCs), whose physiological microenvironment is the same as that of BMMs. No changes in survival and proliferation capabilities of BMSCs cultured with elevated concentrations of the M-CSFRGD variants were observed (Fig 7I). Taken together, these results show that all M-CSFRGD variants are highly efficient in inhibiting osteoclast differentiation and spreading even at a low concentration and have no apparent toxic effects on other primary bone-marrow–residing cells.
Fig 7. Effects of M-CSFRGD variants on murine osteoclast differentiation.

Murine BMMs were cultured for 96 h in a medium containing recombinant mouse M-CSF (20 ng/ml), RANKL (20 ng/ml), and different concentrations of inhibitors. The same medium without inhibitors was used as a positive control. The medium for the negative control was supplemented with recombinant M-CSF. (A) Cells were fixed and stained for TRAP. (B–D) Cells were examined for: (B) number of osteoclasts, (C) number of nuclei within osteoclasts, (D) total surface area, and (E) total TRAP absorbance were normalized to the positive control. The effect of the inhibitors on markers of osteoclast differentiation was assessed using quantitative PCR for (F) NFATc1 and (G) Oscar mRNA expression. (H) The effect of the inhibitor on pre-osteoclast cell survival was assayed by measuring PI incorporation in osteoclasts cultured for 48 h in the presence of 1 μM of each inhibitor. (I) To test whether there is an unspecific toxic effect of the inhibitors, BMSCs were tested for cell viability in the XTT assay in the presence of three different concentrations of inhibitor (50 nM, 1 μM, and 5 μM). Data are means ± SEM of triplicates. A total of 2,340 frames were analyzed for 1,581 osteoclasts and 5,313 nuclei. *p < 0.05, **p < 0.01, ***p < 0.001. Source data and analysis can be found in S4 Data. BMM, bone-marrow–derived monocyte; BMSC, bone-marrow–derived mesenchymal stromal cell; M-CSF, macrophage colony-stimulating factor; PI, propidium iodide; RANKL, receptor activator of the nuclear factor–kappa-B ligand; RGD, Arginine-Glycine-Aspartic acid; TRAP, tartrate-resistant acid phosphatase; XTT, 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide.
Fig 8. Effects of M-CSFRGD variants on human osteoclast differentiation.

Human CD14+ cells were cultured for 96 h in medium containing recombinant human M-CSF (20 ng/ml), murine RANKL (20 ng/ml), and different concentrations of inhibitors. The same medium without inhibitors was used as a positive control. The αMEM medium for the negative control was supplemented with recombinant M-CSF. (A) Cells were fixed and stained for TRAP. (B-D) Cells were examined for (B) number of mature osteoclasts, (C) number of nuclei within osteoclasts, and (D) total surface area. Results were normalized to the positive control. Data are means ± SEM of triplicates. A total of 1,620 frames were analyzed for 477 osteoclasts and 1,897 nuclei. *p < 0.05, **p < 0.01, ***p < 0.001. Source data and analysis can be found in S5 Data. αMEM, alpha Minimum Essential Medium; M-CSF, macrophage colony-stimulating factor; RANKL, receptor activator of the nuclear factor–kappa-B ligand; RGD, Arginine-Glycine-Aspartic acid; TRAP, tartrate-resistant acid phosphatase.
In vivo biodistribution of M-CSFRGD variant 5.6
To evaluate the biodistribution of M-CSFRGD, we chose variant 5.6 because yields of this purified protein were higher than those for purified variants 4.22 and 4.24. The protein was stained with DyLight 680-NHS (N-hydroxysuccinimide) Ester, and 2 nmol of stained variant 5.6 or unconjugated dye were injected subcutaneously (s.c.) into wild-type (WT) C57BL6 mice. The mice were killed 1.5 h and 3 h after injection, their organs were removed, and the accumulation of variant 5.6 in the organs was compared to that in a control untreated mouse. Variant 5.6 accumulated in the kidneys and the bladder as part of the natural clearance and not in other organs, such as the heart, liver, and spleen (Fig 9A, right). Importantly, examination of the epiphysis and diaphysis of a C57BL6 mouse femur (Fig 9A, left) showed that variant 5.6 did indeed reach the bones. The observed accumulation of the unconjugated dye in the liver but not in the kidneys or in the bones served to confirm the specificity of variant 5.6 for bone tissue.
Fig 9. M-CSFRGD proteins inhibit bone resorption in ovariectomized mice.
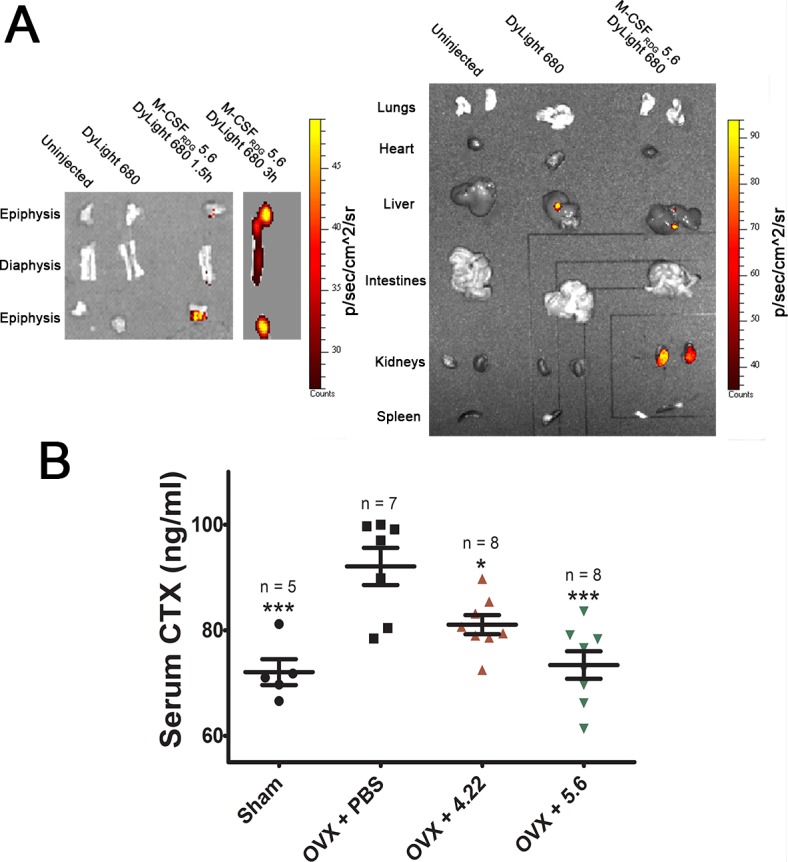
(A) Ex vivo images of 12-weeks-old WT C57BL6 mice organs. The organs were removed and imaged 1.5 h and 3 h after mice were injected s.c. and compared with organs of a mouse that had been injected with unconjugated dye and of a mouse that had not been injected with the proteins (right). To determine whether M-CSFRGD variant 5.6 accumulates in the bones, the epiphysis and diaphysis of the femur of a mouse injected with variant 5.6 were compared with those of the control mice (left). (B) Ten-weeks-old mice were ovariectomized, and starting 2 wk after the surgery, they were injected twice a day with PBS or M-CSFRGD variants 4.22 or 5.6 for 3 d. Thereafter, serum CTX-I levels were determined by ELISA. Data are means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. Source data and its analysis can be found in S6 Data. CTX-I, carboxy-terminal telopeptide of type I collagen; ELISA, enzyme linked immunosorbent assay; M-CSF, macrophage colony-stimulating factor; OVX, ovariectomy-induced bone loss; PBS, phosphate-buffered saline; RGD, Arginine-Glycine-Aspartic acid; s.c., subcutaneously; WT, wild type.
M-CSFRGD variants 4.22 and 5.6 inhibit osteoclast activity in an animal model of osteoporosis
To determine whether variants 4.22 and 5.6 (which gave better production yields than 4.24) inhibit osteoclast activity in vivo, we used the well-established mouse model of ovariectomy-induced bone loss (OVX). Two weeks after the mice had undergone ovariectomy or a sham operation, they were injected s.c. twice a day for 3 d with phosphate-buffered saline (PBS), variant 4.22, or variant 5.6. After the last injection, blood was collected, and serum concentrations of the marker for bone resorption (osteoclast activity), the cross-linked carboxy-terminal telopeptide of type I collagen (CTX-I), were determined using ELISA. Two weeks after the operation, there was a significant increase in serum CTX-I levels in ovariectomized mice injected with PBS as compared to the sham-operated mice. Analysis of the CTX-I levels in mice injected with variants 4.22 and 5.6 showed a significant reduction compared to ovariectomized mice injected with vehicle. Strikingly, serum CTX-I levels of mice that were injected with variant 5.6 were reduced almost to the level of CTX-I serum levels of the sham-operated mice (Fig 9B).
Discussion
A number of studies have demonstrated the crosstalk between biological processes mediated by c-FMS, αvβ3 integrin, and their ligands in the context of osteoclast differentiation, cytoskeleton organization, and bone resorption [14, 16, 19]. Since enhanced osteoclast function is an underlying cause of many bone-related disorders, with osteoclast differentiation and function being dependent on c-FMS and αvβ3 integrin, these two proteins constitute attractive targets for therapeutic intervention. However, since these two receptors regulate common pathways, combination therapy or high drug doses may be needed to fully block the differentiation and resorption processes, with resulting increases in potential side effects and in the cost of therapy. Dual-specific proteins that could bind to and inhibit both c-FMS and αvβ3 integrin would therefore have superior therapeutic potential, as such proteins would offer improved binding avidity and selectivity compared to monospecific protein therapeutics. Nevertheless, despite extensive efforts in the field, there are currently no clinically approved therapeutic agents that are specific for both c-FMS and αvβ3 integrin. The new approach described here, which entails engineering dual-specific therapeutic proteins, is the first to use a natural ligand as a scaffold for engineering high-affinity binding to two therapeutic targets and to apply the engineered bispecific ligands in preclinical studies of osteoporosis.
We have shown in this study that M-CSF is an excellent scaffold for engineering bispecific ligands with affinity and specificity for both c-FMS and αvβ3 integrin. Many integrins recognize the RGD motif in extracellular matrix ligands, including fibronectin, vitronectin, fibrinogen, and osteopontin [38]. Since the RGD motif in natural ligands is typically found in flexible solvent-exposed loops, we mutated either loop 1 or loop 3, both of which are located in the dimerization interface of M-CSFWT and remain accessible in M-CSFRGD monomers [39]. Mutating either one of these loops with an RGD sequence flanked by three residues on each side allows M-CSFRGD to interact with αvβ3 integrin. This interaction does not disrupt the contact with the native c-FMS receptor, as the latter interacts with residues located on the opposite side of the M-CSFRGD ligand. Overall, the interaction area between M-CSFRGD variant 4.22 and αvβ3 integrin is relatively large and scattered, in accordance with the physiological integrin–ligand interaction [40].
The M-CSFRGD variants 4.22, 4.24, and 5.6 had enhanced inhibitory capabilities compared to M-CSFc-FMS and M-CSFαvβ3, thereby highlighting the effects of the dual functionality that we engineered into these proteins. The M-CSFRGD variants were shown to be better inhibitors of osteoclast actin ring formation and osteoclast differentiation than M-CSFc-FMS and M-CSFαvβ3.We showed by SPR and cell-surface–binding assays that the engineered dual-specific M-CSF proteins can bind both αvβ3 integrin and c-FMS receptors, leading to antagonism of immediate signaling events (c-FMS and Akt phosphorylation) and downstream biochemical events regulating osteoclast differentiation. Moreover, the presence of αvβ3 integrin and c-FMS on the surface of the cell enhances M-CSFRGD’s capability to bind it and lead to a stronger inhibition than the monospecific controls.
The three M-CSFRGD proteins, 5.6, 4.22, and 4.24, which differed in the amino acids flanking the RGD motif, also differed in their binding affinities for c-FMS and αvβ3 integrin. These variations in affinities were translated into differences in their biological activities. Variant 5.6 had the highest binding affinity to c-FMS and accordingly showed the highest binding and inhibition of c-FMS and Akt phosphorylation in murine pre-osteoclast cells and the lowest inhibitory effect on actin ring formation. However, variant 5.6 exhibited lower inhibition of murine osteoclast differentiation in vitro than variants 4.22 and 4.24. The latter two variants showed higher binding affinity to αvβ3 integrin and a drastic inhibition of actin ring formation, which may suggest that increased binding to integrin led to a more efficient inhibition of murine osteoclast differentiation in vitro. This notion is supported by our observation that inhibition of murine osteoclast differentiation was higher for M-CSFαvβ3 (the αvβ3-integrin–only binder) than for variant 5.6. In contrast, variant 5.6 showed higher efficiency than M-CSFαvβ3 in inhibiting human osteoclast differentiation in vitro. Moreover, variant 5.6 showed better inhibition of bone resorption in vivo than variant 4.22, which suggests that higher affinity to c-FMS results in stronger inhibition of human osteoclast differentiation in vitro and promotes inhibition of osteoclast activity in mice in vivo. These results could be explained by the fact that variant 5.6 exhibits better thermal stability than variant 4.22, although we cannot rule out the possibility that other differences, such as differences in proteostability, might affect their activities in vivo.
The main goal of this study was to exploit the attributes of M-CSF to engineer a versatile platform for the development of a targeted osteoporosis therapy. By demonstrating that this natural ligand could be engineered to function as a scaffold with the ability to target two receptors at the same time, we showed that engineering of natural agonists to function as receptor antagonists could become an alternative strategy for generating therapeutics not only for osteoporosis but also for other important biomedical targets. We envision that protein variants generated from such efforts could promote the development of the next generation of therapeutics, including, but not limited to, targeted drug delivery agents and selective tissue targeting probes. Similarly, this approach will provide new tools for studying molecular mechanisms and cell signaling pathways—not only those that mediate osteoclast differentiation and function but also pathways that are involved in different pathological processes.
Methods
Ethics statement
This study was carried out according to protocols approved by the Ben-Gurion University Committee for the Ethical Care and Use of Animals in Experiments (permit number: IL32072017). All surgery was performed under anesthesia, and all efforts were made to minimize discomfort, distress, and suffering.
Flow cytometry sorting and analysis
For expressing the M-CSFC31S gene and the two M-CSFRGD libraries on the yeast surface, the yeast culture was grown in the tryptophan-selective SDCAA medium (2% dextrose, 0.67% yeast nitrogen base, 0.5% bacto casamino acids, 1.47% sodium citrate, 0.429% citric acid monohydrate, pH 4.5). This was followed by suspension in SGCAA medium (2% galactose, 0.67% yeast nitrogen base, 0.5% bacto casamino acids, 1.47% sodium citrate, 0.429% citric acid monohydrate) for overnight incubation at 30 °C until the culture reached OD600 = 10.0 (108 cells per ml). A number of cells equal to 10 times the library size was added to 1 ml PBS containing 1% bovine serum albumin (PBSA); the suspension was centrifuged, and the supernatant was removed. For analysis of binding of M-CSFC31S and the M-CSFRGD libraries to c-FMS, yeast cells were incubated with mouse anti-c-myc antibody, 9E10 (Abcam, Cambridge, MA, USA) in a 1:50 ratio and different concentrations of soluble Fc-conjugated human c-FMS (R&D systems, Minneapolis, MN, USA) for 1 h at room temperature. After an additional washing step, cells were labeled with secondary phycoerythrin (PE) anti-mouse (Sigma-Aldrich, St. Louis, MO, USA), and goat anti-human Fc FITC antibodies (Sigma-Aldrich) for 30 min at 4 °C in the dark. For analysis of binding of M-CSFRGD libraries to integrin, the experiment was performed in integrin binding buffer (IBB; 20 mM Tris, pH 7.5, 2 mM CaCl2, 100 mM NaCl, 1 mM MgCl2, and 1 mM MnCl2) + 1% BSA. The yeast cells were incubated with chicken anti-c-myc and different concentrations of soluble human αvβ3 integrin (R&D Systems) for 1 h. After an additional washing step, cells were labeled with secondary PE-anti-chicken and FITC-anti-human β3 integrin (BioLegend, San Diego, CA, USA) antibodies for 30 min in the dark. Labeled cells were washed with 1 ml PBSA or IBB + 1% BSA. A total of five rounds of affinity maturation sorting were performed; in each sort, a diagonal shape was created with the aim to enrich the high protein binders. In each sort, 0.5% to 2% of the population was collected into a new tube containing SDCC medium. The first sort was performed against 500 nM c-FMS (sort 0), and the subsequent sorts against 500 nM αvβ3 integrin (sort 1), 250 nM αvβ3 integrin (sort 2), 100 nM αvβ3 integrin (sort 3), 20 nM αvβ3 integrin (sort 4), and 50 nM of c-FMS (sort 5). The high-affinity binders were enriched using an iCyt Synergy FACS apparatus (Sony Biotechnology, San Jose, CA, USA).
Choosing and identifying high-affinity αvβ3 integrin and c-FMS binders
To identify the best αvβ3 integrin and c-FMS binders, 25 different clones from sort 4 and 25 different clones from sort 5 were tested for binding to 20 nM αvβ3 integrin, as described in the flow-cytometry–sorting section. Then, 10 αvβ3 integrin binding clones from sort 4 and 10 αvβ3 integrin binding clones from sort 5 having the highest affinities to αvβ3 integrin were analyzed for binding to 50 nM c-FMS. The clones with the highest binding affinity for both αvβ3 integrin and c-FMS were selected, and DNA was extracted from these clones using Zymoprep Yeast Plasmid Miniprep I (Zymo Research, Irvine, CA, USA) according to the manufacturer's protocol. The extracted plasmids were incubated with Escherichia coli-competent cells for 30 min on ice and transferred into 0.2-cm gap cuvettes (Bio-Rad, Hercules, CA, USA). The cuvettes were inserted into a Micropulser electroporator (Bio-Rad) and pulsed with 2.5 kV. Immediately, 1 ml warm Luria Broth (LB) medium was added to each cuvette, and the suspensions were incubated at 37 °C for 1 h. The bacteria were seeded on LB agar plates containing 1:1,000 ampicillin and grown overnight at 37 °C. Colonies were moved to LB medium containing ampicillin and grown overnight. The plasmids were extracted from the bacterial culture with HiYield plasmid mini kit (RBC, Bioscience, Taiwan) according to the manufacturer's protocol. The purified plasmids were sequenced to confirm that they contained the desired sequence and to prevent repetitions (The DNA Microarrays and DNA Sequencing Laboratory, National Institute for Biotechnology in the Negev, Ben-Gurion University of the Negev [NIBN, BGU], Beer-Sheva, Israel). To determine the binding specificity to other RGD binding integrins—namely, αvβ5, α4β7, α2β2b, and α5β1 (BioLegend)—the three chosen M-CSFRGD variants (4.22, 4.24, and 5.6) were analyzed for binding to 250 nM of each integrin. To detect integrin binding, M-CSFRGD variants were incubated with APC anti-human CD49d, APC anti-human CD41, FITC anti-human CD49e, or FITC anti-human CD5 (BioLegend) and analyzed with Accuri C6 flow cytometry analyzer (BD Biosciences, San Jose, CA, USA). M-CSFc-FMS, M-CSFαvβ3, and M-CSFRGD variants were produced, purified, and characterized as described in S1 Text.
SPR
Determination of binding of the soluble purified proteins to c-FMS and αvβ3 integrin was performed on a ProteOn XPR36 (Bio-Rad). The M-CSFRGD variants, M-CSFc-FMS, and M-CSFαvβ3 proteins were immobilized on the surface of the chip by using the amine coupling reagents sulfo-NHS, 0.1 M, and EDC (1-ethyl-3-[3dimethylaminopropyl]-carbodiimide, Bio-Rad), 0.4 M. To attach the M-CSFRGD, M-CSFc-FMS, and M-CSFαvβ3 variants covalently to the chip, 1 μg of each protein and 3 μg of BSA in 10 mM sodium acetate buffer (pH 4.0) were used to give 1,141, 900, 1,329, 1,272, and 1,337 response units (RUs) for M-CSFc-FMS and M-CSFαvβ3 variants 4.22, 4.24, and 5.6, respectively. Unbound esters were deactivated with 1 M ethanolamine HCl at pH 8.5, and the temperature was set at 25 °C. Then, the chip was rotated, and the human c-FMS soluble proteins (Sino Biological, China) were allowed to flow over the chip at six different concentrations (0, 12.5, 25, 50, 100, and 200 nM) at a flow rate of 50 μl/min for 490 s, followed by dissociation for 600 s in PBS + 0.005% Tween (PBST). For determining αvβ3 integrin binding, the chip was regenerated using 50 mM NaOH at a flow rate of 100 μl/min, and different concentrations of αvβ3 integrin proteins (0, 12.5, 25, 50, 100, and 200 nM) were allowed to flow over the chip. The interactions obtained were normalized to the initial protein binding RUs to the chip surface. Then, for each concentration, the KD was obtained from the equilibrium binding phase of the sensorgram. To determine the αvβ3 integrin specificity for each M-CSFRGD variant, a new chip was loaded with different RGD binding integrins, as follows: 8.5 μg of α3β1 integrin, 8.5 μg of α4β7 integrin, 6 μg of α5β1 integrin, and 8.5 μg of αvβ3 integrin, as well as 3 μg of BSA as a negative control. The proteins were covalently bound to the chip with 10 mM sodium acetate buffer (pH 4.0) as described above. Then, 1 μM of the M-CSFRGD variant was allowed to flow over the chip at a rate of 50 μl/min for 409 s, followed by 600 s of dissociation with PBS + 1% Tween. The interactions obtained were normalized to the initial protein binding RUs to the chip surface. To achieve statistical significance, we made sure that the χ2 values were at least 10% or lower than the Rmax values.
Computational docking model of αvβ3 integrin, M-CSFRGD, and c-FMS complex
Molecular coordinates for the αvβ3 integrin headpiece were taken from the 1L5G Protein Data Bank (PDB) structure [29] (1–438 of the αv subunit and 55–432 of the β3 subunit). M-CSFWT in complex with c-FMS was obtained from the 4WRL PDB structure [39]. The M-CSFRGD variant 4.22 was created by replacing residues 25–32 of the native protein with the residues QTSRGDSPS by using PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC (De Lano). The M-CSFRGD structure was energy minimized using the Gromacs 4.6.7 package of programs [41]. Next, M-CSFRGD together with the αvβ3 integrin headpiece were subjected to a receptor-ligand docking procedure by using the PatchDock server [28]. To avoid irrelevant structures, potential binding sites both for the αvβ3 integrin receptor and the M-CSFRGD ligand were defined according to PatchDock recommendations. Slight variations in the interaction restraints yielded five experiments, thus resulting in a total of 500 structures. These were clustered with a 0.5-nm cutoff by using Gromacs. The c-FMS receptor was aligned to the representative models, and snapshots were prepared by the VMD program [42].
Cell binding assay
M-CSFc-FMS, M-CSFαvβ3, and two variants of M-CSFRGD (4.22 and 5.6) were labeled with a molar ratio of 1:1 DyLight 488 NHS Ester (Thermo Fisher Scientific) and the purified protein. The solution was incubated for 1 h at room temperature, and the residual unbound dye was washed three times with Vivaspin (GE Healthcare Life Sciences) with a 3,000-Da cutoff. M-CSFRGD variant 4.24 was not tested for direct cell binding because of low protein-purification yields. The following two assays were conducted:
MDA-MB-231 breast cancer cells (ATCC HTB-28) were used for assaying direct cell binding of the purified proteins. Cells were plated at a density of 105 per well in 96-well plates and washed with 1 ml 0.1% PBSA. Cells were then centrifuged at 150g for 5 min, and the supernatant was removed; this step was repeated twice. Then, labeled M-CSFc-FMS, M-CSFαvβ3 or M-CSFRGD protein variants were added in IBB + 1% BSA to the cells at different concentrations (1, 2.5, and 7.5 μM) in a total volume of 100 μl and incubated at 4 °C with gentle agitation for 2 h. For the competition assay for variant 5.6, the cells were incubated with 1 μM 5.6 in the presence of 5 μM M-CSFWT or 10 μM cRGD separately or with both together. For determination of c-FMS expression, the cells were stained with PE-anti human CD115 (BioLegend) at a dilution of 1:50, and for αvβ3 integrin expression, with FITC-anti human β3 integrin (BioLegend) at a dilution of 1:25 for 30 min. Then, the cells were washed twice as described above and analyzed with an Accuri C6 flow cytometry analyzer (BD Biosciences). The mean autofluorescence of the cells was subtracted from the fluorescence reads of all samples. The experiment was repeated three times at each concentration.
Murine BMMs were obtained by killing mice with isoflurane (Piramal, India), followed by flushing the bone marrow from the femurs and tibias of WT C57BL6 mice. The cells were treated with ACK red lysis buffer (Thermo Fisher Scientific) and plated on bacterial culture dishes in αMEM growth medium (Sigma-Aldrich) containing recombinant murine M-CSF (40 ng/ml) (R&D systems). The cells were incubated for 3 d at 37 °C under 5% CO2 to induce monocyte adhesion and proliferation. The plates were washed with PBS, the cells were detached using a cell scraper, and 105 cells were transferred to each well of a 96-well plate. The cells were washed twice with 200 μl 0.1% PBSA. Binding was determined as described for labeled M-CSFRGD proteins. For determination of murine c-FMS expression, anti-mouse CD115 (CSF-1R) (BioLegend) was used at 1:50 concentration and detected with goat anti-rat (PE) antibody (Abcam). For determination of αvβ3 integrin expression, Alexa Fluor 488 anti-mouse/rat CD61 antibody (BioLegend) was used as previously described. Samples were analyzed with an Accuri C6 flow cytometry analyzer (BD Biosciences). The experiment was repeated three times at each concentration.
c-FMS and Akt phosphorylation assay
Murine BMM cells were obtained as described in the previous section. For this assay, 700,000 cells per well were seeded for differentiation with M-CSF (20 ng/ml) and RANKL (20 ng/ml) in a 6-well plate for 48 h held in an incubator at 37 °C. Then, the cells were incubated for 4 h in starvation medium (αMEM growth medium without fetal bovine serum [FBS]) at 37 °C. For the competition assay, cells were washed with PBS, and 1 μM of M-CSFRGD variants and M-CSFc-FMS with M-CSFWT (20 ng/ml) were added to each well for 1 min for c-FMS and 10 min for Akt; the plates were then immediately placed on ice. Cells were lysed with lysis buffer (deoxycholate 0.5%, 25 mM NaF, 10 mM NaPO4, 1 mM Na3VO4×2H2O, 5 mM EDTA, 5 mM EGTA, 100 mM NaCl, 2% Triton X-100, protease inhibitors cocktail, 20 mM para-nitrophenylphosphate [PNPP]) and scraped from the wells, incubated for 10 min on ice, and centrifuged at 14,000 × g at 4 °C for 30 min; the supernatants were then transferred to fresh tubes. To determine whether the variants spontaneously activated c-FMS or Akt, the proteins were added without M-CSFWT, followed by lysis as described. Protein concentrations were determined using a BCA kit (Thermo Fisher Scientific) according to the manufacturer's protocol. To include and analyze a large sample size, proteins from separate gels were compared using the following method. Fifteen μg of three protein samples were boiled, loaded, and separated on each 10% SDS PAGE for c-FMS and 12% SDS PAGE for Akt (for example, a gel was organized in the following way: protein ladder | 4.22 #1| 4.22 #2 | 4.22 #3 | protein ladder | 4.22 #1 | 4.22 #2 | 4.22 #3, with #1 in both lanes representing the same biological sample, #2 representing the same sample, etc.). Proteins were transferred to a PVDF membrane (Bio-Rad, CA, USA) using a Trans-Blot Turbo Transfer System (Bio-Rad, CA, USA). Membranes were cut into three parts, for example: ladder to 4.22 #3 that was blotted to c-FMS (first 4 samples from left to right above), ladder to 4.22 #3 that was blotted to pc-FMS (phosphorylated form of c-FMS), and low Mw part that was blotted to detect actin. In order to reduce the effects of different exposure times and device errors after blotting that was done with the same antibody stock, all the membranes were imaged at the same day with the same developing system (Fusion FX device manufactured by Vilber Lourmat), the same ECL incubation, and the same exposure times. To make sure that the same amount of protein was loaded and that the ECL-developing process was correct, we quantified and normalized each lane to its corresponding actin levels to see that there were no problems with loading or developing (Densitometry values are included in S2 Data).
Every sample was normalized to its corresponding actin densitometric value (that was run on the same gel in the same lane) to exclude the effect of different exposure times. For example, 4.22 #1 pc-FMS was normalized to expression levels (4.22 #1 c-FMS) and to its own actin levels (4.22 #1 actin). Membranes were blocked with 5% BSA in TBS (20 mM Tris, 150 mM NaCl, pH 7.6) + 0.1% Tween 20 (TBST) for 1 h. For detection of phosphorylated receptors, a 1:500 ratio of anti-phospho-M-CSF receptor (Tyr723) (Cell Signaling, MA, USA) and a 1:1,000 ratio of anti-phospho-Akt (Ser473) (Cell Signaling) antibodies were added to 5% BSA in TBST for overnight incubation at 4 °C. For total receptor expression, a 1:1,000 ratio of anti-M-CSF receptor (Cell Signaling) and anti-Akt (Cell Signaling) antibodies were added to 5% BSA in TBST for overnight incubation at 4 °C. Membranes were washed three times for 10 min with PBST. Then, the HRP-conjugated anti-rabbit secondary antibody (Cell Signaling) was added at a ratio of 1:1,000 for 1 h, and the membranes were washed again as described. All antibodies were used from the same stock for each blot. The membranes were developed using ECL-developing reagent (Biological Industries, Israel) according to the manufacturer's protocol and imaged using a Fusion FX device (Vilber Lourmat, Germany). Each membrane went through the same exposure time, which was according to the antibody that was used. Membranes were stripped with stripping buffer (2% sodium dodecyl sulfate [SDS], 62.5 mM Tris HCl [pH 6.8], 0.8% β-mercaptoethanol) at 50 °C for 30 min, and the procedure was repeated again with actin antibody as a loading control (Cell Signaling). The intensity of the protein bands was measured using ImageJ [43] with the analyze gels tool, and the phosphorylated c-FMS or Akt band intensity was normalized to c-FMS or Akt expression, respectively, and to its corresponding loading control on the same membrane (actin). The experiments with M-CSFc-FMS and M-CSFRGD variants 4.22 and 5.6 were repeated three times, and those with M-CSFRGD variant 4.24 were repeated six times.
Osteoclast actin-belt–formation assay
Murine BMMs were obtained as described in the cell binding assay section. For this assay, 7,000 cells per well were seeded for differentiation with M-CSF (20 ng/ml) and RANKL (20 ng/ml) in a 384 μClear plate (Greiner Bio-One, Austria) for 60 h in an incubator at 37 °C. Then, the different inhibitors were added in a concentration of 5 μM for an additional 24 h. Cells were fixed with a fixation solution (paraformaldehyde 3%, Triton X-100 10%, glutaraldehyde 8%) for 15 min at 37 °C. Following fixation, the cells were washed with PBS and incubated with cytoskeleton buffer (10 mM MES, 150 mM NaCl, 5 mM EGTA, 5 mM MgCl2, 5 mM glucose, 100 μM sodium borohydride, pH 6.1) for 15 min. To stain for actin, the cells were washed and incubated with Acti-stain 670 phalloidin (Cytoskeleton, CO, USA) for 45 min at room temperature. Thereafter, cells were washed and incubated with DAPI (Thermo Fisher Scientific) for an additional 15 min. Actin rings were visualized by Operetta (Perkin Elmer, MA, USA). Each condition was repeated five times, and from each well, 20 pictures were obtained. For actin belts, quantification osteoclasts were defined as cells harboring three or more nuclei and were counted in a double-blind manner, and actin belts were determined above an intensity threshold by ImageJ software.
Cell viability assay
Mouse mesenchymal stem cells (MSCs) were obtained by flushing the bone marrow from femurs and tibias of 10-weeks-old mice as described in the cell binding assay section. Cells were cultured in MesenCult MSC Basal Medium with 1/5 MSC stimulatory supplements, 50 U/L penicillin-streptomycin, and 10 mg/mL gentamycin and kept in a Hypoxia Incubator Chamber (Thermo Fisher Scientific, Waltham, MA, USA) under 5% oxygen. Then, 7,000 cells were plated in each well of a 96-well plate and held for 48 h in an incubator at 37 °C in the presence of different concentrations of inhibitors (50 nM, 1 μM, and 5 μM). After 48 h, the XTT reagent (Biological Industries, Israel) was added to the cells according to manufacturer's protocol for 2 h at 37 °C. The plate was rocked gently, and the absorbance was measured at 450 nm and 650 nm for background subtraction with a Synergy 2 multidetection microplate reader (Biotek, Winooski, VT, USA). Each condition was repeated three times, and the blank absorbance measurements and the background absorbance were subtracted from the sample's absorbance.
Cell death assay
Murine BMMs were obtained as described in the cell binding assay section, washed with PBS, and detached with a cell scraper, and 1.5 × 105 cells were plated in each well of a 24-well plate. To induce differentiation, M-CSF (20 ng/μl) and RANKL (20 ng/μl) with 1 μM of M-CSFc-FMS, M-CSFαvβ3, and M-CSFRGD variants and GW2580 (Abcam, Cambridge, MA, USA) were added for 48 h. Then, cells were detached with Accutase (Biological industries, Israel), centrifuged at 300 × g for 5 min, and washed three times with PBSA 0.1%. PI was added, and cells were analyzed with Accuri C6 flow cytometry analyzer.
Cell differentiation assay
Murine BMMs were obtained as described in the cell-binding–assay section, washed with PBS, and detached with a cell scraper, and 2 × 104 cells were plated in each well of a 96-well plate. Human peripheral blood CD14+ monocytes (PromoCell, Germany) were grown in complete αMEM medium for 5 d in the presence of human M-CSF (20 ng/μl). Then, 1 × 104 cells were transferred to each well of a 96-well plate. Osteoclast differentiation was induced by culturing the cells in alpha Minimum Essential Medium (αMEM) containing 10% of FBS, 50,000 units penicillin, 50 mg streptomycin, 20 ng/ml murine M-CSF (Peprotech, Israel) or human M-CSF (R&D systems, Minneapolis, MN, USA), and 20 ng/ml murine RANKL (R&D Systems). To determine the influence of the M-CSFRGD variants, M-CSFc-FMS, and M-CSFαvβ3 on the osteoclasts, the proteins were added to the differentiation medium (with M-CSF and RANKL) in three different concentrations (50 nM, 1 μM, and 5 μM for BMMs and 50 nM, 250 nM, and 1 μM for human CD14+). After 72–96 h, once the cells had differentiated fully, they were fixed in 4% paraformaldehyde and stained using a TRAP staining kit (Sigma-Aldrich, USA) according to the manufacturer's protocol with additional staining of the nuclei with DAPI. As a positive control, PBS was used instead of the inhibitors, and negative controls comprised cells incubated in differentiation medium without RANKL and inhibitors. Osteoclast parameters were obtained by analysis of 20 images from random areas in each well; the osteoclasts were observed with an Olympus ×83 microscope with an automated stage. For murine osteoclasts differentiation, a total of 2,340 frames were analyzed for 1,581 osteoclasts and 5,313 nuclei; for human CD14+, a total of 1,620 frames were analyzed for 477 osteoclasts and 1,897 nuclei. Osteoclasts were defined as TRAP-positive cells harboring three or more nuclei and were counted in a double-blind manner, and the number of nuclei in the osteoclasts and the total osteoclast surface area were determined using ImageJ software. To evaluate the TRAP staining in each well, the plate was measured with a 650 nm Synergy 2 multidetection microplate reader. Three repeats were performed for every condition, and each repeat was normalized to the positive control values.
qPCR
Murine BMMs were obtained as described in the cell binding assay section, washed with PBS, and detached with a cell scraper, and 7.5 × 105 cells were plated in each well of a 6-well plate. Then, osteoclast differentiation was induced as described in the cell differentiation assay section with and without inhibitors at a concentration of 50 nM. When cells reached full differentiation, they were washed once with PBS and lysed, and the mRNA was isolated using TRIzol reagent (Thermo Fisher Scientific). cDNA was prepared using a cDNA Reverse Transcriptase kit (Applied Biosystems) according to manufacturer’s protocol. For each reaction, 100 ng of cDNA was used with a SYBR Green gene expression assay (Applied Biosystems) and analyzed with a LightCycler 480 II analyzer (Roche) for OSCAR (forward primer: CACCTACTGTTGCTATTACC, reverse primer: GAACCTTCGAAACTGATGAC) and NFATc1 (forward primer: CCAGTTCTACTTGGATGATG, reverse primer: GTAAGTTGGGATTTCTGAGTG). Expression levels of genes were normalized to the HPRT (forward primer: CCCCAAAATGGTTAAGGTTG, reverse primer: AGTACTCATTATAGTCAAGGGC) reference gene using the comparative Ct method (ΔΔ Ct) that was compared to the positive control.
In vivo imaging
M-CSFRGD variant 5.6 was labeled with DyLight 680-NHS Ester and was washed rigorously to remove residual dye molecules. Unconjugated dye was incubated with Tris for 30 min at room temperature. C57BL6 mice were anesthetized in an isoflurane chamber and injected s.c. with 2 nmol of labeled protein or unconjugated dye. Then, the mice were killed with isoflurane, and their internal organs were removed and placed on the IVIS platform and imaged with the IVIS system (Perkin Elmer, MA, USA). Similarly, 3 h after injection, C57BL6 mice were killed, and their bones were placed on the IVIS platform and photographed.
Injection of ovariectomized mice with M-CSFRGD
Ten-weeks-old female C57BL6 mice were ovariectomized under complete anesthesia. Two weeks after the operation, the mice were injected s.c. twice a day for 3 d with PBS or 10 mg/kg of M-CSFRGD variants 4.22 or 5.6. Three h after the last injection, the mice were anesthetized with isoflurane, and their blood was collected through a cardiac puncture. Then, blood samples were centrifuged at 4,000 × g for 10 min, and the serum samples were transferred to fresh tubes. To determine the serum CTX-I levels, a RatLaps ELISA (Immunodiagnostic systems, UK) was used according to manufacturer’s protocol. Hemolytic samples were not used.
Statistical analysis
The data from the differentiation assay was analyzed for column statistics with GraphPad Prism version 5.00 for Windows (La Jolla, CA, USA). Data is shown as means ± SEM. Statistical significance was determined by column statistics and ANOVA test analysis. A p value < 0.05 was considered statistically significant.
Supporting information
(A) The amino acid sequence of M-CSF with C31, which is required for dimerization, indicated in red. The two flexible loops in the dimerization interface are colored blue (loop 1, residues 25–32) and green (loop 3, residues 64–71). (B) M-CSFRGD library 1 colored in blue, where residues 25–32 were replaced with an RGD motif having three random amino acids on each side. (C) M-CSFRGD library 2 colored green, where residues 64–71 were replaced with an RGD motif with three random amino acids on each side and C31 was replaced with serine to inhibit disulfide-linked homodimerization. (D) M-CSFαvβ3 is based on the sequence of M-CSFRGD variant 4.22 with two single-point mutations in H9A and H15A, indicated in red, to inhibit binding to c-FMS. (E) M-CSFc-FMS was created by changing the RGD motif on M-CSFRGD variant 4.22 to RDG with the aim to prevent binding to αvβ3 integrin. (F) Sequences of the mutated loop of the three M-CSFRGD clones that were selected after four (4.22 and 4.24) and five (5.6) rounds of the affinity maturation process. M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid; WT, wild type.
(TIF)
YSD M-CSFC31S was analyzed for (A) forward scatter and side scatter and (B) expression using mouse anti-c-myc antibody followed by a secondary PE-labeled anti mouse antibody. (C) The binding of YSD M-CSFC31S to soluble c-FMS-Fc was detected by a goat anti-human Fc-FITC antibody. (D) Cells expressing M-CSFC31S on the yeast cell wall were incubated with 10 different concentrations of c-FMS-Fc (0.5–2000 nM) and were tested for binding by flow cytometry. The curve shows a good fit to a single binding-site curve, and the apparent KD is 20 nM. Source data can be found in S7 Data. FITC, fluorescein isothiocyanate; M-CSF, macrophage colony-stimulating factor; PE, phycoerythrin; YSD, yeast surface display.
(TIF)
The M-CSFRGD library was covalently linked to Aga1p and the yeast cell wall. Binding for c-FMS was determined with c-FMS-Fc recombinant protein and goat anti-human Fc FITC conjugated secondary antibody, and the expression levels were measured with a mouse anti-c-myc primary antibody and PE anti-mouse secondary antibody. For determination of αvβ3 integrin binding, yeast cells were incubated with recombinant αvβ3 integrin and mouse anti-human CD49d FITC secondary antibody, and the expression levels were measured with chicken anti-c-myc primary antibody and PE goat anti-chicken secondary antibody. FITC, fluorescein isothiocyanate; M-CSF, macrophage colony-stimulating factor; PE, phycoerythrin; RGD, Arginine-Glycine-Aspartic acid; YSD, yeast surface display.
(TIF)
M-CSFRGD (A–D) library 1 and (E–H) library 2 were analyzed for (A and E) FSC/SSC, (B and F) expression, (C and G) 100 nM c-FMS binding, and (D and H) 500 nM αvβ3 integrin binding. FACS, fluorescence-activated cell sorting; FSC, forward scatter; M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid; SSC, side scatter.
(TIF)
Yeast-displayed mutant libraries were analyzed, and the living cells population in each sort is represented by a black polygon-shaped gate. The affinity maturation sorting process started with (A) a presorted library followed by (B) sort 1, (C) sort 2, (D) sort 3, (E) sort 4, and (F) sort 5. FACS, fluorescence-activated cell sorting; FSC, forward scatter; SSC, side scatter.
(TIF)
Twenty-five different clones from each of sorts 4 (A) and 5 (C) were tested for binding to 20 nM of αvβ3 integrin, normalized to the lowest binder. (B) The best 15 αvβ3 integrin M-CSFRGD binders from sort 4 and the best 10 αvβ3 integrin M-CSFRGD binders from sort 5 (D) were evaluated for binding to 50 nM of c-FMS, normalized to M-CSFC31S. The chosen clones (4.22, 4.24, and 5.6) are indicated in blue. (E) Variants 4.22, 4.24, and 5.6 were evaluated for integrin specificity by testing their binding to 250 nM of α4β7, αIIbβ3, αvβ5, and α5β1 integrins in comparison with their binding to αvβ3 integrin. Source data can be found in S8 Data. M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid; YSD, yeast surface display.
(TIF)
(A) Size exclusion chromatography of nonglycosylated M-CSFRGD clone 4.22 with high molecular weight standards. Variant 4.22 was eluted at the size of 21 kDa. (B) Mass spectrometry of nonglycosylated variant 5.6. (C) CD spectra of nonglycosylated variant 4.22 (red line), nonglycosylated variant 4.24 (blue line), nonglycosylated variant 5.6 (green line), nonglycosylated M-CSFc-FMS (pink line), and nonglycosylated M-CSFαvβ3 (gray lines). (D) Temperature-dependent CD measurements of unfolded proteins determined at 217 nm normalized to fully denatured proteins. (E) SDS-PAGE for all purified proteins: nonglycosylated M-CSFC31S (lane 1), nonglycosylated variant 4.22 (lane 2), nonglycosylated variant 4.24 (lane 3), nonglycosylated variant 5.6 (lane 4), nonglycosylated M-CSFc-FMS (lane 5), and non-glycosylated M-CSFαvβ3 (lane 6). Source data can be found in S9 Data. CD, circular dichroism; M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid.
(TIF)
Dimerization of the purified proteins was determined by using increasing concentrations of BS3 cross-linker, denaturation, and analysis on SDS-PAGE. M-CSFWT dimerized at all BS3 concentrations, but the three M-CSFRGD variants, M-CSFc-FMS, and M-CSFαvβ3 did not show any dimerization capability. BS3, bis(sulfosuccinimidyl)suberate; M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid; WT, wild type.
(TIF)
To determine the protein KD,app, the RUs at saturation for each protein concentration were plotted, and a fitted curve was created for (A) c-FMS and (B) αvβ3 integrin. Source data and its analysis can be found in S10 Data. RUs, response units.
(TIF)
(A) αvβ3, (B) α3β1, (C) α4β7, and (D) α5β1 integrins were immobilized on the surface of the chip. Thereafter, the three M-CSFRGD variants 4.22 (green), 4.24 (blue), and 5.6 (red) were allowed to flow over the surface of the chip at a concentration of 1 μM. Source data can be found in S11 Data. M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid.
(TIF)
M-CSFC31S is shown in pink, c-FMS in cyan, αv in yellow, and β3 in green. Residues 25–32 of M-CSFC31S are represented in red. M-CSF, macrophage colony-stimulating factor.
(TIF)
M-CSFRGD/αvβ3 integrin interface seen from different angles (A–D). αv in yellow “surf” presentation, β3 in green, M-CSFRGD in pink, and the mutant QTSRGDSPS loop in red. M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid.
(TIF)
αv in yellow, β3 in green, RGD from M-CSFRGD in cyan, and RGD from the crystal in pink. cRGD, cyclic RGD; M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid.
(TIF)
The expression levels of c-FMS and αvβ3 integrin were measured using flow cytometry. The red histograms represent the negative control, and the blue histograms represent receptor expression. Mouse BMMs without differentiation cytokines (t = 0) express c-FMS (A) and αvβ3 integrin (B). MDA-MB-231 breast cancer cell line express c-FMS (C) and αvβ3 integrin (D). BMM, bone-marrow–derived monocyte; MDA-MB-231, MDA Anderson metastatic breast 231.
(TIF)
Murine BMMs were seeded for differentiation for 48 h, followed by incubation of purified M-CSFc-FMS, M-CSFαvβ3, and M-CSFRGD variants without the addition of murine M-CSF. Cells were lysed and subjected to SDS-PAGE to test spontaneous activation of (A) c-FMS and (B) Akt. The aspect ratios of the membranes were changed. BMM, bone-marrow–derived monocyte; M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid.
(TIF)
Differentiated murine BMMs were incubated for additional 24 h without (positive control) or with inhibitors (5 μM) followed by fixation and F-actin and nuclei staining. Cells were able to form a solid actin ring (white arrowheads), scattered actin ring [44] (white arrows) or amorphous actin distribution (barbed arrowheads). Pictures are representatives of 35 images acquired from five different wells per sample. BMM, bone-marrow–derived monocyte; M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid.
(TIF)
(DOCX)
M-CSF, macrophage colony-stimulating factor; SPR, surface plasmon resonance.
(XLSX)
SPR, surface plasmon resonance.
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
SPR, surface plasmon resonance.
(XLSX)
Acknowledgments
The authors thank Dr. Alon Zilka and Hagit Gdalya for their technical assistance.
Abbreviations
- αMEM
alpha Minimum Essential Medium
- BMM
bone-marrow–derived monocyte
- BMSC
bone-marrow–derived mesenchymal stromal cells
- BS3
bis(sulfosuccinimidyl)suberate
- CD
circular dichroism
- cRGD
cyclic RGD
- CTX-I
carboxy-terminal telopeptide of type I collagen
- EDC
1-ethyl-3-(3dimethylaminopropyl)-carbodiimide
- FACS
fluorescence-activated cell sorting
- FBS
fetal bovine serum
- FDA
Food and Drug Administration
- FSC
forward scatter
- LB
Luria Broth
- M-CSFWT
wild-type M-CSF
- M-CSF
macrophage colony-stimulating factor
- MDA-MB-231
MD Anderson metastatic breast 231
- MSC
mesenchymal stem cell
- NF-кB
nuclear factor–kappa B
- NHS
N-hydroxysuccinimide
- OSCAR
osteoclast-associated receptor
- OVX
ovariectomy
- PBS
phosphate-buffered saline
- PBSA
PBS containing 1% bovine serum albumin
- PBST
PBS + 0.005% Tween
- PE
phychoerythrin
- PI
propidium iodide
- PNPP
para-nitrophenylphosphate
- RANKL
receptor activator of nuclear factor–kappa-B ligand
- RGD
Arginine-Glycine-Aspartic acid
- RUs
response units
- s.c.
subcutaneously
- SDS
sodium dodecyl sulfate
- SPR
surface plasmon resonance
- SSC
side scatter
- TRAP
tartrate-resistant acid phosphatase
- WT
wild type
- XTT
2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
- YSD
yeast surface display
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
Israel Science Foundation (grant number 544/15). NL. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. European Research Council (ERC) (grant number 336041). NP. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Melton L.J. 3rd, et al. , How many women have osteoporosis? JBMR Anniversary Classic. JBMR, Volume 7, Number 9, 1992. J Bone Miner Res, 2005. 20(5): p. 886–92. 10.1359/jbmr.2005.20.5.886 [DOI] [PubMed] [Google Scholar]
- 2.Johnell O. and Kanis J.A., An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int, 2006. 17(12): p. 1726–33. 10.1007/s00198-006-0172-4 [DOI] [PubMed] [Google Scholar]
- 3.Stone K.L., et al. , BMD at multiple sites and risk of fracture of multiple types: long‐term results from the Study of Osteoporotic Fractures. Journal of Bone and Mineral Research, 2003. 18(11): p. 1947–1954. 10.1359/jbmr.2003.18.11.1947 [DOI] [PubMed] [Google Scholar]
- 4.Magnusson C., et al. , Breast‐cancer risk following long‐term oestrogen‐and oestrogen‐progestin‐replacement therapy. International Journal of Cancer, 1999. 81(3): p. 339–344. [DOI] [PubMed] [Google Scholar]
- 5.Dick S.E., DeWitt D.E., and Anawalt B.D., Postmenopausal hormone replacement therapy and major clinical outcomes: a focus on cardiovascular disease, osteoporosis, dementia, and breast and endometrial neoplasia. American Journal of Managed Care, 2002. 8(1): p. 95–104. [PubMed] [Google Scholar]
- 6.Adami S. and Zamberlan N., Adverse effects of bisphosphonates. Drug Safety, 1996. 14(3): p. 158–170. [DOI] [PubMed] [Google Scholar]
- 7.Okada N., et al. , Identification of the risk factors associated with hypocalcemia induced by denosumab. Biological and Pharmaceutical Bulletin, 2013. 36(10): p. 1622–1626. [DOI] [PubMed] [Google Scholar]
- 8.Schilcher J. and Aspenberg P., Atypical fracture of the femur in a patient using denosumab–a case report. Acta orthopaedica, 2014. 85(1): p. 6–7. 10.3109/17453674.2014.885355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Novack D.V. and Teitelbaum S.L., The osteoclast: friend or foe? Annu Rev Pathol, 2008. 3: p. 457–84. 10.1146/annurev.pathmechdis.3.121806.151431 [DOI] [PubMed] [Google Scholar]
- 10.Teitelbaum S.L., Bone resorption by osteoclasts. Science, 2000. 289(5484): p. 1504–8. [DOI] [PubMed] [Google Scholar]
- 11.Dai X.M., et al. , Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood, 2002. 99(1): p. 111–20. [DOI] [PubMed] [Google Scholar]
- 12.Rodan S.B. and Rodan G.A., Integrin function in osteoclasts. J Endocrinol, 1997. 154 Suppl: p. S47–56. [PubMed] [Google Scholar]
- 13.Ross F.P., et al. , Interactions between the bone matrix proteins osteopontin and bone sialoprotein and the osteoclast integrin alpha v beta 3 potentiate bone resorption. J Biol Chem, 1993. 268(13): p. 9901–7. [PubMed] [Google Scholar]
- 14.Zou W. and Teitelbaum S.L., Integrins, growth factors, and the osteoclast cytoskeleton. Ann N Y Acad Sci, 2010. 1192: p. 27–31. 10.1111/j.1749-6632.2009.05245.x [DOI] [PubMed] [Google Scholar]
- 15.McHugh K.P., et al. , Mice lacking β3 integrins are osteosclerotic because of dysfunctional osteoclasts. The Journal of clinical investigation, 2000. 105(4): p. 433–440. 10.1172/JCI8905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faccio R., et al. , c-Fms and the alphavbeta3 integrin collaborate during osteoclast differentiation. J Clin Invest, 2003. 111(5): p. 749–58. 10.1172/JCI16924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zou W., et al. , DAP12 couples c-Fms activation to the osteoclast cytoskeleton by recruitment of Syk. Mol Cell, 2008. 31(3): p. 422–31. 10.1016/j.molcel.2008.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duong L.T., et al. , PYK2 in osteoclasts is an adhesion kinase, localized in the sealing zone, activated by ligation of alpha (v) beta3 integrin, and phosphorylated by src kinase. Journal of Clinical Investigation, 1998. 102(5): p. 881 10.1172/JCI3212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faccio R., et al. , Dynamic changes in the osteoclast cytoskeleton in response to growth factors and cell attachment are controlled by beta3 integrin. J Cell Biol, 2003. 162(3): p. 499–509. 10.1083/jcb.200212082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakai H., et al. , Activated c-Fms recruits Vav and Rac during CSF-1-induced cytoskeletal remodeling and spreading in osteoclasts. Bone, 2006. 39(6): p. 1290–1301. 10.1016/j.bone.2006.06.012 [DOI] [PubMed] [Google Scholar]
- 21.Elsegood C.L., et al. , M-CSF induces the stable interaction of cFms with α V β 3 integrin in osteoclasts. The international journal of biochemistry & cell biology, 2006. 38(9): p. 1518–1529. [DOI] [PubMed] [Google Scholar]
- 22.Faccio R., et al. , M-CSF regulates the cytoskeleton via recruitment of a multimeric signaling complex to c-Fms Tyr-559/697/721. J Biol Chem, 2007. 282(26): p. 18991–9. 10.1074/jbc.M610937200 [DOI] [PubMed] [Google Scholar]
- 23.Li W. and Stanley E.R., Role of dimerization and modification of the CSF-1 receptor in its activation and internalization during the CSF-1 response. The EMBO journal, 1991. 10(2): p. 277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deng P., et al. , The role of individual cysteine residues in the processing, structure, and function of human macrophage colony-stimulating factor. Biochemical and biophysical research communications, 1996. 228(2): p. 557–566. 10.1006/bbrc.1996.1698 [DOI] [PubMed] [Google Scholar]
- 25.Elegheert J., et al. , Allosteric competitive inactivation of hematopoietic CSF-1 signaling by the viral decoy receptor BARF1. Nat Struct Mol Biol, 2012. 19(9): p. 938–47. 10.1038/nsmb.2367 [DOI] [PubMed] [Google Scholar]
- 26.Takeshita S., Kaji K., and Kudo A., Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. Journal of Bone and Mineral Research, 2000. 15(8): p. 1477–1488. 10.1359/jbmr.2000.15.8.1477 [DOI] [PubMed] [Google Scholar]
- 27.Belvisi L., et al. , Potent integrin antagonists from a small library of RGD-including cyclic pseudopeptides. Organic letters, 2001. 3(7): p. 1001–1004. [PubMed] [Google Scholar]
- 28.Schneidman-Duhovny D., et al. , PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic acids research, 2005. 33(suppl 2): p. W363–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiong J.-P., et al. , Crystal structure of the extracellular segment of integrin αVβ3 in complex with an Arg-Gly-Asp ligand. Science, 2002. 296(5565): p. 151–155. 10.1126/science.1069040 [DOI] [PubMed] [Google Scholar]
- 30.Elegheert J., et al. , Extracellular complexes of the hematopoietic human and mouse CSF-1 receptor are driven by common assembly principles. Structure, 2011. 19(12): p. 1762–1772. 10.1016/j.str.2011.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Persad S., et al. , Regulation of protein kinase B/Akt-serine 473 phosphorylation by integrin-linked kinase critical roles for kinase activity and amino acids arginine 211 and serine 343. Journal of Biological Chemistry, 2001. 276(29): p. 27462–27469. 10.1074/jbc.M102940200 [DOI] [PubMed] [Google Scholar]
- 32.Pfaff M. and Jurdic P., Podosomes in osteoclast-like cells. Journal of cell science, 2001. 114(15): p. 2775–2786. [DOI] [PubMed] [Google Scholar]
- 33.Jurdic P., et al. , Podosome and sealing zone: specificity of the osteoclast model. European journal of cell biology, 2006. 85(3): p. 195–202. [DOI] [PubMed] [Google Scholar]
- 34.Chabadel A., et al. , CD44 and β3 integrin organize two functionally distinct actin-based domains in osteoclasts. Molecular biology of the cell, 2007. 18(12): p. 4899–4910. 10.1091/mbc.E07-04-0378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horton M., et al. , Arg Gly Asp (RGD) peptides and the anti-vitronectin receptor antibody 23C6 inhibit dentine resorption and cell spreading by osteoclasts. Experimental cell research, 1991. 195(2): p. 368–375. [DOI] [PubMed] [Google Scholar]
- 36.Kim N., et al. , A novel member of the leukocyte receptor complex regulates osteoclast differentiation. Journal of Experimental Medicine, 2002. 195(2): p. 201–209. 10.1084/jem.20011681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ishida N., et al. , Large Scale Gene Expression Analysis of Osteoclastogenesisin Vitro and Elucidation of NFAT2 as a Key Regulator. Journal of Biological Chemistry, 2002. 277(43): p. 41147–41156. 10.1074/jbc.M205063200 [DOI] [PubMed] [Google Scholar]
- 38.Plow E.F., et al. , Ligand binding to integrins. Journal of Biological Chemistry, 2000. 275(29): p. 21785–21788. 10.1074/jbc.R000003200 [DOI] [PubMed] [Google Scholar]
- 39.Felix J., et al. , Structure and assembly mechanism of the signaling complex mediated by human CSF-1. Structure, 2015. 23(9): p. 1621–1631. 10.1016/j.str.2015.06.019 [DOI] [PubMed] [Google Scholar]
- 40.Takagi J., Structural basis for ligand recognition by RGD (Arg-Gly-Asp)-dependent integrins. Biochemical Society Transactions, 2004. 32(3): p. 403–406. [DOI] [PubMed] [Google Scholar]
- 41.Van Der Spoel D., et al. , GROMACS: fast, flexible, and free. Journal of computational chemistry, 2005. 26(16): p. 1701–1718. 10.1002/jcc.20291 [DOI] [PubMed] [Google Scholar]
- 42.Dalke A. and Schulten K., VMD-visual molecular dynamics. William Humphrey Journal of Molecular Graphics, 1996. 14: p. 33–38. [DOI] [PubMed] [Google Scholar]
- 43.Abràmoff M.D., Magalhães P.J., and Ram S.J., Image processing with ImageJ. Biophotonics international, 2004. 11(7): p. 36–42. [Google Scholar]
- 44.Davies J., et al. , The osteoclast functional antigen, implicated in the regulation of bone resorption, is biochemically related to the vitronectin receptor. The Journal of Cell Biology, 1989. 109(4): p. 1817–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) The amino acid sequence of M-CSF with C31, which is required for dimerization, indicated in red. The two flexible loops in the dimerization interface are colored blue (loop 1, residues 25–32) and green (loop 3, residues 64–71). (B) M-CSFRGD library 1 colored in blue, where residues 25–32 were replaced with an RGD motif having three random amino acids on each side. (C) M-CSFRGD library 2 colored green, where residues 64–71 were replaced with an RGD motif with three random amino acids on each side and C31 was replaced with serine to inhibit disulfide-linked homodimerization. (D) M-CSFαvβ3 is based on the sequence of M-CSFRGD variant 4.22 with two single-point mutations in H9A and H15A, indicated in red, to inhibit binding to c-FMS. (E) M-CSFc-FMS was created by changing the RGD motif on M-CSFRGD variant 4.22 to RDG with the aim to prevent binding to αvβ3 integrin. (F) Sequences of the mutated loop of the three M-CSFRGD clones that were selected after four (4.22 and 4.24) and five (5.6) rounds of the affinity maturation process. M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid; WT, wild type.
(TIF)
YSD M-CSFC31S was analyzed for (A) forward scatter and side scatter and (B) expression using mouse anti-c-myc antibody followed by a secondary PE-labeled anti mouse antibody. (C) The binding of YSD M-CSFC31S to soluble c-FMS-Fc was detected by a goat anti-human Fc-FITC antibody. (D) Cells expressing M-CSFC31S on the yeast cell wall were incubated with 10 different concentrations of c-FMS-Fc (0.5–2000 nM) and were tested for binding by flow cytometry. The curve shows a good fit to a single binding-site curve, and the apparent KD is 20 nM. Source data can be found in S7 Data. FITC, fluorescein isothiocyanate; M-CSF, macrophage colony-stimulating factor; PE, phycoerythrin; YSD, yeast surface display.
(TIF)
The M-CSFRGD library was covalently linked to Aga1p and the yeast cell wall. Binding for c-FMS was determined with c-FMS-Fc recombinant protein and goat anti-human Fc FITC conjugated secondary antibody, and the expression levels were measured with a mouse anti-c-myc primary antibody and PE anti-mouse secondary antibody. For determination of αvβ3 integrin binding, yeast cells were incubated with recombinant αvβ3 integrin and mouse anti-human CD49d FITC secondary antibody, and the expression levels were measured with chicken anti-c-myc primary antibody and PE goat anti-chicken secondary antibody. FITC, fluorescein isothiocyanate; M-CSF, macrophage colony-stimulating factor; PE, phycoerythrin; RGD, Arginine-Glycine-Aspartic acid; YSD, yeast surface display.
(TIF)
M-CSFRGD (A–D) library 1 and (E–H) library 2 were analyzed for (A and E) FSC/SSC, (B and F) expression, (C and G) 100 nM c-FMS binding, and (D and H) 500 nM αvβ3 integrin binding. FACS, fluorescence-activated cell sorting; FSC, forward scatter; M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid; SSC, side scatter.
(TIF)
Yeast-displayed mutant libraries were analyzed, and the living cells population in each sort is represented by a black polygon-shaped gate. The affinity maturation sorting process started with (A) a presorted library followed by (B) sort 1, (C) sort 2, (D) sort 3, (E) sort 4, and (F) sort 5. FACS, fluorescence-activated cell sorting; FSC, forward scatter; SSC, side scatter.
(TIF)
Twenty-five different clones from each of sorts 4 (A) and 5 (C) were tested for binding to 20 nM of αvβ3 integrin, normalized to the lowest binder. (B) The best 15 αvβ3 integrin M-CSFRGD binders from sort 4 and the best 10 αvβ3 integrin M-CSFRGD binders from sort 5 (D) were evaluated for binding to 50 nM of c-FMS, normalized to M-CSFC31S. The chosen clones (4.22, 4.24, and 5.6) are indicated in blue. (E) Variants 4.22, 4.24, and 5.6 were evaluated for integrin specificity by testing their binding to 250 nM of α4β7, αIIbβ3, αvβ5, and α5β1 integrins in comparison with their binding to αvβ3 integrin. Source data can be found in S8 Data. M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid; YSD, yeast surface display.
(TIF)
(A) Size exclusion chromatography of nonglycosylated M-CSFRGD clone 4.22 with high molecular weight standards. Variant 4.22 was eluted at the size of 21 kDa. (B) Mass spectrometry of nonglycosylated variant 5.6. (C) CD spectra of nonglycosylated variant 4.22 (red line), nonglycosylated variant 4.24 (blue line), nonglycosylated variant 5.6 (green line), nonglycosylated M-CSFc-FMS (pink line), and nonglycosylated M-CSFαvβ3 (gray lines). (D) Temperature-dependent CD measurements of unfolded proteins determined at 217 nm normalized to fully denatured proteins. (E) SDS-PAGE for all purified proteins: nonglycosylated M-CSFC31S (lane 1), nonglycosylated variant 4.22 (lane 2), nonglycosylated variant 4.24 (lane 3), nonglycosylated variant 5.6 (lane 4), nonglycosylated M-CSFc-FMS (lane 5), and non-glycosylated M-CSFαvβ3 (lane 6). Source data can be found in S9 Data. CD, circular dichroism; M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid.
(TIF)
Dimerization of the purified proteins was determined by using increasing concentrations of BS3 cross-linker, denaturation, and analysis on SDS-PAGE. M-CSFWT dimerized at all BS3 concentrations, but the three M-CSFRGD variants, M-CSFc-FMS, and M-CSFαvβ3 did not show any dimerization capability. BS3, bis(sulfosuccinimidyl)suberate; M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid; WT, wild type.
(TIF)
To determine the protein KD,app, the RUs at saturation for each protein concentration were plotted, and a fitted curve was created for (A) c-FMS and (B) αvβ3 integrin. Source data and its analysis can be found in S10 Data. RUs, response units.
(TIF)
(A) αvβ3, (B) α3β1, (C) α4β7, and (D) α5β1 integrins were immobilized on the surface of the chip. Thereafter, the three M-CSFRGD variants 4.22 (green), 4.24 (blue), and 5.6 (red) were allowed to flow over the surface of the chip at a concentration of 1 μM. Source data can be found in S11 Data. M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid.
(TIF)
M-CSFC31S is shown in pink, c-FMS in cyan, αv in yellow, and β3 in green. Residues 25–32 of M-CSFC31S are represented in red. M-CSF, macrophage colony-stimulating factor.
(TIF)
M-CSFRGD/αvβ3 integrin interface seen from different angles (A–D). αv in yellow “surf” presentation, β3 in green, M-CSFRGD in pink, and the mutant QTSRGDSPS loop in red. M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid.
(TIF)
αv in yellow, β3 in green, RGD from M-CSFRGD in cyan, and RGD from the crystal in pink. cRGD, cyclic RGD; M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid.
(TIF)
The expression levels of c-FMS and αvβ3 integrin were measured using flow cytometry. The red histograms represent the negative control, and the blue histograms represent receptor expression. Mouse BMMs without differentiation cytokines (t = 0) express c-FMS (A) and αvβ3 integrin (B). MDA-MB-231 breast cancer cell line express c-FMS (C) and αvβ3 integrin (D). BMM, bone-marrow–derived monocyte; MDA-MB-231, MDA Anderson metastatic breast 231.
(TIF)
Murine BMMs were seeded for differentiation for 48 h, followed by incubation of purified M-CSFc-FMS, M-CSFαvβ3, and M-CSFRGD variants without the addition of murine M-CSF. Cells were lysed and subjected to SDS-PAGE to test spontaneous activation of (A) c-FMS and (B) Akt. The aspect ratios of the membranes were changed. BMM, bone-marrow–derived monocyte; M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid.
(TIF)
Differentiated murine BMMs were incubated for additional 24 h without (positive control) or with inhibitors (5 μM) followed by fixation and F-actin and nuclei staining. Cells were able to form a solid actin ring (white arrowheads), scattered actin ring [44] (white arrows) or amorphous actin distribution (barbed arrowheads). Pictures are representatives of 35 images acquired from five different wells per sample. BMM, bone-marrow–derived monocyte; M-CSF, macrophage colony-stimulating factor; RGD, Arginine-Glycine-Aspartic acid.
(TIF)
(DOCX)
M-CSF, macrophage colony-stimulating factor; SPR, surface plasmon resonance.
(XLSX)
SPR, surface plasmon resonance.
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
SPR, surface plasmon resonance.
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.