

Abstract
Medulloblastoma (MB) is a malignant pediatric brain tumor with poor prognosis. Signal transducers and activators of transcription-3 (STAT3) is constitutively activated in MB where it functions as an oncoprotein, mediating cancer progression and metastasis. Here, we have delineated the functional role of activated STAT3 in MB, by using a cell permeable STAT3-NH2 terminal domain inhibitor (S3-NTDi) that specifically perturbs the structure/function of STAT3. We have implemented several biochemical experiments using human MB tumor microarray (TMA) and pediatric MB cell lines, derived from high-risk SHH-TP53-mutated and MYC-amplified Non-WNT/SHH tumors. Treatment of MB cells with S3-NTDi leads to growth inhibition, cell cycle arrest and apoptosis. S3-NTDi downregulated expression of STAT3 target genes, delayed migration of MB cells, attenuated epithelial-mesenchymal transition (EMT) marker expressions and reduced cancer stem-cell associated protein expressions in MB-spheres. To elucidate mechanisms, we showed that S3-NTDi induce expression of pro-apoptotic gene, C/EBP-homologous protein (CHOP) and decrease association of STAT3 to the proximal promoter of CCND1 and BCL2. Of note, S3-NTDi downregulated microRNA-21, which in turn, de-repressed Protein Inhibitor of Activated STAT3 (PIAS3), a negative regulator of STAT3 signaling pathway. Furthermore, combination therapy with S3-NTDi and cisplatin significantly decreased highly aggressive MYC-amplified MB cell growth and induced apoptosis by downregulating STAT3 regulated proliferation and anti-apoptotic gene expression. Together, our results revealed an important role of STAT3 in regulating MB pathogenesis. Disruption of this pathway with S3-NTDi, therefore, may serves as a promising candidate for targeted MB therapy by enhancing chemosensitivity of MB cells and potentially improving outcomes in high-risk patients.
Keywords: Medulloblastoma, STAT3, PIAS3, miR-21, MYC
INTRODUCTION
MB is a highly aggressive, malignant tumor of central nervous system (CNS) that accounts for 20–25% of all pediatric CNS tumors. Despite recent advances in surgical techniques and generation of newer chemotherapeutic and targeted agents, 30% of children affected with MB succumb to the disease. Genetically, MB is a heterogeneous disease with four molecular subtypes: Wingless (WNT), Sonic Hedgehog (SHH), and Groups 3 and 4 [1, 2]. Although, no specifically affected cell signaling pathways have been identified for Groups 3 and 4, frequent amplification of the MYC oncogene is consistently found in these groups. c-MYC amplified Group 3 tumors are found to have worse prognosis among all MB groups and no druggable targets have been so far identified, emphasizing the need for new therapeutic interventions. Recently, a new three-tier risk stratification system for MB has been proposed that recommend classification of MB into three subgroups: WNT, SHH and non-WNT/SHH, with supplementation by prognostic marker MYC for non-WNT/SHH tumors [3].
STAT3 is a latent cytoplasmic transcription factor, activated by the interleukin-6 (IL-6) family of cytokines and growth factors [4, 5]. Upon receptor mediated activation, STAT3 is phosphorylated on a single tyrosine residue (Tyr-705) by Janus activated kinases. Phosphorylated STAT3 (pSTAT3) subsequently dimerizes and translocate into the nucleus, where it binds enhancer sequences of its target genes that control fundamental cellular processes, including proliferation, differentiation, survival, and immunity [5–7]. Aberrant constitutive activation of STAT3 is frequently associated with various human cancers including MB, where it plays a deleterious role via prolonged activation of its downstream targets, promoting tumor progression [8]. Activated STAT3 is known to bind MYC promoter, a master regulator of proliferation and induces expression of the MYC gene in cancers [9]. Both MYC and STAT3 are oncoproteins and function as a sequence-specific transcription factors, governing the regulation of target genes integral to tumorigenesis [10]. In addition, activated STAT3 in cancer cells is known to stimulate its own transcription causing an increase in unphosphorylated STAT3 that contributes to tumorigenesis by mechanisms different from phosphorylated STAT3 [11, 12].
STAT3 transcriptional activity under normal conditions is transient and is tightly regulated by the action of its negative regulators, e.g, Suppressor of cytokine signaling 3, Src-homology 2 domain (SH2)-containing phosphatase 2 and Protein Inhibitor of Activated STAT3 (PIAS3) [13, 14]. Importantly, PIAS3, a key cellular inhibitor, has been reported to inhibit STAT3 phosphorylation and DNA binding activity, followed by the suppression of STAT3-mediated gene activation. In some cancer models, it was shown that loss of PIAS3 activity contributes to STAT3 activation and subsequent cancer cell proliferation [15]. Although, mechanisms of PIAS protein mediated regulation of transcription factors are well documented [16], PIAS3 dysregulation in cancers is poorly understood, except, for few studies, that report it might be regulated by noncoding micro-RNAs [17, 18].
Structurally, STAT3 comprises six functional domains of which STAT3 NH2 terminal domain (NTD) is known to play an important role in promoting protein-protein interactions and formation of a stable STAT3 tetramer, required for enhanced transcription of its target genes [19]. Although, several lines of evidence demonstrated that disrupting constitutively activated STAT3 inhibit tumor growth with induction of apoptosis in MB, the role of STAT3 NTD in regulating activated STAT3 functions and promoting MB tumorigenesis is largely unexplored [20, 21]. In this study, by using a cell permeable, penetratin-tagged S3-NTDi [22, 23], we showed attenuation of MB cell growth and survival and increased sensitivity of highly aggressive MYC amplified Groups 3 MB cells to chemotherapy. In addition, we unraveled the underlying mechanisms of activated STAT3 function in MB, in suppression of PIAS3, a STAT3 negative regulator via upregulation of microRNA-21 (miR-21). Overall, our study provides important insights into the positive feedback loop of STAT3/miR-21 on epigenetic downregulation of PIAS3, which in turn promotes persistent STAT3 activation in MB. Thus, STAT3-NTD may serve as a novel target for therapeutic intervention in the treatment of pediatric MB.
MATERIALS AND METHODS
Cell culture and Reagents
MB cell lines DAOY (SHH-TP53-mutated) and D341 (Group 3, Non-WNT/SHH), purchased from ATCC, Rockville MD, were grown in EMEM media with 10% and 20% FBS respectively. HD-MB03 cells (Group 3, Non-WNT/SHH, MYC amplified) purchased from DSMZ, Germany, were grown in RPMI media with 10% FBS. All cells were cultured with 1% Pen-Strep, in a 37 °C incubator with 5% CO2. Cells were authenticated by ATCC with the STR analyses information.
For MB-sphere culture, cells were grown at confluence in adherent conditions. Cells were tripsinized, counted and plated (1 × 104 cells) in ultra-low attachment 6-well plates (Corning Inc., NY, USA) for 7–10 days in serum-free medium containing, EGF 20ng/mL, bFGF 40ng/mL, Heparin 2μg/mL, β-Me 0.1mM, B27 1%, N2 1% and Pen-Strep 1%. Pictures of MB-spheres formed after 7 days were taken in EVOS Cell Imaging Systems.
IL-6 and soluble IL-6 receptor alpha (sIL-6Rα), purchased from PeproTech Inc (Rocky Hill, NJ), were used to stimulate STAT3 activation in MB cells (in STAT3 regulated protein and gene expression studies) to mimic tumor microenvironment. Concentration of IL-6/ sIL-6Rα used was mentioned in figure/legend.
Penetratin-tagged STAT3-NTD inhibitor (S3-NTDi), used in this study was reported earlier (as S3Hel2A2/ST3-H2A2) [22, 23]. S3-NTDi and control peptide (penetratin) were synthesized from Pierce custom peptide service and were resuspended in DMSO and stored at −20o C for treatment of MB cells. Cisplatin, purchased from Sigma-Aldrich (St. Louis, MO), was resuspended in 0.9% NaCl (w/w) and stored at −200C. BP-1–102, purchased from Selleckchem was reconstituted in DMSO and was stored at −200C until used.
Control and STAT3 siRNA (SMARTpool, siGenome) were purchased from Dharmacon (Lafayette, CO). siRNAs were transfected in MB cells using Lipofectamine 2000 following manufacturer’s instructions. Locked nucleic acid-miR-21 inhibitor (LNA-anti-miR21), Product No 4100688–101 (mmu-miR-21a-5p) and LNA-control, Product No 199006–101 (Negative control A) were purchased from Exiqon A/S (Denmark). The LNA control and LNA-miR-21 power inhibitors were added directly into the cell culture media as recommended by the manufacturer.
MTT assays
MB cells were plated at a density of 25000–30,000 cells/well of a 96 well plate in the presence/absence of the inhibitors and MTT assays were performed followed manufacturer’s protocol (ATCC 30–1010K). Briefly, cells in 96 well plates were treated with a S3-NTDi at a concentration/time mentioned in figure/legends. Next day, 10 μl of MTT solution was added in each well and after 4 hours of incubation at 37oC, 100 μl detergent was added to solubilize the cells at room temperature for overnight in the dark. The intensity of the color developed was determined at a 570 nm wavelength using a plate reader (Biotek, Germany).
Cell Cycle Analysis
Cell cycle analysis of DNA by flow cytometry in the absence/presence of S3-NTDi was measured by propidium iodide (PI) staining, following manufacturer’s instruction (Abcam, cat # ab139418, Cambridge, MA). Briefly, 1 × 106 cells were fixed in ice cold 100 % ethanol for 2 h. Cells were washed twice and resuspended in 200 μl of PI+ RNAase for 20–30 min at 370C. After incubation, cells were placed on ice, until analyzed by flow cytometry.
Apoptosis Assays
The ability of S3-NTDi to induce apoptosis in MB cell lines were determined using an Annexin V-FITC double staining kit (cat #130–092-052, MACS, CA USA) and following manufacturer’s instruction.
Western Blot Analysis
Treated MB cells were washed twice with ice-cold PBS and whole cell extracts (WCE) were prepared by lysing cells in modified RIPA buffer and fractionated in 4–20% Tris-Glycine Gels (Bio-Rad, Hercules, CA). Western Blot analysis were performed by transferring proteins to PVDF membranes (Millipore, Bedford, Mass) as previously described [24, 25]. Membranes were probed using the appropriate primary and secondary antibodies, diluted according to the manufacturers’ instructions. Signals were detected by enhanced chemiluninescence and using MyECL imager (ThermoScientific, MA, USA).
Primary antibodies (Abs) used for Western blots are as follows:
STAT3 (sc-483 and sc-482), MYC (9E10, sc-40), BCL2 (sc-7382), CCND1 (sc-753), β-Actin (sc-47778), E-Cadherin (sc-21791), N-Cadherin (sc-271386), Vimentin (sc-6260), CHOP (sc-7351), PIAS3 (sc-46682) and OCT4 (sc-5279) were purchased from Santa Cruz (Dallas, TX).
P-Tyr 705 STAT3 (# 9145), PARP (# 9532), GAPDH (# 5174), Vinculin (# 3901), Cyclophilin (# 2175) were purchased from Cell Signaling (Danvers, MA). Nestin Ab (ABD69) was purchased from Millipore (Billerica, MA).
Quantitative RT-PCR (qRT-PCR)
Total RNA and miRNA were prepared by using trizol reagent (Ambion) and RNA quality and integrity were measured prior to qRT-PCR. 2 μg of total RNA was used for reverse transcription using superscript VILO cDNA kit (Invitrogen). cDNA products was amplified in 10 μl reaction using SYBR Green Super Mix (Applied Biosystems) and validated gene specific primers, purchased from IDT DNA integrated technology, as mentioned. All reactions were processed in QuantStudio 3 Real-Time PCR System and results were analyzed by data analysis software (Applied Biosystems) by normalizing with housekeeping gene (GAPDH, β -Actin, Cyclophilin, Ribo S9) transcript level.
The miScript II RT Kit, miScript Primer Assays and SYBR Green PCR Kit (Qiagen, MD) were used for miRNA detection and quantification.
Cell Migration Assays
MB cell migrations were detected by ‘wound healing” assay by using CytoSelect™ 24-well plates (Cat # CBA-120–5, Cell Biolabs, San Diego, CA) and following manufacturer’s instructions.
Colony Assay
MB cells treated with S3-NTDi overnight, were reseeded at a very low density (~500 cells/well in a 6-well plate). Cells were allowed to grow in normal medium for two weeks, prior to fixing with formaldehyde and staining of colonies with crystal violet solution.
Chromatin Immunoprecipitation (ChIP) Assay
S3-NTDi treated and untreated MB cells were protein-protein cross-linked with Disuccinimidyl glutarate (DSG), followed by protein-DNA cross-linked with formaldehyde, as described previously [26]. Cells were then lysed and sheared chromatin were immunoprecipitated with either STAT3 Ab or IgG, following instructions from Pierce Magnetic ChIP Kit (#26157). Quantitative genomic PCR was performed in QuantStudio 3 Real-Time PCR System, using human Bcl-2 promoter and CCND1 promoter primers (#12924, #12531) from Cell Signaling. Data were corrected for variations in input and expressed as fold change relative to IgG controls.
Confocal Microscopy
MB cells grown on cover slips were treated with inhibitors as described in figure/legends. Cells were stimulated with IL-6/sIL-6Rα prior to fixation and immunofluorescence staining as described previously [27]. Primary antibodies p-Tyr705 STAT3 (cell signaling Ab #9145, used at dilution 1:50) or PIAS3 (Santa Cruz Ab, sc-14017 used at dilution 1:100) were used. Confocal images were taken with a Zeiss LSM 5 Pascal confocal microscope (Carl Zeiss, Oberkochen, Germany) using a 63X objective with a numerical aperture (NA) of 1.4 and appropriate filters in UNMC advanced confocal microscopy facility.
Immunohistochemistry (IHC)
MB brain TMA containing three normal cerebellar tissue and 20 cases of MB were obtained from US Biomax, Derwood, MD. IHC staining was performed with p-Tyr705-STAT3 Ab (cell signaling #9145) and PIAS3 Ab (Abcam # ab58406), following manufacturer’s instructions, at UNMC tissue science facility.
Statistical Analysis
The results are representative of three independent experiments and are expressed as the means ± SD, as indicated. Statistical analysis of the data was performed using the Student’s t test.
RESULTS
Effects of S3-NTDi on MB cell survival, apoptosis and cell cycle.
To investigate the role of STAT3 in MB pathogenesis, we first aimed to determine the extent of activated STAT3 by immunostaining a MB tissue microarray (TMA) slide for phospho-Tyr705 STAT3 Ab (Fig.1A). Representative images show that the frequency of constitutively activated STAT3 was almost undetectable in normal cerebellar tissue, whereas in MB tumors, the presence of positive nuclear staining indicated as brown/dark signals, represent activation of pSTAT3 in rapidly proliferating sub-population of tumor cells. Next, to examine the functional role of STAT3-NTD in MB, we used a highly sensitive cell permeable S3-NTDi, that was previously shown to perturbs specifically the STAT3 signaling pathway and not STAT1 [22, 23]. To assess the effects of S3-NTDi on MB cell lines HD-MB03, D341 and DAOY, we first measured cell viability and its ability to induce apoptosis by MTT assay and Annexin-V staining respectively. We showed that treatment of MB cells with varied concentrations of S3-NTDi for 24 and 48 h, decreased cell growth in a dose and time dependent manner, with a half maximal inhibitory concentrations (IC50) ranging from 10–12 μM (Fig.1B). MTT assay with control peptide has minimal effects on MB cell viability (Fig. S1), as previously observed in case of breast and prostate cancer [22, 28]. Flow cytometric analyses of Annexin-V stained MB cells treated with S3-NTDi revealed a significant induction of apoptotic cell death (Fig.1, C-D). Interestingly, we observed that S3-NTDi treated Group 3 MB cells, HD-MB03 and D341 cells, had more apoptotic cell death than DAOY (Group SHH) [29]. We next investigated the percentages of cells and their DNA content in different phases of the cell cycle. Here, sub-confluent cultures of MB cells were treated with S3-NTDi and were then labeled with PI and analyzed by flow cytometry (Fig. S2). Stacked bar diagram (Fig.1E) shows S3-NTDi caused an increase in the number of MB cells in the S phase of the cell cycle with a corresponding decrease in G1 and a minor change in G2-M phase cells, indicating that S3-NTDi induced cell cycle arrest in the S and G2/M phases as reported earlier in prostate cancer [22].
Figure 1.
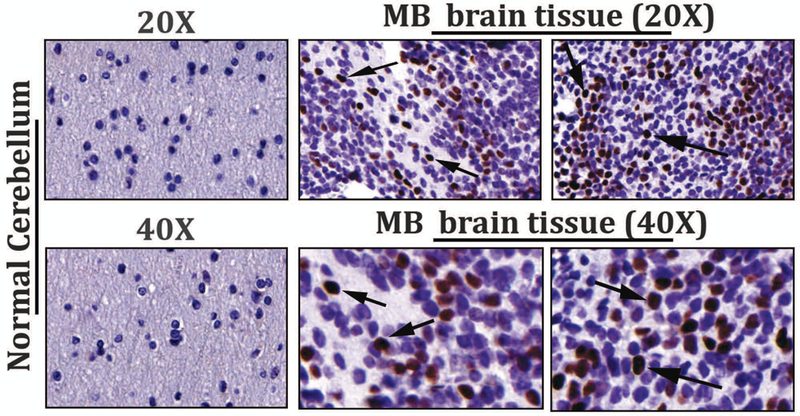
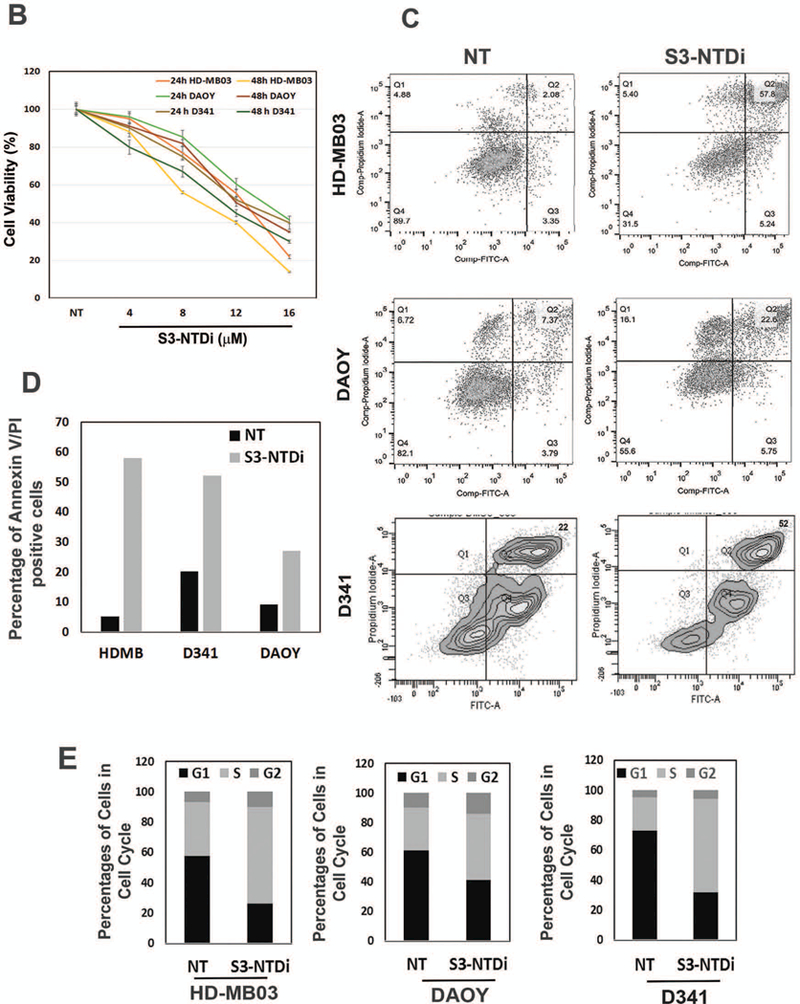
Effects of S3-NTDi on MB cell viability, apoptosis and cell cycle. (A) Representative images of pSTAT3 expression in MB tumor microarrays (TMA), performed by immunohistochemical staining (IHC) staining with p-Tyr705 STAT3 Ab (Cell Signaling, # 9145). Normal cerebellar tissues (20X, top left and 40X, bottom left) and MB tumors (20X, top and 40X, bottom) are shown. Arrowheads show the strong nuclear staining for activated STAT3. (B) MB cell line, DAOY, HD-MB03 and D341 treated either with S3-NTDi at a concentration of 4, 8, 12 and 16 μM or left untreated (NT: non-treated vehicle control). The growth of these cells was determined at 24 and 48 hours using MTT assays. The average absorbance of NT control was regressed against the concentration of the inhibitor and thus allowed us to calculate IC50 for S3-NTDi. The values represent the means ± SD from five wells of a 96-well plates. (C) Each MB cell lines were treated with S3-NTDi at a concentration of 10 μM for overnight. The percentage of cells undergoing apoptosis was determined using annexin-V-FITC apoptosis detection kit. Figure shows representative scatter diagram for the apoptotic cells. NT: non-treated control. (D) Quantification of the apoptotic cells (% Annexin-V/PI double positive) from one of three independent experiments following treatment with S3-NTDi in MB cells were shown. (E) MB cells were treated with 10 μM of S3-NTDi for overnight and cell cycle analysis was performed by labeling cells with PI followed by flow cytometry. The percentage of MB cell populations in the G1, G2, and S phases of cell cycle are shown in the bar diagram. NT: non-treated control.
STAT3-NTD inhibition alters STAT3 regulated downstream target gene/protein expression.
STAT3 activation plays an important role in stimulating cellular proliferation and resisting apoptosis in cancer. To determine if STAT3-NTD inhibition affected protein expression of its downstream targets in MB cells, we performed dose-out experiment. For this purpose, cells were first treated with increasing concentrations of S3-NTDi for 24 h and then stimulated with IL-6 and sIL-6Rα for 20 mins, prior to harvest. We observed STAT3 regulated MYC, CCND1 and BCL2 protein expression were decreased in a dose dependent manner in MB cells (Fig. 2A). To determine if above decreased protein expression was a result of changes in gene expression, we determined the effect of S3-NTDi on STAT3 target gene expression in HD-MB03 cells. As shown by qRT-PCR, STAT3 regulated expression of MYC and CCND1 was downregulated in the presence of S3-NTDi (Fig. 2B), along with other STAT3 target genes (Fig S3). Particularly, we noted that expression of base excision repair protein APE1, a co-regulator of STAT3’s function, was reduced (Fig S3) [25]. Also, we observed that treatment of cells with control peptide did not show any changes in STAT3 regulated gene expression (Fig. S4). To validate the above effects of S3-NTDi on target gene expression, we used a small interfering RNA (siRNA) to knockdown STAT3 in HD-MB03 cells and confirmed the specificity S3-NTDi on STAT3 functions. We showed that downregulation of STAT3 protein level (inset Fig. 2C) decreased expression of STAT3 regulated genes (MYC and BCL2), supports a functional role of S3-NTD in target gene expression (Fig. 2C and Fig S5).
Figure 2.
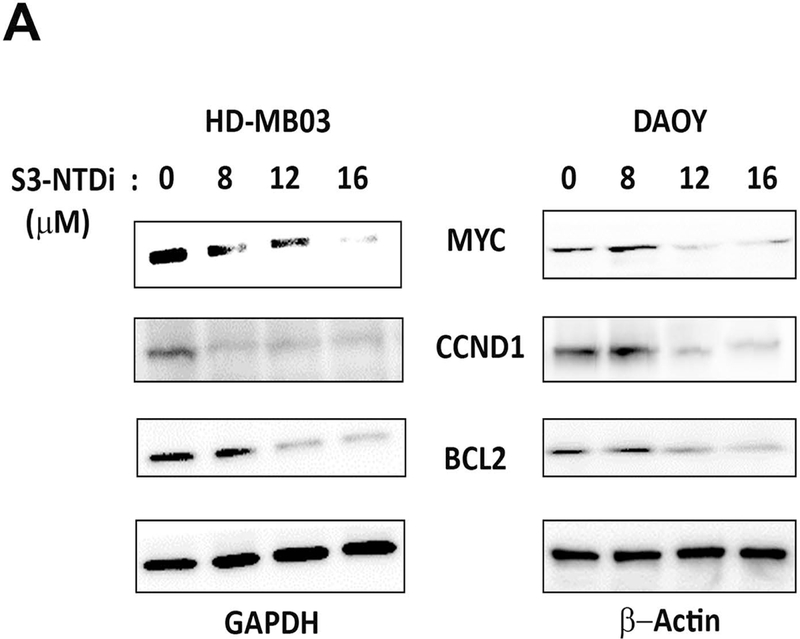
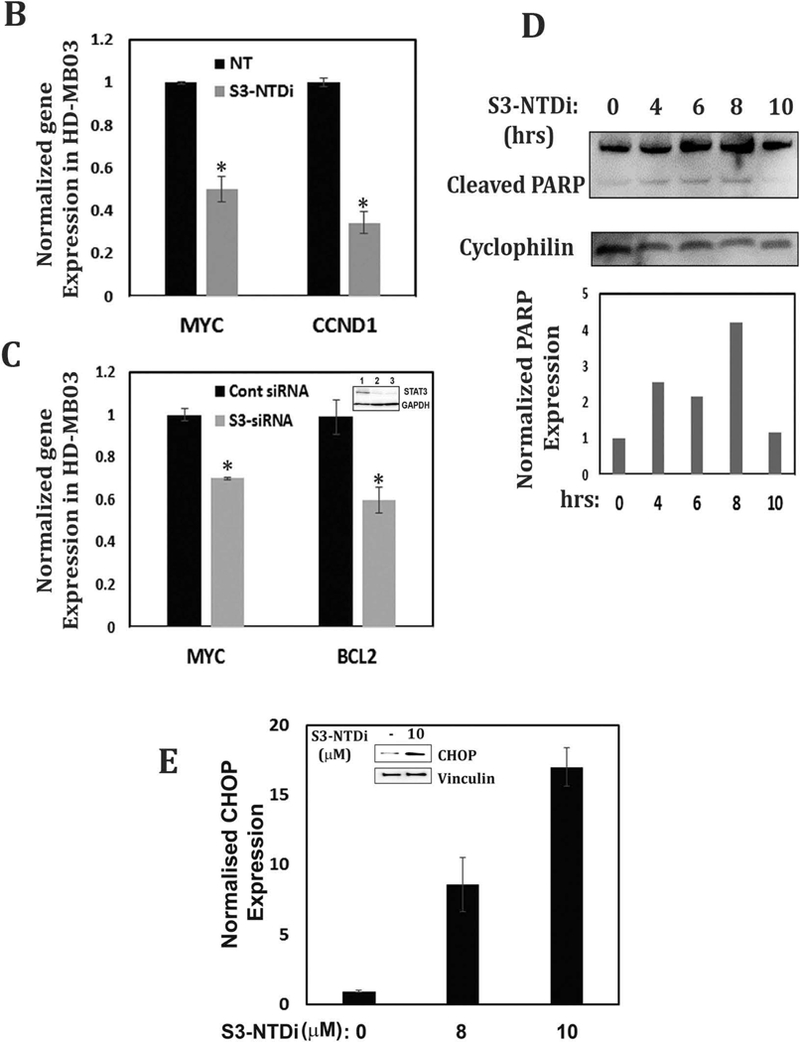
S3-NTDi downregulates expression of STAT3 target genes. (A) HD-MB03 and DAOY cells were treated with increasing concentrations of S3-NTDi (0, 8, 12 and 16 μM) for 24 h. Cells were stimulated with IL-6/sIL-6Rα (50/25 ng/ml) for 20 mins prior to making whole cell extracts (WCE). Expression of MYC, CCND1 and BCl2 were analyzed by Western immunoblot analysis and GAPDH and β-Actin were used as a loading control. (B) HD-MB03 cells were treated with 10 μM S3-NTDi for overnight or left untreated. Cells were stimulated with IL-6/sIL-6Rα for 20 mins prior to harvest. STAT3 regulated gene expressions were analyzed by qRT-PCR. * represents p<0.001, NT: non-treated control. (C) STAT3 activity is knockdown by 75 nM siRNA (SMARTpool) in HD-MB03 cells for 72 h (inset figure) and STAT3 regulated gene expressions was analyzed by qRT-PCR. * represents p<0.005. (D) Expression of PARP cleavage in HD-MB03 cells, treated with 10 μM of S3-NTDi for 0, 4, 6, 8 and 10 h are shown by Western blot. Intensity of cleaved PARP is shown in bar diagram (bottom). (E) HD-MB03 cells were treated with 0, 8 and 10 μM of S3-NTDi overnight. Total RNA isolated from these cells was subjected to qRT-PCR for CHOP expression. The inset figure shows Western blot of CHOP expression. * represents p<0.001.
Next, we determined formation of Poly ADP-ribose polymerase (PARP) cleaved fragments, a hallmark of apoptosis, in the presence of S3-NTDi. We observed a time dependent increase in PARP cleaved fragment in HD-MB03 cells after treatment with S3-NTDi, indicating induction of apoptosis (Fig. 2D). To evaluate if S3-NTDi activates pro-apoptotic gene/protein expression of C/EBP-homologous protein (CHOP), we prepared total RNA and WCE from HD-MB03 cells and performed qRT-PCR and Western blot respectively (Fig. 2E). We found a dose dependent increase of CHOP gene expression in the presence S3-NTDi and a concomitant increase in CHOP protein expression in HD-MB03 (inset Fig. 2E). Together, this result indicated that S3-NTDi downregulated proliferation and anti-apoptotic marker gene expression and induced apoptosis by pro-apoptotic gene (CHOP) expression and by PARP cleavage.
S3-NTDi impedes cell migration and colony formation.
To investigate whether S3-NTDi decreased cancer cell mobility, we conducted in vitro wound healing assays, as many cellular processes of tumor metastasis replicate wound healing steps [30]. Here, we artificially created a gap by a “scratch” in HD-MB03 cell monolayers and serial images of cell migrations were taken over the next 72 h. We observed that non-treated (NT) control cells migrated to fill the gap area completely within 48 h (Fig. 3A), whereas S3-NTDi treated cells took significantly longer time to fill only 15% of the scratch area (Fig. 3B). This indicates that S3-NTDi profoundly affects the migratory properties of MB cells and likely their ability to metastasize.
Figure 3.
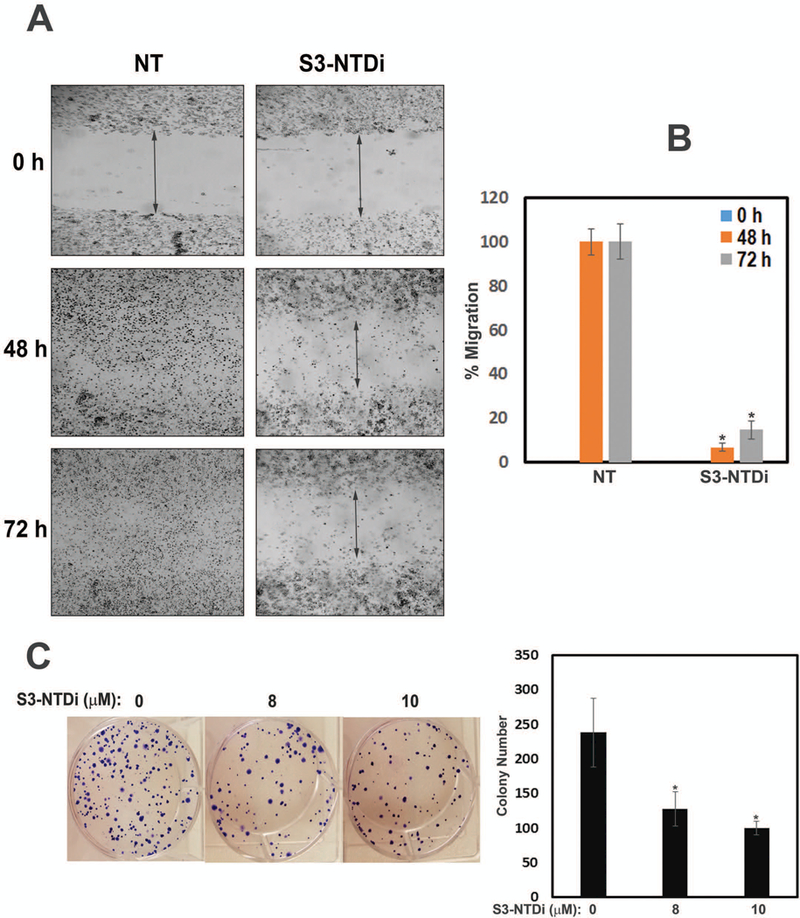
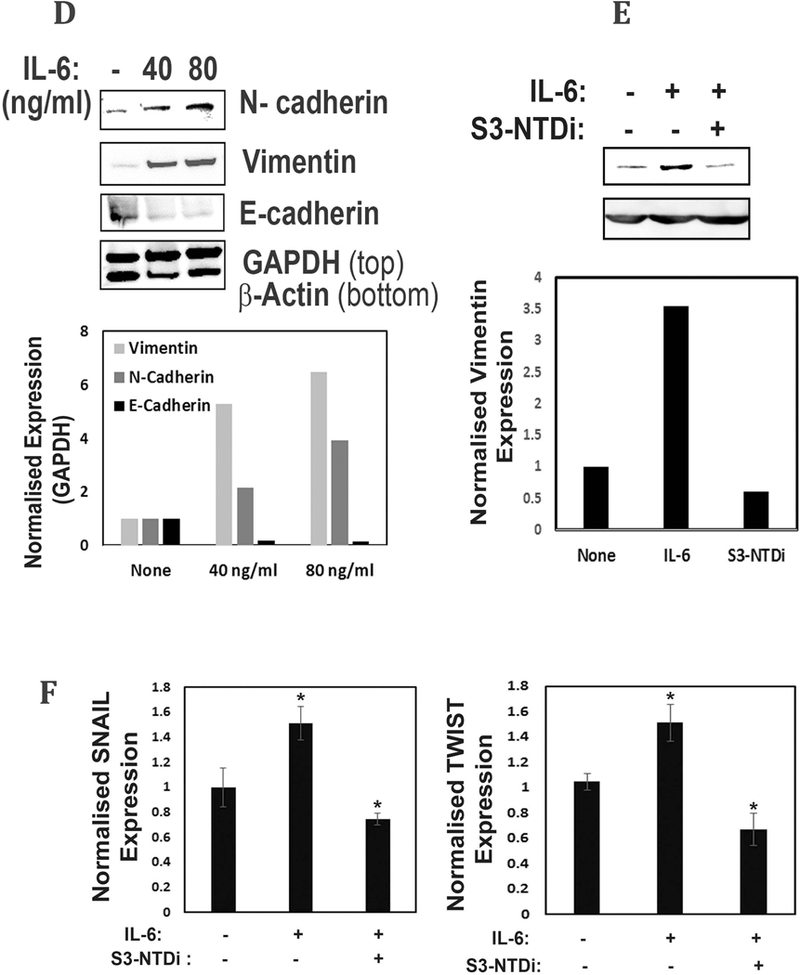
S3-NTDi inhibits MB cell migration, reduced colony formation and IL-6 mediated EMT. (A) Wound healing assays performed by seeding HD-MB03 cells into CytoSelect™ 24-Well assay plates (Cell Biolabs Inc) until a monolayer formed, at which time the inserts were removed and a cell-free gap (0.9mm) is created in which the cell migration was analyzed either in presence of vehicle or 10 μM S3-NTDi. Images of cell migration were taken after every 12 h for 72 h. Representative images taken at 0, 48 and 72 h are shown. NT: non-treated control. (B) The percentage of cells migrated to fill the gap area were calculated according to the manufacture’s instruction. Percent migration is shown in bar diagram. NT: non-treated control, * represents p<0.001 (C) HD-MB03 cells were treated with either 0, 8 or 10 μM S3-NTDi for 8h. Equal numbers of cells were reseeded in 6-well plates and allowed to grow for 2 weeks in normal media. Colonies formed from single cell were fixed with acetic acid/methanol 1:7 (vol/vol) and stained with 0.5% crystal violet solution. Number of colonies counted from three independent experiments is shown in bar diagram (right). * represents p<0.005. (D) HD-MB03 cells were treated with either 0, 40/20 ng/ml of IL-6/sIL-6Rα or 80/40 ng/ml of IL-6/sIL-6Rα and WCE were subjected to Western immunoblots with N-cadherin, Vimentin and E-cadherin Ab. GAPDH and β-Actin were used as a loading control. Bar diagram below shows the quantitation of normalized expression of the proteins. (E) HD-MB03 cells were treated with or without 10 μM S3-NTDi along with 80/40 ng/ml of IL-6/sIL-6Rα for overnight. WCE were then subjected to Western immunoblot with Vimentin Ab. Vinculin was used as loading control. Below shows the band intensity of vimentin normalized with Vinculin. (F) HD-MB03 cells were either treated with10 μM S3-NTDi or left untreated in the presence of IL-6/sIL-6Rα (40/20 ng/ml) for overnight. EMT related transcription factor expressions were measured by qRT-PCR. * represents p<0.005.
We next determined the ability of HD-MB03 cells to sustain proliferation after pretreatment with S3-NTDi, by a colony formation assay (Fig. 3C). S3-NTDi significantly reduced the number of viable colonies as compared to no treatment control, indicating that S3-NTDi affects the ability of single cells to reproduce and to form large, visible colonies.
Role of STAT3-NTD on IL-6 mediated EMT.
IL-6 is a pleiotropic cytokine which is elevated in many types of cancer, enhancing cancer cell proliferation and inducing EMT, a phenotypic conversion prerequisite for tumor invasion and metastasis [31, 32]. To examine if IL-6 mediates EMT changes in MB, we cultured HD-MB03 cells overnight in the presence or absence of IL-6. Fig. 3D shows that IL-6 stimulated HD-MB03 cells markedly increased Vimentin and N-Cadherin expression, about 6 and 4 fold respectively, while at the same time decreasing the expression of E-cadherin (>60%), one of the hallmarks of EMT. We further showed that treatment of HD-MB03 cells with S3-NTDi, downregulated IL-6 induced Vimentin expression, indicating the role of STAT3 in this process, (Fig. 3E). Next, we investigated if IL-6 induced EMT marker protein expressions is mediated via activation of SNAIL and TWIST transcription factors that are known to be regulated by STAT3. For this purpose, we treated IL-6 stimulated HD-MB03 cells with S3-NTDi and gene expression was analyzed by qRT-PCR. Fig. 3F shows that IL-6 stimulation caused induction of SNAIL and TWIST transcription factors as compared to untreated IL-6 control but treatment with S3-NTDi in the presence of IL-6, downregulated its expression, indicating a functional role of STAT3 in IL-6 mediated EMT induction in MB.
S3-NTDi affects MB-sphere formation and marker gene expression.
Induction of EMT in cancer cells plays an important role in promoting self-renewing cancer “stem cell” enrichment and giving rise to tumor spheres [33]. Here, we examined MB-sphere formation in HD-MB03 and DAOY cells when grown in serum free media and in ultra-low adherent tissue culture plates [34]. We found that both DAOY and HD-MB03 cells form varied numbers and sizes of MB-spheres (Fig. 4A), whereas in the presence of S3-NTDi the number of MB-spheres significantly decreased with accumulation of small spheres which eventually disintegrated (Fig S6). We next tested the effect of S3-NTDi on neural stem cell associated marker expression on MB-sphere extracts [35]. Fig. 4B, shows expression of OCT4 and Nestin in DAOY and HD-MB03 cells respectively, in non-treated controls, whereas in the presence of S3-NTDi, expression was reduced, indicating an essential role of STAT3 in sphere formation and maintaining pluripotency gene expression of cancer-initiating MB stem cells.
Figure 4.
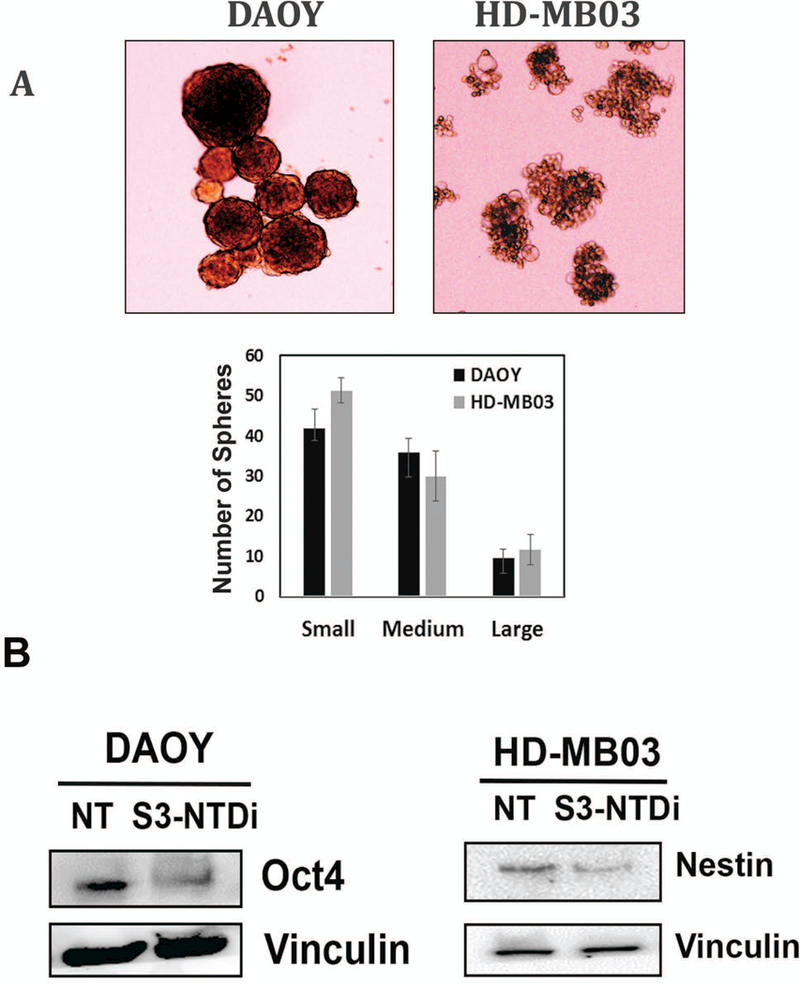
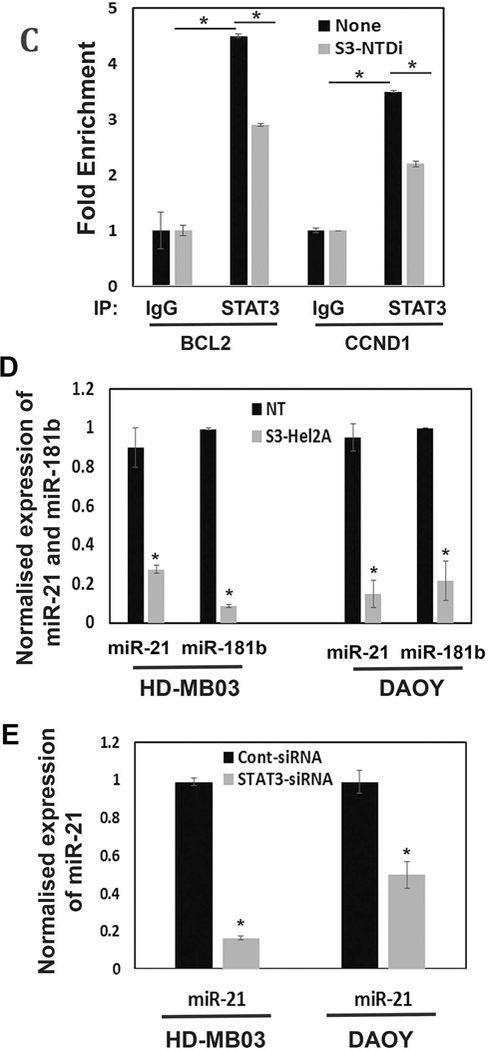
Effect of S3-NTDi on MB-spheres formation, STAT3 target gene promoter binding and miR-21 expression. (A) Representative morphology of MB-spheres from DAOY and HD-MB03 cells grown in serum free media are shown. Small, medium and large spheres from DAOY and HD-MB03 were enumerated and sizes measured and are shown below. (B) DAOY and HD-MB03 MB-spheres were treated with 10 μM S3-NTDi for 24h and Western blot analyses for OCT4 and Nestin expression were analyzed respectively. Vinculin was used as a loading control. NT: non-treated control (C) HD-MB03 cells were treated with10μM S3-NTDi overnight and then treated with IL-6/sIL-6Rα for 20min, prior to protein-protein and protein-DNA crosslinking. STAT3 binding to the BCL2 and CCND1 gene promoters in HD-MB03 cells was demonstrated by ChIP-qPCR. IgG Ab were used as a negative control. SimpleChIP® Human Bcl-2 Promoter Primers (# 12924) and SimpleChIP® Human CCND1 Promoter Primers (#12531) purchased from cell signaling, were used for quantitative genomic PCR. * represents p≤0.005; t-test. (D) HD-MB03 and DAOY cells were either treated with 10 μM S3-NTDi for overnight, or left untreated. Cells were stimulated with IL-6/sIL-6α for 20 mins prior to harvesting miRNAs. Expression of miR-21 and miR-181b were measured by qRT-PCR. NT: non-treated control and * represents p≤0.001 (E) Expression of miR-21 in HD-MB03 and DAOY in the presence of either control siRNA or STAT3 siRNA was measured by qRT-PCR. * represents p≤0.001, t-test.
S3-NTDi affects occupancy of STAT3 on target gene promoters and miRNA expression.
To determine the mechanism by which S3-NTDi downregulates MB cell proliferation and anti-apoptosis, we examined the recruitment of STAT3 to its downstream target gene promoters, CCND1 and BCL2, by a two-step ChIP assay [26]. Fig. 4C shows that in the presence of S3-NTDi, the abundance of STAT3 in CCND1 and BCl2 promoter regions is significantly reduced compared to non-treated control. This suggests that the occupancy of STAT3 to target gene promoters is required for driving transcription and inhibiting STAT3-NTD may impair cooperative DNA binding and thereby inhibit gene expression in MB [19].
Dysregulation of miRNA is known to be involved in STAT3-mediated tumorigenesis. In particular, transcription of miR-21, also consider oncomiR-21, are reported to be elevated in different types of cancer including MB [36]. To confirm this, we performed qRT-PCR and examined if STAT3-NTD inhibition affects miRNA expression in HD-MB03 and DAOY cells. We found that in the presence of S3-NTDi, expression of miR-21 and 181b (also, a STAT3 regulated miRNA) are inhibited significantly, suggesting STAT3’s role in regulating miRNA expression in MB (Fig. 4D). To confirm this directly, miRNA expressions were determined after downregulation of STAT3 by siRNA (Fig. 4E). Although, the presence of STAT3 siRNA markedly knocked down miR-21 expression in both HD-MB03 and DAOY cells, it did not show any inhibitory effects on miR-181b expression (Fig S7), indicating it’s expression may also be regulated by alternative pathways. Overall, this result suggested that elevated STAT3 expression in MB directly activate miR-21 expression, consistent with previous reports [21, 36]. Moreover, inhibition of miR-21 by STAT3 siRNA further supports an inhibitory effect of S3-NTDi on miR-21 in MB.
MiR-21 suppressed PIAS3 expression in MB
miRNAs in cancer are known to modulate transcription/translation of several key regulatory molecules by epigenetic modifications. These include tumor suppressors (e.g. PTEN) and negative regulators, including PIAS3, a direct target of activated STAT3 [14, 17, 18, 37]. For this reason, we first determined the level of PIAS3 expressions in the MB TMA specimen and performed IHC with PIAS3 Ab. We observed positive PIAS3 staining mainly in the nucleus of normal cerebellar tissues, whereas, the MB tumors demonstrated low or negative nuclear staining of PIAS3 (Fig. 5A). This indicated that PIAS3 expression in a subset of MB tumors could be repressed or lost (Fig S8). Next, to validate the result of IHC, we examined PIAS3 expression in HD-MB03 cells in the presence or absence of S3-NTDi by Western blot. We observed that in untreated control cells, expression of PIAS3 was almost undetectable, whereas with S3-NTDi treatment, to our surprise, there was a robust increase in PIAS3 expression with downregulation of pSTAT3 (Fig. 5B). Further, STAT3 inhibitor BP-1–102 (STAT3 SH2 domain inhibitor) did not show any change in PIAS3 expression (Fig S9) [38].
Figure 5.
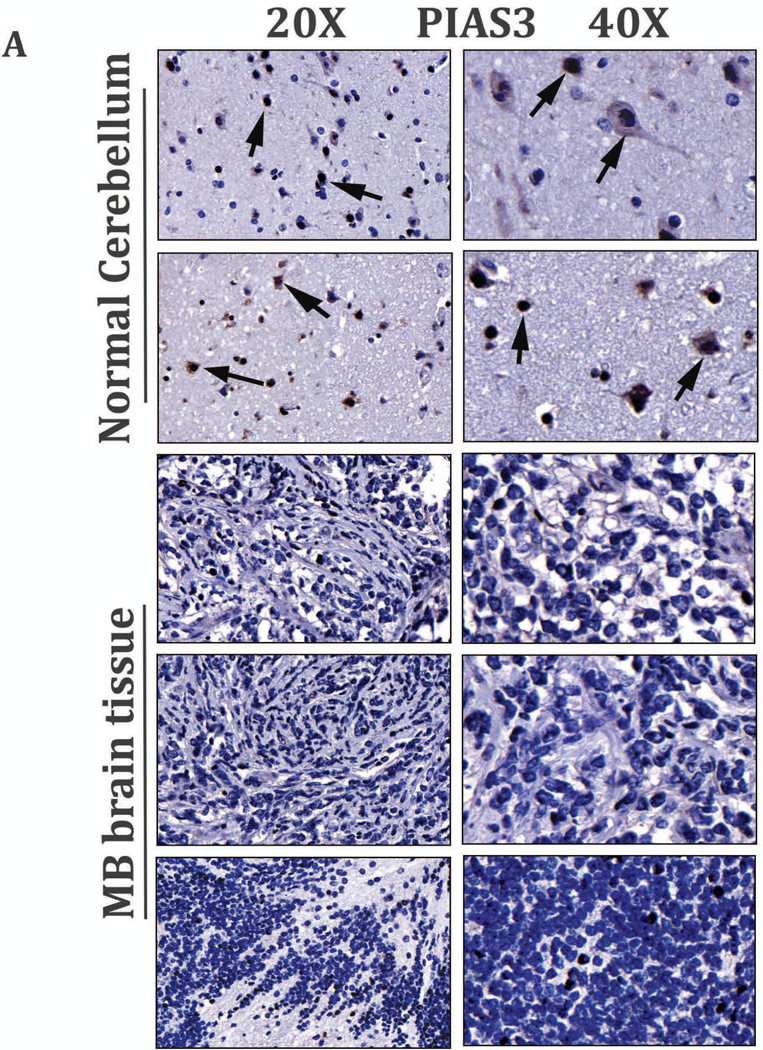
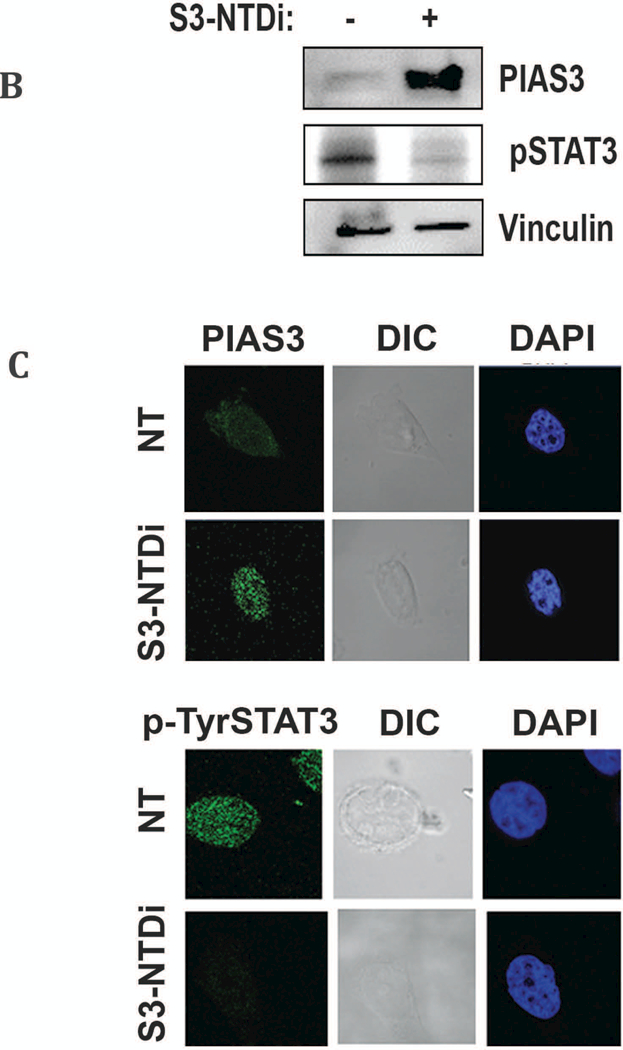
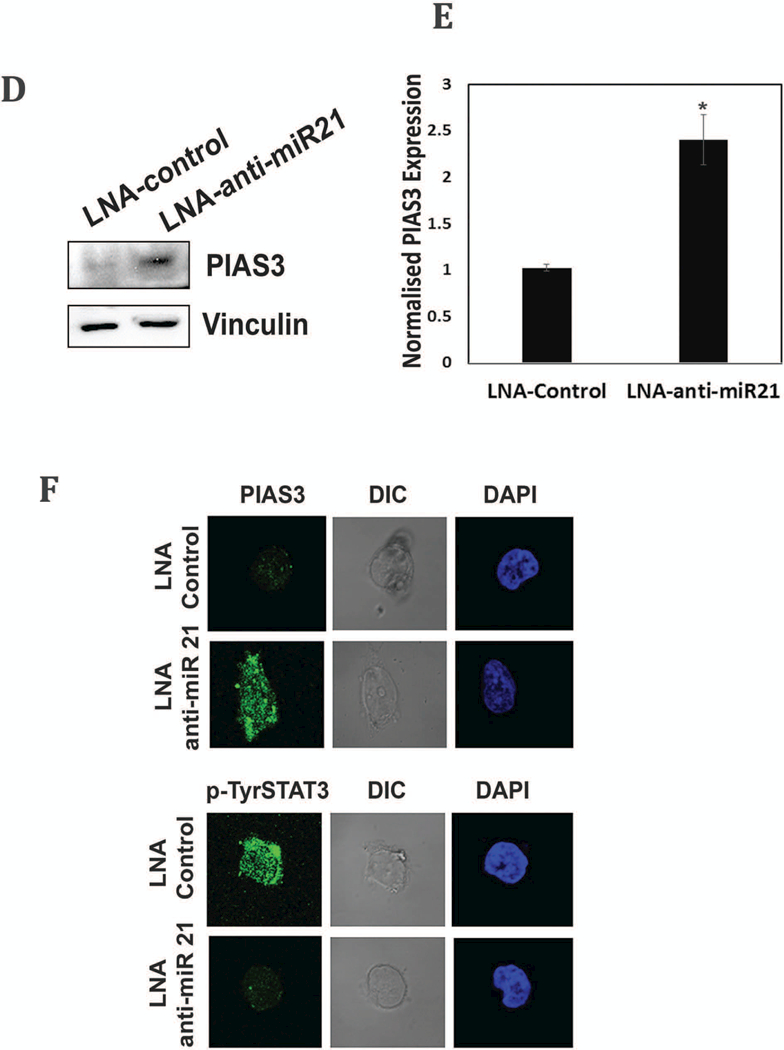
Activation of PIAS3 by S3-NTDi treatment via inhibition of miR-21. (A) Representative images of PIAS3 expression in MB tumor microarray (TMA) were performed by IHC staining with PIAS3 Ab (Abcam, # ab58406). Normal cerebellar tissues (two rows from top, 20X on left and 40X on right) and MB tumors (rows 3–5 from top, 20X, left, top and 40X, right) are shown. Arrowheads show the nuclear staining for PIAS3. (B) Expression of PIAS3 and pSTAT3 in HD-MB03 following treatment of 10 μM S3-NTDi for 24 h are shown by Western blot. Vinculin used as loading control. (C) Expression of PIAS3 (top panel) and p-Tyr705 STAT3 (bottom panel) in HD-MB03 in the presence of 10 μM S3-NTDi is shown by confocal microscopy. NT: non-treated control. (D) HD-MB03 cells were treated either with LNA-control or LNA-anti-miR-21 oligonucleotides (50 nM). PIAS3 expression was detected after 72 h by Western blot and (E) by qRT-PCR. *p<0.001. (F) Expression of PIAS3 (top panel) and p-Tyr705 STAT3 (bottom panel) in HD-MB03 cells in the presence of either LNA-control or LNA-anti-miR-21 is shown by confocal microscopy.
PIAS3 expression was further validated by confocal microscopy (Fig. 5C), after staining cells with PIAS3 Ab (Top). We observed a similar result that in untreated cells, PIAS3 staining in HD-MB03 cells is negligible compared to cells treated with S3-NTDi. We also observed a corresponding decrease in p-Tyr 705 STAT3 expression (bottom) in the presence S3-NTDi compared to DMSO control, indicating negative regulation of pSTAT3 by increased PIAS3 levels.
We next determined if STAT3 mediated activation of miR-21 could modulate PIAS3 expression in MB, we treated HD-MB03 cells either with LNA-anti-miR-21 or LNA-control [39]. Fig. 5D and E show that in Western blot and qRT-PCR, both PIAS3 protein and mRNA expression were increased respectively in HD-MB03 cells, treated with LNA-anti-miR-21 as compared to LNA-control. Further, confocal microscopic analysis confirmed PIAS3 upregulation (Top) and decreased pSTAT3 expression (bottom), in the presence of LNA-anti-miR-21 vs LNA-control (Fig. 5F). Together, these results indicated that PIAS3 is repressed in MB and its expression is inversely correlated with p-Tyr705 STAT3 activation status. Suppression of STAT3 NTD prevents miRNA 21 expression which, in turn, de-repressed PIAS3 and attenuated the STAT3 signaling pathway.
S3-NTDi sensitizes MB cells to chemotherapy
Cisplatin is considered first line chemotherapy for the treatment of MB [40]. STAT3 activation in cancer cells promotes induction of survival signals and anti-apoptosis and this often is associated with resistance to chemotherapy. To evaluate if S3-NTDi can enhance the efficacy of cisplatin therapy for MB, we measured HD-MB03 cell viability by MTT assays. Treatment of cells with 12 μM of S3-NTDi alone for 24 h, resulted in ~60% viable cells whereas cisplatin treated cells at a concentration of 5 μM had ~40 % viability (Fig.6A). However, when combined, MB cell viability dropped significantly to less than ~10% at 24 h and to an undetectable level when treated for 48 h (Fig. 6B). To further validate the effects of this combination regimen on apoptosis, we determined Annexin-V positive cells by flow cytometry. As shown in Fig. 6C, there is 59% and 22% apoptotic cell death in HD-MB03 with S3-NTDi (12 μM) and cisplatin (2.5 μM) respectively, but when used in combination there was a significant increase of apoptosis about ~90%, compared to single agent treatment. However, in DAOY cells, although combination of S3-NTDi and cisplatin did increase the number of apoptotic cells, the effect was not significant compared to S3-NTDi and Cisplatin treatment alone (Fig. S10), suggesting that combination treatment is more effective in MYC amplified Group 3 MB cells. Furthermore, we investigated if S3-NTDi along with cisplatin affects downstream target genes of the STAT3 signaling pathway by qRT-PCR. Fig. 6D shows that the combination of S3-NTDi and cisplatin significantly downregulated expression of MYC, BCL-XL, Survivin, VEGFA, MMP2 and APE1 when compared to single agent treatment. These results indicated that although induction of apoptotic cell death is induced with S3-NTDi and cisplatin alone, the combination treatment sensitizes MB cells to exaggerated apoptosis, in part mediated by STAT3 target gene inhibition and PIAS3 activation.
Figure 6.
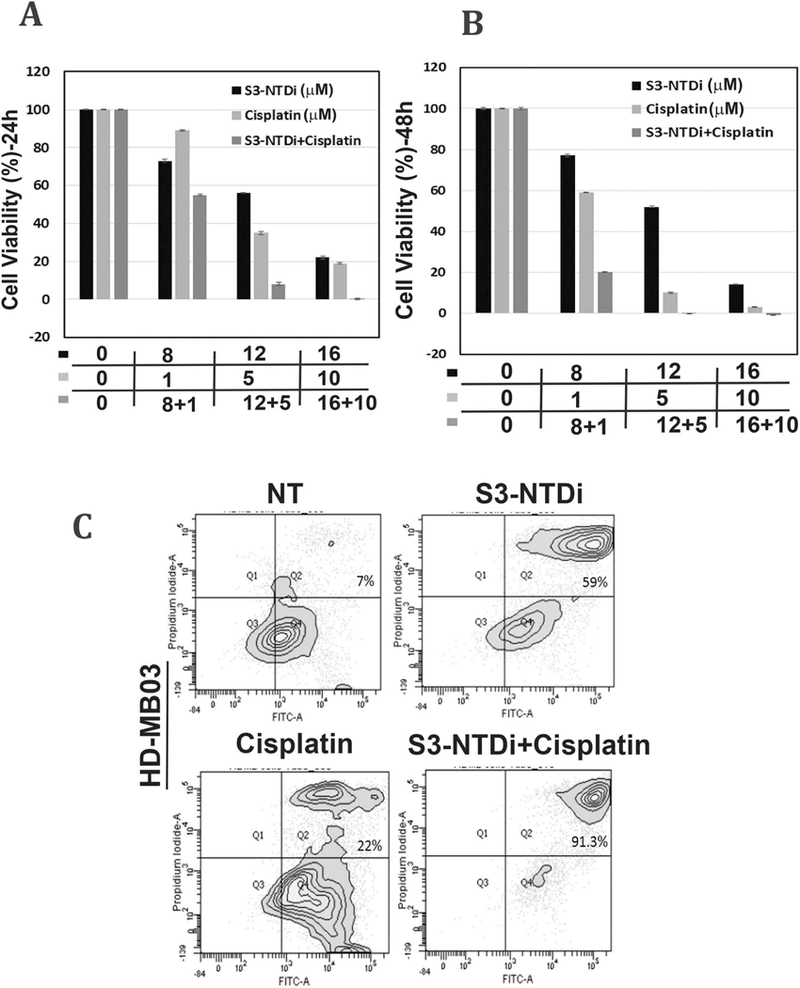
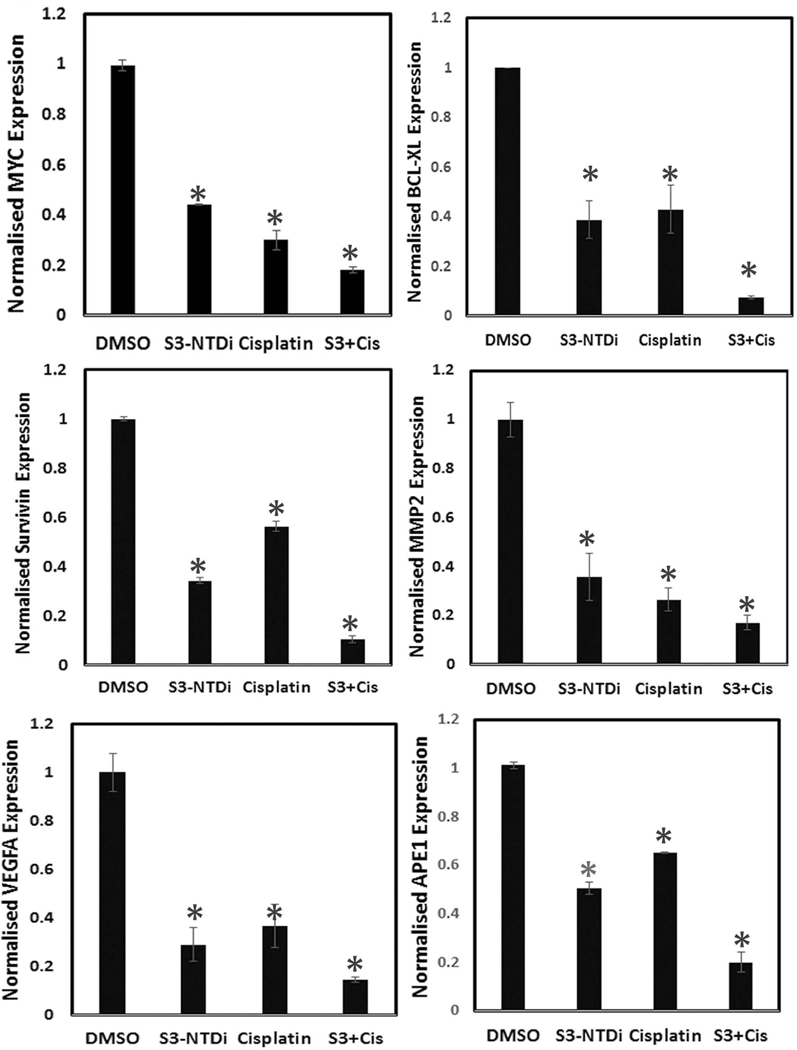
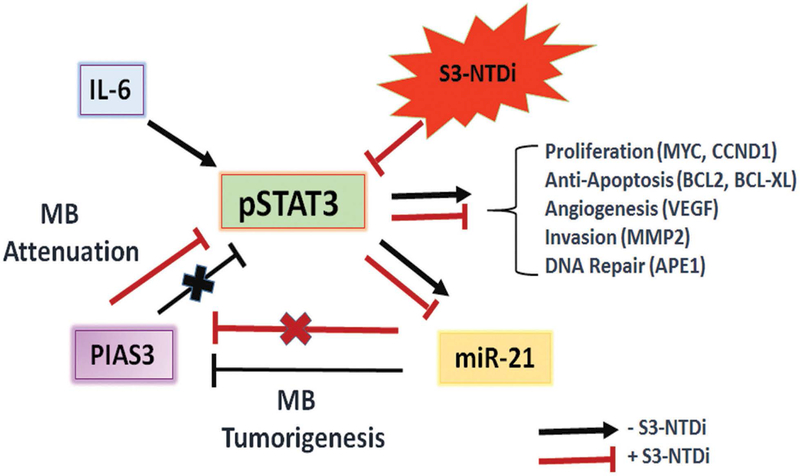
S3-NTDi confers MB cells sensitive to chemotherapy. (A) A representative bar graph shows HD-MB03 cell viability in the presence of either S3-NTDi or cisplatin alone or in combination, at specified concentrations for 24 and (B) 48 h by MTT assay. (C) Representative scatter diagram for the apoptotic cell analyses in HD-MB03, following treatment of either S3-NTDi (12 μM) or cisplatin (2.5 μM) alone or in combination for 24h. NT: non-treated control. (D) STAT3 target gene expression (as mentioned in the figure) were analyzed in HD-MB03 by qRT-PCR either in the presence of S3-NTDi (12 μM) or cisplatin (2.5 μM) alone or in combination. * represents p≤0.005, t-test. NT: non-treated control. (E) Schematic diagram of STAT3 mediated MB pathogenesis. IL6 stimulated pSTAT3 activation in MB upregulates STAT3 target genes that control proliferation, anti-apoptosis, invasion and angiogenesis. STAT3 also directly induces miR-21, which in turn represses PIAS3, a cellular STAT3 inhibitor, forming a positive feedback loop. S3-NTDi downregulates miR-21 expression, thereby activating PIAS3 which negatively regulates activated STAT3 signaling and attenuating MB tumorigenesis.
DISCUSSIONS
Human Group3 MB is characterized by amplification of the MYC oncogene, isochromosome 17q and large cell anaplastic histology and have the worst prognosis among all MB sub-types [41]. Reappearance of tumors after surgical resection is common in MB and often confers resistant to chemotherapy and radiation. Therefore, development of novel therapeutic approaches, by delineating specific molecular pathways that leads to aggressive tumor growth and chemoresistance are highly desirable [2, 42].
STAT3 is constitutively activated in human solid tumors, due to the paracrine effects of secreted IL-6 and growth factors from the tumor microenvironment [43]. Out of six functional domains of STAT3, STAT3-NTD is highly conserved between species and does not share homology with any other proteins and therefore, STAT3-NTD inhibitors offer minimum chances of off-target effects, compared to its other domains [44]. Moreover, STAT3-NTD can fold independently and can undergo several post-translational modifications (PTM), required for protein-protein interactions and recruitment of transcriptional machinery. Previously, we have shown that STAT3-NTD undergoes PTM, by acetylation and by monoubiquitination, required for nuclear complex formation with several transcriptional regulators [24, 25, 45–47]. For these reasons, we have targeted STAT3-NTD in MB by using a cell permeable peptide inhibitor that was previously been shown effective on breast and prostate cancer cells [22, 23]. We provided evidence for the first time that STAT3-NTD inhibition has profound cytotoxic effects in pediatric MB cell lines, derived from high-risk SHH-TP53-mutated (DAOY) and MYC-amplified Non-WNT/SHH tumors (HDMB and D341) [29]. Our data showed that S3-NTDi induces apoptotic cell death (Fig 1C-D), in part, by the induction of pro-apoptotic protein CHOP (Fig 2E), downregulation of BCL2 expression (Fig 2A) and cleavage of PARP (Fig 2D). Our observation that S3-NTDi downregulates a panel of cancer-associated genes in MB cells, indicates suppression of oncogenic role of STAT3. We have also shown that suppression of the STAT3-NTD, inhibited expression of oncogene MYC (Fig 2A-B), a master regulator of proliferation and oncomiR-21 that modulates expression of key regulatory protein (PIAS3) in the STAT3 signal transduction pathway (Fig 4D).
Tumor cell migration and invasion is the first step of cancer metastasis, enabled by EMT [48]. In our study, we showed that S3-NTDi reduces increased mobility of MB cells, in a wound-healing assay (Fig 3A) and downregulates IL-6 induced TWIST and SNAIL expression (Fig 3F), the direct transcriptional repressors of E-cadherin, indicated a crucial role of STAT3 in IL-6 mediated EMT. Such observation were also reported in head and neck tumors, breast cancer and in gastric cancer where, IL-6 in the tumor microenvironment induced EMT by constitutively expressed SNAIL and TWIST [49, 50].
Despite several advances in treatment, the complete eradication of MB tumors is still complicated because of the persistence of cancer “stem cells” or tumor initiating cells that lead to tumor recurrence. A recent report demonstrated that a small subpopulation of MB tumor cells that express CD133, exhibit enhanced self-renewal capacity and are dependent on STAT3 signaling [20]. Others have shown expression of neural stem cell surface markers CD133, Nestin, OCT4 and SOX2 in MB cell lines [35]. Consistent with this, S3-NTDi mediated downregulation of OCT4 and Nestin in MB-sphere extracts (Fig 4B) indicated that this inhibitor not only affects MB-sphere growth but also inhibits neural stem cell marker expression.
One striking observation of this study is that, simultaneous treatment of S3-NTDi and cisplatin synergistically killed MB cells by inducing apoptosis (Fig 6A-C). This suggested that combination of S3-NTDi with cisplatin may promote DNA damaged induced apoptotic cell death by inhibiting DNA repair pathways. Consistent with this, we found that combination treatment significantly downregulated the expression of a key DNA repair protein APE1 (Fig. 6D), which is often overexpressed in tumor cells and is implicated in resistance to many chemotherapeutic drugs, including cisplatin [51, 52]. Our study demonstrates that S3-NTDi may enhances cytotoxic efficiency of cisplatin at nontoxic doses. On the basis of our finding, it appears that use of S3-NTDi along with chemotherapy should be extended to investigate its inhibitory efficacy in in vivo mouse xenograft models.
Finally, we have provided evidence that identifies mechanisms by which S3-NTDi mediates attenuation of STAT3 signaling pathway in MB. First, we showed that S3-NTDi reduces occupancy of STAT3, in target gene promoters of BCL2 and CCND1, thereby inhibiting proliferation and protection from apoptosis (Fig 4C). Second, it inhibited oncomiR-21, involved in gene regulation by epigenetic mechanism (Fig 4D). Third, it upregulated STAT3 negative regulator PIAS3 (Fig 5B-C), (with no effect on tumor suppressor PTEN, Fig S11), and lastly, it downregulated pSTAT3 expression (Fig 5B-C). Although dysregulation of PIAS3 expression is implicated in some cancer models and very recently in MB, its regulation has not previously been described in MB [17, 53]. Our observation of pSTAT3 (Fig 1A) activation and PIAS3 downregulation (Fig 5A) in TMA, supports an inverse correlation between pSTAT3 and PIAS3 expression observed in MB cells. To summarize our results, we propose a model (Fig. 6E) in MB, where STAT3 is activated by tyrosine phosphorylation by secreted cytokines/growth factors from the tumor microenvironment. Activated STAT3 induces expression of multiple tumor promoting genes, involved in cell proliferation (MYC, CCND1), survival (BCL2, BCL2L1, Survin), invasion (MMP2) and angiogenesis (VEGF). STAT3 activation positively induces miR-21 expression, by binding to its promoter [54]. Elevated miR-21 suppresses PIAS3, possibly at the post-transcriptional level. Loss of PIAS3 expression by STAT3/ miR-21 axis, releases negative regulation by PIAS3 on STAT3 signaling pathway, thereby accelerating MB pathogenesis. By suppressing STAT3-NTD, we have established a link between STAT3/miR-21/PIAS3 circuitry that functions in MB. S3-NTDi, thereby, may serve as a novel therapeutic candidate, attenuating STAT3 signaling pathway by activation of PIAS3 and along with chemotherapy at pharmacologically achievable doses, it should therefore benefit pediatric MB patients, improving outcomes and the quality of life.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Fred & Pamela Buffett Cancer Center Support Grant (P30CA036727), Pediatric Cancer Research Group Support, State of Nebraska, LB905 (to DC) and NCI/NIH RO1 grant CA148941 (to KKB).
The authors thank the Flow Cytometry, Tissue Science facilities and Advanced Microscopy Core Facilities of University of Nebraska Medical Center for their help in these studies. The authors thank Fred & Pamela Buffett Cancer Center Support Grant (P30CA036727), Pediatric Cancer Research Group Support, State of Nebraska, LB905 (to DC) and NCI/NIH RO1 grant CA148941(to KKB).
Abbreviations used:
- MB
Medulloblastoma
- STAT3
Signal transducers and activators of transcription-3
- NTD
NH2-Terminal Domain
- MiR-21
microRNA-21
- PIAS3
Protein Inhibitor of Activated STAT3
- S3-NTDi
STAT3-NH2 terminal domain inhibitor
- TMA
Tumor microarray
- EMT
Epithelial-mesenchymal transition
REFERENCES
- 1.Gilbertson RJ, Ellison DW: The origins of medulloblastoma subtypes. Annu Rev Pathol 2008, 3:341–365. [DOI] [PubMed] [Google Scholar]
- 2.Northcott PA, Jones DT, Kool M, Robinson GW, Gilbertson RJ, Cho YJ, Pomeroy SL, Korshunov A, Lichter P, Taylor MD et al. : Medulloblastomics: the end of the beginning. Nat Rev Cancer 2012, 12(12):818–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dasgupta A, Gupta T, Jalali R: Indian data on central nervous tumors: A summary of published work. South Asian J Cancer 2016, 5(3):147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bromberg JF: Activation of STAT proteins and growth control. Bioessays 2001, 23(2):161–169. [DOI] [PubMed] [Google Scholar]
- 5.Darnell JE, Jr., Kerr IM, Stark GR: Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264(5164):1415–1421. [DOI] [PubMed] [Google Scholar]
- 6.Darnell JE Jr.: STATs and gene regulation. Science 1997, 277(5332):1630–1635. [DOI] [PubMed] [Google Scholar]
- 7.Hillmer EJ, Zhang H, Li HS, Watowich SS: STAT3 signaling in immunity. Cytokine Growth Factor Rev 2016, 31:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levy DE, Darnell JE Jr.: Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 2002, 3(9):651–662. [DOI] [PubMed] [Google Scholar]
- 9.Kiuchi N, Nakajima K, Ichiba M, Fukada T, Narimatsu M, Mizuno K, Hibi M, Hirano T: STAT3 is required for the gp130-mediated full activation of the c-myc gene. J Exp Med 1999, 189(1):63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kidder BL, Yang J, Palmer S: Stat3 and c-Myc genome-wide promoter occupancy in embryonic stem cells. PLoS One 2008, 3(12):e3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, Stark GR: Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res 2005, 65(3):939–947. [PubMed] [Google Scholar]
- 12.Srivastava J, DiGiovanni J: Non-canonical Stat3 signaling in cancer. Mol Carcinog 2016, 55(12):1889–1898. [DOI] [PubMed] [Google Scholar]
- 13.Greenhalgh CJ, Hilton DJ: Negative regulation of cytokine signaling. J Leukoc Biol 2001, 70(3):348–356. [PubMed] [Google Scholar]
- 14.Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, Shuai K: Specific inhibition of Stat3 signal transduction by PIAS3. Science 1997, 278(5344):1803–1805. [DOI] [PubMed] [Google Scholar]
- 15.Dabir S, Kluge A, Kresak A, Yang M, Fu P, Groner B, Wildey G, Dowlati A: Low PIAS3 expression in malignant mesothelioma is associated with increased STAT3 activation and poor patient survival. Clinical cancer research : an official journal of the American Association for Cancer Research 2014, 20(19):5124–5132. [DOI] [PubMed] [Google Scholar]
- 16.Schmidt D, Muller S: PIAS/SUMO: new partners in transcriptional regulation. Cell Mol Life Sci 2003, 60(12):2561–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi L, Wan Y, Sun G, Zhang S, Wang Z, Zeng Y: miR-125b inhibitor may enhance the invasion-prevention activity of temozolomide in glioblastoma stem cells by targeting PIAS3. BioDrugs 2014, 28(1):41–54. [DOI] [PubMed] [Google Scholar]
- 18.Wang C, Ba X, Guo Y, Sun D, Jiang H, Li W, Huang Z, Zhou G, Wu S, Zhang J et al. : MicroRNA-199a-5p promotes tumour growth by dual-targeting PIAS3 and p27 in human osteosarcoma. Sci Rep 2017, 7:41456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu T, Yeh JE, Pinello L, Jacob J, Chakravarthy S, Yuan GC, Chopra R, Frank DA: Impact of the N-Terminal Domain of STAT3 in STAT3-Dependent Transcriptional Activity. Mol Cell Biol 2015, 35(19):3284–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garg N, Bakhshinyan D, Venugopal C, Mahendram S, Rosa DA, Vijayakumar T, Manoranjan B, Hallett R, McFarlane N, Delaney KH et al. : CD133+ brain tumor-initiating cells are dependent on STAT3 signaling to drive medulloblastoma recurrence. Oncogene 2017, 36(5):606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao H, Bid HK, Jou D, Wu X, Yu W, Li C, Houghton PJ, Lin J: A novel small molecular STAT3 inhibitor, LY5, inhibits cell viability, cell migration, and angiogenesis in medulloblastoma cells. J Biol Chem 2015, 290(6):3418–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Timofeeva OA, Tarasova NI, Zhang X, Chasovskikh S, Cheema AK, Wang H, Brown ML, Dritschilo A: STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc Natl Acad Sci U S A 2013, 110(4):1267–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Timofeeva OA, Gaponenko V, Lockett SJ, Tarasov SG, Jiang S, Michejda CJ, Perantoni AO, Tarasova NI: Rationally designed inhibitors identify STAT3 N-domain as a promising anticancer drug target. ACS Chem Biol 2007, 2(12):799–809. [DOI] [PubMed] [Google Scholar]
- 24.Ray S, Lee C, Hou T, Boldogh I, Brasier AR: Requirement of histone deacetylase1 (HDAC1) in signal transducer and activator of transcription 3 (STAT3) nucleocytoplasmic distribution. Nucleic Acids Res 2008, 36(13):4510–4520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ray S, Lee C, Hou T, Bhakat KK, Brasier AR: Regulation of signal transducer and activator of transcription 3 enhanceosome formation by apurinic/apyrimidinic endonuclease 1 in hepatic acute phase response. Mol Endocrinol 2010, 24(2):391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nowak DE, Tian B, Brasier AR: Two-step cross-linking method for identification of NF-kappaB gene network by chromatin immunoprecipitation. Biotechniques 2005, 39(5):715–725. [DOI] [PubMed] [Google Scholar]
- 27.Roychoudhury S, Nath S, Song H, Hegde ML, Bellot LJ, Mantha AK, Sengupta S, Ray S, Natarajan A, Bhakat KK: Human Apurinic/Apyrimidinic Endonuclease (APE1) Is Acetylated at DNA Damage Sites in Chromatin, and Acetylation Modulates Its DNA Repair Activity. Mol Cell Biol 2017, 37(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mae M, Langel U: Cell-penetrating peptides as vectors for peptide, protein and oligonucleotide delivery. Curr Opin Pharmacol 2006, 6(5):509–514. [DOI] [PubMed] [Google Scholar]
- 29.Ivanov DP, Coyle B, Walker DA, Grabowska AM: In vitro models of medulloblastoma: Choosing the right tool for the job. J Biotechnol 2016, 236:10–25. [DOI] [PubMed] [Google Scholar]
- 30.Dvorak HF: Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 1986, 315(26):1650–1659. [DOI] [PubMed] [Google Scholar]
- 31.Lee SO, Yang X, Duan S, Tsai Y, Strojny LR, Keng P, Chen Y: IL-6 promotes growth and epithelial-mesenchymal transition of CD133+ cells of non-small cell lung cancer. Oncotarget 2016, 7(6):6626–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalluri R, Weinberg RA: The basics of epithelial-mesenchymal transition. J Clin Invest 2009, 119(6):1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bagheri V, Razavi MS, Momtazi AA, Sahebkar A, Abbaszadegan MR, Gholamin M: Isolation, Identification, and Characterization of Cancer Stem Cells: A Review. J Cell Physiol 2016. [DOI] [PubMed] [Google Scholar]
- 34.Zanini C, Ercole E, Mandili G, Salaroli R, Poli A, Renna C, Papa V, Cenacchi G, Forni M: Medullospheres from DAOY, UW228 and ONS-76 cells: increased stem cell population and proteomic modifications. PLoS One 2013, 8(5):e63748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mehrazma M, Madjd Z, Kalantari E, Panahi M, Hendi A, Shariftabrizi A: Expression of stem cell markers, CD133 and CD44, in pediatric solid tumors: a study using tissue microarray. Fetal Pediatr Pathol 2013, 32(3):192–204. [DOI] [PubMed] [Google Scholar]
- 36.Grunder E, D’Ambrosio R, Fiaschetti G, Abela L, Arcaro A, Zuzak T, Ohgaki H, Lv SQ, Shalaby T, Grotzer M: MicroRNA-21 suppression impedes medulloblastoma cell migration. Eur J Cancer 2011, 47(16):2479–2490. [DOI] [PubMed] [Google Scholar]
- 37.Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K: STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell 2010, 39(4):493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X, Yue P, Page BD, Li T, Zhao W, Namanja AT, Paladino D, Zhao J, Chen Y, Gunning PT et al. : Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc Natl Acad Sci U S A 2012, 109(24):9623–9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nedaeinia R, Sharifi M, Avan A, Kazemi M, Rafiee L, Ghayour-Mobarhan M, Salehi R: Locked nucleic acid anti-miR-21 inhibits cell growth and invasive behaviors of a colorectal adenocarcinoma cell line: LNA-anti-miR as a novel approach. Cancer Gene Ther 2016, 23(8):246–253. [DOI] [PubMed] [Google Scholar]
- 40.Newton HB: Review of the molecular genetics and chemotherapeutic treatment of adult and paediatric medulloblastoma. Expert Opin Investig Drugs 2001, 10(12):2089–2104. [DOI] [PubMed] [Google Scholar]
- 41.Gilbertson RJ: Medulloblastoma: signalling a change in treatment. Lancet Oncol 2004, 5(4):209–218. [DOI] [PubMed] [Google Scholar]
- 42.Ferreira M, Pomeroy SL: Medulloblastoma Subtypes Defined by Gene Expression Analysis 864. Neurosurgery 2006, 59(2):475. [Google Scholar]
- 43.Fisher DT, Appenheimer MM, Evans SS: The two faces of IL-6 in the tumor microenvironment. Semin Immunol 2014, 26(1):38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Timofeeva OA, Tarasova NI: Alternative ways of modulating JAK-STAT pathway: Looking beyond phosphorylation. JAKSTAT 2012, 1(4):274–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ray S, Boldogh I, Brasier AR: STAT3 NH2-terminal acetylation is activated by the hepatic acute-phase response and required for IL-6 induction of angiotensinogen. Gastroenterology 2005, 129(5):1616–1632. [DOI] [PubMed] [Google Scholar]
- 46.Ray S, Zhao Y, Jamaluddin M, Edeh CB, Lee C, Brasier AR: Inducible STAT3 NH2 terminal mono-ubiquitination promotes BRD4 complex formation to regulate apoptosis. Cell Signal 2014, 26(7):1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hou T, Ray S, Brasier AR: The functional role of an interleukin 6-inducible CDK9.STAT3 complex in human gamma-fibrinogen gene expression. J Biol Chem 2007, 282(51):37091–37102. [DOI] [PubMed] [Google Scholar]
- 48.Wu Y, Zhou BP: New insights of epithelial-mesenchymal transition in cancer metastasis. Acta Biochim Biophys Sin (Shanghai) 2008, 40(7):643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yadav A, Kumar B, Datta J, Teknos TN, Kumar P: IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol Cancer Res 2011, 9(12):1658–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu X, Tao P, Zhou Q, Li J, Yu Z, Wang X, Li J, Li C, Yan M, Zhu Z et al. : IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sengupta S, Mantha AK, Song H, Roychoudhury S, Nath S, Ray S, Bhakat KK: Elevated level of acetylation of APE1 in tumor cells modulates DNA damage repair. Oncotarget 2016, 7(46):75197–75209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bapat A, Fishel ML, Kelley MR: Going ape as an approach to cancer therapeutics. Antioxid Redox Signal 2009, 11(3):651–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li C, Li H, Zhang P, Yu LJ, Huang TM, Song X, Kong QY, Dong JL, Li PN, Liu J: SHP2, SOCS3 and PIAS3 Expression Patterns in Medulloblastomas: Relevance to STAT3 Activation and Resveratrol-Suppressed STAT3 Signaling. Nutrients 2016, 9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sawant DV, Wu H, Kaplan MH, Dent AL: The Bcl6 target gene microRNA-21 promotes Th2 differentiation by a T cell intrinsic pathway. Mol Immunol 2013, 54(3–4):435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.