

Abstract
Macrophage migration inhibitory factor (MIF) is a cytokine with pleiotropic actions that is produced by several organs and cell types. Depending on the target cell and the inflammatory context, MIF can engage its two component receptor complex CD74 and CD44 and the chemokine receptors CXCR2/4. MIF is constitutively expressed in renal proximal tubular cells, stored in intracellular preformed pools, and released at a low rate. Recently, a second MIF-like protein (i.e., MIF-2/D-DT) has been characterized in mammals. Our study was aimed at examining the role of MIF-2/D-DT, which mediates tissue protection in the heart, in tubular cell regeneration from ischemia-reperfusion injury. We found that Mif−/−, Mif-2−/−, and Cd74−/− mice had significantly worse tubular injury compared with wild-type (WT) control mice and that treatment with MIF-2/D-DT significantly improved recovery of injured epithelial cells. RNAseq analysis of kidney tissue from the ischemia-reperfusion injury model revealed that MIF-2/D-DT treatment stimulates secretory leukocyte proteinase inhibitor (SLPI) and cyclin D1 expression. MIF-2/D-DT additionally activates of eukaryotic initiation factor (eIF) 2α and activating transcription factor (ATF) 4, two transcription factors involved in the integrated stress response (ISR), which is a cellular stress response activated by hypoxia, nutrient deprivation, and oxygen radicals. MIF-2/D-DT also inhibited apoptosis and induced autophagy in hypoxia-treated mouse proximal tubular (MPT) cells. These results indicate that MIF-2/D-DT is an important factor in tubular cell regeneration and may be of therapeutic utility as a regenerative agent in the clinical setting of ischemic acute kidney injury.
Keywords: MIF, autophagy, apoptosis, ischemia, regeneration, SLPI, ATF4
macrophage migration inhibitory factor (MIF) is a cytokine with pleiotropic actions that is produced by several organs and cell types (6). MIF is a highly conserved protein with a molecular mass of 12.5 kDa and shows extremely high homology between human and mouse proteins (53). Biologically active MIF is a 37.5-kDa homotrimer with unique structural topology that acts by receptor-dependent signaling mechanisms to modulate both innate and adaptive immune responses (3, 5, 18, 51, 54). MIF has been implicated in the pathogenesis of several autoimmune and inflammatory diseases, including sepsis, rheumatoid arthritis, glomerulonephritis, and the acute respiratory distress syndrome (15, 17, 19). Depending on the target cell and the inflammatory context, MIF can engage its two component receptor complex CD74 and CD44 and the chemokine receptors CXCR2/4 (39). MIF is expressed in a wide variety of cells, such as lymphocytes, macrophages, endothelial cells, and epithelial cells (1, 73, 74). Knowledge regarding MIF receptor expression in kidney cells is sparse. Podocytes are known to express CD74 and CXCR4, and tubular cells may express CD74, CXCR4, and CXCR2 (29, 64).
Most cells in the kidney can synthesize MIF, i.e., podocytes, mesangial cells, tubular cells, and endothelial cells (30, 40, 59). MIF is constitutively expressed in tubular cells, stored in intracellular preformed pools, and is released at a low rate (30). Cellular stress, endotoxins, inflammation, and other stimuli increase circulating MIF levels. Initial MIF release following hypoxia stimulus is from intracytoplasmic stores, but with prolonged hypoxia increased MIF synthesis also will occur (65). In an inflammatory injury model of antiglomerular basement membrane-mediated crescentic glomerulonephritis, MIF was expressed in glomerular cells and showed increased abundance in tubular epithelial cells (38). In an urate nephropathy model, MIF mRNA was increased severalfold in renal tubules (35). Infiltrating macrophages and T cells also express MIF, which may play a role in MIF-dependent tubular cell regeneration (48). Moreover, MIF has been shown to induce proliferation and cell survival in neutrophils, human endothelial cells, and fibroblasts (4, 14, 34). The mechanisms that mediate survival involve antiapoptotic effects by p53 and BCL2 and inhibition of the proapoptotic factor Bim (21, 42). The proliferative and cell survival effects of MIF are mediated through interaction with CD74 (67).
In 2011, the observation that MIF receptor (CD74) gene deficiency produced a more severe phenotype in mice than MIF gene deficiency prompted exploration of a second ligand for CD74, leading to the cloning and characterization of the homologous D-dopachrome tautomerase (D-DT) gene product as a second MIF-like protein (i.e., MIF-2/D-DT) in mammals (49). MIF-2 shows 34% sequence identity with MIF and close three-dimensional structural homology, including the presence of the MIF superfamily canonical NH2-terminal proline. In models of cardiac ischemia-reperfusion (I/R) injury, MIF-2/D-DT is expressed regionally and exerts similar tissue protective actions as MIF. Notably, MIF-2/D-DT differs from MIF in lacking the pseudo(E)LR motif necessary for activation of chemokine receptors. Accordingly, it may exert fewer inflammatory actions and a more selective tissue protective action via CD74 activation than MIF.
A central mechanism in mRNA translation control involves phosphorylation of the α-subunit of eukaryotic initiation factor (eIF) 2α, which initiates repression of protein synthesis, while a new gene program is adopted to prevent damage (46). Activation of eIF2α selectively enhances the translation of activating transcription factor (ATF) 4, a transcriptional activator of genes involved in redox status regulation and regulation of apoptosis (24, 72). The eIF2α/ATF4 pathway is referred to as the integrated stress response (ISR) and shares many features with general control nonderepressable 4 translational control in yeast, highlighting its evolutionary conserved role in ameliorating nutritional deficiencies (26, 27). Functional studies of ATF4-dependent gene expression identified target genes involved in gaging cellular redox status, regulation of autophagy, control of apoptosis, and feedback regulation of the ISR (31, 61, 63). A known promoter activated by ATF4 is asparagine synthetase leading to increased asparagine synthetase mRNA synthesis (8). This is a necessary step in the unfolded protein response. ATF4 expression has also been shown to be an important response to combat hypoxia in tumor cells (7).
Secretory leukocyte proteinase inhibitor (SLPI) is a protein of 12 kDa with inhibitory activity against such as cathepsin L, B, and S and antimicrobial activity (25, 70). SLPI is synthesized predominantly in epithelial cells of mucosal surfaces but has also been found in solid organs such as the pancreas and kidney (56, 57). A recent study in human kidney biopsies showed significant abundance of SLPI in kidney tubular epithelial cells (57). The main physiological function of SLPI is probably to buffer extracellular protease-mediated effects by inflammatory cells, but it has also been shown to have a role in stimulating tumor cell proliferation through enhanced cyclin D1 expression (77).
Our study was aimed at examining the role of MIF-2/D-DT, which exerts tissue protection in the heart, in tubular cell regeneration from renal I/R injury. We found that Mif−/−, Mif-2−/−, and Cd74−/− mice had significantly worse tubular injury compared with wild-type (WT) control mice. Moreover, treatment with MIF-2 significantly improved recovery of injured epithelial cells. By RNAseq analysis of kidney tissue from the I/R injury model, we found that MIF-2/D-DT treatment stimulates SLPI and cyclin D1 expression, as well as several genes regulating cell proliferation. These findings were confirmed in a hypoxic proximal tubule cell injury model. Moreover, we found that MIF-2/D-DT stimulates activation of eIF2α and ATF4, two transcription factors involved in the ISR, which is a cellular response activated by hypoxia, nutrient deprivation, and oxygen radicals. MIF-2/D-DT treatment further inhibited apoptosis and induced autophagy. Our results show that MIF-2/D-DT is an important factor in tubular cell regeneration and may have therapeutic utility as a regenerative agent in the clinical setting of ischemic acute kidney injury.
METHODS
Mice
Adult congenic Mif−/−, Mif-2−/−, Cd74−/−, and WT mice on a C57BL/6 background were used in this study (21). All mice were maintained at the Yale Animal Resource Center under pathogen-free conditions, and experiments were performed under a protocol approved by Yale Animal Care and Use Committee (No. 2013–11583).
Renal I/R Injury in Mice
Mice were anesthetized by intraperitoneal injection of ketamine hydrochloride (100 mg/kg) and xylazine (10 mg/kg), and both kidneys were exposed by midline laparotomy incision. Both renal arteries and veins of both kidneys were clamped by surgical clips for 30 min. Successful ischemia was confirmed visually by darkening color of kidneys. After the surgical clips were removed, successful reperfusion was confirmed visually by changing colors of kidneys from dark to pink, and then, 12.5 μg/100 μl PBS of MIF-2/D-DT solution were intraperitoneally injected before the peritoneum and skin were surgically closed. MIF-2/D-DT was produced recombinantly as described and purified free of endotoxin, which was undetectable by the rFactor C fluorescensce assay. MIF-2/D-DT injections were continuously performed every 12 h. Mice were euthanized at 24, 48, and 72 h after I/R surgery, and both kidneys, blood, and urine samples were harvested. Right kidneys were fixed by 4% paraformaldehyde (PFA) and embedded in paraffin, and the left kidneys were stored separately in −80°C without fixation. Sham-operated animals received the same surgical procedure as I/R-treated groups, without clamping or renal arteries. The number of animals per experimental group was six to eight, and equal numbers of male and female mice were used in the experiments.
Evaluation of Kidney Damage
PFA-fixed and paraffin-embedded mice kidney specimens were sectioned at a 3-μm thickness and were stained with periodic acid-Schiff. The percentage of acute tubular injury (%ATI) was assessed by light microscopy by one renal pathologist (G. Moeckel). Criteria for ATI were proximal tubular lumen dilation, intratubular cast formation, brush border loss, cytoplasm vacuolization, and epithelial nuclear drop out (23).
Immunohistochemistry (IHC) assessment of MIF-2/D-DT, cyclin D1, Ki67, CD74, and CD44 were conducted on sections from WT and MIF-1 null (MIF−/−) mice using the following antibodies: MIF-2/D-DT (1:150; clone Y910; gift from D. R. Bucala), cyclin D1 (1:200; CME432; Biocare Medical), Ki67 (1:100; No. 325; Biocare Medical), CD74 (1:200; PA5-22113; Thermo Scientific), and CD44 (1:75; M7082; DAKO). IHC staining for MIF-2/D-DT employed a mouse anti- MIF-2/D-DT monoclonal antibody (IgG1, clone Y910) that was raised by sequential immunization with recombinant mouse and human MIF-2/D-DT. Y910 shows no crossreactivity with denatured or native MIF, as assessed by Western blotting and ELISA against recombinant mouse or human MIF. All procedures were done by the Yale Pathology Tissue Services at Yale University.
Measurement of Serum Blood Urea Nitrate and Cre Levels
Serum/plasma creatinine concentrations were measured using liquid-chromatography/tandem mass spectrometry analysis performed by the Yale O’Brien Kidney Center (Renal Physiology Core) in conjunction with the Yale Mouse Metabolic Phenotyping Center. Serum/plasma albumin concentrations were measured by bromocresol green quantitative colorimetric assay at 550 nm (Stanbio Laboratory, Boerne, TX). Serum/plasma blood urea nitrate concentration were measured by diacetylmonoxime quantitative colorimetric assay at 520 nm (Stanbio Laboratory) (Table 1).
Table 1.
Serum creatinine and BUN values in WT and MIF−/− mice
| Animal | 24-h Creatinine | 48-h Creatinine | 24-h BUN | 48-h BUN |
|---|---|---|---|---|
| WT | 1.5 ± 0.5 | 0.6 ± 0.15 | 38.9 ± 0.6 | 10 ± 1.2 |
| MIF−/− | 1.1 ± 0.5 | 2.1 ± 0.7 | 19.7 ± 0.05 | 89.6 ± 4.3 |
Values are means ± SE; n = 8–9. BUN, blood urea nitrate; WT, wild-type; MIF, migration inhibitory factor.
RNAseq Analysis
RNAseq library prep.
Total RNA from murine kidneys was isolated by the Rneasy Mini Kit (Qiagen), and purity was determined by estimating the A260/A280 and A260/A230 ratios by nanodrop (Thermo Scientific). RNA integrity was determined by Agilent Bioanalyzer 2100 (Agilent Technologies). The RNA sample that had more than a 7.0 RNA integration number was taken up for seq library preparation.
mRNA was purified from ~500 ng of total RNA with oligo-dT beads and sheared by incubation at 94°C. Following first-strand synthesis with random primers, second-strand synthesis was performed with dUTP for generating strand-specific sequencing libraries. The cDNA library was then end repaired and A tailed, and adapters were ligated and second-strand digestion was performed by Uricil-DNA-Glycosylase. Indexed libraries meeting appropriate quality cut-offs for both were quantified by quantitative RT-PCR using a commercially available kit (KAPA Biosystems), and insert size distribution was determined with the LabChip GX or Agilent Bioanalyzer. Samples with a yield of ≥0.5 ng/ul are used for sequencing.
Flow cell preparation and sequencing.
Sample concentrations were normalized to 2 nM and loaded onto Illumina Rapid or High-output flow cells at a concentration yielding 150–250 million passing filter clusters per lane. Samples were sequenced using 75-bp single or paired-end sequencing on an Illumina HiSeq 2500 according to Illumina protocols. The 6-bp index was read during an additional sequencing read that automatically follows the completion of read 1. Data generated during sequencing runs were simultaneously transferred to the YCGA high-performance computing cluster. A positive control (prepared bacteriophage Phi X library) provided by Illumina was spiked into every lane at a concentration of 0.3% to monitor sequencing quality in real time.
Data analysis and storage.
Signal intensities were converted to individual base calls during a run using the system's Real Time Analysis software. Base calls were transferred from the machine's dedicated personal computer to the Yale High Performance Computing cluster via a 1-Gigabit network mount for downstream analysis. Primary analysis, sample demultiplexing and alignment to the human genome, was performed using Illumina's CASAVA 1.8.2 software suite.
Cell Culture and Hypoxic Treatment
Mouse proximal tubular (MPT) cell lines were derived from the proximal tubule of the Immortomouse as previously described (66). MPT cells were obtained from Dr. Lloyd Cantley (Yale University, New Haven, CT), and the absence of mycoplasma was confirmed by DAPI staining. MPT cells were cultured in DMEM-F12 (GIBCO) with 10% FBS (ATLANTA Biologicals), 100 U/ml penicillin and 100 μg/ml streptomycin (GIBCO), and 10 U/ml interferon-γ (IFNγ; Roche) at 33°C. At 70% confluence, cells were maintained in IFNγ-free culture medium at 37°C for 3 days to remove effects of IFNγ; cells then were serum starved overnight at 37°C. To mimic an ischemic condition, cells were cultured in Hanks’ HBSS (GIBCO) medium in a hypoxic incubator (COY Laboratory Products; 0.1% O2-5% CO2-94.9% N2) for 6 h. To mimic ischemic conditions, culture medium was replaced to DMEM/F12 medium with 10% FBS, and cells were cultured in 21% O2 atmosphere at 37°C. At the same time, we added 100 ng/ml MIF-2/D-DT or PBS, and we harvested these cells at different time points. We used non-MIF-2/D-DT treated cells as control and compared the results of following experiments between two groups.
Cell Proliferation Assay
Cell proliferation of MPT cells was assessed by 5-bromo-2′-deoxyuridine (BrdU) and Ki67 staining assays. We purchased BrdU from Sigma-Aldrich. For BrdU assay, 10 μM of BrdU were added to MPT cells for 1 h at the end of the each experiment. After BrdU labeling, cells were fixed by 4% PFA (Boston BioProducts) for 20 min. Then, cellular DNA was denatured by 2 N hydrochloric acid for 30 min. Nonspecific epitopes were blocked by incubating cells for 1 h in blocking solution, PBS with 5% bovine albumin (Sigma), and 1% Triton-X. Cells were incubated with mouse monoclonal anti-BrdU antibody (1:100; sc32323; Santa Cruz Biotechnology) overnight at 4°C. The next day cells were incubated with Texas red anti-mouse IgG antibody (1:300; TI-2000; Vector Laboratories) and DAPI (5 μg/ml) solution for 1 h at room temperature. Cells were mounted with ProLong Gold antifade reagent (Life Technologies).
For Ki67 staining, MPT cells were fixed by 4% PFA for 20 min after each experiment. Then, cells were incubated with the blocking solution for 1 h at room temperature and subsequently incubated with rabbit polyclonal anti-Ki67 antibody (1:300; AB9260; EMD Millipore) overnight at 4°C. Next day, cells were incubated with Texas red anti-mouse IgG antibody (1:300; TI-2000; Vector Laboratories) and DAPI (5 μg/ml) solution for 1 h at room temperature and mounted with ProLong Gold antifade reagent.
Stained cells were observed by fluorescence microscopy (Axiophot El-einsatz 451888; Karl Zeiss). BrdU- and Ki67-positive cell ratios were calculated by dividing BrdU- or Ki67-positive cell numbers by DAPI-positive cell number.
SLPI Transfection
MPT cells were seeded into six-well plates and cultured in IFNγ-free medium for 3 days at 37°C. When cells reached ~70% confluence, they were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. For BrdU assay, 1 × 104 cells were seeded into six-well plates with cover glass slips and cultured in IFNγ-free medium for 3 days at 37°C. Then, cells were transfected using Lipofectamine 2000. Cells were transfected with 4 μg of a p156 pWZL-EGFP-SLPI plasmid (gift from Ramesh Shivdasani; Addgene plasmid no. 11069) or with 4 μg of pWZL-Neo empty vector (Cell Biolabs) using Lipofectamine 2000. After transfection, cells were cultured for 36 h in DMEM/F12 medium with 10% heat-inactivated FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin at 37°C, 21% O2.
Western Blot Analysis
After each treatment, MPT cells were washed twice with ice-cold PBS. Proteins from the whole mice kidney and MPT cells were lysed in RIPA lysis buffer (Sigma-Aldrich) with protease inhibitor cocktail (Roche) containing 2 mM Na3VO4, and 10 mM NaF. After the removal of insoluble materials by centrifugation, the sample protein concentrations were measured by using BCA protein assays (Thermo Scientific). The lysates (20 μg protein per lane) were mixed with sample-loading buffer and separated on SDS-PAGE gels. Proteins then were transferred to nitrocellulose membranes (Bio-Rad) and immunoblotted for MIF-2/D-DT (1:500; clone Y910), MIF1 (1:1,000; clone R102), CD74 (1:500: AF7478; R&D Systems), CXCR4 (1:500; bs-1011R; Bioss Antibodies), phospho-eIF2α (1:1,000: no. 3398; Cell Signaling Technology), eIF2α (1:1,000; no. 5324; Cell Signaling Technology), ATF4 (1:1,000; no. 11815; Cell Signaling Technology), LC3A/B (1:1,000; no. 12741; Cell Signaling Technology), SLPI (1:1,000; AF1735; R&D Systems), cyclin D1 (1:1,000; no. 06–137; Millipore,), hypoxia-inducible fatctor-α (1:1,000; no. 14179; Cell Signaling Technology), cleaved caspase 3 (1:1,000; no. 9664; Cell Signaling Technology), α-tubulin (1:1,000; no. 2144; Cell Signaling Technology), and β-actin (1:1,000; no. 4970; Cell Signaling Technology). Protein bands were identified using ECL (Pierce) and quantified using an ImageJ 1.49v software (National Institute of Health).
Statistical Analysis
All data are presented as means ± SD. Statistical analysis was performed using a two-tailed Student’s t-test or ANOVA with subsequent Fisher’s protected least significant difference test. The following significance levels were used: *P < 0.05, **P < 0.01, ***P < 0.001, #P < 0.05, ##P < 0.01, and ###P < 0.001. All figures were generated from at least three repeated experiments with similar patterns.
RESULTS
Impact of MIF and MIF-2/D-DT on Renal I/R Injury
The effect of MIF or MIF-2/D-DT (i.e., Mif-2−/−) germline gene deletion first was assessed on tubular injury following bilateral renal artery occlusion and reperfusion. Histological evaluation of H&E-stained kidney sections 48 h post-I/R injury showed severe acute tubular injury involving 55 ± 15% of cortical tissue in WT mice (Figs. 1A and 2A). Mif−/− mice showed significantly more extensive tubular injury involving ~85 ± 15% of cortex (Figs. 1C and 2A). Moreover, Mif-2−/− mice showed extensive, severe tubular injury involving over 90 ± 10% of the renal cortex (Fig. 1E). Remarkable was the finding of extensive cast formation in the Mif−/− and Mif-2−/− mice, compared with the WT controls (Fig. 1, C and E, black arrows). Treatment with MIF-2/D-DT dramatically ameliorated the tubular injury score and the degree of tubular cast formation in all three different mouse genotypes (Fig. 1, B, D, and F). Similar to Mif−/− animals, those mice with deletion of the common MIF receptor CD74, showed more severe cortical tubular injury at 48 h after I/R injury (Fig. 2C).
Fig. 1.

Acute tubular injury (ATI) in mouse cortical tissue sections 48 h after ischemia-reperfusion (I/R) injury. A: untreated wild-type (WT) mice showed 40–50% of cortical tissue with diffuse acute tubular injury (tubules with pink casts). B: macrophage migration inhibitory factor-like protein (MIF-2/D-DT) treatment markedly decreased the extend and severity of damaged proximal tubule tissue in WT mice. C: Mif−/− mice showed 70–90% of cortical tissue with severe tubular injury and extensive intraluminal cast formation (black arrow). D: injection of MIF-2/D-DT dramatically ameliorated the degree of ATI in Mif−/− mice. E: Mif-2−/− mice showed an even more severe degree of tubular injury (95% of cortex) with extensive cast formation (black arrow). F: MIF-2/D-DT injections reduced the extend and severity of the tubular injury by 70% in Mif-2−/− mice. A–F: hematoxylin and eosin: ×40. All animal groups (sham or I/R) contained 6–8 animals.
Fig. 2.

Tissue injury scores and serum creatinine concentrations comparing Mif−/− vs WT mice with or without recombinant MIF-2/D-DT treatment. A and C: acute tubular injury following sham surgery or 30-min bilateral renal ischemia in Mif−/− (A, blue bars) or Cd74-null (C, green) mice compared with WT mice. B: serum creatinine levels in mice as in (A) at 24 or 48 h following I/R. D: acute tubular injury following bilateral renal ischemia at 48 or 72 h in mice as in A with administration of MIF-2/D-DT at the time of the release of ischemia and every 12 h thereafter (hashed bars) or left untreated (clear bars). Mif−/− mice showed significant delay in tubular cell regeneration at 48 and 72 h after I/R (blue bars). MIF-2/D-DT treatment significantly improved the tissue injury score in Mif−/− and WT animals (hatched bars). **P < 0.05; ***P < 0.01; ****P < 0.001; n = 6–8 mice in each experimental group.
Serum creatinine levels in WT mice initially increased up to 1.5 mg/dl at 24 h post-I/R injury and then decreased to below 0.8 mg/dl at 48 h post-I/R, consistent with regeneration from injury (Fig. 2B, gray bars). In Mif−/− animals, the serum creatinine levels were similar to WT animals at 24 h (1.1 mg/dl) but increased further to 2.2 mg/dl at 48 h post-I/R injury (Fig. 2B, blue bars), indicative of failed regeneration (Table 1). Treatment with MIF-2/D-DT significantly enhanced tubular cell regeneration and decreased the ATI score up to 30% in WT mice and up to 60% in Mif−/− animals within 48 h after injury (Fig. 2D, hatched bars). Seventy-two hours after I/R the WT mice showed large areas of cortex with tubular regeneration and a diminished ATI scores (Fig. 2D). In WT animals, treatment with MIF-2/D-DT did not further improve tubular injury scores at 72 h. Untreated Mif−/− mice still showed 63% ATI at 72 h, indicative of prolonged injury, while MIF-2/D-DT treated Mif−/− mice showed a decrease in tubular injury that was similar to that observed in WT animals (Fig. 2D, blue hatched bars).
MIF-2/D-DT Enhances Cell Proliferation in Mouse Proximal Tubule (MPT) Cells
We used RNAseq studies from I/R mouse kidneys to assess the effect of MIF depletion and MIF-2/D-DT treatment on gene expression levels. As expected, Mif expression was markedly decreased in Mif−/− mice, compared with WT mice, and MIF-2/D-DT treatment did not stimulate Mif gene expression in the knockout mice (Fig. 3A, left). Surprisingly, MIF-2/Ddt expression levels in Mif−/− mice were almost double those in WT mice and further MIF-2/D-DT treatment did not enhance the Mif-2/Ddt expression in the Mif knockout mice (Fig. 3A, right). This effect on MIF-2/D-DT protein abundance was confirmed by using MIF-2/D-DT IHC stains in mouse tissue sections and Western blot analysis of I/R kidney tissue (Fig. 3D). Furthermore, Mif−/− mice showed higher CD74 and Cxcr4 expression levels, compared with WT mice, which were further increased by MIF-2/D-DT treatment (Fig. 3B). Interestingly, Mif−/− showed almost no expression of the MIF signaling coreceptor CD44, whose expression is strongly MIF dependent (76). CD44 expression was markedly enhanced by MIF-2/D-DT treatment in knockout mice. These effects of MIF-2/D-DT on CD74 and CD44 protein abundance were confirmed using IHC studies in mouse kidney tissue sections. Cxcr2 expression levels were unaffected in knockout mice, with or without MIF-2/D-DT treatment (Fig. 3B). Furthermore, we found significant changes in mRNA of the proliferation genes PCNA and Ki-67. As shown in Fig. 3C, Mif−/− mice showed 60% reduction of PCNA mRNA and 98% reduction of Ki-67 mRNA levels (light gray bars) compared with WT controls (dark gray bars). Treatment of Mif−/− mice with MIF-2/D-DT ameliorated both PCNA and the Ki-67 mRNA reductions (gray bars) significantly (Fig. 3C). The effect of MIF-2/D-DT on MIF-2, CD74 and CD44 Ki-67 abundance was confirmed by IHC (Figs. 4, 5 and 6. Hypoxic MPT cells, followed by MIF-2/D-DT treatment, showed increased BrdU-positive cells at 24 and 36 h after hypoxia, indicative of the stimulatory effect of MIF-2/D-DT on proximal tubule cell proliferation (Fig. 7, A–D).
Fig. 3.

mRNA expression levels of MIF-1, MIF-2/D-DT and its target receptors in WT and Mif−/− mice 48 h after I/R injury. A: Mif−/− mice showed minimal MIF-1 expression compared with WT animals. Treatment with MIF-2/D-DT did not affect MIF-1 expression in Mif−/− mice (left: light gray and gray bars). However, Mif−/− mice showed increased MIF-2/D-DT expression compared with WT mice, which was not further stimulated by MIF-2/D-DT treatment (right: light gray and gray bars). B: RNAseq data revealed that WT mice showed robust expression of Cd74, Cd44, Cxcr2, and Cxcr4 in kidney tissue (dark gray bars). Mif−/− mice showed almost complete lack of Cd44 expression but slightly enhanced Cd74 and Cxcr4 expression levels (gray bars). Treatment with MIF-2/D-DT significantly increased Cd44 expression in Mif−/− mice and also enhanced Cd74 and Cxcr4 expression in Mif−/− animals (light gray bars). C: RNAseq analysis showed markedly decreased PCNA and Ki-67 expression levels in Mif−/− mice (gray bars). MIF-2/D-DT treatment enhanced PCNA and Ki-67 expression levels to 80% and 50% of WT levels respectively (light gray bars). D: Western blot assessment of CD74, CXCR4, D-DT, and MIF1 abundance showed similar results compared with RNAseq data. MIF−/− mice showed enhanced abundance of CD74, CXCR4, and D-DT. MIF-2/D-DT treated mice showed increased abundance in CD74 and CXCR4 compared with WT controls. *P < 0.05, **P < 0.01, ***P < 0.001 compared with WT; #P < 0.05, ##P < 0.01; 6–8 animals per experimental group.
Fig. 4.

Immunohistochemistry staining for MIF-2/D-DT in WT and Mif−/− mice 48 h after I/R or sham operation (Sham = non I/R kidney (control); I/R = bilateral 30 min I/R). WT I/R kidneys show focal cortical interstitial MIF-2/D-DT expression but no expression in glomeruli. Mif−/−I/R mice show strong MIF-2/D-DT expression in glomeruli, parietal cells and in cortical interstitial cells.
Fig. 5.

Immunohistochemistry stain for CD74 (left) and CD44 (right). CD74 showed a cytoplasmic expression pattern in proximal tubule cells, which was stronger in Mif−/− mice compared with WT mice. Intraperitoneal injection with MIF-2/D-DT enhanced CD74 expression in proximal tubule cells. CD44 was not expressed in proximal tubule cells in WT animals with I/R injury. Only very little CD44 was seen in tubular epithelial cells in Mif−/− kidney sections. Injection with MIF-2/D-DT enhanced CD44 expression in injured tubular epithelial cells in Mif−/− mice.
Fig. 6.
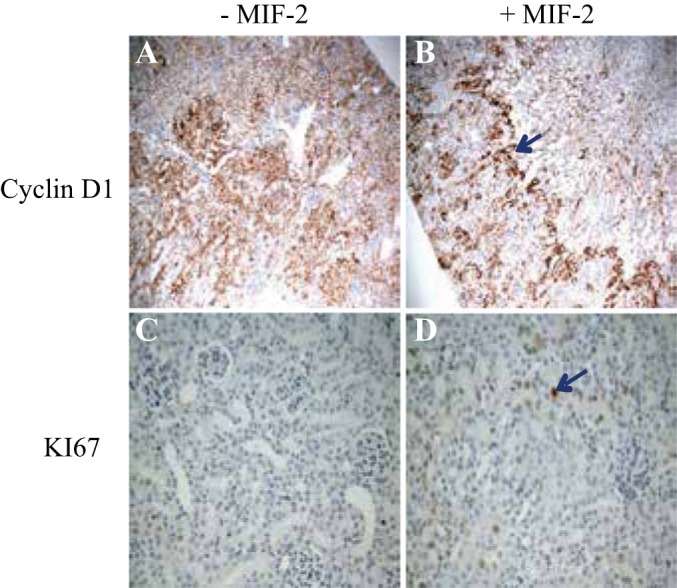
Immunohistochemistry (IHC) staining for cell proliferation markers in mouse kidney tissue 48 h after severe I/R injury. A: IHC for cyclin D1 in cortical tissue of Mif−/− mice without treatment. B: IHC stain for cyclin D1 in Mif−/− mice treated with MIF-2/D-DT. There is robust cyclin D 1 staining in proximal tubule at the cortico-medullary junction (blue arrow). C: IHC stain for Ki-67 in cortical tissue of Mif−/− without treatment. D: IHC staining for Ki-67 in Mif−/− mice, treated with MIF-2/D-DT, shows strong Ki-67 staining in proximal tubule cells (blue arrow); n = 6–8 animals per group.
Fig. 7.

MIF-2/D-DT treatment stimulates proliferation in mouse proximal tubular (MPT) cells. A: immunofluoresence stain for the cell proliferation marker 5-bromo-2′-deoxyuridine (BrdU) in hypoxic MPT cells. Right: BrdU-positive cells at 24 and 36 h posthypoxia with MIF-2/D-DT treatment. Left: untreated controls. B: analysis of BrdU-positive cells with MIF-2/D-DT treatment compared with untreated controls. At 36 h posthypoxia, MIF-2/D-DT-treated cells showed a significant increase in proliferating cell numbers. BrdU-positive cell ratio calculated as described in methods. *P < 0.05; n = 3–4 samples. C: IF stain for Ki-67 in MPT cells 36 h posthypoxia. Right: Ki-67-positive cells after MIF-2/D-DT treatment (purple nuclei). D: analysis of Ki-67-positive cells with MIF-2/D-DT treatment compared with untreated controls. At 36 h posthypoxia, MIF-2/D-DT treated cells showed a significant increase in Ki-67-positive cells. *P < 0.05; n = 3–4 samples.
MIF-2/D-DT Stimulates SLPI Expression in Kidney Tissue and Proximal Tubule Cells
To assess the gene expression patterns in Mif−/− and WT mice kidneys following severe bilateral ischemic injury, we conducted RNAseq analysis in respective kidney tissues 48 h post-I/R injury. RNAseq analysis revealed near 100% reduction in SLPI mRNA levels in Mif−/− mice kidneys compared with WT controls. Treatment with MIF-2/D-DT improved SLPI mRNA levels in Mif−/− mice from nearly 0 to 60% of control levels (Fig. 8A). We then examined the abundance of SLPI in MPT cells, which were treated with or without MIF-2/D-DT following hypoxic injury. MIF-2/D-DT treatment enhanced SLPI abundance at 12, 18, and 24 h posthypoxia (Fig. 8, B and C).
Fig. 8.

A: RNAseq analysis in kidney 48 h post-I/R injury showed near complete absence of secretory leukocyte proteinase (SLPI) mRNA in Mif−/− animals compared with WT controls. Treatment with MIF-2/D-DT restored the SLPI mRNA levels to 60% of WT control levels. **P < 0.01; ***P < 0.001; n = 6–8 animals per group. B and C: treatment of MPT cells with MIF-2/D-DT (100 ng/ml) significantly increased SLPI abundance at 12, 18, and 24 h posthypoxic injury. *P < 0.05, **P < 0.01; n = 3–4 experiments.
Since SLPI has a direct effect on cell cycle regulation (77), we examined the cell cycle regulatory gene expression using RNAseq. In Mif−/− mice, the expression of several cell cycle regulatory genes was decreased compared with WT controls (Fig. 9A, gray bars vs. dark gray bars). Notably, cyclin E1 and cyclin E2 expressions were markedly decreased (gray bars), while treatment with MIF-2/D-DT showed the strongest enhancing effect on cyclin D2 expression in Mif−/− mice (Fig. 9A, light gray bar). The RNAseq data were confirmed by Western blot in I/R kidney tissue showing the effect of MIF-2/D-DT on cyclin D1 abundance (Fig. 9B).
Fig. 9.

Cell cycle related gene expression in WT and Mif−/− mice 48 h after I/R injury. A: compared with WT animals (dark gray bars) Mif−/− mice showed decreased expression levels of cyclin E1, cyclin E2, and cyclin D1 and CDK2-6 (light gray bars) in RNAseq experiments in mouse kidney tissue. Treatment with MIF-2/D-DT enhanced the gene expression of all cyclins, especially cyclin D2 and CDK proteins (gray bars). B: Western blot analysis showed similar pattern for cyclin D1 abundance then RNAseq data and IHC stains (compare Fig. 8). D-DT treatment significantly enhanced cyclin D1 abundance in Mif−/− mice. *P < 0.05, **P < 0.01, ***P < 0.001 (compared with WT). #P < 0.05, ###P < 0.001 (compared with Mif−/− mice); n = 6–8 animals per group.
To examine the relationship between SLPI and cell cycle regulatory proteins in proximal tubule cells, we transfected SLPI containing plasmids into MPT cells and assessed cyclin D1 abundance and the cell proliferation marker BrdU. As shown in Fig. 10A, SLPI transfection markedly enhanced total cyclin D1 abundance in proximal tubule cells (Fig. 10, A and B). Proximal tubule cells transfected with SLPI showed more than double the numbers of BrdU- and Ki-67-positive cells compared with nontransfected controls, indicative of significant increase in proliferating cells (Fig. 10, C and D). These results indicate that SLPI directly stimulates cyclin D1 expression and enhances proliferation in proximal tubule cells. We then performed immunohistochemistry (IHC) staining for cell proliferation markers in mouse kidney tissue 48 h post-I/R injury. IHC stain for cyclin D1 in cortical tissue of Mif−/− mice without treatment showed diffuse weak positivity (Fig. 6A). However, IHC stain for cyclin D1 in Mif−/− mice, previously treated with MIF-2/D-DT, showed strong cyclin D 1 positivity in S3 segments of proximal tubule (Fig. 6B, blue arrow). The IHC stain for Ki-67 in cortical tissue of Mif−/− without MIF-2/D-DT treatment showed weak focal positivity in tubular epithelial cells, while the IHC staining for Ki-67 in Mif−/− mice treated with MIF-2/D-DT showed strong Ki-67 staining in proximal tubule cells (Fig. 6D, blue arrow).
Fig. 10.

Transfection of proximal tubule cells with SLPI. A: MPT cell cultures were transfected with SLPI or empty vector (see detailed materials and methods) and cyclin D1 abundance was analyzed using Western blot (representative blot from 3 different experiments shown). B: SLPI overexpression markedly enhanced the cyclin D1 abundance in MPT cells (*P < 0.05; n = 3–4 experiments). C and D: MPT cells transfected with SLPI showed robust increase in both BrdU-positive cells (C) and Ki-67-positive cells (D) (*P < 0.05; **P < 0.01, ***P < 0.001; n = 3–4 experiments).
MIF-2/D-DT Initiates the Integrated Stress Response, Stimulates Autophagy, and Inhibits Apoptosis in Proximal Tubule Cells
Depression of protein synthesis is characteristic of kidneys suffering from ischemic reperfusion (58). A number of mechanisms can lead to translation initiation inhibition, including stress to the endoplasmic reticulum, which activates specific regulatory pathways leading to the phosphorylation of the eIF2α (62). In our experiments treatment of MPT cells with MIF-2/D-DT resulted in a robust increase in phosphorylation of eIF2α at 30 and 60 min posthypoxia (Fig. 11A, row 2). Moreover, posthypoxic treatment with MIF-2/D-DT showed increased abundance of ATF4 at 60 and 90 min posthypoxia (Fig. 11A, row 4). Recently, a novel regulatory role of the eIF2α-ATF4 pathway has been shown, which consists of fine-tuning the autophagy gene transcription program (2). We therefore examined the effects of MIF-2/D-DT on LC3B-II abundance, a marker for autophagy. LC3B-II, was increased by MIF-2/D-DT treatment at 30, 60, 90, and 120 min after hypoxia (Fig. 11A, row 5). Over the same time period, MIF-2/D-DT treatment decreased cleaved caspase 3 levels in proximal tubule cells, indicative of decreases proximal tubule cell apoptosis (Fig. 11C, 120 and 180 min). The time sequence of the above molecular events is consistent with the concept that MIF-2/D-DT induces phosphorylation of eIF2α, which enhances ATF4 expression leading to activation of autophagy. These results show that MIF2/D-DT initiates a tiered series of molecular reactions that lead to overall tubular cell regeneration.
Fig. 11.

A: effect of MIF-2/D-DT treatment on transcription factors of the integrated stress response (ISR). Cultured MPT cells showed strong activation of eukaryotic initiation factor 2α (eIF2α) at 30 min (2nd row) and of the activating transcription factor 4 (ATF4) at 60, 90, and 120 min of treatment duration (4th row). The autophagy marker LC3B-II was markedly stimulated by MIF-2/D-DT at 30, 60, 90, and 120 min of treatment duration (bottom band, 4th row). Hypoxia-inducible factor-1α (HIF1α) abundance only showed a weak increase after 30 min of MIF-2/D-DT treatment (1st row, 120-kDa band). B: time course of transcription factor stimulation in the ISR response shows eIF2α activation preceding ATF4 and autophagy activation. *P < 0.05; n = 3–4 experiments. C: antiapoptotic effect of MIF-2/D-DT treatment. MPT cells showed significant increase in cleaved caspase 3 levels under hypoxic culture conditions and progressive inhibition of cleaved caspase 3 at 120 and 180 min of MIF-2/D-DT treatment.
DISCUSSION
Ischemic tubular injury is a common clinical problem affecting millions of patients worldwide, and potential therapeutic approaches are hampered by insufficient knowledge of the molecular pathways mediating damage and repair (37). The present study shows that the mediator MIF-2/D-DT initiates strong regenerative effects on proximal tubule cells in a mouse model of severe, bilateral I/R injury (Fig. 12). Mif−/−, Mif-2−/−, and Cd74−/− mice showed more extensive proximal tubular damage and tubular cast formation after I/R injury compared with WT mice. MIF-2/D-DT treatment ameliorated tubular injury in all mouse genotypes tested (Fig. 1). These findings indicate that MIF-2/D-DT and its receptor CD74 constitute an important pathway in tubular epithelial cell repair.
Fig. 12.
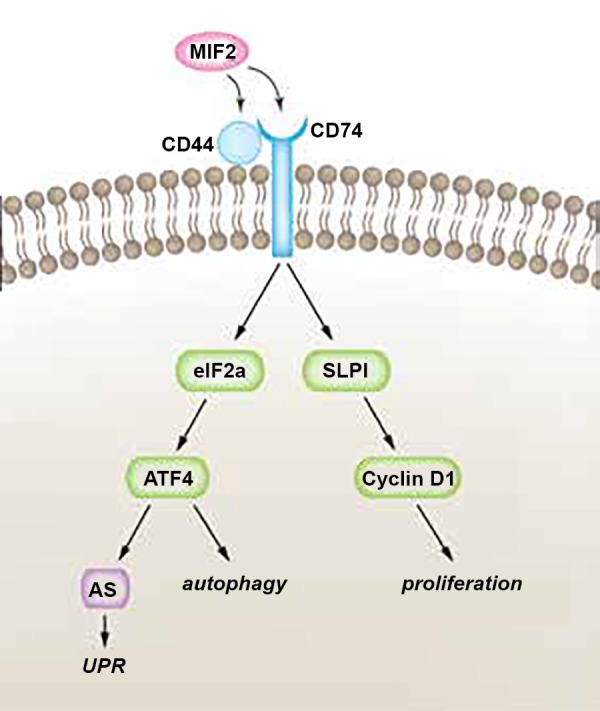
Hypothetical pathway of MIF-2/D-DT-dependent mechanisms in proximal tubule cell regeneration. Our study shows that MIF-2/D-DT treatment enhances expression of its receptors Cd74 and Cd44 in kidney tubular epithelial cells following I/R injury. Moreover, MIF-2/D-DT treatment rapidly (within 1–3 h) activates the transcription factors eIF2α and ATF4, which stimulate cell regeneration through autophagy and the unfolded protein response (UPR). A long-term effect of MIF-2/D-DT treatment (24–36 h) is the stimulation of cell proliferation through SLPI-dependent activation of cyclin D1 and D2. AS, asparagine synthetase.
Our RNAseq data showed that MIF-2/D-DT treatment stimulated the expression of the MIF target receptors CD74, CD44, and CXCR4. Moreover, MIF2/D-DT increased BrdU incorporation into proximal tubule cells in concert with increasing expression of the proliferation genes PCNA and Ki-67 in proximal tubule cells (Figs. 3, 7, and 10). RNAseq data from I/R kidneys further showed that SLPI, a transcription factor that targets cyclin D genes, as well as other cell cycle related genes, including CDK2-6, showed enhanced expression after MIF-2/D-DT treatment in vivo. MIF-2/D-DT treatment also stimulated activation of the transcription factors eIF2α and ATF4, which are important factors in the ISR. In hypoxic proximal tubular cells, MIF-2/D-DT activates markers of autophagy (LC3B-II), while simultaneously inhibiting apoptosis (Fig. 10). Our study shows a tiered effect by MIF-2/D-DT on tubular cell regeneration post-I/R injury by activating autophagy, inhibiting apoptosis, and stimulating cell proliferation.
MIF induces cell proliferation and cell survival in both primary and transformed cell lines (16, 52). The mechanisms that mediate cell survival include CD74 engagement and signal transduction, suppression of p53, and inhibition of apoptosis (4, 14, 28, 32, 34, 42). In a recent study in gastric cancer cells, the relationship among MIF, cyclin D1, and cancer cell proliferation has been shown (41). Our experiments show that MIF-2/D-DT treatment enhances SLPI and cyclin D gene expression within the first 48 h after I/R injury in vivo and that transfection of proximal tubule cells with SLPI induces cyclin D expression in vitro. We conclude that MIF-2/D-DT treatment enhances tubule cell proliferation within the first 24 to 48 h post-I/R injury and that this is an important second phase of the MIF-dependent tubular repair response. Since MIF-2/D-DT abundance was predominantly seen in interstitial cells following I/R injury, local MIF-2/D-DT secretion may contribute to proximal tubular cell repair.
The ISR is an elaborate signaling pathway activated by extrinsic cell stress such as hypoxia, amino acid deprivation, glucose deprivation, and viral infection (24, 63, 75). The common point of intersection between the different stressors that activate the ISR is the phosphorylation of eIF2α (60). Activation of eIF2α causes reduction in global protein synthesis and induces specific gene transcripts, such as ATF4 (45). Dephosphorylation of eIF2α initiates the return to normal protein synthesis (55). The ISR can also induce cell death pathways in case the cell adaptive response does not restore homeostasis. ATF4 is a basic leucine zipper (bZIP) transcription factor that is preferentially translated in response to activation of eIF2α. Increased ATF4 synthesis can activate target genes of the ISR by forming complexes with other bZIP transcription factors, including c-Fos, c-jun, Nrf2, and C/EBP homologous protein (CHOP) (20, 33). Cell death activation by ATF4 is largely mediated through downstream targets such as CHOP (22). Our experiments show an activation of eIF2α within 30 min of MIF-2/D-DT treatment in proximal tubule cells subjected to hypoxic injury. This is followed by a robust increase in ATF4 abundance 30 min later (Fig. 11). Furthermore, eIF2α activation is abolished at 120 min, followed by a decrease in ATF4 abundance 60 min later. At this time, there is evidence of MIF-2-dependent autophagy by increased conversion of LC3B-1 to LC3B-II (Fig. 11A). These findings indicate a rapid MIF-2/D-DT-dependent induction of the I/RS in proximal tubule cells recovering from hypoxic injury. Interestingly, MIF-2/D-DT treatment did not activate CHOP in cultured proximal tubule cells (data not shown).
Autophagy is an autodigestive mechanism that manages the turnover of intracellular organelles and macromolecules through terminal degradation and recycling (13). Early studies examining the role of autophagy in renal I/R injury models suggested a role in tubular cell death (11, 69). More recent studies, however, support a role for autophagy in tubular cell protection from I/R injury (44). Atg5 deficiency in the proximal tubule markedly sensitizes the cells to ischemic injury, which supports the role of autophagy in maintaining tubular integrity and survival during ischemia (36, 43). Autophagy has been shown to disrupt cell-cell junctions (10), which also may be an important step in the regeneration process of proximal tubular cells. MIF was previously found to induce autophagy in hepatocytes (12). More recently, MIF was shown to induce autophagy by inhibiting mechanistic target of rapamycin signaling in endothelial cells (9). In endothelial cells, MIF induced autophagy within 30 min and lasted up to 240 min, a time course quite similar to our data in posthypoxia proximal tubule cells (Fig. 11). We conclude that induction of autophagy represents the early phase of the MIF-dependent tubular cell regeneration process in the mouse I/R model, while cell proliferation represents a response later in the tubular repair process.
Regarding the clinical relevance of the present findings, several studies have demonstrated that MIF/MIF-2/D-DT act as early stress regulating cytokines in the inflammatory response of patients with myocardial I/R, which often is associated with acute kidney injury (68, 71). Autophagy is activated in myocardial I/R injury (47), which, together with the above findings, raises the question of whether elevated blood levels of MIF/MIF-2/D-DT occur in a concentration range that modulates autophagic activity in a beneficial manner. If further validated in clinical studies, the present findings supporting the functional role of MIF/MIF-2/D-DT-dependent autophagy in I/R injury thus may open potential therapeutic approaches for autophagy regulation by MIF/MIF-2/D-DT with relevant clinical implications.
In summary, our experiments show that MIF-2/D-DT is an early response cytokine in the I/R injury repair of the proximal tubule. The data support a model whereby MIF-2/DD-T initiates an integrated ISR by activation of eIF2α, followed by stimulation of ATF4 and induction of autophagy. At a later time point (24–48 h), MIF enhances cell proliferation through SLPI-dependent cyclin D expression, which enhances proliferation of surviving proximal tubule cells, especially in the S3 segment of the outer medulla. The observation that Mif−/− and Mif2−/− mice have significantly worse tubular injury underlines the importance of this pleiotropic cytokine in tubular injury repair.
GRANTS
This work was supported by National Institute of Arthritis and Musculoskeletal and Skin Diseases Grant R01-AR-049610 (to R. Bucala) and the Uehara Memorial Foundation and the Sumitomo Life Welfare and Culture Foundation (to A. Ochi).
DISCLOSURES
R. Bucala is listed as coinventor on a Yale patent describing the tissue protective actions of MIF-2/D-DT.
AUTHOR CONTRIBUTIONS
A.O. and G.W.M. conceived and designed research; A.O., D.C., N.G.M., and M.P. performed experiments; A.O., D.C., W.S., N.G.M., L.A., and G.W.M. analyzed data; A.O., D.C., L.L., N.G.M., M.P., L.A., R.B., and G.W.M. interpreted results of experiments; A.O. and G.W.M. prepared figures; A.O. and G.W.M. drafted manuscript; A.O., W.S., L.L., C.S., R.B., and G.W.M. edited and revised manuscript; R.B. and G.W.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Xin Du and Michelle Wakim for technical support in antibody generation and Gunilla Thulin for surgical expertise performing the bilateral renal artery clamping experiments and the MIF-2/D-DT injections in Mif−/−, Cd74−/−, and WT mice.
REFERENCES
- 1.Aeberli D, Yang Y, Mansell A, Santos L, Leech M, Morand EF. Endogenous macrophage migration inhibitory factor modulates glucocorticoid sensitivity in macrophages via effects on MAP kinase phosphatase-1 and p38 MAP kinase. FEBS Lett : 974–981, 2006. doi: 10.1016/j.febslet.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 2.B’chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res : 7683–7699, 2013. doi: 10.1093/nar/gkt563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bacher M, Metz CN, Calandra T, Mayer K, Chesney J, Lohoff M, Gemsa D, Donnelly T, Bucala R. An essential regulatory role for macrophage migration inhibitory factor in T-cell activation. Proc Natl Acad Sci USA : 7849–7854, 1996. doi: 10.1073/pnas.93.15.7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baumann R, Casaulta C, Simon D, Conus S, Yousefi S, Simon HU. Macrophage migration inhibitory factor delays apoptosis in neutrophils by inhibiting the mitochondria-dependent death pathway. FASEB J : 2221–2230, 2003. doi: 10.1096/fj.03-0110com. [DOI] [PubMed] [Google Scholar]
- 5.Bendrat K, Al-Abed Y, Callaway DJ, Peng T, Calandra T, Metz CN, Bucala R. Biochemical and mutational investigations of the enzymatic activity of macrophage migration inhibitory factor. Biochemistry : 15356–15362, 1997. doi: 10.1021/bi971153a. [DOI] [PubMed] [Google Scholar]
- 6.Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, Manogue KR, Cerami A, Bucala R. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature : 756–759, 1993. doi: 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- 7.Blais JD, Addison CL, Edge R, Falls T, Zhao H, Wary K, Koumenis C, Harding HP, Ron D, Holcik M, Bell JC. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol Cell Biol : 9517–9532, 2006. doi: 10.1128/MCB.01145-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen H, Pan YX, Dudenhausen EE, Kilberg MS. Amino acid deprivation induces the transcription rate of the human asparagine synthetase gene through a timed program of expression and promoter binding of nutrient-responsive basic region/leucine zipper transcription factors as well as localized histone acetylation. J Biol Chem : 50829–50839, 2004. doi: 10.1074/jbc.M409173200. [DOI] [PubMed] [Google Scholar]
- 9.Chen HR, Chuang YC, Chao CH, Yeh TM. Macrophage migration inhibitory factor induces vascular leakage via autophagy. Biol Open : 244–252, 2015. doi: 10.1242/bio.201410322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L, Zhang B, Toborek M. Autophagy is involved in nanoalumina-induced cerebrovascular toxicity. Nanomedicine (Lond) : 212–221, 2013. doi: 10.1016/j.nano.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chien CT, Shyue SK, Lai MK. Bcl-xL augmentation potentially reduces ischemia/reperfusion induced proximal and distal tubular apoptosis and autophagy. Transplantation : 1183–1190, 2007. doi: 10.1097/01.tp.0000287334.38933.e3. [DOI] [PubMed] [Google Scholar]
- 12.Chuang YC, Su WH, Lei HY, Lin YS, Liu HS, Chang CP, Yeh TM. Macrophage migration inhibitory factor induces autophagy via reactive oxygen species generation. PLoS One : e37613, 2012. doi: 10.1371/journal.pone.0037613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crotzer VL, Blum JS. Autophagy and adaptive immunity. Immunology : 9–17, 2010. doi: 10.1111/j.1365-2567.2010.03321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Damico RL, Chesley A, Johnston L, Bind EP, Amaro E, Nijmeh J, Karakas B, Welsh L, Pearse DB, Garcia JG, Crow MT. Macrophage migration inhibitory factor governs endothelial cell sensitivity to LPS-induced apoptosis. Am J Respir Cell Mol Biol : 77–85, 2008. doi: 10.1165/rcmb.2007-0248OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denkinger CM, Metz C, Fingerle-Rowson G, Denkinger MD, Forsthuber T. Macrophage migration inhibitory factor and its role in autoimmune diseases. Arch Immunol Ther Exp (Warsz) : 389–400, 2004. [PubMed] [Google Scholar]
- 16.Denz A, Pilarsky C, Muth D, Rückert F, Saeger HD, Grützmann R. Inhibition of MIF leads to cell cycle arrest and apoptosis in pancreatic cancer cells. J Surg Res : 29–34, 2010. doi: 10.1016/j.jss.2009.03.048. [DOI] [PubMed] [Google Scholar]
- 17.Djudjaj S, Lue H, Rong S, Papasotiriou M, Klinkhammer BM, Zok S, Klaener O, Braun GS, Lindenmeyer MT, Cohen CD, Bucala R, Tittel AP, Kurts C, Moeller MJ, Floege J, Ostendorf T, Bernhagen J, Boor P. Macrophage migration inhibitory factor mediates proliferative GN via CD74. J Am Soc Nephrol : 1650–1664, 2016. doi: 10.1681/ASN.2015020149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donn RP, Ray DW. Macrophage migration inhibitory factor: molecular, cellular and genetic aspects of a key neuroendocrine molecule. J Endocrinol : 1–9, 2004. doi: 10.1677/joe.0.1820001. [DOI] [PubMed] [Google Scholar]
- 19.Donnelly SC, Haslett C, Reid PT, Grant IS, Wallace WA, Metz CN, Bruce LJ, Bucala R. Regulatory role for macrophage migration inhibitory factor in acute respiratory distress syndrome. Nat Med : 320–323, 1997. doi: 10.1038/nm0397-320. [DOI] [PubMed] [Google Scholar]
- 20.Fawcett TW, Martindale JL, Guyton KZ, Hai T, Holbrook NJ. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem J : 135–141, 1999. [PMC free article] [PubMed] [Google Scholar]
- 21.Fingerle-Rowson G, Petrenko O, Metz CN, Forsthuber TG, Mitchell R, Huss R, Moll U, Müller W, Bucala R. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci USA : 9354–9359, 2003. doi: 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galehdar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J Neurosci : 16938–16948, 2010. doi: 10.1523/JNEUROSCI.1598-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goujon JM, Hauet T, Menet E, Levillain P, Babin P, Carretier M. Histological evaluation of proximal tubule cell injury in isolated perfused pig kidneys exposed to cold ischemia. J Surg Res : 228–233, 1999. doi: 10.1006/jsre.1998.5526. [DOI] [PubMed] [Google Scholar]
- 24.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell : 619–633, 2003. doi: 10.1016/S1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 25.Hiemstra PS, Maassen RJ, Stolk J, Heinzel-Wieland R, Steffens GJ, Dijkman JH. Antibacterial activity of antileukoprotease. Infect Immun : 4520–4524, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinnebusch AG. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol Mol Biol Rev : 434–467, 2011. doi: 10.1128/MMBR.00008-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hinnebusch AG. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol : 407–450, 2005. doi: 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- 28.Honda A, Abe R, Yoshihisa Y, Makino T, Matsunaga K, Nishihira J, Shimizu H, Shimizu T. Deficient deletion of apoptotic cells by macrophage migration inhibitory factor (MIF) overexpression accelerates photocarcinogenesis. Carcinogenesis : 1597–1605, 2009. doi: 10.1093/carcin/bgp160. [DOI] [PubMed] [Google Scholar]
- 29.Huber TB, Reinhardt HC, Exner M, Burger JA, Kerjaschki D, Saleem MA, Pavenstädt H. Expression of functional CCR and CXCR chemokine receptors in podocytes. J Immunol : 6244–6252, 2002. doi: 10.4049/jimmunol.168.12.6244. [DOI] [PubMed] [Google Scholar]
- 30.Imamura K, Nishihira J, Suzuki M, Yasuda K, Sasaki S, Kusunoki Y, Tochimaru H, Takekoshi Y. Identification and immunohistochemical localization of macrophage migration inhibitory factor in human kidney. Biochem Mol Biol Int : 1233–1242, 1996. [DOI] [PubMed] [Google Scholar]
- 31.Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR, Wek RC. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol : 1365–1377, 2004. doi: 10.1128/MCB.24.3.1365-1377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jung H, Seong HA, Ha H. Critical role of cysteine residue 81 of macrophage migration inhibitory factor (MIF) in MIF-induced inhibition of p53 activity. J Biol Chem : 20383–20396, 2008. doi: 10.1074/jbc.M800050200. [DOI] [PubMed] [Google Scholar]
- 33.Kilberg MS, Shan J, Su N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol Metab : 436–443, 2009. doi: 10.1016/j.tem.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim JY, Kwok SK, Hur KH, Kim HJ, Kim NS, Yoo SA, Kim WU, Cho CS. Up-regulated macrophage migration inhibitory factor protects apoptosis of dermal fibroblasts in patients with systemic sclerosis. Clin Exp Immunol : 328–335, 2008. doi: 10.1111/j.1365-2249.2008.03637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim YG, Huang XR, Suga S, Mazzali M, Tang D, Metz C, Bucala R, Kivlighn S, Johnson RJ, Lan HY. Involvement of macrophage migration inhibitory factor (MIF) in experimental uric acid nephropathy. Mol Med : 837–848, 2000. [PMC free article] [PubMed] [Google Scholar]
- 36.Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, Rakugi H, Isaka Y. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol : 902–913, 2011. doi: 10.1681/ASN.2010070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lameire NH, Bagga A, Cruz D, De Maeseneer J, Endre Z, Kellum JA, Liu KD, Mehta RL, Pannu N, Van Biesen W, Vanholder R. Acute kidney injury: an increasing global concern. Lancet : 170–179, 2013. doi: 10.1016/S0140-6736(13)60647-9. [DOI] [PubMed] [Google Scholar]
- 38.Lan HY, Mu W, Yang N, Meinhardt A, Nikolic-Paterson DJ, Ng YY, Bacher M, Atkins RC, Bucala R. De Novo renal expression of macrophage migration inhibitory factor during the development of rat crescentic glomerulonephritis. Am J Pathol : 1119–1127, 1996. [PMC free article] [PubMed] [Google Scholar]
- 39.Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, Delohery T, Chen Y, Mitchell RA, Bucala R. MIF signal transduction initiated by binding to CD74. J Exp Med : 1467–1476, 2003. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leung JC, Tang SC, Chan LY, Tsang AW, Lan HY, Lai KN. Polymeric IgA increases the synthesis of macrophage migration inhibitory factor by human mesangial cells in IgA nephropathy. Nephrol Dial Transplant : 36–45, 2003. doi: 10.1093/ndt/18.1.36. [DOI] [PubMed] [Google Scholar]
- 41.Li XJ, Luo Y, Yi YF. P115 promotes growth of gastric cancer through interaction with macrophage migration inhibitory factor. World J Gastroenterol : 8619–8629, 2013. doi: 10.3748/wjg.v19.i46.8619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu L, Chen J, Ji C, Zhang J, Sun J, Li Y, Xie Y, Gu S, Mao Y. Macrophage migration inhibitory factor (MIF) interacts with Bim and inhibits Bim-mediated apoptosis. Mol Cells : 193–199, 2008. [PubMed] [Google Scholar]
- 43.Liu S, Hartleben B, Kretz O, Wiech T, Igarashi P, Mizushima N, Walz G, Huber TB. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy : 826–837, 2012. doi: 10.4161/auto.19419. [DOI] [PubMed] [Google Scholar]
- 44.Livingston MJ, Dong Z. Autophagy in acute kidney injury. Semin Nephrol : 17–26, 2014. doi: 10.1016/j.semnephrol.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol : 27–33, 2004. doi: 10.1083/jcb.200408003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, Kaufman RJ, Ron D, Harding HP. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J : 169–179, 2004. doi: 10.1038/sj.emboj.7600030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma S, Wang Y, Chen Y, Cao F. The role of the autophagy in myocardial ischemia/reperfusion injury. Biochim Biophys Acta : 271–276, 2015. doi: 10.1016/j.bbadis.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 48.Matsumoto K, Kanmatsuse K. Increased production of macrophage migration inhibitory factor by T cells in patients with IgA nephropathy. Am J Nephrol : 455–464, 2001. doi: 10.1159/000046649. [DOI] [PubMed] [Google Scholar]
- 49.Merk M, Zierow S, Leng L, Das R, Du X, Schulte W, Fan J, Lue H, Chen Y, Xiong H, Chagnon F, Bernhagen J, Lolis E, Mor G, Lesur O, Bucala R. The D-dopachrome tautomerase (DDT) gene product is a cytokine and functional homolog of macrophage migration inhibitory factor (MIF). Proc Natl Acad Sci USA : E577–E585, 2011. doi: 10.1073/pnas.1102941108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Metz CN, Bucala R. Role of macrophage migration inhibitory factor in the regulation of the immune response. Adv Immunol : 197–223, 1997. doi: 10.1016/S0065-2776(08)60598-2. [DOI] [PubMed] [Google Scholar]
- 52.Meyer-Siegler KL, Iczkowski KA, Leng L, Bucala R, Vera PL. Inhibition of macrophage migration inhibitory factor or its receptor (CD74) attenuates growth and invasion of DU-145 prostate cancer cells. J Immunol : 8730–8739, 2006. doi: 10.4049/jimmunol.177.12.8730. [DOI] [PubMed] [Google Scholar]
- 53.Mitchell R, Bacher M, Bernhagen J, Pushkarskaya T, Seldin MF, Bucala R. Cloning and characterization of the gene for mouse macrophage migration inhibitory factor (MIF). J Immunol : 3863–3870, 1995. [PubMed] [Google Scholar]
- 54.Morand EF, Bucala R, Leech M. Macrophage migration inhibitory factor: an emerging therapeutic target in rheumatoid arthritis. Arthritis Rheum : 291–299, 2003. doi: 10.1002/art.10728. [DOI] [PubMed] [Google Scholar]
- 55.Novoa I, Zhang Y, Zeng H, Jungreis R, Harding HP, Ron D. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J : 1180–1187, 2003. doi: 10.1093/emboj/cdg112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nyström M, Bergenfeldt M, Ljungcrantz I, Lindeheim A, Ohlsson K. Production of secretory leucocyte protease inhibitor (SLPI) in human pancreatic beta-cells. Mediators Inflamm : 147–151, 1999. doi: 10.1080/09629359990478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ohlsson S, Ljungkrantz I, Ohlsson K, Segelmark M, Wieslander J. Novel distribution of the secretory leucocyte proteinase inhibitor in kidney. Mediators Inflamm : 347–350, 2001. doi: 10.1080/09629350120102389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Plestina S, Gamulin S. Kidney ischaemia-reperfusion injury and polyribosome structure. Nephron : 201–207, 2001. doi: 10.1159/000046068. [DOI] [PubMed] [Google Scholar]
- 59.Rice EK, Nikolic-Paterson DJ, Hill PA, Metz CN, Bucala R, Atkins RC, Tesch GH. Interferon-gamma induces macrophage migration inhibitory factor synthesis and secretion by tubular epithelial cells. Nephrology (Carlton) : 156–161, 2003. doi: 10.1046/j.1440-1797.2003.00152.x. [DOI] [PubMed] [Google Scholar]
- 60.Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest : 1383–1388, 2002. doi: 10.1172/JCI0216784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, Lambin P, van der Kogel AJ, Koritzinsky M, Wouters BG. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest : 127–141, 2010. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rowlands AG, Panniers R, Henshaw EC. The catalytic mechanism of guanine nucleotide exchange factor action and competitive inhibition by phosphorylated eukaryotic initiation factor 2. J Biol Chem : 5526–5533, 1988. [PubMed] [Google Scholar]
- 63.Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I, Harris AL. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene : 4424–4435, 2010. doi: 10.1038/onc.2010.191. [DOI] [PubMed] [Google Scholar]
- 64.Sanchez-Niño MD, Sanz AB, Ihalmo P, Lassila M, Holthofer H, Mezzano S, Aros C, Groop PH, Saleem MA, Mathieson PW, Langham R, Kretzler M, Nair V, Lemley KV, Nelson RG, Mervaala E, Mattinzoli D, Rastaldi MP, Ruiz-Ortega M, Martin-Ventura JL, Egido J, Ortiz A. The MIF receptor CD74 in diabetic podocyte injury. J Am Soc Nephrol : 353–362, 2009. doi: 10.1681/ASN.2008020194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Simons D, Grieb G, Hristov M, Pallua N, Weber C, Bernhagen J, Steffens G. Hypoxia-induced endothelial secretion of macrophage migration inhibitory factor and role in endothelial progenitor cell recruitment. J Cell Mol Med : 668–678, 2011. doi: 10.1111/j.1582-4934.2010.01041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sinha D, Wang Z, Price VR, Schwartz JH, Lieberthal W. Chemical anoxia of tubular cells induces activation of c-Src and its translocation to the zonula adherens. Am J Physiol Renal Physiol : F488–F497, 2003. doi: 10.1152/ajprenal.00172.2002. [DOI] [PubMed] [Google Scholar]
- 67.Starlets D, Gore Y, Binsky I, Haran M, Harpaz N, Shvidel L, Becker-Herman S, Berrebi A, Shachar I. Cell-surface CD74 initiates a signaling cascade leading to cell proliferation and survival. Blood : 4807–4816, 2006. doi: 10.1182/blood-2005-11-4334. [DOI] [PubMed] [Google Scholar]
- 68.Stoppe C, Werker T, Rossaint R, Dollo F, Lue H, Wonisch W, Menon A, Goetzenich A, Bruells CS, Coburn M, Kopp R, Bucala R, Bernhagen J, Rex S. What is the significance of perioperative release of macrophage migration inhibitory factor in cardiac surgery? Antioxid Redox Signal : 231–239, 2013. doi: 10.1089/ars.2012.5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Suzuki C, Isaka Y, Takabatake Y, Tanaka H, Koike M, Shibata M, Uchiyama Y, Takahara S, Imai E. Participation of autophagy in renal ischemia/reperfusion injury. Biochem Biophys Res Commun : 100–106, 2008. doi: 10.1016/j.bbrc.2008.01.059. [DOI] [PubMed] [Google Scholar]
- 70.Taggart CC, Lowe GJ, Greene CM, Mulgrew AT, O’Neill SJ, Levine RL, McElvaney NG. Cathepsin B, L, and S cleave and inactivate secretory leucoprotease inhibitor. J Biol Chem : 33345–33352, 2001. doi: 10.1074/jbc.M103220200. [DOI] [PubMed] [Google Scholar]
- 71.Takahashi M, Nishihira J, Shimpo M, Mizue Y, Ueno S, Mano H, Kobayashi E, Ikeda U, Shimada K. Macrophage migration inhibitory factor as a redox-sensitive cytokine in cardiac myocytes. Cardiovasc Res : 438–445, 2001. doi: 10.1016/S0008-6363(01)00408-4. [DOI] [PubMed] [Google Scholar]
- 72.Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA : 11269–11274, 2004. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wada S, Fujimoto S, Mizue Y, Nishihira J. Macrophage migration inhibitory factor in the human ovary: presence in the follicular fluids and production by granulosa cells. Biochem Mol Biol Int : 805–814, 1997. [DOI] [PubMed] [Google Scholar]
- 74.Wistow GJ, Shaughnessy MP, Lee DC, Hodin J, Zelenka PS. A macrophage migration inhibitory factor is expressed in the differentiating cells of the eye lens. Proc Natl Acad Sci USA : 1272–1275, 1993. doi: 10.1073/pnas.90.4.1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, Bobrovnikova-Marjon E, Diehl JA, Ron D, Koumenis C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J : 2082–2096, 2010. doi: 10.1038/emboj.2010.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yoo SA, Leng L, Kim BJ, Du X, Tilstam PV, Kim KH, Kong JS, Yoon HJ, Liu A, Wang T, Song Y, Sauler M, Bernhagen J, Ritchlin CT, Lee P, Cho CS, Kim WU, Bucala R. MIF allele-dependent regulation of the MIF coreceptor CD44 and role in rheumatoid arthritis. Proc Natl Acad Sci USA : E7917–E7926, 2016. doi: 10.1073/pnas.1612717113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang D, Simmen RC, Michel FJ, Zhao G, Vale-Cruz D, Simmen FA. Secretory leukocyte protease inhibitor mediates proliferation of human endometrial epithelial cells by positive and negative regulation of growth-associated genes. J Biol Chem : 29999–30009, 2002. doi: 10.1074/jbc.M203503200. [DOI] [PubMed] [Google Scholar]