

SUMMARY
Immunoglobulin M (IgM) memory cells undergo differentiation in germinal centers following antigen challenge, but the full effector cell potential of these cells is unknown. We monitored the differentiation of enhanced yellow fluorescent protein (eYFP)- labeled CD11c+ and CD11cneg T-bet+ IgM memory cells after their transfer into naive recipient mice. Following challenge infection, many memory cells differentiated into IgM-producing plasmablasts. Other donor B cells entered germinal centers, down- regulated CD11c, underwent class switch recombination, and became switched memory cells. Yet other donor cells were maintained as IgM memory cells, and these IgM memory cells retained their multi-lineage potential following serial transfer. These findings were corroborated at the molecular level using immune repertoire analyses. Thus, IgM memory cells can differentiate into all effector B cell lineages and undergo self-renewal, properties that are characteristic of stem cells. We propose that these memory cells exist to provide long-term multi-functional immunity and act primarily to maintain the production of protective antibodies.
Graphical Abstract

In Brief
T-bet+ B cells have now been identified in a wide range of immunological contexts. Using a model bacterial infection, Kenderes et al. show that single T-bet+ IgM memory cells exhibit multi-lineage potential and can undergo self-renewal, both properties of stem cells.
INTRODUCTION
Class-switched memory B cells have long been known to be a source of high-affinity antibodies responsible for protective immunity (Good-Jacobson and Shlomchik, 2010; Kurosaki et al., 2015). However, there is now a greater appreciation for a role for unswitched memory cells in long-term immunity (Dogan et al., 2009; Pape et al., 2011). Immunoglobulin M (IgM) memory cells are generated in response to infections and to immunizations with defined antigens (Della Valle et al., 2014; Krishnamurty et al., 2016; Pape et al., 2011). IgM memory cell responses, which can be long lived (Taylor et al., 2012), differ in important ways from those of switched memory B cells. In particular, IgM memory cells retain the capacity to enter germinal centers (GCs) and to undergo class switching and affinity maturation (Dogan et al., 2009), attributes which likely allow greater flexibility in the response to secondary infections (Purtha et al., 2011).
Previous studies of IgM memory cells revealed that, after antigen challenge, IgM memory cells can enter GCs and undergo affinity maturation and class switching to produce high-affinity switched immunoglobulin (swIg) memory B cells (Dogan et al., 2009). This process of differentiation differs from that of swIg memory cells; although swIg memory cells can re-enter GCs, upon challenge, most swIg memory cells differentiate into antibody-secreting cells (ASCs) (Pape et al., 2011). However, in some cases, swIg memory B cells can dominate the secondary GC response and undergo additional affinity maturation (McHeyzer-Williams et al., 2015). Other studies have demonstrated that memory cell fate is not specified by B cell receptor (BCR) signals. CD80/PDL2 double-positive memory B cells rapidly differentiated into ASCs, and double-negative cells preferentially entered into GCs (Zuccarino-Catania et al., 2014). Yet other studies demonstrated that IgM memory B cells could generate secondary antibody responses to malarial infection without switching (Krishnamurty et al., 2016). Thus, a better understanding is needed as to how both IgM and swIg memory cells contribute to secondary immunity.
We identified IgM memory cells in our studies of ehrlichial infection (Yates et al., 2013). The ehrlichiae are obligate intracellular rickettsiae that infect macrophages and dendritic cells (Walker et al., 2004). Even though these pathogens are largely confined to host cells, humoral immunity plays an essential role in host defense (Bitsaktsis et al., 2007; Li et al., 2001). Although we first identified IgM memory cells in our studies by their unique expression of CD11c, we later demonstrated that these cells also express T-bet as well as a number of cell surface markers characteristic of memory B cells, including CD73, PD- L2, and several integrins (Papillion et al., 2017; Yates et al., 2013). A number of additional criteria were used to establish that the CD11c+ T-bet+ B cells identified in our infection model are IgM memory cells: (1) they persist for at least one year post-infection; (2) they are not actively dividing; (3) they do not spontaneously produce IgM or IgG; (4) they do not express markers characteristic of GC cells; and (5) depletion of the CD11c+ B cells ablated secondary IgG responses to antigen, independent of CD11c-negative bone marrow plasma cells (Yates et al., 2013). At least a portion of the population underwent somatic mutation (Yates et al., 2013). High frequencies of IgM memory cells are derived from cells present on day 10 post-infection, co-incident with a large extrafollicular plasma- blast (exfPB) response (Racine et al., 2008).
Our laboratory first identified CD11c+ B cells in studies of exfPBs elicited early during infection; the exfPBs also expressed T-bet (Winslow et al., 2017). The splenic CD11c+ T-bet+ exfPBs generate a robust T-independent neutralizing IgM response, and this process is accompanied by the suppression of GC B cells; development of the latter is delayed until about three weeks post-infection (Racine et al., 2010). Thereafter, IgM and swIg are maintained indefinitely, and both unswitched and switched Ig contribute to long-term immunity. Persistent antibodies maintain long-term immunity, as C57BL/6 mice are completely immune to reinfection (Bitsaktsis et al., 2007). Immunity to reinfection is maintained in the presence of a low-level chronic ehrlichial infection, revealing that IgM memory can be maintained even in the presence of chronic antigen.
CD11c+ T-bet+ B cells have been described in a number of other immunological contexts, including autoimmune diseases (Rubtsov et al., 2013, 2017). These B cells have been characterized as age-related B cells (Hao et al., 2011; Rubtsov et al., 2011), although it is evident now that T-bet+ B cells are also generated early following acute infection with a range of microbial agents (Chang et al., 2017; Knox et al., 2017; Weiss et al., 2009). The T-bet+ IgM memory cells that are the focus of our studies are closely related or identical in phenotype to the T-bet+ B cells described in other experimental models of chronic immunity, suggesting that this subset of B cells shares common functions (Chang et al., 2017; Knox et al., 2017). Thus, studies of T-bet+ memory B cells will likely lead to a better understanding of B cell memory and the function of T-bet+ B cells.
Our objective herein was to address how T-bet+ memory B cells differentiate following challenge infection. Our studies focused on IgM memory cells, because these cells are the major population of memory cells generated during ehrlichial infection. We show that these memory cells exhibit stem cell characteristics: following challenge infection, they can reconstitute all major effector B cell lineages and can self-renew. We propose that the primary function of these IgM memory cells is not to generate a more rapid response to secondary infection but rather to maintain long-term production of protective antibodies. Our studies highlight the importance of this B cell subset as an important component of humoral immunity.
RESULTS
Marking Aicda-Expressing CD11c+ T-Bet+ IgM Memory Cells In Vivo
Our previous studies demonstrated that CD11c+ IgM memory cells elicited during E. muris infection undergo limited somatic mutation and showed that these cells could be irreversibly labeled in (activation-induced cytidine deaminase [AID]-Cre-ERT2 x Rosa26 enhanced yellow fluorescent protein [eYFP]) F1 mice (Papillion et al., 2017). In the (AID-Cre-ERT2 x Rosa26 eYFP) F1 mice, all cells expressing AID at the time of tamoxifen administration irreversibly express eYFP (Dogan et al., 2009). In our studies, few if any cells were labeled in uninfected (AID- Cre-ERT2 x Rosa26 eYFP) F1 mice, indicating that the labeled cells were infection specific (Papillion et al., 2017). Although our previous studies focused on CD11c+ IgM memory cells, eYFP+ B cells detected after tamoxifen administration were found to be more diverse. In addition to CD11c+ T-bet+ IgM memory B cells, smaller populations of differentiated GL7+ GC B cells, as well as CD138+ ASCs, were detected within the eYFP+ B cell population (Figures 1A, top middle panel, and S1A. Nearly all of the GL7- and CD138-double-negative eYFP- labeled B cells expressed IgM (R1; i.e., are memory IgM cells), although low frequencies of swIg cells, also presumably memory cells, were detected (Figure 1A, R4).
Figure 1. Characterization of Aicda-Expressing IgM+ Memory Cells In Vivo.
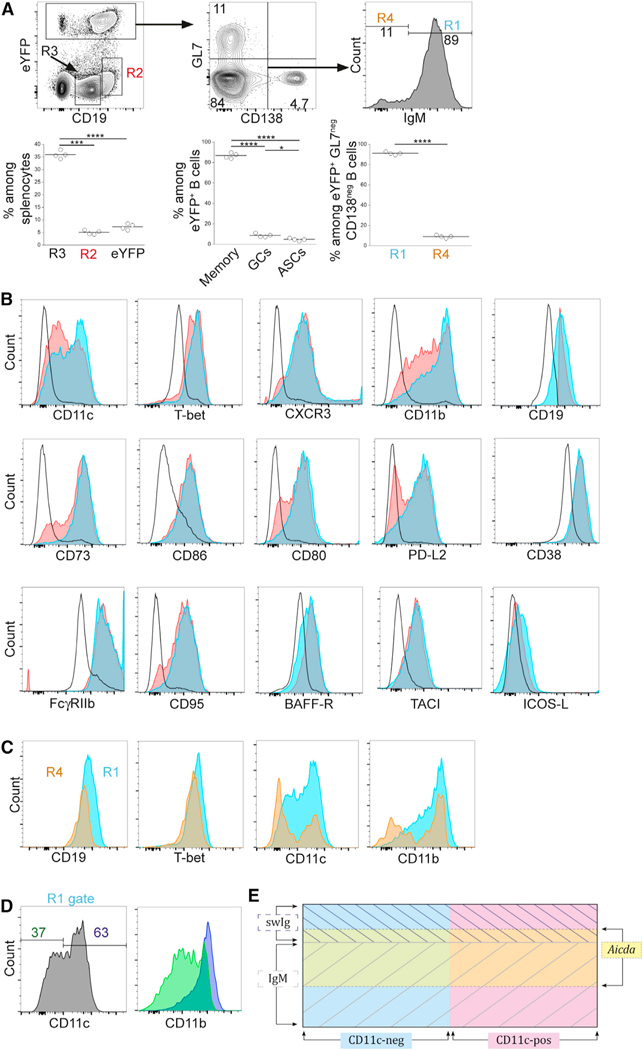
E.-muris-infected (AID-creERT2 × ROSA26-eYFP) F1 mice were administered tamoxifen on days 7 and 10 post-infection, and splenocytes were analyzed on day 70 post-infection.
(A) eYFP+ GL7neg CD138neg IgM+ memory cells (R1), CD19hi B cells (R2), CD19+ follicular B cells (R3), and eYFP+ GL7neg CD138neg IgMneg switched memory cells (R4) were identified. Data from a representative experiment are shown in the plots at the top; the plots at the bottom are aggregate data indicating the frequency of each ofthe populations. *p < 0.05, ***p < 0.001, and ****p < 0.0001.
(B) The B cells identified in the regions defined in (A) were monitored for their expression of a panel of markers previously characterized on IgM memory B cells (Yates et al., 2013). Cells in R1 are shown in blue and R2 in red; R3 cells are indicated with a black line (open histograms).
(C) The expression of the indicated markers was analyzed on eYFP+ GL7neg CD138neg IgM+ memory cells (R4; orange histogram) and eYFP+ GL7neg CD138neg IgMneg memory cells (R1; blue histogram); overlapping cells appear as green.
(D) The expression of CD11b was analyzed in eYFP+ GL7neg CD138neg CD11c+ (purple histogram) and CD11cneg IgM+ memory cells (green histogram).
The data in (A)-(D) are representative of two experiments that used 4 mice per group. (A) Statistical significance was determined using a repeated- measures one-way ANOVA with Tukey’s multiple comparison test for the left (p < 0.0001; F = 0.678; df =11) and middle panels (p < 0.0001; F = 0.0002; df = 11) or a two-tailed paired t test for the data in the right panel (p < 0.0001;t = 59;df = 3). In (C) and (D), **p < 0.01, ***p < 0.001, and ****p < 0.001.(E) A Venn diagram is shown that illustrates the relationships between the various populations that were characterized. CD11c+ and CD11cneg cells and cells expression Aicda are indicated by the colors. IgM and swIg memory cellsare indicated by cross-hatching. See text for details.
The eYFP-labeled IgM memory cells exhibited cell surface marker expression similar to the IgM memory cells described in our previous studies (Yates et al., 2013). However, approximately 40% of the labeled IgM memory cells did not express CD11c (Figure 1B). We had not identified these putative CD11cneg memory cells in our previous studies, which relied on the unique expression of CD11c for memory cell identification (Yates et al., 2013). Also included in the analyses were eYFPneg CD19hi B cells (Figure 1A, R2); these cells exhibited a cell surface phenotype nearly identical to that of the IgM memory cells (Winslow et al., 2017), although GC cells and plasmablasts were not excluded from that population. High expression of CD19, relative to canonical B cells, is characteristic of IgM and swIg memory cells generated during E. muris infection and may indicate that the cells have enhanced signaling capabilities (Li et al., 2017). For comparison, we also analyzed eYFPneg CD19+ cells, which are primarily naive follicular CD19+ B cells (R3). The eYFP+ population was, nevertheless, representative of the IgM memory cells we characterized on the basis of CD11c expression alone, although the approach used here necessarily excluded Aicdaneg cells, as well as any memory cells that did not express CD11c or T-bet. The eYFP+ IgM memory cells (R1) also exhibited high expression of T-bet, CXCR3, CD11b, CD73, CD86, CD80, PD-L2, CD95, BAFF-R, and TACI and similar expression of ICOS-L, relative to CD19+ follicular B cells. The mean fluorescence intensity (MFI) of each of the cell populations shown in Figure 1 is provided in Table 1.
Table 1.
Surface Marker Expression on IgM Memory Cells (MFI)
| Surface Marker | Cell Population |
||||||
|---|---|---|---|---|---|---|---|
| eYFP+ IgM Memory |
CD19 |
CD19hi |
eYFP+swIg Memory |
||||
| MFIa | MFI | Fold Difference | MFI | Fold Difference | MFI | Fold Difference | |
| T-bet | 2,967 | 721 | 4.1b | 2,535 | 1.1b | 1,896 | 1.5b |
| CXCR3 | 978 | 38 | 26b,c | 1,033 | −1.1 | 478 | 2.0 |
| CD11b | 6,071 | 91 | 67b,c | 3,080 | 2.0 | 3,208 | 1.9 |
| CD73 | 4,333 | 94 | 46b | 3,015 | 1.4b | 3092 | 1.4b |
| CD86 | 1,565 | 250 | 6.3b,c | 1,532 | 1.0 | 1,000 | 1.6 |
| CD80 | 1,214 | 57 | 21b | 921 | 1.3b | 805 | 1.5b |
| PD-L2 | 996 | 29 | 34b | 698 | 1.4b | 401 | 2.5b |
| FcγRIIb | 41,645 | 4,697 | 8.9b | 41,449 | 1.0 | 18,700 | 2.2b |
| CD95 | 1,165 | 40 | 29b | 934 | 1.2b | 675 | 1.7b |
| BAFF-R | 1,599 | 757 | 2.1b | 2023 | -1.3b | 997 | 1.6b |
| TACI | 538 | 54 | 10b | 485 | 1.1b | 331 | 1.6b |
| CD19 | 7,018 | 2,602 | 2.7b | 8,086 | -1.2b | 4,271 | 1.6b |
| CD38 | 41,240 | 12,101 | 3.4b | 41,546 | 1.0 | 19,778 | 2.1b |
| ICOS-L | 186 | 78 | 2.4b | 156 | 1.2b | 126 | 1.5b |
MFI values represent the mean determined from the analysis of 4 mice on day 70 post infection.
indicates statistical significance, as determined using a RM one-way ANOVA using Dunnett’s multiple comparisons test.
The data were non-parametric, as determined using a Friedman test with a Dunn’s multiple comparisons test.
SwIg memory cells were also detected and constituted approximately 11% of the eYFP+ memory cells (Figure 1, R4). The IgM and swIg memory cells exhibited similar expression of transcription factors and cell surface markers, including T-bet and CD19, although a higher proportion of the swIg memory cells did not express CD11c and CD11b (Figures 1C and S1B; Table 1). Characterization of CD11c+ and CD11cneg eYFP+ IgM memory cells revealed that the two populations were largely identical, again with the exception of CD11b, which exhibited lower MFI in the CD11cneg B cells (Figures 1D and S1C; Table S1). The memory B cell populations that were identified are summarized in the schematic shown in Figure 1E. The eYFP+ IgM+ B cells, all T-bet+, represent a portion of total memory B cells that expressed Aicda early during infection. The memory B cells can be further subdivided by their expression of CD11c. These analyses extend our previous characterizations of memory cells by revealing additional sub-populations of both IgM and swIg memory cells. Our findings also demonstrate that Aicda expression can provide a means for marking B cells that differentiate in response to infection.
IgM Memory Cells Differentiate upon Reinfection
The availability ofthe eYFP-labeled T-bet+ IgM memory cells allowed us to address how these cells differentiate following reinfection. Challenge infection of mice containing eYFP-labeled IgM memory cells was not productive, as it did not result in an increase in bacterial colonization (Figure S2). These data indicated it was not possibleto studythe differentiation of IgM memory cells in previously infected (AID-Cre-ERT2 x Rosa26 eYFP) F1 mice, due to the presence of pre-existing neutralizing antibodies. To eliminate nonmemory eYFP+ B cells in these studies, IgM memory cells were obtained by sorting for GL7-, CD138-, and IgG-negative eYFP+ B cells (Figure 2A).
Figure 2. IgM Memory Cells Differentiated Only after Challenge Infection.
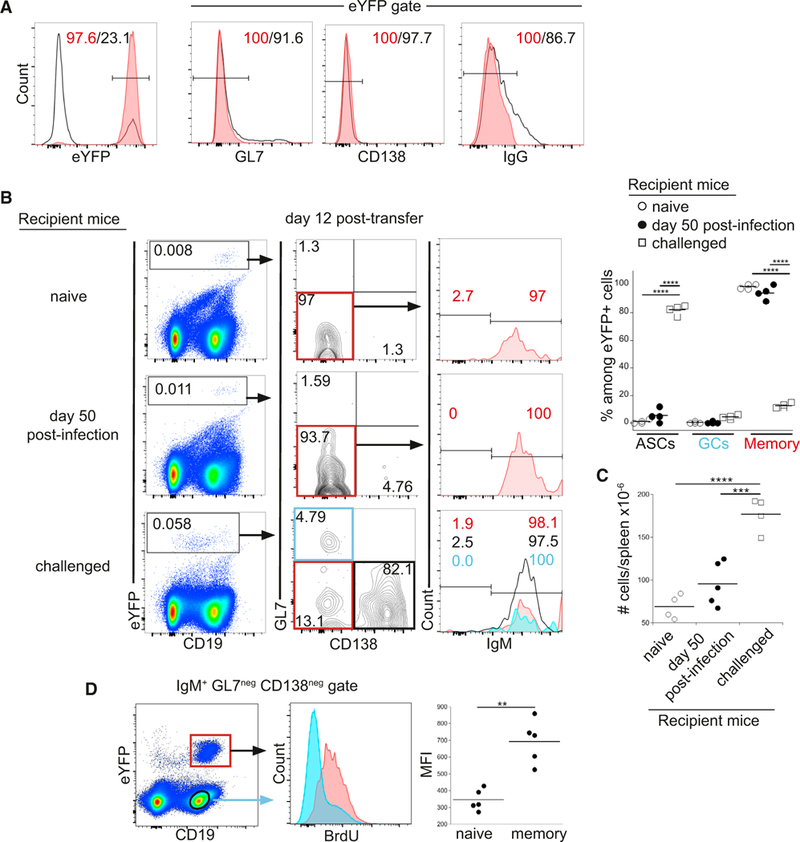
(A) Purification ofIgM memoryBcells. eYFP+GL7neg CD138neg IgGneg splenic B cellswere purified by flow cytometry; representative dot plots show expression of the indicated cell surface markers among the eYFP+ cells prior to (open histograms) and after (red histograms) purification.
(B) Purified IgM memory cells were transferred into naive mice (top panels), mice infected for50 days (middle panels), or naive micethat were challenged at the time of cell transfer; each of the groups of recipient mice were analyzed 12 days post-cell transfer. eYFP+ donor cells (gated in plots on left) were analyzed for expression ofGL7 and CD138 (middle plots). Expression of IgM was analyzed in GL7neg CD138neg memory cells (plots on right; red histograms); analyses ofthe GL7neg CD138+ASCs(bottom right plots; open histogram) and GL7+ CD138neg GC cells(blue histogram) are shown only for the challenged mice. The percentage of eYFP+ cells in each ofthe populations in the recipient mice is quantified in the plot on right. Statistical significance was determined using an ordinary one-way ANOVA (p < 0.0001; F = 948.2; df = 35) and a Holm-Sidak’s multiple comparison test.
(C) Spleen cell numbers in the mice analyzed in (B) are shown. Statistical significance was determined using an ordinary one-way ANOVA (p < 0.0001; F = 28.7; df = 12) and with Tukey’s multiple comparison test. The data in (A)-(C) are representative of one experiment that used 4 or 5 mice per group.
(D) Recipient mice were administered BrdU from day 14to21 following transferofT-cell-depleted splenocytes from infected (AID-Cre-ERT2 × Rosa26 eYFP) F1 mice; the recipient mice were infected immediately following cell transfer. Spleen IgM+, GL7neg and CD138neg, CD19+ B cells were analyzed on day 21 for BrdU incorporation. Aggregate data are shown in the panel on the right. Statistical significance was determined using a two-tailed paired t test (p = 0.0019; t = 7.289; df = 4).
In (C) and (D), **p < 0.01, ***p < 0.001, and ****p < 0.001.
The purified T-bet+ IgM memory cells were first transferred to naive mice to determine whether they underwent differentiation in the absence of infection. We have previously demonstrated that CD11c+ IgM memory cells are largely quiescent (Yates et al., 2013). Similarly, the eYFP+ IgM memory cells did not undergo differentiation following their transfer to naive recipient mice (Figures 2B and S3). We next addressed whether IgM memory cells differentiated following their transfer to mice that had been infected for 50 days. Analysis of donor cells 12 days post-transfer revealed that the IgM memory cells again did not undergo any detectable differentiation (Figure 2B, middle plots). Finally, donor IgM memory cell differentiation was assessed 12 days after their transfer to naive recipient mice that were infected following adoptive transfer. Under these conditions, most of the IgM memory cells differentiated into IgM+ CD138+ ASCs, although small numbers of spleen GL7+ GC B cells and undifferentiated donor cells were also detected (Figure 2B, bottom plots). The exfPB response was observed as early as day 7 post-infection and was maximal on day 12 post-infection, closely mirroring the kinetics of the primary response (Figure S4A; Yates et al., 2013). The lack of an accelerated response from the donor IgM memory cells may have been due to the absence of memory CD4 T cells in the recipient mice. This was not the case, however, as we observed similar secondary response kinetics of IgM memory cells following the transfer of unseparated splenocytes, which included memory CD4 T cells (Figure S4B). IgM memory cells and CD138+ plasmablasts were also detected in the latter studies in inguinal and mesenteric lymph nodes (Figure S4C). Related studies that addressed the fate of swIg memory cells failed to identify switched donor cells 30 days post-transfer; the reason for this outcome is as yet unresolved.
We observed splenomegaly and a productive infection only in the challenged mice (Figure 2C), indicating that IgM memory cells did not undergo differentiation in the absence of infection or in low-level chronically infected mice. These data provide additional evidence that the IgM+ donor cells function as memory cells: they are long lived, quiescent, and respond to challenge infection. To address whether the donor IgM memory cells proliferated following infection, bromodeoxyuridine (BrdU) was administered from day 14 to 21 post-transfer of T-cell-depleted splenocytes obtained from previously infected (AID-Cre-ERT2 x Rosa26 eYFP) F1 mice; donor-derived IgM memory cells were analyzed (after excluding germinal center B cells, switched cells, and plasmablasts). Nearly all of the donor cells had incorporated BrdU, relative to canonical resting CD19+ B cells (Figure 2D), indicating that memory cell differentiation was accompanied by cell division.
Multi-lineage Effector Cell Generation from T-Bet* IgM Memory Cells
To address additional stages of the secondary IgM response, we characterized IgM memory cell differentiation at later times following challenge infection. By day 31 post-transfer, approximately 18% of the donor-derived eYFP+ IgM memory cells exhibited expression of markers characteristic of GC cells (Figure 3A). Among the GL7+ CD38lo GC donor cells, approximately 80% had undergone class switching (i.e., were IgMneg). Among the donor-derived non-GC cells, a population of CD138+ ASCs persisted in the spleen, the majority of which expressed IgM. The non-GC, CD138neg donor-derived cells, formally memory cells, were predominantly IgM+ and expressed CD11c. CD11cneg IgM memory cells were also detected, as well as swIg memory cells (both CD11c+ and CD11cneg). The IgM memory cells recovered after secondary infection largely retained expression of CXCR3, a surrogate marker for T-bet expression (Figure 3B; Koch et al., 2009; Serre et al., 2012). In contrast, GC differentiation was accompanied by a loss CXCR3 expression, indicative of T-bet downregulation.
Figure 3. IgM Memory B Cells Generated All Effector and Memory Subsets following Challenge Infection.
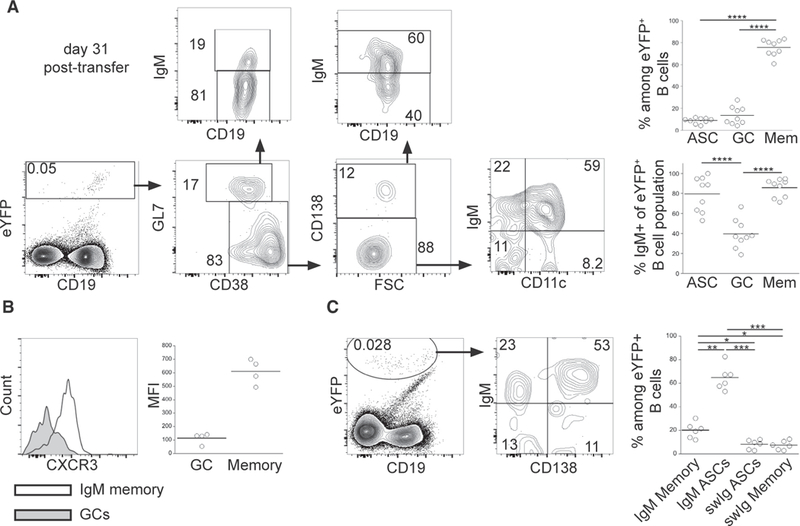
(A) Purified eYFP+ IgM memory cells were transferred into naive mice, the recipient mice were infected, and splenic B cells were analyzed 31 days post-transfer. Representative analyses of differentiated eYFP+ donor-cell-derived B cells are shown; aggregate data are shown in the plots on the right. See text for details. Statistical significance was determined using a repeated-measure (RM) one-way ANOVA with Tukey’s multiple comparison test in upper (p < 0.0001; F = 196; df = 26) and lower (p < 0.0001; F = 125.8; df = 26) panels.
(B) CXCR3 expression among eYFP+ CD38lo GL7+ GC B cells (shaded histogram) and eYFP+CD38hi GL7neg CD138neg memory cells (open histogram) on day 31 post-transfer. The MFI of each of the populations is shown in the plot on the right. Statistical significance was determined using a two-tailed Wilcoxon test (p = 0.125).
(C) IgM and CD138 expression on eYFP+ donor-derived cells in the BM of recipient mice on day 31 post-transfer. Statistical significance was determined using an RM one-way ANOVA with Tukey’s multiple comparison test (p < 0.0001; F = 72.29; df = 23). The data in (A)-(C) are representative of 3 experiments that used 2–4 mice per group. In (A) and (C), *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
We also addressed whether donor-derived cells were detected in the bone marrow (BM), because we previously described a population of IgM BM ASCs in infected mice (Racine et al., 2011). These ASCs are derived from cells present early during infection and are generated independent of CD4 T cell help (Papillion et al., 2017). The BM eYFP+ donor-derived B cells were predominately IgM+ CD138+ ASCs, but some CD138neg IgM+ cells and swIg cells were also detected (Figure 3C). Thus, the IgM memory cells were also capable of generating BM ASCs. These data reveal that a portion of IgM memory cells undergo differentiation following challenge infection and that these cells are able to reconstitute all effector B cell lineages, including spleen and BM ASCs, IgM and swIg GC cells, exfPBs, and swIg memory cells. Furthermore, IgM memory cells were maintained, indicating that the IgM memory cells can self-renew.
CD11c Expression Did Not Influence IgM Memory Cell Differentiation
Because we detected both CD11c+ and CD11cneg eYFP+ IgM memory cells, we next addressed whether these two populations generated different effector cells following challenge infection. For these studies, IgM memory cells were separated on the basis of CD11c expression (Figure 4A). Equal numbers of CD11c+ and CD11cneg cells were transferred into naive mice, and donor cells were analyzed on day 30 post-infection. Both CD11c+ and CD11cneg IgM memory cells exhibited a pattern of differentiation that was characteristic of the unseparated population (Figure 4B). The CD11c+ and CD11cneg IgM memory donor cells interconverted, as both populations were detected following transfer of either donor population. Both populations of donor IgM memory cells gave rise to BM ASCs. We also observed that CD11c+ donor-derived IgM memory cells downregulated CD11c during GC differentiation (Figure 4C). The data reveal that both CD11c+ and CD11cneg IgM memory cells can differentiate into GC cells, spleen and BM IgM, and swIg ASCs, as well as IgM and swIg memory cells, indicating that CD11c expression by itself does not specify IgM memory cell differentiation.
Figure 4. IgM Memory B Cell Differentiation Occurred Independently of CD11c.
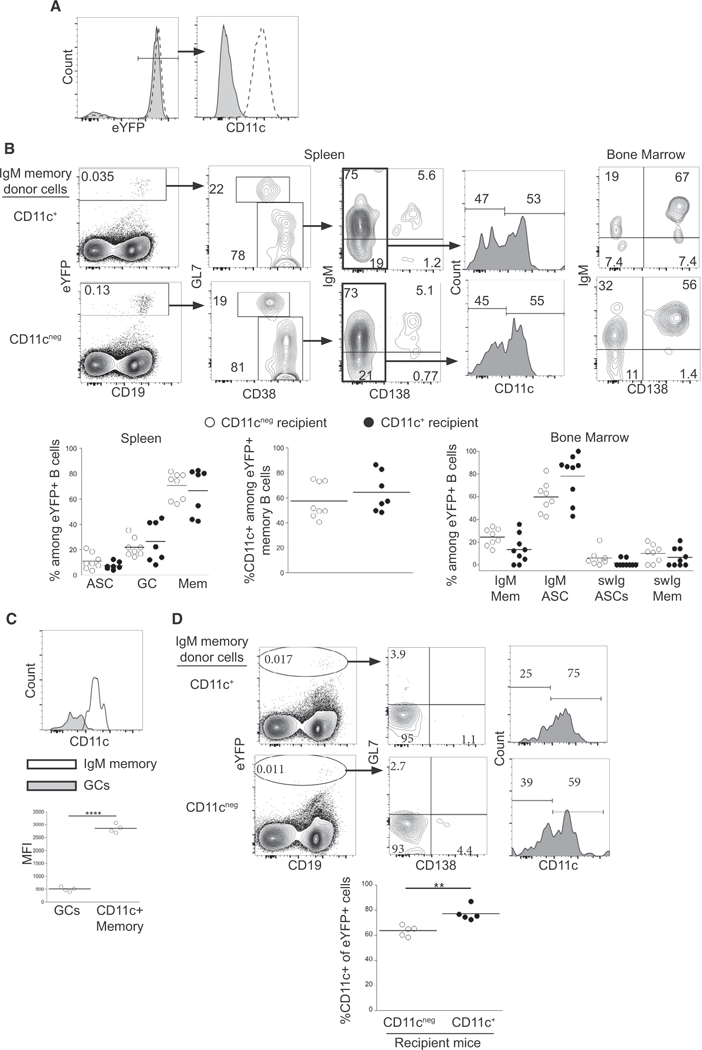
(A) Purification of CD11c+ and CD11cneg IgM memory cells. eYFP+ GL7neg CD138neg IgGneg memory cells were separated on the basis of CD11c expression. The purified CD11c+ (dashed histogram) and CD11cneg (shaded histogram) B cells are shown.
(B) Equal numbers CD11c+ or CD11cneg eYFP+ IgM memory cells were transferred into naive mice, and the recipient mice were infected. Differentiation of eYFP+ cells in the spleen and BM of recipient mice was analyzed on day 30 post-transfer, as in Figure 3. The percentage of eYFP+ cells in each population and the percentage of CD11c+ among the memory cells are quantified in the plots at the bottom. Statistical significance was determined by doing a comparison between recipient mice for each population, using an ordinary one-way ANOVA (p < 0.0001; F = 26.79; df = 51) with Sidak’s multiple comparison test (left panel), two tailed unpaired t test (middle panel; p = 0.413; t = 0.8531; df = 10.3), and a Kruskal-Wallis test (p < 0.0001) with Dunn’s multiple comparison test (right panel).
(C) CD11c expression among eYFP+ CD38lo GL7+ GC B cells and eYFP+ CD38hi GL7neg CD138neg CD11c+ memory cells in mice that received CD11c+ IgM memory cells 30 days post-transfer. MFI values are quantified in the plot below. Statistical significance was determined using a two-tailed paired t test (p < 0.0001; t = 46.5; df = 3).
(D) Steady-state interconversion of IgM memory cells. CD11c+ or CD11cneg eYFP+ IgM memory cells were transferred into recipient mice that had been infected for 47 days, and differentiation of donor cells in the spleen of the recipient mice was analyzed 21 days later. Statistical significance was determined using a two-tailed unpaired t test in the middle panel (p = 0.0031; t = 4.294; df = 7.335).
The data are representative of 2 experiments (A-C), each containing 3 or 4 mice per group, or one experiment with 5 mice per group (D). In (C) and (D), **p < 0.01, ****p < 0.0001.
We also addressed whether interconversion of CD11c expression occurred under steady-state conditions by performing transfer studies of CD11c+ and CD11cneg donor IgM memory cells to chronically infected mice. Both donor populations interconverted in the absence of reinfection, although a bias toward CD11c+ B cells was observed (Figure 4D). Thus, although integrins have been shown to play important roles in B cell localization and migration (Belnoue et al., 2012; Rose et al., 2007), a major role for CD11c in B cell differentiation was not observed.
IgM Memory Cells Retained Their Multi-lineage Potential following Serial Transfer
Because IgM memory cells were maintained following secondary challenge infection, presumably via a process of self-renewal, we next addressed whether these memory cells retained their multi-lineage potential following a second challenge. To address this question, purified eYFP+ IgM memory cells were obtained from mice that had received a primary cell transfer 30 days earlier. The cells were again transferred to naive mice, and the differentiation of the serially transferred IgM memory cells was evaluated on day 30 post-secondary challenge. IgM memory cells were found to undergo differentiation similar to that observed after the primary transfer and challenge. Both IgM and swIg memory, IgM and swIg ASCs, and GC B cells were generated from the serially transferred donor cells (Figure 5A). IgM memory cells also retained their ability to differentiate into IgM+ CD138+ BM ASCs (Figure 5B). Overall, no differences were observed between secondary and tertiary responses of the IgM memory cells. These data indicate that the multi-lineage potential of the IgM memory cells was retained following secondary challenge. Although, for technical reasons, we were unable to monitor cell division in donor-derived IgM memory cells, the serial transfer studies suggest that the memory cells were maintained by a process of self-renewal. Therefore, the T-bet+ IgM memory cells generated during ehrlichial infection, as a population, exhibit stem-cell-like characteristics.
Figure 5. IgM Memory B Cells Retained Their Multi-lineage Potential after Serial Transfer.
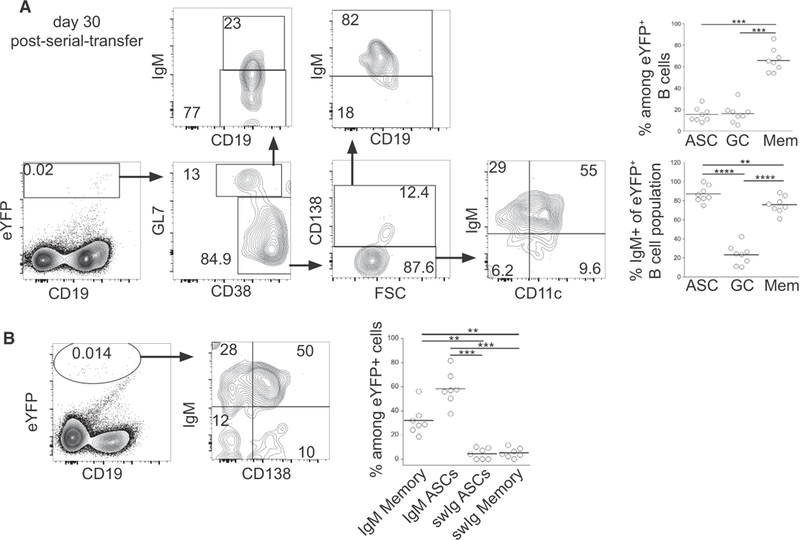
(A) eYFP+ IgM memory cellswere purified from micethat had been recipientsofeYFP+ IgM memory cells 31 days prior; the donor cells were transferred into naive mice, which were subsequently infected. Splenocytes from the recipient mice were analyzed 30 days post-secondary transfer. Representative dot plots illustrating the gating forthe differentiation ofeYFP+ cells are shown (left); aggregate data are shown in the plots on the right. Statistical significance was determined using an RM one-way ANOVA with Tukey’s multiple comparison test for the upper panel (p < 0.0001; F = 57.8; df = 23) and lower panel (p < 0.0001; F = 269.1; df = 23).
(B) Representative dot plots of IgM and CD138 expression on eYFP+ donor cells in the BM of recipient mice day 30 post-secondary transfer. Statistical significance was determined using an RM one-way ANOVA (p = 0.0001; F = 37.16; df = 27) with Tukey’s multiple comparison test. The data are representative of 3 experiments that used 2 or 3 mice per group.
**p < 0.01, ***p < 0.001, and ****p < 0.0001.
Shared Ig Repertoire after IgM Memory Cell Differentiation
The population-based studies shown above suggested that IgM memory cells were multipotent. However, this conclusion requires that a single IgM memory cell clone be capable of repopulating all effector B cell lineages and undergo self-renewal. An alternative possibility is that particular clones of IgM memory cells generate distinct effector cells lineages, for example, on the basis of the affinity of the BCR for antigen or access to T cell help (Gitlin et al., 2014; Shulman et al., 2014). We reasoned that, if identical clones of B cells could be detected in IgM memory cells in each of the differentiated effector cell populations, the data would support the conclusion that individual IgM memory cells were multipotent.
To address the potency of a single B cell, IgM memory cells were purified and transferred into naive mice, and eYFP+ donor-derived cells were purified on day 21 or 27 post-infection; the recovered cells included GC cells, BM ASCs, splenic ASCs, and memory cells. To determine whether the differentiated progeny of the IgM memory cells contained related clones, we performed an analysis of V-region usage of the four differentiated cell populations, as well as the donor IgM memory cells. These data revealed similar V-region usage in all effector population and in the donor cells, indicating that differentiation was not biased by V-region usage (Figure 6A). To address clonal relationships, clones from each of the effector cell populations were first defined on the basis of their CDR3 region sequence. In each of the populations, between 300 and 1,000 different clones were identified. A number of clones were shared between all four populations within each recipient mouse (Figure 6B). These data indicated that a single IgM memory cell clone could differentiate into GC B cells, BM and spleen ASCs, and memory B cells. The number of shared clones represented a small fraction of the total number of clones identified; however, the shared clones were enriched compared to clones that were unique to each population (Figure 6C). Moreover, 30%−50% of the clones in each effector cell population were shared with at least one other donor-derived population within the same recipient mouse (Figure 6D). These shared clones were only observed between populations within a single recipient mouse, however. For example, when clones from a replicate experiment were compared to each of the four effector populations described above, fewer than 3% of clones were shared (see control group in Figure 6D). Comparisons between replicate mice from the same experiment also revealed limited overlap (Figure S5A). Thus, effector B cell clones within a recipient mouse were very closely related, but each recipient contained clonally distinct effector cells.
Figure 6. IgH Chain Repertoire Analyses of Differentiated IgM Memory Cells.
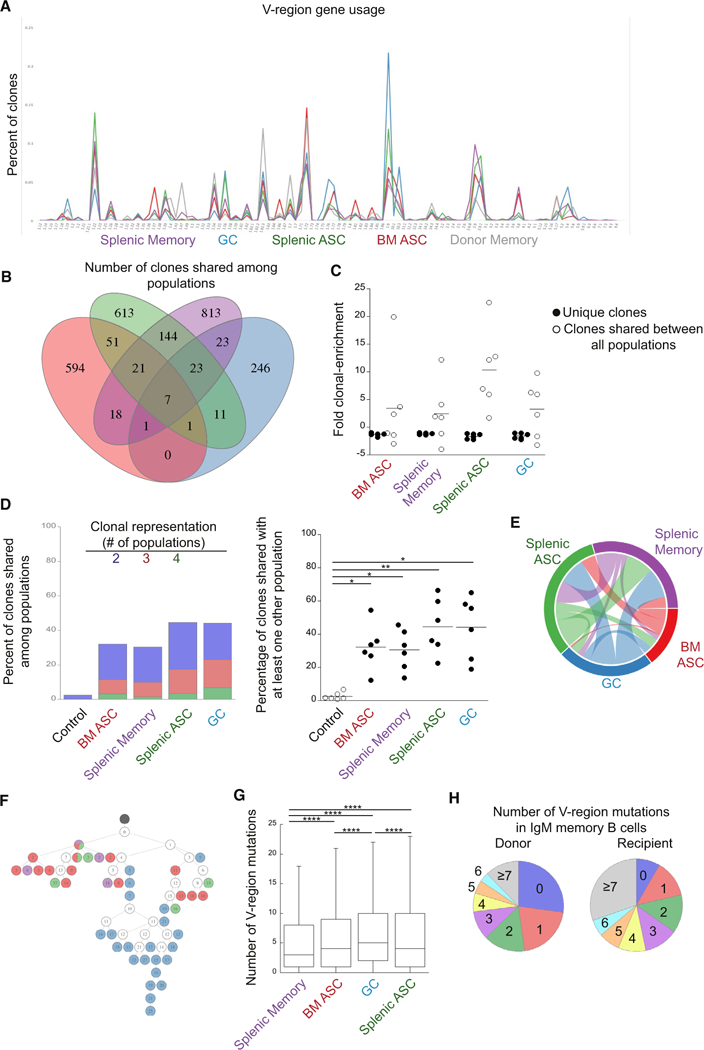
IgSeq analysis was performed on B cell populations generated from transferred IgM memory B cells.
(A) Frequency of IgH V-gene usage in the donor IgM memory population and each IgM memory cell-derived B cell population. V-regions are indicated on the ordinate.
(B) A Venn diagram is shown that illustrates the number of clonotypes shared among B cell populations in a representative mouse. Regions of overlap represent the number of clonotypes shared between the overlapping populations.
(C) Clonal enrichment. The plot on the right shows the fold clonal enrichment (i.e., the clonal frequency divided by the expected frequency; assuming equal representation) of clones unique to each population and clones identified in all effector populations. For frequencies less than one, the negative reciprocal was plotted.
(D) Percent of clonotypes shared among B cell populations. The blue bar segments represent the frequency of clones within each population that were also found in one other population; the red bars represent clones found in two other populations, and the green bars represents clones found in three other populations. The plot on the right shows the percentage of clones shared between at least one other population within an individual mouse. Each mouse was compared to a different recipient mouse from a different time point (control). Statistical significance was determined using a RM one-way ANOVA (p = 0.0008; F = 16.92; df = 29) with Dunnett’s multiple comparisons test.
(E) A Circos plot representing the clonal overlap in pairwise comparisons of the different B cell populations is shown.
(F) A lineage tree illustrating common clones identified in all of the isolated B cell populations. Black circles represent a germline clone, and white circles represent inferred clones. Daughter clones are shown in color, based on the effector population from which they were identified. The number of mutations from germline is indicated by the number shown in each circle.
(G) The number of replacement and silent mutations, from germline, in the V-region of the indicated populations is shown. Statistical significance was determined by an ordinary one-way ANOVA (p < 0.0001; F = 1244; df = 145,175) with Tukey’s multiple comparisons test. ****p < 0.0001.
(H) The percentage of clones with the indicated number of replacement and silent mutations in the V-region of the IgM memory donor population (left) and the IgM memory recipient population on day 27 post-challenge (right) is shown. Outliers have been removed for clarity. All data of the recipient mice are from 2 experiments with 3 mice per group. The donor population is representative of two experiments with three pooled mice per experiment.
Within each recipient mouse, pairwise comparisons illustrated the clonal relationship between the various populations (Figure 6E). For example, splenic ASCs exhibited the most clonal overlap with splenic memory cells and vice versa. The GC B cells exhibited the most clonal overlap with the splenic ASCs and memory cells, and a smaller portion of the clones from the splenic memory and ASC populations were found in the GC populations. Because GC clones overlapped with other populations and a reciprocal overlap was not observed, these data are indicative of an expansion and selection of clones within the GC. The BM ASCs, in contrast, exhibited the least similarity with the other effector cell populations; the largest portion of these clones was shared with splenic ASCs (Figure S5B). Thus, the different effector populations exhibited different degrees of clonal overlap, indicating that some lineages are more closely related, likely because they were derived from the same population. Thus, clones observed in the splenic ASC and memory cell populations likely underwent expansion in GCs. Similarly, the BM ASCs exhibited the most overlap with splenic ASCs, suggesting that the former were derived from the latter, supporting our previous work that demonstrated both spleen and BM ASC populations are T independent (Racine et al., 2011).
We next examined whether the mutational diversity differed between the various effector populations. This analysis was performed by generating lineage trees, based on mutation analysis of clones that were identified in all four effector populations. Clones were defined based on VJ usage, junction region length, and hamming distance. In most cases, daughter clones that were found within the same effector cell population were found to cluster (Figures 6F and S5C). The clustering was accompanied by differing degrees of mutational diversity for each effector population; however, the amount of mutational diversity for a particular effector population varied. Thus, although mutational diversity differed within each clone, the clustering of daughter clones within each effector populations likely reflects diversification within each of the effector populations. However, the frequency of mutations was similar in all of the effector populations (Figure 6G). Moreover, about 8% of the clones in the IgM memory population were unmutated (Figure 6H). The low number of mutations, as well as the percentage of germline sequences in the IgM memory cells, indicated the cells likely express low-affinity receptors. Low affinity could contribute to the stem-cell-like nature of IgM memory cells by allowing for further diversification during their differentiation. Comparison of the percentage of IgM memory clones with germline sequences before and after challenge infection identified a decrease in the percentage of germline sequences, however, suggesting that germline clones are eventually lost from the IgM memory population. These data, along with the cellular studies, indicate that a single IgM memory cell clone can generate all effector and memory B cell lineages.
DISCUSSION
Our studies of T-bet+ IgM memory cells, including both CD11c+ and CD11cneg variants, demonstrate that these cells have the capacity to undergo multi-lineage differentiation following challenge infection. This property distinguishes these memory cells from swlg memory cells, which exhibit a more limited differentiative potential (Dogan et al., 2009; Gitlin et al., 2016; Pape et al., 2011). The two memory cell populations therefore complement one another; swig memory cells produce high-affinity antibodies, whereas the IgM memory cells produce lower affinity antibodies but retain a greater capacity to generate different effector cells. The IgM memory cells, therefore, act as atypical memory cells, similar to atypical memory cells described in humans (Pupovac and Good-Jacobson, 2017).
Differentiation of IgM memory cells in our studies occurred only after challenge infection and in the absence of pre-existing antibody, as the transferred IgM memory cells persisted days in both uninfected recipient mice and in chronically infected seropositive mice. Our data are also consistent with the idea that memory B cell diversification occurs independently of B cell isotype, at least with respect to IgM. Other studies have reported that memory B cells subset on the basis of CD80 and PDL2 expression, independent of BCR isotype, exhibited different fates (Zuccarino-Catania et al., 2014). The IgM memory cells we have characterized all exhibited high expression of both CD80 and PD-L2, indicating that these receptors do not by themselves regulate IgM memory cell differentiation.
We also addressed whether CD11c expression affected IgM memory cell fate, because CD11c is one of the few surface markers differentially expressed among IgM memory B cells. However, CD11c expression did not appear to influence IgM memory cell differentiation and CD11c+ and CD11cneg cells interconverted in vivo. Given the unique expression of CD11c by T-bet+ B cells, it was unexpected that the loss of the integrin would have no apparent effect on function, given its likely role in B cell localization in vivo (Lu and Cyster, 2002). However, it is not known how rapidly interconversion occurs; it is possible, for example, that IgM memory cells interconvert throughout their differentiation, and modulation of receptor expression is important for differentiation.
The differentiation of the IgM memory cells we observed differed from classically defined memory cells in that the kinetics of the response were not accelerated. Instead, the IgM memory response was very similar to the B cell responses we have described during primary ehrlichial infection. For example, most of the donor IgM memory cells differentiated to CD138+ ASCs by day 12 post-infection, data that mimicked the massive exfPB response we reported previously (Racine et al., 2008). Transfer of all splenocytes, including memory T and B cells, did not alter the kinetics of the secondary plas- mablast response, indicating that memory T cells do not influence the rate at which the IgM memory cells differentiate into exfPBs and confirms other studies that have reported that IgM memory cells do not require memory T cells for their differentiation (Zuccarino-Catania et al., 2014). Therefore, we propose that the IgM memory cells’ primary function is not to provide an earlier response to infection but rather to maintain concentrations of neutralizing antibodies sufficient to protect the host from reinfection by maintaining populations of long- lived IgM and IgG ASCs. Antibodies play a major role in protection against the ehrlichiae, as well as many other bacterial and viral infections, so it is not necessary for the host to mount a more rapid secondary response if it can maintain sufficiently high concentrations of neutralizing antibodies. Zinkernagel and others have elegantly articulated this concept (Lanzavec- chia et al., 2006; Zinkernagel, 2012). This paradigm for memory is significant because it challenges dogma that memory cells by nature must respond more rapidly to infection. Earlier cellular responses are necessary only when neutralizing antibodies decline such that the host is once again susceptible to serious infection. This is clearly not the case for immunity to many pathogens, where pre-existing antibodies are paramount to preventing reinfection.
Our interpretation is supported by our findings that the IgM memory cells did not differentiate in the presence of protective antibodies in chronically infected mice. The mechanism whereby this apparent inhibition occurs is not yet known but may involve suppression via Fc receptors for IgM, IgG, or both (Nguyen et al., 2017; Nimmerjahn and Ravetch, 2008). FcγRIIb is expressed at 4-fold higher levels on IgM memory cells. The possibility that pre-existing antibodies suppress IgM memory cells was proposed by Pape and colleagues, who reported that transfer of immune serum suppressed memory cell differentiation (Pape et al., 2011).
The IgM memory cells, as a population, are also distinct in their capacity to both differentiate and to self-renew, a property characteristic of hematopoietic stem cells (HSCs). The latter property was evident from our observations that, even though all or nearly all IgM memory cells proliferated following challenge infection, undifferentiated IgM memory cells were maintained. T memory stem cells with such characteristics have been described in humans (Gattinoni et al., 2017; Graef et al., 2014; Lugli et al., 2013). Similar properties have not been demonstrated for memory B cells, although transcriptional signatures of HSCs have been described (Luckey et al., 2006). Although the studies herein only characterized Aicda-positive memory cells, a significant portion of the IgM memory cells remained unmutated following challenge infection. Retaining germline configuration would allow the IgM memory cells to initiate somatic hypermutation after secondary infection and to undergo diversification similar to naive B cells. This stem-cell-like characteristic of IgM memory cells may allow for more efficient responses to variant pathogens and suggests that the defining feature of IgM memory cells is, instead, their longevity (Taylor et al., 2012), a feature that distinguishes them from naive B cells.
Our studies of the Ig repertoire of the IgM memory cells, prior to and following challenge infection, yielded several important conclusions. First, identical clones were found in each of the differentiated B cell populations analyzed. These studies provided additional evidence that IgM memory cells are multipotent. Although some clones were differentially distributed within the effector cell populations, the most highly represented clones were found in all of the populations analyzed. Thus, individual clones do not appear to be biased in their differentiation to particular effector cell lineages. Second, we observed major differences in the IgM memory repertoire between individual recipient mice, an observation that we attribute to the complexity of the donor IgM memory population. Third, different effector populations displayed varying amounts of clonal overlap. The splenic ASCs and memory were the most closely related, and the BM ASCs were the most distinct. Because the IgSeq analysis provides a ‘‘snapshot’’ of the clones present at the time of analysis, BM cells, which occupy a very different anatomical niche, might be expected to differ more from clones occupying the same biological niche (i.e., the spleen). The BM ASCs exhibited the most clonal overlap with splenic ASCs, suggesting also that the BM cells were derived from splenic ASCs, as has been suggested by our other studies (Racine et al., 2008, 2011). Fourth, the same clone found in each of the effector populations underwent different degrees of mutational diversification. This finding suggests that, whereas a single cell can give rise to multiple effector lineages, the progeny of that cell can undergo independent diversification. However, analysis of multiple lineage trees and the mutation frequencies indicated that mutational diversity was not favored by a particular lineage. Finally, the low number of mutations detected in the IgM memory cells suggests that IgM produced by these cells is of low affinity. Lower affinity, broadly reactive antibodies may more effectively recognize closely related pathogens than high-affinity antibodies produced by swIg memory cells.
Our studies also have important implications for our knowledge of T-bet+ B cells, as we have shown here that this emerging B cell subset can differentiate as both IgM and swIg memory B cells. Indeed, T-bet+ cells observed in autoimmunity, aging, and chronic infections might be functionally equivalent by the common feature that they are maintained under conditions of low-level chronic antigen stimulation (Winslow et al., 2017). In a model of Systemic Lupus erythematosus (SLE), conditional deletion of T-bet in B cells was associated with reduced activation of B cells and mitigated kidney damage (Rubtsova et al., 2017). These latter studies support the notion that T-bet+ B cells act to maintain long-term antibody; however, when autoreactive, these may be to the detriment of the host. Our data also demonstrate that T-bet memory cells are fully functional and suggest they may serve similar functions in long-term antibody responses and memory cell maintenance in other infection models, aging, and in autoimmunity. The function of T-bet in long-term IgM memory cells is not yet known but will be addressed in ongoing studies.
Our work demonstrates that both CD11c+ and CD11cneg T-bet+ IgM memory cells can differentiate into ASCs, enter GCs, and generate class-switched and IgM memory cells. The IgM memory cells self-renewed after challenge infection, and they retained ability to differentiate into multiple lineages after secondary challenge. These findings indicate that IgM memory cells are formally stem cells, similar to those described in CD8 memory T cells (Graef et al., 2014). Because the kinetics of the IgM memory response mirrored those observed during primary infection, we also propose that T-bet+ IgM memory cells act as memory stem cells to maintain long-term humoral immunity to pathogens.
STAR ★ METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
- EXPERIMENTAL MODEL AND SUBJECT DETAILS
-
○Mice
-
○
- METHOD DETAILS
-
○Infections and tamoxifen treatment
-
○Flow cytometry
-
○BrdU administration and staining
-
○Adoptive transfer of FACS-purified cells
-
○Heavy chain Ig repertoire analysis
-
○
- QUANTIFICATION AND STATISTICAL ANALYSIS
-
○Statistical analyses
-
○
DATA AND SOFTWARE AVAILABILITY
STAR ★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE-conjugated anti-CD38 (Clone 90) | eBioscience | Cat#5010337 |
| APC-eFlour780-conjugated anti-CD11c (N418) | eBioscience | Cat#5016143 |
| eflour660-conjugated anti-GL7 (GL7) | eBioscience | Cat#501124407 |
| Brilliant Violet 421-conjugated anti-CD138 (281–2) | 3iolegend | Cat#142507; RRID: AB_11204257 |
| Alexa fluor 700-conjugated anti-CD19 (6D5) | 3iolegend | Cat#115527; RRID: AB_493734 |
| Brilliant Violet 605-conjugated anti-CXCR3 (CXCR3–176) | 3iolegend | Cat#353729; RRID: AB_2562628 |
| PE-conjugated anti-CD95 (15A7) | eBioscience | Cat#501122253 |
| PE-conjugated anti-ICOS-L (HK5.3) | eBioscience | Cat#5011056 |
| PE-conjugated anti-TACI (ebio8F10–3) | eBioscience | Cat#5011004 |
| PE-conjugated anti-CD73 (TY/23) | BD Biosciences | Cat#550741; RRID: AB_393860 |
| PE-conjugated anti-CD11b (M1/70) | BD Biosciences | Cat#553311; RRID: AB_394775 |
| PE-conjugated anti-BAFF-R (7H22-E16) | BD Biosciences | Cat#565783 |
| PE-conjugated anti-CD86 (GL-1) | BD Biosciences | Cat#553692; RRID: AB_394994 |
| PE-conjugated anti-PD-L2 (TY25) | BD Biosciences | Cat#557796 |
| PE-conjugated anti-CD80 (16–10A1) | BD Biosciences | Cat#562504 |
| PE-conjugated anti-CD16/32 (2.4G2) | BD Biosciences | Cat#553145; RRID: AB_394660 |
| PE-conjugated anti-CD138 (281–2) | BD Biosciences | Cat#553714; RRID: AB_395000 |
| Biotinylated IgG1 (A85–1) | BD Biosciences | Cat#553441; RRID: AB_394861 |
| Biotinylated IgG3 (R40–82) | BD Biosciences | Cat#553401; RRID: AB_394838 |
| Biotinylated IgG2b | Southern Biotech | Cat#1090–05 |
| PE-conjugated anti-BrdU (Bu20a) | Biolegend | Cat#339811; RRID: AB_1626188 |
| PE-Cy7-conjugated anti-IgM (R6–60.2) | BD Parmigen | Cat#552867; RRID: AB_394500 |
| Brilliant Violet 421-conjugated streptavidin | BD Biosciences | Cat#563259 |
| Bacterial and Virus Strains | ||
| Ehrlichia muris | N/A | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| QIAzol | QIAGEN | Cat#79306 |
| Critical Commercial Assays | ||
| TetrocDNA Synthesis Kit | Bioline | Cat#Bio-65042 |
| EasySep Mouse CD90.2 Positive selection Kit II | Stem Cell | Cat#18951 RF |
| AxyPrep | Fisher Scientific | Cat# 14–223-227 |
| Ampure | Beckman Coulter | N/A |
| Deposited Data | ||
| IgH repertoire sequencing files | NCBI BioProject database | PRJNA473804 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 | The Jackson Laboratory | Stock# 000664 |
| B6.Cg-Gt(Rosa)26Sortm3(CAG-eYFP)Hze/J | The Jackson Laboratory | Stock# 007903 |
| AID-Cre-ERT2 | Dr. Jean-Claude Weill, ISERM, Paris, France | N/A |
| Oligonucleotides | ||
| Nextera XT indexing primers | Illumina | FC-131–1024 |
| Ig Cμ, Cγ1, Cγ2b, Cγ2c, and Cγ3 region primers | Tiller et al., 2009 | N/A |
| Software and Algorithms | ||
| FlowJo | Tree Star | N/A |
| Diva | BD Bioscience | N/A |
| Pear | Zhang et al., 2014 | N/A |
| pRESTO | Vander Heiden et al., 2014 | N/A |
| MiXCR | Bolotin et al., 2015 | N/A |
| VDJTools Software | Shugay et al., 2015 | N/A |
| IgBlast | Ye etal., 2013 | N/A |
| Changeo | Gupta et al., 2015 | N/A |
| SHazaM | Gupta et al., 2015 | N/A |
| Alakazam | Gupta et al., 2015 | N/A |
| Other | ||
| MiSeq Reagent Kit v3 | Illumina | MS-102–3001 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6 and B6.Cg-Gt(Rosa)26Sortm3(CAG-eYFP)Hze/J mice were obtained from The Jackson Laboratory (Bar Harbor, ME). The AID- Cre-ERT2 mice, provided by Dr. Jean-Claude Weill, ISERM, Paris, France, were bred to the Rosa26 strain to generate F1 mice. Mice that carried the eYFP transgene were identified by PCR-based genotyping provided by Mouse Genotype (Escondido, CA). For all experiments, female mice of at least 6 weeks of age were used and randomly assigned to experimental groups. All mice were bred, grouped housed with littermates of the same sex and maintained under microisolator conditions at Upstate Medical University (Syracuse, NY) in accordance with institutional guidelines for animal welfare.
METHOD DETAILS
Infections and tamoxifen treatment
Mice were infected, intraperitoneally, with 5×104 copies of E. muris, as described previously (Bitsaktsis et al., 2007). Tamoxifen was dissolved in peanut oil at a concentration of 20 mg/ml, and 0.5 mL was administered via oral gavage.
Flow cytometry
Spleens were disrupted, and erythrocytes removed by treatment with ACK lysis buffer (Quality Biological). The cells were treated with anti-CD16/32 (2.4G2) prior to incubation with the antibodies. For most experiments the following antibodies were used: PE-conjugated anti-CD38 (clone 90), APC-eFlour780-conjugated anti CD11c (N418), eflour660-conjugated anti-GL-7 (GL7), PE-Cy7-conju- gated anti-IgM (R6–60.2), Brilliant Violet 421-conjugated anti-CD138 (281–2), Alexafluor 700-conjugated anti-CD19 (6D5), and Brilliant Violet 605-conjugated CXCR3 (CXCR3–173). For cell surface phenotyping, the following PE-conjugated antibodies were used: CD95 (15A7), ICOS-L (HK5.3), TACI (ebio8F10–3), CD73 (TY/11.8), CD11b (M1/70), BAFF-R (7H22-E16), CD86 (GL-1), PD-L2 (TY25), CD80 (16–10A1), and CD16/32 (2.4G2). The cells were stained at 4°C for 20 min, washed, and analyzed without fixation. Unstained cells were used to establish the flow cytometer voltage settings, and single-color positive control samples were used to adjust compensation. Data were acquired on a BD Fortessa flow cytometer, using Diva software (BD bioscience), and were analyzed using FlowJo software (Tree Star).
BrdU administration and staining
To assess proliferation, Mice were administered BrdU (0.8 mg), by i.p. injection, and were maintained on BrdU in drinking water (0.8 mg/mL, plus 10% dextrose), ad libitum, for 7 days. Splenocytes were stained for surface markers, and fixed using a BD Cytofix/Cytoperm solution, for 30 minutes at 4°C. BD Perm/Wash solution was added to the cells prior to pelleting. Cells were then resuspended in 10% DMSO in BD Perm/Wash solution for 10 minutes at 4°C. Cells were then re-fixed with BD Cytofix/Cytoperm solution for 5 minutes at 4°C. DNase I in PBS was then added to the cells for 1 hour at 37°C. The cells were then incubated with PE-conjugated anti-BrdU (Bu20a) diluted in BD perm/wash solution for 30 minutes at room temperature then washed and analyzed as above.
Adoptive transfer of FACS-purified cells
To obtain donor B cells, spleen cells from infected (AID-Cre-ERT2 x Rosa26 eYFP) F1mice were disrupted, and erythrocytes removed by treatment with ACK lysis buffer (Quality Biological). The spleens were then depleted of T cells by magnetic bead selection, using a T cell Enrichment Kit (StemCell Technologies). The T cell depleted spleens were stained with the following antibodies: eFlour660-con- jugatedanti-GL-7 (GL-7), PE-conjugated anti-CD138 (281–2), biotinylated IgG1 (A85–1), IgG3 (R40–82), and IgG2b, followed by strep- tavidin Brilliant Violet 421. IgM memory cells were purified by sorting on eYFP+, GL7neg, CD138neg, IgGneg cells, using a FACS Aria cell sorter (BD Bioscience). Samples of sorted populations were analyzed on the FACS Aria to ensure purity of the sorted cells. In each case the resulting population was greater than 95% pure. Following flow cytometric purification, cells were resuspended at 1–10 × 106 cells/mL, for primary transfer, or 0.5–1.0×105 cells/mL, for secondary transfer experiments. The cells were administered via the retro-orbital sinus. Where indicated, recipient mice were administered 250 μg of doxycycline intraperitoneally, daily, from day −1 today 7 post-transfer.
Heavy chain Ig repertoire analysis
Donor IgM memory cells were purified using flow cytometry; 1×105 cells were resuspended in QIAzol (QIAGEN), and the remaining cells were transferred to recipient mice. Following transfer, splenic eYFP+CD38loGL7+B cells, eYFP+ CD38hi GL7neg CD138+ cells, eYFP+ CD38hi GL7neg CD138neg cells, and eYFP+ CD138+ BM cells, were sorted into QIAzol. mRNA was isolated and reverse trancribed using a TetrocDNA Synthesis Kit (Bioline), using primers specific for the Ig Cm, Cg1, Cg2b, Cg2c, and Cg3 regions. Ig transcripts were amplified using the MSVHE and C-outer primers described by Tiller et al. (Tiller et al., 2009). Illumina adaptor overhang sequences were added, using MSVHE and C-inner primers, concatenated to the forward and reverse adaptor overhang sequences, respectively. The samples were purified using either AxyPrep (Fisher Scientific), or Ampure magnetic beads (Beckman Coulter), and were indexed using Nextera XT indexing primers (Illumina). Indexed samples were Ampure bead-purified, and pooled. The pooled samples were sequenced paired end, using an Illumina MiSeq platform, with a MiSeq Reagent Kit v3 (2 × 300; Illumina). The paired-end reads were merged using Pear (Zhang et al., 2014), and pRESTO (Vander Heiden et al., 2014) was used to filter reads by a q score of 20 and to mask primer sequences. Identical reads were collapsed and filtered to obtain only those reads with at least two duplicates, using pRESTO (Vander Heiden et al., 2014). The reads were aligned and assembled into clones using MiXCR (Bolotin et al., 2015). VDJTools Software (Shugay et al., 2015) was used to determine VH segment usage and clonal overlap between samples. For lineage trees and mutational analysis, filtered and masked reads were aligned using IgBlast (Ye et al., 2013), and were assembled into clones using Changeo and SHazaM (Gupta et al., 2015). Lineage trees were constructed using Alakazam (Gupta et al., 2015).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses
Statistical analyses were performed using Prism 7 software (Graphpad). A minimum p value of < 0.05 was used to establish statistical significance; the following symbols were used throughout to indicate statistical significance: *: p < 0.05, **: p < 0.01, ***: p < 0.001, and ****: p < 0.0001. Differences that were not statistically significant were not indicated with symbols. The statistical tests performed on the data are indicated in the figure legends along with sample size (n) indicating the number of animals used. All data summary plots have a line indicating the mean of that dataset. All data was tested for normality prior to selection of the statistical test.
Supplementary Material
Highlights.
T-bet+ IgM memory cells are generated in response to ehrlichial infection
The memory cells can generate all effector B cell populations and self-renew
Multi-lineage potential is a property of single cells
The memory cells act to maintain long-term neutralizing antibodies
ACKNOWLEDGMENTS
The authors gratefully acknowledge excellent technical assistance provided by the Upstate Medical University Flow Cytometry Core and Molecular Core Facilities. We thank Lisa Phelps and Karen Gentile for excellent technical support. We thank Alice Bernat for assistance with the graphic in Figure 1, Drs. F. Middleton and D. Amberg (Upstate Medical University), and the Nappi Family Foundation for support for the IgSeq studies. We also thank Dr. Jean-Claude Weill (INSERM) for generously providing the AID-Cre-ERT2 mice and Drs. L. Leadbetter (University of Texas Health Science Center), R. King (University ofAlabama at Birmingham), and E. Schererfor helpful comments. This work was supported bythe NIAID and by US Department of Health and Human Services grant R01AI114545 to G.M.W.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
The accession number for the Ig-sequencing data reported in this paper is NCBI SRA: SRP149354.
Supplemental Information includes five figures and one table can be found with this article online at https://doi.org/10.1016/j.celrep.2018.06.074.
REFERENCES
- Belnoue E, Tougne C, Rochat AF, Lambert PH, Pinschewer DD, and Siegrist CA (2012). Homing and adhesion patterns determine the cellular composition ofthe bone marrowplasmacell niche. J. Immunol. 188,1283–1291. [DOI] [PubMed] [Google Scholar]
- Bitsaktsis C, Nandi B, Racine R, MacNamara KC, and Winslow G (2007). T-Cell-independent humoral immunity is sufficient for protection against fatal intracellular ehrlichia infection. Infect. Immun. 75, 4933–4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Pu- tintseva EV, and Chudakov DM (2015). MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods 12, 380–381. [DOI] [PubMed] [Google Scholar]
- Chang LY, Li Y, and Kaplan DE (2017). Hepatitis C viraemia reversibly maintains subset of antigen-specific T-bet+ tissue-like memory B cells. J. Viral Hepat. 24, 389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DellaValle L, Dohmen SE, Verhagen OJ, Berkowska MA, Vidarsson G, and Ellen van der Schoot C. (2014). The majority of human memory B cells recognizing RhD and tetanus resides in IgM+ B cells. J. Immunol. 193, 1071–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan I, Bertocci B, Vilmont V, Delbos F, Megret J, Storck S, Reynaud CA, and Weill JC (2009). Multiple layers of B cell memory with different effector functions. Nat. Immunol. 10, 1292–1299. [DOI] [PubMed] [Google Scholar]
- Gattinoni L, Speiser DE, Lichterfeld M, and Bonini C (2017). T memory stem cells in health and disease. Nat. Med. 23, 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin AD, Shulman Z, and Nussenzweig MC (2014). Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature 509, 637–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin AD, von Boehmer L, Gazumyan A, Shulman Z, Oliveira TY, and Nussenzweig MC (2016). Independent roles of switching and hypermutation in the development and persistence of B lymphocyte memory. Immunity 44, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good-Jacobson KL, and Shlomchik MJ (2010). Plasticityand heterogeneity in the generation of memory B cells and long-lived plasma cells: the influence of germinal center interactions and dynamics. J. Immunol. 185, 3117–3125. [DOI] [PubMed] [Google Scholar]
- Graef P, Buchholz VR, Stemberger C, Flossdorf M, Henkel L, Schie- mann M, Drexler I, Hӧifer T, Riddell SR, and Busch DH (2014). Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity 41, 116–126. [DOI] [PubMed] [Google Scholar]
- Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, and Kleinstein SH (2015). Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31, 3356–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, O’Neill P, Naradikian MS, Scholz JL, and Cancro MP (2011). A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood 118, 1294–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox JJ, Buggert M, Kardava L, Seaton KE, Eller MA, Canaday DH, Robb ML, Ostrowski MA, Deeks SG, Slifka MK, et al. (2017). T-bet+ B cells are induced by human viral infections and dominate the HIV gp140 response. JCI Insight 2, 92943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, and Campbell DJ (2009). The transcription factor T-bet controls regulatoryT cell homeostasis and function during type 1 inflammation. Nat. Immunol. 10, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurty AT, Thouvenel CD, Portugal S, Keitany GJ, Kim KS, Holder A, Crompton PD, Rawlings DJ, and Pepper M (2016). Somatically hypermutated Plasmodium-specific IgM(+) memory B cells are rapid, plastic, early responders upon Malaria rechallenge. Immunity 45, 402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki T, Kometani K, and Ise W (2015). Memory B cells. Nat. Rev. Immunol. 15, 149–159. [DOI] [PubMed] [Google Scholar]
- Lanzavecchia A, Bernasconi N, Traggiai E, Ruprecht CR, Corti D, and Sallusto F (2006). Understanding and making use of human memory B cells. Immunol. Rev. 211, 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JS, Yager E, Reilly M, Freeman C, Reddy GR, Reilly AA, Chu FK, and Winslow GM (2001). Outer membrane protein-specific monoclonal antibodies protect SCID mice from fatal infection by the obligate intracellular bacterial pathogen Ehrlichia chaffeensis. J. Immunol. 166, 1855–1862. [DOI] [PubMed] [Google Scholar]
- Li X, Ding Y, Zi M, Sun L, Zhang W, Chen S, and Xu Y (2017). CD19, from bench to bedside. Immunol. Lett. 183, 86–95. [DOI] [PubMed] [Google Scholar]
- Lu TT, and Cyster JG (2002). Integrin-mediated long-term B cell retention in the splenic marginal zone. Science 297, 409–412. [DOI] [PubMed] [Google Scholar]
- Luckey CJ, Bhattacharya D, Goldrath AW, Weissman IL, Benoist C, and Mathis D (2006). Memory T and memory B cells share a transcriptional program of self-renewal with long-term hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 103, 3304–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugli E, Dominguez MH, Gattinoni L, Chattopadhyay PK, Bolton DL, Song K, Klatt NR, Brenchley JM, Vaccari M, Gostick E, et al. (2013). Superior T memory stem cell persistence supports long-lived T cell memory. J. Clin. Invest. 123, 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHeyzer-Williams LJ, Milpied PJ, Okitsu SL, and McHeyzer-Williams MG (2015). Class-switched memory B cells remodel BCRs within secondary germinal centers. Nat. Immunol. 16, 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TT, Klӓsener K, Zϋrn C, Castillo PA, Brust-Mascher I, Imai DM, Bevins CL, Reardon C, Reth M, and Baumgarth N (2017). The IgM receptor FcμR limits tonic BCR signaling by regulating expression ofthe IgM BCR. Nat. Immunol. 18, 321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn F, and Ravetch JV (2008). Fcgamma receptorsas regulatorsof immune responses. Nat. Rev. Immunol. 8, 34–47. [DOI] [PubMed] [Google Scholar]
- Pape KA, Taylor JJ, Maul RW, Gearhart PJ, and Jenkins MK (2011). Different B cell populations mediate early and late memory during an endogenous immune response. Science 331, 1203–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papillion AM, Kenderes KJ, Yates JL, and Winslow GM (2017). Early derivation of IgM memory cells and bone marrow plasmablasts. PLoS ONE 12, e0178853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pupovac A, and Good-Jacobson KL (2017). An antigen to remember: regulation of B cell memory in health and disease. Curr. Opin. Immunol. 45, 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purtha WE, Tedder TF, Johnson S, Bhattacharya D, and Diamond MS (2011). Memory B cells, but not long-lived plasma cells, possess antigen specificities for viral escape mutants. J. Exp. Med. 208, 2599–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine R, Chatterjee M, and Winslow GM (2008). CD11cexpression identifies a population of extrafollicular antigen-specific splenic plasmablasts responsible for CD4 T-independent antibody responses during intracellular bacterial infection. J. Immunol. 181, 1375–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine R, Jones DD, Chatterjee M, McLaughlin M, Macnamara KC, and Winslow GM (2010). Impaired germinal center responses and suppression of local IgG production during intracellular bacterial infection. J. Immunol. 184,5085–5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine R, McLaughlin M, Jones DD, Wittmer ST, MacNamara KC, Woodland DL, and Winslow GM (2011). IgM production by bone marrow plasmablasts contributes to long-term protection against intracellular bacterial infection. J. Immunol. 186, 1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose DM, Alon R, and Ginsberg MH (2007). Integrin modulation and signaling in leukocyte adhesion and migration. Immunol. Rev. 218, 126–134. [DOI] [PubMed] [Google Scholar]
- Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, and Marrack P (2011). Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood 118, 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov AV, Rubtsova K, Kappler JW, and Marrack P (2013). TLR7 drives accumulation of ABCs and autoantibody production in autoimmune- prone mice. Immunol. Res. 55, 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsova K, Rubtsov AV, Thurman JM, Mennona JM, Kappler JW, and Marrack P (2017). B cells expressing the transcription factor T-bet drive lupus-like autoimmunity. J. Clin. Invest. 127, 1392–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre K, Cunningham AF, Coughlan RE, Lino AC, Rot A, Hub E, Moser K, Manz R, Ferraro A, Bird R, et al. (2012). CD8 T cells induce T-bet-dependent migration toward CXCR3 ligands by differentiated B cells produced during responses to alum-protein vaccines. Blood 120, 4552–4559. [DOI] [PubMed] [Google Scholar]
- Shugay M, Bagaev DV,Turchaninova MA, Bolotin DA, Britanova OV, Putintseva EV, Pogorelyy MV, Nazarov VI, Zvyagin IV, Kirgizova VI, et al. (2015). VDJtools: unifying post-analysis of T cell receptor repertoires. PLoS Comput. Biol 11,e1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman Z, Gitlin AD, Weinstein JS, Lainez B, Esplugues E, Flavell RA, Craft JE, and Nussenzweig MC (2014). Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science 345, 1058–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JJ, Pape KA, and Jenkins MK (2012).Agerminal center-independent pathway generates unswitched memory B cells early in the primary response. J. Exp. Med. 209, 597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiller T, Busse CE, and Wardemann H (2009). Cloning and expression of murine Ig genes from single B cells. J. Immunol. Methods 350, 183–193. [DOI] [PubMed] [Google Scholar]
- Vander Heiden JA, Yaari G, Uduman M, Stern JN, O’Connor KC, Hafler DA, Vigneault F, and Kleinstein SH (2014). pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics 30, 1930–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DH, Ismail N, Olano JP, McBride JW, Yu XJ, and Feng HM (2004). Ehrlichia chaffeensis: a prevalent, life-threatening, emerging pathogen. Trans. Am. Clin. Climatol. Assoc. 115, 375–382, discussion 382–384. [PMC free article] [PubMed] [Google Scholar]
- Weiss GE, Crompton PD, Li S, Walsh LA, Moir S, Traore B, Kayentao K, Ongoiba A, Doumbo OK, and Pierce SK (2009). Atypical memory B cells are greatly expanded in individuals living in a malaria-endemic area. J. Immunol. 183,2176–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow GM, Papillion AM, Kenderes KJ, and Levack RC (2017). CD11c+ T-bet+ memory B cells: Immune maintenance during chronic infection and inflammation? Cell. Immunol. 321, 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates JL, Racine R, McBride KM, and Winslow GM (2013). T cell- dependent IgM memory B cells generated during bacterial infection are required for IgG responses to antigen challenge. J. Immunol. 191, 1240–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Ma N, Madden TL, and Ostell JM (2013). IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 41, W34–W40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kobert K, Flouri T, and Stamatakis A (2014). PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkernagel RM, (2012). Immunological memory s protective immunity. Cell. Mol. Life Sci 69, 1635–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, Good-Jacobson KL, and Shlomchik MJ (2014). CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat. Immunol. 15, 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.