

Abstract
Acute intermittent porphyria (AIP), an autosomal dominant disorder due to the half-normal activity of hydroxymethylbilane synthase (HMBS), is characterized by acute neurovisceral attacks that are precipitated by factors that induce heme biosynthesis. Molecular diagnosis is the most sensitive and specific diagnostic test for AIP, and importantly, it permits the identification of asymptomatic family members for genetic counseling and avoidance of precipitating factors. Here, we report the identification of 40 novel HMBS mutations, including 11 missense, 4 nonsense, 16 small insertions or deletions, 8 consensus splice-site mutations, and a complex insertion-deletion mutation in unrelated individuals with AIP. Prokaryotic expression of the missense mutations demonstrated that all mutants had ≤ 5% of expressed wildtype activity, except for c.1039G>C (p.A347P), which had 51% residual HMBS activity but was markedly thermolabile. Of note, the mutation c.612G>T (p.Q204H) altered the last nucleotide of exon 10, which resulted in an alternative HMBS transcript with an in-frame 9 base-pair deletion at the 3-terminus of exon 10 (encoding protein Q204HΔ3). When expressed, Q204HΔ3 and an in-frame three base-pair deletion (c.639_641delTGC) had no detectable HMBS activity. Western blot analyses and mapping of the missense mutations on the human HMBS crystal structure revealed that mutations near the active site or at the dimerization interface resulted in stably expressed proteins, while most that altered surface residues resulted in unstable proteins, presumably due to improper protein folding. These studies identified novel pathogenic HMBS mutations and expanded the molecular heterogeneity of AIP.
Keywords: hydroxymethylbilane synthase, acute intermittent porphyria, acute hepatic porphyria, heme biosynthetic disorders, molecular diagnosis
INTRODUCTION
Acute intermittent porphyria (AIP; OMIM#176000) is an autosomal dominant disorder due to the half-normal activity of hydroxymethylbilane synthase (HMBS; EC 2.5.1.61), the third enzyme in the heme biosynthetic pathway. It is the most common of the four acute hepatic porphyrias, which also include hereditary coproporphyria (HCP; OMIM#121300), variegate porphyria (VP; OMIM#176200), and the ultra-rare 5′-aminolevulinic acid dehydratase deficiency porphyria (ADP), which are due to mutations in the coproporphryinogen oxidase (CPOX), protoporphyrinogen oxidase (PPOX), and 5-aminolevulinic acid dehydratase (ALAD) genes, respectively (Anderson KE, 2001; Puy et al., 2010). While most individuals (>90%) with the acute hepatic porphyrias are clinically latent, symptomatic individuals experience life-threatening acute neurovisceral attacks that typically begin with excruciating abdominal pain often associated with hypertension, tachycardia, constipation, motor weakness and seizures. These attacks are precipitated by various factors, including certain drugs, fasting, and hormonal changes, all of which induce the hepatic expression of 5′-aminolevlulinic acid synthase 1 (ALAS1; EC 2.3.1.37), the first and rate-limiting enzyme of the heme biosynthetic pathway (Granick, 1963, 1966; Sassa and Granick, 1970). When hepatic ALAS1 is induced, the respective deficient enzymatic activity becomes rate-limiting, leading to insufficient heme production. This results in derepression and further induction of ALAS1 expression and the consequential hepatic accumulation of the putative neurotoxic porphyrin precursors, 5’-aminolevulinic acid (ALA) and porphobilinogen (PBG) (only ALA accumulates in ADP), which are secreted into plasma and excreted through the urine.
The biochemical diagnosis of AIP is often challenging, making mutation-based diagnoses even more important. This is particularly true of patients who previously had an acute attack but are currently asymptomatic, as their plasma and urinary ALA and PBG levels typically return to normal. For such cases, molecular diagnosis of the genes for the three autosomal dominant porphyrias, AIP, HCP, and VP is the most sensitive and specific method (Puy et al., 2010; Whatley and Badminton, 2013). Importantly, molecular diagnosis permits the detection of asymptomatic family members so that these individuals can be counseled to avoid factors that may precipitate acute attacks.
To date, over 400 mutations causing AIP have been reported in the Human Gene Mutation Database (HGMD) (Stenson et al., 2017). With the exception of founder mutations in the Netherlands and Scandinavian countries (Gu et al., 1993; Floderus et al., 2002; Kauppinen and von und zu Fraunberg, 2002; Mykletun et al., 2014) and those mutations that occur at CpG mutational hotspots, most AIP families have a ‘private’ mutation. Here, we report the identification of 40 novel molecular lesions in 43 unrelated individuals who had an acute attack and whose blood samples were sent to the Mount Sinai Porphyrias Diagnostic Laboratory for molecular diagnosis. All novel missense mutations and an in-frame small deletion were evaluated by prokaryotic expression to assess their pathogenicity. One missense mutation had > 50% residual HMBS activity and was further assessed by thermostability assays. In addition, western blot and molecular modeling analyses were performed to investigate how each of the missense mutations affected HMBS protein stability and structure.
MATERIALS AND METHODS
DNA isolation and Mutation Analyses
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation and with the Helsinski Declaration of 1975, as revised in 2000 (5). This article does not contain any studies with animal subjects performed by any of the authors. Blood samples were collected in EDTA from 43 unrelated individuals who were referred by their physicians due to elevated ALA and/or PBG levels or clinical symptoms compatible with an acute hepatic porphyria. All patients provided informed consent for genetic testing, and the study was approved by the Icahn School of Medicine Institution Review Board. Genomic DNA was isolated using the Gentra Purgene DNA Isolation kit (Qiagen, Germantown, MA, USA), and the entire HMBS, CPOX, and PPOX genes were amplified by polymerase chain reaction (PCR). For the HMBS gene, two overlapping amplicons were amplified: the shorter amplicon was 638 bp and included 157 bp of 5′-UTR and exon 1 and the longer amplicon was 6,325 bp and included exons 2 to 15 and 266 bp of 3′-UTR. PCR conditions used were 95°C for 5 min, 15 cycles of 95°C for 15 sec, 62°C (gradient to 55°C, in 0.5°C intervals) for 30 sec, 72°C for 3 min, followed by 19 cycles of 95°C for 15 sec, 55°C for 30 sec, 72°C for 3 min, and 72°C for 5 min. Methods for amplifying and sequencing the CPOX and PPOX genes are provided in Supplementary methods. Amplicons were purified using the Qiagen PCR purification kit and their exonic and flanking regions were sequenced bi-directionally using an ABI 3730xl DNA analyzer. The sequences of all primers used for PCR amplification and sequencing of the HMBS gene are provided in Supplementary Table 1.
RNA Isolation and reverse transcription (RT)-PCR
Whole blood was collected in RNAgard tubes (Biomatrica, San Diego, CA, USA) and total RNA was isolated using the BioMaxi Blood RNA Purification Kit (Biomatrica, San Diego, CA, USA). The full length ~1.4 kb HMBS transcript was PCR amplified using primers 5′-GGAGACCAGGAGTCAGACTGTAGGA-3′ (forward) and 5′-AGGCTGTTGCTT GGACTTCTCTAAAG-3′ (reverse) and the following thermocycling conditions: 95°C for 10 min, 40 cycles of 95°C for 15 sec, 55°C for 15 sec, 72°C for 30 sec, 72°C for 10 min. The same primers were used to sequence the amplicons.
Generation and Expression of HMBS Constructs in E. Coli
The full-length human HMBS cDNA was cloned into the pkk vector and designated pkk-WT (Chen et al., 1994). Mutant constructs pkk-L30P (c.89T>C), pkk-F77L (c.231T>G), pkk-A112P (c.334G>C), pkk-A122D (c.365C>A), pkk-T145l (c.434C>T), pkk-L154P (c.461T>C), pkk-L170P (c.509T>C), pkk-Q204H Δ 3 (c.604_612del9), pkk-V224E (c.671T>A), pkk-D240G (c.719A>G), pkk-A347P (c.1039G>C), and pkk-639del3 (c.639_641delTGC) were generated by site-directed mutagenesis (Agilent, Santa Clara, CA, USA) using primers provided in Supplementary Table 2. The constructs were confirmed by sequencing and transformed into BL21 E. coli (Promega, Madison, WI, USA), which were cultured overnight in LB medium with 0.2% (w/v) glucose. A 1:100 dilution was freshly cultured on the second day until OD600 reached 0.5–0.8. Inoculates were then induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and incubated for an additional 3 hr. The cells were pelleted by centrifugation at 1,000 × g for 20 min and resuspended in 0.1 M Tris buffer, pH 8.0, containing 0.1 mM dithiothreitol (DTT) and Roche protease inhibitor (Indianapolis, Indiana, USA). Samples were frozen at −80°C overnight, thawed, and then sonicated for 2 min (10 sec on and 5 sec off). The lysates were centrifuged at 21,000 × g for 25 min at 4°C, and the supernatants were collected and stored at −80°C in the dark until use.
HMBS Enzyme Activity and Thermostability Assays
HMBS enzyme activities were determined in the supernatants as previously described (Chen et al., 1994, Song et al., 2009, Chen et al., 2016) and normalized for total protein concentrations, which were determined using the Bio-Rad DC Protein Assay kit (Philadelphia, PA, USA). All assays were performed in duplicate, and the mean values were used to calculate the percent of wildtype activity. For semi-quantitative estimate of the Cross-Reacting Protein (CRIM)-specific activity of individual expressed mutant HMBS enzymes, the relative HMBS and GAPDH protein amounts were determined from the Western Blot using densitometric analyses (Fiji ImageJ software; Schindelin et al., 2012), and the ratios of the two proteins were used to normalize the expressed HMBS activity (i.e the CRIM-specific HMBS activities). For thermostability assays, wildtype and mutant lysates were incubated at 65°C and aliquots were taken at 0 and 90 min and placed on ice. HMBS enzyme activities for each sample were then determined.
Western Blot Analysis
Protein lysates (~3 μg) were mixed with loading buffer (62.5 mM Tris-HCl, 2.5 % SDS, 0.002 % Bromophenol Blue, 5% β-mercaptoethanol, 10 % glycerol, pH 6.8) and denatured, separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred to a polyvinylidene difluoride (PVDF) membrane. The membranes were blocked with 5% non-fat dry milk in buffer (50 mM Tris, 150 mM NaCl, pH 7.6, and 0.05% Tween-20) for 1 hr at room temperature and incubated with anti-HMBS antibody (Abcam, cat# ab129092, Cambridge, MA, USA) for 1 hr at 4°C (1:30,000 dilution), and then incubated with secondary HRP-conjugated anti-rabbit IgG antibodies (1:100,000 dilution) for 20 min at room temperature. Signals were detected using the Chemiluminescence HRP substrate (Millipore, Burlington, MA, USA). The membranes were stripped and re-probed with anti-GAPDH (Thermo Fisher Scientific, Waltham, MA, USA) at 1:5,000 dilution and secondary HRP-conjugated anti-mouse IgG antibodies (1:50,000 dilution) for loading normalization.
Mapping of the Missense Mutations on the Human HMBS Crystal Structure
The 2.2-angstrom crystal structure of human HMBS, (PDB 3ECR) (Song et al., 2009) was viewed in PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrodinger, LLC). The structure has the dipyrromethane cofactor bound at the active site (Song et al., 2009). The molecular mutations were made using PyMOL, and the distances from the active site to the different mutations were determined using the distance feature of the software. All graphical representations were made using PyMOL.
RESULTS
Mutation Detection of the HMBS Gene
Genomic DNA was isolated from 43 unrelated individuals with either elevated urinary ALA or PBG levels or with clinical symptoms consistent with an acute hepatic porphyria. Each patient’s HMBS gene was PCR-amplified and sequenced for all fifteen exonic and adjacent intronic regions. Molecular analysis of each patient’s PPOX and CPOX genes revealed normal sequences other than known common single nucleotide polymorphisms. The 40 novel HMBS mutations identified included 11 missense, 4 nonsense, 6 small duplication/insertions, 10 small deletions, 8 consensus splice-site mutations, and a complex insertion-deletion mutation. None of the identified missense mutations occurred at a CpG dinucleotide site. As shown in Table 1, which summarizes the 40 mutations and patient information, two of the mutations [c.612 G>T (p.Q204H) and c. 612+1 G>A] were identified in more than one unrelated proband.
Table 1.
Novel HMBS Mutations & Clinical Information
| Nucleotide Change | Amino Acid Change | Gender | Number of Unrelated Probands | Elevation of Urinary PBG and/or ALA | Reduced Erythrocyte HMBS Enzyme Activity |
|---|---|---|---|---|---|
| c.89T>C | p.Leu30Pro | F | 1 | + | |
| c.231T>G | p.Phe77Leu | F | 1 | + | |
| c.334G>C | p.Ala112Pro | F | 1 | NA | |
| c.365C>A | p.Ala122Asp | F | 1 | + | |
| c.434C>T | p.Thr145Ils | F | 1 | + | |
| c.461T>C | p.Leu154Pro | F | 1 | NA | |
| c.509T>C | p.Leu170Pro | F | 1 | + | |
| c.612G>T | p.Gln204His | M/F | 2 | + | |
| c.671T>A | p.Val224Glu | F | 1 | + | |
| c.719A>G | p.Asp240Gly | F | 1 | NA | |
| c.1039G>C | p.Ala347Pro | F | 1 | + | + |
| c.208A>T | p.Lys70X | F | 1 | + | |
| c.633C>A | p.Cys211X | M | 1 | + | |
| c.831C>A | p.Tyr277X | F | 1 | NA | |
| c.996C>T | p.Gln332X | F | 1 | NA | |
| c.33_33+1delGG | F | 1 | + | ||
| c.107_111delACAG | F | 1 | NA | ||
| c.143delG | F | 1 | + | ||
| c.439_445delTCCCTGC | F | 1 | + | ||
| c.478delC | M | 1 | + | ||
| c.630_633delATGC | F | 1 | NA | ||
| c.639_641delTGC | M | 1 | − | ||
| c.850_860del11 | F | 1 | + | ||
| c.940_941delCA | F | 1 | + | ||
| c.1084delT | F | 1 | + | ||
| c.632dupG | F | 1 | − | ||
| c.633dupC | F | 1 | + | + | |
| c.720_745dup26 | F | 1 | + | ||
| c.730_731dupCT | F | 1 | − | ||
| c.863_864insT | F | 1 | NA | ||
| c.905_906insC | F | 1 | + | ||
| c.266+1G>A | F | 1 | NA | ||
| c.267-1G>C | F | 1 | NA | ||
| c.612+1G>A | M/F | 3 | NA | ||
| c.651+1G>A | F | 1 | + | ||
| c.825+3delA | F | 1 | NA | ||
| c.826-2A>T | F | 1 | + | ||
| c.825+2dupT | F | 1 | NA | ||
| c.913-1G>C | F | 1 | + | ||
| c.578_581delTGCAGinsA, c.604_612+28del37 | M | 1 | NA |
NA. Not available
Of interest, the c.612G>T (p.Q204H) missense mutation occurred at the last nucleotide of exon 10. RT-PCT analysis of mRNA isolated from the patient’s blood sample indicated that this mutation resulted in an aberrantly spliced HMBS transcript, which used an alternative splice donor site that was nine nucleotides upstream of the wildtype donor site and resulted in an in-frame deletion of nine nucleotides. This transcript encodes a mutant protein designated Q204HΔ 3 in which the three amino acids, V202, G203, and Q204, were deleted (Figure 1).
Figure 1.
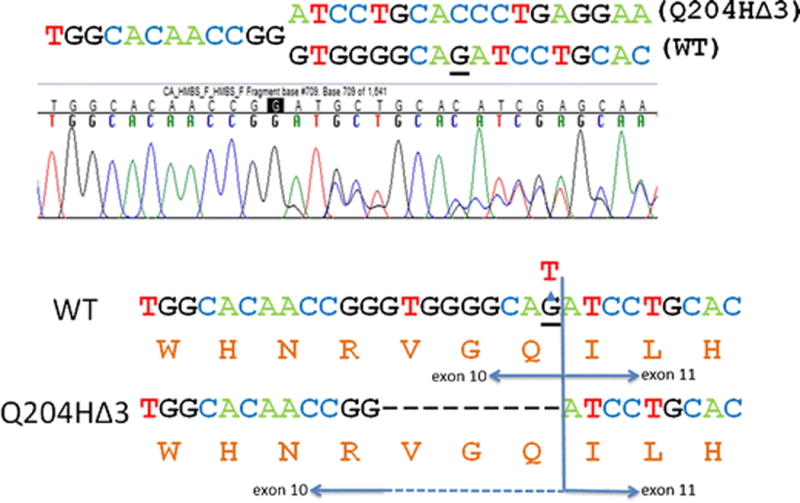
The c.612G>T mutation leads to aberrant splicing. DNA sequencing chromatogram showing two different HMBS cDNA transcripts from a patient heterozygous for the c.612G>T mutation (top panel). Diagram depicting the expected protein sequences from the wildtype and mutant transcripts (bottom panel). c.612 is underlined.
The Effects of HMBS Missense and In-frame Deletion Mutations on HMBS Enzyme Activity and Thermostability
The 11 missense mutations, including p.Q204HΔ 3, and the in-frame deletion mutation (c.639_641delTGC) were expressed in E. coli to determine HMBS activities. With the exception of c.1039G>C (p.A347P), all mutants retained ≤5% of the expressed wildtype HMBS activity, confirming their pathogenicity (Table 2). The p.A347P mutant had ~51% of the expressed wildtype activity (Table 2).
Table 2.
Prokaryotic Expression of HMBS Missense and In-frame Deletion Mutations
| Mutation | Mean % Expressed WT Activity |
|---|---|
| WT | 100 |
| p.L30P | 0.2 |
| p.F77L | 0.1 |
| p.A112P | 0.1 |
| p.A122D | 0.2 |
| p.T145I | 0.4 |
| p.L154P | 0.1 |
| p.L170P | BLD** |
| p.Q204HΔ3 | BLD** |
| p.V224E | 0.9 |
| p.D240G | 5 |
| p.A347P | 51 |
| c.639_641delTGC* | BLD** |
This mutation leads to an in-frame deletion of a single amino acid
BLD: Below the limits of detection
Thermostability assays showed that the p.A347P mutant protein retained little, if any, HMBS activity after a 90 min incubation (65°C), while the expressed wildtype enzyme retained ~60% of its initial activity, thus indicating that the p.A347P protein was highly thermolabile (Figure 2).
Figure 2.
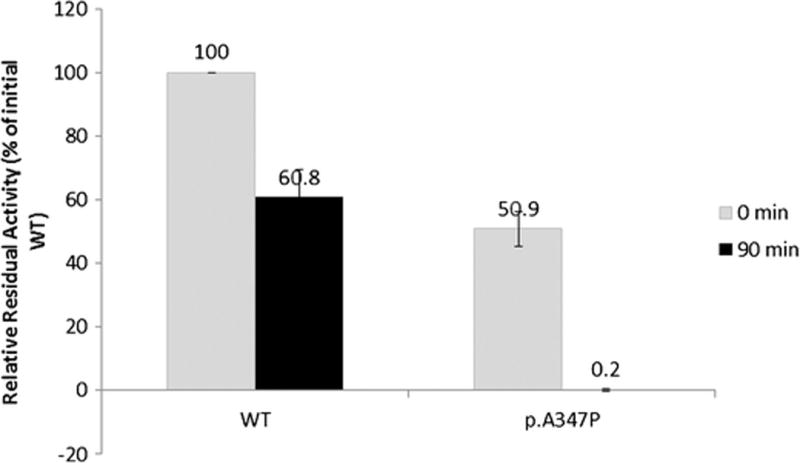
The p.A347P mutant protein is thermolabile. The thermostability of the WT and p.A347P enzymes was assessed by comparing their respective enzyme activity to the initial WT activity prior to thermoinactivation. The experimental results reflect the means of four independent experiments and the error bars denote 1 standard deviation.
The Effects of HMBS Missense Mutations on HMBS Protein Stability and Structure
Western blot analyses showed that the mutations encoding p.L30P, p.F77L, p.T145I, p.L170P, and p.A347P proteins were expressed at levels similar to that of expressed wildtype, whereas the mutations encoding p.A112P, p.A122D, p.L154P, p.V224E and p.D240G were expressed at significantly reduced protein levels (Figure 3).
Figure 3.
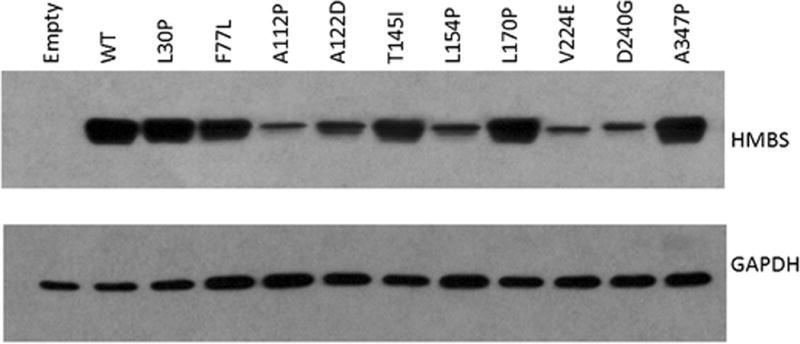
Western blot of HMBS missense mutants. The stability of the expressed mutant proteins were compared to that of wildtype by western blot analysis.
Mapping the missense mutations on the crystal structure of the human HMBS enzyme showed that residues L30, T145 and L170 were all within 5.5 Å of the dipyrromethane cofactor (Figure 4, top panel), which is attached to the active site cysteine residue (Jordan and Warren, 1987). L30 and L170 reside on an α-helix, and the substitution to proline likely disrupts the helical structure. In contrast, the remaining residues (F77, A112, A122, L154, V224, D240, A347) all were located on the surface of the enzyme, far from the dipyrromethane cofactor and the active site (Figure 4, bottom panel). Notably, the F77 residue was located at the dimer interface (data not shown). Four residues (A112, A122, V224 and D240) were located in β-sheet structures whereas F77, L154 and A347 were on α-helices (Figure 4, bottom panel).
Figure 4.
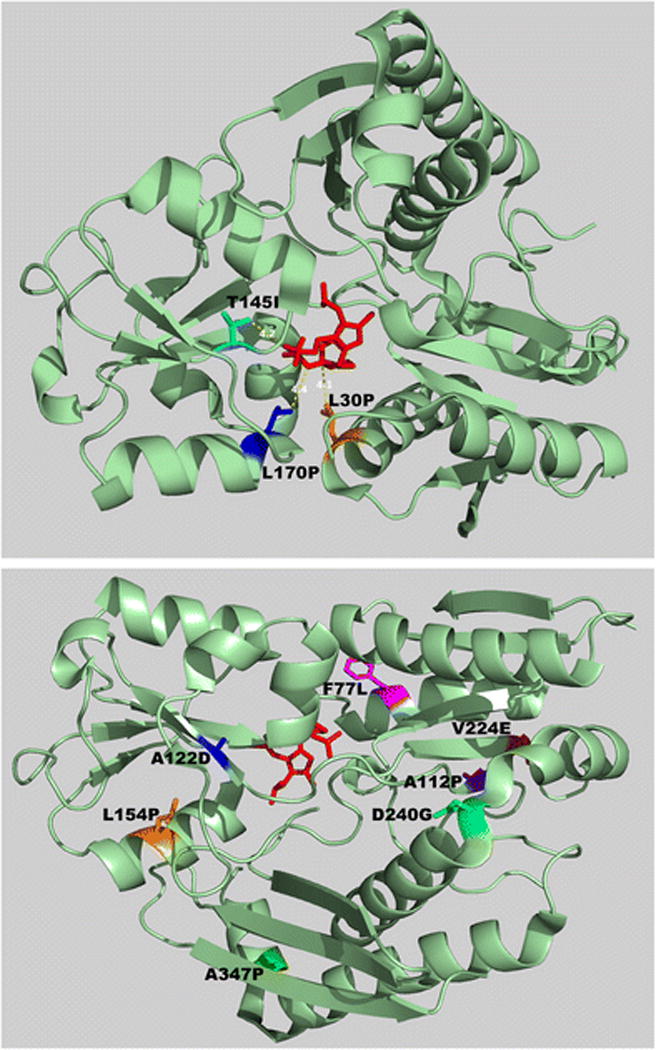
Mapping of the missense mutations on a crystal structure of the human HMBS protein. Three mutations altered amino acids close to the dipyrromethane co-factor/active site (top panel), while the others disrupted residues on the surface of the protein (bottom panel). In the top panel, the distance between each of the mutated residues and the dipyrromethane co-factor are shown.
DISCUSSION
The diagnosis of AIP is usually significantly delayed due to the non-specific presenting symptoms of the disorder (Anderson et al., 2005) and the fact that it is a rare disease with an estimated prevalence of 1 in ~2,000 individuals but a penetrance of only ~1% (Chen et al., 2016). Once AIP is suspected, however, establishing the diagnosis is fairly straightforward, particularly during an acute attack when abnormal porphyrin precursor (i.e. ALA and PBG) and porphyrin intermediates can be documented (Anderson et al., 2001; Anderson et al, 2005). Alternatively, AIP may be diagnosed by demonstration of decreased HMBS enzymatic activity in erythrocytes (Anderson et al., 2005). However, this method is problematic, as there is significant overlap in the range of HMBS activities between affected individuals with half-normal activity and low normal range (Aarsand et al., 2006) and ~3% of AIP heterozygotes have mutations in exon 1 (Whatley and Badminton, 2005), which is not expressed in the erythroid form of the protein due to alternative splicing in erythroid cells (Mustajoki and Desnick, 1985; Grandchamp et al., 1987). Thus, mutation analysis of the HMBS gene has become the most useful diagnostic test, as it is highly specific and sensitive and permits the identification of AIP patients who are in between attacks as well as asymptomatic family members.
Over the past 10 years, our laboratory has received requests from physicians of >1,200 unrelated individuals for AIP testing. Of those with pathogenic HMBS mutations, here, we report the identification of 40 novel HMBS mutations from 43 unrelated individuals with elevated ALA and/or PBG or symptoms compatible with an acute hepatic porphyria. Consistent with previous reports (De Siervi et al. 1999; Schuurmans et al., 2001; Floderus et al., 2002; Hift and Meissner, 2005; von und zu Fraunberg et al., 2005; Bonkovsky et al., 2014), the majority of the patients in our study were female, with a female to male ratio of 38/5 (Table 1). With the exception of the c.639_641delTGC mutation, which was in-frame but expressed no residual HMBS activity in vitro (Table 2), and the c.1084delT mutation, all deletions, insertions, and the complex indel mutation were frame-shift mutations predicted to result in premature stop codons. Consequentially, these mutations either triggered nonsense mediated decay (NMD) and were degraded at the mRNA level, or they generated truncated non-functional HMBS polypeptide chains. The c.1084delT mutation disrupted the stop codon and was predicted to result in no-go decay (van Hoof et al., 2002) of the transcript or a mutant HMBS protein that was 180 amino acids longer than the normal enzyme. Notably, all eight splice site mutations altered the consensus sequence and were predicted to result in aberrant splicing patterns using two splice site prediction programs, CRYP-SKIP (Divina et al., 2009) and RegRNA 2.0 (Chang et al., 2013).
Among the 11 missense mutations, of particular interest was the c.612G>T (p.Q204H) mutation, which occurred at the last nucleotide of exon 10. CRYP-SKIP (Divina et al., 2009) and RegRNA 2.0 (Chang et al., 2013) both identified an upstream alternative donor splice site at c.604_605GT in the presence of the c.612G>T substitution. RT-PCR studies on the patient’s isolated total RNA confirmed the use of this upstream splice site, resulting in an alternative HMBS transcript with an in-frame deletion of the last nine nucleotides (Fig. 1). The resultant protein lacking amino acids V202, G203, and Q204 (Q204HΔ3), had no residual enzyme activity (Table 2). Interestingly, the expressed Q204H protein retained ~80% of wildtype HMBS activity when the alternative splicing error is not present (data not shown), indicating that a glutamine to histidine substitution at this position has minimal impact on the enzymatic activity. These findings underscore the importance of performing both qualitative and quantitative RNA studies when mutations are at or near exon/intron boundaries to assess whether normal splicing occurs and at what frequency.
With the exception of the mutation encoding p.A347P, which retained 51% of wildtype activity, all of the expressed mutations had <5% of expressed wildtype HMBS activity (Table 2). The p.L30P, p.F77L, p.T145I, and p.L170P mutant proteins were detected on Western blots at levels that were comparable to expressed wildtype enzyme levels (Fig. 3), suggesting that they were relatively structurally stable at 37°C but catalytically inactive. When the HMBS activities were normalized against relative HMBS protein content (designated as semi-quantitative CRIM-specific HMBS activity), as determined by the Western blot analyses (Fig. 3), all four mutations had < 1% of CRIM-specific wildtype activity, indicating that these mutations completely abolished the enzyme’s catalytic activity. In line with this finding, the L30, T145 and L170 residues were in close proximity to the dipyrromethane cofactor/active site (Fig. 4 top panel). Thus, the p.L30P, p.T145I, and p.L170P mutations presumably disrupted the active site architecture, or the formation and/or binding of the dipyrromethane cofactor to the apo-HMBS enzyme. The F77 residue was located at the dimer interface, suggesting that the p.F77L mutation interfered with dimer formation but had no impact on the monomeric enzyme’s stability. In contrast, protein expression of the mutations encoding p.A112P, p.A122D, p.L154P, p.V224E, and p.D240G were markedly decreased relative to expressed wildtype activity (Fig. 3), suggesting that these alterations disrupted structural elements that are critical for proper HMBS folding. The CRIM-specific HMBS activities were ~4, 3, 2, 19, and 55% of the expressed wildtype enzyme for the p.A112P, p.A122D, p.L154P, p.V224E, and p.D240G mutant enzymes, respectively. Thus, these mutant proteins presumably decreased not only the stability of the enzyme but also the catalytic activity. Interestingly, all of these residues were located at the surface of the HMBS enzyme structure, away from the dipyrromethane cofactor/active site (Fig. 4, bottom). The A347 residue also was located on the enzyme surface, at the end of the α-helix in domain 1 before the active-site loop (Fig 4, bottom). The p.A347P enzyme protein was unique among the surface residue mutations in that it was stable at 37°C but was highly thermolabile at 65°C (Fig. 2, Fig. 3), suggesting that this alteration had less of an impact on overall protein folding. Notably, the p.A347P mutant protein had a CRIM-specific activity comparable to the expressed wildtype enzyme, indicating that this mutation decreased the enzyme’s stability but had no impact on its catalytic ability.
To date, a total of 414 HMBS mutations have been reported in the Human Gene Mutation Database (version 2017.v3). These studies increased the total number of HMBS mutations to 454 (a 9.7% increase) (Table 3). In vitro studies of the 11 missense mutations and one in-frame deletion confirmed the pathogenicity of these lesions. These studies facilitated the molecular diagnoses of AIP family members and further expanded the molecular heterogeneity of this acute hepatic porphyria.
Table 3.
Expansion of the Molecular Heterogeneity of AIP
| Mutation Type | Missense | Nonsense | Splicing | Regulatory | Small Deletion | Small Insertion | Small Indels | Gross Insertion/Deletion | Complex | Total |
|---|---|---|---|---|---|---|---|---|---|---|
| HGMD (2017 v3) | 136 | 33 | 91 | 7 | 81 | 41 | 8 | 15 | 2 | 414 |
| This study | 11 | 4 | 8 | – | 10 | 6 | – | – | 1 | 40 |
| Total | 147 | 37 | 99 | 7 | 91 | 47 | 8 | 15 | 3 | 454 |
| % of increase from HGMD 2017.V3 | 7.5 | 10.8 | 8.1 | – | 11 | 12.8 | – | – | 33.3 | 8.8 |
Supplementary Material
Acknowledgments
We thank Vendenii Zaikov for his excellent technical assistance. This work was funded in part by Career Development Awards K01 DK087971 (to M.Y.) and K23 DK095946 (to M.B.) from the National Institutes of Health (NIH) and a cooperative grant (U54 Dk083909) for the Porphyrias Consortium, which is a part of the NIH Rare Diseases Research Network (RDCRN) and supported through collaboration between the NIH Office of Rare Diseases Research (ORDR) at the National Center for Advancing Translational Science (NCATS) and National Institutes of Diabetes and Digestive and Kidney Diseases (NIDDK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
All authors confirm independence from the sponsors and that the content of the article has not been influenced by the sponsors.
Footnotes
Ethics approval: These studies were approved by the Icahn School of Medicine Institutional Review Board.
Patient Consent Statement: All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study
Animal Rights: This article does not contain any studies with animal subjects performed by any of the authors.
Competing interest statement: Manisha Balwani, Robert Desnick and John Phillips are consultants for Alnylam Pharmaceuticals and Recordati Rare Diseases. Manisha Balwani receives clinical trial grants and Robert Desnick has received research grants from Alnylam Pharmaceuticals and Recordati Rare Diseases. Robert Desnick and Makiko Yasuda are inventors of intellectual property licensed to Alnylam Pharmaceuticals. Brenden Chen, Constanza Solis-Villa, Angelika Erwin, and Irina Nazrenko declare that they have no conflict of interest
Author Contributions: B.C., C.V.S., A.L.E., M.B., I.N., J.D.P, R.J.D. and M.Y. designed the studies; B.C., C.V.S., A.L.E., I.N. and J.D.P performed experiments; B.C., C.V.S, M.B., J.D.P, R.J.D. and M.Y. analyzed data; B.C., R.J.D. and M.Y. wrote the manuscript.
References
- Aarsand AK, Petersen PH, Sandberg S. Estimation and application of biological variation of urinary delta-aminolevulinic acid and porphobilinogen in healthy individuals and in patients with acute intermittent porphyria. Clin Chem. 2006;52:650–656. doi: 10.1373/clinchem.2005.060772. [DOI] [PubMed] [Google Scholar]
- Anderson KE, Sassa S, Bishop DF, Desnick RJ. Disorders of Heme Biosynthesis: X-Linked Sideroblastic Anemia and the Porphyrias. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th. New York: McGraw-Hill; 2001. pp. 2961–3062. [Google Scholar]
- Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142:439–450. doi: 10.7326/0003-4819-142-6-200503150-00010. [DOI] [PubMed] [Google Scholar]
- Bonkovsky HL, Maddukuri VC, Yazici C, et al. Acute porphyrias in the USA: features of 108 subjects from porphyrias consortium. Am J Med. 2014;127:1233–1241. doi: 10.1016/j.amjmed.2014.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TH, Huang HY, Hsu JB, et al. An enhanced computational platform for investigating the roles of regulatory RNA and for identifying functional RNA motifs. BMC Bioinformatics. 2013;14(Suppl 2):S4. doi: 10.1186/1471-2105-14-S2-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Astrin KH, Lee G, et al. Acute intermittent porphyria: identification and expression of exonic mutations in the hydroxymethylbilane synthase gene. An initiation codon missense mutation in the housekeeping transcript causes “variant acute intermittent porphyria” with normal expression of the erythroid-specific enzyme. J Clin Invest. 1994;94:1927–1937. doi: 10.1172/JCI117543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Solis-Villa C, Hakenberg J, et al. Acute Intermittent Porphyria: Predicted Pathogenicity of HMBS Variants Indicates Extremely Low Penetrance of the Autosomal Dominant Disease. Hum Mutat. 2016;37:1215–1222. doi: 10.1002/humu.23067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Siervi A, Rossetti MV, Parera VE, et al. Acute intermittent porphyria: biochemical and clinical analysis in the Argentinean population. Clin Chim Acta. 1999;288:63–71. doi: 10.1016/s0009-8981(99)00139-4. [DOI] [PubMed] [Google Scholar]
- Divina P, Kvitkovicova A, Buratti E, et al. Ab initio prediction of mutation-induced cryptic splice-site activation and exon skipping. Eur J Hum Genet. 2009;17:759–765. doi: 10.1038/ejhg.2008.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floderus Y, Shoolingin-Jordan PM, Harper P. Acute intermittent porphyria in Sweden. Molecular, functional and clinical consequences of some new mutations found in the porphobilinogen deaminase gene. Clin Genet. 2002;62:288–297. doi: 10.1034/j.1399-0004.2002.620406.x. [DOI] [PubMed] [Google Scholar]
- Grandchamp B, De Verneuil H, Beaumont C, et al. Tissue-specific expression of porphobilinogen deaminase. Two isoenzymes from a single gene. Eur J Biochem. 1987;162:105–110. doi: 10.1111/j.1432-1033.1987.tb10548.x. [DOI] [PubMed] [Google Scholar]
- Granick S. Induction of the synthesis of delta-aminolevulinic acid synthetase in liver parenchyma cells in culture by chemical that induce acute porphyria. J Biol Chem. 1963;238:2247–2249. [PubMed] [Google Scholar]
- Granick S. The induction in vitro of the synthesis of delta-aminolevulinic acid synthetase in chemical porphyria: a response to certain drugs, sex hormones, and foreign chemicals. J Biol Chem. 1966;241:1359–1375. [PubMed] [Google Scholar]
- Gu XF, de Rooij F, Lee JS, et al. High prevalence of a point mutation in the porphobilinogen deaminase gene in Dutch patients with acute intermittent porphyria. Hum Genet. 1993;91:128–130. doi: 10.1007/BF00222712. [DOI] [PubMed] [Google Scholar]
- Hift RJ, Meissner PN. An analysis of 112 acute porphyric attacks in Cape Town, South Africa: Evidence that acute intermittent porphyria and variegate porphyria differ in susceptibility and severity. Medicine (Baltimore) 2005;84:48–60. doi: 10.1097/01.md.0000152454.56435.f3. [DOI] [PubMed] [Google Scholar]
- Jordan PM, Warren MJ. Evidence for a dipyrromethane cofactor at the catalytic site of E. coli porphobilinogen deaminase. FEBS Lett. 1987;225:87–92. doi: 10.1016/0014-5793(87)81136-5. [DOI] [PubMed] [Google Scholar]
- Kauppinen R, von und zu Fraunberg M. Molecular and biochemical studies of acute intermittent porphyria in 196 patients and their families. Clin Chem. 2002;48:1891–1900. [PubMed] [Google Scholar]
- Mustajoki P, Desnick RJ. Genetic heterogeneity in acute intermittent porphyria: characterisation and frequency of porphobilinogen deaminase mutations in Finland. Br Med J (Clin Res Ed) 1985;291:505–509. doi: 10.1136/bmj.291.6494.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mykletun M, Aarsand AK, Stole E, et al. Porphyrias in Norway. Tidsskr Nor Laegeforen. 2014;134:831–836. doi: 10.4045/tidsskr.13.0649. [DOI] [PubMed] [Google Scholar]
- Puy H, Gouya L, Deybach JC. Porphyrias. Lancet. 2010;375:924–937. doi: 10.1016/S0140-6736(09)61925-5. [DOI] [PubMed] [Google Scholar]
- Sassa S, Granick S. Induction of-aminolevulinic acid synthetase in chick embryo liver cells in cluture. Proc Natl Acad Sci U S A. 1970;67:517–522. doi: 10.1073/pnas.67.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuurmans MM, Schneider-Yin X, Rufenacht UB, et al. Influence of age and gender on the clinical expression of acute intermittent porphyria based on molecular study of porphobilinogen deaminase gene among Swiss patients. Mol Med. 2001;7:535–542. [PMC free article] [PubMed] [Google Scholar]
- Song G, Li Y, Cheng C, et al. Structural insight into acute intermittent porphyria. FASEB J. 2009;23:396–404. doi: 10.1096/fj.08-115469. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136:665–677. doi: 10.1007/s00439-017-1779-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hoof A, Frischmeyer PA, Dietz HC, Parker R. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science. 2002;22:2262–2264. doi: 10.1126/science.1067272. [DOI] [PubMed] [Google Scholar]
- von und zu Fraunberg M, Pischik E, Udd L, et al. Clinical and biochemical characteristics and genotype-phenotype correlation in 143 Finnish and Russian patients with acute intermittent porphyria. Medicine (Baltimore) 2005;84:35–47. doi: 10.1097/01.md.0000152455.38510.af. [DOI] [PubMed] [Google Scholar]
- Whatley SD, Badminton MN. Acute Intermittent Porphyria. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews(R) Seattle (WA): 2005. [Google Scholar]
- Whatley SD, Badminton MN. Role of genetic testing in the management of patients with inherited porphyria and their families. Ann Clin Biochem. 2013;50:204–216. doi: 10.1177/0004563212473278. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.