

Abstract
Selectivity of kinase inhibitors, or the lack thereof, continues to be an intensely debated topic in drug discovery research. Especially, type I inhibitors, which represent most of the currently available kinase inhibitors, are often thought to lack selectivity because they target the largely conserved adenosine triphosphate-binding site in kinases. Herein, we present a large-scale analysis of potential selectivity among multikinase inhibitors, covering 141 human kinases and more than 10 000 qualifying compounds. By design, the analysis was focused on type I inhibitors and carried out at the level of systematically generated kinase pairs sharing inhibitors. Kinase pair category- and compound-based selectivity profiles identified in part highly selective inhibitors for many kinases. Sets of inhibitors associated with kinase pairs frequently contained nonselective as well as increasingly selective compounds. Selectivity of inhibitors did not result from gatekeeper residues settings or phylogenetic distance of kinases. Rather, it was most likely attributable to subtle differences between binding regions in kinases. Taken together, the results of our study reveal that many multikinase inhibitors are more selective than one might assume.
1. Introduction
Inhibitors of human kinases are among the most intensely investigated compounds in drug development.1−5 Most currently available kinase inhibitors target the adenosine triphosphate (ATP) (cofactor)-binding site that is largely conserved across the human kinome.6,7 Accordingly, ATP-site-directed kinase inhibitors are expected to be promiscuous and lack selectivity, as indicated by a number of kinase inhibitor profiling studies.8−11 Therefore, attempts have been made to discover other types of inhibitors that target different regions in kinases and act by different mechanisms.12,13 ATP-site-directed (type I) inhibitors bind to the so-called “DFG-in” conformation of the activation loop near the catalytic site, i.e., the active form of the kinase. In addition, type II inhibitors bind to the inactive “DFG-out” conformation of the activation segment, occupying pockets adjacent to the ATP-binding site that are less conserved.13 Thus, type II inhibitors are expected to be more selective than type I inhibitors. Furthermore, there are type III and IV inhibitors that bind to regions outside the ATP-binding site and act by allosteric mechanisms.13 Only a limited number of allosteric kinase inhibitors has been reported thus far, but these types of inhibitors might indeed be most selective.14−16
However, the often assumed lack of selectivity of type I inhibitors continues to be debated17 and expected selectivity differences between type I and II inhibitors are subject to further investigation. For example, profiling experiments using type II inhibitors have shown that these inhibitors are often active against many kinases.13 Furthermore, although subsets of highly promiscuous type I inhibitors have been identified18 and promiscuity of kinase inhibitors has become a hallmark for successful cancer treatment,2 there is also evidence for selectivity of ATP-site-directed inhibitors. For example, although a number of kinase inhibitor profiling experiments have indicated a lack of selectivity of type I inhibitors,8−11 others have revealed selectivity patterns.19,20 In addition, type I inhibitors are also capable of acting by different mechanisms.21 Furthermore, on the basis of high-confidence activity data, 76% of publicly available kinase inhibitors were found to be annotated with a single kinase.22 When activity data confidence criteria were iteratively lowered, no notable increase in kinase inhibitor promiscuity was detected,23 suggesting that promiscuity was not a general rule. Of course, it has long been known that the ATP-binding site in kinases has some sequence variation, in particular, at the “gatekeeper” position,7 where the presence of smaller or larger residues differentiates between classes of type I inhibitors. However, whether or not the gatekeeper is the only factor responsible for inhibitor differentiation within the ATP-binding site is currently unknown. Other subtle differences might also play a role. Clearly, the issue of kinase inhibitor selectivity is still not fully explored.
Herein, we present a systematic analysis of selectivity among multikinase inhibitors on the basis of currently available activity data. Selectivity profiles were generated for sets of inhibitors shared by kinases. The profiles revealed significant potency variations of subsets of inhibitors and identified compounds with selectivity for given kinases over others.
2. Materials and Methods
2.1. Compounds, Targets, and Activity Data
Inhibitors of human protein kinases were assembled from ChEMBL version 23.24 Compounds with activity in assays detecting direct interactions (target relationship type “D”) with human protein kinases at the highest confidence level (confidence score 9) were selected. As potency measurements, IC50 values were considered. The amount of available Ki values was too small for a meaningful statistical analysis. If multiple IC50 values were available for a compound, the final potency annotation was calculated as the geometric mean of these values, provided all fell within the same order of magnitude (otherwise, the compound was disregarded). Approximate measurements associated with “>”, “<”, or “∼” were not taken into account. On the basis of these criteria, 40 627 inhibitors with activity against 274 human kinases were obtained. From this compound pool, inhibitors were selected that were active against at least two kinases, yielding a final set of 10 367 inhibitors with activity against 266 human kinases. ChEMBL target identifiers of these kinases were mapped to UniProt,25 and kinases were assigned to families and groups (of families) according to Manning et al.6 and Miranda-Saavedra et al.26
2.2. Protein Kinase Pairs
The selected multikinase inhibitors were used to systematically form compound-based target pairs. Two kinases were paired if they shared at least 10 inhibitors. Given this constraint, a total of 596 pairs were obtained that included 141 kinases and 10 060 inhibitors. Kinase pairs were assigned to three different categories: same family, i.e., both kinases belonged to the same family (132 pairs); different families, i.e., both kinases belonged to different families within the same kinase group (262 pairs); and different groups, i.e., both kinases belonged to different groups (202 pairs). Kinases in pairs from the same family, different families, and different groups were increasingly distant (unrelated). For each pair, compound selectivity was assessed by calculating the logarithmic potency difference (ΔpIC50) for each inhibitor.
2.3. Gatekeeper Residue and Binding-Site Comparison
The kinase–ligand interaction fingerprints and structures (KLIFS)27,28 database defines a kinase “binding pocket” for type I–IV inhibitors as a set of 85 discontinuous residues. This sequence segment, which contains the gatekeeper residue at position 45, can be extracted for human kinases from KLIFS on the basis of UniProt identifiers using the 3D-e-Chem-VM engine.29 For kinase pairs, gatekeeper residues were compared and sequence identity over the 85-residue segment was calculated as an indicator of binding-site resemblance. Phylogenetic trees of the human kinome were drawn with Kinome Render.30
3. Results and Discussion
3.1. Qualifying Kinase Inhibitors
Figure 1 shows the distribution of inhibitors over all 596 pairs of kinases sharing at least 10 compounds, yielding a median value of 18 inhibitors per pair. Hence, kinase pairs were associated with sufficient numbers of inhibitors for a systematic assessment of selectivity profiles. The pairs involved 141 kinases distributed across the human kinome and 10 060 multikinase inhibitors from ChEMBL.
Figure 1.
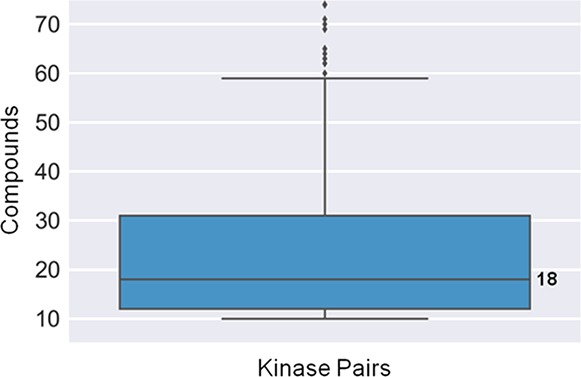
Distribution of compounds over kinase pairs. The boxplot reports the distribution of inhibitors over kinase pairs, yielding a median value of 18 inhibitors per pair. Boxplots report the smallest value (bottom line), first quartile (lower boundary of the box), median value (thick line), third quartile (upper boundary of the box), largest value (top line), and outliers (points below the smallest or above the largest value).
Mapping of type II kinase inhibitor signature fragments13 indicated that less than 1% of kinase inhibitors available in ChEMBL were type II inhibitors.18 Thus, although it is not exactly known how many type II, or rare type III/IV, inhibitors are currently available in ChEMBL, for all practical considerations, our analysis was focused on type I multikinase inhibitors.
3.2. Global Selectivity
Potency differences of inhibitors against kinases forming pairs were calculated as a measure of selectivity. The larger the potency difference was, the more selective an inhibitor was for one kinase over the other. Initially, the global potency difference distribution was determined. Figure 2 (left) shows that average potency differences for all inhibitors associated with a pair were rather small, with a median ΔpIC50 value of 0.64 (i.e., well within 1 order of magnitude). At a first glance, this was what one might expect for largely nonselective inhibitors. However, the picture changed when only the inhibitor with largest potency difference from each pair was considered, as also shown in Figure 2 (right). In this case, the distribution yielded a median ΔpIC50 of 2.37, a difference of more than 2 orders of magnitude (100-fold), and a third quartile difference of 3 orders of magnitude. Thus, for individual inhibitors, a global tendency of selectivity emerged. Systematically enumerating pairs of kinases sharing inhibitors ensured that all possible selectivity relationships were taken into account. The union of pairwise relationships was expected to reveal general selectivity trends, if they existed.
Figure 2.

Compound potency differences for kinase pairs. Boxplots report the distribution of potency differences of inhibitors for paired kinases as the mean potency difference of all inhibitors (left) or the largest potency difference (most selective compounds; right). The distributions yield ΔpIC50 median values of 0.64 (left) and 2.37 (right).
The global selectivity tendency was also observed at the level of different kinase pair categories. Figure 3a shows the distribution of potency differences for the three pair categories in different formats. In all three cases, the median difference for all compounds fell within the same order of magnitude and exceeded 2 orders of magnitude for the most selective compounds.
Figure 3.

Compound potency differences for pair categories. (a) Distributions of ΔpIC50 values (left) for all versus the most selective inhibitors according to Figure 2 for kinase pairs from the same family (blue, 132 pairs), different families (green, 262 pairs), and different groups (red, 202 pairs). In addition, a comparison of ΔpIC50 median values is shown (right). (b) Selectivity profiles for the three pair categories that record the potency differences of the most selective inhibitor for each pair (in the order of increasing potency differences from left to right).
3.3. Pair Category-Based Selectivity Profiles
The global selectivity tendency was further corroborated by pair category-based selectivity profiles shown in Figure 3b. These profiles were generated by recording the largest inhibitor potency difference for each pair and ordering the pairs by increasing ΔpIC50 values. In each case, more than half of the kinase pairs had one or more inhibitors with a potency difference exceeding 2 orders of magnitude. Furthermore, in each case, potency differences exceeding 4 or even 5 orders of magnitude were observed for multiple pairs. For kinases from different groups, 55% of the pairs had inhibitor(s) with potency differences of more than 2 orders of magnitude and 22% of more than 3 orders of magnitude.
3.4. Compound-Based Selectivity Profiles
Detailed views of inhibitor selectivity were provided by compound-based selectivity profiles. Figure 4 (left) shows exemplary profiles for kinase pairs from the same family, different families, and different groups. Kinases from each pair had the same gatekeeper residue. In these profiles, potency values of all inhibitors are compared for kinases of a pair and inhibitors are ordered according to increasing potency differences. In addition, Figure 4 shows the least and most selective inhibitor for each pair (middle) and the location of paired kinases on a phylogenetic tree representing the human kinome (right). For each pair, the most selective inhibitor displayed a potency difference of more than 4 or 5 orders of magnitude.
Figure 4.

Compound-based selectivity profiles. Left: exemplary compound selectivity profiles for kinase pairs belonging to different categories. For each inhibitor, the potency against the two kinases is compared. From the left to the right, inhibitors are ordered according to increasing potency differences. On the lower right of each graph, gatekeeper residues of the kinase pair are reported (e.g., “M|M”). Middle: comparison of the least and most selective inhibitors for each pair. Right: kinases forming each pair are mapped onto a phylogenetic tree of the human kinome to illustrate their category relationships.
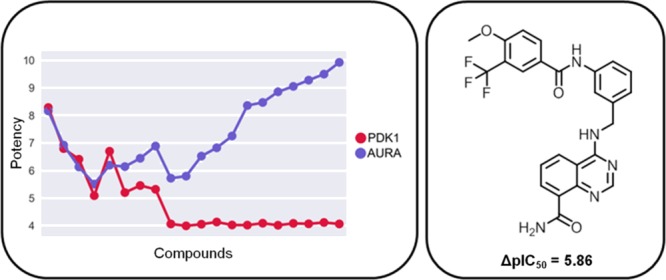
The selectivity profiles revealed in part striking differences in relative potencies between inhibitors. Compounds shared by the closely related protein kinase C eta type (PKCh) and protein kinase C theta type (PKCt) were generally slightly more potent against PKCh, preserving relative potency differences. However, two notable exceptions were detected, where potency against PKCh decreased sharply. In one of these cases, the inhibitor was essentially inactive against PKCh but retained high potency against PKCt, resulting in high selectivity for PKCt. The profile for macrophage colony-stimulating factor 1 receptor kinase (FMS) and tyrosine-protein kinase Lck (LCK) contained six inhibitors with comparable potency and four others with increasing potency differences and selectivity for FMS over LCK. Moreover, for the distantly related 3-phosphoinositide-dependent protein kinase 1 (PDK1) and aurora kinase A (AurA), there were five inhibitors with the same potency against both kinases, three with relatively small potency differences, and 12 others that were essentially inactive against PDK1 but increasingly potent against AurA, yielding a subset of selective AurA inhibitors. The most selective compound had a potency difference of nearly 6 orders of magnitude. Many other profiles revealing similar selectivity relationships were obtained. Thus, many inhibitors shared by pairs of 141 human kinases were highly selective, a rather unexpected finding.
3.5. Comparison of Gatekeeper Residues, Binding Regions, and Compound Selectivity
In light of these findings, we further investigated whether there might be straightforward explanations for the observed selectivity trends. Therefore, for all kinase pairs, combinations of gatekeeper residues were determined. For each gatekeeper combination, the number of pairs associated with inhibitor(s) having a ΔpIC50 of at least 2 orders of magnitude (selectivity criterion) was identified and compared to the number of pairs not meeting this selectivity criterion. The results are shown in Figure 5a. For most gatekeeper combinations, including conserved and different residues, more pairs with selective than nonselective inhibitors were available. Hence, conservation of gatekeeper residues did not preclude compound selectivity, as also illustrated in Figure 4, and for all gatekeeper combinations represented by multiple kinase pairs, selective inhibitors were available.
Figure 5.

Gatekeeper residues, binding pocket similarity, and compound selectivity. (a) Histograms compare the number of kinase target pairs for each observed combination of gatekeeper residues (top, conserved residues; bottom, different residues), for which one or more selective (red) or no selective (gray) inhibitors were available. As a selectivity criterion, a potency difference of at least 2 orders of magnitude (ΔpIC50 ≥ 2) was applied. (b) Swarm plot (i.e., a boxplot in which all individual data points are displayed) capturing distributions of binding pocket similarity (sequence identity over the 85-residue segment) of kinases in pairs belonging to different categories to the presence (red) or absence (gray) of selective inhibitors. Individual data points on the X-axis are centered on the not displayed boxplot whisker for each category and depart from the central position if additional points have the same binding pocket similarity value. The percentage of kinase pairs with selective inhibitors (“selective pairs”) is given for each category. (c) Distribution of compounds over kinase pairs in different categories. In addition, the proportion of selective inhibitors is given.
Furthermore, binding pocket similarity was calculated for all kinase pairs with selective inhibitors and others, as shown in Figure 5b. As expected, the similarity of binding regions decreased with increasing phylogenetic distances of paired kinases. However, pairs with selective and nonselective inhibitors were widely distributed over the entire similarity range, including all three pair categories. Hence, there was no detectable correlation between similarities of binding regions and the presence or absence of selective inhibitors. As shown in Figure 5b, even kinases with highly similar binding regions shared inhibitors that were selective. In addition, for each category, the percentage of kinase pairs for which selective inhibitors were available is provided. More than half of the kinase pairs in each category had selective inhibitors. However, there was no detectable correlation between the frequency of pairs with selected inhibitors and phylogenetic distance. Taken together, these findings indicated that rather subtle structural and/or property differences between kinases were largely responsible for the selectivity of shared inhibitors.
Figure 5c shows the distribution of shared inhibitors over kinase pairs from different categories. The number of shared inhibitors decreased with increasing phylogenetic distance between kinases in pairs. For each category, the proportion of selective unique inhibitors was also calculated. As expected, the percentage of selective inhibitors increased with increasing phylogenetic distance, as also shown in Figure 5c.
4. Conclusions
In this study, we have analyzed potential selectivity of multikinase inhibitors on a large scale based on currently available compound activity data. Previous studies have focused on kinase inhibitor selectivity profiling to identify new chemical probes for orphan receptors or compounds active against still little explored therapeutically relevant kinases.31,32 Our analysis was facilitated by systematically generating pairs of 141 qualifying human kinases with increasing phylogenetic distances that shared 10 or more inhibitors, providing a new reference frame for selectivity analysis. Contrary to our initial expectations, pair category- and compound-based selectivity profiles introduced herein revealed the presence of subsets of in part highly selective inhibitors for the majority of kinase pairs, providing extensive kinase coverage. Because the analysis was based on a statistically significant sample of more than 10 000 multikinase inhibitors, the detected selectivity trends were sound. Some striking observations were made at the level of compound-based selectivity profiles. In many instances, sets of inhibitors associated with kinase pairs contained subsets of nonselective compounds and others that were increasingly selective. These observations were of particular interest because the analysis was intrinsically focused on type I kinase inhibitors, which are often (but not always) thought to lack selectivity. We have also shown that observed inhibitor selectivity was not attributable to well-known kinase features, such as gatekeeper constellations or phylogenetic distances. It follows that selectivity determinants in kinases are likely to result from subtle differences that are far from being obvious, which should provide ample opportunities for future research. Clearly, although much progress has been made in recent years in rationalizing kinase inhibition and underlying mechanisms of actions, especially at the structural level, the jury on kinase inhibitor selectivity and its possible molecular origins is still out there. To support further exploration of kinase inhibitor selectivity, our kinase pair and inhibitor data set is made freely available as an open access deposition.33
Acknowledgments
The authors thank A. Kooistra for additional information about 3D-e-Chem-VM, R. Kunimoto for support with kinome representations, and M. Vogt for helpful discussions.
Author Contributions
The study was carried out and the manuscript was written with contributions of all authors. All authors have approved the final version of the manuscript.
The authors declare no competing financial interest.
References
- Cohen P. Protein Kinases-the Major Drug Targets of the Twenty-First Century?. Nat. Rev. Drug Discovery 2002, 1, 309–315. 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- Knight Z. A.; Lin H.; Shokat K. M. Targeting the Cancer Kinome through Polypharmacology. Nat. Rev. Cancer 2010, 10, 130–137. 10.1038/nrc2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinase Drug Discovery; Ward R. A., Goldberg F. W., Eds.; RSC: Cambridge, U.K., 2011. [Google Scholar]
- Simmons D. L. Targeting Kinases: A New Approach to Treating Inflammatory Rheumatic Diseases. Curr. Opin. Pharmacol. 2013, 13, 426–434. 10.1016/j.coph.2013.02.008. [DOI] [PubMed] [Google Scholar]
- Laufer S.; Bajorath J. New Frontiers in Kinases: Second Generation Inhibitors. J. Med. Chem. 2014, 57, 2167–2168. 10.1021/jm500195x. [DOI] [PubMed] [Google Scholar]
- Manning G.; Whyte D. B.; Martinez R.; Hunter T.; Sudarsanam S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Noble M. E.; Endicott J. A.; Johnson L. N. Protein Kinase Inhibitors: Insights into Drug Design from Structure. Science 2004, 303, 1800–1805. 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- Fabian M. A.; Biggs W. H. 3rd; Treiber D. K.; Atteridge C. E.; Azimioara M. D.; Benedetti M. G.; Carter T. A.; Ciceri P.; Edeen P. T.; Floyd M.; Ford J. M.; Galvin M.; Gerlach J. L.; Grotzfeld R. M.; Herrgard S.; Insko D. E.; Insko M. A.; Lai A. G.; Lélias J. M.; Mehta S. A.; Milanov Z. V.; Velasco A. M.; Wodicka L. M.; Patel H. K.; Zarrinkar P. P.; Lockhart D. J. A Small Molecule-Kinase Interaction Map for Clinical Kinase Inhibitors. Nat. Biotechnol. 2005, 23, 329–336. 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. A Quantitative Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2008, 26, 127–132. 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Cheng A. C.; John Eksterowicz J.; Geuns-Meyer S.; Sun Y. Analysis of Kinase Inhibitor Selectivity Using a Thermodynamics-Based Partition Index. J. Med. Chem. 2010, 53, 4502–4510. 10.1021/jm100301x. [DOI] [PubMed] [Google Scholar]
- Metz J. T.; Johnson E. F.; Soni N. B.; Merta P. J.; Kifle L.; Hajduk P. J. Navigating the Kinome. Nat. Chem. Biol. 2011, 7, 200–202. 10.1038/nchembio.530. [DOI] [PubMed] [Google Scholar]
- Gavrin L. K.; Saiah E. Approaches to Discover Non-ATP Site Kinase Inhibitors. Med. Chem. Commun. 2013, 4, 41–51. 10.1039/C2MD20180A. [DOI] [Google Scholar]
- Zhao Z.; Wu H.; Wang L.; Liu Y.; Knapp S.; Liu Q.; Gray N. S. Exploration of Type II Binding Mode: A Privileged Approach for Kinase Inhibitor Focused Drug Discovery?. ACS Chem. Biol. 2014, 9, 1230–1241. 10.1021/cb500129t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohren J. F.; Chen H.; Pavlovsky A.; Whitehead C.; Zhang E.; Kuffa P.; Yan C.; McConnell P.; Spessard C.; Banotai C.; Mueller W. T.; Delaney A.; Omer C.; Sebolt-Leopold J.; Dudley D. T.; Leung I. K.; Flamme C.; Warmus J.; Kaufman M.; Barrett S.; Tecle H.; Hasemann C. A. Structures of Human MAP Kinase Kinase 1 (MEK1) and MEK2 Describe Novel Noncompetitive Kinase Inhibition. Nat. Struct. Mol. Biol. 2004, 11, 1192–1197. 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]
- Adrián F. J.; Ding Q.; Sim T.; Velentza A.; Sloan C.; Liu Y.; Zhang G.; Hur W.; Ding S.; Manley P.; Mestan J.; Fabbro D.; Gray N. S. Allosteric Inhibitors of Bcr-Abl-Dependent Cell Proliferation. Nat. Chem. Biol. 2006, 2, 95–102. 10.1038/nchembio760. [DOI] [PubMed] [Google Scholar]
- Ashwell M. A.; Lapierre J. M.; Brassard C.; Bresciano K.; Bull C.; Cornell-Kennon S.; Eathiraj S.; France D. S.; Hall T.; Hill J.; Kelleher E.; Khanapurkar S.; Kizer D.; Koerner S.; Link J.; Liu Y.; Makhija S.; Moussa M.; Namdev N.; Nguyen K.; Nicewonger R.; Palma R.; Szwaya J.; Tandon M.; Uppalapati U.; Vensel D.; Volak L. P.; Volckova E.; Westlund N.; Wu H.; Yang R. Y.; Chan T. C. Discovery and Optimization of a Series of 3-(3-Phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amines: Orally Bioavailable, Selective, and Potent ATP-Independent Akt Inhibitors. J. Med. Chem. 2012, 55, 5291–5310. 10.1021/jm300276x. [DOI] [PubMed] [Google Scholar]
- Levitzki A. Tyrosine Kinase Inhibitors: Views of Selectivity, Sensitivity, and Clinical Performance. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 161–185. 10.1146/annurev-pharmtox-011112-140341. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Furtmann N.; Bajorath J. Current Compound Coverage of the Kinome. J. Med. Chem. 2015, 58, 30–40. 10.1021/jm5008159. [DOI] [PubMed] [Google Scholar]
- Anastassiadis T.; Deacon S. W.; Devarajan K.; Ma H.; Peterson J. R. Comprehensive Assay of Kinase Catalytic Activity Reveals Features of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2011, 29, 1039–1045. 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. Comprehensive Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Müller S.; Chaikuad A.; Gray N. S.; Knapp S. The Ins and Outs of Selective Kinase Inhibitor Development. Nat. Chem. Biol. 2015, 11, 818–821. 10.1038/nchembio.1938. [DOI] [PubMed] [Google Scholar]
- Dimova D.; Bajorath J. Assessing Scaffold Diversity of Kinase Inhibitors Using Alternative Scaffold Concepts and Estimating the Scaffold Hopping Potential for Different Kinases. Molecules 2017, 22, 730 10.3390/molecules22050730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpfe D.; Tinivella A.; Rastelli G.; Bajorath J. Promiscuity of Inhibitors of Human Protein Kinases at Varying Data Confidence Levels and Test Frequencies. RSC Adv. 2017, 7, 41265–41271. 10.1039/C7RA07167A. [DOI] [Google Scholar]
- Gaulton A.; Bellis L. J.; Bento A. P.; Chambers J.; Davies M.; Hersey A.; Light Y.; McGlinchey S.; Michalovich D.; Al-Lazikani B.; Overington J. P. ChEMBL: A Large-Scale Bioactivity Database for Drug Discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt: A Hub for Protein Information. Nucleic Acids Res. 2015, 43, D204–D212. 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda-Saavedra D.; Barton G. J. Classification and Functional Annotation of Eukaryotic Protein Kinases. Proteins 2007, 68, 893–914. 10.1002/prot.21444. [DOI] [PubMed] [Google Scholar]
- van Linden O. P. J.; Kooistra A. J.; Leurs R.; de Esch I. J. P.; de Graaf C. KLIFS: A Knowledge-Based Structural Database To Navigate Kinase-Ligand Interaction Space. J. Med. Chem. 2014, 57, 249–277. 10.1021/jm400378w. [DOI] [PubMed] [Google Scholar]
- Kooistra A. J.; Kanev G. K.; van Linden O. P. J.; Leurs R.; de Esch I. J. P.; de Graaf C. KLIFS: A Structural Kinase-Ligand Interaction Database. Nucleic Acids Res. 2016, 44, D365–D371. 10.1093/nar/gkv1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire R.; Verhoeven S.; Vass M.; Vriend G.; de Esch I. J. P.; Lusher S. J.; Leurs R.; Ridder L.; Kooistra A. J.; Ritschel T.; de Graaf C. 3D-e-Chem-VM: Structural Cheminformatics Research Infrastructure in a Freely Available Virtual Machine. J. Chem. Inf. Model. 2017, 57, 115–121. 10.1021/acs.jcim.6b00686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier M.; Chénard T.; Barker J.; Najmanovich R. Kinome Render: A Stand-Alone and Web-Accessible Tool to Annotate the Human Protein Kinome Tree. PeerJ 2013, 1, e126 10.7717/peerj.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins J. M.; Fedele V.; Szklarz M.; Abdul Azeez K. R.; Salah E.; Mikolajczyk J.; Romanov S.; Sepetov N.; Huang X. P.; Roth B. L.; Al Haj Zen A.; Fourches D.; Muratov E.; Tropsha A.; Morris J.; Teicher B. A.; Kunkel M.; Polley E.; Lackey K. E.; Atkinson F. L.; Overington J. P.; Bamborough P.; Müller S.; Price D. J.; Willson T. M.; Drewry D. H.; Knapp S.; Zuercher W. J. Comprehensive Characterization of the Published Kinase Inhibitor Set. Nat. Biotechnol. 2016, 34, 95–103. 10.1038/nbt.3374. [DOI] [PubMed] [Google Scholar]
- Miduturu C. V.; Deng X.; Kwiatkowski N.; Yang W.; Brault L.; Filippakopoulos P.; Chung E.; Yang Q.; Schwaller J.; Knapp S.; King R. W.; Lee J. D.; Herrgard S.; Zarrinkar P.; Gray N. S. High-Throughput Kinase Profiling: A More Efficient Approach toward the Discovery of New Kinase Inhibitors. Chem. Biol. 2011, 18, 868–879. 10.1016/j.chembiol.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://doi.org/10.5281/zenodo.1148959.