
Figure 1. Novel domesticated PiggyBac transposases in Paramecium.
(A) Domain organization of the PiggyBac transposase (PB) from T. ni and of Paramecium PiggyBac-related proteins (Pgm and PgmLs). The Pfam domain DDE_Tnp_1_7 is shown as a bipartite orange domain, with the RNase H fold corresponding to its right part (conserved catalytic D residues are indicated by vertical bars). The DDE_Tnp_1-like zinc ribbon is in grey. Id: % of amino acid identity; sim: % of similarity. (B) Protein sequence alignment of the residues surrounding the three catalytic aspartic acids (DDD). Following secondary structure prediction, sequence alignments were adjusted manually, using the expected position of the three catalytic D residues in the first and fourth β strands and immediately downstream of the fourth α helix of the RNase H fold domain, respectively (Hickman et al., 2010). ‘?' indicates that the expected α4 helix could not be predicted using the PSIPRED secondary structure prediction software.
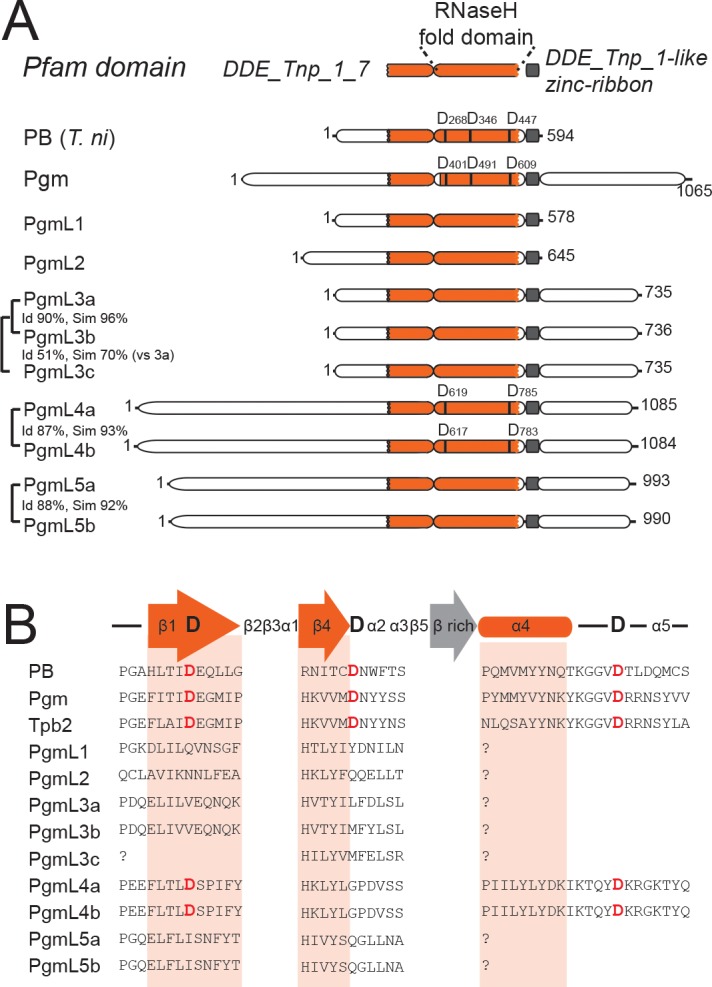
Figure 1—figure supplement 1. MUSCLE alignment of the cysteine-rich domains of ciliate domesticated PB transposases and other PB transposases.

Figure 1—figure supplement 2. Maximum Likelihood tree of ciliate domesticated PB transposases and other PB transposases.
