

Abstract
Using activity-based protein profiling (ABPP), functional proteins can be interrogated in their native environment. Despite their pharmaceutical relevance, G protein-coupled receptors (GPCRs) have been difficult to address through ABPP. In the current study, we took the prototypical human adenosine A2A receptor (hA2AR) as the starting point for the construction of a chemical toolbox allowing two-step affinity-based labeling of GPCRs. First, we equipped an irreversibly binding hA2AR ligand with a terminal alkyne to serve as probe. We showed that our probe irreversibly and concentration-dependently labeled purified hA2AR. Click-ligation with a sulfonated cyanine-3 fluorophore allowed us to visualize the receptor on SDS-PAGE. We further demonstrated that labeling of the purified hA2AR by our probe could be inhibited by selective antagonists. Lastly, we showed successful labeling of the receptor in cell membranes overexpressing hA2AR, making our probe a promising affinity-based tool compound that sets the stage for the further development of probes for GPCRs.
Introduction
The adenosine receptors, belonging to the family of G protein-coupled receptors (GPCRs), have been coined adenosine A1, A2A, A2B, and A3. These receptors are widely distributed through the human body and are considered promising targets for a wide range of diseases.1 Regadenoson, a selective human adenosine A2A receptor (hA2AR) agonist used to increase vasodilation during cardiac imaging, has been approved by the FDA, exemplifying the potential therapeutic applications for the hA2AR. Likewise, hA2AR antagonists are currently being pursued as potential treatment of Parkinson’s disease2 and as adjuvants in cancer immunotherapy.3
The hA2AR was one of the first GPCRs for which a crystal structure was elucidated.4 However, the challenges in structural biology of GPCRs, including the low expression level in native tissue and inherent poor protein stability,5 still exist. To overcome these obstacles, covalent probes have been developed as useful pharmacological tools. Such probes, also named affinity labels, represent compounds that feature a reactive cross-linking moiety, which can irreversibly and specifically bind to a receptor. For example, an irreversible antagonist was used to stabilize the adenosine A1 receptor for cocrystallization, resulting in the visualization of key amino acids important for ligand–receptor binding.6
The design of covalent probes for GPCRs generally follows a similar strategy, which is to incorporate a warhead in a high-affinity, reversibly binding ligand. Based on the type of warhead used, two categories of irreversible ligands can be discerned: photoaffinity and chemoreactive ligands.7,8 Whereas in the former type a photoreactive warhead is employed, the latter is equipped with an electrophilic chemical moiety capable of binding nucleophilic residues in the target protein. A commonly used warhead is aryl sulfonyl fluoride, which is capable of covalently binding to many nucleophilic amino acid residues, such as serine, threonine, lysine, and cysteine.9 This warhead has been incorporated in several reported covalent ligands for the adenosine receptors, including FSCPX,10 FSPTP,11 fluorosulfonyl-functionalized pyrimidine derivatives,12 and LUF7445.13 Likewise, fluorescent tags have been incorporated into adenosine receptor ligands to visualize the receptor, which yielded, e.g., FITC-ADAC,14 MRS5422,15 and NBD-NECA.16 However, fluorescent moieties are of significant size, and a priori derivatization of a ligand with such a group may negatively affect receptor affinity. Here two-step affinity-based probes (AfBPs) might be a better alternative, as a reporter tag is added after the reactive ligand has bound its target.17
Interestingly, from the field of activity-based protein profiling (ABPP), combined with click chemistry, many techniques have emerged that could potentially be applied to GPCRs using our covalent ligand. Normally in ABPP, an irreversible ligand is equipped with a ligation handle and after binding to the protein of interest is paired with a clickable fluorophore. In this way, via a Huisgen 1,3-dipolar cycloaddition, a stable triazole-linked product is formed, effectively attaching a fluorescent label to the protein.18−20 Currently, this technique serves as a tool to profile the activities of drug targets (currently mainly enzymes) in native biological systems. One-step labeling, where the reporter group is preattached to the probe, has been applied on GPCRs previously.21−23 Moreover, similar two-step labeling strategies have been applied for other targets.24,25 However, due to their low abundance, GPCRs are difficult to address with this otherwise promising technique. Within the entire GPCR family with over 800 members, until recently, only the mGlu5 receptor had been the subject of this approach, albeit with limited success.26 Very recently, the type 2 cannabinoid receptor (CB2R) has been probed with a two-step photoaffinity probe, leading to great insights into receptor localization and target engagement.27
In this study, we describe our efforts to obtain a clickable affinity-based probe, with an electrophilic warhead, as a logical extension of our previous research on the successful design of a covalent antagonist of hA2AR, compound 1 (LUF7445).13 We used the antagonist ZM241385 as the starting point in our design efforts and synthesized a series of fluorosulfonyl derivatives with diverse linker lengths (compounds 1–3, Figure 1). The most potent ligand, with low nanomolar affinity, was retained for further structural modification and was equipped with an alkyne-click handle, resulting in probe 4, as shown in Figure 1. We then validated that the ligand’s binding to the receptor was wash-resistant. Additionally, we demonstrated the ligand’s covalent labeling capacity for purified receptors via a bioorthogonal copper-catalyzed azide–alkyne ligation reaction with a fluorescent moiety, sulfonated cyanine 3 ((E)-2-((E)-3-(1-(6-((3-azidopropyl)amino)-6-oxohexyl)-3,3-dimethyl-5-sulfo-3H-indol-1-ium-2-yl)allylidene)-3,3-dimethyl-1-(3-sulfopropyl)indoline-5-sulfonate). Finally, this probe was able to profile the presence of hA2AR in a relatively complex biological sample. Hence, this is one of the first AfBPs for a GPCR and may set the stage for similar probes to facilitate target discovery and bioanalysis of GPCRs associated with human disease.
Figure 1.

Chemical structures of the hA2AR antagonists examined in this study. The lead compound ZM241385, a selective hA2AR antagonist, inspired the design of covalent antagonist 1.13 In the current study, the effect of the linker length between scaffold and warhead on affinity was further examined, yielding compound 2 and, preferably, compound 3. The affinity-based probe 4 was then synthesized from compound 3, bearing an alkyne ligation-handle and a fluorosulfonyl electrophilic warhead. The electrophilic warhead is in red and the click-ligation handle is in blue.
Results and Discussion
Chemistry
Our research group has been evaluating structural modifications of triazolotriazine derivatives based on the selective adenosine A2A antagonist 4-(2-(7-amino-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-5-ylamino)ethyl)phenol (ZM241385), to obtain a covalent ligand for the hA2AR. The rational design of this covalent ligand originated from a reported hA2AR crystal structure (PDB: 4EIY) in complex with ZM241385.4 In it, the ligand binding pocket demonstrated a deep, planar, and narrow cavity embracing the aromatic core and furan ring of ZM241385. Therefore, an extension of the hydroxyphenethylamine moiety into the extracellular domain of the receptor offered us the playground for integration of the electrophilic reactive groups. Our earlier covalent antagonist, compound 1 (Figure 1), in which the 4-hydroxyphenylethylamine side chain in ZM241385 was replaced with a similar side chain harboring an electrophilic fluorosulfonyl moiety, was recognized by hA2AR with an apparent pKi of 8.99.13 To optimize the irreversible binding potential of our compound, with our current aim of developing an AfBP in mind, an exploration of linker length was performed, varying the linker between the fluorosulfonyl warhead moiety and the aromatic recognition element from three to five carbon atoms. To this end, compounds 2 and 3 were synthesized as detailed in Scheme 1. The synthesis starts from 2-(furan-2-yl)-5-(methylsulfonyl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-7-amine 5, synthesized as previously reported,13 and involves a linear sequence comprising aromatic substitution with either commercially available mono-Boc-protected butyldiamine or pentyldiamine and subsequent Boc-deprotection toward intermediates 8 and 9. Introduction of the fluorosulfonylbenzoyl warhead proceeded with low yields due to difficult purification, providing ligands 2 and 3 in 4% and 2% yield, respectively.
Scheme 1. Synthesis of Compounds 2–4.

Reagents and conditions: (a) tert-butyl (4-aminobutyl)carbamate or tert-butyl (5-aminopentyl)carbamate, DiPEA, MeCN, 70–85 °C, 46–74%; (b) TFA, quant; (c) 4-fluorosulfonylbenzoyl chloride, DiPEA, MeCN, 70 °C, 2–4%; (d) Boc2O, DCM, quant; (e) PPh3, CBr4, 90%; (f) propargylamine, DiPEA, 46%; (g) 4-fluorosulfonylbenzoyl chloride, DiPEA, MeCN, quant; (h) i. 5, TFA, DCM, ii. DiPEA, MeCN, 70 °C, 45%.
The synthetic route toward probe 4 (LUF7487, Figure 1) is depicted in Scheme 1. First, the amino group of 5-aminopentanol was protected with a Boc group and the hydroxyl was converted to a bromide using an Appel reaction, providing intermediate 12. Nucleophilic substitution of the bromide with propargylamine afforded amine 13, which was acylated with 4-fluorosulfonylbenzoyl chloride to give Boc-protected bifunctional spacer 14 uneventfully. Finally, in a two-step process, the spacer was deprotected and coupled to scaffold 5, to provide probe 4 in 45% yield.
Biology
To assess the affinity for the hA2AR, compounds 2 and 3 were tested in [3H]ZM241385 displacement experiments (n = 3), which demonstrated a concentration-dependent inhibition of radioligand binding to hA2AR overexpressed in HEK293 cells. To better understand the time-dependent binding characteristics of these compounds, we then carried out displacement assays performed with two different incubation times. Representative graphs for these experiments are given in Figure 2a and 2b, in which the concentration-dependent inhibition of specific [3H]ZM241385 binding shifted to the left with an incubation time extension from 0.5 h (standard) to 3 h. As detailed in Table 1, the affinities of both compound 2 and 3 significantly increased by approximately 5-fold to subnanomolar values with longer incubation times. In other words, both designed covalent ligands became more potent in displacing the radioligand [3H]ZM241385 from the receptor over time. Similarly to 1,13 this pronounced affinity increase may be attributed to an irreversible binding nature of the compounds, leading to a higher receptor occupancy with a longer incubation time. It should be kept in mind that due to the covalent nature of the interaction, affinity values can only be apparent as no dynamic equilibrium can be reached.
Figure 2.
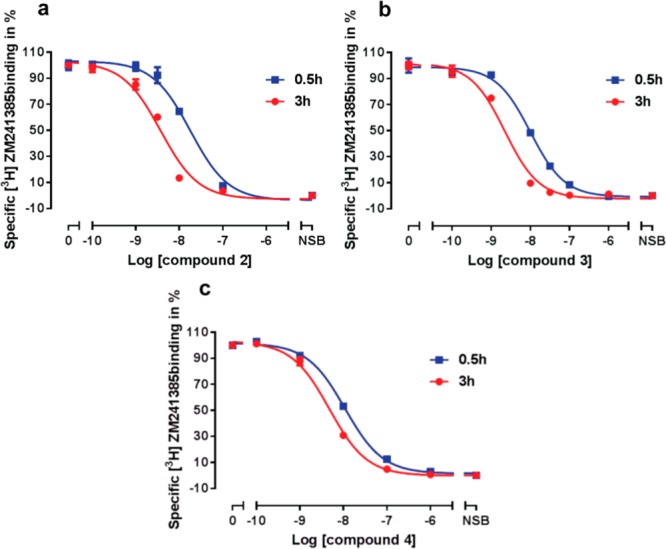
Displacement of specific [3H]ZM241385 binding from HEK293 cell membranes stably expressing the hA2AR receptor at 25 °C by compound 2 (a), 3 (b), and 4 (c) with an incubation time of 0.5 h (blue curve) and 3 h (red curve), respectively. Representative graphs are from one experiment performed in duplicate.
Table 1. (Apparent) Affinities of Synthesized Ligands for the Human Adenosine A2A Receptora.
| compoundb | pKic (0.5 h) | pKid (3 h) | pKi shifte |
|---|---|---|---|
| 1f | 8.27 ± 0.04 | 8.99 ± 0.01*** | 0.72 |
| 2 | 8.20 ± 0.13 | 9.05 ± 0.07*** | 0.85 |
| 3 | 8.56 ± 0.03 | 9.21 ± 0.01*** | 0.65 |
| 4 | 8.41 ± 0.02 | 8.82 ± 0.02*** | 0.41 |
Data are expressed as means ± SEM of three separate experiments each performed in duplicate. ***P < 0.001 compared with the pKi values in displacement experiments with a 0.5 h incubation time; Student’s t test.
For all the designed covalent antagonists, pKi values can only be apparent, as true equilibrium cannot be reached.
Affinity, expressed as pKi value, determined from displacement of specific [3H]ZM241385 binding from the hA2AR at 25 °C during a 0.5 h incubation.
Affinity, expressed as pKi value, determined from displacement of specific [3H]ZM241385 binding from the hA2AR at 25 °C during a 3 h incubation.
Affinity shift was calculated as [pKi (3 h) – pKi (0.5 h)].
Data previously reported provided for comparison.13
Compound 3 inhibited the specific [3H]ZM241385 binding to the hA2AR with a pKi of 9.21, compared to the affinity of compound 2 (pKi = 9.05 ± 0.07) and 1 (pKi = 8.99 ± 0.01). Thus, the extension of the linker to five carbon atoms slightly increased the apparent affinity. This could be caused by more steric freedom, allowing the fluorosulfonyl group to orient toward the adjacent nucleophilic residue in the receptor binding site compared to ligands with a shorter linker. A similar example is an electrophilic probe for the cannabinoid CB1 receptor, 7′-NCS-1′,1′-DMH-Δ8-THC, in which lengthening the C-3 alkyl side chain to seven carbons resulted in a significantly improved affinity.28 Above all, high affinity is a key requirement for the development of irreversible ligands, as it increases the presence of the chemoreactive moiety in proximity to a nucleophilic residue in the binding site, thereby improving receptor occupancy and causing a decrease in nonspecific binding to other unrelated targets. As we anticipated a greater demand for steric freedom for the incorporation of the alkyne group and the subsequent ligation between the alkyne moiety and a bulky fluorescent dye, we retained the preferable five-carbon atom linker length for the design of our probe.
Inspired by the most promising compound 3, we incorporated the alkyne click-handle to afford a novel covalent probe, compound 4 (LUF7487, Figure 1). As detailed in Table 1, affinity-based probe 4 demonstrated a high affinity, displacing [3H]ZM241385 with an apparent pKi value of 8.82. Under these conditions 4 was at least 10-fold selective over human A1 and A3 receptors (SI Table S1). In a time-dependent study, probe 4 generated a significant increase in specific [3H]ZM241385 displacement over time (Table 1). In analogy to the covalent ligand 3, the designed probe was markedly influenced by prolonged incubation times (Figure 2c), suggesting an increasing level of covalent binding over time. However, compared to 3, the slight decrease in affinity may be attributed to the incorporation of the click handle, possibly leading to a steric hindrance in the ligand–receptor complex and/or the formation of a covalent bond between the warhead and other nucleophilic residues.
To better understand the receptor–ligand binding nature, the novel affinity-based probe was then evaluated for its covalent nature by determining its capacity to irreversibly block [3H]ZM241385 to hA2AR binding sites. Membranes overexpressing hA2AR were pretreated with probe 4 or ZM241385 at the indicated concentration (IC50 or 0.3 fold IC50) for 3 h, followed by a three-cycle washing step to remove the noncovalently bound material. The membranes pretreated with probe 4 (Figure 3a) at increasing concentrations revealed a concomitant decline in specific [3H]ZM241385 binding, which was reduced from 65 ± 2% to 43 ± 2%. However, membranes pretreated with the reversible antagonist ZM241385 (Figure 3b) at increasing concentrations showed no decrease in specific [3H]ZM241385 binding, proving that the washing procedure was extensive enough to remove all noncovalently binding compound. Meanwhile, the affinity of unlabeled ZM241385 was not influenced significantly by the preincubation and washing procedure, indicating that the extensive washing did not damage the membrane integrity or alter the membrane binding sites (SI Table S2). Therefore, it could be concluded that the concentration-dependent decrease in specific [3H]ZM241385 binding observed with probe 4 resulted from an irreversible occupancy of the hA2A receptor binding pocket. Similar results have been obtained on other GPCRs, e.g., for the adenosine A1 receptor irreversible antagonist FSCPX29,30 and the covalent histamine H4 receptor partial agonist VUF14480,31 although these compounds lack the alkyne moiety to perform a click chemistry approach.
Figure 3.
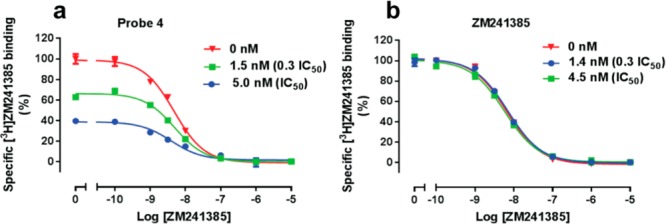
Probe 4 irreversibly binds to hA2AR HEK293 cell membranes stably expressing hA2AR, and they were preincubated with probe 4 (a) or ZM241385 (b) at the indicated comparable concentrations. Pretreated membranes were washed three times extensively before further displacement studies of specific [3H]ZM241385 binding from the hA2AR at 25 °C by nonlabeled ZM241385 were performed. Representative graphs are from three independent experiments performed in duplicate with error bars representing SEM values.
Fluorescent Labeling of the hA2AR
Having shown that the designed probe 4 meets the requirement of covalent binding, we then set out to evaluate its ability to function as an affinity-based probe. Purified hA2AR was first incubated with the alkyne-containing probe 4 to ensure formation of a covalent probe–hA2AR adduct. Then all samples were subjected to a copper(I)-catalyzed sulfonated cyanine 3-azide (Cy3-azide) attachment to the terminal alkyne.32,33 The subsequent fluorescence scanning of a SDS-PAGE showed that in the presence of fluorescent dye Cy3-azide (Figure 4a), probe 4 was concentration-dependently incorporated into a fixed amount of purified hA2AR, while in the absence of probe, little fluorescence intensity was detected. Importantly, Western blot analysis using the purified hA2AR receptor and specific antihistidine antibodies unambiguously validated that the labeling band was hA2AR (Figure 4a). Interestingly, a second band was observed in both affinity labeling results and Western blots, most likely resulting from posttranslationally modified receptors,34 as has been shown previously on CB2R.27 Quantification of the fluorescence intensity of the main labeling bands in the hA2AR is reflected in the concentration–effect curve in Figure 4b. This revealed that clickable probe 4 labeled hA2AR with a pEC50 value of 6.10 ± 0.04, resulting in a maximal labeling achieved with 10 μM probe 4 when incubated with 0.1 mg mL–1 of purified hA2AR. Collectively, these data demonstrate that probe 4 can be used as an affinity-based probe for purified hA2AR.
Figure 4.

Concentration-dependent affinity labeling of purified, His-tagged hA2AR by probe 4. (a) Purified hA2AR material was incubated with the indicated concentrations of probe 4 or vehicle (1% DMSO) and subjected to click chemistry ligation with Cy3-azide, followed by SDS-PAGE separation and in-gel fluorescence scanning (left). The blotted membranes were probed with antihistidine antibody, wherein bands corresponding to purified hA2AR molecular weight (∼47 kDa) were evident in all samples (right). (b) Quantification of fluorescence intensity from purified hA2AR labeled by probe 4 clicked to Cy3-azide. Representative graphs are from three independent experiments, with errors bars representing SEM values. In-gel fluorescence of the hA2AR band at ∼47 kDa was normalized to the corresponding hA2AR immunoreactivity in each sample.
To further characterize our affinity-based probe, we then investigated whether competitive antagonists could inhibit the labeling of purified receptors by probe 4. We chose to evaluate reversible antagonist ZM241385 and irreversible compound 1, at saturating concentrations (10 μM, i.e., 10 times higher than the concentration of the clickable probe 4). Purified hA2AR, preincubated with the competitors and subsequently treated as mentioned previously to incorporate the sulfonated cyanine 3 fluorophore, showed little if any fluorescence intensity of labeling bands under these conditions. This revealed that both a reversible and an irreversible antagonist competed with probe 4 (Figure 5a, left panel) for the same binding site at the hA2AR, which was available at identical amounts in all conditions (as evidenced by His-tagging: Figure 5a, right panel). Theoretically, both reversible and irreversible ligands inhibit affinity labeling, provided that they target the same receptor binding site and are present in a sufficient concentration. Of note, in practice, this is not always easily observed, as in the competition between reversible ligand and covalently binding probe there is an inherent bias toward the irreversible pathway, hindering the interaction between the receptor and a reversible ligand. For instance, in the few other studies where an AfBP has been used on GPCRs it was found that a reversible mGlu5 negative allosteric modulator, MPEP, could not inhibit the tandem photoaffinity labeling of purified mGlu5, whereas on CB2R, inhibition of labeling by various competitors was observed.26,27 Apparently, this was less of a problem on the hA2AR. Our results demonstrate that the developed AfBP system can serve as an effective chemical tool for profiling the purified hA2AR in vitro, prompting us to further evaluate the potency and selectivity of probe 4 in profiling the activity of the adenosine A2A receptor in more complex biological samples.
Figure 5.
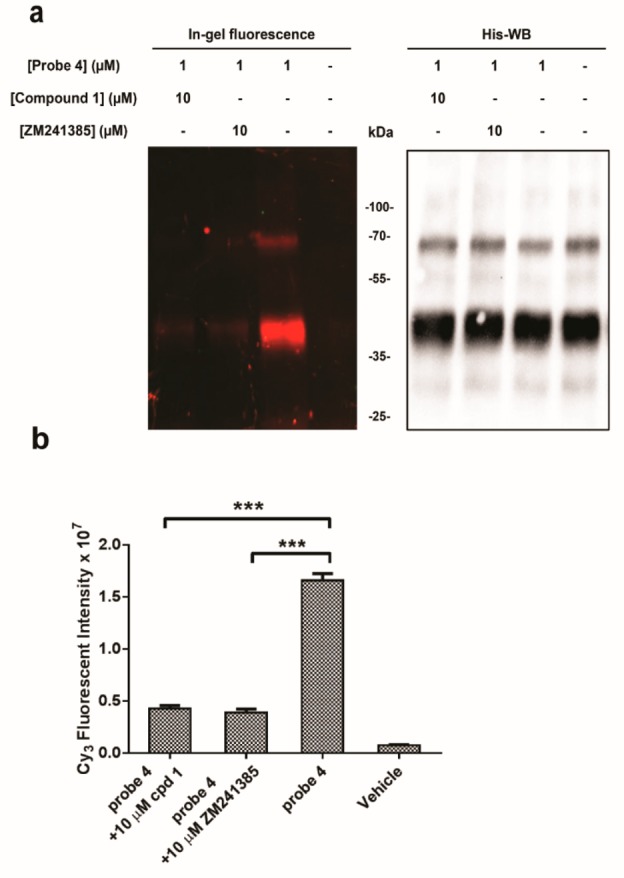
Competitive affinity labeling of the purified hA2AR by probe 4 (a) Affinity labeling of purified hA2AR by probe 4 (1 μM) is inhibited by preincubation with either compound 1 (10 μM) or ZM241385 (10 μM) (left). The blotted membranes were probed with antihistidine antibody, wherein bands corresponding to purified hA2AR molecular weight (∼47 kDa) were evident in all samples (right). (b) Quantification of fluorescence intensity from pretreated purified hA2AR labeled by probe 4 clicked to Cy3-azide. Representative graphs are from three independent experiments, with errors bars representing SEM values. ***P < 0.001 compared with the fluorescent intensity of purified hA2AR labeled by probe 4 (1 μM); Student’s t test. In-gel fluorescence of the hA2AR band at ∼47 kDa was normalized to the corresponding hA2AR immunoreactivity in each sample.
We further explored the ability of probe 4 to label hA2AR in cell membranes prepared from HEK293 cells, which were transiently transfected with N-terminally FLAG-tagged and C-terminally His-tagged human adenosine A2A receptors (FLAG-hA2AR-His). Therefore, FLAG-hA2AR-His cell membranes were incubated with probe 4 at room temperature for 1 h, followed by click ligation to Cy3-azide treatment. As detailed in Figure 6, a band corresponding to the molecular weight of the FLAG-hA2AR-His was observed upon fluorescent SDS-PAGE scanning, which was then validated by Western blot using specific anti-FLAG antibodies. In these initial proof-of-concept experiments we highlighted the versatility of probe 4, which can be efficiently used to label the adenosine A2A receptor in cell membrane samples.
Figure 6.
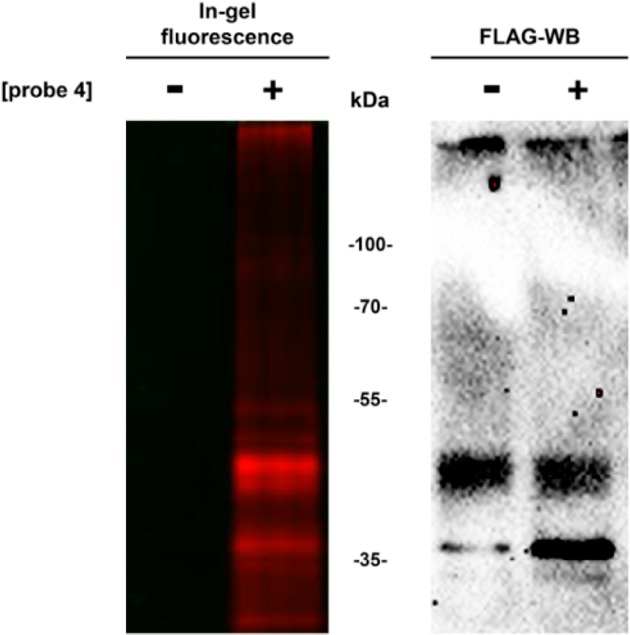
Affinity labeling of hA2AR in HEK293 cell membranes transiently expressing FLAG-tagged hA2AR using probe 4. (a) Cell membranes overexpressing FLAG-tagged hA2AR were incubated with either 1 μM probe 4 or vehicle (1% DMSO) and then subjected to click chemistry ligation with Cy3-azide, followed by SDS-PAGE separation and in-gel fluorescence scanning (left). The blotted membranes were probed with anti-FLAG antibody, wherein bands corresponding to the hA2AR molecular weight (∼50 kDa) are evident in all samples.
Background signals caused by nonspecific labeling of abundant proteins in the complex proteomes may sometimes confound the analysis of on-target labeling of low expression proteins such as GPCRs. Thus, we utilized cell membranes transiently transfected with FLAG-hA2AR-His, which have a relatively high level of receptor expression. Additionally, instead of premixing the copper sulfate and sodium ascorbate reagents, we slightly altered the click procedure by adding the copper sulfate last to achieve efficient and selective labeling of the A2A receptors.35,36 Although we were able to decrease the strong background signals, a significant nonspecific labeling was still observed. Several explanations may be put forward, such as a low efficiency of the click reaction between the fluorescent dyes and labeled receptors, nonspecific protein binding of the probe due to the inherently reactive warhead, and the sensitivity of the used detection method. Hence, further technological refinement should help us in achieving better labeling of endogenously expressed GPCRs, e.g., in human tissues as has been shown recently on CB2R.27 The monitoring of endogenous GPCR expression and target engagement in human cells holds promise for future GPCRs studies.
Conclusion
Starting from a selective antagonist, ZM241385, we designed and synthesized a series of covalent ligands using the electrophilic nature of sulfonyl fluorides, eventually yielding probe 4, the first affinity-based probe for the hA2AR. We successfully demonstrated a concentration-dependent labeling of purified receptor by probe 4 via an experimental two-step labeling strategy, which could be inhibited by both reversible and irreversible competing ligands. Additionally, probe 4 displayed target selectivity in cell membranes overexpressing the hA2AR, indicating that it may become a useful pharmacological tool to identify the hA2AR in living organisms for target validation or to assess receptor subtype distribution. In this strategy a probe depicts the native binding with less perturbation, which bridges the chemical biology study with molecular pharmacology to better investigate receptor–ligand interactions.
In future research, different tags may be introduced; for instance a biotin-tag would allow for streptavidin-mediated receptor enrichment followed by LC/MS analysis. Similarly, the approach developed in this study may be applied to other GPCRs, such as the other adenosine receptor subtypes.
Experimental Section
Chemistry
All solvents and reagents were purchased from commercial sources and were of analytical grade. 1H NMR spectra were recorded on a Bruker AV 400 liquid spectrometer (1H NMR, 400 MHz) at ambient temperature. Chemical shifts are reported in parts per million (ppm) and are designated by δ. Coupling-constants are reported in hertz (Hz) and are designated as J. Analytical purity of the final compounds was determined by high pressure liquid chromatography (HPLC) with a Phenomenex Gemini 3 μ C18 110A column (50 × 4.6 mm, 3 μm), measuring UV absorbance at 254 nm. Sample preparation and HPLC method was as follows: 0.5 mg of compound was dissolved in 1 mL of a 1:1:1 mixture of CH3CN/H2O/tBuOH and eluted from the column within 15 min, with a three-component system of H2O/CH3CN/1% TFA in H2O, decreasing polarity of the solvent mixture in time from 80/10/10 to 0/90/10. All compounds showed a single peak at the designated retention time and are at least 95% pure. Liquid chromatography–mass spectrometry (LC–MS) analyses were performed using Thermo Finnigan Surveyor – LCQ Advantage Max LC-MS system and a Gemini C18 Phenomenex column (50 × 4.6 mm, 3 μm). The sample preparation was the same as for HPLC analysis. The elution method was set up as follows: 1–4 min isocratic system of H2O/CH3CN/1% TFA in H2O, 80:10:10; from the fourth min, a gradient was applied from 80:10:10 to 0:90:10 within 9 min, followed by 1 min of equilibration at 0:90:10 and 1 min at 80:10:10. Thin-layer chromatography (TLC) was routinely performed to monitor the progress of reactions, using aluminum-coated Merck silica gel F254 plates. Purification by column chromatography was achieved by use of Grace Davison Davisil silica column material (LC60A 30–200 μm). Solutions were concentrated using a Heidolph Laborota W8 2000 efficient rotary evaporation apparatus and by a high vacuum on a Binder APT line vacuum drying oven.
4-((4-((7-Amino-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-5-yl)amino)butyl)carbamoyl)benzenesulfonyl Fluoride (2)
Previously synthesized N5-(4-aminobutyl)-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazine-5,7-diamine 8 (TFA salt, 250 mg, 0.40 mmol, 1.0 equiv) was suspended in acetonitrile (10 mL) and purged with N2. Then DiPEA (0.42 mL, 2.4 mmol, 6.0 equiv) was added after which 4-fluorosulfonylbenzoyl chloride (134 mg, 0.60 mmol, 1.5 equiv) was added last and the mixture was heated to 70 °C for 7 h and then stirred at room temperature for another 17 h. A flash column (MTBE + 1% AcOH → 90% MTBE + 10% EtOAc + 1% AcOH), a subsequent preparative TLC (1:1 MTBE:EtOAc + 1% MeOH), and an extraction using acetonitrile (10 mL) and petroleum ether (4 × 10 mL) afforded the product as a white solid (8 mg, 0.017 mmol, 4% yield). 1H NMR (DMSO-d6, 400 MHz): δ 8.29–8.18 (m, 5H), 7.75 (s, 1H), 7.40 (br s, 2H), 7.05 (d, J = 3.2 Hz, 1H), 6.73 (m, 1H), 6.62 (s, 1H), 3.55–3.46 (m, 4H), 1.75–1.74 (m, 4H). HPLC: 96.5%, RT 7.478 min. LC-MS: [ESI + H]+: 475.20
4-((5-((7-Amino-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-5-yl)amino)pentyl)carbamoyl)benzenesulfonyl Fluoride (3)
N5-(5-Aminopentyl)-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazine-5,7-diamine 9 (TFA salt, 674 mg, 0.85 mmol, 1 equiv) was suspended in acetonitrile (5 mL) . 4-Fluorosulfonylbenozyl chloride (208 mg, 0.94 mmol, 1.1 equiv) was added, along with DiPEA (0.8 mL, 5 mmol, 5.8 equiv). The mixture was heated at 70 °C under N2 atmosphere for 2.5 h. A flash column (DCM → 60% DCM, 40% EtOAc) with subsequent preparative TLC (100% EtOAc) was used to obtain the title compound as a colorless solid (9 mg, 0.018 mmol, 2% yield). 1H NMR (C3D6O, 400 MHz): δ 8.27–8.16 (m, 4H), 7.73 (dd, J = 1.7, 0.8 Hz, 1H), 7.39 (s, 2H), 7.05 (d, J = 3.2 Hz, 1H), 6.80–6.63 (m, 1H), 6.62 (dd, J = 3.4, 1.8 Hz, 1H), 3.52–3.38 (m, 4H), 1.78–1.62 (m, 4H), 1.56–1.44 (m, 2H). HPLC: 100%, RT 7.637 min, LC-MS:[ESI + H]+: 489.00
4-((5-((7-Amino-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-5-yl)amino)pentyl)(prop-2-yn-1-yl)carbamoyl)benzenesulfonyl Fluoride (4)
tert-Butyl (5-(4-(fluorosulfonyl)-N-(prop-2-yn-1-yl)benzamido)pentyl)carbamate 14 (586 mg, 1.38 mmol, 1 equiv) was dissolved in DCM (10 mL). To this solution was added TFA (10 mL). After 2 min, the solvents were removed in vacuo. This crude intermediate was suspended in acetonitrile (10 mL), and 2-(furan-2-yl)-5-(methylsulfonyl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-7-amine 5 (386 mg, 1.38 mmol, 1 equiv) was added, along with DiPEA (2 mL, 11.0 mmol, 8 equiv). The reaction mixture was heated at 70 °C for 2 h. Then the reaction mixture was concentrated and purified by a flash column (EtOAc) to yield a yellow solid (330 mg, 0.62 mmol, 45%). 1H NMR (DMSO-d6, 353 K, 400 MHz,) δ 8.17 (d, J = 8.3 Hz, 2H), 7.93–7.73 (m, 5H), 7.12 (t, J = 5.3 Hz, 1H), 7.03 (d, J = 3.3 Hz, 1H), 6.64 (dd, J = 3.0, 1.5 Hz, 1H), 4.17 (s, 2H), 3.41 (s, 2H), 3.29 (d, J = 6.2 Hz, 2H), 3.14 (s, 1H), 1.73–1.61 (m, 2H), 1.61–1.44 (m, 2H), 1.40–1.23 (m, 2H) ppm. HPLC: 95.772%, RT: 8.117 min, MS: [ESI + H]+: 527.20
tert-Butyl (4-((7-Amino-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-5-yl)amino)butyl)carbamate (6)
2-(Furan-2-yl)-5-(methylsulfonyl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-7-amine 5 (435 mg, 1.55 mmol, 1.0 equiv), synthesized as previously reported,13 was suspended in acetonitrile to yield a 0.1 M solution. tert-Butyl (4-aminopropyl)carbamate (0.33 mL, 1.71 mmol, 1.1 equiv) was added, followed by the addition of N,N-diisopropylethylamine (1.08 mL, 6.21 mmol, 4 equiv). The mixture was heated at 85 °C for 29 h and stirred at rt for another 18 h. A flash column (DCM:EtOAc, 0% → 90% EtOAc) was used to purify the crude mixture. This gave a yellowish solid (444 mg, 1.14 mmol, 74% yield). 1H NMR (DMSO-d6, 400 MHz): δ 8.49–7.92 (m, 2H), 7.86 (s, 1H), 7.51 (t, J = 5.9 Hz, rotamer 1, 0.3H), 7.44 (t, J = 5.7 Hz, rotamer 2, 0.7H), 7.07–7.00 (m, 1H), 6.87–6.76 (m, 1H), 6.67 (dd, J = 3.2, 1.7 Hz, 1H), 3.29–3.19 (m, 2H), 2.92 (d, J = 6.5 Hz, 2H), 1.48 (d, J = 7.2 Hz, 2H), 1.44–1.29 (m, 11H).
tert-Butyl (5-((7-Amino-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-5-yl)amino)pentyl)carbamate (7)
2-(Furan-2-yl)-5-(methylsulfonyl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-7-amine 5 (280 mg, 1.0 mmol, 1.0 equiv) and commercially available tert-butyl (5-aminopentyl)carbamate (0.2 mL, 1.0 mmol, 1.1 equiv) were put in a microwave tube and dissolved in acetonitrile (1.5 mL). DIPEA (0.3 mL, 1.7 mmol) was added, and the tube was charged with a stirring bar, sealed, and heated at 70 °C for 1.5 h. After 1.5 h, HPLC analysis indicated full conversion. The mixture was concentrated, and EtOAc (50 mL) and HCl (1 M in H2O, 50 mL) were added for extraction. The organic layer was washed with H2O (50 mL) and brine (50 mL). After drying over MgSO4, the solvent was removed in vacuo to give the title compound as a yellow foam (186 mg, 0.46 mmol, 46% yield). 1H NMR (DMSO-d6, 400 MHz,) δ 8.48–7.96 (m, 2H), 7.86 (s, 1H), 7.48 (t, J = 5.1 Hz, rotamer, 0.38H), 7.41 (t, J = 5.7 Hz, rotamer, 0.62H), 7.10–7.01 (m, 1H), 6.77 (t, J = 5.0 Hz, 1H), 6.67 (dd, J = 3.0, 1.7 Hz, 1H), 3.28–3.17 (m, 2H), 2.90 (d, J = 6.6 Hz, 2H), 1.57–1.44 (m, 2H), 1.44–1.21 (m, 13H).
N5-(4-Aminobutyl)-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazine-5,7-diamine (8)
TFA (4.3 mL, 57 mmol, 50 equiv) was added to the suspension of Boc-protected amine 6 (444 mg, 1.14 mmol, 1 equiv) in DCM (equal volume as TFA). Solvents were removed under reduced pressure after completion of the reaction (5 min). This gave the product as brown oil (899 mg, 1.13 mmol, quantitative yield). Products were confirmed by 1H NMR first and then stored under N2 until use. 1H NMR (DMSO-d6, 400 MHz): δ 8.49–8.05 (m, 3H (R-NH3+), 7.88 (dd, J = 1.7, 0.7 Hz, 1H), 7.62 (br s, 3H (R-NH3+)), 7.52 (t, J = 6.0 Hz, 1H), 7.09–7.02 (m, 1H), 6.68 (dd, J = 3.4, 1.7 Hz, 1H), 3.35–3.23 (m, 2H), 2.87–2.76 (m, 2H), 1.63–1.50 (m, 4H).
N5-(5-Aminopentyl)-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazine-5,7-diamine (9)
TFA (3 mL, 40 mmol, 50 equiv) was added to the suspension of Boc-protected amine 7 (324 mg, 0.8 mmol, 1 equiv) in DCM. Once the reaction was completed, the solvent was removed and the mixture was coevaporated twice with water and dried using high vacuum. This gave a brown oil (556 mg, 0.8 mmol, quantitative yield) as a TFA salt. The crude product was used without further purification.
tert-Butyl (5-Hydroxypentyl)carbamate (11)
5-Amino-1-pentanol 10 (4.2 mL, 38.8 mmol) was dissolved in DCM (20 mL). Di-tert-butyl dicarbonate (8.4 g, 38.8 mmol) was slowly added as a solid. The reaction was left stirring at rt for 18 h, and then the solvent was removed to give a yellow oil (8.83 g, quantitative yield, some t-BuOH left). 1H NMR (CDCl3, 400 MHz) δ 4.57 (s, 1H), 3.65 (t, J = 6.5 Hz, 2H), 3.13 (t, J = 6.5 Hz, 2H), 1.67–1.35 (m, 15H (under water peak)).
tert-Butyl (5-Bromopentyl)carbamate (12)
tert-Butyl (5-hydroxypentyl)carbamate 11 (8.83 g, 38.8 mmol, 1eq) and PPh3 (15.3 g, 58.2 mmol, 1.5 equiv) were dissolved in THF (120 mL). A solution of CBr4 (19.3 g, 58.2 mmol, 1.5 equiv) in THF (40 mL) was added over 2 h using a syringe pump. After 3 h at room temperature, the reaction mixture was filtered and the filtrate was concentrated. This crude product was dissolved in DCM (∼5 mL) and purified by flash column chromatography (100% PE → 90% PE + 10% EtOAc). This gave the product as a colorless oil (9.31 g, 35.0 mmol, 90% yield). 1H NMR (400 MHz, CDCl3) δ 4.54 (s, 1H), 3.41 (t, J = 6.7 Hz, 2H), 3.13 (d, J = 5.9 Hz, 2H), 1.97–1.80 (m, 2H), 1.58–1.36 (m, 13H) ppm. 13C NMR (101 MHz, CDCl3) δ 40.5, 33.8, 32.5, 29.4, 28.6, 25.5.
tert-Butyl (5-(Prop-2-yn-1-ylamino)pentyl)carbamate (13)
Propargylamine (1 mL, 15 mmol, 3 equiv) was dissolved in acetonitrile (10 mL). To this stirred solution was added a solution of tert-butyl (5-bromopentyl)carbamate 12 (798 mg, 3 mmol, 1 equiv) and DiPEA (1 mL, 6 mmol, 2 equiv) in acetonitrile (18 mL) using a syringe pump. Afterward the solvent was removed and the product purified by flash column chromatography (EtOAc). This gave a yellowish oil (331 mg, 1.38 mmol, 46% yield) with EtOAc as an impurity. 1H NMR (CDCl3, 400 MHz) δ 4.54 (s, 1H), 3.44 (d, J = 2.0 Hz, 2H), 3.12 (q, J = 6.4 Hz, 2H), 2.70 (t, J = 7.1 Hz, 2H), 2.22 (t, J = 2.2 Hz, 1H), 1.55–1.34 (m, 15H) ppm.
tert-Butyl (5-(4-(Fluorosulfonyl)-N-(prop-2-yn-1-yl)benzamido)pentyl)carbamate (14)
tert-Butyl (5-(prop-2-yn-1-ylamino)pentyl)carbamate 13 (664 mg, 1.38 mmol, 1 equiv) was dissolved in acetonitrile (10 mL), and 4-fluorosulfonyl benzoyl chloride (338 mg, 1.52 mmol, 1.1 equiv) was added and followed by the addition of DiPEA (0.75 mL, 4.14 mmol, 3 equiv). Once the reaction was completed, the solvent was removed and the crude mixture purified by flash column chromatography (DCM + 5% MTBE → DCM + 7.5% MTBE). This yielded a yellow oil (586 mg, 1.52 mmol, quantitative yield). 1H NMR (DMSO-d6, 332 K, 400 MHz) δ 8.19 (d, J = 8.0 Hz, 2H), 7.77 (d, J = 7.9 Hz, 2H), 6.37 (s, 1H), 4.16 (s, 2H), 3.37 (s, 2H), 3.15 (s, 1H), 2.90 (s, 2H), 1.61 (s, 2H), 1.44–1.29 (m, 9H), 1.29–1.14 (m, 4H), 1.11 (d, J = 16.6 Hz, 2H).
Biology
The radioligand [3H]ZM241385 with a specific activity of 50 Ci mmol–1 was purchased from ARC Inc. (St. Louis, MO). Unlabeled ZM241385 was a kind gift from Dr. S. M. Poucher (Astra Zeneca, Macclesfield, UK). 5′-N-Ethylcarboxamidoadenosine (NECA) was purchased from Sigma-Aldrich (Steinheim, Germany). Adenosine deaminase (ADA) was purchased from Sigma-Aldrich Chemie N.V. Bicinchoninic acid (BCA) and BCA protein assay reagent were obtained from Pierce Chemical Company (Rockford, IL). Human embryonic kidney (HEK) 293 cells stably expressing the hA2A receptor (hA2AR-WT) were kindly provided by Dr. J. Wang (Biogen/IDEC, Cambridge, MA). The purified hA2A receptor material was kindly provided by Dr. Niek Dekker and Dr. Euan Gordon (AstraZeneca). All other chemicals were of analytical grade and obtained from standard commercial sources.
Cell Culture, Transfection, and Membrane Preparation
We followed the procedures reported previously.13,37 Briefly, HEK293 cells were grown as monolayers in Dulbecco’s modified Eagle’s medium supplemented with 2 mM glutamine, 10% newborn calf serum, 50 μg mL–1 streptomycin, and 50 IU mL–1 penicillin at 37 °C and 7% CO2 atmosphere. Cells were subcultured twice a week at a ratio of 1:20 on 10 cm ⌀ culture plates. The cells were transfected with pcDNA3.1(−) plasmid containing the hA2AR with N-terminal FLAG and C-terminal His tags (FLAG-hA2AR-His4) using the calcium phosphate precipitation method (1 μg of plasmid DNA), followed by a 48-h incubation, as previously described.38 Stably transfected hA2AR-WT cells were grown in the same medium but with the addition of G-418 (500 mg mL–1). Both transiently transfected cells and stably transfected hA2AR-WT cells were detached from the plates by scraping them into PBS and centrifuged to remove PBS buffer. The pellets were resuspended in ice-cold Tris-HCl buffer (50 mM, pH 7.4) and then homogenized. The cell membrane suspensions were centrifuged at 100 000g at 4 °C for 20 min, after which the procedure was repeated one more time. After this, the same Tris-HCl buffer was used to resuspend the pellet, and adenosine deaminase was added to break down endogenous adenosine. HEK293 cells stably expressing hA2AR were grown as monolayers in the same culture medium and detached from plates by the same treatment for membrane preparation. Both membranes were stored in 250 μL aliquots at −80 °C until further use. Membrane protein concentrations were measured using the BCA method.39
[3H]ZM241383 Radioligand Displacement Assay
Radioligand displacement experiments were performed as previously described.13 hA2AR-WT cell membrane aliquots containing 10 μg of protein were incubated in a total volume of 100 μL of assay buffer to obtain an assay window of approximately 3000 DPM of receptor-specific radioligand binding. Nonspecific binding was determined in the presence of 100 μM NECA and represented less than 10% of the total binding. Briefly, to each tube were added 25 μL of cell membranes (10 μg of protein), 25 μL of radioligand [3H]ZM241383, 25 μL of assay buffer [25 mM Tris-HCl, pH 7.4 at 25 °C, supplemented with 5 mM MgCl2 and 0.1% (w/v) CHAPS], and 25 μL of the indicated compounds in increasing concentrations in the same assay buffer. The mixture was incubated at 25 °C for 60 min to reach equilibrium. Incubations were terminated by rapid vacuum filtration to separate the bound and free radioligand through 96-well GF/B filter plates using a PerkinElmer Filtermate-harvester (PerkinElmer, Groningen, Netherlands). Filters were subsequently washed three times with 2 mL of ice-cold buffer (25 mM Tris-HCl, pH 7.4, supplemented with 5 mM MgCl2). The filter-bound radioactivity was determined by scintillation spectrometry using a P-E 1450 Microbeta Wallac Trilux scintillation counter (PerkinElmer).
Heterologous Displacement Binding of Probe 4 and ZM241385 to hA2AR-WT Cell Membranes
To assess the irreversible binding level, cell membranes stably expressing hA2AR were incubated with either 50 mM Tris-HCl (pH = 7.4) or two concentrations (0.3 IC50 and IC50) of probe 4 or ZM241385 for 3 h at 25 °C on an Eppendorf Thermomixer. Subsequently, the mixture was centrifuged at 16 100g at 4 °C for 5 min, and the supernatant was removed, followed by a resuspension of the pellet in 1 mL of assay buffer and spun again for 5 min at 16 100g at 4 °C. This washing procedure was repeated three times. The 50 μL aliquots of these pretreated membranes were incubated with 25 μL of radioligand [3H]ZM241383 and 25 μL of a concentration range (100 pM to 1 μM) of unlabeled ZM241385 for 1 h at 25 °C. Incubation was terminated as described under [3H]ZM241385 radioligand displacement assay.
Expression and Purification of Wild-Type hA2AR
The gene coding for hA2AR (residues 1–316) was synthesized by Genscript and cloned into pPICZb with an N-terminal α-factor signal sequence from Saccharomyces cerevisiae (MRFPSIFTAVLFAASSLAAPVNTTEDETAQIPAAVIGYSDLEDFDVAVLPSNSTNNGLLINTTIASIAAEEGVSLERLVPRGS), followed by hA2AR and a C-terminus biotinylation domain from Propionibacterium shermanii (TSEFENLYQGQFGGGTG APAPAAGGAGGKAGEGEIPALAGTVSKILVEGDTVKAGQVLVLEAMKMEEINA PTDGKVEKVLKERDAVQGQGLIKI) for enhanced expression40 and a decaHis tag (GHHHHHHHHHGS).
The receptor was expressed in Pichia pastoris SMD1168 at 3 L scale in a fermentor essentially as described,41 except that dissolved oxygen was maintained at 25%, and 2.5% DMSO and 10 mM theophylline were included in the fermentation media. Approximately 200 g of wet cells were harvested per liter. Cells (200 g) were resuspended using a Turax in 600 mL ice-cold lysis buffer (50 mM HEPES pH 7.4, 200 mM NaCl, Complete EDTA free protease inhibitor tablets (Roche) at 1/50 mL). Cells were lysed by a single passage through a Constant Cell system at 30 kpsi with extensive cooling. Cell debris was removed by centrifugation at 1000g for 10 min at 4 °C. Membranes were collected by ultracentrifugation at 100 000g for 45 min at 4 °C. Membrane pellet was resuspended in buffer to a total protein concentration of 20 mg mL–1 (final volume of 180 mL) and stored at −80 °C.
Membranes (20 mL) were resuspended in 200 mL of solubilization buffer (25 mM HEPES, pH7.4, 300 mM NaCl, 20% glycerol, 1% DDM/0.1% CHS, Complete tablets (1/50 mL), 200 μM theophylline). The suspension was incubated for 2 h at 4 °C on a rolling table, prior to centrifugation for 30 min at 100 000g to remove unsolubilized material. Imidazole was added to a final concentration of 15 mM, and the clarified solution was loaded on a 5 mL HisTrap crude column at 2.5 mL min–1. The column was washed with 100 mL buffer A (25 mM HEPES, 25 mM imidazole pH 7.4, 300 mM NaCl, 10% glycerol, 0.05% DDM/0.0005% CHS, 100 μM theophylline) to which imidazole was added to final concentration of 25 mM to reduce nonspecific binding, followed by stepwise washes with increasing concentrations of imidazole in this buffer (50 mM and 75 mM), and hA2AR was eluted in 25 mM HEPES pH 7.4, 300 mM NaCl, 10% glycerol, 0.05% DDM/0.0005%CHS, 300 mM imidazole, 100 μM theophylline. Fractions were analyzed on SDS-PAGE, and those containing hA2AR were pooled and concentrated to 2.5 mL using a 50 kDa filter. High concentrations of imidazole are harmful to hA2AR, and the buffer was changed to buffer A on a PD10 G25 column. The eluted fraction was further concentrated to 0.5 mL and loaded on a Superdex-200 10/30 column running in 25 mM NaPi pH 7.2, 100 mM NaCl, 10 μM LMNG, 500 μM caffeine. Fractions were analyzed on SDS-PAGE. hA2AR eluted as single peak at expected position for the detergent–protein complex (around 80 kDa). Fractions were pooled and concentrated on a 50 kDa filter to final volume of 0.4 mL and stored at −80 °C. Protein concentration was determined using absorbance measurement against buffer A (Abs280(0.1%) = 1.05). Final concentration was 7 mg mL–1 with a total of ∼2 mg hA2AR.
Affinity-Based Protein Labeling Assay on Purified hA2AR with Probe 4
For purified hA2AR, both affinity labeling and click reactions were performed on ice, unless indicated otherwise. Purified hA2AR was diluted to a concentration of 0.1 mg mL–1 in assay buffer (25 mM HEPES pH 7.5, 100 mM NaCl, and 10 μM LMNG). The 38 μL samples were incubated with 2 μL of probe 4 at indicated concentrations or vehicle control (1% DMSO) for 1 h. To initiate the click reaction, 5.6 mM CuSO4 (2.5 μL/reaction, from a 100 mM stock solution in water) was mixed vigorously with 33 mM sodium ascorbate (1.5 μL/reaction, freshly made as a 1 M stock solution in water) to obtain a yellow mixture, followed by the immediate addition of 1.1 mM THPTA (0.5 μL/reaction, from a 100 mM stock solution in water) and 4.4 μM fluorescent tag Cy3-azide (0.5 μL/reaction, from a 400 μM stock solution in DMSO). The reaction mixtures were incubated for 1 h and quenched with 15 μL 4×SDS loading buffer. Proteins in the mixture were separated by SDS-PAGE on 10% polyacrylamide gels. In-gel fluorescence was detected with a ChemiDoc MP system (605/50 filter). Proteins were transferred from gel to a PVDF membrane by Trans-BlotTurbo (BioRad). Then the membrane was washed in 20 mL of TBS for 10 min on a roller bench, followed by a three times wash with TBST (PBS with 0.1% Tween-20). Afterward, the membrane was blocked in 5% (w/v) nonfat milk for 1 h at room temperature and probed with rabbit-anti-His antibody (Rockland)(1:1000 [v/v] dilution in blocking buffer) overnight at 4 °C, washed three times again with TBST, and incubated with goat-antirabbit IgG-HRP (1:5000 in 5% milk in TBST; Santa Cruz) for 1 h at room temperature. After two wash cycles in TBST and one in TBS, the blot was developed in the dark using a 10 mL luminal solution, with 100 μL of ECL enhancer and 3 μL of H2O2. Chemiluminescence was visualized with ChemiDoc XRS (BioRad).
Competitive Labeling Assays in Purified hA2AR by Probe 4
Prior to the two-step labeling experiment, purified hA2AR was diluted to a concentration of 0.1 mg mL–1 in assay buffer and incubated with 10 μM compound 1, ZM241385, or vehicle control (1% DMSO) for 1 h on ice, followed by labeling with 1 μM probe 4 for 0.5 h on ice. Samples were then subjected to the click chemistry procedure using the protocol described above.
Affinity-Based Protein Labeling of Membranes Transiently Overexpressing FLAG-hA2AR-His
FLAG-hA2AR-His membranes were diluted to a concentration of 1 mg mL–1 in 50 mM Tris-HCl (pH = 7.4 at 25 °C). Either 2 μL of probe 4 at indicated concentrations (0.1 μM, 0.3 μM, 1 μM, and 3 μM) or vehicle control (1% DMSO) was added to 38 μL samples for 1 h incubation at room temperature. Then all samples were subjected to the click chemistry conjugation reaction. The click reagents were added in the following sequence: 4.4 μM fluorescent Cy3-azide (0.5 μL/reaction, 400 μM stock in DMSO) was added to the mixture followed by 33 mM sodium ascorbate (1.5 μL/reaction, freshly made in 1 M stock in water) and 1.1 mM THPTA (0.5 μL/reaction, 100 mM stock in water). Finally, 5.6 mM CuSO4 (2.5 μL/reaction, 100 mM stock in water) was added to start and run the cycloaddition reaction for 1 h at room temperature. Then the reaction was quenched with 15 μL 4×SDS loading buffer and protein material denatured for 30 min at 37 °C. Proteins (60 μL sample) were separated by SDS-PAGE on 10% polyacrylamide gels. In-gel fluorescence was detected with the ChemiDoc MP system (605/50 filter). Proteins were transferred from gel to a PVDF membrane by Trans-BlotTurbo (BioRad). Then the membrane was washed in 20 mL of TBS for 10 min on a roller bench, followed by a three times wash with TBST (PBS with 0.1% Tween-20). Then the membrane was blocked in 5% (w/v) nonfat milk and incubated with mouse-anti-FLAG (Sigma) (1:5000 [v/v] dilution in blocking buffer) as primary antibody. Thereafter, the membrane was washed in TBST three times and incubated with goat-antimouse HRP (Sigma) (1:5000 [v/v] dilution in blocking buffer) as secondary antibody. After two wash cycles in TBST and one in TBS, the blot was developed in the dark using a 10 mL luminal solution, with 100 μL of ECL enhancer and 3 μL of H2O2. Chemiluminescence was imaged using a ChemiDoc XRS (BioRad).
Acknowledgments
Dr. Julien Louvel who has recently passed away is acknowledged for his conceptual contribution regarding this study and his dedication to the medicinal chemistry in our research group. We are grateful to Dr. Hui Deng for helpful discussions and technical assistance. We also thank Dr. R. Liu for determining the selectivity profile of compound 4. Xue Yang is supported by the Chinese Scholarship Council (CSC).
Glossary
Abbreviations Used
- ADA
adenosine deaminase
- BCA
bicinchoninic acid
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
- Cy3-azide
sulfonated cyanine 3 dye azide
- DiPEA
diisopropylethylamine
- ECL
enhanced chemiluminescence
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- LMNG
lauryl maltose neopentyl glycol
- MTBE
methyl tert-butyl ether
- NaPi
sodium phosphate buffer
- NECA
5′-N-ethylcarboxamidoadenosine
- TBS
Tris-buffered saline
- TBST
Tris-buffered saline with 0.05% Tween
- THPTA
tris(3-hydroxypropyl triazolylmethyl)amine
- TFA
trifluoroacetic acid
- PVDF
polyvinylidene difluoride
- ZM241385
4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5-ylamino]ethyl)phenol
Supporting Information Available
. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b00860.
Author Contributions
X.Y. performed the radioligand binding assay, SDS-gel based assay, and data analysis. T.J.M.M. and C.D.J. synthesized compounds for this study. M.S. and M.V.D.S. did the early affinity-based labeling assay optimization. All the work mentioned above was performed under the supervision of M.V.D.S., L.H.H., D.V.D.E., and A.P.I. N.D. designed and performed adenosine A2A receptor purifications. E.G. performed the receptor expression and purifications. M.V.D.S., L.H.H., D.V.D.E., and A.P.I. contributed to the experimental design, results and discussion, and assay optimizations. A.P.I. initiated the project and conceptualized this study. X.Y., N.D., L.H.H., D.V.D.E., and A.P.I. wrote the original draft of the manuscript with contributions from all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Fredholm B. B.; IJzerman A. P.; Jacobson K. A.; Linden J.; Muller C. E. International union of basic and clinical pharmacology. LXXXI. nomenclature and classification of adenosine receptors-an update. Pharmacol. Res. 2011, 63, 1–34. 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzschild M. A.; Agnati L.; Fuxe K.; Chen J. F.; Morelli M. Targeting adenosine A2A receptors in Parkinson’s disease. Trends Neurosci. 2006, 29, 647–654. 10.1016/j.tins.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Adams J. L.; Smothers J.; Srinivasan R.; Hoos A. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discovery 2015, 14, 603–622. 10.1038/nrd4596. [DOI] [PubMed] [Google Scholar]
- Jaakola V. P.; Griffith M. T.; Hanson M. A.; Cherezov V.; Chien E. Y.; Lane J. R.; IJzerman A. P.; Stevens R. C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217. 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum D. M.; Rasmussen S. G. F.; Kobilka B. K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glukhova A.; Thal D. M.; Nguyen A. T.; Vecchio E. A.; Jorg M.; Scammells P. J.; May L. T.; Sexton P. M.; Christopoulos A. Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 2017, 168, 867–877. 10.1016/j.cell.2017.01.042. [DOI] [PubMed] [Google Scholar]
- Weichert D.; Gmeiner P. Covalent molecular probes for class A G protein-coupled receptors: advances and applications. ACS Chem. Biol. 2015, 10, 1376–1386. 10.1021/acschembio.5b00070. [DOI] [PubMed] [Google Scholar]
- Jorg M.; Scammells P. J. Guidelines for the synthesis of small-molecule irreversible probes targeting G protein-coupled receptors. ChemMedChem 2016, 11, 1488–1498. 10.1002/cmdc.201600066. [DOI] [PubMed] [Google Scholar]
- Narayanan A.; Jones L. H. Sulfonyl fluorides as privileged warheads in chemical biology. Chem. Sci. 2015, 6, 2650–2659. 10.1039/C5SC00408J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas M.; Shryock J. C.; Scammells P. J.; Ruble J.; Baker S. P.; Belardinelli L. A novel irreversible antagonist of the A1 adenosine receptor. Mol. Pharmacol. 1996, 50, 196–205. [PubMed] [Google Scholar]
- Shryock J. C.; Snowdy S.; Baraldi P. G.; Cacciari B.; Spalluto G.; Monopoli A.; Ongini E.; Baker S. P.; Belardinelli L. A2A-Adenosine receptor reserve for coronary vasodilation. Circulation 1998, 98, 711–718. 10.1161/01.CIR.98.7.711. [DOI] [PubMed] [Google Scholar]
- Baraldi P. G.; Cacciari B.; Moro S.; Romagnoli R.; Ji X.; Jacobson K. A.; Gessi S.; Borea P. A.; Spalluto G. Fluorosulfonyl- and bis-(β-chloroethyl)amino-phenylamino functionalized pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives: Irreversible antagonists at the human A3 adenosine receptor and molecular modeling studies. J. Med. Chem. 2001, 44, 2735–2742. 10.1021/jm010818a. [DOI] [PubMed] [Google Scholar]
- Yang X.; Dong G.; Michiels T. J. M.; Lenselink E. B.; Heitman L.; Louvel J.; IJzerman A. P. A covalent antagonist for the human adenosine A2A receptor. Purinergic Signalling 2017, 13, 191–201. 10.1007/s11302-016-9549-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K. A.; Ukena D.; Padgett W.; Kirk K. L.; Daly J. W. Molecular probes for extracellular adenosine receptors. Biochem. Pharmacol. 1987, 36, 1697–1707. 10.1016/0006-2952(87)90056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kecskes A.; Tosh D. K.; Wei Q.; Gao Z. G.; Jacobson K. A. GPCR ligand dendrimer (GLiDe) conjugates: Adenosine receptor interactions of a series of multivalent xanthine antagonists. Bioconjugate Chem. 2011, 22, 1115–1127. 10.1021/bc1005812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macchia M.; Salvetti F.; Bertini S.; Di Bussolo V.; Gattuso L.; Gesi M.; Hamdan M.; Klotz K. N.; Laragione T.; Lucacchini A.; Minutolo F.; Nencetti S.; Papi C.; Tuscano D.; Martini C. 7-Nitrobenzofurazan (NBD) derivatives of 5′-N-ethylcarboxamidoadenosine (NECA) as new fluorescent probes for human A3 adenosine receptors. Bioorg. Med. Chem. Lett. 2001, 11, 3023–3026. 10.1016/S0960-894X(01)00610-2. [DOI] [PubMed] [Google Scholar]
- Blair J. A.; Rauh D.; Kung C.; Yun C. H.; Fan Q. W.; Rode H.; Zhang C.; Eck M. J.; Weiss W. A.; Shokat K. M. Structure-guided development of affinity probes for tyrosine kinases using chemical genetics. Nat. Chem. Biol. 2007, 3, 229–238. 10.1038/nchembio866. [DOI] [PubMed] [Google Scholar]
- Speers A. E.; Adam G. C.; Cravatt B. F. Activity-based protein profiling in vivo using a copper(I)-catalyzed azide–alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003, 125, 4686–4687. 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- Kolb H. C.; Sharpless K. B. The growing impact of click chemistry on drug discovery. Drug Discovery Today 2003, 8, 1128–1137. 10.1016/S1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- Speers A. E.; Cravatt B. F. Profiling enzyme activities in vivo using click chemistry methods. Chem. Biol. 2004, 11, 535–546. 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Blex C.; Michaelis S.; Schrey A. K.; Furkert J.; Eichhorst J.; Bartho K.; Gyapon Quast F.; Marais A.; Hakelberg M.; Gruber U.; Niquet S.; Popp O.; Kroll F.; Sefkow M.; Schulein R.; Dreger M.; Koster H. Targeting G protein-coupled receptors by capture compound mass spectrometry: A case study with sertindole. ChemBioChem 2017, 18, 1639–1649. 10.1002/cbic.201700152. [DOI] [PubMed] [Google Scholar]
- Grunbeck A.; Sakmar T. P. Probing G protein-coupled receptor-ligand interactions with targeted photoactivatable cross-linkers. Biochemistry 2013, 52, 8625–8632. 10.1021/bi401300y. [DOI] [PubMed] [Google Scholar]
- Burgermeister W.; Nassal M.; Wieland T.; Helmreich E. J. A carbene-generating photoaffinity probe for beta-adrenergic receptors. Biochim. Biophys. Acta, Biomembr. 1983, 729, 219–228. 10.1016/0005-2736(83)90488-1. [DOI] [PubMed] [Google Scholar]
- Tam E. K.; Li Z.; Goh Y. L.; Cheng X.; Wong S. Y.; Santhanakrishnan S.; Chai C. L.; Yao S. Q. Cell-based proteome profiling using an affinity-based probe (AfBP) derived from 3-deazaneplanocin A (DzNep). Chem. - Asian J. 2013, 8, 1818–1828. 10.1002/asia.201300303. [DOI] [PubMed] [Google Scholar]
- Cheng X. M.; Li L.; Uttamchandani M.; Yao S. Q. A tuned affinity-based staurosporine probe for in situ profiling of protein kinases. Chem. Commun. 2014, 50, 2851–2853. 10.1039/c4cc00184b. [DOI] [PubMed] [Google Scholar]
- Gregory K. J.; Velagaleti R.; Thal D. M.; Brady R. M.; Christopoulos A.; Conn P. J.; Lapinsky D. J. Clickable photoaffinity ligands for metabotropic glutamate receptor 5 based on select acetylenic negative allosteric modulators. ACS Chem. Biol. 2016, 11, 1870–1879. 10.1021/acschembio.6b00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soethoudt M.; Stolze S. C.; Westphal M. V.; van Stralen L.; Martella A.; van Rooden E. J.; Guba W.; Varga Z. V.; Deng H.; van Kasteren S. I.; Grether U.; IJzerman A. P.; Pacher P.; Carreira E. M.; Overkleeft H. S.; Ioan-Facsinay A.; Heitman L. H.; van der Stelt M. Selective Photoaffinity Probe That Enables Assessment of Cannabinoid CB2 Receptor Expression and Ligand Engagement in Human Cells. J. Am. Chem. Soc. 2018, 140, 6067–6075. 10.1021/jacs.7b11281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picone R. P.; Fournier D. J.; Makriyannis A. Ligand based structural studies of the CB1 cannabinoid receptor. J. Pept. Res. 2002, 60, 348–356. 10.1034/j.1399-3011.2002.21069.x. [DOI] [PubMed] [Google Scholar]
- Jorg M.; Glukhova A.; Abdul-Ridha A.; Vecchio E. A.; Nguyen A. T.; Sexton P. M.; White P. J.; May L. T.; Christopoulos A.; Scammells P. J. Novel irreversible agonists acting at the A1 adenosine receptor. J. Med. Chem. 2016, 59, 11182–11194. 10.1021/acs.jmedchem.6b01561. [DOI] [PubMed] [Google Scholar]
- van Muijlwijk-Koezen J. E.; Timmerman H.; van der Sluis R. P.; van de Stolpe A. C.; Menge W. M. P. B.; Beukers M. W.; van der Graaf P. H.; de Groote M.; IJzerman A. P. Synthesis and use of FSCPX, an irreversible adenosine A1 antagonist, as a ’receptor knock-down’ tool. Bioorg. Med. Chem. Lett. 2001, 11, 815–818. 10.1016/S0960-894X(01)00069-5. [DOI] [PubMed] [Google Scholar]
- Nijmeijer S.; Engelhardt H.; Schultes S.; van de Stolpe A. C.; Lusink V.; de Graaf C.; Wijtmans M.; Haaksma E. E. J.; de Esch I. J. P.; Stachurski K.; Vischer H. F.; Leurs R. Design and pharmacological characterization of VUF14480, a covalent partial agonist that interacts with cysteine 983.36 of the human histamine H4 receptor. Br. J. Pharmacol. 2013, 170, 89–100. 10.1111/bph.12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worrell B. T.; Malik J. A.; Fokin V. V. Direct evidence of a dinuclear copper intermediate in Cu(I)-catalyzed azide-alkyne cycloadditions. Science 2013, 340, 457–460. 10.1126/science.1229506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himo F.; Lovell T.; Hilgraf R.; Rostovtsev V. V.; Noodleman L.; Sharpless K. B.; Fokin V. V. Copper(I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]
- Barrington W. W.; Jacobson K. A.; Stiles G. L. Glycoprotein nature of the A2-adenosine receptor-binding Subunit. Mol. Pharmacol. 1990, 38, 177–183. [PMC free article] [PubMed] [Google Scholar]
- Krysiak J. M.; Kreuzer J.; Macheroux P.; Hermetter A.; Sieber S. A.; Breinbauer R. Activity-based probes for studying the activity of flavin-dependent oxidases and for the protein target profiling of monoamine oxidase inhibitors. Angew. Chem., Int. Ed. 2012, 51, 7035–7040. 10.1002/anie.201201955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong V.; Presolski S. I.; Ma C.; Finn M. G. Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angew. Chem., Int. Ed. 2009, 48, 9879–9883. 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D.; Mulder-Krieger T.; IJzerman A. P.; Heitman L. H. Functional efficacy of adenosine A2A receptor agonists is positively correlated to their receptor residence time. Br. J. Pharmacol. 2012, 166, 1846–1859. 10.1111/j.1476-5381.2012.01897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J; Fritsch E. F.; Maniatis T.. Expression of Cloned Genes in Cultured Mammalian Cells. In Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: New York, 1990; Vol. 343, pp 604–605. [Google Scholar]
- Smith P. K.; Krohn R. I.; Hermanson G. T.; Mallia A. K.; Gartner F. H.; Provenzano M. D.; Fujimoto E. K.; Goeke N. M.; Olson B. J.; Klenk D. C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Andre N.; Cherouati N.; Prual C.; Steffan T.; Zeder-Lutz G.; Magnin T.; Pattus F.; Michel H.; Wagner R.; Reinhart C. Enhancing functional production of G protein-coupled receptors in Pichia pastoris to levels required for structural studies via a single expression screen. Protein Sci. 2006, 15, 1115–1126. 10.1110/ps.062098206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohri A. B.; Hillertz P.; Eriksson P. O.; Meuller J.; Dekker N.; Snijder A. Thermodynamic studies of ligand binding to the human homopentameric glycine receptor using isothermal titration calorimetry. Mol. Membr. Biol. 2013, 30, 169–183. 10.3109/09687688.2012.696733. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.