

Abstract
A barrier to our understanding of how various cell types and signals contribute to synaptic circuit function is the lack of relevant models for studying the human brain. One emerging technology to address this issue is the use of three dimensional (3D) neural cell cultures, termed 'organoids' or 'spheroids', for long term preservation of intercellular interactions including extracellular adhesion molecules. However, these culture systems are time consuming and not systematically generated. Here, we detail a method to rapidly and consistently produce 3D cocultures of neurons and astrocytes from human pluripotent stem cells. First, pre-differentiated astrocytes and neuronal progenitors are dissociated and counted. Next, cells are combined in sphere-forming dishes with a Rho-Kinase inhibitor and at specific ratios to produce spheres of reproducible size. After several weeks of culture as floating spheres, cocultures ('asteroids') are finally sectioned for immunostaining or plated upon multielectrode arrays to measure synaptic density and strength. In general, it is expected that this protocol will yield 3D neural spheres that display mature cell-type restricted markers, form functional synapses, and exhibit spontaneous synaptic network burst activity. Together, this system permits drug screening and investigations into mechanisms of disease in a more suitable model compared to monolayer cultures.
Keywords: Developmental Biology, Issue 138, Stem Cells, Neurons, Astrocytes, Synapses, Organoid, Coculture, Bioreactor, Multielectrode array
Introduction
Astrocytes are a highly abundant glial cell type within the central nervous system (CNS) with a variety of functional responsibilities beyond structural support. Through secretion of soluble synaptogenic factors and extracellular matrix (ECM) components, astrocytes aid in the establishment and clustering of mature synapses during development1. They also play a critical role in maintaining the health and plasticity of synapses through extracellular signaling2,3,4,5, and contribute to long-term stability of homeostatic environments by regulating extracellular potassium and glutamate, as well as the secretion of energy substrates and ATP6,7,8. Finally, they can contribute to neurotransmission by influencing extrasynaptic currents9, and can indirectly influence activity through other cell types such as promoting myelination10. Importantly, because abnormality or dysfunction of astrocytes can lead to many neurodevelopmental syndromes and adult neuropathology, there is an obvious need to include astrocytes alongside neurons within engineered neural networks in order for an improved model of the endogenous brain environment. An integral characteristic of astrocytes is their ability to form dynamic interactions with neuronal synapses1,11,12. In the absence of glia, neurons form a limited number of synapses, which in general also lack functional maturity13.
Human astrocytes display morphological, transcriptional, and functional characteristics — such as increased size and complexity of branching, as well as species-specific genes — that are not recapitulated in rodents12,14,15. As a result, studies utilizing human pluripotent stem cell (hPSC)-derived neural cells have become widely accepted as a means of examining CNS-related diseases in vitro while developing novel therapies, injury models, and culture paradigms16,17. Furthermore, hPSCs permit the study of human synapse formation and function without the need for primary tissue18,19.
A barrier to our understanding of how various cell types and signals contribute to synaptic circuit function is the lack of relevant models of the human brain. There is a need for an appropriate platform to recapitulate its synaptic networks with high fidelity and reproducibility. Recently, interest has emerged in the production of 3D culture systems (broadly known as 'organoids,' 'spheroids,' or 'mini brains')20 to model complex three-dimensional (3D) structures at the cellular and macro levels. 3D culture systems retain ECM and cell-cell interactions that are normally absent or limited during typical 2D coculture paradigms21,22. An abundance of techniques exist for culturing 3D neural spheroids23,24,25; however, many require lengthy culture periods (months to years) for spontaneous development and layer preservation, with the user exhibiting very little control over the output.
Here, we illustrate a systematic method to rapidly and consistently bioengineer neural interactions among multiple cell types (pre-differentiated neurons and astrocytes) derived from hPSCs by assembling cells into sphere cocultures ('asteroids')26 that recapitulate human-specific morphological complexities in 3D. This high-density neural system generates evenly-dispersed neural subtypes that take on mature properties over time and can be screened or assayed in a high-throughput manner. We demonstrate for the first time that human astrocytes induce synaptic network burst activity in these 3D cocultures. In addition, this protocol is easily adaptable to generate spheres of different sizes, to utilize cells specified to different regional identities of the CNS, and to study interactions of multiple other cell types as desired.
Protocol
1. Cell Culture and Reagent Preparation
NOTE: The protocols in this section are written in the order in which they appear in the differentiation protocol (section 2). See the Table of Materials for materials and catalog numbers.
- Prepare coated plates for cell culture.
- Dilute extracellular matrix (ECM) coating solution with DMEM/F12 media to prepare a 1 mg/mL stock solution. Aliquot the diluted ECM stock solution into 30 conical tubes of 3 mL each and immediately store in -20 °C. For a working solution, resuspend 3 mL of ECM stock into 33 mL of pre-chilled DMEM/F12 media, bringing the total volume to 36 mL for a concentration of 80 µg/mL.
- Coat 1 mL per well of a 6-well cell culture plate with ECM working solution. Add an additional 1 mL DMEM/F12 per well (if necessary) to ensure that the surface of the well is fully covered. Allow the coated 6-well cell culture plates to sit at room temperature for at least 1 h before use. NOTE: Coated plates may be stored in the incubator or at 4 °C for up to 2 weeks.
- Prepare media formulations, growth factors, and small molecules.
- To prepare 500 mL of human pluripotent stem cell (hPSC) medium27, add 20x and 500x supplements according to manufacturers’ instructions.
- To prepare 500 mL of neural medium (NM), add heparin to a final concentration of 2 mg/mL, 1x of antibiotic/antimycotic solution, 1x B27 supplement or 1x N2 supplement to a 500 mL bottle of DMEM/F12 with L-glutamine supplement as previously detailed28.
- To prepare a 10 mM (1,000x) stock solution of Rho-Kinase Inhibitor Y27632 (Y), add 10 mg of powder into 3 mL of phosphate buffered saline (PBS). Filter sterilize, aliquot, and store at -20 °C. Use at a 10 µM working concentration in media.
- To prepare a 20 mM (10,000x) stock solution of SB431542, add 10 mg of powder into 1.3 mL of dimethyl sulfoxide (DMSO). To prepare a 2 mM (1,000x) stock solution of DMH1, add 10 mg of powder into 13.1 mL of DMSO. Filter sterilize, aliquot, and store each solution at -20 °C. Use each at a 2 µM working concentration in media (NM + SB431542 + DMH1). NOTE: This protocol recommends 2 µM working concentrations of both SB431542 and DMH1 based upon our observations and reports from others29 stating that this concentration effectively promotes neural induction from hPSCs. Higher concentrations have also been reported30. Additionally, with alternative media, it has also been shown that small molecules are not necessary for high neural conversion31. Thus, working concentrations will vary depending on alternative cell culture environments, and optimal concentrations should be tested by individual researchers.
- To prepare individual 100 µg/mL (10,000x) stock solutions of epidermal growth factor (EGF) and fibroblast growth factor-2 (FGF2), add 1 mg of each into 10 mL of PBS + 0.1% BSA. Aliquot EGF and FGF2 stock solutions into 50 µL aliquots and store at -80 °C. Use each at a 10 ng/mL working concentration in media; e.g., add 5 µL EGF and 5 µL FGF2 to 50 mL of NM (NM + EGF + FGF2).
- To prepare a stock solution of doxycycline hydrochloride (Dox), first dissolve powder in DMSO to 100 mg/mL and store at -20 °C. Further dilute it to make a 2 mg/mL (1,000x) stock solution in PBS and store at -20 °C. Use at 2 µg/mL working concentration in media; e.g., add 50 µL Dox to 50 mL of NM (NM + Dox). Note: Do not re-freeze solutions after thawing.
2. Generation of Neural Subtypes from Human Induced Pluripotent Cells (hPSCs)
NOTE: All cell cultures should be maintained in an incubator with 5% CO2 at 37 °C. These cultures are maintained at room oxygen levels, though lower levels may be utilized.
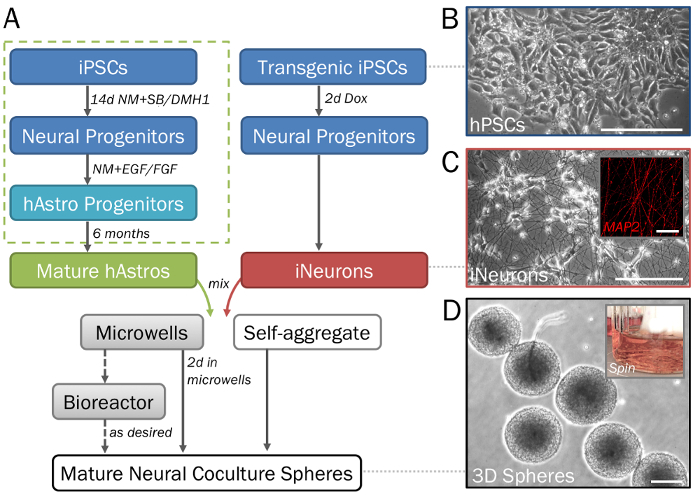
- Generate and maintain astrocyte progenitors (hAstros) from hPSCs. NOTE: This section provides a brief and simplified differentiation protocol. For a detailed description (with embryoid body formation, rosette selection and regional patterning steps) refer to the previously described protocol28 (Figure 1A, dotted green box).
- Seed clusters of hPSCs (Figure 1B) onto ECM-coated 6-well plates (see step 1.1) with 2 mL of hPSC medium + Y per well. Feed the stem cells every day until they are about 50% confluent and split as needed.
- To split hPSCs, add 1 mL of passaging reagent to wells and aspirate after 1 min. Incubate cells at room temperature for 5 min, add 1 mL of hPSC medium, and split aggregates 1:10 onto new ECM-coated wells in 2 mL of hPSC medium + Y per well.
- Maintain the stem cells until they are about 50% confluent, then change media to NM + SB431542 + DMH1 to induce neural differentiation (day 0). When cells are about 95% confluent, split 1:6 into new ECM-coated wells in the same medium.
- On day 14, dissociate the cells with detachment solution and transfer to a non-coated flask with Y to promote formation of aggregates. NOTE: The addition of Y is utilized to promote cell survival and sphere formation but is not included during every feed.
- For the generation of neuronal progenitors and neurons (hNeurons), use these day-14 cells as described below. Alternatively, utilize these cells at later time points from monolayer or sphere cultures before neuronal maturation occurs or gliogenesis commences. NOTE: By default, this protocol produces dorsal-cortical astrocytes. However, astrocyte subtypes can be regionally specified by the addition of patterning morphogens if desired28,32,33. Retinoic acid (RA) may be added to caudalize cells into spinal cord phenotypes, while sonic hedgehog (SHH), smoothened agonist (SAG), or purmorphamine will produce ventral phenotypes.
- For the generation of astrocyte progenitors and astrocytes in spontaneously-formed 3D aggregates (hAstrospheres), switch to NM + EGF + FGF2 and feed weekly (or as needed to ensure stable pH) as previously detailed34.
- Gently dissociate hAstro aggregates with detachment solution when dark centers appear and remove spheres that spontaneously attach. Break spheres gently once a week to maintain sphere health and to avoid necrotic cores. NOTE: Do not exceed 5 min of treatment with detachment solution.
- After 4-6 months of expansion, confirm cell identity and either freeze in cryopreservation medium (according to manufacturer’s instructions) or alternative storage conditions for long-term preservation in liquid nitrogen, or use immediately for experimentation.
- To thaw a frozen stock of hAstrospheres, quickly thaw a vial at room temperature, transfer the contents to an empty 15 mL conical tube, and centrifuge at 300 x g for 1 min. Carefully aspirate supernatant, wash with DMEM/F12, and repeat for a total of two wash steps. Add to a T25 flask with a total volume of 6 mL of NM + EGF + FGF2 + Y (or scale up as needed).
- Generate and maintain inducible neurons (iNeurons) from hPSCs.
- Seed transgenic hPSCs (with a stable doxycycline-inducible neurogenin 2 transgene35) on ECM-coated 6-well plates with 2 mL of hPSC medium + Y per well (as described above for non-transgenic lines). Feed everyday with 2 mL of media per well until they are ready to lift at 70% confluency. Split cells as described in step 2.1.2.
- When cells are about 35% confluent, add NM + Dox to induce differentiation into iNeurons. Maintain as a monolayer culture with NM + Dox for 2 days before proceeding to step 3.2. NOTE: Cells will transition into neuronal progenitors but will not yet extend neurites (Figure 1C).
3. Preparation and Maintenance of 3D Sphere Cocultures
Dissociate hAstros from 3D aggregates in suspension (as described in step 2.1.5.1).
- Dissociate hNeurons or iNeurons from a 2D monolayer.
- Remove media from 6-well plate and add 500 µL of detachment solution to each well containing monolayer cultures. Incubate at 37 °C for 5 min. Gently add 2-3 mL DMEM/F12 to remove attached cells.
- Collect cells and media in a 15 mL conical tube and centrifuge at 300 x g for 1 min. Aspirate the supernatant, add 1 mL of fresh media, and pipette up and down gently with a 1,000 µL micropipette to achieve a single cell suspension.
- Form 3D spheres using microwell culture plates.
- Prepare plate by adding 0.5 mL of anti-adherence rinsing solution to each well of the microwell plate. Centrifuge plate at 2,000 x g for 5 min. Aspirate rinsing solution, add 1 mL of DMEM/F12 to each well to wash, and aspirate again. NOTE: Always ensure that the centrifuge is balanced during use.
- Count each cell type using a hemocytometer or automated cell counter. Add desired ratio of dissociated hAstros and hNeurons or iNeurons to each well in a total volume of 2 mL of NM + Y (include Dox if iNeurons are utilized). Centrifuge plate at 100 x g for 3 min and return to the incubator. NOTE: 24-well microwell plates (see the Table of Materials) contain 300 microwells per well. Add a minimum of 6 x 105 cells (for spheres of 2 x 103 cells each) and a maximum of 6 x 106 cells (for spheres of 2 x 104 cells each) in 2 mL of media per well. For viable spheres of a specific density within this range, use a defined quantity of cells and divide by 300 to calculate the number of cells per sphere. Alternative sphere forming methods and tools may also be utilized if desired. Follow manufacturer’s instructions for acceptable ranges of cell densities.
- Allow cells to self-assemble into densely packed spheres within microwell plates over the next 2 days (Figure 2A). Replace 50% media if the culture is yellowing. NOTE: Exchange only half media and pipette gently when removing and adding media in order not to disturb spheres. Spheres may lift up from microwells and fuse together with higher force.
- After 2 days, gently remove spheres from microwells with a 1,000 µL micropipette (Figure 1D). Lightly apply force to the bottom of microwells with media to remove any additional adhered spheres. Let spheres settle in a 15 mL conical tube, aspirate the old media, and add fresh media.
- Set up the spinner flask bioreactor system for forming 3D spheres.
- Clean the surface of a magnetic stir plate with 70% ethanol, place it into a cell culture incubator. Autoclave individual spinner flasks to sterilize.
- Add spheres with a minimum of 50–60 mL media to each spinner flask (Figure 1D, inset). Place the flask on the magnetic stir plate set at 60 rpm. Culture spheres in spinner flasks until they are ready for data collection. NOTE: A minimum of 3 weeks in culture is needed for synapse formation. If the spinner flask bioreactor system is not desired, spheres may be cultured in stationary conditions in cell culture flasks or dishes. Spheres may also be embedded in ECM or hydrogel.
4. Measurement of Live Synaptic Physiology with Multielectrode Arrays (MEAs)
- Prepare MEAs for cell culture.
- Clean surface of each MEA with 1 mL of detergent for 1 h. Rinse with sterile deionized (DI) water, rinse with 70% ethanol to sterilize, and then air dry in biosafety cabinet under UV light. NOTE: MEAs can be stored in DI water at 4 °C under sterile conditions until use.
- Prepare a 0.5 mg/mL solution of poly-ornithine (PLO) by dissolving 50 mg of PLO into 100 mL of boric acid buffer. Coat the surface of each MEA with 1 mL of PLO to render the surface hydrophilic and incubate at 37 °C for a minimum of 4 h. Remove the PLO, wash the surface with DI water, add 1 mL of ECM (see step 1.1.1), and incubate at 37 °C for a minimum of 4 h.
- Remove ECM and place spheres in 1.5 mL of media on a MEA surface, ensuring that the spheres are positioned on top of the electrode array (Figures 3A, 3A’). Allow to adhere for 2 days. Change half of the media every 2–3 days (or more often if needed), ensuring that the spheres are not disturbed. NOTE: Addition of growth factors may improve culture survival and maturation.
- Measure electrical activity of neural spheres.
- Set up a multielectrode array (MEA) system with a temperature-controlled headstage. Create a software program with a 5 Hz high-pass filter, and a 200 Hz low-pass filter, and a spike threshold of 5x standard deviation (SD).
- Place the MEA on headstage and record spontaneous electrical activity (Figure 3B, B’). Save raw data including spike frequency and amplitude for statistical analysis. NOTE: A perfusion system may be utilized with the MEA to add pharmacological agents or to increase flow of fresh media for long-term recording. Additional post-processing of spikes may be performed as desired36,37.
5. Measurement of Synaptic Density with Immunocytochemistry
- Prepare the reagents.
- To make a 500 mL stock solution of 4% paraformaldehyde (PFA), add 400 mL of 1x PBS to a glass beaker or a stir plate in a ventilated hood. Add stir bar and heat to 60 °C while stirring; do not boil. Add 20 g of PFA powder to the heated PBS solution and raise the pH to 6.9 by slowly adding 1 N NaOH dropwise. NOTE: 4% PFA solution can be aliquoted and stored at 4 °C for up to one month.
- Add 20 g or 30 g of sucrose to 100 mL of PBS to make 20% and 30% solutions of sucrose, respectively.
- Add 1 mL of detergent to 10 mL of PBS to prepare a 10% stock solution. Invert several times to homogenize.
- Add 250 µL of 10% detergent stock solution (final concentration of 0.25%) and 500 µL each of goat and donkey serums (final concentration of 5% each) to 10 mL PBS to prepare the primary blocking buffer.
- Add 100 µL each of goat and donkey serums (final concentration of 1% each) to 10 mL PBS to prepare the secondary blocking buffer.
- Prepare the samples.
- Transfer the spheres into a 15 mL conical tube, allow them to settle at the bottom of the tube, and aspirate the old media. Rinse spheres with 500 µL of PBS, let settle, and aspirate PBS. Add 500 µL of 4% PFA (or enough to cover spheres) and incubate at 4 °C for 30 min.
- Aspirate the PFA and gently wash twice with PBS. Add 20% sucrose solution and incubate at 4 °C for several hours or overnight. Carefully aspirate, add 30% sucrose solution, and incubate at 4 °C for several hours or overnight. NOTE: Samples may be stored at 4 °C in either PBS or sucrose for up to 1 week before proceeding to the next step. If not slicing, maintain as 3D spheres (Figure 4A-C) in a 1.5 mL tube and proceed to step 5.3.
- If slicing the spheres, carefully transfer spheres to an embedding cryogenic mold on top of dry ice. Aspirate the 30% sucrose solution and slowly pour tissue embedding solution into the mold until it fully covers the sample, avoiding air bubbles. Wait until solution freezes and store at -80 °C until cryostat slicing.
- Using a cryostat at -20 °C, slice the embedded blocks into 30 µm sections (or as desired) and transfer to glass slides. Store at -80 °C until ready to stain.
- Immunostain the spheres.
- Outline the edges of the slides with a hydrophobic PAP pen to prevent liquid spillage. Prepare primary and secondary blocking solutions with antibodies (see the Table of Materials). Add 500 µL of primary blocking buffer to each slide or tube and incubate at room temperature for 30 min.
- Remove the liquid, add 500 µL of fresh primary blocking buffer with diluted primary antibodies to each slide or tube, and incubate overnight at 4 °C. Remove primary antibody solution and wash 3x with PBS for 10 min each.
- Add 500 µL of secondary blocking buffer with diluted secondary antibodies to each slide or tube and incubate at 4 °C for 1 h. Remove secondary antibody solution and wash 3x with PBS for 10 min each. NOTE: A list of recommended antibodies is provided in the Table of Materials. Other antibodies or dyes may be used as desired. Do not expose fluorescent samples to light (step 5.3.3 onward).
- Add 50 µL of mounting solution dropwise to cover the surface and carefully place a coverslip over the slide, avoiding bubbles. Allow the slides to dry at room temperature for 24 h and store at 4 °C.
- Image the slides.
- Use a confocal or two-photon fluorescent microscope with a 63X oil immersion objective to image pre- and post-synaptic protein abundance and colocalization within sphere slices (Figure 4D). Use a 20X or 40X objective to visualize morphological features of the hAstros.
- Open fluorescence image files with ImageJ software. Count pre- and post- synaptic puncta manually using the Cell Counter plug-in, using an unbiased counting method as previously detailed38, or with alternative methods.
Representative Results
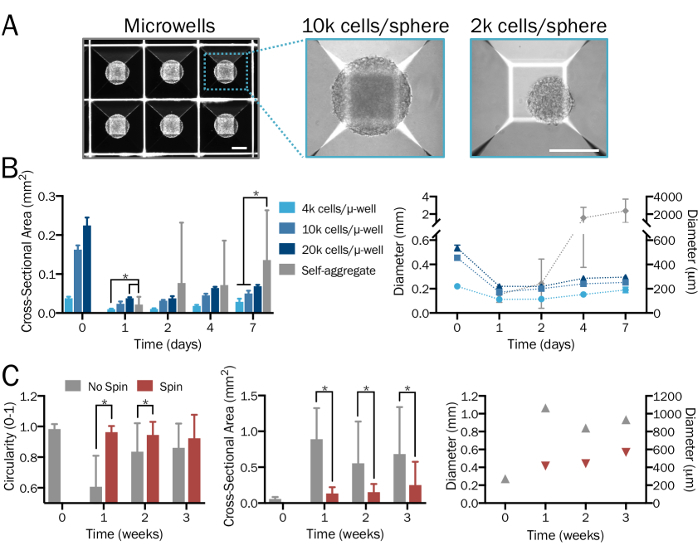
When performed properly, this protocol will produce defined populations of functional cocultures of astrocytes28,33,34 and neurons35 generated from hPSCs (Figure 1A-1C), as detailed previously26 and described here in steps 2.1–2.2. This stepwise procedure, with the use of microwell plates, is expected to yield 3D neural spheres of consistent size and shape (step 3.3; Figure 1D) with evenly dispersed cells and without significant signs of cell death. A range of starting cell densities, with a desired ratio of cell types, will produce spheres of varying sizes.In the absence of microwell plates, cells will combine to form significantly larger and nonuniform aggregates whose diameters (> 2 mm) surpass the limit of diffusion (Figure 2A-2B). It is anticipated that the use of a spinner flask or equivalent bioreactor will maintain uniformity among spheres and reduce fusion (step 3.4; Figure 2C). However, while spinner flasks allow for culture of spheres for weeks, their use is not necessary.
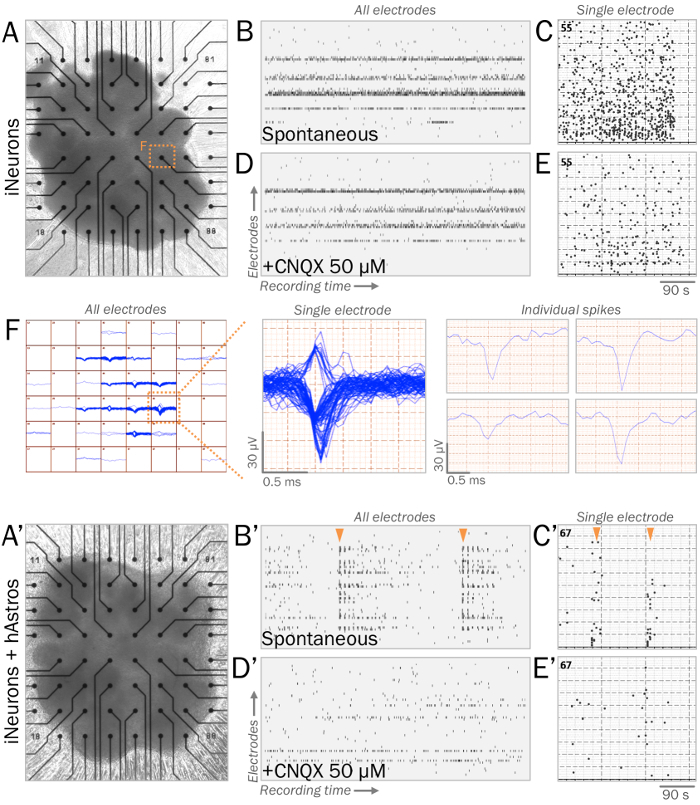
To stabilize cocultures for electrophysiological analysis, ECM-mimicking substrates allow spheres to readily adhere while maintaining their 3D structure (Figure 3A, 3A'). During recordings, healthy spheres of iNeurons placed on MEAs will spontaneously elicit voltage spikes greater than ± 40 µV with consistent firing frequencies (steps 4.1–4.2; Figure 3B-C, 3F). Coculture spheres are expected to display greater network connectivity with the presence of hAstros, resulting in an increase of synchronous network bursts of spikes (Figure 3B'-3C'). The application of CNQX35,39,40, a postsynaptic AMPA receptor antagonist, reduces network burst synchrony in coculture spheres (Figure 3D'-3E').
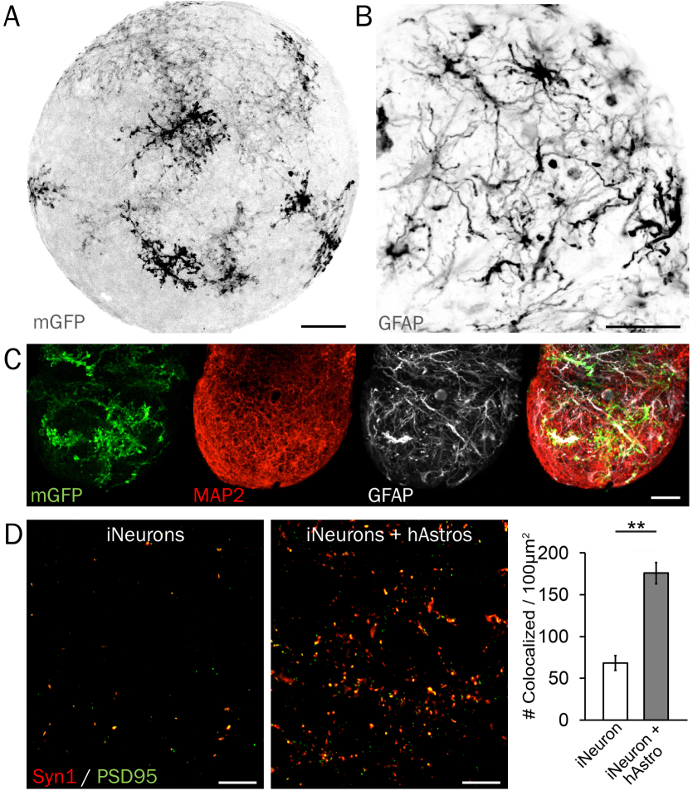
Neural cell-type restricted protein markers visually demonstrate the evenly dispersed arrangement and maturity of astrocytes and neurons within 3D spheres. A maximum projection of astrocyte morphology and branching is shown in Figure 4A in a representative 3D sphere with hAstros sparsely labeled with membrane-bound GFP. Mature hAstros express markers including GFAP (Figure 4B), S100B, and Glt134, whereas hNeurons and iNeurons express microtubule-associated protein 2 (MAP2; Figure 4C) and tubulin beta 3 (Tuj1/TUBB3). Finally, though iNeurons express pre- and post-synaptic proteins including Synapsin 1 (Syn1) and Homer or PSD95, respectively, synaptic density is significantly enhanced by the presence of hAstros in coculture (Figure 4D)26. If available, the alternate use of brain clearing techniques and light sheet microscopy will enable rapid imaging of intact spheres.
Taken together, the expression of mature neural markers along with spontaneous electrical activity confirm the success of the protocol detailed above in producing 3D cocultures of functional synaptic microcircuits.
Figure 1: Stepwise depiction of the differentiation and formation of 3D neural spheres derived from hPSCs. (A) Timeline of key steps in the protocol. (B) Pure populations of neural cells can be generated from human induced pluripotent stem cells (hPSCs). For the generation of astrocyte progenitors (dotted green box), see step 2.1 as well as Ref28. (C) Inducible neurons (iNeurons; see section 2.2) generated from transgenic hPSCs via induced overexpression of neurogenin 2 demonstrate neuronal morphology on 2D ECM (day 7) and are positive for MAP2 (inset). (D) Spheres removed from microwell plates (see steps 3.1–3.3) demonstrate consistent size for high-throughput screening. Spheres may be cultured in a spinner flask bioreactor (inset; see step 3.4) if desired to prevent fusion. Scale bar = 50 µm (C, inset). Scale bars = 200 µm (A, D). Please click here to view a larger version of this figure.
Figure 2: Systematic and reproducible generation of bioengineered 3D neural spheres. (A) Microwell (µ-well) culture plates are used to form 3D neural spheres of desired density and neuron-to-astrocyte ratios in a systematic and user-specified manner, yielding consistent size and shape. Images are shown 1 day after sphere formation. Scale bars = 200 µm. (B) A range of starting cell densities (from 4 x 103 to 2 x 104 cells per microwell) produces spheres of varying sizes. In the absence of microwell plates, hPSCs combine to form significantly larger and nonuniform aggregates whose diameters surpass the limit of diffusion after a week (n = 6-14 spheres per group and time point). (C) iNeuron spheres cultured in a spinner flask at 80 rpm exhibited less fusion and thus were significantly smaller, more uniform, and exhibited greater circularity compared to those in stationary culture over a period of three weeks (n = 6-51 spheres per group and time point). Plots represent mean ± SD; * indicates significance (p < 0.05) between groups, determined using two-tailed t-tests. Please click here to view a larger version of this figure.
Figure 3: Representative live synaptic physiology of neural spheres on multielectrode arrays (MEAs). (A, A') MEAs are utilized to measure live synaptic physiology of neural spheres. Spheres of iNeurons (A) or cocultures of iNeurons and hAstros (A') can be cultured on MEA surfaces with ECM-mimicking substrates (see step 4.1). (B, B') Raster plots of representative spontaneous electrical activity measured across all electrodes (vertical axis) during a recording window (horizontal axis). After 4 weeks of culture in neurophysiological basal medium41, iNeuron spheres (B) displayed spontaneous spikes, while coculture spheres (B') revealed increased network bursts of synchronous firing patterns (orange arrows). (C, C') Histogram of spikes measured during MEA recording windows from representative electrodes. (D, D') Raster plots of spontaneous electrical activity measured across all electrodes after the application of 50 µM CNQX, an AMPA receptor antagonist (same time scale as B, B'). CNQX eliminated the presence of synchronous bursts of spikes observed in cocultures (D'). (E, E') Histogram of spikes measured during MEA recording windows from representative electrodes after 50 µM CNQX application (same time scale as C, C'). (F) Map of spike tracings of iNeuron spheres from all electrodes over time (left). Demonstrative single electrode displaying multiple clustered traces over time (middle); individual spike traces can be sorted and analyzed individually (right). Please click here to view a larger version of this figure.
Figure 4: Characteristic immunohistochemistry of synaptic microcircuits. (A) A membrane-bound GFP (mGFP) reporter is useful for examining the arrangement and morphology of hAstros in 3D neural spheres. (B) hAstros differentiated from hPSCs are GFAP-positive. (C) Spheres containing hAstros and iNeurons can be visualized and analyzed using cell type-restricted protein markers such as GFAP and MAP2. Scale bars = 50 µm. (D) Representative images of pre- and post-synaptic densities (Syn1 and PSD95, respectively) in spheres of iNeurons without (left) and with (right) cocultured hAstros. A significantly increased density of colocalized Syn1 and PSD95 was observed in coculture spheres compared to iNeuron spheres at day 35, demonstrating the ability of hAstros to induce synapse formation (n = 3 independent replicates each; data reprinted from Ref26 with permission). Scale bars = 10 µm. Plot represents mean ± SEM; significant differences (* indicates p < 0.05; ** indicates p < 0.01) between groups were determined using two-tailed t-tests. Please click here to view a larger version of this figure.
Discussion
In this protocol, we describe a systematic method for the production of 3D spheres of neural cocultures. The spheres are composed of astrocytes and neurons, which are derived independently from hPSCs. Though not the focus of this protocol, the generation of pure populations of astrocytes from hPSCs28 is a critical step and can be technically challenging if performed without prior experience. This first step in the generation of these synaptic microcircuits should be performed with meticulous timing and attention to detail. A limitation of the use of hPSC-derived astrocytes is the lengthy differentiation process; however, the production of large quantities of cells that can be frozen for future use and thawed at the time of experimentation (see step 2.1.5) eliminates the need to begin the process from the initial hPSC stage. Though the iNeurons generated from transgenic hPSCs are synaptogenic and have the ability to exhibit spontaneous postsynaptic electrical currents, both characteristics are notably enhanced by the presence of glia12,35— including the hAstros detailed in this protocol. Thus, it should be noted that the choice of cell type, number, and developmental stage or maturity is a critical component of this protocol, and adjustments may result in variations in electrical activity or synapse visualization. However, this protocol also permits manipulation of cell density or ratios in a manner that reflects the flexibility of possible conditions or outcomes from the individual researcher.
As described above, we take advantage of bioengineering tools to produce an alternative to neural organoid methods42,43, with the caveat that this system does not recapitulate the self-organization and layering phenotypes of organoids that may be desired20,44. The simultaneous use of microwell culture plates26 and a spinner flask bioreactor45,46 ensure reproducibility, thus streamlining not only the production but also the examination and analysis of neural microcircuits with a variety of cell culture assays. It should be emphasized that although the use of a spinner flask or equivalent bioreactor is optional, a stationary culture of spheres in close contact may result in their fusing together, thus limiting nutrients and oxygen beyond the limit of diffusion in the center of spheres47. Notably, the use of 3D printed mini-bioreactors have been reported as a higher throughput approach compared to large bioreactors46.
Traditional tools to measure synaptic formation include physiological methods such whole-cell patch clamping, MEAs36,48,49, and calcium imaging50. Here, we choose to describe the use of MEAs as they provide a simple and high-throughput examination of electrical activity in the spheres on a brief time scale. However, other techniques may also be utilized.
A limitation of the use of microwell plates in this protocol is the maximum number of spheres (~300) and cells per sphere (~2 x 104) that are permitted in each well. Alternative sphere formation tools may be used for an increased number; however, a higher number of cells will be required at the starting point. The 24-well format was chosen here for its ability to generate larger spheres that can be handled for analysis and visible by eye, while limiting cell death within the center of the spheres. Overall, the addition of this step to the protocol will ensure a robust and scalable production of uniform 3D spheres.
Our protocol also boasts the flexibility of adjusting the number and ratio of each cell type as well as the possibility of introducing other cell types into the 3D spheres in a controlled, defined manner. The use of region-specific neuronal and astrocyte subtypes permits the study of synaptic microcircuits of different regions of the central nervous system. These coculture spheres can also be fused together51 to study cellular migration and long range signaling. The addition of oligodendrocytes, endothelial cells, or microglia could prove these 3D spheres to be an informative brain model with increased complexity, as a promising future direction for this method. Finally, in addition to disease and injury modeling, this system permits the study of therapeutic approaches, such as cellular replacement therapy or drug development, to enhance neuroregeneration.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We would like to thank Dr. Erik Ullian (UCSF) for intellectual input on the design of these procedures, Dr. Michael Ward (NIH) for technical advice on iNeuron differentiation, and Saba Barlas for preliminary image analysis.
References
- Ullian EM, Christopherson KS, Barres BA. Role for Glia in Synaptogenesis. Glia. 2004;47:209–216. doi: 10.1002/glia.20082. [DOI] [PubMed] [Google Scholar]
- Baldwin KT, Eroglu C. Molecular mechanisms of astrocyte-induced synaptogenesis. Current Opinion in Neurobiology. 2017;45:113–120. doi: 10.1016/j.conb.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AV, et al. Astrocyte-encoded positional cues maintain sensorimotor circuit integrity. Nature. 2014;509(7499):189–194. doi: 10.1038/nature13161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan S, et al. Synaptic Integration of Adult-Born Hippocampal Neurons Is Locally Controlled by Astrocytes. Neuron. 2015;88:957–972. doi: 10.1016/j.neuron.2015.10.037. [DOI] [PubMed] [Google Scholar]
- Clarke LE, Barres BA. Emerging roles of astrocytes in neural circuit development. Nat Rev Neuroscience. 2013;14(5):311–321. doi: 10.1038/nrn3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung G, Sibille J, Zapata J, Rouach N. Activity-Dependent Plasticity of Astroglial Potassium and Glutamate Clearance. Neural Plasticity. 2015. p. 109106. [DOI] [PMC free article] [PubMed]
- Ghezali G, Dallerac G, Rouach N. Perisynaptic astroglial processes dynamic processors of neuronal information. Brain Struct Funct. 2016;221:2427–2442. doi: 10.1007/s00429-015-1070-3. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Nedergaard M. Functions of Astrocytes and their Potential As Therapeutic Targets. Neurotherapeutics. 2010;7:338–353. doi: 10.1016/j.nurt.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pál B. Astrocytic Actions on Extrasynaptic Neuronal Currents. Frontiers in Cellular Neuroscience. 2015;9:474. doi: 10.3389/fncel.2015.00474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiray H, Lindsay SL, Hosseinzadeh S, Barnett SC. The multifaceted role of astrocytes in regulating myelination. Experimental Neurology. 2016;283:541–549. doi: 10.1016/j.expneurol.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen NJ, Eroglu C. Cell Biology of Astrocyte-Synapse Interactions. Neuron. 2017;96(3):697–708. doi: 10.1016/j.neuron.2017.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krencik R, van Asperen JV, Ullian EM. Human astrocytes are distinct contributors to the complexity of synaptic function. Brain Research Bulletin. 2017;129:66–73. doi: 10.1016/j.brainresbull.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of Synapse Number by Glia. Science. 2001;291:657–662. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- Oberheim Bush NA, Nedergaard M. Do Evolutionary Changes in Astrocytes Contribute to the Computational Power of the Hominid Brain? Neurochemical Research. 2017;42(9):2577–2587. doi: 10.1007/s11064-017-2363-0. [DOI] [PubMed] [Google Scholar]
- Han X, et al. Forebrain Engraftment by Human Glial Progenitor Cells Enhances Synaptic Plasticity and Learning in Adult Mice. Cell Stem Cell. 2013;12(3):342–353. doi: 10.1016/j.stem.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Nagata N, Kurokawa H, Yamanaka S. iPS cells: a game changer for future medicine. The EMBO Journal. 2014;33(5):409–417. doi: 10.1002/embj.201387098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology a decade of progress. Nature Reviews Drug Discovery. 2017;16(2):115–130. doi: 10.1038/nrd.2016.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodla MC, Mumaw J, Stice SL. Role of astrocytes, soluble factors, cells adhesion molecules and neurotrophins in functional synapse formation: implications for human embryonic stem cell derived neurons. Stem Cell Res Ther. 2010. pp. 251–260. [DOI] [PubMed]
- Krencik R, Ullian EM. A cellular star atlas: using astrocytes from human pluripotent stem cells for disease studies. Frontiers in Cellular Neuroscience. 2013;7:1–10. doi: 10.3389/fncel.2013.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca SP. The rise of three-dimensional human brain cultures. Nature. 2018;553(7689):437–445. doi: 10.1038/nature25032. [DOI] [PubMed] [Google Scholar]
- Huch M, Knoblich JA, Lutolf MP, Martinez-arias A. The hope and the hype of organoid research. Development. 2017;144:938–941. doi: 10.1242/dev.150201. [DOI] [PubMed] [Google Scholar]
- Mason JO, Price DJ. Building Brains in a Dish: Prospects for Growing Cerebral Organoids from Stem Cells. Neuroscience. 2016;334:105–118. doi: 10.1016/j.neuroscience.2016.07.048. [DOI] [PubMed] [Google Scholar]
- Kelava I, Lancaster MA. Dishing out mini-brains: Current progress and future prospects in brain organoid research. Developmental Biology. 2016;420(2):199–209. doi: 10.1016/j.ydbio.2016.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelava I, Lancaster MA. Stem Cell Models of Human Brain Development. Cell Stem Cell. 2016;18(6):736–748. doi: 10.1016/j.stem.2016.05.022. [DOI] [PubMed] [Google Scholar]
- Sloan SA, et al. Human Astrocyte Maturation Captured in 3D Cerebral Cortical Spheroids Derived from Pluripotent Stem Cells. Neuron. 2017. pp. 779–790. [DOI] [PMC free article] [PubMed]
- Krencik R, et al. Systematic three-dimensional coculture rapidly recapitulates interactions between human neurons and astrocytes. Stem Cell Reports. 2017;9(6):1745–1753. doi: 10.1016/j.stemcr.2017.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, et al. Chemically defined conditions for human iPSC derivation and culture. Nature Methods. 2011;8(5):424–429. doi: 10.1038/nmeth.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krencik R, Zhang S-C. Directed differentiation of functional astroglial subtypes from human pluripotent stem cells. Nature Protocols. 2011;6(11):1710–1717. doi: 10.1038/nprot.2011.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z-W, et al. Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nature Communications. 2015;6:6626. doi: 10.1038/ncomms7626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely MD, et al. DMH1, a highly selective small molecule BMP inhibitor promotes neurogenesis of hiPSCs: Comparison of PAX6 and SOX1 expression during neural induction. ACS Chemical Neuroscience. 2012;3(6):482–491. doi: 10.1021/cn300029t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann ES, Estevez-Silva MC, Ashton RS. Defined Human Pluripotent Stem Cell Culture Enables Highly Efficient Neuroepithelium Derivation Without Small Molecule Inhibitors. Stem Cells. 2014;32:1032–1042. doi: 10.1002/stem.1622. [DOI] [PubMed] [Google Scholar]
- Eggan K, Kawada J, Kaneda S, Kirihara T, Maroof A. Generation of a Motor Nerve Organoid with Human Stem Cell-Derived Neurons. Stem Cell Reports. 2017;9:1441–1449. doi: 10.1016/j.stemcr.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krencik R, Weick JP, Liu Y, Zhang Z-J, Zhang S-C. Specification of transplantable astroglial subtypes from human pluripotent stem cells. Nature Biotechnology. 2011;29(6):528–534. doi: 10.1038/nbt.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krencik R, et al. Dysregulation of astrocyte extracellular signaling in Costello syndrome. Science Translational Medicine. 2015;7(286):286. doi: 10.1126/scitranslmed.aaa5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, et al. Scalable Production of iPSC-Derived Human Neurons to Identify Tau- Lowering Compounds by High-Content Screening. Stem Cell Reports. 2017;9(4):1221–1233. doi: 10.1016/j.stemcr.2017.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin H, Maccione A, Marinaro F, Zordan S, Nieus T, Berdondini L. Electrical Responses and Spontaneous Activity of Human iPS-Derived Neuronal Networks Characterized for 3-month Culture with 4096-Electrode Arrays. Frontiers in Neuroscience. 2016;10 doi: 10.3389/fnins.2016.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapucu FE, Mäkinen ME, Tanskanen JMA, Ylä-Outinen L, Narkilahti S, Hyttinen JAK. Joint analysis of extracellular spike waveforms and neuronal network bursts. Journal of Neuroscience Methods. 2016;259:143–155. doi: 10.1016/j.jneumeth.2015.11.022. [DOI] [PubMed] [Google Scholar]
- Ippolito DM, Eroglu C. Quantifying Synapses: an Immunocytochemistry-based Assay to Quantify Synapse Number. Journal of Visualized Experiments. 2010;45:2–9. doi: 10.3791/2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78(5):785–798. doi: 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odawara A, Katoh H, Matsuda N, Suzuki I. Physiological maturation and drug responses of human induced pluripotent stem cell-derived cortical neuronal networks in long-term culture. Scientific reports. 2016;6:26181. doi: 10.1038/srep26181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardy C, Hurk , et al. Neuronal medium that supports basic synaptic functions and activity of human neurons in vitro. PNAS. 2015;112(25):E2725–E2734. doi: 10.1073/pnas.1504393112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monzel AS, et al. Derivation of Human Midbrain-Specific Organoids from Neuroepithelial Stem Cells. Stem Cell Reports. 2017;8:1144–1154. doi: 10.1016/j.stemcr.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nature Protocols. 2014;9(10):2329–2340. doi: 10.1038/nprot.2014.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, Heo I, Clevers H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends in Molecular Medicine. 2018;23(5):393–410. doi: 10.1016/j.molmed.2017.02.007. [DOI] [PubMed] [Google Scholar]
- Lancaster MA, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501(7647):373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell. 2016;165(5):1238–1254. doi: 10.1016/j.cell.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, et al. Derivation of Cortical Spheroids from Human Induced Pluripotent Stem Cells in a Suspension Bioreactor. Tissue Engineering Part A. 2016. pp. 1–46. [DOI] [PubMed]
- Obien MEJ, Deligkaris K, Bullmann T, Bakkum DJ, Frey U. Revealing neuronal function through microelectrode array recordings. Frontiers in Neuroscience. 2015;9(JAN):423. doi: 10.3389/fnins.2014.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales CM, Rolston JD, Potter SM. How to Culture, Record and Stimulate Neuronal Networks on Micro-electrode Arrays (MEAs) Journal of Visualized Experiments. 2010. pp. 1–7. [DOI] [PMC free article] [PubMed]
- Shigetomi E, Patel S, Khakh BS. Probing the Complexities of Astrocyte Calcium Signaling. Trends in Cell Biology. 2016;26(4):300–312. doi: 10.1016/j.tcb.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagley JA, Reumann D, Bian S, Lévi-strauss J, Knoblich JA. Fused cerebral organoids model interactions between brain regions. Nat Methods. 2017;14(7) doi: 10.1038/nmeth.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]