

Abstract
Cardioprotection is a common goal of new therapeutic strategies in patients with coronary artery disease and/or left ventricular dysfunction. Myocardial damage following ischaemia/reperfusion injury lead to left ventricular adverse remodelling through many mechanisms arising from different cell types in different myocardial districts, namely the border and remote zone. Cardioprotection must face this complex, dynamic network of cooperating units. In this scenario, thyroid hormones can represent an effective therapeutic strategy due to the numerous actions and regulating mechanisms carried out at the level of the myocytes, interstitium and the vasculature, as well as to the activation of different pro-survival intracellular pathways involved in cardioprotection.
Keywords: Cardioprotection, acute myocardial infarction, left ventricular remodelling, heart failure, thyroid hormone, low T3 syndrome, hypothyroidism
Cardioprotection includes all methods and mechanisms that lead to the reduction in infarct size, and is thus involved in the evolution of post-ischaemic heart failure (HF). This is a growing research issue since ischaemic heart disease is the leading cause of morbidity and mortality worldwide. A key challenge for these studies is the unravelling of cardioprotection complexities such as the mechanisms leading to myocardial damage, the cascade of activated cellular signalling pathways, and the cellular and extracellular districts involved, which are strictly interconnected. This liaison between the heart and thyroid hormone (TH) adds complexity as it arises from the numerous TH effects on the heart, starting with cardiac differentiation in the transition from foetal to postnatal growth, during which THs induce transcriptional programming leading to the typical gene expression profile of the adult heart, as well as the maintenance of cardiovascular homeostasis directly through genomic and non-genomic actions and indirectly by TH regulating effects on other systemic pathways. Recent evidence highlights multiple actions of the TH system on the heart that can have a relevant role in cardioprotection, including the regulation of different intracellular pro-survival pathways, the preservation of mitochondrial function and morphology, the antifibrotic and pro-angiogenic effect, and also the potential induction of cell regeneration and growth. This review mainly focuses on these new actions of TH on the heart in relation to cardioprotection.
Definition of Cardioprotection
According to Heusch,[1] cardioprotection is a highly concerted spatio-temporal programme in which different factors with different pathophysiological mechanisms are involved, at different times, in minimising irreversible ischaemic damage and favouring the functional recovery of the injured myocardium. In this context intracellular pro-survival pathways are activated in a complex cross-talking system. Taken as whole, cardioprotection can actually be defined as a complex dynamic network of cooperating units, which is characterised by global properties independent of the details of the units in the absence of cooperation.[2] This suggests the need to take into account the intertwisted actions of the large number of components to understand the network of cardioprotection in order to give effectiveness to the therapeutic approaches. There are three main stages of myocardial damage:
in the acute phase, coronary occlusion is the ‘primum movens’ of myocardial damage, causing ischaemic injury;
this is followed by coronary revascularisation, i.e. percutaneous coronary angiography that causes reperfusion injury;
in the chronic phase, when left ventricular dysfunction develops, the post-ischaemic remodelling process is another dynamic mechanism influencing myocyte function and survival.
Furthermore, the complexity of cardioprotection lies in the activation of the molecular mechanisms of cytoprotection, including activation of heat shock proteins (HSP), protein kinase C (PKC), extracellular signal-regulated kinases (ERK), protein kinase B (AKT), p38 mitogen-activated protein kinase (p38MAPK), as well as the stimulation of cell growth, angiogenesis and metabolic adaptation. In addition, the maintenance of mitochondrial integrity is an emerging aspect of cardioprotection. Mitochondria participate in the regulation of myocardial calcium flux, myocyte cell death reactive oxygen species (ROS) generation and antioxidant response.[3] The mechanisms of mitochondrial injury are different during ischaemic/reperfusion (I/R):
The activation of the proapoptotic B-cell lymphoma 2 (BCL-2) proteins leads to mitochondrial outer membrane permeabilization, the release of cytochrome complex, caspase activation and apoptosis.[4]
Oxidative stress can lead to a sudden increase in inner mitochondrial membrane permeability that is attributable to the opening of the so-called permeability transition pore (PTP), whose opening is accompanied by the release of ROS and calcium.[5,6] This can culminate in the activation of calcium-dependent proteases (calpains) and lipases (cPLA2), inducing necrotic cell death.[7,8]
In the chronic phase, the activation of the neuroendocrine system, gathering renin-angiotensin-aldosterone and natriuretic peptides in the sympathetic autonomic nervous system, as well as prompting the inflammatory system, is initially protective and adaptive to haemodynamic changes induced by reduced cardiac output, conferring resistance to myocardial hypoxic injury.[9] However, when these systems are continuously activated, their initial protective mechanisms become at first less effective, then maladaptive and dangerous for the entire body and heart, contributing to myocardial damage and progression of HF syndrome.[10–12]
Thyroid System in Patients with Acute Myocardial Infarction
In the clinical setting of acute myocardial infarction (AMI) the most frequent alteration of TH metabolism is low triiodothyronine (T3) syndrome.[13] This occurs within 12 hours from the onset of symptoms, reaching the nadir at 72 hours. Low T3 syndrome is associated with a larger myocardial infarction (MI) and intense pro-inflammatory and stress response[14,15] and, similarly to higher post-ischaemic levels of reverse T3, the TH inactive metabolite is considered an independent predictor of short-term and long-term mortality.[16] Additionally, a large amount of clinical data support the clinical and prognostic role of altered thyroid metabolism in HF patients.[17–22] T3 circulating levels were higher in patients with New York Heart Association (NYHA) class I and II with respect to patients in NYHA class III and IV with high brain natriuretic peptide (BNP) levels and lower left ventricular ejection fraction (LVEF).[18,20,23]
Thyroid System in Patients with Heart Failure
Low T3 syndrome and subclinical hypothyroidism have been associated with a worse prognosis in patients with HF. In particular, the prognostic power of low T3 syndrome was independent and additive with respect to conventional clinical and cardiac variables, such as LVEF. Furthermore, the negative prognostic power is enhanced in patients with higher BNP concentration both in acute decompensated and chronic compensated HF.[23,24] Moreover, in patients with clinically stable HF, short-term synthetic T3 replacement therapy significantly improved neuroendocrine profiles and ventricular performance, characterised by an increase in stroke volume and reduction in the plasma circulating levels of noradrenaline, N-terminal pro-B-type natriuretic peptide (NT-proBNP) and aldosterone.[25] According to the clinical data, experimental evidence shows that abnormal TH metabolic patterns, such as hypothyroidism and low T3 syndrome, can cause several histological, molecular and structural abnormalities within the myocardium that can be reversed after normalisation of the TH metabolic profile.[26]
Thyroid System and Cardioprotection
A large amount of experimental evidence highlights that TH can effectively play a role in the complex scenario of cardioprotection (see Table 1),[27–43] and that this role is multifaceted due to the numerous actions and regulating mechanisms mediated directly or indirectly by the TH system (see Figure 1). In fact, TH-mediated actions are carried out at the level of the myocytes, the interstitium and the vasculature. TH also plays an important role in orchestrating the activation and function of different pro-survival intracellular pathways that are involved in cardioprotection. Furthermore, TH influences the neuroendocrine hormonal pathways activated in HF, as well as the inflammatory system.
Table 1: Experimental Conditions in which Thyroid Hormone Exert a Cardioprotective Effect.
| Cardiac Disease Model | Mode of Thyroid Hormone Administration and Dose | Timing and Duration of Thyroid Hormone Administration | Extent | References |
|---|---|---|---|---|
| Post-ischaemic HF (rat) |
T3 (0.42 µg/mg) T4 (1.7 µg/mg) in rat chow |
24 h post-ischaemia for 2 weeks |
|
27 |
|
T3 (0.42 µg/mg) T4 (1.7 µg/mg) in rat chow |
24 h post-ischaemia for 13 weeks |
|
28 |
|
|
T3 (0.42 µg/mg) and T4 (1.7 µg/mg) in rat chow |
13 weeks post-ischaemia for 2 weeks |
Positive LV reshaping |
29 |
|
|
T3 at 3 µg/kg/day T3 at 6 µg/kg/day T3 at 60 µg/kg/day Constant subcutaneous infusion |
1 week post-infarction for up to 9 weeks |
|
30 |
|
|
T3 (1.2 µg/kg/day) Constant subcutaneous infusion |
72 h post-infarction for 4 weeks |
|
31 |
|
|
T4 3.3 mg pellet 60-day sustained release form Subcutaneously |
Immediately following infarction for 8 weeks |
|
32 |
|
|
T4 3.3 mg pellet 60-day sustained release form Subcutaneously |
2 weeks post-infarction for 2 month |
|
33 |
|
|
T3 about 6 μg/kg/day In drinking water |
Immediately following surgery for 2 months |
|
34 |
|
| IR in perfused heart (rat) |
T4 (25 μg/100 g/day) Subcutaneously |
Pre-treatment for 2 weeks before I/R |
|
35 |
|
T3 (40 μg/l) In reperfusion medium |
At reperfusion |
|
36 |
|
|
T3 60 nM or T4 60 nM or T4 400 nM In reperfusion medium |
At reperfusion |
|
37 |
|
| Myocardial IR (rat) |
T3 (6 µg/kg/day) Constant subcutaneous infusion |
At 24 h from infarction for 48 h |
|
38 |
|
T3 (6 µg/kg/day) Constant subcutaneous infusion |
At 24 h from infarction for 48 h |
|
39 |
|
| Diabetes mellitus (rat) |
T3 (0.03 μg/ml) In drinking water |
At 1 month from diabetes mellitus induction for 2 months |
|
40 |
| Hypothyroidism (rat) |
T3 (14 μg/kg/day) |
After hypothyroidism establishment every 24 h for 36 h and 72 h |
|
41 |
| IR, cardiopulmonary bypass (piglet) |
T3 0.6 μg/kg bolus followed by T3 continuous infusion (0.2 μg/kg per hour) |
At reperfusion for 8 h |
|
42 |
|
T3 0.6 μg/kg bolus |
At 80 min and 110 min from reperfusion |
|
43 |
HF = Heart failure; IR = ischaemia and reperfusion; LV = left ventricle; T3 = triiodothyronine; T4 = thyroxine.
Figure 1: A Synopsis of the Thyroid Hormone-mediated Cardioprotection and the Relative Mechanisms to Limit Myocardial Damage in the Acute Phase of Myocardial Infarction, Ischaemia/Reperfusion Injury and in the Chronic Phase, Adverse Left Ventricular Remodelling.
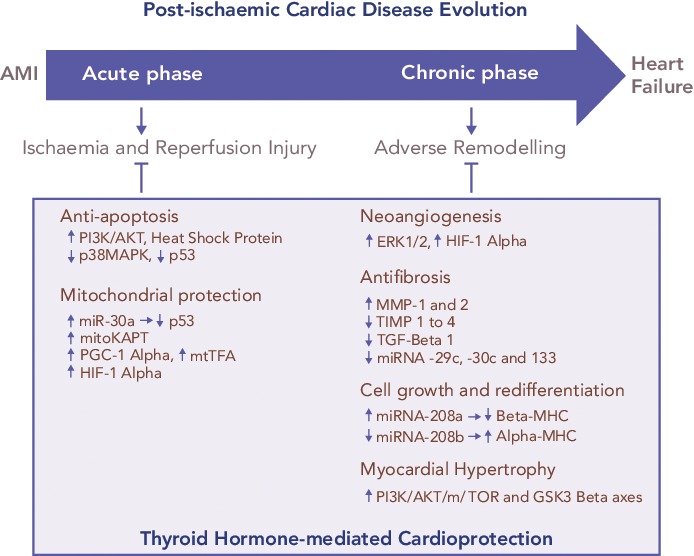
AMI = acute myocardial infarction; AKT = protein kinase B; ERK1/2 = extracellular signal-regulated kinases; GSK3 beta = mitochondrial adenosine Glycogen Synthase Kinase 3 beta; HIF-1 alpha = hypoxia-inducible factor 1-alpha; MHC = myosin heavy chain; mitoKATP = mitochondrial adenosine triphosphate-dependent potassium; MMP-1 = matrix metalloproteinases; mtTFA = mitochondrial transcription factor A; mTOR = mammalian target of rapamycin; PGC-1 alpha = peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PI3K = phosphatidylinositol 3-kinase; TGF-beta 1 = transforming growth factor beta-1; TIMP 1 = tissue inhibitors of MMPs.
Thyroid System and Pro-survival Intracellular Pathways
THs regulate the activation and function of the phosphatidylinositol 3-kinase (PI3K)/Akt and PKC signalling cascade, the expression, phosphorylation and translocation of HSP70 and HSP27;[36,44] and the suppression of p38MAPK signalling.[35] In particular, T3 treatment for 3 days after AMI reduced myocyte apoptosis in the border area of infarction via AKT signalling activation and also through decreased p38MAPK activation.[32,36] Importantly, TH has a dose-dependent effect on AKT phosphorylation that can be mild[45] and beneficial at low TH dose, while further induction of AKT signalling by higher doses of TH can be accompanied by increased mortality and activation of ERK, a kinase that has been associated with pathological remodelling.[36] In addition, 2 weeks of thyroxine (T4) administration increased HSP70 expression and decreased p38MAPK activation in response to ischaemia, changes that closely resemble ischaemic preconditioning.[36] The same treatment led to an increase in the basal expression and phosphorylation of HSP27, and earlier and sustained redistribution of HSP27 from the cytosol-membrane to the cytoskeleton-nucleus cellular fraction.[35] Such changes might help to protect the myocardium against ischaemic insult, resulting in the improvement of post-ischaemic functional recovery.
Thyroid System and Mitochondria
The TH system has multiple actions to protect mitochondria. Among them, the TH is an important regulator of the tumour suppressor p53. This protein network is activated under stress conditions such as AMI and enhances the mitochondrial pathway of cell death.[46] p53 expression is blunted by microRNA 30a (miR-30a) through direct targeting.[47] In the post-ischaemic setting the miR-30a levels drop, which contributes to p53 accumulation, enhanced mitochondrial dysfunction and bcl-2-like protein 4 (BAX) activation, with the result of extended myocardial cell loss. In the post-ischaemic setting, T3 treatment counteracts the decrease in miR-30a levels, thus limiting the activation of p53 and the cascade leading to mitochondrial injury and cell death in the border zone of MI.[38]
Translationally, in patients with de novo post-ischaemic HF within 1 year of AMI, the levels of p53-responsive microRNAs (miR-192, miR-194 and miR-34a) were elevated in the early phase of AMI and were associated with increased left ventricular diastolic dimension.[48] Moreover, T3 can protect mitochondrial integrity through a mitochondrial adenosine triphosphate-dependent potassium pathway, and by increasing the expression of the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1 alpha) and the mitochondrial transcription factor A (mtTFA) in the border zone of infarction.[31] These are key intracellular signals that control mitochondrial activity and biogenesis, and whose overexpression limits post-ischaemic left ventricular (LV) remodelling and preserves cardiac performance.[49,50] Further T3 treatment preserves the expression of hypoxia-inducible factor 1-alpha (HIF-1 alpha), whose protective effect against reperfusion injury is mediated by inhibiting the mitochondrial opening of the PTP.[7,8,51,52]
Thyroid System and Foetal Recapitulation
During the evolution of HF, a foetal profile of gene expression is observed. This condition known as foetal recapitulation, is characterised by an isoform switch from the fast contracting alpha-myosin heavy chain to the slow beta-myosin heavy chain, by a decrease in sarco/endoplasmic reticulum calcium ATPase (SERCA)/phospholamban (PLB) ratio, and also by the preference of glucose metabolism over fatty acids.[51] At its onset in response to stress conditions, such as hypoxia or ischaemia and reperfusion injury, this mechanism may be beneficial to lower energy expenditure and oxygen consumption of the diseased myocardium. It is now understood that it may become maladaptive if continuously maintained.[52,53] Altered expression of the TH receptor (TR) isoforms seems to contribute to the reactivation of the foetal transcriptional profile.[54] Increased beta-myosin isoform expression, as well as impairment of calcium handling and cell contraction, were associated with the overexpression of unliganded TH receptor alpha 1 (TR alpha 1) in neonatal cardiomyocytes.[55,56] Inhibition of T3 binding to TR alpha 1 prevented the differentiation of cardiac embryonic cells.[57] Furthermore, phenylephrine administration in the absence of TH induced a switch of myosin isoform expression to a foetal pattern, which was associated to the redistribution of TR alpha 1 from the cytosolic to the nuclear compartment.[57] The inhibition of the mammalian target of rapamycin (mTOR) signalling abolished this TR alpha 1 response and favoured cell atrophy.[58] Recent evidence indicates that myosin isoform switching is under the control of a complex network of microRNA, including miR-208a and b and miRNA 499, that also presides over cardiac hypertrophy mechanisms.[59] miR-208 family b is a cardiac-specific miRNA enclosed within the myosin heavy chain (MHC) genes. Therefore, the expression of miR-208a and miR-208b is associated with the expression of the MHC alpha and MHC beta, respectively.[59] T3 induces MHC alpha up-regulation and vice-versa downregulation of MHC beta.[30] Deregulation of TH signalling in cardiac disease leads to inhibition of alpha-MHC and miRNA-208a expression, while in vitro treatment with THs significantly upregulates alpha-MHC and miRNA-208a and reduces beta-MHC and miRNA-208b expression, as well as miRNA-499.[60] Based on this, physiological TH concentration seems necessary to guarantee adequate miRNA levels and to avoid foetal myosin isoform switching.
Thyroid System and Myocardial Interstitium
Besides cardiomyocytes, other myocardial cell types contribute to cardioprotection including fibroblast and endothelial cells that play a key role for the maintenance of cardiac architecture and function, and are involved in the pathophysiological evolution of HF. The function of these cells is critical for the synthesis and degradation of extracellular matrix components, as well as for the regulation of cell proliferation, migration, differentiation and apoptosis.[61,62] In the post-ischaemic wound healing process, an interstitial remodelling occurs due to abnormal synthesis and deposition of collagen along with the dysregulation of matrix metalloproteinases (MMPs) and their inhibitors, the tissue inhibitors of MMPs (TIMPs).[63] It has been demonstrated that MMPs increase whereas TIMPs reduce their activity following MI. A temporal and spatial pattern of MMPs and TIMPs activation has been documented where MMP-1,2,3,7,9 activate early, and MMP-8,13 and 14 later following AMI.[64] TIMPs 1–3 expression is also reduced in both the remote and border infarcted zone, whereas TIMP-4 is decreased only in the border infarcted zone.[65] TH treatment with both T3 and T4 is paralleled to a reduction of the interstitial fibrosis in animal models of ischaemic and non-ischaemic HF, and this effect may be related, at least in part, to the effect of TH on MMP and TIMP activity.[66,67] T3-dependent cardiac hypertrophy was not accompanied by interstitial fibrosis but an increase of MMP-2 and TIMP-2 expression was evidenced.[67] Similarly, in rats, cardiac hypertrophy induced by T3 treatment has been associated with an increase of MMP-1 activity, a reduction of collagen I and III and a decrease in TIMP-l and 4 expression.[68] More recently, in long-term T4 treated MI rats, a reduction of collagen deposition in the LV non-infarcted area has been reported along with a tendency towards increased MMP-2 and TIMPs-1–4 expression.[32] The antifibrotic effect of T3 is further suggested by the evidence that early T3 replacement in a rat model of ischaemia/reperfusion reduced the scar size while improving long-term cardiac performance, and these effects were associated with the inhibition of the profibrotic transforming growth factor beta-1 (TGF-beta 1) signalling cascade and with the maintenance of the antifibrotic miRNA-29c, 30c and 133 expression.[39]
Thyroid System and Neo-angiogenesis
THs exert well-documented pro-angiogenic effects. Accordingly, chronic hypothyroidism is associated with rarefaction of small arterioles within the myocardium with consequent impaired coronary vasodilation. This alteration is reversed by T3 administration that prompts the proliferation of vascular smooth muscle cells, pericytes and endothelial cells.[41,69] This pro-angiogenic action is mediated by several molecular mechanisms and starts as a non-genomic action at the plasma membrane of the endothelial cells through the interaction with the integrin alpha V beta 3.[26] The transduction of the TH signal is driven by the mitogen-activated protein kinase ERK1/2 with the consequent transcription of pro-angiogenic genes, such as basic fibroblastic growth factor (bFGF) and vascular endothelial growth factor (VEGF).[70] Another molecular cascade implicated in the T3 pro-angiogenic effect is triggered by the expression of HIF-1 alpha, which is induced by the activation of P13K signalling through the binding of TH with cytoplasmic TR beta.[71,72] T3 induced angiogenesis has been documented in several experimental rat models of cardiovascular disease, including ischaemia, hypertension and diabetic cardiomyopathy.[32,66,73] In a rat model of post-ischaemic HF, T3 supplementation to correct the low T3 state favoured a better retention of capillary density in the border zone in association with HIF-1 alpha stabilisation and TR alpha 1 upregulation.[72]
Thyroid System and Myocardial Hypertrophy
Post-ischaemic LV remodelling is the final result of molecular, subcellular, cellular and interstitial processes leading to changes in cardiomyocytes, extracellular matrix and vasculature within the peri-infarcted region (border zone) and remote region.[74] Myocardial hypertrophy is one of the adaptive mechanisms involved in left ventricular remodelling and is influenced by TH dyshomeostasis. Hypothyroidism induces abnormal myocyte growth characterised by cell lengthening from the addition of sarcomeres in series, a change specific to dilated HF.[75] In the study by Tang et al.,[75] chronic hypothyroidism in rats, treated with propylthiouracil (PTU) for 1 year, caused progressive systolic dysfunction and heart dilatation associated with myocyte lengthening due to series sarcomere addition, which is a typical myocyte remodelling in HF[76] Similarly, myocyte atrophy of myocytes characterised by an increase in length:width ratio occurred after 4 weeks treatment with PTU in rats. These changes were reversible 6 weeks after discontinuing PTU treatment.[77]
Conversely, TH treatment favours physiological hypertrophy through the activation of PI3K/AKT/mTOR and GSK3 beta axes, and through genomic regulation of specific target genes that encode both structural and functional proteins.[78] As evidenced by a histological study, TH treatment of rats with hypertension and dilated HF, induced growth in myocyte transverse dimensions only, a change that reduced systolic wall stress.[79] Furthermore, angiotensin type 1 receptor (AT1R) is a determinant of cardiac hypertrophy induced by hyperthyroidism, and also mediates TH induction of cardiac miR-208a and reduction of cardiac miR-208b levels in hyperthyroid rats. These data strongly suggest that AT1R might have an important regulatory role in cardiac muscle strength and contractility, influencing the efficiency of the cardiac function in hyperthyroidism.[80]
Thyroid System and Neuroendocrine Activation
In general, TH modulates the sympathetic and plasma renin-angiotensin-aldosterone axis, and natriuretic peptide increasing their release and function.[81] At the level of cardiac myocytes T3 promotes BNP gene transcription and regulates the cardiac beta-adrenergic receptor-adenylate cyclase system by controlling the rate of transcription of the beta-1 adrenergic receptor gene. The net effect is the increase in BNP and catecholamines.[82,83] In hyperthyroid rats the levels of norepinephrine in the cardiac muscle increased significantly, whereas it was undetectable in hypothyroid rats. Accordingly, increased and decreased NT-proBNP levels have been observed in patients with hyperthyroidism or hypothyroidism, respectively.[84] Furthermore, in patients with HF and low T3 syndrome dobutamine infusion was associated with the TH metabolic normalisation and this, in turn, with short-term haemodynamic and neurohormonal improvement.[85] Similarly, the same results have been obtained in the same patients through continuous L-T3 infusion for 3 days.[25]
Thyroid System and Inflammation
Plasma cytokines, in particular interleukin 6 (IL-6) and tumour necrosis alpha (TNF-alpha), are elevated in AMI and HF, and this is associated with the severity of the clinical status and a worse outcome.[86,87] A cross-talk between TH and inflammation has been documented in experimental and human studies showing that administration of IL-6 in animals caused T3 decrease, due to the reduction in peripheral conversion of T4 into T3 following the inhibition of the 5’deiodinase activity.[88–92]
Conclusion
Cardioprotection can be considered an example of complexity applied to the human biological system, which is comparable to a non-linear, dynamic and intertwisted network in which small changes can result in important consequences and big changes may result in no or small consequences. In this complex dynamic network, the TH system may be a newly identified player orchestrating the different molecular, tissue and cellular elements involved both in the acute phase, when ischaemic/reperfusion injuries occur, and in the chronic phase, when the post-ischaemic remodelling process evolves. Notwithstanding the large amount of experimental data showing the potential effective role of TH on cardioprotection, there are still few clinical data available, in particular in the acute setting of AMI. It is important to underpin that the goal of TH treatment in patients with AMI and HF should be to restore and maintain euthyroidism in those patients with altered peripheral TH metabolism, and to avoid potentially dangerous pharmacological hyperthyroidism, as shown in two studies using TH replacement therapy in cardiac patients. The Coronary Drug Project (CDP), carried out in the early 1970s,[93] demonstrated adverse outcomes, particularly with respect to the pro-arrhythmic effects of D-T4 (the inactive form of thyroxine). Patients were given 6 mg/day D-T4, which is equivalent to 225 μg of L-T4, corresponding to more than double the endogenous production of T4, which is ~ 90–100 μg/day.[94] It was later found that the D-T4 preparation used in the CDP was contaminated with a high level of active L-T4,[95] which is the active form of thyroxine. Therefore, the cumulative dose of the D-T4 and L-T4 being administered was equivalent to several times the L-T4 dose that would be given to a patient to correct overt hypothyroidism. Furthermore in the study by Goldman et al., the TH analogue 3,5 diiodothyropropionic acid (DITPA) was administered to patients with HF.[96] No improvement on outcome was observed, but rather fatigue was more frequent in the DITPA group, in association with weight loss, reduction in serum cholesterol and increase in heart rate, all signs and symptoms suggesting thyrotoxicosis. Therefore, several questions need to be answered, including: the type of hormone to administer, T3 or T4 or both; the dosage and duration of the treatment to reach the goal that is the restoration of euthyroidism; the starting point of the treatment, for example before, during or late after the revascularisation procedure; the patient selection, i.e. patients with altered TH metabolism; and finally the clinical, functional and prognostic targets to assess the effective benefit of TH treatment.
Acknowledgments
We are grateful to Karin J Tyack for the English revision of the manuscript.
References
- 1.Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res. 2015;116:674–99. doi: 10.1161/CIRCRESAHA.116.305348. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 2.West BJ, Geneston EL, Grigolini P. Maximizing information exchange between complex networks. Physics Reports. 2008;468:1–99. doi: 10.1016/j.physrep.2008.06.003. DOI: [DOI] [Google Scholar]
- 3.Marín-García J, Goldenthal MJ. Mitochondrial centrality in heart failure. Heart Fail Rev. 2008;13:137–50. doi: 10.1007/s10741-007-9079-1. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 4.Baines CP. The cardiac mitochondrion: nexus of stress. Annu Rev Physiol. 2010;72:61–80. doi: 10.1146/annurev-physiol-021909-135929. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 5.Brady NR, Hamacher-Brady A, Gottlieb RA. Proapoptotic BCL-2 family members and mitochondrial dysfunction during ischemia/reperfusion injury, a study employing cardiac HL-1 cells and GFP biosensors. Biochim Biophys Acta. 2006;1757:667–78. doi: 10.1016/j.bbabio.2006.04.011. PMID: [DOI] [PubMed] [Google Scholar]
- 6.Zorov DB, Filburn CR, Klotz LO et al. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–14. doi: 10.1084/jem.192.7.1001. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Assaly R, de Tassigny Ad, Paradis S et al. Oxidative stress, mitochondrial permeability transition pore opening and cell death during hypoxia-reoxygenation in adult Cardiomyocytes. Eur J Pharmacol. 2012;675:6–14. doi: 10.1016/j.ejphar.2011.11.036. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Dorado D, Ruiz-Meana M, Inserte J et al. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc Res. 2012;94:168–80. doi: 10.1093/cvr/cvs116. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 9.Gerdes AM, Kellerman SE, Moore JA et al. Structural remodelling of cardiac myocytes in patients with ischemic cardiomyopathy. Circulation. 1992;86:426–30. doi: 10.1161/01.cir.86.2.426. PMID: [DOI] [PubMed] [Google Scholar]
- 10.Daly PA, Sole MJ. Myocardial catecholamines and the pathophysiology of heart failure. Circulation. 1990;82(2 Suppl):I35–43. PMID: [PubMed] [Google Scholar]
- 11.Wong GH, Goeddel DV. Induction of manganous superoxide dismutase by tumor necrosis factor: possible protective mechanism. Science. 1988;242:941–4. doi: 10.1126/science.3263703. PMID: [DOI] [PubMed] [Google Scholar]
- 12.Finkel MS, Oddis CV, Jacob TD et al. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. 1992;257:387–9. doi: 10.1126/science.1631560. PMID: [DOI] [PubMed] [Google Scholar]
- 13.Li L, Guo CY, Yang J et al. Negative association between free triiodothyronine level and international normalized ratio in euthyroid subjects with acute myocardial infarction. Acta Pharmacol Sin. 2011;32:1351–6. doi: 10.1038/aps.2011.118. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friberg L, Werner S, Eggetsen G, Ahnve S. Rapid down-regulation of thyroid hormones in acute myocardial infarction: is it cardioprotective in patients with angina? Arch Intern Med. 2002;162:1388–94. doi: 10.1001/archinte.162.12.1388. PMID: [DOI] [PubMed] [Google Scholar]
- 15.Kimur T, Kotajima N, Kanda T et al. Correlation of circulating interleukin-10 with thyroid hormone in acute myocardial infarction. Res Commun Mol Pathol Pharmacol. 2001;110:53–8. PMID: [PubMed] [Google Scholar]
- 16.Friberg L, Drvota V, Bjelak AH et al. Association between increased levels of reverse triiodothyronine and mortality after acute myocardial infarction. Am J Med. 2001;111:699–703. doi: 10.1016/s0002-9343(01)00980-9. PMID: [DOI] [PubMed] [Google Scholar]
- 17.Iervasi G, Pingitore A, Landi P et al. Low-T3 syndrome: a strong prognostic predictor of death in patients with heart disease. Circulation. 2003;107:708–13. doi: 10.1161/01.cir.0000048124.64204.3f. PMID: [DOI] [PubMed] [Google Scholar]
- 18.Pingitore A, Landi P, Taddei MC et al. Triiodothyronine levels for risk stratification of patients with chronic heart failure. Am J Med. 2005;118:132–6. doi: 10.1016/j.amjmed.2004.07.052. PMID: [DOI] [PubMed] [Google Scholar]
- 19.Iervasi G, Molinaro S, Landi P et al. Association between increased mortality and mild thyroid dysfunction in cardiac patients. Arch Intern Med. 2007;167:1526–32. doi: 10.1001/archinte.167.14.1526. PMID: [DOI] [PubMed] [Google Scholar]
- 20.Chen P, Li S, Lei X et al. Free triiodothyronine levels and short-term prognosis in chronic heart failure patients with type 2 diabetes. Am J Med Sci. 2015;350:87–94. doi: 10.1097/MAJ.0000000000000524. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 21.Ning N, Gao D, Triggiani V et al. Prognostic Role of Hypothyroidism in Heart Failure: A Meta-Analysis. Medicine (Baltimore) 2015;94:e1159. doi: 10.1097/MD.0000000000001159. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li X, Yang X, Wang Y et al. The prevalence and prognostic effects of subclinical thyroid dysfunction in dilated cardiomyopathy patients: a single-center cohort study. J Card Fail. 2014;20(7):506–12. doi: 10.1016/j.cardfail.2014.05.002. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 23.Passino C, Pingitore A, Landi P et al. Prognostic value of combined measurement of brain natriuretic peptide and triiodothyronine in heart failure. J Card Fail. 2009;15(1):35–40. doi: 10.1016/j.cardfail.2008.08.008. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 24.Chuang CP, Jong YS, Wu CY, Lo HM. Impact of triiodothyronine and N-terminal pro-B-type natriuretic peptide on the long-term survival of critically ill patients with acute heart failure. Am J Cardiol. 2014;113:845–50. doi: 10.1016/j.amjcard.2013.11.039. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 25.Pingitore A, Galli E, Barison A et al. Acute effects of triiodothyronine (T3) replacement therapy in patients with chronic heart failure and low-T3 syndrome: a randomized, placebo-controlled study. J Clin Endocrinol Metab. 2008;93(4):1351–8. doi: 10.1210/jc.2007-2210. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 26.Gerdes AM. Restoration of thyroid hormone balance: a game changer in the treatment of heart failure? Am J Physiol Heart Circ Physiol. 2015;308:H1–10. doi: 10.1152/ajpheart.00704.2014. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 27.Pantos C, Mourouzis I, Markakis K et al. Thyroid hormone attenuates cardiac remodeling and improves hemodynamics early after acute myocardial infarction in rats. Eur J Cardiothorac Surg. 2007;32(2):333–9. doi: 10.1016/j.ejcts.2007.05.004. PMID: [DOI] [PubMed] [Google Scholar]
- 28.Pantos C, Mourouzis I, Markakis K et al. Long-term thyroid hormone administration reshapes left ventricular chamber andimproves cardiac function after myocardial infarction in rats. Basic Res Cardiol. 2008;103:308–18. doi: 10.1007/s00395-008-0697-0. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 29.Pantos C, Mourouzis I, Tsagoulis N et al. Thyroid hormone at supra-physiological dose optimizes cardiac geometry and improvescardiac function in rats with old myocardial infarction. J Physiol Pharmacol. 2009;60:49–56. PMID: [PubMed] [Google Scholar]
- 30.Henderson KK, Danzi S, Paul JT et al. Physiological replacement of T3 improves left ventricular function in an animal model of myocardial infarction-induced congestive heart failure. Circ Heart Fail. 2009;2:243–52. doi: 10.1161/CIRCHEARTFAILURE.108.810747. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 31.Forini F, Lionetti V, Ardehali H et al. Early long-term L-T3 replacement rescues mitochondria and prevents ischemic cardiac remodelling in rats. J Cell Mol Med. 2011;15(3):514–24. doi: 10.1111/j.1582-4934.2010.01014.x. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen YF, Weltman NY, Li X et al. Improvement of left ventricular remodeling after myocardial infarction with eight weeks L-thyroxine treatment in rats. J Transl Med. 2013;11:40. doi: 10.1186/1479-5876-11-40. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Dedkov EI, Lee B 3rd et al. Thyroid hormone replacement therapy attenuates atrial remodeling and reduces atrial fibrillation inducibility in a rat myocardial infarction-heart failure model. J Card Fail. 2014;20:1012–9. doi: 10.1016/j.cardfail.2014.10.003. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rajagopalan V, Zhang Y, Ojamaa K et al. Safe Oral Triiodo-L-Thyronine Therapy Protects from Post-Infarct Cardiac Dysfunction and Arrhythmias without Cardiovascular Adverse Effects. PLoS One. 2016;11:e0151413. doi: 10.1371/journal.pone.0151413.eCollection2016. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pantos C, Malliopoulou V, Paizis I et al. Thyroid hormone and cardioprotection: study of p38 MAPK and JNKs during ischaemia and at reperfusion in isolated rat heart. Mol Cell Biochem. 2003;242:173–80. PMID: [PubMed] [Google Scholar]
- 36.Pantos C, Mourouzis I, Saranteas T et al. Thyroid hormone improves postischaemic recovery of function while limiting apoptosis: a new therapeutic approach to support hemodynamics in the setting of ischaemia-reperfusion? Basic Res Cardiol. 2009;104:69–77. doi: 10.1007/s00395-008-0758-4. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 37.Pantos C, Mourouzis I, Saranteas T et al. Acute T3 treatment protects the heart against ischemia reperfusion injury via TR alpha 1 receptor. Mol Cell Biochem. 2011;353:235–41. doi: 10.1007/s11010-011-0791-8. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 38.Forini F, Kusmic C, Nicolini G et al. Triiodothyronine prevents cardiac ischemia/reperfusion mitochondrial impairment and cell loss by regulating miR30a/p53 axis. Endocrinology. 2014;155(11):4581–90. doi: 10.1210/en.2014-1106. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 39.Nicolini G, Forini F, Kusmic C et al. Early and Short-term Triiodothyronine Supplementation Prevents Adverse Postischemic Cardiac Remodeling: Role of Transforming Growth Factor-beta 1 and Antifibrotic miRNA Signaling. Mol Med. 2015;21(1):900–11. doi: 10.2119/molmed.2015.00140. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weltman NY, Ojamaa K, Schlenker EH et al. Low-dose T3 replacement restores depressed cardiac T3 levels, preserves coronary microvasculature and attenuates cardiac dysfunction in experimental diabetes mellitus. Mol Med. 2014;20:302–12. doi: 10.2119/molmed.2013.00040. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Savinova OV, Liu Y, Aasen GA et al. Thyroid hormone promotes remodeling of coronary resistance vessels. PLoS One. 2011;6(9):e25054. doi: 10.1371/journal.pone.0025054. DOI: PMCID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Files MD, Kajimoto M, O‘Kelly Priddy CM et al. Triiodothyronine facilitates weaning from extracorporeal membrane oxygenation by improved mitochondrial substrate utilization. J Am Heart Assoc. 2014;3:e000680. doi: 10.1161/JAHA.113.000680. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olson AK, Bouchard B, Ning XH et al. Triiodothyronine increases myocardial function and pyruvate entry into the citric acid cycle after reperfusion in a model of infant cardiopulmonary bypass. Am J Physiol Heart Circ Physiol. 2012;302:H1086–93. doi: 10.1152/ajpheart.00959.2011. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rybin V, Steinberg SF. Thyroid hormone represses protein kinase C isoform expression and activity in rat cardiac myocytes. Circ Res. 1996;79:388–98. doi: 10.1161/01.res.79.3.388. PMID: [DOI] [PubMed] [Google Scholar]
- 45.Kehat I, Davis J, Tiburcy M et al. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ. Res. 2011;108:176–83. doi: 10.1161/CIRCRESAHA.110.231514. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–10. doi: 10.1038/35042675. PMID: [DOI] [PubMed] [Google Scholar]
- 47.Li J, Donath S, Li Y et al. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010;6(1):e1000795. doi: 10.1371/journal.pgen.1000795. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matsumoto S, Sakata Y, Suna S et al. Circulating p53-responsive microRNAs are predictive indicators of heart failure after acute myocardial infarction. Circ Res. 2013;113:322–6. doi: 10.1161/CIRCRESAHA.113.301209. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 49.Ikeuchi M, Matsusaka H, Kang D et al. Overexpression of mitochondrial transcription factor a ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation. 2005;112:683–90. doi: 10.1161/CIRCULATIONAHA.104.524835. PMID: [DOI] [PubMed] [Google Scholar]
- 50.Garnier A1, Fortin D, Deloménie C Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol. 2003. 551. pp. 491–501. PMID: [DOI] [PMC free article] [PubMed]
- 51.Taegtmeyer S, Sen S, Vela D. Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann N Y Acad Sci. 2010;1188:191–8. doi: 10.1111/j.1749-6632.2009.05100.x. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pantos C, Mourouzis I, Dimopoulos A et al. Enhanced tolerance of the rat myocardium to ischemia and reperfusion injury early after acute myocardial infarction. Basic Res Cardiol. 2007;102:327–33. doi: 10.1007/s00395-007-0645-4. PMID: [DOI] [PubMed] [Google Scholar]
- 53.Pantos C, Mourouzis I, Saranteas T et al. Thyroid hormone receptors alpha1 and beta1 are downregulated in the post-infarcted rat heart: consequences on the response to ischaemia-reperfusion. Basic Res Cardiol. 2005;100:422–32. doi: 10.1007/s00395-005-0545-4. PMID: [DOI] [PubMed] [Google Scholar]
- 54.Mourouzis I, Forini F, Pantos C, Iervasi G. Thyroid hormone and cardiac disease: from basic concepts to clinical application. J Thyroid Res. 2011;2011:958626. doi: 10.4061/2011/958626. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kinugawa K, Jeong MY, Bristow MR, Long CS. Thyroid hormone induces cardiac myocyte hypertrophy in a thyroid hormone receptor alpha1-specific manner that requires TAK1 and p38 mitogen-activated protein kinase. Mol Endocrinol. 2005;19:1618–28. doi: 10.1210/me.2004-0503. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tavi P, Sjögren M, Lunde PK et al. Impaired Ca2+ handling and contraction in cardiomyocytes from mice with a dominant negative thyroid hormone receptor alpha1. J Mol Cell Cardiol. 2005;38:655–63. doi: 10.1016/j.yjmcc.2005.02.008. PMID: [DOI] [PubMed] [Google Scholar]
- 57.Pantos C, Xinaris C, Mourouzis I et al. Thyroid hormone receptor alpha 1: a switch to cardiac cell “metamorphosis”? J Physiol Pharmacol. 2008;59:253–69. PMID: [PubMed] [Google Scholar]
- 58.Pantos C, Mourouzis I, Galanopoulos G et al. Thyroid hormone receptor alpha1 downregulation in postischemic heart failure progression: the potential role of tissue hypothyroidism. Horm Metab Res. 2010;42:718–24. doi: 10.1055/s-0030-1255035. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 59.van Rooij E, Quiat D, Johnson BA et al. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev Cell. 2009;17:662–73. doi: 10.1016/j.devcel.2009.10.013. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Callis TE, Pandya K, Seok HY et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest. 2009;119:2772–86. doi: 10.1172/JCI36154. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Collen A, Hanemaaijer R, Lupu F et al. Membrane-type matrix metalloproteinase-mediated angiogenesis in a fibrin-collagen matrix. Blood. 2003;101:1810–7. doi: 10.1182/blood-2002-05-1593. PMID: [DOI] [PubMed] [Google Scholar]
- 62.Givvimani S, Tyagi N, Sen U et al. MMP-2/TIMP-2/TIMP-4 versus MMP-9/TIMP-3 in transition from compensatory hypertrophy and angiogenesis to decompensatory heart failure. Arch Physiol Biochem. 2010;116:63–72. doi: 10.3109/13813451003652997. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev. 2007;87:1285–342. doi: 10.1152/physrev.00012.2007. PMID: [DOI] [PubMed] [Google Scholar]
- 64.Lindsey ML, Zamilpa R. Temporal and spatial expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases following myocardial infarction. Cardiovasc Ther. 2012;30:31–41. doi: 10.1111/j.1755-5922.2010.00207.x. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilson EM, Moainie SL, Baskin JM et al. Region- and type-specific induction of matrix metalloproteinases in post-myocardial infarction remodeling. Circulation. 2003;107:2857–63. doi: 10.1161/01.CIR.0000068375.40887.FA. PMID: [DOI] [PubMed] [Google Scholar]
- 66.Weltman NY, Pol CJ, Zhang Y et al. Long-term physiological T3 supplementation in hypertensive heart disease in rats. Am J Physiol Heart Circ Physiol. 2015;309:H1059–65. doi: 10.1152/ajpheart.00431.2015. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ziegelhöffer-Mihalovicová B, Briest W, Baba HA et al. The expression of mRNA of cytokines and of extracellular matrix proteins in triiodothyronine-treated rat hearts. Mol Cell Biochem. 2003;247:61–8. doi: 10.1023/a:1024153003249. PMID: [DOI] [PubMed] [Google Scholar]
- 68.Ghose Roy S, Mishra S, Ghosh G, Bandyopadhyay A. Thyroid hormone induces myocardial matrix degradation by activating matrix metalloproteinase-1. Matrix Biol. 2007;26:269–79. doi: 10.1016/j.matbio.2006.12.005. PMID: [DOI] [PubMed] [Google Scholar]
- 69.Liu Y, Sherer BA, Redetzke RA, Gerdes AM. Regulation of arteriolar density in adult myocardium during low thyroid conditions. Vascul Pharmacol. 2010;52:146–50. doi: 10.1016/j.vph.2009.10.003. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 70.Balzan S, Del Carratore R, Nicolini G et al. Proangiogenic effect of TSH in human microvascular endothelial cells through its membrane receptor. J Clin Endocrinol Metab. 2012;97:1763–70. doi: 10.1210/jc.2011-2146. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 71.Eckle T, Köhler D, Lehmann R et al. Hypoxia-inducible factor-1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation. 2008;118:166–75. doi: 10.1161/CIRCULATIONAHA.107.758516. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 72.Moeller LC, Dumitrescu AM, Refetoff S. Cytosolic action of thyroid hormone leads to induction of hypoxia-inducible factor-1alpha and glycolytic genes. Mol Endocrinol. 2005;19:2955–63. doi: 10.1210/me.2004-0542. PMID: [DOI] [PubMed] [Google Scholar]
- 73.Makino A, Suarez J, Wang H et al. Thyroid hormone receptor-beta is associated with coronary angiogenesis during pathological cardiac hypertrophy. Endocrinology. 2009;150:2008–15. doi: 10.1210/en.2008-0634. DOI: PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pfeffer MA. Left ventricular remodeling after acute myocardial infarction. Annu Rev Med. 1995;46:455–66. doi: 10.1146/annurev.med.46.1.455. PMID: [DOI] [PubMed] [Google Scholar]
- 75.Tang YD, Kuzman JA, Said S et al. Low thyroid function leads to cardiac atrophy with chamber dilatation, impaired myocardial blood flow, loss of arterioles, and severe systolic dysfunction. Circulation. 2005;112:3122–30. doi: 10.1161/CIRCULATIONAHA.105.572883. PMID: [DOI] [PubMed] [Google Scholar]
- 76.Gerdes AM, Kellerman SE, Moore JA et al. Structural remodeling of cardia myocytes in patients with ischemic cardiomyopathy. Circulation. 1992;86:426–30. doi: 10.1161/01.cir.86.2.426. PMID: [DOI] [PubMed] [Google Scholar]
- 77.Liu Z, Gerdes AM. Influence of hypothyroidism and the reversal of hypothyroidism on hemodynamics and cell size in the adult rat heart. J Mol Cell Cardiol. 1990;22:1339–48. doi: 10.1016/0022-2828(90)90979-c. PMID: [DOI] [PubMed] [Google Scholar]
- 78.Dillmann WH. Biochemical basis of thyroid hormone action in the heart. Am J Med. 1990;88:626–30. doi: 10.1016/0002-9343(90)90530-q. PMID: [DOI] [PubMed] [Google Scholar]
- 79.Thomas TA, Kuzman JA, Anderson BE et al. Thyroid hormones induce unique and potentially beneficial changes in cardiac myocyte shape in hypertensive rats near heart failure. Am J Physiol Heart Circ Physiol. 2005;288:H2118–22. doi: 10.1152/ajpheart.01000.2004. PMID: [DOI] [PubMed] [Google Scholar]
- 80.Diniz GP, Takano AP, Barreto-Chaves ML. MiRNA-208a and miRNA-208b are triggered in thyroid hormone-induced cardiac hypertrophy - role of type 1 Angiotensin II receptor (AT1R) on miRNA-208a/alpha-MHC modulation. Mol Cell Endocrinol. 2013. 374. pp. 117–24. DOI: PMID: [DOI] [PubMed]
- 81.Iervasi G, Sabatino L, Nicolini G. Iervasi G, Pingitore A. Thyroid and Heart Failure. Italy: Springer-Verlag; 2009. Low Triiodothyronine Syndrome as a Powerful Predictor of Death in Heart Failure. pp. 179–190. [Google Scholar]
- 82.Liang F, Webb P, Marimuthu A et al. Triiodothyronine increases brain natriuretic peptide (BNP) gene transcription and amplifies endothelin-dependent BNP gene transcription and hypertrophy in neonatal rat ventricular myocytes. J Biol Chem. 2003;278(17):15073–83. doi: 10.1074/jbc.M207593200. PMID: [DOI] [PubMed] [Google Scholar]
- 83.Mano T, Sakamoto H, Fujita K et al. Effects of thyroid hormone on catecholamine and its metabolite concentrations in rat cardiac muscle and cerebral cortex. Thyroid. 1998;8(4):353–8. doi: 10.1089/thy.1998.8.353. PMID: [DOI] [PubMed] [Google Scholar]
- 84.Ozmen B, Ozmen D, Parildar Z et al. Serum N-terminal-pro-B-type natriuretic peptide (NT-pro-BNP) levels in hyperthyroidism and hypothyroidism. Endocr Res. 2007;32(1-2):1–8. doi: 10.1080/07435800701670047. PMID: [DOI] [PubMed] [Google Scholar]
- 85.D‘Aloia A, Vizzardi E, Bugatti S et al. Effect of short-term infusive dobutamine therapy on thyroid hormone profile and hemodynamic parameters in patients with acute worsening heart failure and low-triiodothyronine syndrome. J Investig Med. 2012;60(6):907–10. doi: 10.231/JIM.0b013e31825cec9c. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 86.Kimur T, Kotajima N, Kanda T et al. Correlation of circulating interleukin-10 with thyroid hormone in acute myocardial infarction. Res Commun Mol Pathol Pharmacol. 2001;110(1-2):53–8. PMID: [PubMed] [Google Scholar]
- 87.Paraskevaidis IA, Parissis JT, Th Kremastinos D. Anti-inflammatory and antiapoptotic effects of levosimendan in decompensated heart failure: a novel mechanism of drug-induced improvement in contractile performance of the failing heart. Curr Med Chem Cardiovasc Hematol Agents. 2005;3:243–7. doi: 10.2174/1568016054368232. PMID: [DOI] [PubMed] [Google Scholar]
- 88.Boelen A, Platvoet-Ter Schiphorst MC, Wiersinga WM. Association between serum interleukin-6 and serum 3,5,3‘-triiodothyronine in nonthyroidal illness. J Clin Endocrinol Metab. 1993;77(6):1695–9. doi: 10.1210/jcem.77.6.8263160. PMID: [DOI] [PubMed] [Google Scholar]
- 89.Torpy DJ, Tsigos C, Lotsikas AJ et al. Acute and delayed effects of a single-dose injection of interleukin-6 on thyroid function in healthy humans. Metabolism. 1998;47:1289–93. doi: 10.1016/s0026-0495(98)90338-9. PMID: [DOI] [PubMed] [Google Scholar]
- 90.Bartalena L, Bogazzi F, Brogioni S et al. Role of cytokines in the pathogenesis of the euthyroid sick syndrome. Eur J Endocrinol. 1998;138:603–14. doi: 10.1530/eje.0.1380603. PMID: [DOI] [PubMed] [Google Scholar]
- 91.Lubrano V, Pingitore A, Carpi A, Iervasi G. Relationship between triiodothyronine and proinflammatory cytokines in chronic heart failure. Biomed Pharmacother. 2010;64:165–9. doi: 10.1016/j.biopha.2009.09.001. DOI: PMID: [DOI] [PubMed] [Google Scholar]
- 92.Kimura T, Kanda T, Kotajima N et al. Involvement of circulating interleukin-6 and its receptor in the development of euthyroid sick syndrome in patients with acute myocardial infarction. Eur J Endocrinol. 2000;143:179–84. doi: 10.1530/eje.0.1430179. PMID: [DOI] [PubMed] [Google Scholar]
- 93.Findings leading to further modifications of its protocol with respect to dextrothyroxine. The coronary drug project research group. JAMA. 1972;220:996–1008. The coronary drug project. PMID: [PubMed] [Google Scholar]
- 94.Pilo A, Iervasi G, Vitek F et al. Thyroidal and peripheral production of 3,5,3‘-triiodothyronine in humans by multicompartmental analysis. Am J Physiol. 1990;258(4 Pt 1):E715–26. doi: 10.1152/ajpendo.1990.258.4.E715. PMID: [DOI] [PubMed] [Google Scholar]
- 95.Young WF Jr, Gorman CA, Jiang NS et al. L-thyroxine contamination of pharmaceutical D-thyroxine: probable cause of therapeutic effect. Clin Pharmacol Ther. 1984;36:781–7. doi: 10.1038/clpt.1984.257. PMID: [DOI] [PubMed] [Google Scholar]
- 96.Goldman S, McCarren M, Morkin E et al. DITPA (3,5-diiodothyropropionic acid), a thyroid hormone analog to treat heart failure: phase II trial Veterans Affairs cooperative study. Circulation. 2009;119:3093–100. doi: 10.1161/CIRCULATIONAHA.108.834424. DOI: PMID: [DOI] [PubMed] [Google Scholar]