

ABSTRACT
Pharmacologic inhibition of KDM1A (Lysine Demethylase 1A) induces differentiation in certain subtypes of acute myeloid leukemia. Our recent studies reveal this is dependent upon drug-induced disruption of the GFI1 (Growth Factor Independent 1) transcription repressor complex, leading to activation of enhancers distributed close to genes controlling monocytic lineage differentiation.
Keywords: GFI1, LSD1, AML, epigenetics, methylation, acetylation, leukemia
Article
KDM1A (Lysine Demethylase 1A; best known as Lysine Specific Demethylase 1 or LSD1) is known to be a flavin adenine dinucleotide (FAD)-dependent histone demethylase with activity versus mono- and dimethyl histone H3 lysine 4 (H3K4), as well as a number of other targets. It was originally identified as a core component of the REST corepressor 1 (RCOR1) and histone deacetylase (HDAC) transcription corepressor complex.1 Recently there has been much interest in the potential of LSD1 as a therapeutic target in cancer. This has arisen from the observation that the gene is highly expressed in some poor prognosis sub-groups of prostate, lung, brain, breast and other cancers. Furthermore, genetic knockdown of LSD1 promotes differentiation in some types of acute myeloid leukaemia (AML).2 The non-selective monoamine oxidase inhibitor tranylcypromine was the first drug reported to inhibit LSD1, through a suicide inactivation mechanism of action involving covalent binding of the FAD cofactor. Derivatives of tranylcypromine have since been developed that exhibit much higher potencies and specificities. Screening of a large cell line panel with the LSD1 inhibitor GSK2879552 revealed particular sensitivities of AML and small cell lung cancer cells,3 and a related inhibitor (OG86) induced differentiation of murine and human AML cells in vitro and in vivo.4 Indeed, a first-in-man phase 1 trial of ORY1001 (from Oryzon Genomics) demonstrates that, in keeping with pre-clinical data, LSD1 inhibitors promote blast cell differentiation in patients with AML associated with translocations targeting the Mixed Lineage Leukaemia gene (MLL).5,6 With clinical trials ongoing in leukaemia and in other disease settings, an appreciation of the mechanism of action of LSD1 inhibitors is essential.
The assumption has been that inhibitor-induced AML cell differentiation is the result of the blockade of LSD1’s histone demethylase activity. Indeed current LSD1 inhibitors have been developed with the specific aim of maximising the inhibition of LSD1’s enzymatic activity. Countering this assumption, we found that when an MLL-translocated AML cell line (THP1) was treated with the tranylcypromine-derivative LSD1 inhibitor OG86, rapid and extensive changes in transcription were observed that did not correlate with genome-wide changes in the LSD1-demethylation targets mono- and dimethyl H3K4 at LSD1 binding sites on chromatin.7 Furthermore, in knockdown and rescue experiments, the LSD1 K661A catalytic mutant was equally capable of rescuing the clonogenic potential of LSD1 knockdown cells as the wild type protein. These experimental findings suggested that a non-catalytic function of LSD1 might be targeted by LSD1 inhibitors to promote differentiation in AML.
In fact, through bioinformatics analyses we discovered that LSD1 inhibition in AML cells mimics knockdown of the transcription repressor GFI1 (Growth Factor Independent 1) which is known to associate with LSD1 though its N-terminal SNAG (SNAIL/GFI1) domain and to be dependent upon that association for its repressive activity.8 By chromatin immunoprecipitation with next generation sequencing (ChIPseq) we found a significant association of GFI1, LSD1 and RCOR1 on chromatin, with co-localization of the strongest GFI1, LSD1 and RCOR1 binding peaks genome-wide. Multiple distinct inhibitors of LSD1, both reversible and irreversible, disrupt the LSD1:GFI1 interaction leading to release of LSD1 with its binding partner RCOR1 (i.e. the CoREST complex) from GFI1 and chromatin, thus abrogating GFI1 activity (Figure 1). To provide functional evidence that the protein:protein interaction of LSD1 with GFI1 is the most significant target of OG86 rather than the demethylase activity we expressed a doxycycline-regulated construct in which the GFI1 DNA-binding domain was fused directly to LSD1 or the K661A catalytically inactive LSD1, such that the two were no longer separable upon treatment of cells with drug. We observed that the expression of either prevented the pro-differentiation effect induced by several unrelated inhibitors, confirming that the physical interaction of GFI1:LSD1 is the relevant target of LSD1 inhibitor activity in AML cells. Through integrated ChIPseq analyses we identified 1,560 enhancers genome-wide where GFI1, LSD1 and RCOR1 co-localise. These were distributed close to genes coding for key regulators of monocyte/macrophage differentiation such as IRF8 (Interferon Regulatory Factor 8), KLF4, (Kruppel-like Factor 4) and MEF2C (Myocyte Enhancer Factor 2C) and were concomitantly bound by the myeloid transcription activators SPI1 (Spleen Focus Forming Virus Proviral Integration Oncogene 1) and CEBPA (CCAAT Enhancer Binding Protein Alpha) (Figure 1). Treatment of AML cells with LSD1 inhibitor targeted these enhancers for rapid activation by histone acetylation leading to up regulation of transcription of subordinate genes within hours.
Figure 1.
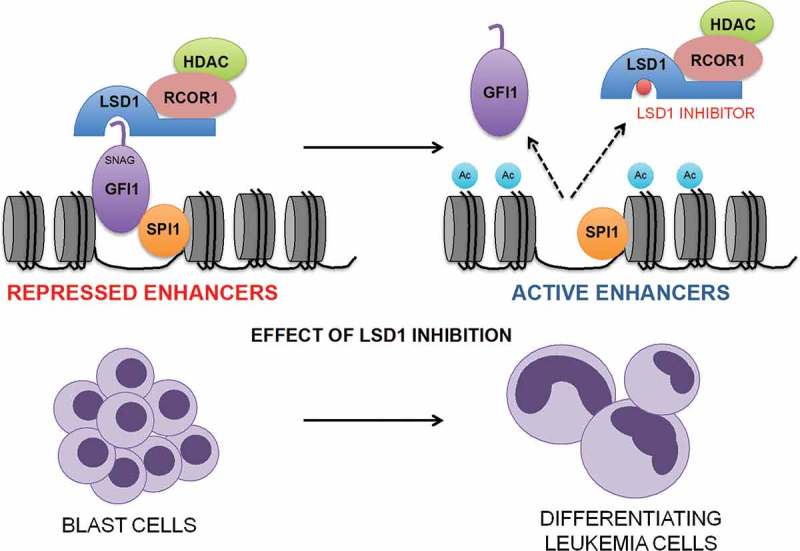
Disruption of the GFI1 (Growth Factor Independent 1) transcription repressor complex by inhibitors of KDM1A (Lysine Demethylase 1A; best known as Lysine Specific Demethylase 1 or LSD1). Inhibitors of LSD1 disrupt the GFI1 transcription repressor complex leading to its release from chromatin followed by activation of enhancers distributed close to genes controlling monocytic lineage differentiation. RCOR1 = REST corepressor 1; HDAC = histone deacetylase; SPI1 = Spleen Focus Forming Virus Proviral Integration Oncogene 1; SNAG = SNAIL/GFI1; Ac = acetyl.
Our findings are consistent with the recently published findings of Cusan and colleagues9 who explored changes in chromatin dynamics during the treatment of MLL-AF9-driven murine and patient-derived leukemias. Drug treated cells exhibited global gains in chromatin accessibility with acquisition of SPI1 and CEBPA motif signatures at LSD1 inhibitor-induced dynamic sites, perhaps consistent with the differentiation process. Interestingly, they also demonstrated that genetic reduction of Spi1 or genetic deletion of Cebpa in MLL-AF9 cells generates resistance of these leukemias to LSD1 inhibition. This is likely in keeping with our finding that GFI1-bound enhancers were pre-loaded with SPI1 and CEBPA: these genetic data suggest that the presence of SPI1 and CEBPA at GFI1-bound loci may be essential for enhancer activation following disruption of the GFI1 transcription repressor complex due to LSD1 inhibition.
All together our data demonstrate that LSD1 has two activities: a demethylase activity and a scaffolding activity. Unexpectedly, inhibitors of LSD1 target both. In the setting of MLL-translocated AML, it is the scaffolding activity which is most important because drug resistance is conferred to leukaemia cells through expression of a constitutively active GFI1 transcription repressor fusion protein. Indeed our data further highlight the significance of GFI1 as a therapeutic target in this molecular subtype of AML. Given the observation that drug-induced separation of GFI1:LSD1 while substantial was not complete, development of LSD1 inhibitors that confer maximal physical disruption of LSD1 from SNAG-domain transcription repressors may possibly prove more effective clinically.
Funding Statement
This work was supported by Cancer Research UK grant number C5759/A20971.
Disclosures of potential conflicts of interest
No conflicts of interest disclosed.
References
- 1.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Pa C, Ra C, Shi Y.. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. PMID:15620353 doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 2.Maiques-Diaz A, Somervaille TC.. LSD1: biologic roles and therapeutic targeting. Epigenomics. 2016;8:1103–1116. PMID: 27479862 doi: 10.2217/epi-2016-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mohammad HP, Smitherman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, Schneck JL, Carson KD, Liu Y, Butticello M, et al. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell. 2015;28:57–69. PMID:26175415 doi: 10.1016/j.ccell.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 4.Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, Ciceri F, Blaser JG, Greystoke BF, Jordan AM, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21:473–487. PMID: 22464800 doi: 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 5.Somervaille TC, Salamero O, Montesinos P, Willekens C, Perez Simon J, Pigneux A, Recher C, Popat R, Molinero C, Mascaro C, et al. Safety, pharmacokinetics (PK), pharmacodynamics (PD) and premliminary activity in acute leukemia of Ory-1001, a first-in-class inhibitor of lysine specific demethylase 1A (LSD1/KDM1A): initial results from a first-in-human phase 1 study. Blood. 2016;128:4026 Paper presented at the 58th Annual Meeting of the American Hematology Association doi: 10.1182/blood-2016-06-724161. [DOI] [Google Scholar]
- 6.Maes T, Mascaro C, Tirapu I, Estiarte A, Ciceri F, Lunardi S, Guibourt N, Perdones A, Lufino MMP, Somervaille TCP, et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell. 2018;33(3):495–511. PMID: 29502954 doi: 10.1016/j.ccell.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Maiques-Diaz A, Spencer GJ, Lynch JT, Ciceri F, Williams EL, Amaral FMR, Wiseman DH, Harris WJ, Li Y, Sahoo S, et al. Enhancer activation by pharmacological displacement of LSD1 from GFI1 induces differentiation in acute myeloid leukemia. Cell Rep. 2018;22:3641–3659. PMID:29590629 doi: 10.1016/j.celrep.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saleque S, Kim J, Rooke HM, Orkin SH. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol Cell. 2007;27:562–572. PMID:17707228 doi: 10.1016/j.molcel.2007.06.039. [DOI] [PubMed] [Google Scholar]
- 9.Cusan M, Cai SF, Mohammad HP, Krivtsov A, Chramiec A, Loizou E, Witkin MD, Smitheman KN, Tenen DG, Ye M, et al. LSD1 inhibition experts it anti-leukemic effect by recommissioning PU.1- and C/EBPA-dependent enhancers in AML. Blood. 2018;February 16 published online doi: 10.1182/blood-2017-09-807024. [DOI] [PMC free article] [PubMed] [Google Scholar]