

Abstract
Over two decades ago, the proteasome was considered a risky or even untenable therapeutic target. Today proteasome inhibitors are a mainstay in the treatment of multiple myeloma (MM) and have sales in excess of three billion US dollars annually. More importantly, the availability of proteasome inhibitors has greatly improved the survival and quality of life for patients with MM. Despite the remarkable success of proteasome inhibitor therapies to date, the potential for improvement remains and the development and optimal use of proteasome inhibitors as anticancer agents continues to be an active area of research. In this review, we briefly discuss the features and limitations of the three proteasome inhibitor drugs currently used in the clinic and provide an update on current efforts to develop next-generation proteasome inhibitors with the potential to overcome the limitations of existing proteasome inhibitor drugs.
Introduction
The proteasome is a large multi-protease complex and is responsible for the controlled degradation of more than 80% of cellular proteins (1). As such, the proteasome plays a key role in maintaining cellular protein homeostasis and regulates numerous biological processes, such as cell survival, DNA repair, apoptosis, signal transduction, and antigen presentation. Structurally, the 20S mammalian proteasome consists of a cylinder made of four stacked rings: two identical outer α-rings and two identical inner β-rings, each containing seven distinct but related subunits (Figure 1). In mammalian proteasomes, each β-ring harbors three catalytic β-subunits (β1, β2 and β5) which display different substrate preferences, referred to as caspase-like (C-L), trypsin-like (T-L) and chymotrypsin-like (CT-L) activities, respectively (2). The active sites of these catalytic subunits face inward, accepting peptide substrates from the proteasome’s hollow inner chamber. By controlling which proteins enter its inner chamber, the proteasome is able to degrade proteins in a highly-regulated fashion (3). Proteins targeted for proteasome-mediated degradation are typically tagged by the covalent attachment of polyubiquitin chains (“ubiquitination”) before being recognized and degraded by the proteasome complex. The concerted action of ubiquitination by a series of enzymes and proteolysis by the proteasome complex is collectively known as the ubiquitin-proteasome system (UPS). Over the past three decades, the UPS has been extensively explored as a target for drug discovery (4, 5), culminating in the remarkable clinical success of proteasome inhibitor (PI) drugs in the treatment of hematological malignancies including multiple myeloma (MM). Although a great amount of effort has been made to develop agents which target other UPS components such as ubiquitin ligases and deubiquitinases, to date only the proteasome has been successfully exploited as a therapeutic target to treat human disease.
Figure 1.
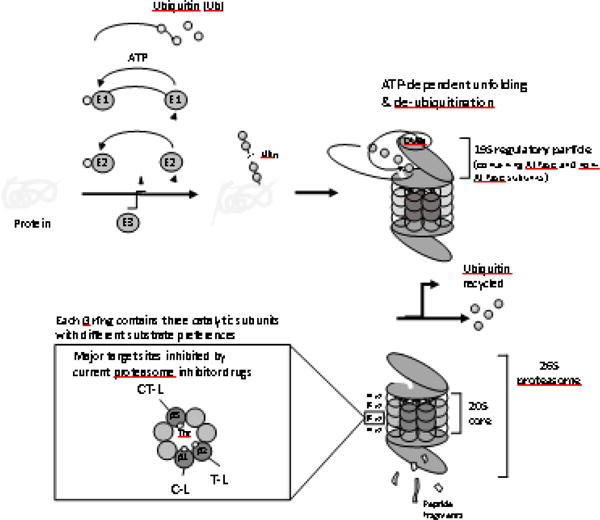
The structure and function of 26S proteasome in the ubiquitin-proteasome system (UPS). Proteins targeted for proteasome-mediated degradation are typically tagged by the covalent attachment of polyubiquitin chains of at least 4 ubiquitin (Ub) moieties (“ubiquitination”). This ubiquitination is carried out by the concerted action of three distinct enzymes, E1 (Ub activation), E2 (Ub conjugation), and E3 (Ub ligation). Subsequently, ubiquitinated proteins are recognized, unfolded and de-ubiquitinated by the lid of 26S proteasome (19S regulatory particles composed of ATPase and non-ATPase subunits). The proteolysis takes place at the inner chamber inside the 20S core, generating short peptide fragments of typically 2 to 24 amino acid residues. The 20S core consists of two outer α rings and two inner β rings, each containing seven distinct subunits. Each β ring harbors three catalytic β-subunits (β1, β2 and β5) which display different substrate preferences and their activities are commonly referred to as caspase-like (C-L), trypsin-like (T-L) and chymotrypsin-like (CT-L) activities, respectively. Among the three catalytic β-subunits, β5 subunit is the major target of current proteasome inhibitor drugs via their interactions with the catalytic threonine (Thr) residue.
Following the clinical success of proteasome-targeted therapies for cancer treatment, much effort has been made to address the limitations associated with existing PI drugs. Like almost all cancer therapeutics, cancer resistance, either acquired or de novo, is a major hurdle for PI drugs. So far, various resistance mechanisms have been reported for PI drugs in preclinical and clinical settings (6, 7) but remain unsettled. In recent years, there have been increasing attempts to design novel PIs that can overcome resistance or bypass cross-resistance to existing PI drugs (8). In addition, PI drugs have shown exquisite efficacy in treating MM and other hematological malignancies, but not solid cancers (9). The lack of therapeutic efficacy of PI drugs against solid cancers has often been attributed in part to their poor pharmacokinetic (PK) profiles including their short circulation time and insufficient distribution to proteasome targets located in solid tumor tissues (10). Moreover, our understanding remains limited on how the kinetics (both the magnitude and duration) and mode of proteasome inhibition can impact the pharmacodynamic (PD, such as efficacy and safety) profiles of PI drugs. Moving forward, an enhanced the understanding of the PKs and PDs of PI drugs and of the relationship between them is needed. In this review, we provide a brief overview of three clinically used PI drugs for cancer therapy focusing on PK/PD considerations and also summarize current efforts to develop next-generation PI drugs.
Proteasome inhibitor drugs in clinical use
Currently, three PIs are in clinical use, bortezomib (BTZ, Velcade®, the first-in-class PI drug with US FDA approval in 2003), carfilzomib (CFZ, Kyprolis®, the second-in-class PI drug with US FDA approval in 2012) and ixazomib (IXZ, Ninlaro®, the first oral PI drug with US FDA approval in 2015) (Figure 2). Although these PI drugs have brought tremendous improvements to the treatment of MM, earlier efforts to develop therapeutics targeting the proteasome had received considerable skepticism. This skepticism was not unreasonable, given the fundamental roles and abundant presence of the proteasome in all types of cells. Despite such skepticism, early preclinical results in models of human cancer were very promising, especially for MM and other hematological malignancies (11, 12). Propelled by exemplary academic-industrial partnerships, BTZ was successfully developed as the first-in-class PI drug with record efficiency in drug development and became a blockbuster drug in cancer therapy (13). The clinical success of BTZ has prompted the development of CFZ and IXZ soon after. Below is a brief account of discovery and development efforts of these clinically used PI drugs.
Figure 2.
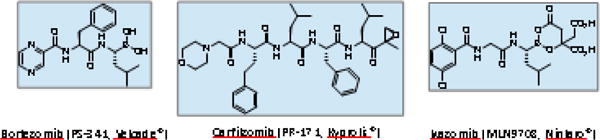
Structures of proteasome inhibitors in clinical use
Bortezomib (BTZ, PS-341, Velcade®): Rise of proteasome inhibitors as an anticancer agent
The earliest efforts to identify specific PIs began in the late 1980’s (14, 15). These early inhibitors were used to probe the function of the proteasome itself and to examine its biological role within the cell. The path towards PIs as therapeutic agents began with research into the role of the UPS in muscle wasting. Goldberg et al. proposed that upregulation of the UPS could explain the muscle wasting phenomenon observed in conditions such as sepsis, cancer, and burn injuries (16). They further suggested that muscle wasting could be treated with PIs by suppressing excessive proteolysis of muscle proteins. In subsequent efforts, a highly potent PI, PS-341, now known as BTZ, was identified (17). Pre-clinical studies soon revealed that BTZ is highly effective against various types of cancers (12, 18).
Structurally, BTZ is a dipeptide boronic acid that forms a coordinate covalent bond with the catalytic threonine residue of the proteasome’s β5 and β1 subunits (19). As a result, BTZ displays a potent inhibitory effect on the CT-L activity and to a lesser extent on the C-L activity of the 20S proteasome (20) (Table 1). In addition to its high affinity binding to the proteasome, BTZ also demonstrated nanomolar cytotoxic potencies against a variety of cancer cell lines, in particular, those derived from MM (12, 21). These in vitro findings also translated into promising in vivo efficacies in mouse xenograft models of both hematological and non-hematological malignancies (12, 18, 22).
Table 1.
Proteasome inhibitors in clinical use or under clinical development: their interactions with the proteasome target
| Drug name | Pharmacophore | Binding mode | IC50 (nM)
|
||
|---|---|---|---|---|---|
| CT-L | C-L | T-L | |||
| Carfilzomib | epoxyketone | irreversible | 2~31 7.92 |
14.5~401 532 |
12001 5902 |
| Bortezomib | boronic acid | reversible | 5.1~5.73 | 2,400 | 3,6004 |
| Ixazomib | boronic acid | reversible | 55 2.8~4.11 |
405 311 |
>10 0005 35001 |
| Oprozomib | epoxyketone | irreversible | 363 | ||
| Delanzomib | boronic acid | reversible | 3.86 | 20~506 | > 1006 |
| Marizomib | β-lactone | irreversible | 3.52 2.67 |
4302 4307 |
282 217 |
Calu-6 cells were treated with PIs for 1 hr. Proteasome-Glo assay (75)
Purified human erythrocyte 20S proteasomes (100)
Purified human 20S proteasomes (84)
Purified human 20S proteasomes (56)
MM.1S cells were treated with ixazomib for 3 hrs and harvested. Cell extracts were then analyzed for CT-L (Chymotrypsin-like), C-L (Caspase-like), and T-L (Trypsin-like) activity assay (74)
RPMI-8226 MM cell lysates (91)
Rabbit 20S proteasome (99)
Prompted by strong preclinical data, several early phase clinical trials had investigated BTZ for its safety and tolerability in over 200 cancer patients by late 2001 (23). BTZ was relatively well tolerated with adverse events consisting of low-grade fever, fatigue, thrombocytopenia, and in some patients, peripheral neuropathy. BTZ soon received US FDA fast-track approval for the treatment of relapsed and refractory MM in 2003, based on the outstanding efficacy results from the phase II open-label SUMMIT trial (24). BTZ’s clinical efficacy was further proven in combination with other therapeutic agents, leading to a full US FDA approval in 2005 as a second-line MM therapy (25) and in 2008 as a first-line therapy for patients with newly diagnosed MM (26). BTZ also received approval for use in patients with previously-treated mantle cell lymphoma from the US FDA in 2014 and from the European Medicines Agency in 2012 (27). Today BTZ is commonly used as a first-line agent in combination with other anti-myeloma agents, for example, immunomodulatory agents such as thalidomide or lenalidomide, cytotoxic drugs like melphalan, and glucocorticoids such as dexamethasone or prednisone. BTZ has also served as a proof-of-concept paving the way for two additional US FDA-approved PI drugs. While a number of clinical trials have investigated the possibility of extending the therapeutic effects of BTZ beyond MM, the results so far have been disappointing (10, 28).
BTZ is currently formulated for intravenous or subcutaneous injections (as a lyophilized powder with mannitol). An earlier study explored the possibilty of oral administration (18), but this approach was not further pursued due to low bioavailability (~11% in mice (29)). BTZ was shown to have rapid and wide biodistribution profiles in preclinical studies (12). Interestingly, a recent publication reported that the biodistribution of BTZ in various tissues is impacted by the tissue density of the proteasome which BTZ tightly and reversibly binds to (30). This study further demonstrated that saturation of proteasome binding sites at high doses of BTZ can contribute to non-dose-proportional PK behaviors of BTZ. Similar to these preclinical findings, the results from a phase I clinical trial also indicated that BTZ displays a large volume of distribution (> 400 L) in patients with solid cancers (31). Subsequent clinical trials reported similar findings on the PK profiles of BTZ (detailed reviews available (32), Table 2). When the metabolism of BTZ was investigated using human liver microsomes, BTZ was converted to pharmacologically inactive metabolites primarily via oxidative deboronation, mediated by multiple cytochrome P450 enzymes (CYPs) with their relative contribution in the following order, CYP3A4, CYP2C19, CYP2D6, CYP1A2 and CYP2C9 (33, 34). Consistent with these results, the systemic exposure of BTZ was increased and decreased with co-administration of ketoconazole (a CYP3A4 inhibitor) and rifampicin (a potent CYP3A4 inducer), respectively (35, 36). On the other hand, co-administration of omeprazole (a CYP2C19 inhibitor) had only a minimal impact on the PK profiles of BTZ in patients with advanced solid cancers (37). Given the importance of hepatic metabolism in the elimination of BTZ, patients with hepatic dysfunction may require dose adjustment, but no guideline or recommendation is available yet. In preclinical studies, the majority of the radio-labeled BTZ was excreted into bile duct (~66%) with the remainder excreted into the urine (12). In a clinical study, patients with renal impairment responded to BTZ therapy similar to those with normal renal function (38).
Table 2.
Clinically used dosing regimens and pharmacokinetic (PK) parameters reported for FDA-approved proteasome inhibitor drugs.
| Drug name | Current clinical dosing regimens | Tested dosing regimens and reported PK parameters
|
References | ||
|---|---|---|---|---|---|
| Tested dosing regimens | PK parameters | Notable characteristics | |||
| Bortezomib (Velcade®, PS-341) | 1.3 mg/m2 IV on days 1, 4, 8 & 11 of 21-day cycles | 1.45 mg/m2, IV (C1D1) | CL, 75.3 (51.2) L/h; Vss, 416 (158) L; t1/2, 8.68 (4.16) h | Phase I trials in patients with advanced solid cancers; dose-proportionality in PK parameters not established | (31) |
|
| |||||
| 1.6 – 2.0 mg/m2, IV (C1D1) | CL, 63.7-112 (29.8-126) L/h; Vss, 696-979 (357-473) L; t1/2, 10.4-14.8 (4.96-10.4) h | ||||
|
| |||||
| 1.3 mg/m2, IV. single dose (C1D1) vs. multiple doses (C1D11, C1D3, & C3D11) |
single dose: CL, 111.6 (73.6) L/h; Vss, 1540 (2730) L; t1/2, 11.5 (12.7) h multiple doses: CL, 18.2-28.0 (9.2-19.8) L/h; Vss, 1613-2213 (1125-2730) L; t1/2, 75.6-108.6 (34.6-64.8) h |
Upon repeated dosing, CL decreased while the systemic exposure and t1/2 increased. | (132) | ||
|
| |||||
| 1.0 mg/m2 IV (C1D11) vs. 2.5 mg/m2 SC (C1D11) |
SC: Cmax, 20.4 (8.87) ng/mL; Tmax, 30 (5–60) min; AUClast, 155 (56.8) ng·h/mL IV: Cmax, 223 (101) ng/mL; Tmax, 2 (2–5) min; AUClast, 151 (42.9) ng37·h/mL |
Phase III study in patients with RRMM. Equivalent systemic exposure between SC and IV groups. | (49, 133) | ||
|
| |||||
| Carfilzomib (Kyprolis®, PR-171) | 20 mg/m2 on days 1 & 2; if tolerated, escalated to 27 mg/m2 (IV infusion, 2-10 min) or 56 mg/m2 (IV infusion, 30 min) on day 8 of cycle 1; followed by tolerated dose on days 9, 15 & 16 of a 28-day cycle and next cycles (additional variations possible in subsequent cycles) |
20 mg/m2, IV (C1D1) | CL, 659 (353) L/h; Vss, 108 (71) L; t1/2, 0.66 (0.48) h | Phase I trial in patients with RRMM. | (65) |
|
| |||||
| 20 mg/m2, 2-10 min IV infusion on D1, 2, 8, 9, 15 & 16 |
D1: CL, 146 (22) L/h D16: CL, 136 (53) L/h |
CL exceeded hepatic blood flow. | (70) | ||
|
| |||||
| 2-10 min IV infusion. 20 mg/m2 on D1 & 2 ➔27 and 36 mg/m2 on D8, 9, 15 & 16 |
20 mg/m2 (D1): CL, 263 (398) L/h; Vss, 27.7 (48.6) L; t1/2, 0.44 (0.15-2.20) h 20 mg/m2 (D16): CL, 136 (52.8) L/h; Vss, 7.75 (3.77) L; t1/2, 1.10 (1.00-1.13) h 27 mg/m2 (D16): CL, 150 (30.9) L/h; Vss, 11.1 (4.45) L; t1/2, 0.35 (0.26-0.92) h |
Phase I/II trials in patients with advanced solid cancers. Rapid systemic CL, large Vss and very short elimination half-lives. |
(69) | ||
|
| |||||
| 30 min IV infusion. 20 mg/m on D1 & 2 ➔ 36, 45, 56 or 70 mg/m2 on D8, 9, 15 & 16 |
20 mg/m2 (C1D1): CL, 143 (56.6†) L/h; t1/2, 0.837 h 27 mg/m2 (C2D16): CL, 102 L/h; t1/2, 0.973 h 56 mg/m2 (C2D16) : CL, 118 (27.7†) L/h; t1/2, 0.875 h |
Phase I trial in patients with RRMM. Comparable PK properties between 30 min and 2-10 min infusion. | (134) | ||
|
| |||||
| 30-min IV infusion. 20 mg/m on D1 ➔ 45, 56, 70 or 88 mg/m2 on D8 & 15 |
20 mg/m2 (D1): CL, 146 (30.4†) L/h; t1/2, 0.64 (0.193-1.29) h; AUClast, 260 (27.6†) ng·h/mL 70 mg/m2 (D15): CL, 131 (28.6†) L/h; t1/2, 0.95 (0.572-1.29) h; AUClast, 1030 (20.5†) ng·h/mL 88 mg/m2 (D15): CL, 138 (34.3†) L/h; t1/2, 0.848 (0.648-0.952) h; AUClast, 1190 (29.1†) ng·h/mL |
Phase I/II trials in patients with RRMM. Dose-proportional increase in AUC. | (135) | ||
|
| |||||
| Ixazomib (Ninlaro®, MLN9708) | 4 mg orally administered on days 1, 8, & 15 of 28-day cycles | 0.24-3.95 mg/m2 on D1, 8 & 15 |
D1: Tmax, 1 (0.5-8.0) h D15: t1/2, 3.6-11.3 days |
Rapid absorption and long terminal half-lives. 2.23 mg/m2 is equivalent to 4.0 mg |
(79) |
|
|
|
||||
| 0.24-2.23 mg/m2 on D1, 4, 8 & 11 of 21-d cycles |
2 mg/m2 (D1): Tmax, 0.65 (0.25-3.97) h 2 mg/m2 (D11): Tmax, 1 (0.5-23.6) h; t1/2, 3.3-7.4 days |
(76) | |||
|
| |||||
| 4 mg on D1, 8 & 15 | CL, 2.0 (4.9‡) L/h; BA, 60%; Tmax, 1.5 (0.3-8) h | Results from population PK modelling. | (136) | ||
|
| |||||
| 4 mg on D1, 8 & 15 | Model parameter: CL, 1.86 L/h; BA, 58%; | Combination treatment with lenalidomide & dexamethasone in RRMM | (81) | ||
Abbreviations: IV, intravenous; SC, subcutaneous; CL, clearance; Vss, volume of distribution at steady-state; t1/2, terminal half-life; Cmax, maximum plasma concentration; Tmax, time to Cmax; AUClast, area under the concentration–time curve from time 0 to the last time point; BA, bioavailability; RRMM, refractory or relapsed multiple myeloma; D, Day(s); C, cycle(s) Values reported as means (standard deviation, % coefficient of variation(†) or % standard error(‡)) except for Tmax and t1/2, which are expressed as median (range).
Being the first-in-class PI drug, BTZ also became the first to be explored for the relationship between proteasomal inhibition (both the magnitude and duration) and anticancer efficacy in vivo. When the PK/PD profiles were compared in mouse xenograft models which responded differently to BTZ, the results indicated that both drug penetration and proteasome inhibition were much attenuated in mice carrying poorly perfused xenograft tumors which did not respond to BTZ treatment (39). These findings were applied to the development of next-generation PI drugs as well as novel drug delivery systems. For example, in order to modulate the magnitude and duration of proteasome inhibition by BTZ, several groups investigated the potential utility of nanoformulations including the design of prodrugs or bone-targeting moieties (40–42). However, the results from these efforts have yet to be translated into clinical application.
Despite the remarkable clinical success achieved by BTZ, several limitations have emerged. Like many other cancer therapies, a subset of patients responds to BTZ therapy while others do not. Even those who initially respond to BTZ therapy almost inevitably develop resistance over time (43). The median duration of clinical response was typically about 12 months (44, 45). The mechanisms underlying cancer resistance to BTZ have been actively investigated, yielding various potential strategies to overcome resistance including the development of PI drugs based on novel structural scaffolds (46). In addition to drug resistance, BTZ therapy is associated with the severe adverse effect of peripheral neuropathy, which was later attributed to its off-target interactions with a serine protease (HtrA2/Omi) involved in neuronal survival (47, 48). This dose-limiting toxicity of BTZ was substantially alleviated by administering the drug via subcutaneous injection (49) or by implementing once-weekly dosing (50, 51). These issues prompted the development of next-generation PIs with more favorable safety profiles and fewer off-target interactions.
Carfilzomib (CFZ, PR-171, Kyprolis®): Novel mode of proteasome inhibition
The second-in-class PI drug CFZ (Kyprolis®, developed by Proteolix/Onyx Pharmaceuticals and now available through Amgen) received its fast-track US FDA approval in 2012, based on its efficacy and safety results in patients with relapsed and refractory MM (52). The development of CFZ was initiated by the identification of the proteasome as the major target of the natural product epoxomicin (53). The design and synthesis of a biotinylated chemical probe led to the discovery that the epoxyketone group of epoxomicin covalently binds to the proteasome with an exceptional selectivity over other types of proteases. Subsequent efforts were made to build a library of epoxomicin analogs and identified a lead candidate, YU-101, based on their potent anticancer activities (54, 55). Later, YU-101 was further modified to yield CFZ which displayed very promising preclinical results (56).
Structurally, CFZ is a tetrapeptide harboring an epoxyketone as its pharmacophore and it forms an irreversible, covalent bond with proteasome catalytic subunits, predominantly β5 (Table 1). The exquisite selectivity of CFZ toward the proteasome is achieved by the formation of two covalent bonds, one with the catalytic Thr1Oγ nucleophile and a second with the adjacent Thr1N amino group. Based on high-resolution co-crystal structures between the proteasome and various epoxyketone-based inhibitors, the formation of a 1, 4-oxazepano adduct has been identified between the epoxyketone of these inhibitors and the catalytic threonine residue within the β5 active site (57, 58). Due to this proteasome-selective mechanism of action, CFZ has afforded much improved safety profiles. Additionally, the irreversible nature of the interaction between CFZ and the proteasome allows it to achieve sustained and durable proteasome inhibition, which may contribute to its efficacy even in the presence of resistance to BTZ (59). Of note, the irreversible modification of the proteasome target by CFZ or other peptide epoxyketones have also been exploited to develop activity-based probes (ABPs) that allow for covalent labeling of functional proteasomes or profiling of proteasome activity under diseased conditions or in response to cellular stimuli (60). Such ABPs may be potentially used as diagnostics to detect disease or monitor response to therapy (61–63).
In 2005, phase I clinical trials with CFZ began and successfully identified the phase II recommended doses and dosing schedules that were further investigated in subsequent clinical trials (64, 65). From early on, it was observed that a subset of patients who did not respond to BTZ-based therapy could still benefit from CFZ. Recently completed phase III clinical trials provided further evidence that CFZ-containing regimens can be effective against relapsed MM, including those patients who relapsed after receiving prior therapies including BTZ (66, 67). In particular, the phase III ENDEAVOR trial was a head-to-head comparison of CFZ and BTZ in patients with relapsed or refractory MM (67). In this trial, CFZ was shown to be superior to BTZ in extending overall survival of patients in the relapsed setting. In addition to its superior efficacy, the CFZ-containing regimen showed much improved safety profiles, especially in terms of peripheral neurotoxicity. While cardiovascular events were observed in CFZ-treated patients, no evidence was found of cumulative cardiac injury or ventricular dysfunction in the CFZ group. With these outstanding outcomes, CFZ is now part of a standard of care for relapsed or refractory MM and will likely evolve as part of frontline therapy in the near future.
When the PK profiles of CFZ were initially assessed in rats, the results indicated very rapid clearance, short circulation time (plasma half-lives less than 1 h) and wide biodistribution (56, 68). At all dose levels tested, the clearance of CFZ exceeded rat hepatic flow. In line with these in vivo results, CFZ was found to be rapidly metabolized in rat hepatocytes, but also in rat blood and in homogenates prepared from other tissues (68). The major metabolites of CFZ were peptide fragments and the diol of CFZ, formed via peptidases and epoxide hydrolases, respectively. Similar to these preclinical results, early phase clinical trials also indicated that CFZ displays very short half-lives (12 ~ 40 min), rapid systemic clearance (116 ~ 263 L/h) and large volumes of distribution at steady state (9 ~ 28 L) at all dose levels tested (11, 15, 20 and 27 mg/m2) (65, 69) (Table 2). Plasma clearance of CFZ in humans also exceeded hepatic blood flow, further indicating a considerable contribution of extrahepatic mechanism to the overall elimination of CFZ (70). Consistent with in vitro results showing only minor roles of CYP-mediated metabolism or renal excretion in the overall disposition of CFZ, the PK profiles of CFZ were not impacted by co-administration with CYP inhibitors or inducers (70) or by renal impairment (71, 72).
Along with its structural and mechanistic differences from BTZ, CFZ offers a treatment option with greatly reduced risk of peripheral neuropathy. CFZ treatment is associated with different types of adverse effects including cardiovascular complications, hypertension, and heart failure, but overall these adverse effects are reversible and manageable with careful monitoring (73). CFZ shares several adverse events with BTZ such as anemia, fatigue, and diarrhea. One potential downside of CFZ is its poor aqueous solubility. Despite the incorporation of a N-terminal morpholine ring to improve solubility, CFZ remains practically insoluble and the current formulation requires the use of a 50-fold excess of a β-cyclodextrin derivative to prepare an injectable solution. As with BTZ, CFZ is not suitable for oral administration and is susceptible to drug resistance in clinical use. These problems have prompted the development of additional next-generation PIs.
Ixazomib (IXZ, MLN9708, Ninlaro®): First oral proteasome inhibitor drug
With both BTZ and CFZ being administered only via intravenous or subcutaneous injection, there has been an unmet need for orally available PI drugs. In 2015, IXZ (Ninlaro®, Takeda Pharmaceuticals Limited) received its US FDA approval as the first orally bioavailable PI drug. Based on the promising efficacy observed in preclinical studies, IXZ rapidly advanced to clinical trials (74, 75). IXZ, orally administered once a week (4 mg on days 1, 8, and 15 of 28-day cycles) in combination with lenalidomide plus dexamethasome, has now been approved in 40 countries including USA and the EU for the treatment of MM patients who have received one prior therapy, based on the superior results in clinical trials (76, 77). IXZ also displayed a good safety profile with no significant inhibitory effect on HtrA2/Omi, a non-proteasomal target of BTZ previously linked to peripheral neuropathy (74, 77, 78). IXZ is currently being investigated in several clinical trials as a single agent and in combination with other agents against multiple types of cancer (https://clinicaltrials.gov).
Structurally, IXZ is a capped dipeptide boronic acid and preferentially and reversibly inhibits the CT-L activity of the proteasome as well as the C-L and T-L activities at high concentrations with potencies similar to BTZ (74). However, the dissociation half-life of IXZ was significantly shorter than that of BTZ (18 vs. 110 min), which may account for the faster recovery of proteasome activity (IXZ vs. BTZ, 69 vs. 20%) in cell-based assays and its larger volume of distribution in mice (IXZ vs. BTZ, 20.2 vs. 4.3 L/kg) (75). Although not examined, some of these differences may have contributed to the improved safety profiles of IXZ over BTZ, despite sharing the boronic acid residue as their pharmacophore.
For oral administration, IXZ is formulated as a citrate ester prodrug (MLN9708) which is rapidly hydrolyzed to the pharmacologically active form (MLN2238) under physiological conditions (75). In phase I clinical trials, orally administered IXZ was rapidly absorbed (mean Tmax, 0.5 ~ 1 h) and had a long terminal half-life (mean T1/2, 3.3 ~ 7.4 days in twice-weekly dosing; 3.3 ~ 11.3 days in weekly dosing) (76, 79) (Table 2). When tested using recombinant CYP enzymes in vitro, IXZ was metabolized by multiple CYPs at concentrations exceeding those observed clinically and deemed unlikely to incur potential drug-drug interactions (80). Yet, co-administration with rifampin, a strong CYP3A inducer, led to substantial changes in the PK profiles of IXZ (Cmax and AUC decreased by 54% and 74%, respectively) (80). Overall, the PK profiles of IXZ showed dose-proportional behaviors. Using the compiled clinical data from 755 patients treated with IXZ, Gupta et al. conducted population PK analyses and reported the following average estimates for PK parameters: absolute bioavailability (58%), volume of distribution (543 L), terminal phase half-life (9.5 days), and systemic clearance (1.86 L/h) (81). Systemic exposure to IXZ was affected by moderate or severe hepatic impairment (82), but not by renal impairment (81). While IXZ has the potential to greatly improve the quality of life for patients with MM, its therapeutic advantages over BTZ or CFZ have yet to be investigated in randomized clinical trials.
Proteasome inhibitors in clinical and pre-clinical development
Following the huge clinical success of existing PI drugs, there have been extensive efforts to develop PIs with improved efficacy and pharmaceutical properties. Towards the goal of developing additional FDA-approved PIs, a number of PIs have been identified over the years but only three PIs are currently under evaluation in clinical trials (Figure 3).
Figure 3.
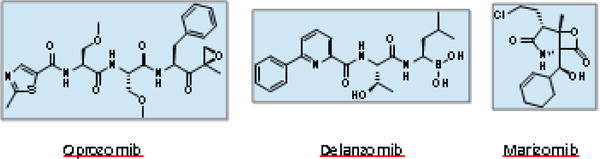
Structures of proteasome inhibitors undergoing clinical trials
Oprozomib (OPZ, ONX-0912, PR-047)
Oprozomib (OPZ) is a structural homologue of CFZ and is currently being investigated in several clinical trials including a multicenter phase Ib/II trial for patients with MM. The development of OPZ was conceived with the intent of developing an orally available PI drug by modifying the chemical structure of CFZ. During preclinical development, in addition to standard 20S proteasome inhibition assays, compounds were evaluated for their ability to kill cells expressing the P-glycoprotein efflux transporter from an early stage (83). In vivo inhibitory assays of tissue CT-L activity in mice following oral dosing were also utilized, as were in vitro metabolic stability assays using mouse and human liver microsomes. With guidance from these assays, CFZ was truncated to a tripeptide epoxyketone and its three amino acid residues were subsequently optimized to yield OPZ, a compound which maintained its selective inhibitory effect on the CT-L activity of purified human 20S proteasomes and which had an antitumor efficacy equivalent to CFZ in mouse xenograft models (84).
Following these initial findings, the therapeutic potential of OPZ was further examined in various in vitro and in vivo models. When tested using two different human MM cell line models, OPZ showed proteasome inhibitory potency similar to CFZ. OPZ was effective in decreasing the viability of MM cells in vitro and effectively suppressed the growth of in vivo xenograft tumors containing human MM cells (85). Subsequent to these positive preclinical results, OPZ has advanced to early stage clinical development and the results from the published phase Ib/II clinical trials have indicated efficacy in patients with hematologic malignancies; overall response rates of 25% and 27.3% were observed in patients with MM relapsed after receiving BTZ- and CFZ-based therapies, respectively (86, 87). During the phase Ib trial, adverse events such as nausea and vomiting were also noted and these likely arise from high concentrations of OPZ in the gastrointestinal tract, potentially resulting in proteasome inhibition in non-targeted tissues (88). To alleviate such side effects, an extended release formulation of OPZ is currently being utilized for the phase Ib/2 clinical study (86).
Being developed as an orally available PI drug, the intestinal absorption profiles of OPZ were investigated in preclinical species. The absolute oral bioavailability of OPZ was assessed to be as high as 39% in rodents and dogs (84) and OPZ was found to be rapidly absorbed from the duodenum and jejunum of rodents and dogs (Tmax: 2~3 min). Once OPZ reaches systemic circulation, it is rapidly cleared via hepatic and extrahepatic metabolism, displaying a plasma half-life of less than 1 h typically (89). Using liver microsomes, microsomal epoxide hydrolase (mEH) was found to be the major enzyme responsible for the metabolic clearance of OPZ (90). However, the expression of mEH is not limited to the liver, but is found in many other tissues. As was noted for CFZ, the plasma clearance of OPZ (~210 mL/min/kg in rats) was found to exceed hepatic blood flow. These results indicate significant extrahepatic contribution to the metabolism of OPZ (90).
Successful development of OPZ may yield a second orally-available PI therapy. Initial results from a phase I clinical trial however indicated that OPZ may have minimal efficacy in patients with solid cancers (89). Similar to other PI drugs, novel drug delivery systems may be implemented to alter the pharmacokinetics of OPZ in vivo and broaden its therapeutic utility.
Delanzomib (CEP-18770)
Delanzomib is a reversible and orally bioavailable structural analogue of BTZ with the boronic acid as its pharmacophore (Figure 3). Delanzomib mainly inhibits the CT-L activity of the proteasome and to a lesser extent the C-L activity (91). Delanzomib displayed slightly reduced potency against a panel of MM and solid cancer cell lines as compared to BTZ, but was more selective to cancerous cells over normal epithelial cells than BTZ (91). Potent anti-MM efficacy was observed via both intravenous and oral administration of delanzomib as a single agent and in combination with other anti-myeloma agents (e.g., BTZ, melphalan, or dexamethasone plus lenalidomide) in mice bearing xenograft tumors composed of human MM cells (91–93). In an initial phase I clinical trial (0.1 ~ 1.8 mg/m2, intravenously administered on days 1, 4, 8 and 11 in 21-day cycle), no significant peripheral neuropathy was observed in patients with advanced solid cancers or MM. However, severe skin toxicity was observed as a dose-limiting side effect in a number of patients (53%, any grade; 31%, Grade ≥3) (94). In a separate phase I/II clinical trial, a higher dose and more frequent dosing schedule (2.1 mg/m2, intravenously administered on days 1, 8, and 15 in 28-day cycle) was investigated, but led to no significant improvement in clinical efficacy. As a result, further development of delanzomib for MM therapy was discontinued (95). The reasons for apparent inconsistencies between preclinical and clinical studies remain unclear. However, a possible opportunity remains in identifying specific patient populations or alternative diseases for which delanzomib therapy could prove useful. For example, delanzomib could be a potent antiangiogenic agent or an inhibitor of RANKL-induced osteoclastogenesis based on available in vitro data (91).
One notable feature of delanzomib is its long duration of proteasomal inhibition in tumoral tissues when evaluated in mice carrying MM xenografts, potentially indicating enhanced distribution to tumor tissues and/or slow dissociation from tumor proteasome target sites. After intravenous administration at the maximum tolerated dose, delanzomib achieved a greater magnitude of proteasome inhibition and a slower recovery of tumoral CT-L activity than BTZ. The extent of proteasomal inhibition in tumoral tissues exceeded 50% in delanzomib-treated mice at 72 h post-dose (91). This is in contrast to clinical observations in which maximal proteasome inhibition (54% at 1.8 mg/m2) was achieved within 1 h and recovered to the baseline within 24 h in the dose range tested (0.4 ~ 1.8 mg/m2) (94).
With regards to its PK profiles, delanzomib was quite comparable to BTZ in preclinical species (29). Delanzomib was slowly eliminated (T1/2, 71 h and 86 h in rats following intravenous and oral administration, respectively; 15 h and 53 h in mice following intravenous and oral administration, respectively). Delanzomib is highly protein-bound across species (mouse, rat, dog: 99.9%; human: 99.8%), and was found to have oral bioavailability of 54% and 39% in rats and mice, respectively (29). In a clinical trial, intravenously administered delanzomib showed a multi-exponential decay with a rapid initial distribution phase, followed by a slow elimination phase (mean T1/2, 34 ~ 100 h) with a large volume of distribution (mean Vd, 55~ 106 L/m2) in the dose ranges tested (0.40 ~ 1.5 mg/m2) (94). The low microsomal stability of delanzomib (T1/2, > 40 min in mouse, dog, and human; 15 min in rat) suggests the involvement of phase I metabolism, but delanzomib itself did not inhibit major CYPs at concentrations up to 30 μM (94).
Marizomib (NPI-0052, Salinosporamide A)
Marizomib (salinosporamide A) is a natural product derived from marine actinomycete bacteria (Salinospora tropica) and is currently under development as a novel orally active PI (96). Unlike other peptide-based PIs, marizomib has a β-lactone-γ-lactam bicyclic ring structure without a linear peptide backbone (97, 98). Marizomib irreversibly inhibits proteasome activities at nanomolar concentrations (preferentially inhibiting the CT-L activity, followed by the T-L activity and to a much lesser extent the C-L activity) in MM cells and purified proteasomes (Table 1) (99–101). In in vivo studies with intravenously administered marizomib, proteasome activities were irreversibly inhibited and slowly recovered. The time courses of recovery varied among various tissues, with inhibition persisting for as long as 72 h in blood (100, 102).
Marizomib more effectively induced apoptosis in tumor cells from MM and chronic lymphocytic leukemia (CLL) patients, while displaying a lower toxicity to normal cells than BTZ (100, 103). Additionally, marizomib was highly potent in MM cells from patients who were refractory to BTZ and was found to act synergistically with BTZ and lenalidomide in vitro and in vivo (100, 104). While the overall response rate of marizomib as a single agent was merely 11% in phase I clinical trials (105), the response rate substantially improved to 53% when combined with pomalidomide and low-dose dexamethasone in patients with refractory or relapsed MM, without significantly increasing the incidence of adverse events (106). Marizomib has however been associated with CNS adverse events (e.g. visual and auditory hallucination, unsteady gait, confusion), suggesting its ability to penetrate the blood-brain barrier (107). As a single or combination agent with a weekly or twice weekly dosing schedule, marizomib is being investigated in phase I/II trials for use in a broad range of advanced hematological malignancies including MM and refractory lymphoma, as well as solid cancers (https://clinicaltrials.gov).
When marizomib was orally administered using a formulation intended for intravenous administration, the bioavailability was 30 ~ 40% in monkeys (98). In a phase I clinical trial (0.075 ~ 0.6 mg/m2, intravenous infusion over 120 min on days 1, 4, 8, 11 in 21-day cycle,), marizomib showed a short half-life (2 ~ 33 min) with a large volume of distribution (18 ~ 129 L) and rapid clearance (54 ~ 1339 L/h) (105), which indicates the involvement of extrahepatic clearance in the overall elimination of marizomib. Careful examination may be warranted to gain a better understanding of PK/PD profiles of marizomib in normal vs. tumor tissues. Overall, marizomib displayed dose-proportional PK profiles (98, 105), but detailed information on its metabolism and excretion is not available yet.
In another interesting line of investigation, marizomib induced apoptosis in glioma cells with minimal cytotoxic effects on normal neuronal cells (108). Although intravenously administered marizomib (0.15 mg/kg) did not result in any significant proteasomal inhibition in the brain of mice and rats (102), orally administered marizomib (0.55 mg/m2 twice weekly and 0.64 mg/m2 weekly) effectively inhibited proteasome activities in the CNS of rats and monkeys (108). Using 3H-labeled marizomib, it was shown that marizomib concentrations in the CNS were approximately one-third of the steady-state blood concentration. These results suggest that marizomib can penetrate the blood-brain barrier in multiple species, providing a rationale for further exploring its potential to treat brain cancer (108). Marizomib is currently being assessed in a phase III trial for the treatment of malignant glioblastoma in combination with temozolomide and radiotherapy (NCT03345095, https://clinicaltrials.gov).
Drug resistance (acquired or de novo): Major hurdles in improving PI therapy
Common in many cancer therapies, the issues of drug resistance also pose major hurdles for PI therapies. MM patients who initially respond to PI therapy almost inevitably develop resistance over time (acquired resistance). Once patients relapse with MM refractory to PI-based therapy, there are currently few effective treatment options left. While a subset of MM patients responds well to PI therapy, others do not (de novo resistance). Several potential mechanisms for resistance to PI therapy have been proposed using cell-based model systems. Yet, those mechanisms await further validation in patients with MM and also in patients with solid cancers. For the lack of clinical benefits of PI therapy for solid cancers, it has been postulated that active PI drugs may have insufficient access to the proteasome target located in solid cancer cells (related to the PK issues). This possibility was supported in part by the preclinical results showing effective tumor growth suppression following direct intratumoral injection of PI drugs (12, 109). In addition, intravenous dosing of BTZ was effective in mice harboring highly perfused xenograft tumors, but not poorly perfused ones (39). Alternatively, it was also proposed that solid cancer cells may be inherently less sensitive to PI therapy than MM cells known for their elevated levels of proteotoxic stress or ER stress (110, 111). To tease out why patients with solid cancers do not benefit from PI therapy, it would be necessary to develop PI drugs that can afford sufficient access to the proteasomes in solid cancer cells and/or to develop targeted drug delivery systems.
Current understanding of resistance mechanisms for PI drugs, although not complete, has provided important platforms to screen for PI drugs that can potentially overcome resistance to existing PI drugs. Several reports observed the presence of mutations in the PSMB5 gene encoding the β5 catalytic subunit from cancer cell line models resistant to BTZ and low levels of Xbp1, a key regulator of one arm of the unfolded protein response (UPR), in primary cells isolated from MM patients following BTZ therapy (83, 112–114). For cancer cell line models resistant to CFZ and epoxomicin, the upregulation of P-glycoprotein was reported to be causally linked to drug resistance (115, 116). This information provided important guidance during the development of another epoxyketone-based PI, OPZ (83, 84). The screening and optimization processes for OPZ and related compounds included the testing in cell lines expressing P-glycoprotein.
Development strategies for next-generation proteasome inhibitors
As discussed above, the discovery of next-generation PIs with improved PK/PD profiles could improve clinical outcomes for MM patients (especially those with resistance to existing PI therapy) and extend therapeutic benefits to patients with solid cancers where existing PI drugs have proved largely ineffective. To achieve this goal, the following development strategies have been actively explored. Given that comprehensive reviews are already available on the first two strategies, we focused on the recent efforts to develop non-peptide-based PIs.
Immunoproteasome-selective inhibitors
The immunoproteasome (iP) is a variant of the constitutive proteasome in which the constitutive catalytic subunits β1, β2 and β5 are replaced by their respective inducible counterparts β1i, β2i and β5i, under inflammatory conditions and certain pathological states including cancer. By targeting the iP, it may achieve more selective inhibition of the proteasomal activity in cancer cells, thereby widening the therapeutic window. Although iP inhibitors have been studied in the preclinical setting, to date none have entered clinical trials (117). As the iP is strongly implicated in inflammatory pathways, iP-selective inhibitors are currently being investigated as potential anti-inflammatory agents. Detailed reviews on iP inhibitors are already available (118–120)).
Peptide-based proteasome inhibitors
The vast majority of existing PIs utilize a peptide backbone and an active warhead that interacts with the catalytic Thr residues of β-subunits with different mechanisms of action (e.g., aldehydes, vinyl sulfones or esters, boronates, epoxyketones, β-lactones). With the successful clinical development of the peptide boronates (BTZ and IXZ) and epoxyketone (CFZ), intense efforts have been underway to further refine the structure-activity relationship (SAR) and to identify compounds with optimal pharmacological profiles among peptide-based proteasome inhibitors. For further information on peptide-based PIs, comprehensive reviews are already available (121, 122).
Non-peptide-based proteasome inhibitors
From one of the earliest efforts to identify structurally-novel PIs via high-throughput screening, PI-083 was identified as a non-peptide PI (Figure 4) (123). Utilizing a 2-chloro-1,4-naphthoquinone scaffold, PI-083 preferentially inhibited the CT-L activity of the 20S proteasome (IC50: 1.0 μM) and inhibited T-L and C-L activities at slightly higher concentrations (IC50: 4.5 μM for both). When tested against a panel of 10 solid cancer cell lines, PI-083 exerted cytotoxic effects with IC50 values ranging from 1.7 to 11 μM. PI-083 was also effective in suppressing in vivo tumor growth in mouse xenograft models at a dose of 1 mg/kg twice weekly. Based on docking results and the compound’s SAR, it is postulated that PI-083 may act as a covalent PI with the chlorinated 2-carbon undergoing nucleophilic attack by the proteasome’s catalytic threonine residue (124). Recovery of proteasome activity following incubation with PI-083 was slow, with only partial recovery of activity after 18 h. Attempts to improve PI-083’s inhibitory potency were generally unsuccessful and the SAR was highly sensitive to modification.
Figure 4.
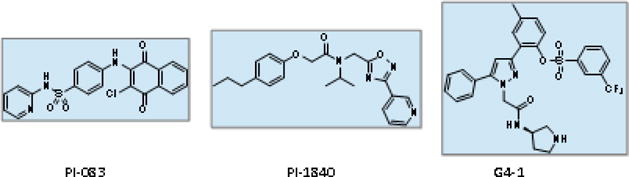
Structures of non-peptide proteasome inhibitors
A subsequent report from the same group identified PI-1840 (Figure 4), a structurally-unrelated non-peptide compound which potently and selectively inhibited the CT-L activity of the 20S constitutive proteasome (IC50: 27 nM) (125). PI-1840 showed no appreciable inhibition of 20S proteasome T-L or C-L activity and had an IC50 value of greater than 1 μM against the CT-L activity of the iP. Analysis via mass spectrometry and dialysis confirmed that PI-1840 acts as a fully-reversible inhibitor. A panel of solid cancer cell lines displayed varying degrees of sensitivity to PI-1840 (IC50: 2.2 ~ 45.2 μM), and the cytotoxic potency appeared to correlate with the degree of proteasome inhibition achieved by PI-1840. When tested in mice bearing MDA-MB-231 human breast cancer xenografts, PI-1840 (150 mg/kg daily via intraperitoneal injection) effectively suppressed tumor growth, in contrast to no appreciable suppression in the control groups that received either BTZ (1 mg/kg twice weekly via intraperitoneal injection) or the vehicle only. No observable toxicity was noted in animals receiving high doses of PI-1840. The safety profiles observed with PI-1840 may be related to its high degree of selectivity for the constitutive β5 subunit relative to the iP subunit β5i and its lack of inhibition of T-L or C-L activities. Given that the existing PI drugs tend to target both β5 and β5i subunits with relatively low selectivity, it awaits further investigations to determine whether the selective inhibition of β5 by PI-1840 may be advantageous or disadvantageous in terms of anticancer efficacy. The PK profiles of PI-1840 have not yet been published.
Another non-peptide PI dubbed G4-1, based on a tri-substituted pyrazole scaffold, was reported by our own research group (Figure 4) (126). Identified via the combination of structure-based virtual screening and in vitro kinetic assays, G4-1 inhibits both β5 and β5i catalytic activities with IC50 values of 1.6 and 2.4 μM, respectively. β1 and β1i subunits (C-L activity) were also inhibited at low micromolar concentrations, with minimal inhibition of T-L activity. G4-1 exerted cytotoxic effects against a variety of solid cancer and MM cell lines, regardless of acquired resistance to BTZ and CFZ. Further structural analyses indicated that G4-1 is a reversible, non-covalent inhibitor. As expected from its non-peptide-based structure, G4-1 displayed much improved in vitro metabolic stability over BTZ or CFZ when tested using mouse and human liver microsomes. In a mouse xenograft model of human prostate cancer, G4-1 (5 mg/kg, twice-weekly) was effective in suppressing tumor growth with no overt signs of toxicity. Additional PK or PD profiles of G4-1 have not yet been published.
In addition to those described above, there have been several other recent reports of efforts to develop non-peptide PIs but further investigations are still needed to validate their mode of interaction with the proteasome, their extent of interaction with non-proteasomal targets and their in vivo efficacy. While there is also a body of research covering peptide-based non-covalent PIs, such as those described by Blackburn et al. (127, 128), it is expected that these compounds will be susceptible to the same rapid, often extrahepatic, clearance as existing peptide-based PIs. Peptide-based PIs may also be less likely to penetrate poorly-perfused tumors due to either their physiochemical properties or their interactions with efflux transporters (129). Moving forward, significant research efforts will be required to identify non-peptide PIs which display optimal PK/PD profiles and suitability for clinical use.
Conclusion
With the successful development of BTZ, CFZ, and IXZ, proteasome inhibition has been firmly established as an effective treatment strategy for hematological malignancies and for MM, in particular. While these PI drugs have dramatically improved outcomes for numerous patients with MM, extensive clinical data also indicate that there remains much room for improvement, especially with regards to drug resistance, rapid metabolic inactivation and short circulation time, dose-limiting toxicities and poor efficacy in other cancer types. To improve upon existing PI drugs, a number of next-generation PIs are currently being investigated in multiple clinical trials. With data accumulating from new PI drug candidates, it has become increasingly evident that the clinical efficacy of PI drugs is impacted not only by their inhibitory potency, but also by the mode, extent and duration of proteasome inhibition. Moving forward, it is critical to carefully examine the PK and PD profiles of PI drug candidates in order to successfully to bridge the gap between initial preclinical results and eventual clinical outcomes. So far, early clinical data with next-generation PI drug candidates suggest that novel approaches including previously unexplored structural scaffolds may be needed to address the limitations and to expand the utility of existing PI drugs. In addition, alternative targets in the UPS (other than the catalytic subunits of the proteasome) have presented promising therapeutic potential. Preclinical evaluation of compounds targeting other broadly acting components of the UPS is underway (130, 131). In particular, deubiquitinases (DUBs), an essential component in the UPS, have emerged as a novel target in cancer therapy, especially for cancers refractory to PI drugs. Further investigations are ongoing to develop therapeutic agents targeting non-proteasomal components of the UPS, on their own or in combination with PI drugs. With continuing efforts, it is hoped that next-generation PIs with improved PK/PD profiles and novel therapeutic agents targeting the UPS will eventually be developed to treat patients with MM as well as those with other types of cancer.
Acknowledgments
We would like to thank the National Institutes of Health (R01 CA188354 to K.B.K.), the National R&D Program for Cancer Control, Ministry of Health and Welfare, Republic of Korea (No.1520250 to W.L.) and Creative-Pioneering Researchers Program through Seoul National University (to W.L.) for financially supporting this work.
Abbreviations
- CT-L
chymotrypsin-like
- T-L
trypsin-like
- C-L
caspase-like
- UPS
ubiquitin proteasome system
- PI
proteasome inhibitor
- MM
multiple myeloma
- BTZ
bortezomib
- CFZ
carfilzomib
- IXZ
ixazomib
- CYPs
cytochrome P450 enzymes
- OPZ
oprozomib
- CNS
central nervous system
- iP
immunoproteasome
- PK
pharmacokinetic
- PD
pharmacodynamic
- CLL
chronic lymphocytic leukemia
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
All authors have read the journal’s policy on disclosure of potential conflicts of interest and declare no competing financial interests. In addition, all authors have read and approved the journal's authorship agreement.
References
- 1.Schmidt M, Finley D. Regulation of proteasome activity in health and disease. Biochim Biophys Acta. 2014;1843(1):13–25. doi: 10.1016/j.bbamcr.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dick TP, Nussbaum AK, Deeg M, Heinemeyer W, Groll M, Schirle M, et al. Contribution of proteasomal beta-subunits to the cleavage of peptide substrates analyzed with yeast mutants. J Biol Chem. 1998;273(40):25637–46. doi: 10.1074/jbc.273.40.25637. [DOI] [PubMed] [Google Scholar]
- 3.Rabl J, Smith DM, Yu Y, Chang SC, Goldberg AL, Cheng Y. Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol Cell. 2008;30(3):360–8. doi: 10.1016/j.molcel.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang X, Dixit VM. Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res. 2016;26(4):484–98. doi: 10.1038/cr.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen P, Tcherpakov M. Will the ubiquitin system furnish as many drug targets as protein kinases? Cell. 2010;143(5):686–93. doi: 10.1016/j.cell.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 6.Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol. 2017;14(7):417–33. doi: 10.1038/nrclinonc.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nikesitch N, Ling SC. Molecular mechanisms in multiple myeloma drug resistance. J Clin Pathol. 2016;69(2):97–101. doi: 10.1136/jclinpath-2015-203414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buac D, Shen M, Schmitt S, Kona FR, Deshmukh R, Zhang Z, et al. From bortezomib to other inhibitors of the proteasome and beyond. Curr Pharm Des. 2013;19(22):4025–38. doi: 10.2174/1381612811319220012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roeten MSF, Cloos J, Jansen G. Positioning of proteasome inhibitors in therapy of solid malignancies. Cancer Chemother Pharmacol. 2018;81(2):227–43. doi: 10.1007/s00280-017-3489-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang Z, Wu Y, Zhou X, Xu J, Zhu W, Shu Y, et al. Efficacy of therapy with bortezomib in solid tumors: a review based on 32 clinical trials. Future Oncol. 2014;10(10):1795–807. doi: 10.2217/fon.14.30. [DOI] [PubMed] [Google Scholar]
- 11.Weinstein JN, Myers TG, O’Connor PM, Friend SH, Fornace AJ, Jr, Kohn KW, et al. An information-intensive approach to the molecular pharmacology of cancer. Science. 1997;275(5298):343–9. doi: 10.1126/science.275.5298.343. [DOI] [PubMed] [Google Scholar]
- 12.Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, et al. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res. 1999;59(11):2615–22. [PubMed] [Google Scholar]
- 13.Sanchez-Serrano I. Success in translational research: lessons from the development of bortezomib. Nat Rev Drug Discov. 2006;5(2):107–14. doi: 10.1038/nrd1959. [DOI] [PubMed] [Google Scholar]
- 14.Orlowski M. The multicatalytic proteinase complex, a major extralysosomal proteolytic system. Biochemistry. 1990;29(45):10289–97. doi: 10.1021/bi00497a001. [DOI] [PubMed] [Google Scholar]
- 15.Vinitsky A, Michaud C, Powers JC, Orlowski M. Inhibition of the chymotrypsin-like activity of the pituitary multicatalytic proteinase complex. Biochemistry. 1992;31(39):9421–8. doi: 10.1021/bi00154a014. [DOI] [PubMed] [Google Scholar]
- 16.Mitch WE, Goldberg AL. Mechanisms of muscle wasting. The role of the ubiquitin-proteasome pathway. N Engl J Med. 1996;335(25):1897–905. doi: 10.1056/NEJM199612193352507. [DOI] [PubMed] [Google Scholar]
- 17.Adams J, Behnke M, Chen S, Cruickshank AA, Dick LR, Grenier L, et al. Potent and selective inhibitors of the proteasome: dipeptidyl boronic acids. Bioorg Med Chem Lett. 1998;8(4):333–8. doi: 10.1016/s0960-894x(98)00029-8. [DOI] [PubMed] [Google Scholar]
- 18.Teicher BA, Ara G, Herbst R, Palombella VJ, Adams J. The proteasome inhibitor PS-341 in cancer therapy. Clin Cancer Res. 1999;5(9):2638–45. [PubMed] [Google Scholar]
- 19.Groll M, Berkers CR, Ploegh HL, Ovaa H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure. 2006;14(3):451–6. doi: 10.1016/j.str.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 20.Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets. 2011;11(3):239–53. doi: 10.2174/156800911794519752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61(7):3071–6. [PubMed] [Google Scholar]
- 22.Nawrocki ST, Sweeney-Gotsch B, Takamori R, McConkey DJ. The proteasome inhibitor bortezomib enhances the activity of docetaxel in orthotopic human pancreatic tumor xenografts. Mol Cancer Ther. 2004;3(1):59–70. [PubMed] [Google Scholar]
- 23.Adams J. Development of the Proteasome Inhibitor PS-341. Oncologist. 2002;7(1):9–16. doi: 10.1634/theoncologist.7-1-9. [DOI] [PubMed] [Google Scholar]
- 24.Kane RC, Bross PF, Farrell AT, Pazdur R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist. 2003;8(6):508–13. doi: 10.1634/theoncologist.8-6-508. [DOI] [PubMed] [Google Scholar]
- 25.Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352(24):2487–98. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- 26.San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359(9):906–17. doi: 10.1056/NEJMoa0801479. [DOI] [PubMed] [Google Scholar]
- 27.Teicher BA, Anderson KC. CCR 20th anniversary commentary: In the beginning, there was PS-341. Clin Cancer Res. 2015;21(5):939–41. doi: 10.1158/1078-0432.CCR-14-2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caravita T, de Fabritiis P, Palumbo A, Amadori S, Boccadoro M. Bortezomib: efficacy comparisons in solid tumors and hematologic malignancies. Nat Clin Pract Oncol. 2006;3(7):374–87. doi: 10.1038/ncponc0555. [DOI] [PubMed] [Google Scholar]
- 29.Dorsey BD, Iqbal M, Chatterjee S, Menta E, Bernardini R, Bernareggi A, et al. Discovery of a potent, selective, and orally active proteasome inhibitor for the treatment of cancer. J Med Chem. 2008;51(4):1068–72. doi: 10.1021/jm7010589. [DOI] [PubMed] [Google Scholar]
- 30.Zhang L, Mager DE. Physiologically-based pharmacokinetic modeling of target-mediated drug disposition of bortezomib in mice. J Pharmacokinet Pharmacodyn. 2015;42(5):541–52. doi: 10.1007/s10928-015-9445-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papandreou CN, Daliani DD, Nix D, Yang H, Madden T, Wang X, et al. Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. J Clin Oncol. 2004;22(11):2108–21. doi: 10.1200/JCO.2004.02.106. [DOI] [PubMed] [Google Scholar]
- 32.Leveque D, Carvalho MC, Maloisel F. Review. Clinical pharmacokinetics of bortezomib. In Vivo. 2007;21(2):273–8. [PubMed] [Google Scholar]
- 33.Uttamsingh V, Lu C, Miwa G, Gan LS. Relative contributions of the five major human cytochromes P450, 1A2, 2C9, 2C19, 2D6, and 3A4, to the hepatic metabolism of the proteasome inhibitor bortezomib. Drug Metab Dispos. 2005;33(11):1723–8. doi: 10.1124/dmd.105.005710. [DOI] [PubMed] [Google Scholar]
- 34.Pekol T, Daniels JS, Labutti J, Parsons I, Nix D, Baronas E, et al. Human metabolism of the proteasome inhibitor bortezomib: identification of circulating metabolites. Drug Metab Dispos. 2005;33(6):771–7. doi: 10.1124/dmd.104.002956. [DOI] [PubMed] [Google Scholar]
- 35.Venkatakrishnan K, Rader M, Ramanathan RK, Ramalingam S, Chen E, Riordan W, et al. Effect of the CYP3A inhibitor ketoconazole on the pharmacokinetics and pharmacodynamics of bortezomib in patients with advanced solid tumors: a prospective, multicenter, open-label, randomized, two-way crossover drug-drug interaction study. Clin Ther. 2009;31(Pt 2):2444–58. doi: 10.1016/j.clinthera.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 36.Hellmann A, Rule S, Walewski J, Shpilberg O, Feng H, van de Velde H, et al. Effect of cytochrome P450 3A4 inducers on the pharmacokinetic, pharmacodynamic and safety profiles of bortezomib in patients with multiple myeloma or non-Hodgkin’s lymphoma. Clin Pharmacokinet. 2011;50(12):781–91. doi: 10.2165/11594410-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 37.Quinn DI, Nemunaitis J, Fuloria J, Britten CD, Gabrail N, Yee L, et al. Effect of the cytochrome P450 2C19 inhibitor omeprazole on the pharmacokinetics and safety profile of bortezomib in patients with advanced solid tumours, non-Hodgkin’s lymphoma or multiple myeloma. Clin Pharmacokinet. 2009;48(3):199–209. doi: 10.2165/00003088-200948030-00006. [DOI] [PubMed] [Google Scholar]
- 38.Kaygusuz I, Toptas T, Aydin F, Uzay A, Firatli-Tuglular T, Bayik M. Bortezomib in patients with renal impairment. Hematology. 2011;16(4):200–8. doi: 10.1179/102453311X13025568941880. [DOI] [PubMed] [Google Scholar]
- 39.Williamson MJ, Silva MD, Terkelsen J, Robertson R, Yu L, Xia C, et al. The relationship among tumor architecture, pharmacokinetics, pharmacodynamics, and efficacy of bortezomib in mouse xenograft models. Mol Cancer Ther. 2009;8(12):3234–43. doi: 10.1158/1535-7163.MCT-09-0239. [DOI] [PubMed] [Google Scholar]
- 40.Ashley JD, Stefanick JF, Schroeder VA, Suckow MA, Kiziltepe T, Bilgicer B. Liposomal bortezomib nanoparticles via boronic ester prodrug formulation for improved therapeutic efficacy in vivo. J Med Chem. 2014;57(12):5282–92. doi: 10.1021/jm500352v. [DOI] [PubMed] [Google Scholar]
- 41.Shen S, Du XJ, Liu J, Sun R, Zhu YH, Wang J. Delivery of bortezomib with nanoparticles for basal-like triple-negative breast cancer therapy. J Control Release. 2015;208:14–24. doi: 10.1016/j.jconrel.2014.12.043. [DOI] [PubMed] [Google Scholar]
- 42.Swami A, Reagan MR, Basto P, Mishima Y, Kamaly N, Glavey S, et al. Engineered nanomedicine for myeloma and bone microenvironment targeting. Proc Natl Acad Sci U S A. 2014;111(28):10287–92. doi: 10.1073/pnas.1401337111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348(26):2609–17. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 44.Vij R, Wang M, Kaufman JL, Lonial S, Jakubowiak AJ, Stewart AK, et al. An open-label, single-arm, phase 2 (PX-171-004) study of single-agent carfilzomib in bortezomib-naive patients with relapsed and/or refractory multiple myeloma. Blood. 2012;119(24):5661–70. doi: 10.1182/blood-2012-03-414359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar S, Rajkumar SV. Many facets of bortezomib resistance/susceptibility. Blood. 2008;112(6):2177–8. doi: 10.1182/blood-2008-07-167767. [DOI] [PubMed] [Google Scholar]
- 46.McConkey DJ, Zhu K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Updat. 2008;11(4-5):164–79. doi: 10.1016/j.drup.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 47.Arastu-Kapur S, Anderl JL, Kraus M, Parlati F, Shenk KD, Lee SJ, et al. Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: a link to clinical adverse events. Clin Cancer Res. 2011;17(9):2734–43. doi: 10.1158/1078-0432.CCR-10-1950. [DOI] [PubMed] [Google Scholar]
- 48.Voortman J, Checinska A, Giaccone G. The proteasomal and apoptotic phenotype determine bortezomib sensitivity of non-small cell lung cancer cells. Mol Cancer. 2007;6:73. doi: 10.1186/1476-4598-6-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moreau P, Pylypenko H, Grosicki S, Karamanesht I, Leleu X, Grishunina M, et al. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority study. Lancet Oncol. 2011;12(5):431–40. doi: 10.1016/S1470-2045(11)70081-X. [DOI] [PubMed] [Google Scholar]
- 50.Bringhen S, Larocca A, Rossi D, Cavalli M, Genuardi M, Ria R, et al. Efficacy and safety of once-weekly bortezomib in multiple myeloma patients. Blood. 2010;116(23):4745–53. doi: 10.1182/blood-2010-07-294983. [DOI] [PubMed] [Google Scholar]
- 51.Reeder CB, Reece DE, Kukreti V, Chen C, Trudel S, Laumann K, et al. Once-versus twice-weekly bortezomib induction therapy with CyBorD in newly diagnosed multiple myeloma. Blood. 2010;115(16):3416–7. doi: 10.1182/blood-2010-02-271676. [DOI] [PubMed] [Google Scholar]
- 52.Herndon TM, Deisseroth A, Kaminskas E, Kane RC, Koti KM, Rothmann MD, et al. U.s. Food and Drug Administration approval: carfilzomib for the treatment of multiple myeloma. Clin Cancer Res. 2013;19(17):4559–63. doi: 10.1158/1078-0432.CCR-13-0755. [DOI] [PubMed] [Google Scholar]
- 53.Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc Natl Acad Sci U S A. 1999;96(18):10403–8. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elofsson M, Splittgerber U, Myung J, Mohan R, Crews CM. Towards subunit-specific proteasome inhibitors: synthesis and evaluation of peptide alpha′,beta′-epoxyketones. Chem Biol. 1999;6(11):811–22. doi: 10.1016/s1074-5521(99)80128-8. [DOI] [PubMed] [Google Scholar]
- 55.Kim KB, Myung J, Sin N, Crews CM. Proteasome inhibition by the natural products epoxomicin and dihydroeponemycin: insights into specificity and potency. Bioorg Med Chem Lett. 1999;9(23):3335–40. doi: 10.1016/s0960-894x(99)00612-5. [DOI] [PubMed] [Google Scholar]
- 56.Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007;67(13):6383–91. doi: 10.1158/0008-5472.CAN-06-4086. [DOI] [PubMed] [Google Scholar]
- 57.Schrader J, Henneberg F, Mata RA, Tittmann K, Schneider TR, Stark H, et al. The inhibition mechanism of human 20S proteasomes enables next-generation inhibitor design. Science. 2016;353(6299):594–8. doi: 10.1126/science.aaf8993. [DOI] [PubMed] [Google Scholar]
- 58.Carmony K, Lee W, Kim KB. High-Resolution Snapshots of Proteasome Inhibitors in Action Revise Inhibition Paradigms and Inspire Next-Generation Inhibitor Design. Chembiochem. 2016;17(22):2115–7. doi: 10.1002/cbic.201600488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Redic K. Carfilzomib: a novel agent for multiple myeloma. J Pharm Pharmacol. 2013;65(8):1095–106. doi: 10.1111/jphp.12072. [DOI] [PubMed] [Google Scholar]
- 60.Hewings DS, Flygare JA, Wertz IE, Bogyo M. Activity-based probes for the multicatalytic proteasome. FEBS J. 2017;284(10):1540–54. doi: 10.1111/febs.14016. [DOI] [PubMed] [Google Scholar]
- 61.Kraus M, Ruckrich T, Reich M, Gogel J, Beck A, Kammer W, et al. Activity patterns of proteasome subunits reflect bortezomib sensitivity of hematologic malignancies and are variable in primary human leukemia cells. Leukemia. 2007;21(1):84–92. doi: 10.1038/sj.leu.2404414. [DOI] [PubMed] [Google Scholar]
- 62.Ruckrich T, Kraus M, Gogel J, Beck A, Ovaa H, Verdoes M, et al. Characterization of the ubiquitin-proteasome system in bortezomib-adapted cells. Leukemia. 2009;23(6):1098–105. doi: 10.1038/leu.2009.8. [DOI] [PubMed] [Google Scholar]
- 63.Kammerl IE, Dann A, Mossina A, Brech D, Lukas C, Vosyka O, et al. Impairment of Immunoproteasome Function by Cigarette Smoke and in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2016;193(11):1230–41. doi: 10.1164/rccm.201506-1122OC. [DOI] [PubMed] [Google Scholar]
- 64.O′Connor OA, Stewart AK, Vallone M, Molineaux CJ, Kunkel LA, Gerecitano JF, et al. A phase 1 dose escalation study of the safety and pharmacokinetics of the novel proteasome inhibitor carfilzomib (PR-171) in patients with hematologic malignancies. Clin Cancer Res. 2009;15(22):7085–91. doi: 10.1158/1078-0432.CCR-09-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alsina M, Trudel S, Furman RR, Rosen PJ, O′Connor OA, Comenzo RL, et al. A phase I single-agent study of twice-weekly consecutive-day dosing of the proteasome inhibitor carfilzomib in patients with relapsed or refractory multiple myeloma or lymphoma. Clin Cancer Res. 2012;18(17):4830–40. doi: 10.1158/1078-0432.CCR-11-3007. [DOI] [PubMed] [Google Scholar]
- 66.Stewart AK, Rajkumar SV, Dimopoulos MA, Masszi T, Spicka I, Oriol A, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372(2):142–52. doi: 10.1056/NEJMoa1411321. [DOI] [PubMed] [Google Scholar]
- 67.Dimopoulos MA, Goldschmidt H, Niesvizky R, Joshua D, Chng WJ, Oriol A, et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): an interim overall survival analysis of an open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18(10):1327–37. doi: 10.1016/S1470-2045(17)30578-8. [DOI] [PubMed] [Google Scholar]
- 68.Yang J, Wang Z, Fang Y, Jiang J, Zhao F, Wong H, et al. Pharmacokinetics, pharmacodynamics, metabolism, distribution, and excretion of carfilzomib in rats. Drug Metab Dispos. 2011;39(10):1873–82. doi: 10.1124/dmd.111.039164. [DOI] [PubMed] [Google Scholar]
- 69.Papadopoulos KP, Burris HA, 3rd, Gordon M, Lee P, Sausville EA, Rosen PJ, et al. A phase I/II study of carfilzomib 2-10-min infusion in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;72(4):861–8. doi: 10.1007/s00280-013-2267-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Z, Yang J, Kirk C, Fang Y, Alsina M, Badros A, et al. Clinical pharmacokinetics, metabolism, and drug-drug interaction of carfilzomib. Drug Metab Dispos. 2013;41(1):230–7. doi: 10.1124/dmd.112.047662. [DOI] [PubMed] [Google Scholar]
- 71.Badros AZ, Vij R, Martin T, Zonder JA, Kunkel L, Wang Z, et al. Carfilzomib in multiple myeloma patients with renal impairment: pharmacokinetics and safety. Leukemia. 2013;27(8):1707–14. doi: 10.1038/leu.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Quach H, White D, Spencer A, Ho PJ, Bhutani D, White M, et al. Pharmacokinetics and safety of carfilzomib in patients with relapsed multiple myeloma and end-stage renal disease (ESRD): an open-label, single-arm, phase I study. Cancer Chemother Pharmacol. 2017;79(6):1067–76. doi: 10.1007/s00280-017-3287-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Korde N, Roschewski M, Zingone A, Kwok M, Manasanch EE, Bhutani M, et al. Treatment With Carfilzomib-Lenalidomide-Dexamethasone With Lenalidomide Extension in Patients With Smoldering or Newly Diagnosed Multiple Myeloma. JAMA Oncol. 2015;1(6):746–54. doi: 10.1001/jamaoncol.2015.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chauhan D, Tian Z, Zhou B, Kuhn D, Orlowski R, Raje N, et al. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin Cancer Res. 2011;17(16):5311–21. doi: 10.1158/1078-0432.CCR-11-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010;70(5):1970–80. doi: 10.1158/0008-5472.CAN-09-2766. [DOI] [PubMed] [Google Scholar]
- 76.Richardson PG, Baz R, Wang M, Jakubowiak AJ, Laubach JP, Harvey RD, et al. Phase 1 study of twice-weekly ixazomib, an oral proteasome inhibitor, in relapsed/refractory multiple myeloma patients. Blood. 2014;124(7):1038–46. doi: 10.1182/blood-2014-01-548826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moreau P, Masszi T, Grzasko N, Bahlis NJ, Hansson M, Pour L, et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N Engl J Med. 2016;374(17):1621–34. doi: 10.1056/NEJMoa1516282. [DOI] [PubMed] [Google Scholar]
- 78.Kumar SK, LaPlant B, Roy V, Reeder CB, Lacy MQ, Gertz MA, et al. Phase 2 trial of ixazomib in patients with relapsed multiple myeloma not refractory to bortezomib. Blood Cancer J. 2015;5:e338. doi: 10.1038/bcj.2015.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kumar SK, Bensinger WI, Zimmerman TM, Reeder CB, Berenson JR, Berg D, et al. Phase 1 study of weekly dosing with the investigational oral proteasome inhibitor ixazomib in relapsed/refractory multiple myeloma. Blood. 2014;124(7):1047–55. doi: 10.1182/blood-2014-01-548941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gupta N, Hanley MJ, Venkatakrishnan K, Bessudo A, Rasco DW, Sharma S, et al. Effects of Strong CYP3A Inhibition and Induction on the Pharmacokinetics of Ixazomib, an Oral Proteasome Inhibitor: Results of Drug-Drug Interaction Studies in Patients With Advanced Solid Tumors or Lymphoma and a Physiologically Based Pharmacokinetic Analysis. J Clin Pharmacol. 2018;58(2):180–92. doi: 10.1002/jcph.988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gupta N, Diderichsen PM, Hanley MJ, Berg D, van de Velde H, Harvey RD, et al. Population Pharmacokinetic Analysis of Ixazomib, an Oral Proteasome Inhibitor, Including Data from the Phase III TOURMALINE-MM1 Study to Inform Labelling. Clin Pharmacokinet. 2017;56(11):1355–68. doi: 10.1007/s40262-017-0526-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gupta N, Hanley MJ, Venkatakrishnan K, Perez R, Norris RE, Nemunaitis J, et al. Pharmacokinetics of ixazomib, an oral proteasome inhibitor, in solid tumour patients with moderate or severe hepatic impairment. Br J Clin Pharmacol. 2016;82(3):728–38. doi: 10.1111/bcp.12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Verbrugge SE, Assaraf YG, Dijkmans BA, Scheffer GL, Al M, den Uyl D, et al. Inactivating PSMB5 mutations and P-glycoprotein (multidrug resistance-associated protein/ATP-binding cassette B1) mediate resistance to proteasome inhibitors: ex vivo efficacy of (immuno)proteasome inhibitors in mononuclear blood cells from patients with rheumatoid arthritis. J Pharmacol Exp Ther. 2012;341(1):174–82. doi: 10.1124/jpet.111.187542. [DOI] [PubMed] [Google Scholar]
- 84.Zhou HJ, Aujay MA, Bennett MK, Dajee M, Demo SD, Fang Y, et al. Design and Synthesis of an Orally Bioavailable and Selective Peptide Epoxyketone Proteasome Inhibitor (PR-047) J Med Chem. 2009 doi: 10.1021/jm801329v. [DOI] [PubMed] [Google Scholar]
- 85.Chauhan D, Singh AV, Aujay M, Kirk CJ, Bandi M, Ciccarelli B, et al. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood. 2010;116(23):4906–15. doi: 10.1182/blood-2010-04-276626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rajan AM, Kumar S. New investigational drugs with single-agent activity in multiple myeloma. Blood Cancer J. 2016;6(7):e451. doi: 10.1038/bcj.2016.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vij R, Savona M, Siegel D, Kaufman J, Badros A, Ghobrial I, et al. Clinical Profile of Single-Agent Oprozomib in Patients (Pts) with Multiple Myeloma (MM): Updated Results from a Multicenter, Open-Label, Dose Escalation Phase 1b/2 Study. Blood. 2014;124:34. [Google Scholar]
- 88.Ghobrial I, Savona M, Vij R, Siegel D, Badros A, Kaufman J, et al. Final Results from a Multicenter, Open-Label, Dose-Escalation Phase 1b/2 Study of Single-Agent Oprozomib in Patients with Hematologic Malignancies. Blood. 2016;128:2110. [Google Scholar]
- 89.Infante JR, Mendelson DS, Burris HA, 3rd, Bendell JC, Tolcher AW, Gordon MS, et al. A first-in-human dose-escalation study of the oral proteasome inhibitor oprozomib in patients with advanced solid tumors. Invest New Drugs. 2016;34(2):216–24. doi: 10.1007/s10637-016-0327-x. [DOI] [PubMed] [Google Scholar]
- 90.Wang Z, Fang Y, Teague J, Wong H, Morisseau C, Hammock BD, et al. In Vitro Metabolism of Oprozomib, an Oral Proteasome Inhibitor: Role of Epoxide Hydrolases and Cytochrome P450s. Drug Metab Dispos. 2017 doi: 10.1124/dmd.117.075226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Piva R, Ruggeri B, Williams M, Costa G, Tamagno I, Ferrero D, et al. CEP-18770: A novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood. 2008;111(5):2765–75. doi: 10.1182/blood-2007-07-100651. [DOI] [PubMed] [Google Scholar]
- 92.Sanchez E, Li M, Steinberg JA, Wang C, Shen J, Bonavida B, et al. The proteasome inhibitor CEP-18770 enhances the anti-myeloma activity of bortezomib and melphalan. Br J Haematol. 2010;148(4):569–81. doi: 10.1111/j.1365-2141.2009.08008.x. [DOI] [PubMed] [Google Scholar]
- 93.Sanchez E, Li M, Li J, Wang C, Chen H, Jones-Bolin S, et al. CEP-18770 (delanzomib) in combination with dexamethasone and lenalidomide inhibits the growth of multiple myeloma. Leuk Res. 2012;36(11):1422–7. doi: 10.1016/j.leukres.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 94.Gallerani E, Zucchetti M, Brunelli D, Marangon E, Noberasco C, Hess D, et al. A first in human phase I study of the proteasome inhibitor CEP-18770 in patients with advanced solid tumours and multiple myeloma. Eur J Cancer. 2013;49(2):290–6. doi: 10.1016/j.ejca.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 95.Vogl DT, Martin TG, Vij R, Hari P, Mikhael JR, Siegel D, et al. Phase I/II study of the novel proteasome inhibitor delanzomib (CEP-18770) for relapsed and refractory multiple myeloma. Leuk Lymphoma. 2017:1–11. doi: 10.1080/10428194.2016.1263842. [DOI] [PubMed] [Google Scholar]
- 96.Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora. Angew Chem Int Ed Engl. 2003;42(3):355–7. doi: 10.1002/anie.200390115. [DOI] [PubMed] [Google Scholar]
- 97.Groll M, Huber R, Potts BC. Crystal structures of Salinosporamide A (NPI-0052) and B (NPI-0047) in complex with the 20S proteasome reveal important consequences of beta-lactone ring opening and a mechanism for irreversible binding. J Am Chem Soc. 2006;128(15):5136–41. doi: 10.1021/ja058320b. [DOI] [PubMed] [Google Scholar]
- 98.Potts BC, Albitar MX, Anderson KC, Baritaki S, Berkers C, Bonavida B, et al. Marizomib, a proteasome inhibitor for all seasons: preclinical profile and a framework for clinical trials. Curr Cancer Drug Targets. 2011;11(3):254–84. doi: 10.2174/156800911794519716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Macherla VR, Mitchell SS, Manam RR, Reed KA, Chao TH, Nicholson B, et al. Structure-activity relationship studies of salinosporamide A (NPI-0052), a novel marine derived proteasome inhibitor. J Med Chem. 2005;48(11):3684–7. doi: 10.1021/jm048995+. [DOI] [PubMed] [Google Scholar]
- 100.Chauhan D, Catley L, Li G, Podar K, Hideshima T, Velankar M, et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell. 2005;8(5):407–19. doi: 10.1016/j.ccr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 101.Manam RR, McArthur KA, Chao TH, Weiss J, Ali JA, Palombella VJ, et al. Leaving groups prolong the duration of 20S proteasome inhibition and enhance the potency of salinosporamides. J Med Chem. 2008;51(21):6711–24. doi: 10.1021/jm800548b. [DOI] [PubMed] [Google Scholar]
- 102.Singh AV, Palladino MA, Lloyd GK, Potts BC, Chauhan D, Anderson KC. Pharmacodynamic and efficacy studies of the novel proteasome inhibitor NPI-0052 (marizomib) in a human plasmacytoma xenograft murine model. Br J Haematol. 2010;149(4):550–9. doi: 10.1111/j.1365-2141.2010.08144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ruiz S, Krupnik Y, Keating M, Chandra J, Palladino M, McConkey D. The proteasome inhibitor NPI-0052 is a more effective inducer of apoptosis than bortezomib in lymphocytes from patients with chronic lymphocytic leukemia. Mol Cancer Ther. 2006;5(7):1836–43. doi: 10.1158/1535-7163.MCT-06-0066. [DOI] [PubMed] [Google Scholar]
- 104.Chauhan D, Singh AV, Ciccarelli B, Richardson PG, Palladino MA, Anderson KC. Combination of novel proteasome inhibitor NPI-0052 and lenalidomide trigger in vitro and in vivo synergistic cytotoxicity in multiple myeloma. Blood. 2010;115(4):834–45. doi: 10.1182/blood-2009-03-213009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Harrison SJ, Mainwaring P, Price T, Millward MJ, Padrik P, Underhill CR, et al. Phase I Clinical Trial of Marizomib (NPI-0052) in Patients with Advanced Malignancies Including Multiple Myeloma: Study NPI-0052-102 Final Results. Clin Cancer Res. 2016;22(18):4559–66. doi: 10.1158/1078-0432.CCR-15-2616. [DOI] [PubMed] [Google Scholar]
- 106.Spencer A, Harrison S, Zonder J, Badros A, Laubach J, Bergin K, et al. A phase 1 clinical trial evaluating marizomib, pomalidomide and low-dose dexamethasone in relapsed and refractory multiple myeloma (NPI-0052-107): final study results. Br J Haematol. 2018;180(1):41–51. doi: 10.1111/bjh.14987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pliquett F, Heinitz J. Changes in passive electric tissue properties of experimental tumors during carcinogenesis. Z Exp Chir Transplant Kunstliche Organe. 1989;22(1):38–44. [PubMed] [Google Scholar]
- 108.Di K, Lloyd GK, Abraham V, MacLaren A, Burrows FJ, Desjardins A, et al. Marizomib activity as a single agent in malignant gliomas: ability to cross the blood-brain barrier. Neuro Oncol. 2016;18(6):840–8. doi: 10.1093/neuonc/nov299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Petrocca F, Altschuler G, Tan SM, Mendillo ML, Yan H, Jerry DJ, et al. A genome-wide siRNA screen identifies proteasome addiction as a vulnerability of basal-like triple-negative breast cancer cells. Cancer Cell. 2013;24(2):182–96. doi: 10.1016/j.ccr.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bianchi G, Oliva L, Cascio P, Pengo N, Fontana F, Cerruti F, et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood. 2009;113(13):3040–9. doi: 10.1182/blood-2008-08-172734. [DOI] [PubMed] [Google Scholar]
- 111.Ling SC, Lau EK, Al-Shabeeb A, Nikolic A, Catalano A, Iland H, et al. Response of myeloma to the proteasome inhibitor bortezomib is correlated with the unfolded protein response regulator XBP-1. Haematologica. 2012;97(1):64–72. doi: 10.3324/haematol.2011.043331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Franke NE, Niewerth D, Assaraf YG, van Meerloo J, Vojtekova K, van Zantwijk CH, et al. Impaired bortezomib binding to mutant beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia. 2012;26(4):757–68. doi: 10.1038/leu.2011.256. [DOI] [PubMed] [Google Scholar]
- 113.Suzuki E, Demo S, Deu E, Keats J, Arastu-Kapur S, Bergsagel PL, et al. Molecular mechanisms of bortezomib resistant adenocarcinoma cells. PLoS One. 2011;6(12):e27996. doi: 10.1371/journal.pone.0027996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kale AJ, Moore BS. Molecular mechanisms of acquired proteasome inhibitor resistance. J Med Chem. 2012;55(23):10317–27. doi: 10.1021/jm300434z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ao L, Wu Y, Kim D, Jang ER, Kim K, Lee DM, et al. Development of peptide-based reversing agents for p-glycoprotein-mediated resistance to carfilzomib. Mol Pharm. 2012;9(8):2197–205. doi: 10.1021/mp300044b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gutman D, Morales AA, Boise LH. Acquisition of a multidrug-resistant phenotype with a proteasome inhibitor in multiple myeloma. Leukemia. 2009;23(11):2181–3. doi: 10.1038/leu.2009.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ettari R, Zappala M, Grasso S, Musolino C, Innao V, Allegra A. Immunoproteasome-selective and non-selective inhibitors: A promising approach for the treatment of multiple myeloma. Pharmacol Ther. 2018;182:176–92. doi: 10.1016/j.pharmthera.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 118.Miller Z, Lee W, Kim KB. The immunoproteasome as a therapeutic target for hematological malignancies. Curr Cancer Drug Targets. 2014;14(6):537–48. doi: 10.2174/1568009614666140723113139. [DOI] [PubMed] [Google Scholar]
- 119.Kisselev AF, Groettrup M. Subunit specific inhibitors of proteasomes and their potential for immunomodulation. Curr Opin Chem Biol. 2014;23:16–22. doi: 10.1016/j.cbpa.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Basler M, Mundt S, Bitzer A, Schmidt C, Groettrup M. The immunoproteasome: a novel drug target for autoimmune diseases. Clin Exp Rheumatol. 2015;33(4 Suppl 92):S74–9. [PubMed] [Google Scholar]
- 121.Micale N, Scarbaci K, Troiano V, Ettari R, Grasso S, Zappala M. Peptide-based proteasome inhibitors in anticancer drug design. Med Res Rev. 2014;34(5):1001–69. doi: 10.1002/med.21312. [DOI] [PubMed] [Google Scholar]
- 122.Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol. 2012;19(1):99–115. doi: 10.1016/j.chembiol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kazi A, Lawrence H, Guida WC, McLaughlin ML, Springett GM, Berndt N, et al. Discovery of a novel proteasome inhibitor selective for cancer cells over non-transformed cells. Cell Cycle. 2009;8(12):1940–51. doi: 10.4161/cc.8.12.8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lawrence HR, Kazi A, Luo Y, Kendig R, Ge Y, Jain S, et al. Synthesis and biological evaluation of naphthoquinone analogs as a novel class of proteasome inhibitors. Bioorg Med Chem. 2010;18(15):5576–92. doi: 10.1016/j.bmc.2010.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kazi A, Ozcan S, Tecleab A, Sun Y, Lawrence HR, Sebti SM. Discovery of PI-1840, a novel noncovalent and rapidly reversible proteasome inhibitor with anti-tumor activity. J Biol Chem. 2014;289(17):11906–15. doi: 10.1074/jbc.M113.533950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Miller Z, Kim KS, Lee DM, Kasam V, Baek SE, Lee KH, et al. Proteasome inhibitors with pyrazole scaffolds from structure-based virtual screening. J Med Chem. 2015;58(4):2036–41. doi: 10.1021/jm501344n. [DOI] [PubMed] [Google Scholar]
- 127.Blackburn C, Gigstad KM, Hales P, Garcia K, Jones M, Bruzzese FJ, et al. Characterization of a new series of non-covalent proteasome inhibitors with exquisite potency and selectivity for the 20S beta5-subunit. Biochem J. 2010;430(3):461–76. doi: 10.1042/BJ20100383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ge Y, Li A, Wu J, Feng H, Wang L, Liu H, et al. Design, synthesis and biological evaluation of novel non-peptide boronic acid derivatives as proteasome inhibitors. Eur J Med Chem. 2017;128:180–91. doi: 10.1016/j.ejmech.2017.01.034. [DOI] [PubMed] [Google Scholar]
- 129.Otvos L, Jr, Wade JD. Current challenges in peptide-based drug discovery. Front Chem. 2014;2:62. doi: 10.3389/fchem.2014.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Deshaies RJ. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 2014;12:94. doi: 10.1186/s12915-014-0094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Skaar JR, Pagan JK, Pagano M. SCF ubiquitin ligase-targeted therapies. Nat Rev Drug Discov. 2014;13(12):889–903. doi: 10.1038/nrd4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Reece DE, Sullivan D, Lonial S, Mohrbacher AF, Chatta G, Shustik C, et al. Pharmacokinetic and pharmacodynamic study of two doses of bortezomib in patients with relapsed multiple myeloma. Cancer Chemother Pharmacol. 2011;67(1):57–67. doi: 10.1007/s00280-010-1283-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Moreau P, Karamanesht II, Domnikova N, Kyselyova MY, Vilchevska KV, Doronin VA, et al. Pharmacokinetic, pharmacodynamic and covariate analysis of subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma. Clin Pharmacokinet. 2012;51(12):823–9. doi: 10.1007/s40262-012-0010-0. [DOI] [PubMed] [Google Scholar]
- 134.Papadopoulos KP, Siegel DS, Vesole DH, Lee P, Rosen ST, Zojwalla N, et al. Phase I study of 30-minute infusion of carfilzomib as single agent or in combination with low-dose dexamethasone in patients with relapsed and/or refractory multiple myeloma. J Clin Oncol. 2015;33(7):732–9. doi: 10.1200/JCO.2013.52.3522. [DOI] [PubMed] [Google Scholar]
- 135.Berenson JR, Cartmell A, Bessudo A, Lyons RM, Harb W, Tzachanis D, et al. CHAMPION-1: a phase 1/2 study of once-weekly carfilzomib and dexamethasone for relapsed or refractory multiple myeloma. Blood. 2016;127(26):3360–8. doi: 10.1182/blood-2015-11-683854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gupta N, Zhao Y, Hui AM, Esseltine DL, Venkatakrishnan K. Switching from body surface area-based to fixed dosing for the investigational proteasome inhibitor ixazomib: a population pharmacokinetic analysis. Br J Clin Pharmacol. 2015;79(5):789–800. doi: 10.1111/bcp.12542. [DOI] [PMC free article] [PubMed] [Google Scholar]