

Abstract
Antimicrobial and anti-proliferative meleagrin and oxaline are roquefortine C-derived alkaloids produced by fungi of the genus Penicillium. Tandem O-methylations complete the biosynthesis of oxaline from glandicoline B through meleagrin. Currently, little is known about the role of these methylation patterns in the bioactivity profile of meleagrin and oxaline. To establish the structural and mechanistic basis of methylation in these pathways, crystal structures were determined for two late-stage methyltransferases in the oxaline and meleagrin gene clusters from Penicillium oxalicum and Penicillium chrysogenum. The homologous enzymes OxaG and RoqN were shown to catalyze penultimate hydroxylamine O-methylation to generate meleagrin in vitro. Crystal structures of these enzymes in the presence of methyl donor S-adenosylmethionine revealed an open active site, which lacks an apparent base indicating that catalysis is driven by proximity effects. OxaC was shown to methylate meleagrin to form oxaline in vitro, the terminal pathway product. Crystal structures of OxaC in a pseudo-Michaelis complex containing sinefungin and meleagrin, and in a product complex containing S-adenosyl-homocysteine and oxaline, reveal key active site residues with His313 serving as a base that is activated by Glu369. These data provide structural insights into the enzymatic methylation of these alkaloids that include a rare hydroxylamine oxygen acceptor, and can be used to guide future efforts towards selective derivatization and structural diversification and establishing the role of methylation in bioactivity.
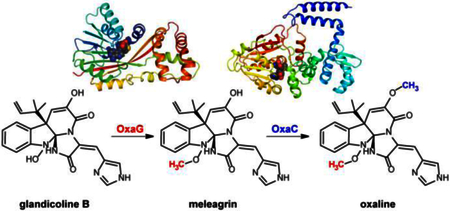
Enzymatic activity and crystal structures of the methyltransferases involved in the biosynthesis of fungal natural products meleagrin and oxaline.
INTRODUCTION
Prenylated indole alkaloids constitute a large class of fungal natural products with complex structures that often present a diverse range of bioactivities including anticancer, antibiotic, anti-parasitic, and insecticidal.1, 2 Within this class are the roquefortine C-derived alkaloids. Roquefortine C has been reported in at least 30 fungal strains.3–5 This alkaloid possesses neurotoxic and antimicrobial activities, most likely through inactivation of cytochrome P450s.6, 7 Frequently isolated from strains producing roquefortine C are glandicolines, meleagrin, and oxaline.8–10 These compounds derive from roquefortine C,11 and share a unique triazaspirocyclic skeleton (Fig. 1).12 The methylation pattern is a key difference among these metabolites; while glandicolines are unmethylated, meleagrin is singly methylated at its hydroxylamine oxygen, whereas oxaline contains a second methyl group at the enol oxygen (Fig. 1).
Fig. 1. Roquefortine C derived alkaloids.

Glandicolines, meleagrins, and oxalines are derived from roquefortine C, whose production has been reported in at least 30 fungal strains.
Currently, information is lacking to establish the importance of the differing methylation patterns with respect to biological activities of meleagrin and oxaline, since only a subset of the compounds were examined in most studies. For instance, oxaline has an IC50 value of 8.7 μM against human T cell leukemia Jurkat cells, while that for the enol-reduced neoxaline was 43.7 μM, indicating that oxaline was a more potent inhibitor of tubulin polymerization.13 When examined as a candidate for treatment of c-Met dependent metastatic and invasive breast malignancies,14 meleagrin and analogs arrested the cell cycle at the G2/M phase and demonstrated cytoxicity for tumor cell lines with IC50 values ranging from 1.8 to 6.7 μM.15 Although no comparison in bioactivity between meleagrin and oxaline or glandicoline B was reported in these studies, the IC50 values clustered in the low μM range, indicating that additional development will likely be required for these compounds to attract further interest as anticancer agents. However, it is important to note that natural modifications were capable of modulating the potency of these inhibitors.16
Additionally, glandicoline B displayed antibacterial activity against Staphylococcus aureus, Escherichia coli and Micrococcus luteus.17 Furthermore, meleagrin, glandicoline A, oxaline, and a panel of semisynthetic derivatives were assessed as inhibitors against the bacterial FabI target. No significant differences were observed between meleagrin and oxaline, which displayed IC50 values of 30–50 μM against E. coli and S. aureus FabI. Notably, methylation of the secondary amide nitrogen at the triazaspiro-center increased inhibitor potency.18
The biosynthesis of the roquefortine C and meleagrin has been explored by genetic disruptions19 and a combination of gene silencing20 and disruption.21 The meleagrin biosynthetic gene cluster has been identified in P. chrysogenum, and catalytic roles have been assigned for the corresponding gene products. More recently, our group identified a homologous gene cluster for the biosynthesis of oxaline in P. oxalicum F30 (Fig. 2).22 Therefore, we sought to perform direct biochemical characterization of key biosynthetic steps in the construction of the unique triazaspirocyclic core system from roquefortine C, and the additional tailoring steps that furnish oxaline. While numerous bacterial derived natural product methyltransferases have been investigated,23 to the best of our knowledge no examples of natural product methyltransferases from fungal pathways had been structurally characterized at the outset of this study. The unique catalytic requirements for an N-OH methyltransferase are unknown for any system, as no such transformation has been studied. Recently, the S-methyltransferases, TmtA and GtmA, have been reported from the gliotoxin biosynthetic pathway.24, 25
Fig. 2. Oxaline biosynthesis.

(A) Gene cluster encoding the biosynthesis of oxaline (3) in P. oxalicum F30. (B) Two methyltranferases, OxaG and OxaC, catalyze late-stage tailoring of glandicoline B (1) to oxaline.
We report here the biochemical activity and crystal structure of three methyltransferases in the biosynthesis of meleagrin and oxaline. These data provided structural and mechanistic insights into the enzymatic methylation of these roquefortine C-derived alkaloids. Our results expand the understanding of enzymatic hydroxylamine methylation and will facilitate future efforts towards derivatization of complex small molecules through site specific, late-stage methylation.
RESULTS AND DISCUSSION
Reconstitution of oxaline biosynthesis from glandicoline B.
We recently reported the oxaline gene cluster from P. oxalicum F30,22 which is homologous to the gene cluster of meleagrin from P. chrysogenum.19, 20 OxaG has 73% sequence identity with RoqN. We reasoned that OxaG and RoqN were responsible for hydroxylamine O-methylation of glandicoline B to generate meleagrin, as it has been demonstrated previously by gene disruption19 and silencing20 in P. chrysogenum. The second methyltransferase, OxaC, lacks a homologous enzyme in P. chrysogenum. Thus, we reasoned that OxaC catalyzes the C9 enol methylation that differentiates oxaline from meleagrin. OxaG and OxaC were cloned from a P. oxalicum F30 cDNA library and heterologously expressed in E. coli. As expected, glandicoline B (1) was converted to meleagrin (2) in the presence of OxaG and SAM (Fig. 3). Low levels of oxaline (3) were also detected after overnight incubation suggesting that the enzyme has a limited capacity to perform a second methylation reaction. Methylation of meleagrin to oxaline was efficiently performed by OxaC in the presence of SAM, resulting in the reconstituted enzymatic synthesis of oxaline from glandicoline B (Fig. 3). Incubation of OxaC with glandicoline B and SAM led to a singly methylated product with the same m/z as meleagrin but a different chromatographic mobility. Preparative-scale enzymatic reactions with OxaC and glandicoline B were performed to generate sufficient quantities of this methylated product for structural characterization. After chromatographic separation, NMR analysis of the product confirmed the expected C9 enol methylation of glandicoline B to generate a previously undescribed metabolite, glandicoline C (4). Purified 4 is not converted to oxaline by OxaG. This suggests that OxaC is regulated in vivo to prevent the accumulation of 4. Steady-state kinetic assays were performed under initial velocity conditions to determine the kinetic constants for the native methylation reactions of OxaC (Table 1). Steady-state kinetic analysis of OxaG was impeded by the high concentration of enzyme (30 μM) required to observe significant product formation on short timescale (0–20 min). Nonetheless, the measured catalytic efficiency (kcat/KM) of 3.3 × 101 M−1·s−1 was substantially lower than that for OxaC of 4.2× 105 M−1·s−1. This drastic difference is brought about primarily by the low turnover number for OxaG, also suggesting that in vivo regulation is required to ensure proper timing of methylation.
Fig. 3. OxaC and OxaG enzymatic activity.

HPLC traces of authentic standards of glandicoline B (1) and meleagrin (2) incubated with OxaC or OxaG and SAM. The traces indicate that OxaG catalyzes methylation of 1 to generate 2, while OxaC catalyzes methylation of both 1 and 2 forming a new metabolite glandicoline C (4) and 3, respectively. The singly methylated product 4 is not a substrate for OxaG.
Table 1:
X-ray Data Collection and Refinement Statistics
| OxaC | OxaC-MLG | OxaC-OXA | RoqN | OxaG | |
|---|---|---|---|---|---|
| PDB | 5W7P | 5W7S | 5W7R | 5W7M | 5W7K |
| Wavelength (Å) | 0.979 | 1.03 | 1.03 | 0.979 | 0.979 |
| Resolution range | 40.4 – 2.40 (2.49 – 2.40) | 46.68 – 2.95 (3.05 – 2.95) | 41.93 – 2.50 (2.59 – 2.50) | 47.01 – 1.70 (1.76 – 1.70) | 49.27 – 1.99 (2.06 – 1.99) |
| Space group | I 4 2 2 | I 4 2 2 | I 4 2 2 | C 2 2 21 | C 1 2 1 |
| Unit cell (Å) | 162.2 162.2 91.7 | 163.0 163.0 91.3 | 162.9 162.9 90.0 | 49.1 100.8 130.1 | 121.5 36.2 126.2 90° 102.8° 90° |
| Total reflections | 590672 (55718) | 193557 (17961) | 349191 (34123) | 232270 (22847) | 238594 (21483) |
| Unique reflections | 24189 (2380) | 13227 (1248) | 21298 (2097) | 35677 (3433) | 37043 (3459) |
| Multiplicity | 24.4 (23.4) | 14.6 (14.4) | 16.4 (16.3) | 6.5 (6.7) | 6.4 (6.2) |
| Completeness (%) | 99.9 (100.0) | 99.5 (96.4) | 99.9 (99.3) | 99.2 (96.9) | 99.2 (94.3) |
| Mean I/sigma(I) | 27.08 (2.29) | 16.37 (1.69) | 22.28 (1.68) | 20.63 (2.02) | 9.26 (1.79) |
| Wilson B-factor | 65.6 | 76.9 | 59.1 | 31.4 | 35.9 |
| R-merge | 0.078 (1.28) | 0.180 (1.84) | 0.120 (1.95) | 0.060 (0.659) | 0.122 (0.959) |
| R-meas | 0.079 (1.31) | 0.186 (1.91) | 0.124 (2.01) | 0.065 (0.714) | 0.133 (1.05) |
| CC1/2 | 1 (0.838) | 0.999 (0.779) | 0.999 (0.559) | 0.999 (0.88) | 0.997 (0.827) |
| R-work | 0.195 (0.316) | 0.191 (0.361) | 0.219 (0.340) | 0.182 (0.313) | 0.221 (0.326) |
| R-free | 0.238 (0.367) | 0.251 (0.466) | 0.249 (0.392) | 0.222 (0.328) | 0.262 (0.363) |
| Number of atoms | 3102 | 3076 | 3084 | 2420 | 4380 |
| protein | 3067 | 3037 | 3041 | 2216 | 4261 |
| solvent | 35 | 7 | 10 | 204 | 118 |
| RMS(bonds)(Å) | 0.008 | 0.010 | 0.010 | 0.008 | 0.008 |
| RMS(angles)(deg) | 1.02 | 1.53 | 1.41 | 0.93 | 0.98 |
| Average B-factor | 82.0 | 83.2 | 70.7 | 36.4 | 50.4 |
| Ramachandran favored (%) | 96.68 | 90.49 | 94.59 | 99.26 | 97.21 |
| allowed (%) | 3.06 | 9.00 | 4.90 | 0.74 | 2.79 |
| outliers (%) | 0.26 | 0.51 | 0.51 | 0.00 | 0.0 |
Crystal structure of hydroxylamine methyltransferases, RoqN and OxaG.
In order to study the structural basis of the first biosynthetic methylation reaction, which generates meleagrin, we sought to determine the crystal structure of OxaG. Diffraction-quality, single crystals of native and selenomethioninyl OxaG were obtained after screening and refinement. However, we were unable to obtain a phasing solution using single-wavelength anomalous diffraction (SAD), likely due to the presence of pseudotranslational symmetry in the lattice. This led us to pursue a crystal structure for RoqN, which shares 73% identity/84% similarity with OxaG and catalyzes the same reaction in vitro (Fig. S1). Crystals of selenomethionyl RoqN showed no lattice pathologies and the structure was solved using SAD (Fig. 4). This model enabled us to determine the structure of OxaG by molecular replacement. A non-crystallographic two-fold symmetry axis in the C2 unit cell was nearly parallel to the crystallographic two-fold symmetry axis, giving rise to pseudo-translational symmetry between two non-equivalent subunits in the OxaG asymmetric unit (Fig. S2). The refined structures for RoqN and OxaG are highly similar with an RMSD = 0.67 Å. The only notable difference is in the orientation of the surface-exposed loop between α9 and β7 near the C-terminus (Fig. S3).
Fig. 4. RoqN crystal structure.

(A) Cartoon representation of RoqN shown in rainbow coloring. Bound SAH is shown as spheres. (B) Surface representation colored by atom of RoqN monomer. The active site is large and open to solvent. (C) Alignment of RoqN (green) and OxaC (gray) methyltransferases. Differences between the acceptor binding domains are apparent, while the SAM binding domains are conserved.
RoqN and OxaG are class I methyltransferases with the characteristic α/β fold of many small-molecule methyltransferases (Fig. 4). A canonical SAM binding domain is present, and the SAH product was bound in both structures (Fig. 5A). The canonical DXGXGXG motif is present and contacts the cofactor ribose. OxaG and RoqN are structurally similar to the gliotoxin S-methyltransferase TmtA,24 with an RMSD = 1.68 Å (Fig. S4A).
Fig. 5. RoqN substrates and modeling.

(A) Bound SAH in the RoqN active site. The electron density (3.0σ) was calculated from coefficients of the form Fo - Fc where SAH was omitted from the phase calculation and refinement. (B) Glandicoline B (white) was modeled into the RoqN active site (green) using Autodock Vina. SAM was modeled manually using the electron density for SAH followed by real space refinement. The lowest energy dock (−9.4 kcal/mol, shown) gave a close distance (3.6Å) between the donor methyl and the acceptor hydroxylamine in the expected near linear arrangement. The conserved Tyr20 is not positioned to activate the acceptor hydroxylamine. (C) Space-filling representation shows the open nature of the RoqN active site.
The cofactor position was used to locate the active site, which surprisingly is large and widely exposed to the solvent (Fig. 4B). Importantly, neither crystal structure had interpretable electron density for the fifteen N-terminal amino acids. The first modeled residues form a significant portion of the putative active site (Fig. 5B). While it is tempting to speculate that the disordered N-terminal region could function as a lid or mobile element that is ordered upon substrate binding, no evidence to support this hypothesis was observed in the crystal structures. The crystal packing in this N-terminal region for both the OxaG and RoqN structures, which crystallized in different space groups, was closely inspected; however no lattice contacts were observed that would obstruct movement of the N-terminal amino acids about the active site. Irrespective of the position of the disordered N-terminal residues, the active site cavity is significantly larger than the glandicoline B acceptor (Fig. 5C).
Despite attempts at co-crystallization and soaking with glandicoline B in both OxaG and RoqN, no change in the N-terminal region and no electron density for the methyl acceptor was observed. This led us to employ Autodock VINA to examine the potential binding modes of the methyl acceptor. Given the large size of the active site, several nearly isoenergetic binding states were found that positioned glandicoline B in multiple orientations in the active site. The most energetically favored state (−8.9 kcal/mol) placed the acceptor N-OH within 3.6 Å of the donor methyl group (Fig. 5B), and in the proper alignment for an SN2 methyltransfer reaction. This binding mode was dominated by nonpolar interactions as the reverse prenyl moiety and indole ring of the substrate were buried in a hydrophobic pocket composed of Ile26, Trp157, Ile230 and Trp277 (Fig. 5).
The molecular docking experiments did not support the role of Tyr20 as an active site base in the reaction. This conserved residue was the only basic amino acid proximal to the methyl donor. To interrogate the role of Tyr20, we generated OxaG/Y20A and Y20F. These mutants showed comparable specific activity to wild-type OxaG, suggesting that an active site base is not required for catalysis. Instead, the enzyme presumably drives catalysis by positioning the methyl donor near the acceptor. This indicates that the hydroxylamine acceptor possesses enough intrinsic nucleophilicity to react with the SAM donor. The homologous methyltransferase TmtA also lacks an active site base, yet the corresponding Tyr20 is present in TmtA. Although this residue is not appropriately positioned to serve as an active site base in the methylation of gliotoxin, it was still important for catalysis as the alanine mutant was inactive24. This is likely due to disruption of the hydrogen bond from Tyr20 to the SAM terminal carboxylate (Fig. S4B). A water molecule forms the analogous hydrogen bond in both OxaG and RoqN, and OxaG/Y20A is active, consistent with no catalytic role for Tyr20.
A final feature regarding substrate binding is the lack of recognition of glandicoline C (4) by RoqN/OxaG. In the docked structure that places the N-OH in position for catalysis, a close contact between Trp162 and the enol hydroxyl group (Fig. 5) indicates that this residue forms an important recognition element as methylation of the hydroxyl group would be expected to perturb this binding mode. In fact, molecular docking with 4 showed a different binding mode as the most favorable, and the energy was higher (−8.2 kcal/mol) than that for glandicoline B.
In summary, OxaG and RoqN belong to the CoQ/UbiE family of small-molecule methyltransferases that catalyze a previously uncharacterized N-OH methylation reaction. The overall OxaG and RoqN structure was found to be similar to the recently reported gliotoxin TmtA and GtmA S-methyltransferases from A. fumigatus.24, 25 Active sites of OxaG and RoqN are unique in their large size and solvent accessibility. Molecular docking revealed several possible binding modes for the methyl acceptor, with the most favored state showing glandicoline B buried in a hydrophobic pocket of the enzyme. The conserved residue Tyr20 does not serve as an active site base and was not required for catalysis.
Crystal structure of OxaC.
The second methylation from glandicoline B through meleagrin generates oxaline, which is catalyzed by OxaC, a methyltransferase unique to the oxaline gene cluster. The structure of OxaC was determined by SAD from the selenomethioninyl protein. The overall structure of OxaC is a typical class I methyltransferase fold, with the SAM donor in the C-terminal domain and the N-terminal domain making up the dimerization domain and acceptor binding site (Fig. 6). Several structural homologues were identified using the DALI server.26 The closest matches were mitomycin 7-O-methyltransferase, MmcR, and carminomycin 4-O-methyltransferase, DnrK, both of which had an RMSD of 2.2 Å with OxaC.27, 28 Considering the alignments with structural homologues, a unique feature of OxaC is a pair of N-terminal helices that comprise a 4-helix bundle in the protein dimer. In OxaC crystals, the dimer forms through a crystallographic symmetry operator. The two subunits are highly interdigitated and bury 4697 Å2 surface area (PISA). The contacts within this 4-helix bundle are predominantly hydrophobic and a significant amount of the total buried surface area is contributed by this region. The role of this 4-helix bundle is currently unknown. The OxaC homologue MmcR lacks these N-terminal helices, yet dimerizes in a similar fashion, as was observed in several structural homologues of OxaC.27, 28 A construct in which the N-terminal two helices were removed from OxaC was insoluble in E. coli, suggesting that they are required for proper folding and oligomerization.
Fig. 6. OxaC complex structures.

(A) Cartoon representation of OxaC monomer shown in rainbow. SAH is shown as spheres. (B) OxaC dimer depicted as a hybrid of cartoon and surface representation. The two N-terminal helices (blue) form a four-helix bundle in the dimer. (C) Pseudo-Michaelis complex with OxaC·sinefungin (SFG)·meleagrin. (D) Product complex with OxaC·S-adenosyl-L-homocysteine (SAH)·oxaline. The electron density (Fo - Fc, 2.5σ contours) was calculated from a structure where the acceptor was omitted from the phase calculation and refinement.
Structures were obtained for a pseudo-Michaelis complex composed of the SAM analog sinefungin and the natural substrate meleagrin (Fig. 6C), and a product complex containing SAH and oxaline (Fig. 6D). The electron density places meleagrin close (2.7 Å) to the methyl donor. The substrate is deeply buried in the enzyme active site and binds in a predominantly hydrophobic environment (Fig. S5). The indole ring and reverse prenyl groups of the substrate lie closely packed against Val150, Ile155, Met193, Val207, Ala210, Trp357, Leu362, Ile365 of the acceptor binding domain. The substrate appears to access the active site through a channel that extends to the enzyme surface near the dimer interface.
As with other methyltransferases in this class, OxaC uses acid/base activation for catalysis through a highly conserved Glu/His dyad (Fig. 7). In OxaC, Glu369 is appropriately placed to activate His313 for the deprotonation of the meleagrin O9 group. Additionally an aspartate residue (Asp314) forms a hydrogen bond with O9 (Fig. 7). To probe its role in catalysis we prepared a D314A mutant in OxaC and observed no significant decrease in specific activity, which led us to conclude that this residue plays a minor role in substrate activation. We observed little conformational difference between the product complex and the pseudo-Michaelis complex (Fig. 7B). This suggests that little conformational movement occurs in the protein during catalysis. Thus, we propose a mechanism for OxaC in which the substrate is activated for nucleophilic attack predominantly by His313 (Fig. 7C). A homologous active site configuration is present in caffeic acid O-methyltransferase from ryegrass (Lolium perenne) (Fig. 7D)29. In that study, the authors similarly determined that a His313/Glu326 dyad was required for catalysis, while Asp267 was shown to interact with phenolic oxygens on the substrate in some cases.
Fig. 7. OxaC active site architecture and mechanism.

(A) Meleagrin (yellow) is ideally positioned for SN2 attack of the donor methyl. (B) The product oxaline (green) shows little displacement after methylation. (C) CoMT (pink) shows homologous active site organization. The phenolic acceptor sinapaldehyde (cyan) is similarly activated by His and Asp residues. All distances are labeled in Ångstroms. (D) Proposed OxaC mechanism. Glu369 primes the active site base His313 for deprotonation of the enol. Asp314 likely plays a role in acceptor positioning.
In summary, the crystal structure of OxaC reveals a canonical class I methyltransferase fold that forms a dimeric assembly tethered by a unique four helix bundle. Structures of a pseudo-Michaelis complex and a product complex provide clear snapshots of O-methylation of meleagrin. The acceptor lies deeply buried in a hydrophobic pocket. A catalytic dyad composed of Glu369 and His313 activates O9 of meleagrin for nucleophilic attack of the SAM donor to form oxaline with little conformational movement in the protein backbone.
Together these data provide structural and mechanistic insights into three fungal natural product methyltransferases. While their respective acceptor substrates have the same core scaffold, the enzymes differ markedly in their three dimensional structure and catalytic efficiency. OxaG and RoqN have a large, solvent exposed active site and a turnover number that is four orders of magnitude lower than OxaC, which by contrast appears finely tuned for O9 methylation of meleagrin and glandicoline B. These studies will guide future efforts that rely on site specific methylation to expand chemical diversity of complex natural products, either through mutasynthesis,30 or through semisynthetic and chemoenzymatic derivatization.22
METHODS
Production of P. oxalicum F30 cDNA library.
P. oxalicum was statically cultivated in 20% artificial seawater with 12.0 g/L glucose, 6.0 g/L starch, 12.0 g/L soytone, 3.0 g/L peptone, 0.18 g/L meat extract, 3.0 g/L yeast extract at 28 °C for 14 days. RNA was isolated using the TRIzol® Reagent (Ambion RNA, Life Technologies). Reverse transcription PCR (RT-PCR) was performed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) using Oligo (dT) primer (Life Technologies).
Production of P. chrysogenum cDNA library.
P. chrysogenum was statically cultivated in 22 g/L corn steep liquor, 40 g/L glucose at 28 °C for 14 days. RNA and cDNA were prepared as above.
Isolation of glandicoline B from Penicillium glandicola IBT 21529.
Isolation of glandicoline B was performed according to the procedures described by Smedsgaard et al.31.
Cloning of methyltransferases.
All constructs were introduced into a pET28 vector modified to contain a Tobacco Etch Virus (TEV) protease cleavage site following an N-terminal 8X His tag using Quikchange cloning strategy.32 Active site mutations were generated using single primer Quikchange mutagenesis (Agilent). Primer sequences are listed in Table S1.
Methyltransferase overexpression and purification.
All enzymes were overexpressed in E. coli BL21 (DE3) and purified in the same manner. Cells were resuspended in lysis buffer (10 mM HEPES pH 7.6, 50 mM NaCl, 10% glycerol, 0.2 mM TCEP, 0.1 mM PMSF) and lysed by the addition of lysozyme, Benzonase (Merck kGaA, 1 kU), and MgSO4 (5 mM) followed by sonication. The lysate was clarified by centrifugation followed by purification using NiNTA agarose. Enzymes were concentrated in a 10 kDa MWCO Amicon Ultra centrifugal concentrator (Millipore) and exchanged into storage buffer (10 mM HEPES pH 7.6, 50 mM NaCl, 0.2 mM TCEP) using a PD-10 desalting column (GE Healthcare). The purified enzymes were flash frozen in liquid nitrogen, and stored at −80 °C. Selenomethioninyl (SeMet) protein was produced by metabolic inhibition33 and purified in the same manner. All proteins were screened for initial crystallization conditions using the MCSG Suite (Microlytic).
Enzyme kinetics.
The steady state kinetic constants of OxaC were determined using an HPLC-based assay. Samples were analyzed using a ZORBAX SB-Phenyl column (4.6×150 mm, 5 μm) using a linear gradient of 15–45% MeCN in ddH2O + 0.1% formic acid over 7 min (2.5 mL/min flow rate). Initial velocities were fit by non-linear regression to the Michaelis-Menten equation using GraphPad Prism Version 6.01 software to determine the kinetic constants kcat and KM. Reactions were initiated by the addition of 100 μL of varied concentrations of substrate in 100% DMSO to 4.1 mL of 0.9 nM OxaC, 0.5 mM SAM, 50 mM Na/K/PO4 pH 8.0(23 °C). 1.0 mL aliquots were quenched by plunging into liquid N2 at various time points (5–20 min). Four time points were taken in each time course. All reactions were performed in duplicate. The frozen aliquots were lyophilized to dryness over 24 h and resuspended in 80 μL MeOH. Similar procedures for OxaG were used under the following reaction conditions: OxaG reactions were initiated by the addition of varied concentrations of glandicoline B in 100% DMSO to 30 μM OxaG, 50 mM Na/K/PO4 pH 8.0, 10% glycerol (23 °C). Aliquots were quenched by the addition of three volumes MeOH at various time points to achieve less than 20% substrate consumption over the reaction time course.
X-ray data collection.
Diffraction data was collected on GM/CA beamlines at the Advanced Photon Source and data was integrated and scaled in XDS34.
Crystallization of OxaG.
Crystals of both native and SeMet OxaG were grown at 20 °C by sitting drop vapor diffusion in Intelli-Plate™ 96–2 shallow well plates (Hampton Research) by combining 1 μL OxaG at 11 mg/mL and 1 mM SAM in storage buffer with 1 μL of well solution composed of 23% PEG3350, 2% MPD, 260 mM MgCl2, 50 mM BisTris pH 6.7. Droplets were nucleated after 2.5 h using a cat’s whisker charged OxaG crystals from an earlier crystallization event. The crystals were cryoprotected by the direct addition of 10 μL of a cryoprotectant solution composed of 24% PEG 3350, 25% MPD, 260 mM MgCl2, 50 mM BisTris pH 6.7, 50 mM NaCl, 10 mM HEPES pH 7.6, 1 mM SAM to the crystallization droplet. Crystals were flash frozen in liquid nitrogen. The structure of OxaG was solved in Molrep35 by using the RoqN structure as search model. The initial model was subjected to iterative rounds of building and refinement using Coot,36 Refmac,37 and Phenix38.
Crystallization of RoqN.
Crystals of both native and SeMet RoqN were grown at 20 °C by sitting drop vapor diffusion in Intelli-Plate™ 96–2 shallow well plates (Hampton Research) by combining 1 μL RoqN at 12 mg/mL and 1 mM SAM in storage buffer with 1 μL of well solution composed of 19% PEG 3350, 2% ethylene glycol, 200 mM Mg(C2H3O2)2. Droplets were immediately nucleated using a cat whisker charged with RoqN crystals from an earlier crystallization event. The crystals were cryoprotected by the direct addition of 10 μL of a cryoprotectant solution composed of 20% PEG 3350, 23% ethylene glycol, 200 mM Mg(C2H3O2)2, 50 mM NaCl, 10 mM HEPES pH 7.6, 1 mM SAM to the crystallization droplet. Crystals were flash frozen in liquid nitrogen. RoqN was solved by single anomalous dispersion (SAD) in the CCP4 software package39 by using the Crank2 pipeline40 in the default configuration. The initial model was subjected to iterative rounds of building and refinement using Coot36 and Refmac 37.
Crystallization of OxaC.
Crystals of both native and SeMet OxaC were grown at 20 °C by sitting drop vapor diffusion in Intelli-Plate™ 24–4 plates (Hampton Research) by combining 2 μL OxaC at 14 mg/mL and 1 mM SAM in storage buffer with 2 μL of well solution composed of 1 M Li2SO4, 400 mM (NH4)2SO4, 20 mM sodium citrate, 18.75% glycerol. The crystals were flash frozen directly from mother liquor in liquid nitrogen. Complex structures were obtained by co-crystallization in the same conditions by combining protein solution with 1 mM meleagrin and 1.6 mM SAM for the product complex and 1 mM meleagrin and 3.125 mM sinefungin for the pseudo-Michaelis complex. OxaC was solved by single anomalous dispersion (SAD) in the CCP4 software package39 by using the Crank2 pipeline40 in the default configuration. The initial model was subjected to iterative rounds of building and refinement using Coot36 and Refmac37.
Enzymatic production of glandicoline C.
Glandicoline C was generated under the following reaction conditions: 10 μM OxaC (8.3 mg), 0.5 mM SAM (14.4 mg), 0.25 mM glandicoline B (2.0 mg), 50 mM Na/K/PO4 pH 7.8, 5% DMSO in 18.8 mL deionized H2O for 2 h at 21 °C in a 9 dram glass vial. Solid NaCl was added to saturation and the reaction was extracted 3 times with 10 mL EtOAc. The combined organic fractions were washed twice with an equal volume of brine, dried over Na2SO4 and concentrated by rotary evaporation. The product was suspended in 0.5 mL MeOH and purified by reverse phase HPLC using a linear gradient of 15–55% MeCN in H2O + 0.1% formic acid over 30 min (3.0 mL/min flow rate) on a Phenomenex Luna Phenyl Hexyl column (5μm, 250 × 10 mm). Elution fractions were lyophilized to give 1.2 mg glandicoline C as a pale yellow solid (92% conversion, 58% isolated yield).
Glandicoline C (4): yellow solid; [α]26D −23.8 (c 0.08, MeOH); UV (MeOH) λmax(log ε) 289 sh (3.41), 343 (3.87) nm; IR (KBr) νmax: 3279, 2925, 1699, 1674, 1630, 1589, 1460, 1353, 1316, 1244, 1232, 1109, 1041 cm−1; 1H NMR and 13C NMR data are provided in Table S2. HRMS (ESI-TOF): m/z [M + H]+ calcd for C23H24N5O4 434.1823, found 434.1829.
Supplementary Material
Table 2.
Steady state kinetic constants.
| Enzyme | Substrate | kcat (s−1) | KM (μM) | kcat/KM (M−1∙s−1) |
|---|---|---|---|---|
| OxaC | glandicoline B | 0.67 ± 0.04 | 4.1 ± 0.5 | 1.6 × 105 |
| OxaC | meleagrin | 1.3 ± 0.2 | 3.1 ± 0.9 | 4.2 × 105 |
| OxaG | glandicoline B | 0.0012 ± 0.0001 | 36 ± 11 | 3.3 × 101 |
ACKNOWLEDGEMENTS
We ae grateful to Dr. Hector Henrique Ferreira Koolen (CBA, Manaus, Brazil) for a gift of glandicoline B.
Funding Sources: This work was supported by, FAPESP (grants 2012/50026–3, 2013/50228–8 and 2014/05670–7) awarded to R.G.S.B, a post-doctoral fellowship (CAPES, BEX 4498/14–3) awarded to S.R., NSF CHE 1220121, and NIH grants CA070375 (R.M.W, D.H.S.) and GM118101 (D.H.S.), and the Hans W. Vahlteich Professorship (D.H.S.).
Footnotes
ASSOCIATED CONTENT
Supporting Information
Full experimental methods, supplementary figures and tables, and NMR spectra
The authors declare that they have no conflicts of interest with the contents of this article.
REFERENCES
- 1.Li SM, Nat Prod Rep, 2010, 27, 57–78. [DOI] [PubMed] [Google Scholar]
- 2.Tibrewal N and Tang Y, Annu Rev Chem Biomol, 2014, 5, 347–366. [DOI] [PubMed] [Google Scholar]
- 3.Polonsky J, Merrien MA and Scott PM, Ann Nutr Aliment, 1977, 31, 963–968. [PubMed] [Google Scholar]
- 4.Ohmomo S, Utagawa T and Abe M, Agr Biol Chem Tokyo, 1977, 41, 2097–2098. [Google Scholar]
- 5.Ohmomo S, Sato T, Utagawa T and Abe M, Agr Biol Chem Tokyo, 1975, 39, 1333–1334. [Google Scholar]
- 6.Aninat C, André F and Delaforge M, Food Addit Contam, 2005, 22, 361–368. [DOI] [PubMed] [Google Scholar]
- 7.Kopp-Holtwiesche B and Rehm HJ, J Environ Pathol Toxicol Oncol, 1990, 10, 41–44. [PubMed] [Google Scholar]
- 8.Nagel DW, Pachler KGR, Steyn PS, Wessels PL, Gafner G and Kruger GJ, J Chem Soc, Chem Commun, 1974, 0, 1021–1022. [Google Scholar]
- 9.Qu P, Wu ZY and Zhu WM, Acta Crystallogr Sect E Struct Rep Online, 2012, 68, o1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Overy DP, Nielsen KF and Smedsgaard J, Journal of Chemical Ecology, 2005, 31, 2373–2390. [DOI] [PubMed] [Google Scholar]
- 11.Reshetilova TA, Vinokurova NG, Khmelenina VN and Kozlovskii AG, Microbiology, 1995, 64, 27–29. [Google Scholar]
- 12.Gober CM, Carroll PJ and Joullie MM, Mini Rev Org Chem, 2016, 13, 126–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koizumi Y, Arai M, Tomoda H and Omura S, Biochim Biophys Acta, 2004, 1693, 47–55. [DOI] [PubMed] [Google Scholar]
- 14.Mady MS, Mohyeldin MM, Ebrahim HY, Elsayed HE, Houssen WE, Haggag EG, Soliman RF and El Sayed KA, Bioorg Med Chem, 2015, DOI: 10.1016/j.bmc.2015.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du L, Feng T, Zhao B, Li D, Cai S, Zhu T, Wang F, Xiao X and Gu Q, J Antibiot (Tokyo), 2010, 63, 165–170. [DOI] [PubMed] [Google Scholar]
- 16.Gomes NG, Lefranc F, Kijjoa A and Kiss R, Mar Drugs, 2015, 13, 3950–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koolen HHF, Soares ER, da Silva FMA, de Almeida RA, de Souza ADL, de Medeiros LS, Rodrigues E and de Souza AQL, Quim Nova, 2012, 35, 771–774. [Google Scholar]
- 18.Zheng CJ, Sohn MJ, Lee S and Kim WG, PloS one, 2013, 8, e78922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia-Estrada C, Ullan RV, Albillos SM, Fernandez-Bodega MA, Durek P, von Dohren H and Martin JF, Chemistry & biology, 2011, 18, 1499–1512. [DOI] [PubMed] [Google Scholar]
- 20.Ali H, Ries MI, Nijland JG, Lankhorst PP, Hankemeier T, Bovenberg RA, Vreeken RJ and Driessen AJ, PloS one, 2013, 8, e65328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ries MI, Ali H, Lankhorst PP, Hankemeier T, Bovenberg RA, Driessen AJ and Vreeken RJ, J Biol Chem, 2013, 288, 37289–37295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newmister SA, Gober CM, Romminger S, Yu F, Tripathi A, Parra LL, Williams RM, Berlinck RG, Joullie MM and Sherman DH, J Am Chem Soc, 2016, 138, 11176–11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liscombe DK, Louie GV and Noel JP, Nat Prod Rep, 2012, 29, 1238–1250. [DOI] [PubMed] [Google Scholar]
- 24.Duell ER, Glaser M, Le Chapelain C, Antes I, Groll M and Huber EM, ACS Chem Biol, 2016, 11, 1082–1089. [DOI] [PubMed] [Google Scholar]
- 25.Dolan SK, Bock T, Hering V, Owens RA, Jones GW, Blankenfeldt W and Doyle S, Open Biol, 2017, 7, 160292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holm L and Laakso LM, Nucleic Acids Res, 2016, 44, W351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jansson A, Koskiniemi H, Mantsala P, Niemi J and Schneider G, J Biol Chem, 2004, 279, 41149–41156. [DOI] [PubMed] [Google Scholar]
- 28.Singh S, Chang A, Goff RD, Bingman CA, Gruschow S, Sherman DH, Phillips GN Jr. and Thorson JS, Proteins, 2011, 79, 2181–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Louie GV, Bowman ME, Tu Y, Mouradov A, Spangenberg G and Noel JP, Plant Cell, 2010, 22, 4114–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ouchaou K, Maire F, Salo O, Ali H, Hankemeier T, van der Marel GA, Filippov DV, Bovenberg RA, Vreeken RJ, Driessen AJ and Overkleeft HS, Chembiochem, 2015, 16, 915–923. [DOI] [PubMed] [Google Scholar]
- 31.Smedsgaard J, Journal of Chromatography A, 1997, 760, 264–270. [DOI] [PubMed] [Google Scholar]
- 32.Chen GJ, Qiu N, Karrer C, Caspers P and Page MG, Biotechniques, 2000, 28, 498–500, 504–495. [DOI] [PubMed] [Google Scholar]
- 33.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL and Clardy J, J Mol Biol, 1993, 229, 105–124. [DOI] [PubMed] [Google Scholar]
- 34.Kabsch W, Acta Crystallogr D Biol Crystallogr, 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vagin A and Teplyakov A, Acta Crystallogr D Biol Crystallogr, 2010, 66, 22–25. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P and Cowtan K, Acta Crystallogr D Biol Crystallogr, 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 37.Murshudov GN, Vagin AA and Dodson EJ, Acta Crystallogr D Biol Crystallogr, 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- 38.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC and Zwart PH, Acta Crystallogr D Biol Crystallogr, 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A and Wilson KS, Acta Crystallogr D Biol Crystallogr, 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ness SR, de Graaff RA, Abrahams JP and Pannu NS, Structure, 2004, 12, 1753–1761. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.