

ABSTRACT
Unresolved ER stress is involved in the onset and progression of several obesity-related metabolic disorders, including dyslipidemia and insulin resistance. Different epigenetic modifications may regulate ER stress response and consequently disease risks. These epigenetic phenomena encompass DNA and histone methylation patterns in ER stress genes and downstream signaling molecules, as well as microRNA expression. Our results suggest potential associations of methylation signatures at ER regulatory genes in white blood cells with an abdominal/central obesity marker (waist circumference), dyslipidemia, and insulin resistance. Interestingly, most of these genes were implicated in ER stress, as revealed by pathway enrichment analysis. Together, these findings add knowledge into the current understanding of relationships between obesity and accompanying complications with epigenetics and ER stress. Here, we comment about the implication of ER stress in central/abdominal adiposity, dyslipidemia, and insulin resistance, with an emphasis on the role that epigenetics may play on these pathological processes.
KEYWORDS: abdominal obesity, dyslipidemia, Epigenetics, ER stress, insulin resistance
Introduction
Overweight and obesity are two pathological conditions characterized by excessive general body fat accumulation, with important adverse effects on health status [1]. In particular, increased central fat distribution, also known as abdominal obesity, is significantly associated with metabolic disturbances, including insulin resistance (IR), dyslipidemia, and inflammation [2]. Indeed, waist circumference (WC), an anthropometric marker of central/abdominal adiposity, is an accepted criterion for the diagnosis of metabolic syndrome [3]. Moreover, the measurement of WC has been suggested to assess obesity-related mortality in adults in addition to body mass index (BMI), as revealed by meta-regression analyses [4].
The relationship between excessive adiposity and metabolic dysfunctions involve multiple cellular mechanisms. One of them appears to be endoplasmic reticulum (ER) stress [5,6] which results from ER protein folding disruption, leading to abnormal accumulation of misfolded proteins within this organelle [7]. As a consequence, eukaryotic cells activate the unfolded protein response (UPR) aimed to restore ER homeostasis by increasing both degradation of misfolded proteins and, concomitantly, reducing protein translation [8]. However, under chronic ER stress, UPR trigger a set of prodeath signals such as mitochondrial-mediated apoptosis and autophagy that are involved in several obesity-related metabolic disorders [9].
Besides environmental factors, different epigenetic modifications may regulate ER stress response and consequently disease risks [10]. These included DNA and histone methylation patterns in or near ER stress gene promoters and interconnected downstream signaling molecules [10–12]. Also, microRNA (miRNA) expression constitutes a fine-tuning mechanism for optimal ER activity during stress conditions [13,14]. Thus, epigenetic phenomena may help to explain, at least in part, inter-individual differences in the susceptibility to developing excessive adiposity and associated comorbidities [15–17]. Moreover, these insights may lead to the search for epigenetic biomarkers to predict metabolic risk and to personalize therapeutic strategies for obesity prevention and management [18,19]. Here, we give a general overview about the potential implication of ER stress in abdominal adiposity, dyslipidemia, and IR, with an emphasis on the role that epigenetics play on these pathological processes.
Obesity, ER stress, dyslipidemia, and insulin resistance
Early experiments using cell culture and mouse models have demonstrated that excessive adiposity results in chronic ER stress, particularly in liver and adipose tissues [20]. Further studies have indicated a significant increase of ER stress in adipose tissue of obese subjects [21,22]. In turn, this adverse cellular event appears to be involved in the development of IR and progression to type 2 diabetes mellitus, where multiple mechanisms have been hypothesized [23]. In this context, ER stress can disrupt insulin receptor signaling through enhanced activation of c-Jun N-terminal kinase (JNK) and subsequent serine-phosphorylation of the insulin receptor substrate-1 (IRS-1), leading to decreased insulin action [20]. Moreover, it has been reported that ER stress state induces lipolysis in adipocytes, which primarily could respond to an adaptive response regulating energy homeostasis [24]. Nevertheless, sustained ER stress triggers an accelerated free fatty acid efflux from adipocytes to the bloodstream and other tissues, thus eventually contributing to lipotoxicity, dyslipidemia, and IR [24]. In addition, obesity-induced ER stress has been linked to autophagy, chronic inflammation, and β-cell apoptosis, all of which are key components implicated in the pathogenesis of IR and metabolic syndrome [25,26]. Furthermore, ER-stress-induced IR may also be the result of increased lipogenesis in the liver and the subsequent intracellular lipid accumulation as well as upregulation of gluconeogenic gene expression [23]. Interestingly, caloric restriction reduced ER stress and improved hepatic insulin action by suppressing JNK-mediated IRS-1 serine-phosphorylation in obese mice [27]. Also, exercise training ameliorated ER stress and IR in high-fat-induced obese rats [28]. Furthermore, markers of ER stress in adipose tissue significantly decreased after surgery-induced weight loss [29].
Epigenetics of ER stress and obesity phenotypes
In a previous study [10], 15 differentially methylated CpG sites within ER regulatory genes in white blood cells that were associated with general obesity (BMI) in an adult population: cg08188400 (MAP2K7), cg20541779 (CASP12), cg24776411 (EIF2AK1), cg14190817 (HSPA5), cg21376454 (ERN1), cg06666486 (EIF2AK1), cg03211481 (DNAJC1), cg18357645 (OS9), cg05801879 (MBTPS1), cg20964082 (ERO1LB), cg17300868 (NFE2L2), cg03384128 (EIF2AK4), cg02712587 (EIF2AK4), cg04972384 (SELS), and cg02240686 (EIF2AK2). In addition to BMI, methylation signatures at cg20964082 (ERO1LB), cg17300868 (NFE2L2), cg05801879 (MBTPS1), and cg03384128 (EIF2AK4) similarly correlated with total fat mass [10].
In a new analysis, methylation patterns at the abovementioned 15 CpGs also correlated with an abdominal obesity marker such as WC (Fig. 1). Interestingly, obesity-associated genes affected key cellular processes in the ER including protein recognition (DNAJC1, HSPA5), protein targeting (ERO1LB, OS9), and ER-induced degradation (SELS). More importantly, most of them were implicated in ER stress, including the master stress sensors ERN1, EIF2AK1, EIF2AK2, EIF2AK4, and downstream regulators such as MAP2K7, CASP12, MBTPS1, and NFE2L2, as revealed by pathway enrichment analysis. In agreement with our findings, a transgenerational set of epigenetic modifications (mainly reduced accumulation of methylated histones in Lxrα/ Nr1h3 and Ero1-α gene promoters) led to up-regulation of lipogenesis and ER stress pathways in the liver of C57BL/6 mice fed a high-fat diet [11]. Additionally, the combination of offspring exposure to maternal obesity and the consumption of an obesogenic diet in mice triggered altered UPR signaling rhythmicity, cellular apoptosis, and hypermethylation of GRP78, a chaperone protein and a master regulator of ER homeostasis [12]. Furthermore, treatment of hepatocytes from high-fat diet obese mice with the specialized proresolving lipid mediator maresin 1 (MaR1) protected cells from lipotoxic and hypoxia-induced endoplasmic reticulum stress via specific miRNA signatures targeting both protein folding and apoptosis [30]. Also, MaR1 ameliorated liver steatosis by reducing lipogenesis, while promoting fatty acid oxidation and autophagy [31].
Figure 1.
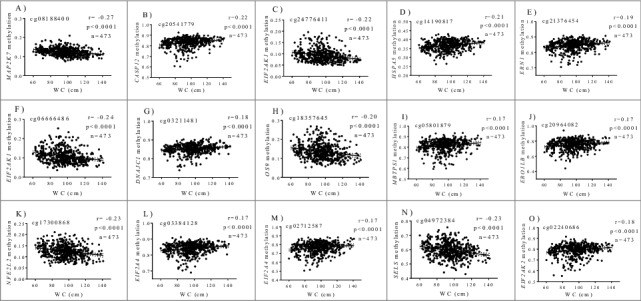
Correlations between methylation levels (beta values) at ER stress genes and WC after adjustments for age and sex. (A) cg08188400, MAP2K7 (B) cg20541779, CASP12 (C) cg24776411, EIF2AK1 (D) cg14190817, HSPA5 (E) cg21376454, ERN1 (F) cg06666486, EIF2AK1 (G) cg03211481, DNAJC1 (H) cg18357645, OS9 (I) cg05801879, MBTPS1 (J) cg20964082, ERO1LB (K) cg17300868, NFE2L2 (L) cg03384128, EIF2AK4 (M) cg02712587, EIF2AK4 (N) cg04972384, SELS (O) cg02240686, EIF2AK2.
It is important to highlight that methylation signatures in blood cells concerning obesity features have been consistently reflected in other tissues. For instance, a methylation map in blood leukocytes mirrored methylation marks found in subcutaneous adipose tissue, which efficiently discriminated obesity from non-obesity status [32]. Moreover, gene methylation parallelisms between white blood cells and oral mucosa samples in relation to overweight and insulin sensibility have been reported [33].
Epigenetics of ER stress, dyslipidemia, and insulin resistance
In a previous investigation [10], statistically significant associations between methylation patterns at cg20964082 (ERO1LB) and cg17300868 (NFE2L2) and insulin and HOMA-IR index were found, whereas cg05801879 (MBTPS1) and cg03384128 (EIF2AK4) were specifically associated with serum triglyceride levels. In a new approach, the methylation status of the aforementioned 4 CpG sites was compared by categories of metabolic markers (Fig. 2). In this sense, increased concentrations of insulin and HOMA-IR index were accompanied by lower methylation levels at cg20964082 (ERO1LB) and cg17300868 (NFE2L2), while a reduced methylation at cg05801879 (MBTPS1), and cg03384128 (EIF2AK4) was found in subjects with higher serum triglyceride levels (Fig. 2). Consistently, impaired glucose homeostasis was found in F2 mice progeny from male founder obesity, which was partially influenced by hepatic ER stress in a sex-specific manner as well as by altered DNA methylation at the Nr1h3 locus, a nuclear factor involved in metabolic syndrome [34]. In vitro and in vivo experiments revealed that ER stress plays an important role in the increase of hepatic glucose production in obesity and diabetes partially by decreasing STAT3 deacetylation [35]. In addition, exposure to the high-fat diet led to hyperacetylation of proteins involved in gluconeogenesis, liver injury and the ER stress response [36].
Figure 2.
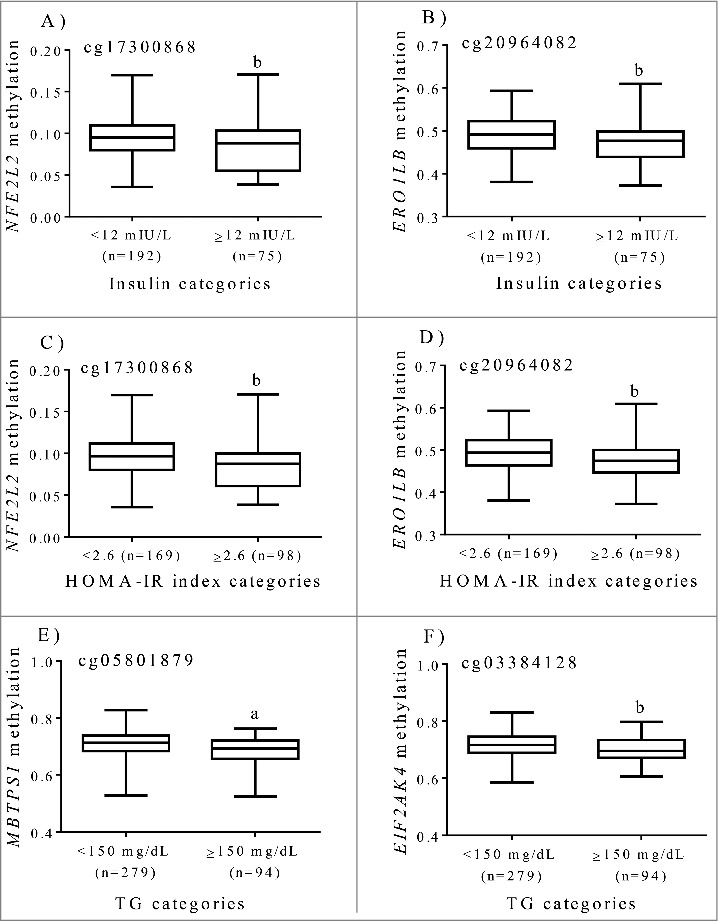
Tenth, 25th, 50th, 75th, and 90th percentiles of methylation levels (beta values) at ER regulatory genes by categories of insulin, HOMA-IR and triglycerides after adjustments for age and sex in subjects with excessive weight. (A) cg17300868, NFE2L2 (B) cg20964082, ERO1LB (C) cg17300868, NFE2L2 (D) cg20964082, ERO1LB (E) cg05801879, MBTPS1 (F) cg03384128, EIF2AK4. a p < 0.0001. b p < 0.05.
Nevertheless, no differences in the methylation status of CpG islands located in the NFE2L2 gene promoter (encoding Nrf2) were found between normal and gestational diabetic mellitus cells [37]. Notably, demethylation of the KEAP1 gene promoter, a negative regulator of NFE2L2, was evidenced in some diabetic complications such as cardiomyopathy and cataracts [38,39]. Consequently, decreased NFE2L2 activity by KEAP1-mediated proteosomal degradation may lead to transcription repression of antioxidant enzymes and alteration of redox-balance up on diabetes. Moreover, hyperglycemia altered the transcriptional function of NFE2L2 to promote antioxidant gene expression through enhance histone methylation at crucial binding element regions [40].
Of note, the administration of sodium butyrate, a known activator of NFE2L2, increased Nrf2 expression at the transcriptional level to ameliorate diabetic nephropathy in mice, possibly by the epigenetic inhibition of histone deacetylase activity [41]. Also, upregulation of NFE2L2 by increasing miR-200a protected against diabetic nephropathy in mice treated with an analogue of curcumin [42]. Similarly, miR-708 was potently upregulated by triggering ER stress in pancreatic islets of ob/ob mice, whose overexpression suppressed β-cell proliferation and induced β-cell apoptosis [43]. Additionally, it was demonstrated that a modest increase in maternal dietary fat in mice programmed triglyceride storage in the liver of their offspring at the adult stage that was controlled by the eIF2α kinase Gcn2 (Eif2ak4), which stimulated epigenetic modification (trimethylation of lysine 4 of histone 3) at the Pparγ2 gene in the neonatal liver [44]. Although the Gcn2-dependent perinatal programming of liver triglyceride storage was proposed as an early nutrition adaptation, this event could predispose individuals to develop liver steatosis in adults. Interestingly, novel regulatory functions of miR-128-2 in cellular cholesterol homeostasis and induction of ER stress response were reported [45]. Likewise, miRNA-induced mitochondrial dysfunction and ER stress are engaged in diverse lipid and lipoprotein disorders by deregulating genes involved in control of intracellular lipid synthesis, fatty acid oxidation, and lipoprotein assembly [46].
Because ER stress is related to inflammation [47], associations between methylation levels at ER stress genes and serum concentrations of inflammatory markers were evaluated in a subsample of the MENA cohort (n=80). A modest positive correlation between HSPA5 methylation at cg14190817 and circulating C-reactive protein concentration was found (r=0.24, p=0.030), supporting a role of HSPA5 as a possible biomarker for inflammatory states [48].
Concluding remarks
Growing scientific evidence support the involvement of ER stress in the onset and progression of obesity-associated chronic diseases (Fig. 3). At the molecular level, ER stress response can be influenced by several epigenetic modifications. Together, our results suggest that methylation status at genes integrating the ER network could be an epigenetic mechanism underlying fat deposition, lipid metabolism, and insulin resistance. These findings contribute to the current understanding of relationships between obesity, epigenetics, and ER stress. Furthermore, this knowledge may favor the identification of potential ER targets as well as the implementation of precision medicine and nutrition strategies aimed to prevent and control excessive adiposity and related metabolic syndrome complications by targeting the epigenome.
Figure 3.
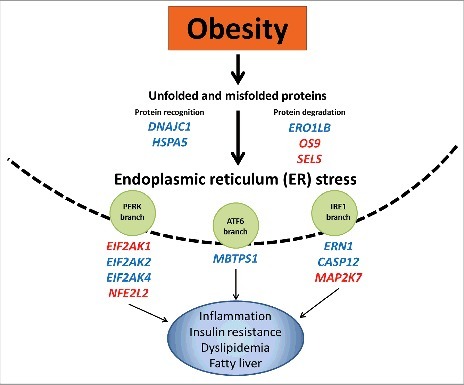
Epigenetic regulation of endoplasmic reticulum stress in obesity and associated metabolic diseases. Obesity induces a chronic activation of the unfolded protein response and consequently ER stress. Differentially methylated ER regulatory genes play a pivotal role in obesity-induced ER stress, leading to the development of metabolic disturbances. Hypomethylated (red color); hypermethyated (blue color).
Funding Statement
This work was supported by grants from the Government of Navarra (PT024), CIBERobn (CB12/03/30002) and MINECO (AGL2013-45554-R and BFU 2015-65937-R). O. R. L. was supported by a postdoctoral grant from National Council of Science and Technology (CONACyT, No. CVU. 444175), Mexico, in collaboration with the PhD program in Molecular Biology in Medicine of the University of Guadalajara (CONACyT, PNPC 000091), Mexico, and the University of Navarra (LE/97), Spain.
Disclosure of potential conflicts of interest
No potential conflicts of interest to declare.
References
- [1].World Health Organization (WHO) Fact sheet: Obesity and overweight. 2017. Available from: http://www.who.int/mediacentre/factsheets/fs311/en/.
- [2].Engin A. The Definition and Prevalence of Obesity and Metabolic Syndrome. Adv Exp Med Biol. 2017;960:1–17. doi: 10.1007/978-3-319-48382-5_1. [DOI] [PubMed] [Google Scholar]
- [3].Smith U. Abdominal obesity: a marker of ectopic fat accumulation. J Clin Invest. 2015;125(5):1790–1792. doi: 10.1172/JCI81507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Carmienke S, Freitag MH, Pischon T, et al.. General and abdominal obesity parameters and their combination in relation to mortality: a systematic review and meta-regression analysis. Eur J Clin Nutr. 2013;67(6):573–585. [DOI] [PubMed] [Google Scholar]
- [5].Pagliassotti MJ, Kim PY, Estrada AL, et al.. Endoplasmic reticulum stress in obesity and obesity-related disorders: An expanded view. Metabolism. 2016;65(9):1238–1246. doi: 10.1016/j.metabol.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yilmaz E. Endoplasmic Reticulum Stress and Obesity. Adv Exp Med Biol. 2017;960:261–276. doi: 10.1007/978-3-319-48382-5_11. [DOI] [PubMed] [Google Scholar]
- [7].Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173–194. doi: 10.1146/annurev-pathol-012513-104649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hiramatsu N, Chiang WC, Kurt TD, et al.. Multiple Mechanisms of Unfolded Protein Response-Induced Cell Death. Am J Pathol. 2015;185(7):1800–1808. doi: 10.1016/j.ajpath.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chakrabarti A, Chen AW, Varner JD. A review of the mammalian unfolded protein response. Biotechnol Bioeng. 2011;108(12):2777–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ramos-Lopez O, Riezu-Boj JI, Milagro FI, et al.. DNA methylation signatures at endoplasmic reticulum stress genes are associated with adiposity and insulin resistance. Mol Genet Metab. 2018;123(1):50–58. doi: 10.1016/j.ymgme.2017.11.011. [DOI] [PubMed] [Google Scholar]
- [11].Li J, Huang J, Li JS, et al.. Accumulation of endoplasmic reticulum stress and lipogenesis in the liver through generational effects of high fat diets. J Hepatol. 2012;56(4):900–907. doi: 10.1016/j.jhep.2011.10.018. [DOI] [PubMed] [Google Scholar]
- [12].Soeda J, Cordero P, Li J, et al.. Hepatic rhythmicity of endoplasmic reticulum stress is disrupted in perinatal and adult mice models of high-fat diet-induced obesity. Int J Food Sci Nutr. 2017;68(4):455–466. doi: 10.1080/09637486.2016.1261086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chitnis NS, Pytel D, Bobrovnikova-Marjon E, et al.. miR-211 is a prosurvival microRNA that regulates chop expression in a PERK-dependent manner. Mol Cell. 2012;48(3):353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gupta A, Hossain MM, Read DE, et al.. PERK regulated miR-424(322)-503 cluster fine-tunes activation of IRE1 and ATF6 during Unfolded Protein Response. Sci Rep. 2015;5:18304. doi: 10.1038/srep18304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Milagro FI, Martínez JA. Epigenetics of obesity and weight loss. Endocrinol Nutr. 2013;60(Suppl):12–14. [DOI] [PubMed] [Google Scholar]
- [16].Martínez JA, Milagro FI, Claycombe KJ, et al.. Epigenetics in adipose tissue, obesity, weight loss, and diabetes. Adv Nutr. 2014;5(1):71–81. doi: 10.3945/an.113.004705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Goni L, Milagro FI, Cuervo M, et al.. Single-nucleotide polymorphisms and DNA methylation markers associated with central obesity and regulation of body weight. Nutr Rev. 2014;72(11):673–690. doi: 10.3945/an.113.004705. [DOI] [PubMed] [Google Scholar]
- [18].Goni L, Cuervo M, Milagro FI, et al.. Future Perspectives of Personalized Weight Loss Interventions Based on Nutrigenetic, Epigenetic, and Metagenomic Data. J Nutr. 2016;146(4):905S–912S. doi: 10.3945/jn.115.218354. [DOI] [PubMed] [Google Scholar]
- [19].Ramos-Lopez O, Milagro FI, Allayee H, et al.. Guide for Current Nutrigenetic, Nutrigenomic, and Nutriepigenetic Approaches for Precision Nutrition Involving the Prevention and Management of Chronic Diseases Associated with Obesity. J Nutrigenet Nutrigenomics. 2017;10(1–2):43–62. doi: 10.1159/000477729. [DOI] [PubMed] [Google Scholar]
- [20].Ozcan U, Cao Q, Yilmaz E, et al.. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- [21].Sharma NK, Das SK, Mondal AK, et al.. Endoplasmic reticulum stress markers are associated with obesity in nondiabetic subjects. J Clin Endocrinol Metab. 2008;93(11):4532–4541. doi: 10.1210/jc.2008-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Boden G, Duan X, Homko C, et al.. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes. 2008;57(9):2438–2444. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Salvadó L, Palomer X, Barroso E, et al.. Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol Metab. 2015;26(8):438–448. doi: 10.1016/j.tem.2015.05.007. [DOI] [PubMed] [Google Scholar]
- [24].Deng J, Liu S, Zou L, et al.. Lipolysis response to endoplasmic reticulum stress in adipose cells. J Biol Chem. 2012;287(9):6240–6249. doi: 10.1074/jbc.M111.299115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kawasaki N, Asada R, Saito A, et al.. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep. 2012;2:799. doi: 10.1038/srep00799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Su J, Zhou L, Kong X, et al.. Endoplasmic reticulum is at the crossroads of autophagy, inflammation, and apoptosis signaling pathways and participates in the pathogenesis of diabetes mellitus. J Diabetes Res. 2013;2013:193461. doi: 10.1155/2013/193461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tsutsumi A, Motoshima H, Kondo T, et al.. Caloric restriction decreases ER stress in liver and adipose tissue in ob/ob mice. Biochem Biophys Res Commun. 2011;404(1):339–344. doi: 10.1016/j.bbrc.2010.11.120. [DOI] [PubMed] [Google Scholar]
- [28].da Luz G, Frederico MJ, da Silva S, et al.. Endurance exercise training ameliorates insulin resistance and reticulum stress in adipose and hepatic tissue in obese rats. Eur J Appl Physiol. 2011;111(9):2015–2023. doi: 10.1007/s00421-010-1802-2. [DOI] [PubMed] [Google Scholar]
- [29].Gregor MF, Yang L, Fabbrini E, et al.. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes. 2009;58(3):693–700. doi: 10.2337/db08-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rius B, Duran-Güell M, Flores-Costa R, et al.. The specialized proresolving lipid mediator maresin 1 protects hepatocytes from lipotoxic and hypoxia-induced endoplasmic reticulum stress. FASEB J. 2017;31(12):5384–5398. doi: 10.1096/fj.201700394R. [DOI] [PubMed] [Google Scholar]
- [31].Laiglesia LM, Lorente-Cebrián S, Martínez-Fernández L, et al.. Maresin 1 mitigates liver steatosis in ob/ob and diet-induced obese mice. Int J Obes (Lond). 2017; doi: 10.1038/ijo.2017.226. [DOI] [PubMed] [Google Scholar]
- [32].Crujeiras AB, Diaz-Lagares A, Sandoval J, et al.. DNA methylation map in circulating leukocytes mirrors subcutaneous adipose tissue methylation pattern: a genome-wide analysis from non-obese and obese patients. Sci Rep. 2017;7:41903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].San-Cristobal R, Navas-Carretero S, Milagro FI, et al.. Gene methylation parallelisms between peripheral blood cells and oral mucosa samples in relation to overweight. J Physiol Biochem. 2016;73(3):465–474. [DOI] [PubMed] [Google Scholar]
- [34].Park JH, Yoo Y, Cho M, et al.. Diet-induced obesity leads to metabolic dysregulation in offspring via endoplasmic reticulum stress in a sex-specific manner. Int J Obes (Lond). 2018;42(2):244–251. doi: 10.1038/ijo.2017.203. [DOI] [PubMed] [Google Scholar]
- [35].Kimura K, Yamada T, Matsumoto M, et al.. Endoplasmic reticulum stress inhibits STAT3-dependent suppression of hepatic gluconeogenesis via dephosphorylation and deacetylation. Diabetes. 2012;61(1):61–73. doi: 10.2337/db10-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kendrick AA, Choudhury M, Rahman SM, et al.. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J. 2011;433(3):505–514. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cheng X, Chapple SJ, Patel B, et al.. Gestational diabetes mellitus impairs Nrf2-mediated adaptive antioxidant defenses and redox signaling in fetal endothelial cells in utero. Diabetes. 2013;62(12):4088–4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Liu ZZ, Zhao XZ, Zhang XS, et al.. Promoter DNA demethylation of Keap1 gene in diabetic cardiomyopathy. Int J Clin Exp Pathol. 2014;7(12):8756–8762. [PMC free article] [PubMed] [Google Scholar]
- [39].Palsamy P, Ayaki M, Elanchezhian R, et al.. Promoter demethylation of Keap1 gene in human diabetic cataractous lenses. Biochem Biophys Res Commun. 2012;423(3):542–548. doi: 10.1016/j.bbrc.2012.05.164. [DOI] [PubMed] [Google Scholar]
- [40].Mishra M, Zhong Q, Kowluru RA. Epigenetic modifications of Nrf2-mediated glutamate-cysteine ligase: implications for the development of diabetic retinopathy and the metabolic memory phenomenon associated with its continued progression. Free Radic Biol Med. 2014;75:129–139. doi: 10.1016/j.freeradbiomed.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dong W, Jia Y, Liu X, et al.. Sodium butyrate activates NRF2 to ameliorate diabetic nephropathy possibly via inhibition of HDAC. J Endocrinol. 2017;232(1):71–83. doi: 10.1530/JOE-16-0322. [DOI] [PubMed] [Google Scholar]
- [42].Wu H, Kong L, Tan Y, et al.. C66 ameliorates diabetic nephropathy in mice by both upregulating NRF2 function via increase in miR-200a and inhibiting miR-21. Diabetologia. 2016;59(7):1558–1568. doi: 10.1007/s00125-016-3958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rodríguez-Comas J, Moreno-Asso A, Moreno-Vedia J, et al.. Stress-Induced MicroRNA-708 Impairs β-Cell Function and Growth. Diabetes. 2017;66(12):3029–3040. doi: 10.2337/db16-1569. [DOI] [PubMed] [Google Scholar]
- [44].Xu X, Hu J, McGrath BC, et al.. GCN2 in the brain programs PPARγ2 and triglyceride storage in the liver during perinatal development in response to maternal dietary fat. PLoS One. 2013;8(10):e75917. doi: 10.1371/journal.pone.0075917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Adlakha YK, Khanna S, Singh R, et al.. Pro-apoptotic miRNA-128-2 modulates ABCA1, ABCG1 and RXRα expression and cholesterol homeostasis. Cell Death Dis. 2013;4:e780. doi: 10.1038/cddis.2013.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Christian P, Su Q. MicroRNA regulation of mitochondrial and ER stress signaling pathways: implications for lipoprotein metabolism in metabolic syndrome. Am J Physiol Endocrinol Metab. 2014;307(9):E729–E737. doi: 10.1152/ajpendo.00194.2014. [DOI] [PubMed] [Google Scholar]
- [47].Grootjans J, Kaser A, Kaufman RJ, et al.. The unfolded protein response in immunity and inflammation. Nat Rev Immunol. 2016;16(8):469–484. doi: 10.1038/nri.2016.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Xiao F, Tan JZ, Xu XY, et al.. Increased levels of HSPA5 in the serum of patients with inflammatory myopathies-preliminary findings. Clin Rheumatol. 2015;34(4):715–720. doi: 10.1007/s10067-015-2911-4. [DOI] [PubMed] [Google Scholar]