

Abstract
Background
The expected increase in global surface temperature due to climate change may have a tremendous effect on the structure and function of the anaerobic food web in flooded rice field soil. Here, we used the metatranscriptomic analysis of total RNA to gain a system-level understanding of this temperature effect on the methanogenic food web.
Results
Mesophilic (30 °C) and thermophilic (45 °C) food web communities had a modular structure. Family-specific rRNA dynamics indicated that each network module represents a particular function within the food webs. Temperature had a differential effect on all the functional activities, including polymer hydrolysis, syntrophic oxidation of key intermediates, and methanogenesis. This was further evidenced by the temporal expression patterns of total bacterial and archaeal mRNA and of transcripts encoding carbohydrate-active enzymes (CAZymes). At 30 °C, various bacterial phyla contributed to polymer hydrolysis, with Firmicutes decreasing and non-Firmicutes (e.g., Bacteroidetes, Ignavibacteriae) increasing with incubation time. At 45 °C, CAZyme expression was solely dominated by the Firmicutes but, depending on polymer and incubation time, varied on family level. The structural and functional community dynamics corresponded well to process measurements (acetate, propionate, methane). At both temperatures, a major change in food web functionality was linked to the transition from the early to late stage. The mesophilic food web was characterized by gradual polymer breakdown that governed acetoclastic methanogenesis (Methanosarcinaceae) and, with polymer hydrolysis becoming the rate-limiting step, syntrophic propionate oxidation (Christensenellaceae, Peptococcaceae). The thermophilic food web had two activity stages characterized first by polymer hydrolysis and followed by syntrophic oxidation of acetate (Thermoanaerobacteraceae, Heliobacteriaceae, clade OPB54). Hydrogenotrophic Methanocellaceae were the syntrophic methanogen partner, but their population structure differed between the temperatures. Thermophilic temperature promoted proliferation of a new Methanocella ecotype.
Conclusions
Temperature had a differential effect on the structural and functional continuum in which the methanogenic food web operates. This temperature-induced change in food web functionality may not only be a near-future scenario for rice paddies but also for natural wetlands in the tropics and subtropics.
Electronic supplementary material
The online version of this article (10.1186/s40168-018-0546-9) contains supplementary material, which is available to authorized users.
Keywords: rRNA, mRNA, Biopolymer degradation, CAZymes, Syntrophy, Formyltetrahydrofolate synthetase, Methanogenesis, Methanosarcina, Methanocella
Background
Methane (CH4) is a potent greenhouse gas. On a molecule basis, it is 72 times more effective than carbon dioxide in trapping heat radiation over a 20-year period [1]. Wetland rice farming is a major source of atmospheric methane, accounting for 10% of the global CH4 budget (e.g., [2]). The methanogenic degradation pathway of organic matter in submerged rice paddies and anoxic wetlands follows common principles and involves a microbial food web composed of different functional guilds of the domains Bacteria and Archaea [3]. These microbial guilds participate in a cascade of anaerobic degradation steps that involve polymer hydrolysis, fermentation, syntrophic conversion of fatty acids, homoacetogenesis, and methanogenesis [4]. Initially, methanogenic degradation may be accomplished by anaerobic respiration (e.g., NO3−, Mn(IV), Fe(III) and SO42−) but the alternative terminal electron acceptors are completely used up during the first few days under anoxic conditions [4–6].
The methanogenic community in rice field soil produces CH4 over a wide range up to 55 °C, with temperature being the key factor controlling CH4 production [7, 8]. Temperature was found not only to affect the rates of methane production but also the composition and function of methanogenic communities in rice field soils in Italy, China, Japan, Philippines, and Thailand [9–14]. Changes in both the pathways of CH4 production and the composition of the methanogenic community are only gradual at a mesophilic temperature range of up to 37 °C. Within this temperature range, methane production is characterized by acetoclastic and hydrogenotrophic methanogenesis [10]; however, the communities were shown to markedly change their methanogenic activities at temperatures higher than 40–42 °C [7, 14–16]. At these thermophilic temperatures, methane production will be dominated solely by hydrogenotrophic methanogenesis [7]. The same temperature-induced change was shown to also occur in Chinese natural wetlands [15]. Although the in situ temperature in paddy soil is generally lower than or around 30 °C [17], present-day temperatures above 40 °C are reached in the rice fields of southeastern China [18]. Moreover, the expected increase in global surface temperature by 2–4 °C by 2050 [1] makes temperature effects on structure and function of soil microbial communities an important research issue [19].
No study has yet been performed to gain a system-level understanding of the temperature effect on the methanogenic food web in rice field soil. In particular, the knowledge of microbial populations involved in polymer hydrolysis is limited. Previous studies were mostly based on total DNA and coupled the analysis of PCR amplicons with T-RFLP analysis or sequencing. Thus, each of these fingerprinting studies could focus only on a particular aspect of the temperature effect on community composition and structure [10, 14, 15, 20]. Along with process measurements, these studies showed that acetate and propionate are the key intermediates of the polymer breakdown in rice field soil. While syntrophic oxidation of propionate is actively occurring at low temperatures [21–23], syntrophic oxidation of acetate is thermodynamically feasible only at thermophilic temperatures [20]. Members of Thermoacetogenium and other Thermoanaerobacteraceae, but also Symbiobacterium, were shown to contribute to syntrophic acetate oxidation in rice field soil at 50 °C, with Methanocellaceae as the hydrogenotrophic partner [20, 24].
Here, we applied rRNA- and mRNA-based metatranscriptomics to assess the effect of temperature on the structural and functional organization of the anaerobic food web in rice field soil. The objectives were to (i) identify the microbial populations and functional genes involved in key metabolic processes and (ii) elucidate structure-function relationships that determine the anaerobic decomposition of plant polymers at mesophilic (30 °C) and thermophilic (45 °C) temperatures. In particular, we aimed to identify structural and functional links between polymer breakdown, syntrophy, and methanogenesis. Field conditions are known to be very complex, given that CH4 is produced from three different sources of organic matter: rice straw, root exudates, and soil organic matter. Their relative contributions change with time of the season [25–28]. Rice straw is the major source in the early season, while root exudates become important with the development of rice plants [25–28]. Hence, the effect of temperature on polymer breakdown and methanogenic pathways cannot be determined unambiguously under heterogenous field conditions. Therefore, we studied the impact of temperature on food web functionality under controlled laboratory conditions, with rice straw as the major carbon source. Anoxic paddy soil slurries were incubated for 30 days. Triplicate sampling for process measurements and random cDNA library construction (total of 30 libraries) were made after 5, 11, 16, 23, and 30 days of incubation.
Results
Co-occurrence networks
Two correlation networks were constructed based on the 16S rRNA reads assigned to each family-level group (Fig. 1a, b). The correlation networks consisted of 70 nodes (30 °C) and 42 nodes (45 °C) of significant (FDR corrected, P ≤ 0.01) and robust (0.6 ≤ ρ ≤ 0.9) positive correlations (Additional file 1: Tables S1 and S2). Modularity indices were 0.57 (30 °C) and 0.47 (45 °C), indicating that both correlation networks had a modular structure [29]. Three highly complex modules were identified for both temperatures 30 °C (LM1, 2, 3; Fig. 1a) and 45 °C (HM1, 2, 3; Fig. 1b). Their taxonomic composition was linked to abundance information (Fig. 1c).
Fig. 1.

Co-occurrence network analyses for 30 °C (a) and 45 °C (b). Nodes were colored by modularity class with labeled family names. Each connection (edge) between two nodes stands for a strong (Spearman’s 0.6 ≤ ρ ≤ 0.9) and significant (FDR corrected P value < 0.01) correlation. The size of each node is proportional to the number of connections (its degree). Designation of the major modules (LM, low-temperature module; HM, high-temperature module) is shown in red bold color. (c) Temporal dynamics of dominant families (> 1%) in different modules at 30 °C and 45 °C
LM1 and HM1 were composed of taxa belonging to a variety of bacterial classes. Both modules contained members of the Bacilli, Clostridia, Actinobacteria, Alphaproteobacteria, and Deltaproteobacteria (Additional file 1: Tables S1 and S2). The relative abundance of these mostly minor populations decreased with incubation time. Particular population dynamics were observed for Clostridium cluster XVIII (30 °C) and Bacillaceae (45 °C) (Additional file 1: Figures S1 and S2). The abundance of Clostridium cluster XVIII was stable for the first 11 days at 30 °C but rapidly decreased thereafter. Bacillaceae rapidly declined in abundance during the first 11 days of incubation at 45 °C but showed a second peak abundance on day 16.
LM2 and HM2 had in common that the peak abundance of most members was around day 16 and both modules contained acetoclastic methanogens (Methanosarcinaceae and Methanotrichaceae [formerly Methanosaetaceae]). However, LM2 and HM2 differed in module structure. LM2 was characterized by the dominant presence of the Methanosarcinaceae and, to a lesser extent, Ruminococcaceae. Instead, HM2 was characterized by the dominant presence of Clostridium cluster I, Ruminococcaceae, Halobacteroidaceae, Lachnospiraceae, and Limnochordaceae. Unlike all other abundant family-level groups co-occurring in HM2, Halobacteroidaceae showed a distinct peak abundance on day 9. Methanogens (Methanosarcinaceae, Methanotrichaceae) were only of minor abundance (< 0.5%) (Fig. 1c and Additional file 1: Figures S1 and S2).
LM3 and HM3 had family-level groups in common that are known to be fermenters, syntrophs, or hydrogenotrophic methanogens. Among these were Christensenellaceae, Syntrophomonadaceae, Heliobacteriaceae, Thermoanaerobacteraceae, Methanobacteriaceae, and Methanocellaceae. Despite the similarities in taxonomic composition, LM3 and HM3 significantly differed in module structure (as previously observed for LM2/HM2). In addition to the Methanocellaceae, the most abundant family-level groups in LM3 were the Christensenellaceae and, to a lesser extent, Planctomycetaceae and Solibacteraceae subgroup 3 (Acidobacteria) (Additional file 1: Tables S1 and S2). The abundance of Methanocellaceae increased to 3.2% (LM3) and 19% (HM3) till day 30 (Fig. 1c and Additional file 1: Figures S1 and S2). Heliobacteriaceae and Thermoanaerobacteraceae were dominant members of HM3 but differed in their temporal dynamics. Heliobacteriaceae had the peak abundance around day 23 (17.3%) and decreased thereafter (7.6% [day 30]). Thermoanaerobacteraceae showed a highly significant co-occurrence correlation (P < 0.0009) with the Methanocellaceae and a peak abundance (14.5%) on day 30. Other taxa that showed peak abundances on day 30 were Syntrophomonadaceae (1.2%) and Methanobacteriaceae (2.4%) (Fig. 1c).
The three highly complex LM modules were distinctly separated, but they collectively represented only a major subset of total mesophilic food web community. A few abundant family-level groups did not cluster with LM1, LM2, or LM3. These were Clostridium cluster I, Bacillaceae, Peptococcaceae, and Lachnospiraceae. These families were grouped into the low-complexity modules LM4 to LM6. Collectively, their total rRNA abundance ranged from 34.1% (day 11) to 33.9% (day 30) (Fig. 1c and Additional file 1: Tables S1 and S2). At thermophilic temperature, all the family-level groups were affiliated with one of the three HM modules, including Bacillaceae and Peptococcaceae (HM1) as well as Clostridium cluster I and Lachnospiraceae (HM2). Unlike LM2/LM3, HM2 and HM3 were connected by a strong co-occurrence link. This link was related to the central network position of the Christensenellaceae between HM2 and HM3.
Functional gene expression
NMDS plots on rRNA and mRNA level confirmed that temperature (P < 0.001) and incubation time (P < 0.001) had significant effects on community composition (Additional file 1: Figure S3). The phylum-level composition between total rRNA and mRNA agreed well at both temperatures 30 °C and 45 °C; however, some discrepancies were evident on family level. In particular, methanogen mRNA was significantly overrepresented relative to methanogen rRNA (Additional file 1: Figure S4).
The temporal dynamics of community-wide mRNA expression significantly differed between 30 and 45 °C (Fig. 2). At mesophilic temperature, bacterial and archaeal mRNA showed a low, but nearly constant, expression level throughout incubation. Collectively, their proportion of total RNA ranged between 0.04 and 0.79%. At thermophilic temperature, bacterial and archaeal mRNA expression changed with incubation time. Bacterial mRNA expression peaked on day 11, decreased towards day 16, and increased again gradually until day 30. Archaeal mRNA abundance slightly increased during the first 16 days, but its expression level greatly increased during the later stage. The proportion of bacterial and archaeal mRNA in total RNA ranged from 0.70% to 2.85% (bacteria) and 0.01% to 1.39% (archaea). Thus, community-wide mRNA expression was significantly increased relative to 30 °C.
Fig. 2.
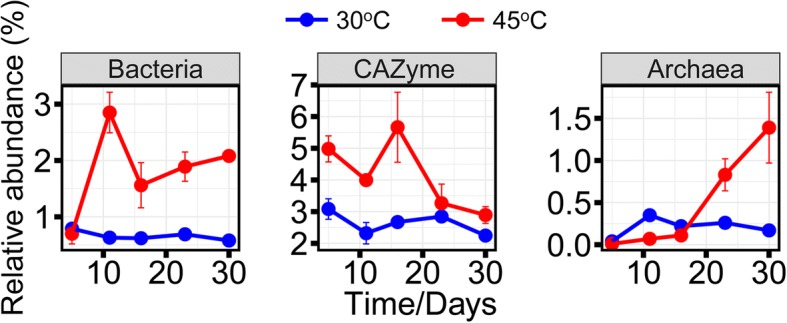
The percentage proportion of bacterial and archaeal mRNA in total RNA reads and CAZyme transcripts in total mRNA reads
The temporal expression dynamics of carbohydrate-active enzymes (CAZymes) also significantly differed between 30 and 45 °C. At mesophilic temperature, the proportion of CAZyme transcripts in total mRNA stably ranged between 2.24 and 3.08% throughout incubation time. At thermophilic temperature, their proportion in total mRNA was as high as 2.89% to 5.66%, with peak abundance on day 16. Thereafter, the CAZyme transcript level steadily decreased till day 30.
Plant polymer breakdown
Totally, 92,508 mRNA reads were identified to encode CAZymes. Of these, 75,438 reads were linked to enzymes involved in the degradation of cellulose, xylan, other hemicelluloses, and chitin (Additional file 1: Table S3). These enzymes were primarily categorized into glycosyl hydrolases (GHs) and carbohydrate-binding modules (CBMs) (Fig. 3).
Fig. 3.

Expression dynamics of GH/CBM transcripts affiliated with particular phyla (> 0.5%) (a) and temporal dynamics of gene expression related to the breakdown of cellulose, xylan, other hemicelluloses, and chitin (b). The corresponding mRNA transcript numbers are shown in Tables S3 and S4
Both temperature and incubation time had a significant impact on the taxonomic composition of the GH/CBM transcript pool. At mesophilic temperature, various microbial phyla contributed to the expression of GHs and CBMs. The majority of GH/CBM transcripts was affiliated with the Firmicutes; however, their relative abundance steadily decreased with incubation time, while that of the Bacteroidetes and Ignavibacteriae increased. At thermophilic temperature, the phylum-level diversity of GH/CBM transcripts was reduced and nearly exclusively related to the Firmicutes (Fig. 3a). On family level, the assignment rate of GH/CBM transcripts depended on the polymer, sampling time point, and temperature, and varied between 57 and 89% (Additional file 1: Table S3). The bacterial composition of the GH/CBM transcripts greatly differed for each of the polymers between 30 and 45 °C, but all the abundant family-level groups (cutoff ≥ 0.5% of total GH/CBM transcripts) belonged to the Firmicutes (Additional file 1: Figure S5).
Key enzymes involved in rice straw degradation are cellulase, endo-1,4-beta-xylanase, and alpha- and beta-galactosidases (Fig. 3b and Additional file 1: Table S4). Transcripts affiliated with four family-level groups were detected to contribute to the production of all the key enzymes, with transcripts affiliated with Methanosarcinaceae being predominant at 30 °C, and Clostridiaceae, Ruminococcaceae, and Paenibacillaceae being most abundant in the GH/CBM transcript pool at 45 °C (Additional file 1: Figure S5). The potential expression of GH transcripts by Methanosarcinaceae was supported by querying 88 methanogen genome sequences against dbCAN. Of a total of 214,249 coding genes, 2691 were functionally annotated to encode GHs, with some of them showing 100% sequence identity to GH families linked to the breakdown of complex polysaccharides (Additional file 1: Figure S5 and Additional file 2).
Acetate, propionate, hydrogen, and methane
At mesophilic temperature, acetate reached its peak concentration (8.4 mM) at day 5 or shortly thereafter and then steadily decreased towards days 16 (1.6 mM) and 30 (0.5 mM) (Fig. 4). Propionate strongly accumulated from day 5 (0.6 mM) onwards, with a peak concentration between days 16 (4.8 mM) and 23 (4.7 mM). Thereafter, its concentration rapidly decreased within 7 days to 0.3 mM on day 30. At thermophilic temperature, acetate accumulated to significantly higher concentrations than at 30 °C. It rapidly accumulated to 11.4 mM on day 5 and reached its peak concentration (17.7 mM) on day 16. Thereafter, the acetate concentration gradually decreased to 4.6 mM on day 30. The propionate concentration reached a steady state between production and consumption already around day 5 (1.2 mM). Thereafter, its concentration ranged between 1 and 1.3 mM until day 30.
Fig. 4.

Dynamics of acetate, propionate, H2, and CH4, and Gibbs free energy (ΔG) of different metabolic pathways during the 30-day incubation at 30 °C and 45 °C (means ± SE, n = 3)
The temperature-dependent concentration patterns of acetate and propionate were governed by the Gibbs free energies (ΔG). The ΔG values indicated that syntrophic acetate oxidation was feasible at 45 °C, but not at 30 °C. By contrast, syntrophic propionate oxidation was permissive at both temperatures, but only after 5 days of incubation at mesophilic temperature. As a consequence, the turnover of acetate and propionate significantly (P < 0.001) differed over the 30-day incubation period between 30 and 45 °C (Fig. 4).
The ΔG values for acetoclastic and hydrogenotrophic methanogenesis were mostly negative at both temperatures 30 °C and 45 °C. At mesophilic temperature, CH4 production started around day 5 (0.15 kPa) and increased significantly to 24.5 kPa (P < 0.001, day 11), 27.5 kPa (day 16), 42.4 kPa (day 23), and 48.5 kPa (day 30). Significant CH4 production at thermophilic temperature started around day 11, with greatest production rates between days 23 (9.5 kPa) and 30 (26.0 kPa). The H2 partial pressure at 30 °C was significantly (P < 0.001) higher until day 11 than at 45 °C. Thereafter, the H2 partial pressure had the same nearly undetectable level regardless of temperature (30 °C or 45 °C).
Syntrophy
The gene encoding formyltetrahydrofolate synthetase (fhs, EC 6.3.4.3) has been widely used as a biomarker for acetogenesis [30–33]. It is a unique pathway gene in reductive acetogenesis (acetyl-CoA pathway); however, formyltetrahydrofolate synthetase is also able to catalyze the reverse reaction, oxidizing acetate into H2 and CO2 [34, 35]. Thus, depending on the thermodynamic conditions, fhs transcripts are indicative of either reductive acetogenesis or syntrophic acetate oxidation.
The relative expression level of fhs was significantly increased (P < 0.001) at 45 °C (926 reads, 0.17% [total mRNA]) relative to 30 °C (83 reads; 0.06% [total mRNA]). The temperature-dependent difference in fhs abundance was most significant on day 23. The good correspondence between acetate turnover, Gibbs free energy calculations, and temperature-dependent fhs transcript dynamics confirmed that syntrophic acetate oxidation occurred at 45 °C, but not at 30 °C (Figs. 4 and 5).
Fig. 5.

Temporal abundance dynamics of formyltetrahydrofolate synthetase (fhs) transcripts at 30 °C and 45 °C (means ± SE, n = 3) (a) and their taxonomic composition (b). For taxonomic analysis, all fhs reads obtained for either 30 °C or 45 °C were combined to a single dataset due to the low number of transcripts obtained for each time point (n = total number of fhs reads that were taxonomically assigned)
Due to limited read numbers, we merged the fhs transcripts of all samplings to a single dataset for taxonomic identification. The vast majority of fhs transcripts at thermophilic temperature could not be assigned on family level, except those affiliated with the Thermoanaerobacteraceae (Fig. 5). Their fhs transcript abundance significantly increased (P < 0.01) with incubation time and showed a peak abundance on day 23 (Fig. 5a). The combined analysis of the rRNA and mRNA dynamics revealed a temporal co-occurrence of a 16S rRNA population assigned on family level to the Heliobacteriaceae and the fhs transcript pool (Figs. 1c and 5). A closer inspection showed that this rRNA population is comprised of rRNA reads assigned to Heliobacteriaceae and “unclassified Firmicutes” (31.7% and 68.3% on day 23, respectively). On genus level, the “unclassified Firmicutes” rRNA reads were assigned to Hydrogenispora with only a single described species (H. ethanolica) for which no genome sequence is available yet. This species belongs to the monophyletic Firmicutes clade OPB54 (Fig. 1c) that, except for H. ethanolica, consists only of environmental 16S rRNA gene sequences. Thus, co-occurrence in the peak abundances of this 16S rRNA-based population and the fhs transcript pool let us conclude that in addition to Thermoanaerobacteraceae, members of the Heliobacteriaceae and, in particular, the Firmicutes clade OPB54 contribute to syntrophic acetate oxidation.
Unlike syntrophic acetate oxidation, there is no specific marker gene that is indicative of syntrophic propionate oxidation. Therefore, we extracted the mRNA reads affiliated with two different types of syntrophic propionate oxidation pathways. The transcripts related to these two pathways made approximately 2.5% (30 °C) and 1.0% (45 °C) of total mRNA. The bacterial populations involved in propionate metabolism greatly differed on family level between 30 and 45 °C. At mesophilic temperature, the mRNA profiles were dominated by the Symbiobacteriaceae and Peptococcaceae. Their mRNA abundances showed opposing temporal dynamics. Most of the transcripts detected at 45 °C were affiliated with Thermoanaerobacteraceae, Ruminococcaceae, and Peptococcaceae (Additional file 1: Figure S6).
Methanogenesis
At mesophilic temperature, acetoclastic Methanosarcinaceae dominated the early incubation stage. Their abundance decreased between days 11 and 16. Concomitantly, the abundance of H2/CO2-utilizing Methanocellaceae increased. These opposing abundance dynamics were most evident on mRNA level (Fig. 6 and Additional file 1: Figure S7) and related to a transient decrease in CH4 production (Fig. 4). Transcripts of individual methanogen proteins showed the same temporal abundance patterns, including methyl coenzyme M reductase (McrA) (Additional file 1: Figures S7 and S8), acetyl-CoA decarbonylase/synthase complex (ACDS), and 5, 10-methylenetetrahydromethanopterin reductase (Mer)/coenzyme F420 hydrogenase (Frh). The latter two enzyme complexes are characteristic of either acetoclastic (ACDS) or hydrogenotrophic (Mer/Frh) methanogenesis (Additional file 1: Figures S7 and S8).
Fig. 6.
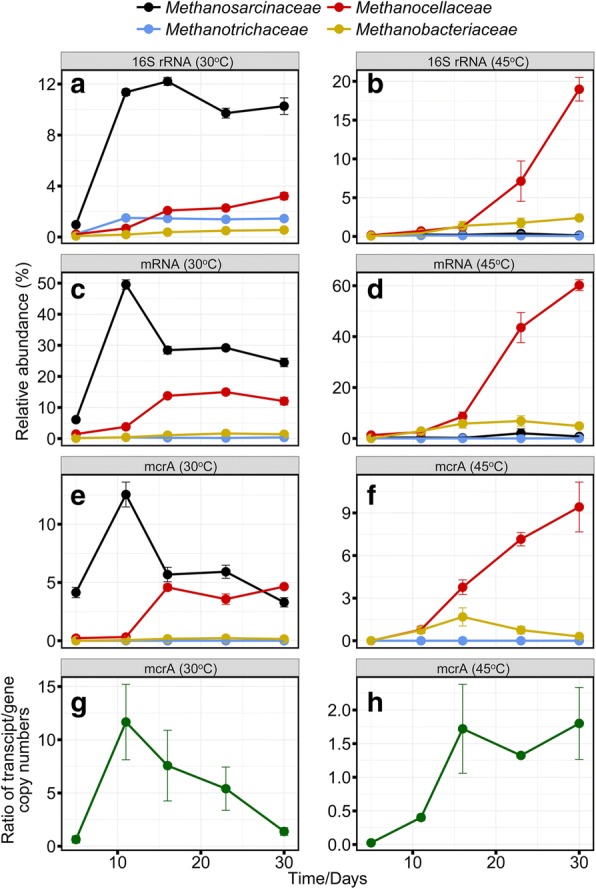
Family-level dynamics of archaeal 16S rRNA (a, b), methanogenic mRNA (c, d) and mcrA transcripts (e, f) in metatranscriptomic analysis (means ± SE, n = 3) and the temporal dynamics of the mcrA transcript/gene ratio (g, h) as determined by qPCR (gene) and RT-qPCR (transcripts) (means ± SE, n = 3). Results are shown for 30 °C and 45 °C
At thermophilic temperature, the pool of methanogen mRNA was dominated by transcripts indicative of hydrogenotrophic methanogenesis (up to 99.96 ± 0.06%) (Additional file 1: Table S5) and related to the Methanocellaceae. Members of the family Methanobacteriaceae were also detected, albeit with low abundance. According to their mcrA transcript dynamics, the greatest activity of the Methanobacteriaceae was around day 16 (Fig. 6f).
Near full-length 16S rRNA sequences of the Methanocellaceae clustered into three phylogenetically distinct populations (Fig. 7). Methanocella_2 was prevalent at 30 °C and 45 °C; however, temperature had a significant effect on the intrafamily rRNA dynamics of Methanocella_1 and Methanocella_3. Methanocella_1 was prevalent at 30 °C, but not at 45 °C. The Methanocella_1 population steadily decreased in abundance from 50.8% (day 5) to a low level on day 30. By contrast, Methanocella_3 was undetectable at 30 °C but rapidly increased in abundance from < 0.5% (day 5) to 46.9% (day 16) at 45 °C. Thereafter, the intrafamily rRNA abundance of this yet uncultured Methanocellaceae population did not significantly change until day 30 (Fig. 7).
Fig. 7.

Temporal dynamics of distinct populations within the family Methanocellaceae. The analysis is based on nearly full-length 16S rRNA sequences assembled by EMIRGE (means ± SE, n = 3) (a). Phylogenetic tree recovered by EMIRGE from archaeal 16S rRNA reads of the metatranscriptomic datasets. Ribo-contigs affiliated with the Methanocellaceae and assembled in this study are shown in color: L (30 °C), H (45 °C), with numbers representing the incubation time point. The outgroup is defined by bacterial reference sequences (b)
At 30 °C, the dynamics of the mcrA transcript to gene ratio corresponded closely with the relative abundance changes of both total mRNA and mcrA transcripts from Methanosarcinaceae (Fig. 6). At 45 °C, the dynamics of the mcrA transcript to gene ratio corresponded well with the relative abundance changes of mcrA transcripts from Methanocellaceae and, in addition, showed a nearly perfect correspondence to the intrafamily abundance dynamics of the Methanocella_3 population (compare Figs. 6h and 7a).
Discussion
We tested the impact of two contrasting temperatures on the anaerobic food web in rice field soil. The mesophilic temperature (30 °C) relates to the present-day conditions in most rice paddies, while the thermophilic temperature (45 °C) represents a scenario that may occur in various tropical and subtropical rice paddies in the near future due to global warming. Rice straw was added as the major source of carbon in CH4 production. The incorporation of rice straw into the soil strongly enhances the emission of CH4 from rice fields [14, 36, 37]. Methanogenic activity in paddy soil without rice straw is low [26, 38]. While the following text discusses the differential effects of temperature on the anaerobic food web, a detailed excursus on polymer breakdown, including a critical assessment of the expression of GH transcripts by methanogenic archaea, is made in the Additional file 3.
Differential temperature effect on the structure and function of the anaerobic food web
The mesophilic and thermophilic food web communities had a similar modular structure consisting each of three complex modules. Corresponding network modules (LM2/HM2; LM3/HM3) had various family-level groups in common. In particular, the affiliation of methanogenic families to either LM2/HM2 (Methanosarcinaceae, Methanotrichaceae) or LM3/HM3 (Methanocellaceae, Methanobacteriaceae) suggested that at both temperatures, the food web communities were governed by similar metabolic interactions. A thorough inspection of the LM and HM networks, however, revealed that along the time axis, temperature had a differential effect on the structural and functional continuum in which the food web operates. This is most evidenced by the rRNA dynamics of family-level groups affiliated with each particular module. These taxon-specific dynamics indicated that each module has a particular functional role within the food web. Corresponding modules, however, differ in their functional role. LM2 is characterized by acetoclastic methanogenesis, whereas HM2 is specified by polymer hydrolysis and primary fermentation. Both LM3 and HM3 are characterized by hydrogenotrophic methanogenesis, but CH4 production is linked to syntrophic oxidation of different intermediates (propionate [30 °C] vs. acetate [45 °C]). Common to both temperatures is that the first module (LM1, HM1) represents the early incubation stage. Their members showed no co-occurrence with methanogens and, except for Clostridium cluster XVIII (LM1) and Bacillaceae (HM1), were mostly of minor abundance. These populations may have been members of the starting community or initially proliferated by utilizing alternative electron acceptors, such as Fe(III), nitrate, sulfur, or sulfate. In anoxic paddy soil, alternative electron acceptors are completely used up during the first few days upon flooding [5].
The gene expression dynamics of Bacteria and Archaea significantly differed between the two temperatures, thereby providing another line of evidence for a differential temperature effect on the anaerobic food web (Fig. 2). At mesophilic temperature, bacterial and archaeal mRNAs were expressed at low but constant level throughout incubation time, with no particular peak abundances. By contrast, their mRNA expression at thermophilic temperature was characterized by strong abundance dynamics. Community-wide transcription showed two distinct activity stages characterized first by polymer hydrolysis and fermentation (days 5–16) and then followed by syntrophic acetate oxidation coupled to hydrogenotrophic methanogenesis (days 16–30). Syntrophy as the predominant process during the later stage is further evidenced by the co-occurrence link between the increasing mRNA expression activities of Bacteria and Archaea.
Temperature significantly affects polymer breakdown in rice field soil. The strongly differing CAZyme transcript dynamics provide first-time molecular evidence. Previous enzymatic research has shown that at mesophilic temperature, the hydrolytic release of easily degradable polymer substances occurs over the first 30 days of incubation and becomes increasingly the rate-limiting step in microbial activity after 14 days [39, 40]. The occurrence of polymer hydrolysis over the complete 30-day incubation period is well supported by the abundance dynamics of plant polymer degraders (discussed below). Apparently, this process is characterized by a low but constant expression level of CAZyme transcripts. At thermophilic temperature, the expression level of CAZyme transcripts was significantly increased. The GH/CBM transcript abundance peaked between days 11 and 16, in good agreement with the temporal expression dynamics of total bacterial mRNA. Thereafter, the CAZyme transcript level steadily decreased, suggesting that syntrophic acetate oxidizers were not majorly involved in polymer hydrolysis. The temporal separation of polymer breakdown and syntrophic acetate oxidation may have several reasons. On the one hand, it is well established that thermophilic conditions generally result in an increase in polymer degradation rates, which is attributed to increased hydrolytic activity [41]. On the other hand, the metabolic interaction between acetate oxidizers and Methanocellaceae may be characterized by growth rates that involve a substantial delay time until critical sizes of syntrophic populations are established, leading to net consumption of acetate.
Gradual plant polymer breakdown drives acetoclastic methanogenesis and syntrophic propionate oxidation at mesophilic temperature
The gradual decomposition of plant polymers over the complete incubation period of 30 days is corroborated by the persistent presence of family-level groups involved in polymer hydrolysis. These groups belonged to the low-complexity modules LM4, LM5, and LM6 (i.e., Bacillaceae, Clostridiaceae, Lachnospiraceae, Peptococcaceae). Other family-level groups were affiliated with LM2 (i.e., Methanosarcinaceae, Ruminococcaceae) but were also abundantly present during the late incubation stage. On CAZyme transcript level, the Firmicutes abundance, however, steadily decreased till day 30. Concomitantly, Proteobacteria, Planctomycetes, Bacteroidetes, and Ignavibacteriae increased in abundance (Fig. 3). Relative to their contribution to total bacterial mRNA, non-Firmicutes were significantly overrepresented in the GH/CBM transcript pool during the later stage (Fig. 3 and Additional file 1: Figure S4). This is most evident for the phylum Bacteroidetes that, after Firmicutes, contributed second most to the bacterial GH/CBM transcript pool. Bacteroidetes are well-known for their important role in polymer degradation [42, 43]. Conclusively, the degradation of plant polymers at 30 °C involves a complex functional interaction of diverse bacterial taxa, in which the contribution of non-Firmicutes populations strongly increased with incubation time (Fig. 3). One may speculate that the abundance increase in non-Firmicutes is causally linked to the previous finding that the easily degradable polymer substances become gradually exhausted during the later incubation stage [40].
Methanosarcinaceae and, to a lesser extent, Methanotrichaceae determine the functional role of LM2. It has been shown that Ruminococcus produces increased amounts of acetate in the presence of methanogens. This redirection of the fermentation process may explain the temporal co-occurrence of Methanosarcinaceae and Ruminococcaceae. Acetoclastic methanogenesis as the dominant process of CH4 production during the early stage is well confirmed by the RNA dynamics, with concurrent peak abundances of rRNA, total mRNA, and transcripts encoding particular methanogenic enzymes (MCR, CODH) (Fig. 6 and Additional file 1: Figures S7 and S8) on day 11. The subsequent shift from acetoclastic methanogenesis towards increasing contribution of hydrogenotrophic methanogenesis to total CH4 production, however, is clearly more pronounced on mRNA level than on rRNA level, reinforcing the view that mRNA is more responsive to environmental change than rRNA [44]. The reduced but steady level of methanosarcinal mRNA during the later stage relates well to the previous result that polymer hydrolysis becomes increasingly the rate-limiting step in microbial activity after 14 days of incubation [40]. Thus, ongoing acetoclastic methanogenesis is presumably driven by acetate produced through rate-limiting polymer breakdown and syntrophic oxidation of propionate [23]. The mcrA gene is present only in one or two copies in methanogen genomes [45, 46], thereby making it possible to relate the temporal dynamics in its transcript to gene ratio to changes in cellular expression level. Thus, it is evident that the significant abundance increase in methanosarcinal mcrA transcripts between days 5 and 11 is primarily related to a strong increase in their cellular expression level, in addition to growth effects and the increase in methanogen population size (Fig. 6g). The mcrA transcript to gene ratio values are similar to those previously observed for highly active acetoclastic methanogens in peatlands [46] and Chinese paddy soils [47].
LM3 is characterized by syntrophic interaction between propionate oxidizers and H2/CO2-utilizing Methanocellaceae. We identified three bacterial families to be involved in propionate metabolism: Christensenellaceae (rRNA level) as well as Symbiobacteriaceae and Peptococcaceae (mRNA level). Christensenellaceae and methanogens significantly co-occur in the gut microbiome of human individuals with low body mass index [48]. The same group of authors also showed that in the gut of mice, elevated levels of propionate and butyrate are linked to the co-occurrence of Christensenellaceae and hydrogenotrophic methanogens. In our study, Christensenellaceae also showed a significant co-occurrence pattern with the Methanocellaceae (Fig. 1); however, their functional role still needs to be elucidated. The two known pathways of propionate degradation are (i) methyl-malonyl-coenzyme A (CoA) pathway and (ii) disproportionation of two propionate molecules to acetate and butyrate followed by syntrophic β-oxidation. The expression of mRNA affiliated with these two pathways by Christensenellaceae could not be confirmed.
Members of the Bacteroidetes and Firmicutes can operate the methyl-malonyl-coenzyme pathway in the reverse direction to produce propionate [49, 50]. Linking propionate turnover with the dynamics of propionate metabolism-related mRNA, it is reasonable to assume that Symbiobacteriaceae were primarily involved in propionate production (Fig. 4 and Additional file 1: Figure S6). Members of this family have been detected in paddy soils [51] and shown to be capable of producing propionate [52]. Most likely, Peptococcaceae were the major players in syntrophic propionate oxidation (Additional file 1: Figure S6). Their mRNA expression dynamics corresponded well to those of the Methanocellaceae. Propionate-oxidizing Peptococcaceae were previously detected with high abundance in paddy soil [21, 23] and Arctic peat [33].
The anaerobic food web at thermophilic temperature is a two-stage process driven by polymer breakdown and syntrophic acetate oxidation
The decomposition of plant polymers only involved members of the Firmicutes, which is well supported by their overwhelming contribution to total bacterial mRNA (Fig. 3 and Additional file 1: Figure S4). Clostridiaceae and Ruminococcaceae were dominantly affiliated with HM2 and the central players in polymer breakdown. These two families contributed most to the expression of key enzymes, such as cellulase, endo-1,4-beta-xylanase, alpha-galactosidase, and beta-galactosidase. Their rRNA dynamics corresponded well with the transcript dynamics of cellulase and, to a lesser extent, endo-1,4-beta-xylanase. The abundance of both transcript types peaked on day 16. Their similar expression dynamics suggest that the degradation of cellulose and xylan is highly interrelated [53, 54].
HM3 was characterized by the co-occurrence of syntrophic acetate oxidizers and Methanocellaceae (Fig. 1b, c). The contribution of Thermoanaerobacteraceae to acetate oxidation was confirmed by their rRNA-based co-occurrence with Methanocellaceae and, in addition, fhs transcript analysis (Figs. 1b,c and 5). Several genera collectively contributed to the abundance increase of the Thermoanaerobacteraceae in co-occurrence with the Methanocellaceae (data not shown). These genera are represented by the thermotolerant Tepidanaerobacter acetatoxydans [55] and Syntrophaceticus schinkii [56], and the thermophilic Thermacetogenium phaeum [20]. Members of these genera are known for their ability to oxidize acetate syntrophically [20, 41, 42].
However, the greatest contribution to syntrophic acetate oxidation was made by unknown or yet uncharacterized populations that had been grouped in fhs transcript analysis as “unclassified Firmicutes” or even “unclassified Bacteria”. The rRNA dynamics of Heliobacteriaceae and OPB54 agreed perfectly with the relative fhs gene expression dynamics (Figs. 1 and 5). Heliobacteriaceae as a possible candidate for contributing to syntrophic acetate oxidation was previously mentioned in a single report [57]. OPB54 is considered to represent a new but undescribed taxon within the Firmicutes at the rank of a new order or even class [58]. On rRNA gene level, the abundant presence of OPB54 in methanogenic thermophilic reactors (55 °C) has been observed, but their functional role remained elusive. Given that no genomic information is available for OPB54, its members may be responsible for the expression of a large proportion of fhs transcripts categorized as unclassified Bacteria, Firmicutes, or Clostridia.
Hydrogenotrophic methanogenesis was overwhelmingly dominated by Methanocellaceae, as confirmed by analysis of total RNA, mRNA, and mcrA transcripts. This increase in methanogenic activity was related to a strong proliferation of Methanocella_3 (Fig. 7). Methanocellaceae are known to have two temperature optima at ~ 41 °C and 50 °C [59], but temperature preference under resource competition has not yet been related to a particular phylogenetic clustering. Our results indicate a relationship between phylogeny and temperature preference, with Methanocella_1 populations preferring mesophilic conditions. The strong proliferation of Methanocella_3 at 45 °C may suggest that this population represents a new Methanocella ecotype with a third previously unknown temperature optimum around 45 °C. Methanocellaceae outcompeted Methanobacteriaceae during the later incubation period (Fig. 6), possibly due to their ability to lower the partial pressure of hydrogen below the level that can be utilized by Methanobacterium spp. [14, 60]. The nearly perfect correspondence between the temporal dynamics of mcrA transcript to gene ratio (Fig. 6h) and intrafamily rRNA abundance of Methanocella_3 (Fig. 7a) suggests a functional link between the increase in Methanocella_3 population size and the mcrA expression level of its cells.
Conclusions
The anaerobic food web is driven by the functional interplay of a complex microbial community (Fig. 8). Some taxa were newly identified as potential members of particular functional guilds, such as Methanosarcinaceae (polymer hydrolysis), Christensenellaceae (propionate metabolism), and Heliobacteriaceae and members of clade OPB54 (syntrophic acetate oxidation). Despite the similarities in modular structure and taxonomic composition, temperature and, in consequence, thermodynamic constraints had a significant impact on all functional processes. At both temperatures, food web functionality was directly linked to polymer hydrolysis and the transition from early to late stage. The temperature-induced change in functionality may not only be a realistic near-future scenario for rice paddies but also for methanogenic wetlands in the tropics and subtropics; however, more research is necessary to elucidate the impact of the climate change-induced change in anaerobic food web functionality on CH4 emission. In particular, flux measurements in the field will be required to gain accurate estimates whether and how this change in food web functionality will affect the source strength of flooded rice fields and other methanogenic wetlands for atmospheric CH4.
Fig. 8.
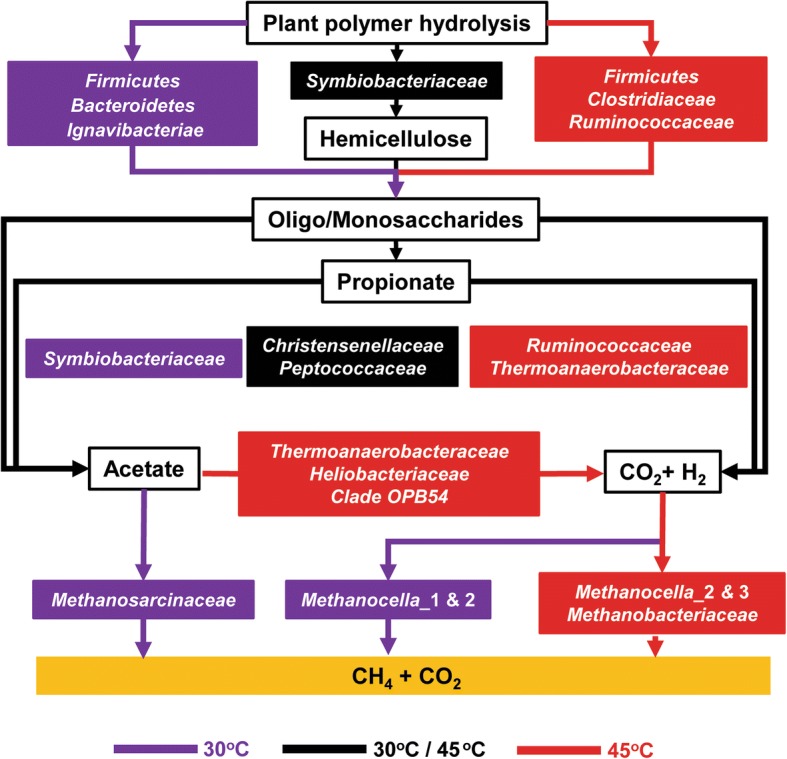
Hypothetical model of the anaerobic food web in rice field soil. The temperature-dependent contribution of particular microbial groups is indicated by different colors. The figure is adapted from Fig. 1 in [4]
Methods
Anoxic incubations
Soil was collected from rice fields at the Italian Rice Research Institute (IRRI) in Vercelli, Italy in June 2013. The soil was air-dried and stored as dry lumps at room temperature. Slurry microcosms were set up by filling 125 mL bottles with 40 g dry soil, 0.5 g rice straw (1–2 cm pieces), and 40 mL of autoclaved water. This amount of rice straw is commonly used in paddy soil slurry studies [14, 18, 61]. It corresponds to 37.5 t ha−1, i.e., about three times higher than under field conditions [62]. The thoroughly mixed slurries were sealed with butyl rubber stoppers and flushed with N2 to establish anoxic conditions. The bottles were then incubated at either 30 °C or 45 °C for 30 days. All bottles were preincubated for 4 days to initiate methanogenic activity [10, 14, 63]. At both temperatures, slurry materials beneath the top layer were collected at days 5, 11, 16, 23, and 30. The samples were immediately shock-frozen using liquid nitrogen and then kept for molecular analysis at − 80 °C. In addition, gas samples (0.1 mL) and liquid samples (0.5 mL) were taken from the same set of slurries for process measurements. All analyses are based on triplicate slurries, destructively sampled at each time point. Acetate concentration and methane formation were monitored as described previously [64]. The same set of slurries was used for chemical (acetate, propionate, and gas measurements) and molecular analyses.
Calculations of Gibbs free energies
Gibbs free energy (ΔG) of acetoclastic methanogenesis, hydrogenotrophic methanogenesis, syntrophic acetate oxidation, and syntrophic propionate oxidation was calculated based on the concentrations of gases and dissolved compounds in the slurries incubated at either 30 °C or 45 °C [65]. The Gibbs free energy of different pathways was calculated by using the actually measured partial pressures and concentrations according to the Nernst equation [65].
Syntrophic acetate oxidation:
Syntrophic propionate oxidation:
Acetoclastic methanogenesis:
Hydrogenotrophic methanogenesis:
∆H0 of each reaction (1–4) is 270.6, 205.1, 17.7, and − 252.9 KJ mol−1, respectively. In addition, RB of each reaction (1–4) is 0.723, 0.447, 0.314, and -0.41 KJ mol-1 K-1 , respectively.
Nucleic acid extraction
The FastDNA ®SPIN Kit for Soil (MP Biomedicals, Solon, OH) was used for extraction of total DNA. Total RNA was extracted from slurries using a previously described method [66]. Briefly, fresh soil (0.5 g) was mixed with the same volume of glass beads and suspended in 700 μl of TPM buffer (0.5 M Tris pH 7.0, 1.7% polyvinylpyrrolidone, 0.2 M MgCl2). The mixture was shaken at 6.0 m s−1 for 45 s followed by centrifugation at 20,000×g for 4 min. The pellet was resuspended in 700 μl of PBL buffer (0.05 M Tris pH 7.0, 0.05 M Na2EDTA, 0.1% SDS w/v, 6% v/v phenol) and the lysis procedure was repeated as described above. Supernatants were purified by two-step phase extraction with phenol-chloroform-isoamylalcohol and chloroform-isoamylalcohol. RNA was precipitated with precooled isopropanol and resuspended in 50 μl of TE buffer (10 mM Tris-HCl, 1 mM EDTA [pH 8.0]). RNA extracts were treated with DNase I (Ambion) and purified using the RNA Clean and Concentrator kit (ZymoResearch) according to the manufacturer’s instructions. The integrity of the purified RNA was checked by Bio-Rad Experion™ and RNA HighSens Chips (Bio-Rad).
RNA-Seq
Thirty samples of total RNA (corresponding to three replicate slurries × 5 time points × 2 temperatures [30 °C, 45 °C]) were subjected to cDNA synthesis using the NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (New England Biolabs, USA) according to the manufacturer’s instructions. cDNA yield and integrity were determined by fluorometry (Qubit®, ThermoFisher, USA) and automated high-resolution electrophoresis (Experion, Bio-Rad, USA). The thirty cDNA libraries were sequenced on an Illumina HiSeq platform in paired-end mode (2 × 250 bp) at the Max Planck Genome Centre Cologne. Illumina RNA-Seq resulted in 133,049,210 reads, ranging from 3,450,734 to 5,841,253 reads per cDNA library (Additional file 1: Table S6).
Bioinformatic processing of total RNA
Paired-end reads were merged using fastq_mergepairs function (fastq_minovlen 20, fastq_minmergelen 250) in USEARCH7. Quality filtering was performed using the USEARCH7 fastq_filter function with a maximum expected error threshold of 0.5 [67]. Analysis of 16S rRNA and putative mRNA reads was carried out with a customized pipeline as described previously [44].
Briefly, both 16S and 18S rRNA were extracted from total RNA by SortMeRNA 2.0 [68]. A total of 77,665,958 quality-filtered reads was obtained from total RNA, of which 34,458,629 and 1,361,405 reads were derived from 16S rRNA and 18S rRNA, respectively (Additional file 1: Table S6). USEARCH7 and QIIME (version 1.9.1) [69] were used as the overall framework for rRNA-derived reads. OTU clustering was done using the UCLUST algorithm integrated into USEARCH and an OTU threshold of 95% sequence identity. Using a custom PYTHON script, the resulting OTU table was converted to match the particular specifications of QIIME. Taxonomic assignments were done using the RDP classifier [70] implemented in QIIME with SILVA SSU (release 128) as the reference database. Due to the low proportion of eukaryal 18S rRNA, our study focused on bacterial and archaeal 16S rRNA. In particular, fungi were nearly not detectable. They contributed between 0.03 and 0.3% to total SSU rRNA reads (Additional file 1: Tables S7 and S8).
For mRNA analysis, reads mapping to rRNA and non-coding small RNA were filtered by SortMeRNA 2.0, using SILVA (release 128) and RFAM as reference databases. The remaining reads were considered putative mRNA. We identified only a low proportion of mRNA reads (0.39–3.20%) to be derived from Eukaryota (Additional file 1: Table S9). Like for SSU rRNA, we therefore extracted only bacterial and archaeal mRNA reads through MEGAN6 Ultimate Edition [71] for downstream analysis. These were a total of 2,245,249 quality-filtered reads (Additional file 1: Table S10). Taxonomic assignment and functional annotation of the mRNA reads were carried out using the UBLAST algorithm implemented in USEARCH7 applying an e value cutoff of 1e-5, maxhits 50, and maxaccepts 50 for database searches against NCBI’s non-redundant protein database (release 2017). MEGAN6 Ultimate Edition was used for parsing and downstream analysis of the UBLAST output. Taxonomy assignments were made by the “lowest common ancestor” (LCA) method using the following parameters: minimum support 1, minimum bit score 150, top percent 10 [72]. A total of 1,242,931 reads had a taxonomic hit in the NCBI’s non-redundant protein database (Additional file 1: Table S10). Taxonomically assigned mRNA was functionally annotated according to SEED subsystems and KEGG categories (release 2017). For in-depth sub-transcriptome analysis, mRNA reads related to particular functional genes and pathways were extracted from MEGAN6 and blasted against NCBI’s non-redundant protein database. UBLAST searches were carried out with an e value threshold of 1e-10, followed by the analysis in MEGAN6 Ultimate Edition.
Assembly of full-length Methanocellaceae 16S rRNA sequences
All the Illumina reads affiliated with 16S rRNA of the Methanocellaceae were extracted by Python scripts and used to reconstruct near full-length 16S rRNA sequences via 60 iterations in EMIRGE. The abundance estimate of assembled reads per 16S rRNA sequence is NormPrior, which is a length-normalized estimate determined by the EMIRGE algorithm [73]. Each 16S rRNA sequence assembled by EMIRGE is the result of Illumina reads > 97% similarity. Assemblies were done separately for each sequence dataset (= replicate slurry) rather than pooling the datasets together. Totally, 152 full-length 16S rRNA sequences were produced across all replicate samples. Phylogenetic analysis of assembled 16S rRNA was done with ARB software (http://www.arb-home.de/; version 2016; [74]) as previously described [14]. Sequences were aligned using SINA webaligner (https://www.arb-silva.de/aligner/) and imported into the SILVA database (nr99 (release 128)) obtained from the SILVA homepage [75]. Then, the alignment was checked and corrected manually by ARB_EDIT. Phylogenetic placement was performed relative to euryarchaeal reference sequences. A 16S rRNA-based tree was constructed for sequences having a length > 1200 bp. All 152 nearly full-length Methanocellaceae 16S rRNA sequences were added to this tree using the ARB parsimony tool, which allows the addition of sequences to phylogenetic trees without changing global tree topologies [76].
Analysis of carbohydrate-active enzymes (CAZymes)
CAZyme-affiliated mRNA reads were identified by querying total mRNA datasets against dbCAN, a database for carbohydrate-active enzyme annotation (release 2017) [77]. Database search was done with DIAMOND [78] in blastx mode applying the default e value cutoff of 1e-3. Functional CAZyme modules (e.g., cellulose degradation) were defined by grouping related enzymatic functions based on their enzyme commission numbers. A mapping file for functional annotation was created using all available entries in dbCAN. The mapping file was stored as indexed sqlite database object and queried using a custom Python script. The resulting annotations are based on matching dbCAN tophits for mRNA reads queried against the formatted mapping file and defined CAZyme modules.
Subsets of mRNA reads linked to CAZyme functions of interest were obtained using custom Python scripts. These subsets were taxonomically assigned by applying the strategy outlined by Menzel and colleagues [79], which relies on identifying maximum exact matches for query sequences in reference databases using the backward search algorithm implemented in the Burrows-Wheeler transform [80, 81]. Prokaryotic genomes from NCBI RefSeq and NCBI nr were used as reference databases.
qPCR and RT-qPCR of mcrA
Numbers of genes and transcripts encoding methyl coenzyme reductase A (mcrA) were quantified with primer set mlas-mod-f/mcrA-rev-r [82]. Total RNA was randomly reverse transcribed using the GoScript Reverse Transcription System (Promega, Mannheim, Germany). Standard curves were constructed using PCR amplicons (mcrA genes/transcripts, calibration range from 10 to 108 copies). The analyses were performed using SYBR Green-based assays [44]. qPCR was carried out using a CFX Connect Real-Time PCR detection system (Bio-Rad). Like for RNA-Seq, qPCR and RT-qPCR of mcrA were each performed from 30 samples. All reactions were carried out in three technical replicates. The PCR efficiency was 89% (R2 > 0.99).
Statistical analyses
Non-metric multidimensional scaling (NMDS) plots were generated in the R (version 3.5.0) software (R Development Core Team, 2014) using the vegan package (version 2.4.4) [83] with the metaMDS function and Bray–Curtis similarity matrices on square-root transformed data. Analyses of Similarity (ANOSIM) of Bray–Curtis similarity were performed to test whether community differences between the (i) two temperatures and (ii) incubation time points in the NMDS ordinations are statistically significant [84].
Co-occurrence networks were constructed for 30 °C and 45 °C using pipelines developed previously [85]. These were based on time-dependent changes in taxon-specific rRNA abundances. Ribosomal RNA reads were clustered into OTUs at 95% sequence identity and all the OTUs assigned on family level were retained for further analysis. Family-level groups with less than 0.5‰ of total reads were discarded. Spearman correlation coefficient 0.6 ≤ ρ ≤0.9 and false discovery rate (FDR) corrected P value ≤ 0.01 were used for co-occurrence network construction. The number of nodes and edges, average path length, and networks were visualized by Gephi software [86].
Additional files
This document contains supporting Figures S1-S8. and Tables S1-S10. The file is published online as Supporting Information. (PDF 1430 kb)
Corroborating GH expression by methanogens through blasting their genome sequences (download from NCBI database) against dbCAN database. (XLSX 378 kb)
Detailed excursus on anaerobic polymer breakdown and the temperature-dependent expression of CAZymes. (PDF 87 kb)
Acknowledgements
We thank Ralf Conrad for critical readings of the manuscript and valuable discussions. We also thank Rehab Abdallah for helpful comments.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft through the collaborative research center SFB 987.
Availability of data and materials
The Illumina RNA-Seq data generated during this study have been deposited at the NCBI Sequence Read Archive under the BioProject number PRJNA412498 with accession number SRR6122723 to SRR6122752. The assembled Methanocellaceae 16S rRNA sequences have been submitted to the NCBI database with accession numbers MG977525–MG977676. The custom PYTHON scripts used in our study are available at http://github.com/wegnerce/peng_et_al_2018.
Authors’ contributions
JP and WL designed the study. QB contributed to the RNA extraction. JP carried out the whole cDNA library preparation and all other experimental research. JP and CEW conducted data analyses. JP did network analysis and fatty acids measurement with significant support from PL. JP, CEW, and WL wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.IPCC. Climate Change 2013: The Physical Science Basis. In: Stocker TF, Qin D, Plattner GK, Tignor M, Allen SK, Boschung J, Nauels A, Xia Y, Bex V, Midgley PM, editors. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge, & New York: Cambridge University Press; 2013.
- 2.Tian HQ, Lu CQ, Ciais P, Michalak AM, Canadell JG, Saikawa E, Huntzinger DN, Gurney KR, Sitch S, Zhang BW, Yang J, Bousquet P, Bruhwiler L, Chen GS, Dlugokencky E, Friedlingstein P, Melillo J, Pan SF, Poulter B, Prinn R, Saunois M, Schwalm CR, Wofsy SC. The terrestrial biosphere as a net source of greenhouse gases to the atmosphere. Nature. 2016;531(7593):225. doi: 10.1038/nature16946. [DOI] [PubMed] [Google Scholar]
- 3.Ferry JG. Fundamentals of methanogenic pathways that are key to the biomethanation of complex biomass. Curr Opin Biotech. 2011;22(3):351–357. doi: 10.1016/j.copbio.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conrad R. Contribution of hydrogen to methane production and control of hydrogen concentrations in methanogenic soils and sediments. FEMS Microbiol Ecol. 1999;28(3):193–202. doi: 10.1111/j.1574-6941.1999.tb00575.x. [DOI] [Google Scholar]
- 5.Achtnich C, Bak F, Conrad R. Competition for electron-donors among nitrate reducers, ferric iron reducers, sulfate reducers, and methanogens in anoxic paddy soil. Biol Fert Soils. 1995;19(1):65–72. doi: 10.1007/BF00336349. [DOI] [Google Scholar]
- 6.Megonigal JP, Hines ME, Visscher PT. Anaerobic metabolism: linkages to trace gases and aerobic processes. In: Schlesinger WH, editor. Biogeochemistry. Oxford: Elsevier-Pergamon; 2004. pp. 317–424. [Google Scholar]
- 7.Conrad R, Klose M, Noll M. Functional and structural response of the methanogenic microbial community in rice field soil to temperature change. Environ Microbiol. 2009;11(7):1844–1853. doi: 10.1111/j.1462-2920.2009.01909.x. [DOI] [PubMed] [Google Scholar]
- 8.Yao H, Conrad R. Effect of temperature on reduction of iron and production of carbon dioxide and methane in anoxic wetland rice soils. Biol Fert Soils. 2000;32(2):135–141. doi: 10.1007/s003740000227. [DOI] [Google Scholar]
- 9.Conrad R, Klose M, Lu YH, Chidthaisong A. Methanogenic pathway and archaeal communities in three different anoxic soils amended with rice straw and maize straw. Front Microbiol. 2012;3:4 10.3389/fmicb.2012.00004. [DOI] [PMC free article] [PubMed]
- 10.Fey A, Conrad R. Effect of temperature on carbon and electron flow and on the archaeal community in methanogenic rice field soil. Appl Environ Microbiol. 2000;66(11):4790–4797. doi: 10.1128/AEM.66.11.4790-4797.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krüger M, Frenzel P, Kemnitz D, Conrad R. Activity, structure and dynamics of the methanogenic archaeal community in a flooded Italian rice field. FEMS Microbiol Ecol. 2005;51(3):323–331. doi: 10.1016/j.femsec.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 12.Ramakrishnan B, Lueders T, Dunfield PF, Conrad R, Friedrich MW. Archaeal community structures in rice soils from different geographical regions before and after initiation of methane production. FEMS Microbiol Ecol. 2001;37(2):175–186. doi: 10.1111/j.1574-6941.2001.tb00865.x. [DOI] [Google Scholar]
- 13.Watanabe T, Cahyani VR, Murase J, Ishibashi E, Kimura M, Asakawa S. Methanogenic archaeal communities developed in paddy fields in the Kojima Bay polder, estimated by denaturing gradient gel electrophoresis, real-time PCR and sequencing analyses. Soil Sci Plant Nutr. 2009;55(1):73–79. doi: 10.1111/j.1747-0765.2008.00334.x. [DOI] [Google Scholar]
- 14.Peng JJ, Lu Z, Rui J, Lu YH. Dynamics of the methanogenic archaeal community during plant residue decomposition in an anoxic rice field soil. Appl Environ Microbiol. 2008;74(9):2894–2901. doi: 10.1128/AEM.00070-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu Y, Fu L, Lu YH, Hugenholtz F, Ma K. Effect of temperature on the structure and activity of a methanogenic archaeal community during rice straw decomposition. Soil Biol Biochem. 2015;81:17–27. doi: 10.1016/j.soilbio.2014.10.031. [DOI] [Google Scholar]
- 16.Wu XL, Friedrich MW, Conrad R. Diversity and ubiquity of thermophilic methanogenic archaea in temperate anoxic soils. Environ Microbiol. 2006;8(3):394–404. doi: 10.1111/j.1462-2920.2005.00904.x. [DOI] [PubMed] [Google Scholar]
- 17.Schütz MM, Hansen LB, Steuernagel GR, Reneau JK, Kuck AL. Genetic parameters for somatic cells, protein, and fat in milk of holsteins. J Dairy Sci. 1990;73(2):494–502. doi: 10.3168/jds.S0022-0302(90)78697-3. [DOI] [Google Scholar]
- 18.Rui JP, Peng JJ, Lu YH. Succession of bacterial populations during plant residue decomposition in rice field soil. Appl Environ Microbiol. 2009;75(14):4879–4886. doi: 10.1128/AEM.00702-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davidson EA, Janssens IA. Temperature sensitivity of soil carbon decomposition and feedbacks to climate change. Nature. 2006;440(7081):165–173. doi: 10.1038/nature04514. [DOI] [PubMed] [Google Scholar]
- 20.Liu FH, Conrad R. Thermoanaerobacteriaceae oxidize acetate in methanogenic rice field soil at 50°C. Environ Microbiol. 2010;12(8):2341–2354. doi: 10.1111/j.1462-2920.2010.02289.x. [DOI] [PubMed] [Google Scholar]
- 21.Gan YL, Qiu QF, Liu PF, Rui JP, Lu YH. Syntrophic oxidation of propionate in rice field soil at 15 and 30°C under methanogenic conditions. Appl Environ Microbiol. 2012;78(14):4923–4932. doi: 10.1128/AEM.00688-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu PF, Conrad R. Syntrophobacteraceae-affiliated species are major propionate-degrading sulfate reducers in paddy soil. Environ Microbiol. 2017;19(4):1669–1686. doi: 10.1111/1462-2920.13698. [DOI] [PubMed] [Google Scholar]
- 23.Lueders T, Pommerenke B, Friedrich MW. Stable-isotope probing of microorganisms thriving at thermodynamic limits: syntrophic propionate oxidation in flooded soil. Appl Environ Microbiol. 2004;70(10):5778–5786. doi: 10.1128/AEM.70.10.5778-5786.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rui JP, Qiu QF, Lu YH. Syntrophic acetate oxidation under thermophilic methanogenic condition in Chinese paddy field soil. FEMS Microbiol Ecol. 2011;77(2):264–273. doi: 10.1111/j.1574-6941.2011.01104.x. [DOI] [PubMed] [Google Scholar]
- 25.Watanabe A, Takeda T, Kimura M. Evaluation of origins of CH4 carbon emitted from rice paddies. J Geophys Res-Atmos. 1999;104(D19):23623–23629. doi: 10.1029/1999JD900467. [DOI] [Google Scholar]
- 26.Ji Y, Liu PF, Conrad R. Response of fermenting bacterial and methanogenic archaeal communities in paddy soil to progressing rice straw degradation. Soil Biol Biochem. 2018;124:70–80. doi: 10.1016/j.soilbio.2018.05.029. [DOI] [Google Scholar]
- 27.Tokida T, Adachi M, Cheng WG, Nakajima Y, Fumoto T, Matsushima M, Nakamura H, Okada M, Sameshima R, Hasegawa T. Methane and soil CO2 production from current-season photosynthates in a rice paddy exposed to elevated CO2 concentration and soil temperature. Glob Chang Biol. 2011;17(11):3327–3337. doi: 10.1111/j.1365-2486.2011.02475.x. [DOI] [Google Scholar]
- 28.Yuan Quan, Pump Judith, Conrad Ralf. Partitioning of CH4 and CO2 Production Originating from Rice Straw, Soil and Root Organic Carbon in Rice Microcosms. PLoS ONE. 2012;7(11):e49073. doi: 10.1371/journal.pone.0049073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newman MEJ. Modularity and community structure in networks. Proc Natl Acad Sci U S A. 2006;103(23):8577–8582. doi: 10.1073/pnas.0601602103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lovell CR, Leaphart AB. Community-level analysis: key genes of CO2-reductive acetogenesis. Environ Microbiol. 2005;397:454–469. doi: 10.1016/S0076-6879(05)97028-6. [DOI] [PubMed] [Google Scholar]
- 31.Mosbaek F, Kjeldal H, Mulat DG, Albertsen M, Ward AJ, Feilberg A, Nielsen JL. Identification of syntrophic acetate-oxidizing bacteria in anaerobic digesters by combined protein-based stable isotope probing and metagenomics. ISME J. 2016;10(10):2405–2418. doi: 10.1038/ismej.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tveit A, Schwacke R, Svenning MM, Urich T. Organic carbon transformations in high-Arctic peat soils: key functions and microorganisms. ISME J. 2013;7(2):299–311. doi: 10.1038/ismej.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tveit AT, Urich T, Frenzel P, Svenning MM. Metabolic and trophic interactions modulate methane production by Arctic peat microbiota in response to warming. Proc Natl Acad Sci U S A. 2015;112(19):2507–2516. doi: 10.1073/pnas.1420797112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hori T, Sasaki D, Haruta S, Shigematsu T, Ueno Y, Ishii M, Lgarashi Y. Detection of active, potentially acetate-oxidizing syntrophs in an anaerobic digester by flux measurement and formyltetrahydrofolate synthetase (FTHFS) expression profiling. Microbiology. 2011;157:1980–1989. doi: 10.1099/mic.0.049189-0. [DOI] [PubMed] [Google Scholar]
- 35.Xu KW, Liu H, Du GC, Chen J. Real-time PCR assays targeting formyltetrahydrofolate synthetase gene to enumerate acetogens in natural and engineered environments. Anaerobe. 2009;15(5):204–213. doi: 10.1016/j.anaerobe.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 36.Glissmann K, Conrad R. Fermentation pattern of methanogenic degradation of rice straw in anoxic paddy soil. FEMS Microbiol Ecol. 2000;31(2):117–126. doi: 10.1111/j.1574-6941.2000.tb00677.x. [DOI] [PubMed] [Google Scholar]
- 37.Liu C, Lu M, Cui J, Li B, Fang CM. Effects of straw carbon input on carbon dynamics in agricultural soils: a meta-analysis. Glob Chang Biol. 2014;20(5):1366–1381. doi: 10.1111/gcb.12517. [DOI] [PubMed] [Google Scholar]
- 38.Wegner CE, Liesack W. Microbial community dynamics during the early stages of plant polymer breakdown in paddy soil. Environ Microbiol. 2016;18(9):2825–2842. doi: 10.1111/1462-2920.12815. [DOI] [PubMed] [Google Scholar]
- 39.Fey A, Conrad R. Effect of temperature on the rate limiting step in the methanogenic degradation pathway in rice field soil. Soil Biol Biochem. 2003;35(1):1–8. doi: 10.1016/S0038-0717(02)00175-X. [DOI] [Google Scholar]
- 40.Glissmann K, Conrad R. Saccharolytic activity and its role as a limiting step in methane formation during the anaerobic degradation of rice straw in rice paddy soil. Biol Fert Soils. 2002;35(1):62–67. doi: 10.1007/s00374-002-0442-z. [DOI] [Google Scholar]
- 41.Carrere H, Dumas C, Battimelli A, Batstone DJ, Delgenes JP, Steyer JP, Ferrer I. Pretreatment methods to improve sludge anaerobic degradability: a review. J Hazard Mater. 2010;183(1–3):1–15. doi: 10.1016/j.jhazmat.2010.06.129. [DOI] [PubMed] [Google Scholar]
- 42.Kadnikov Vitaly V., Mardanov Andrey V., Podosokorskaya Olga A., Gavrilov Sergey N., Kublanov Ilya V., Beletsky Alexey V., Bonch-Osmolovskaya Elizaveta A., Ravin Nikolai V. Genomic Analysis of Melioribacter roseus, Facultatively Anaerobic Organotrophic Bacterium Representing a Novel Deep Lineage within Bacteriodetes/Chlorobi Group. PLoS ONE. 2013;8(1):e53047. doi: 10.1371/journal.pone.0053047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomas F, Hehemann JH, Rebuffet E, Czjzek M, Michel G. Environmental and gut Bacteroidetes: the food connection. Front Microbiol. 2011;2:93 10.3389/fmicb.2011.00093. [DOI] [PMC free article] [PubMed]
- 44.Peng JJ, Wegner CE, Liesack W. Short-term exposure of paddy soil microbial communities to salt stress triggers different transcriptional responses of key taxonomic groups. Front Microbiol. 2017;8:400. 10.3389/fmicb.2017.00400. [DOI] [PMC free article] [PubMed]
- 45.Alvarado A, Montanez-Hernandez LE, Palacio-Molina SL, Oropeza-Navarro R, Luevanos-Escareno MP, Balagurusamy N. Microbial trophic interactions and mcrA gene expression in monitoring of anaerobic digesters. Front Microbiol. 2014;5:597. 10.3389/fmicb.2014.00597. [DOI] [PMC free article] [PubMed]
- 46.Freitag TE, Prosser JI. Correlation of methane production and functional gene transcriptional activity in a peat soil. Appl Environ Microbiol. 2009;75(21):6679–6687. doi: 10.1128/AEM.01021-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma K, Conrad R, Lu YH. Responses of methanogen mcrA genes and their transcripts to an alternate dry/wet cycle of paddy field soil. Appl Environ Microbiol. 2012;78(2):445–454. doi: 10.1128/AEM.06934-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, Spector TD, Clark AG, Ley RE. Human genetics shape the gut microbiome. Cell. 2014;159(4):789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Macy JM, Probst I. The biology of gastrointestinal bacteroides. Annu Rev Microbiol. 1979;33:561–594. doi: 10.1146/annurev.mi.33.100179.003021. [DOI] [PubMed] [Google Scholar]
- 50.Marchandin H, Teyssier C, Campos J, Jean-Pierre H, Roger F, Gay B, Carlier JP, Jumas-Bilak E. Negativicoccus succinicivorans gen. nov., sp nov., isolated from human clinical samples, emended description of the family Veillonellaceae and description of Negativicutes classis nov., Selenomonadales ord. nov. and Acidaminococcaceae fam. nov. in the bacterial phylum Firmicutes. Int J Syst Evol Micr. 2010;60:1271–9. [DOI] [PubMed]
- 51.Yuan Q, Hernandez M, Dumont MG, Rui JP, Scavino AF, Conrad R. Soil bacterial community mediates the effect of plant material on methanogenic decomposition of soil organic matter. Soil Biol Biochem. 2018;116:99–109. doi: 10.1016/j.soilbio.2017.10.004. [DOI] [Google Scholar]
- 52.Shiratori-Takano H., Akita K., Yamada K., Itoh T., Sugihara T., Beppu T., Ueda K. Description of Symbiobacterium ostreiconchae sp. nov., Symbiobacterium turbinis sp. nov. and Symbiobacterium terraclitae sp. nov., isolated from shellfish, emended description of the genus Symbiobacterium and proposal of Symbiobacteriaceae fam. nov. INTERNATIONAL JOURNAL OF SYSTEMATIC AND EVOLUTIONARY MICROBIOLOGY. 2014;64(Pt 10):3375–3383. doi: 10.1099/ijs.0.063750-0. [DOI] [PubMed] [Google Scholar]
- 53.Penttila PA, Varnai A, Pere J, Tammelin T, Salmen L, Siika-aho M, Viikari L, Serimaa R. Xylan as limiting factor in enzymatic hydrolysis of nanocellulose. Biores Technol. 2013;129:135–141. doi: 10.1016/j.biortech.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 54.Simmons Thomas J., Mortimer Jenny C., Bernardinelli Oigres D., Pöppler Ann-Christin, Brown Steven P., deAzevedo Eduardo R., Dupree Ray, Dupree Paul. Folding of xylan onto cellulose fibrils in plant cell walls revealed by solid-state NMR. Nature Communications. 2016;7:13902. doi: 10.1038/ncomms13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Westerholm M, Roos S, Schnürer A. Tepidanaerobacter acetatoxydans sp. nov., an anaerobic, syntrophic acetate-oxidizing bacterium isolated from two ammonium-enriched mesophilic methanogenic processes. Syst Appl Microbiol. 2011;34(4):260–266. doi: 10.1016/j.syapm.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 56.Westerholm M, Roos S, Schnürer A. Syntrophaceticus schinkii gen. nov., sp. nov., an anaerobic, syntrophic acetate-oxidizing bacterium isolated from a mesophilic anaerobic filter. FEMS Microbiol Lett. 2010;309(1):100–104. doi: 10.1111/j.1574-6968.2010.02023.x. [DOI] [PubMed] [Google Scholar]
- 57.Noll M, Klose M, Conrad R. Effect of temperature change on the composition of the bacterial and archaeal community potentially involved in the turnover of acetate and propionate in methanogenic rice field soil. FEMS Microbiol Ecol. 2010;73(2):215–225. doi: 10.1111/j.1574-6941.2010.00883.x. [DOI] [PubMed] [Google Scholar]
- 58.Liu PF, Yang YX, Lu Z, Lu Y. Response of a rice paddy soil methanogen to syntrophic growth as revealed by transcriptional analyses. Appl Environ Microbiol. 2014;80(15):4668–4676. doi: 10.1128/AEM.01259-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fey A, Chin KJ, Conrad R. Thermophilic methanogens in rice field soil. Environ Microbiol. 2001;3(5):295–303. doi: 10.1046/j.1462-2920.2001.00195.x. [DOI] [PubMed] [Google Scholar]
- 60.Lu F, Bize A, Guillot A, Monnet V, Madigou C, Chapleur O, Mazeas L, He PJ, Bouchez T. Metaproteomics of cellulose methanisation under thermophilic conditions reveals a surprisingly high proteolytic activity. ISME J. 2014;8(1):88–102. doi: 10.1038/ismej.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weber S, Lueders T, Friedrich MW, Conrad R. Methanogenic populations involved in the degradation of rice straw in anoxic paddy soil. FEMS Microbiol Ecol. 2001;38(1):11–20. doi: 10.1111/j.1574-6941.2001.tb00877.x. [DOI] [Google Scholar]
- 62.Schütz H, Holzapfel-Pschorn A, Conrad R, Rennenberg H, Seiler W. A 3-year continuous record on the influence of daytime, season, and fertilizer treatment on methane emission rates from an Italian rice paddy. J Geophys Res-Atmos. 1989;94(D13):16405–16416. doi: 10.1029/JD094iD13p16405. [DOI] [Google Scholar]
- 63.Noll M, Frenzel P, Conrad R. Selective stimulation of type I methanotrophs in a rice paddy soil by urea fertilization revealed by RNA-based stable isotope probing. FEMS Microbiol Ecol. 2008;65(1):125–132. doi: 10.1111/j.1574-6941.2008.00497.x. [DOI] [PubMed] [Google Scholar]
- 64.Conrad R, Klose M. Dynamics of the methanogenic archaeal community in anoxic rice soil upon addition of straw. Eur J Soil Sci. 2006;57(4):476–484. doi: 10.1111/j.1365-2389.2006.00791.x. [DOI] [Google Scholar]
- 65.Conrad R, Wetter B. Influence of temperature on energetics of hydrogen metabolism in homoacetogenic, methanogenic, and other anaerobic bacteria. Arch Microbiol. 1990;155(1):94–98. doi: 10.1007/BF00291281. [DOI] [Google Scholar]
- 66.Mettel C, Kim Y, Shrestha PM, Liesack W. Extraction of mRNA from soil. Appl Environ Microbiol. 2010;76(17):5995–6000. doi: 10.1128/AEM.03047-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Edgar RC, Flyvbjerg H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics. 2015;31(21):3476–3482. doi: 10.1093/bioinformatics/btv401. [DOI] [PubMed] [Google Scholar]
- 68.Kopylova E, Noe L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28(24):3211–3217. doi: 10.1093/bioinformatics/bts611. [DOI] [PubMed] [Google Scholar]
- 69.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Tumbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huson Daniel H., Beier Sina, Flade Isabell, Górska Anna, El-Hadidi Mohamed, Mitra Suparna, Ruscheweyh Hans-Joachim, Tappu Rewati. MEGAN Community Edition - Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLOS Computational Biology. 2016;12(6):e1004957. doi: 10.1371/journal.pcbi.1004957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Radax R, Rattei T, Lanzen A, Bayer C, Rapp HT, Urich T, Schleper C. Environ Microbiol. 2012;14(5):1308–1324. doi: 10.1111/j.1462-2920.2012.02714.x. [DOI] [PubMed] [Google Scholar]
- 73.Miller Christopher S, Baker Brett J, Thomas Brian C, Singer Steven W, Banfield Jillian F. EMIRGE: reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biology. 2011;12(5):R44. doi: 10.1186/gb-2011-12-5-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ludwig W. ARB: a software environment for sequence data. Nucleic Acids Research. 2004;32(4):1363–1371. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig WG, Peplies J, Glöckner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35(21):7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lueders T, Friedrich M. Archaeal population dynamics during sequential reduction processes in rice field soil. Appl Environ Microbiol. 2000;66(7):2732–2742. doi: 10.1128/AEM.66.7.2732-2742.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yin YB, Mao XZ, Yang JC, Chen X, Mao FL, Xu Y. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40(1):445–451. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12(1):59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 79.Menzel Peter, Ng Kim Lee, Krogh Anders. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nature Communications. 2016;7:11257. doi: 10.1038/ncomms11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Burrows M, Wheeler DJ. A block-sorting lossless data compression algorithm. Technical Report 124. Palo Alto: Digital Equipment Corporation; 1994. [Google Scholar]
- 81.Ferragina P, Manzini G. Proceedings of the 41st Annual Symposium on Foundations of Computer Science (FOCS 2000), IEEE Computer Society. 2000. Opportunistic data structures with applications; pp. 390–398. [Google Scholar]
- 82.Angel R, Claus P, Conrad R. Methanogenic archaea are globally ubiquitous in aerated soils and become active under wet anoxic conditions. ISME J. 2012;6(4):847–862. doi: 10.1038/ismej.2011.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H. Vegan: Community ecology package. R package version 2.2-0;2014.
- 84.Clark KR. Non-parametric multivariate analyses of changes in community structure. Aust J Ecol. 1993;18:117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x. [DOI] [Google Scholar]
- 85.Williams RJ, Howe A, Hofmockel KS. Demonstrating microbial co-occurrence pattern analyses within and between ecosystems. Front Microbiol. 2014;5:358 10.3389/fmicb.2014.00358. [DOI] [PMC free article] [PubMed]
- 86.Bastian M, Heymann S, Jacomy M. Gephi: an open source software for exploring and manipulating networks. San Jose: International AAAI Conference on Weblogs and Social Media; 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This document contains supporting Figures S1-S8. and Tables S1-S10. The file is published online as Supporting Information. (PDF 1430 kb)
Corroborating GH expression by methanogens through blasting their genome sequences (download from NCBI database) against dbCAN database. (XLSX 378 kb)
Detailed excursus on anaerobic polymer breakdown and the temperature-dependent expression of CAZymes. (PDF 87 kb)
Data Availability Statement
The Illumina RNA-Seq data generated during this study have been deposited at the NCBI Sequence Read Archive under the BioProject number PRJNA412498 with accession number SRR6122723 to SRR6122752. The assembled Methanocellaceae 16S rRNA sequences have been submitted to the NCBI database with accession numbers MG977525–MG977676. The custom PYTHON scripts used in our study are available at http://github.com/wegnerce/peng_et_al_2018.