

Abstract
Plasmacytoid dendritic cells (pDC) are the major source of type I interferons (IFN‐I) during viral infections, in response to triggering of endosomal Toll‐like receptors (TLRs) 7 or 9 by viral single‐stranded RNA or unmethylated CpG DNA, respectively. Synthetic ligands have been used to disentangle the underlying signaling pathways. The adaptor protein AP3 is necessary to transport molecular complexes of TLRs, synthetic CpG DNA, and MyD88 into endosomal compartments allowing interferon regulatory factor 7 (IRF7) recruitment whose phosphorylation then initiates IFN‐I production. High basal expression of IRF7 by pDC and its further enhancement by positive IFN‐I feedback signaling appear to be necessary for robust cytokine production. In contrast, we show here that in vivo during mouse cytomegalovirus (MCMV) infection pDC produce high amounts of IFN‐I downstream of the TLR9‐to‐MyD88‐to‐IRF7 signaling pathway without requiring IFN‐I positive feedback, high IRF7 expression, or AP3‐driven endosomal routing of TLRs. Hence, the current model of the molecular requirements for professional IFN‐I production by pDC, established by using synthetic TLR ligands, does not strictly apply to a physiological viral infection.
Keywords: IRF7, mouse cytomegalovirus, plasmacytoid dendritic cells, type I interferons, viral infection
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction
Introduction
Type I and III interferons (IFN‐I/III) are cytokines produced early in vivo during viral infections and playing a major role in host resistance. Both cytokine families induce the expression of numerous interferon‐stimulated genes (ISG) involved in diverse biological functions, including the direct inhibition of viral replication and the activation of innate and adaptive immunity (Tomasello et al, 2014). IFN‐I can act on all cell types since their receptor is ubiquitously expressed, whereas IFN‐III selectively targets epithelial cells due to their selective expression of the receptor for these cytokines.
Upon sensing of viral replication, through triggering of cytosolic receptors for RNA or DNA, virtually any infected cell type can produce IFN‐I/III. However, many viruses have evolved to interfere with this detection, expressing genes able to delay, blunt, or prevent IFN‐I/III production by infected cells. As a likely countermeasure to this immune evasion strategy, vertebrates have evolved professional IFN‐I/III‐producing cells, corresponding to specialized mononuclear phagocytes that are refractory to viral infection, thus escaping cell‐intrinsic effects of viral immunoevasion genes, but that can engulf and recognize viral material to produce high levels of cytokines (Tomasello et al, 2014). The first identified professional IFN‐I‐producing cells were human plasmacytoid dendritic cells (pDC) (Cella et al, 1999; Siegal et al, 1999). pDC are well conserved along warm‐blooded vertebrate evolution, based on both their professional IFN‐I production and their global gene expression profile (Asselin‐Paturel et al, 2001; Dalod et al, 2002; Robbins et al, 2008; Vu Manh et al, 2015). Activated pDC also produce IFN‐III (Yin et al, 2012) and pro‐inflammatory cytokines and chemokines (Swiecki & Colonna, 2015).
pDC are the major early source of IFN‐I/III in vivo during viral infections, as shown in mice and macaques (Malleret et al, 2008; Tomasello et al, 2014). In mice, pDC can promote resistance to systemic acute infection by the herpes simplex virus 2 (HSV‐2) (Swiecki et al, 2013). However, how their professional IFN‐I/III production precisely contributes to this protection remains to be deciphered. pDC role in shaping antiviral innate and adaptive immunity might supersede their contribution to the induction of IFN‐I‐dependent cell‐intrinsic antiviral defenses, as recently proposed in various mouse models of viral infections (Swiecki et al, 2013; Cocita et al, 2015; Brewitz et al, 2017). During chronic viral infections in mice, macaques, and humans, pDC production of, and response to, IFN‐I can play protective or deleterious roles (Tomasello et al, 2014). The tumor microenvironment inhibits pDC production of IFN‐I/III and polarizes their functions toward immunosuppression, which can be reversed in mice upon intratumoral administration of oncolytic viruses or synthetic virus‐type stimuli (Fonteneau et al, 2013; Le Mercier et al, 2013). IFN‐I production by pDC contributes to several autoimmune or inflammatory diseases (Tomasello et al, 2014). Hence, understanding the mechanisms controlling IFN‐I/III production specifically in pDC should help developing innovative treatments against viral infections, cancer, and autoimmune/inflammatory diseases.
pDC are mainly activated upon triggering of endosomal TLR7 and TLR9 by their respective ligand pairs, uridine‐containing single‐stranded RNA and guanosine (Shibata et al, 2016; Zhang et al, 2016), unmethylated CpG DNA (Kadowaki et al, 2001), and 5′‐xCx DNA (Ohto et al, 2018). To be active, TLR7/9 need UNC93B1‐dependent routing from ER to dedicated endosomes where they are cleaved to expose their ligand‐binding domain. Their triggering then initiates a MyD88‐to‐IRF7 signaling pathway leading to high‐level production of all IFN‐I/III subtypes (Honda et al, 2005a,b; Swiecki & Colonna, 2015). Studies in Irf5‐KO mice suggested that pDC production of pro‐inflammatory cytokines is mainly IRF5‐dependent (Takaoka et al, 2005). This differential involvement of IRF7 versus IRF5 in the production of different cytokines by mouse pDC has been associated with signaling from different endosomes (Sasai et al, 2010). The signaling cascade leading to IFN‐I production is proposed to be initiated in dedicated endosomes to which TLR7/9 are sorted by AP3‐dependent mechanisms for pre‐association with MyD88 and IRF7. Upon TLR triggering, this allows fast and efficient activation of IRF7 by the NF‐kB pathway kinases IKKα and IKKβ (Hoshino et al, 2006; Pauls et al, 2012; Hayashi et al, 2018). In most cells, IRF7 expression is barely detectable at steady state, but significantly increased in response to IFN‐I via positive feedback (Marie et al, 1998; Sato et al, 1998). However, pDC constitutively express high levels of IRF7 which is commonly accepted as being a key mechanism endowing them with their professional IFN‐I/III production (Kerkmann et al, 2003; Honda et al, 2005b; Prakash et al, 2005; O'Brien et al, 2011; Kim et al, 2014; Swiecki & Colonna, 2015). High constitutive IRF7 expression by pDC has been proposed to result from tonic responses to very low homeostatic IFN‐I levels, based on in vitro experiments (Taniguchi & Takaoka, 2001; Honda et al, 2005b; Prakash et al, 2005; O'Brien et al, 2011). However, whether in vivo pDC professional IFN‐I production necessitates positive feedback from these cytokines is controversial. It may depend on the precise nature of the TLR7/9 ligands used. It is critical when mouse pDC are stimulated in vivo with synthetic TLR7/9 ligands (Asselin‐Paturel et al, 2005). It might not be necessary in mice infected with vesicular stomatitis virus (VSV) (Barchet et al, 2002) but appears to boost systemic IFN‐I production during mouse cytomegalovirus (MCMV) infection (Dalod et al, 2002). IFN‐I modulation of their own production by pDC in vivo has only been investigated in mice genetically deficient for the expression of the IFN‐I receptor (Ifnar1‐KO animals), where the responses of all cells to the cytokines are abrogated leading to enhanced viral replication and perturbed immune responses. Hence, further experiments are required to rigorously investigate whether, and how, cell‐intrinsic IFN‐I responses modulate pDC functions in vivo. More generally, because many studies aiming at deciphering how the production of IFN‐I/III is controlled specifically in pDC have been conducted in vitro or with synthetic TLR ligands, it is crucial to examine whether the same mechanisms operate in vivo during a viral infection. To address these questions, we combined MCMV infection as a physiological model of the interactions between a virus and its natural host, with gene expression profiling and functional studies on ex vivo isolated pDC, comparing results between WT and mutant mixed bone marrow chimera (MBMC) mice. Our results show that in vivo production of IFN‐I by mouse pDC required triggering of the canonical TLR9‐to‐MyD88‐to‐NFKB/IRF7 signaling pathway both during MCMV infection and upon CpG DNA (CpG) administration, whereas positive IFN‐I feedback signaling and AP3 function were only essential in response to the later stimulation. Thus, our study suggests that physiological viral infections likely trigger additional activation pathways in pDC as compared to stimulation with synthetic TLR ligands, allowing to bypass some of the molecular requirements identified in the later stimulation condition.
Results
pDC are activated under combined and distinctive instruction by TLR, IFN‐I, and IFN‐γ, during MCMV infection in vivo
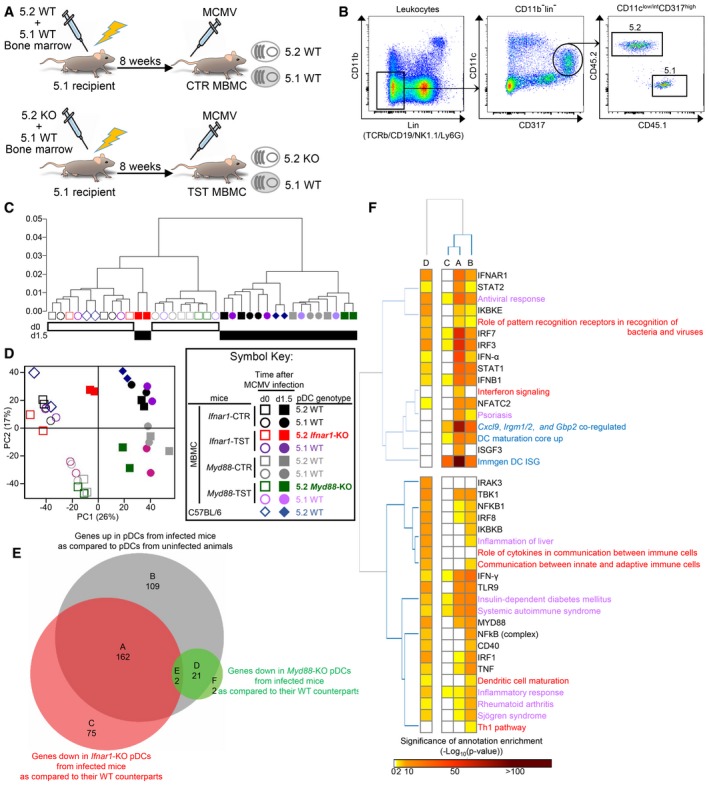
Intraperitoneal injection of mice with MCMV leads to a rapid and robust production of IFN‐I by splenic pDC, in a TLR7/9‐ and MyD88‐dependent manner (Dalod et al, 2002, 2003; Krug et al, 2004; Delale et al, 2005; Zucchini et al, 2008a,b). It also induces pDC phenotypic maturation under IFN‐I instruction (Dalod et al, 2003). Hence, we used this model of interaction between a virus and its natural mammalian host to more broadly investigate the respective roles of MyD88‐ and IFN‐I‐dependent signaling pathways in shaping pDC activation under a physiological viral infection. Because MyD88 and the receptor for IFN‐I (IFNAR) are expressed in many cell types, their genetic inactivation affects many processes during MCMV infection (Krug et al, 2004; Delale et al, 2005; Strobl et al, 2005; Baranek et al, 2012; Cocita et al, 2015; Madera & Sun, 2015; Puttur et al, 2016). Thus, to address the cell‐intrinsic role of MyD88‐ and IFN‐I‐dependent signaling pathways in pDC, we generated MBMC mice (Fig 1A). Recipient CD45.1 mice were lethally irradiated and reconstituted with a 1:1 mixture of bone marrow (BM) cells from WT CD45.1 and Ifnar1‐KO or Myd88‐KO CD45.2 donor animals, to generate test mice (TST MBMC). Congenic CD45.1+ mice differ from the CD45.2+ C57BL/6 strain for several gene polymorphisms and immune parameters (Waterstrat et al, 2010). Therefore, we included as controls mice engrafted with a mixture of WT CD45.1 and WT CD45.2 BM cells (CTR MBMC) (Fig 1A). MBMC were infected with MCMV at least 8 weeks after BM graft. Splenic pDC, identified as lineage− CD11b− CD11clow/int CD317high cells (Figs 1B and EV1), were isolated 1.5 days after infection, at the peak of IFN‐I production, sorted into CD45.1+ WT versus CD45.2+ Ifnar1‐KO or Myd88‐KO populations (Fig 1B), and used for pangenomic gene expression profiling by microarrays (Fig 1C–F). Our strategy for pDC phenotypic identification was validated by two independent analyses. First, we showed that over 94% of the gated cells expressed high levels of SiglecH (Fig EV1), another cell surface marker selectively expressed on mouse pDC. Second, gene expression profiling showed that the cell populations sorted as lineage− CD11b− CD11clow/int CD317high cells harbored a transcriptional signature typical of bona fide pDC, likewise to the pDC sorted by the ImmGen consortium (Fig EV2; Appendix Fig S1). They lacked any evidence of contamination by other immune cell types, namely B cells, T cells, NK cells, and other lymphoid cells; cDC1s, cDC2s, monocytes, macrophages, and other myeloid cells (Fig EV2; Appendix Fig S1). To ensure that the transcriptomic responses observed in MBMC pDC were not strongly affected by BM transplantation, we also sorted splenic pDC from non‐irradiated C57BL/6 animals.
Figure 1. IFN‐I, Myd88, and IFN‐γ activate distinct sets of genes in mouse pDC during MCMV infection.
-
AGeneration of mixed bone marrow chimera (MBMC) mice; CTR, control; TST, test. Recipient CD45.1 (5.1) mice were irradiated and then reconstituted with equal proportions of bone marrow cells isolated from indicated mice. Mice were used at least 8 weeks after reconstitution.
-
BGating strategy. pDC were identified as lineage (TCRb, CD19, Ly6G, NK1.1)− CD11b− CD11clow/int CD317high cells. 5.2 versus 5.1 cells were finally discriminated within the pDC gate.
-
C, D5.2+ pDC were isolated by sorting from MBMC reconstituted with BM cells from WT B6 (CTR), Ifnar1 −/−‐, or Myd88 −/−‐deficient (TST) mice. Their 5.1+ WT counterparts were isolated from the same mice. Two independent experiments were performed for each experimental condition (i.e., with Ifnar1‐CTR and –TST MBMC on the one hand, versus Myd88‐CTR and TST MBMC on the other hand). Samples from uninfected animals are shown as empty symbols and those from MCMV‐infected mice as filled symbols. Altogether, this led to 14 biological replicates for WT pDC, irrespective of their CD45 allotypic status and of the type of MBMC they originated from, both for uninfected and MCMV‐infected MBMC. pDC isolated from uninfected or MCMV‐infected C57BL/6 mice were included as internal control. Unsupervised hierarchical clustering (HC) and principal component analysis (PCA) were performed on all the probe sets of the microarrays. For HC, Pearson correlation distance and Ward's method linkage were used.
-
EVenn diagram showing the overlap between the lists of genes significantly upregulated in WT pDC of infected mice (n = 14) as compared to WT pDC of uninfected animals (n = 14) (gray circle), or downregulated in Ifnar1‐KO (n = 2) (red circle) or Myd88‐KO (n = 2) (green circle) pDC isolated from infected TST MBMC, as compared to WT pDC (n = 6) isolated from the same TST or from their matched CTR infected MBMC.
-
FEnrichment of the gene modules identified in (E) for independent gene sets previously established from the analysis of other microarray datasets (blue font), belonging to specific signaling pathways (red font), associated with specific functions or diseases (violet font) or regulated by the indicated upstream regulators (black font), as assessed by using Ingenuity Pathway Analysis (IPA). Data are represented as a heatmap, with gene modules identified in panel (E) as columns and gene sets as rows. The colored cells indicate the significance of the enrichment of each gene module for each gene set, as computed by IPA analysis and according to the color scale shown below the heatmap.
Figure EV1. Expression of CD317 and SiglecH on pDC isolated from uninfected versus MCMV‐infected C57BL/6 or Ifnar1‐KO mice.
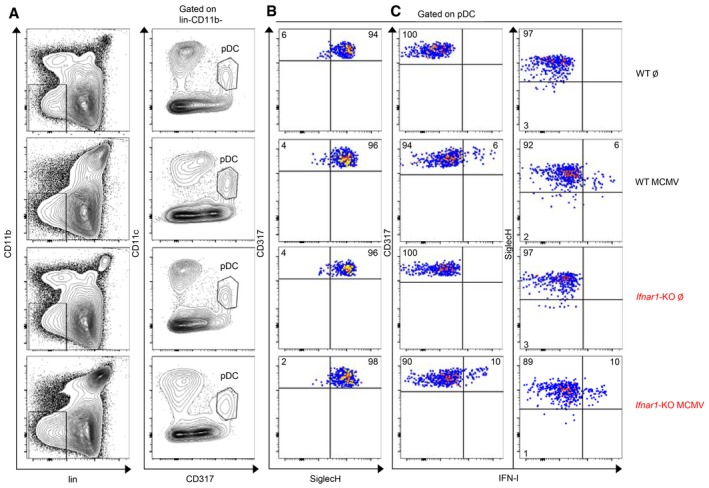
- pDC were identified, as shown in Fig 1B, as lineage (CD3, CD19, Ly6G, NK1.1)− CD11b− CD11clow/int CD317high cells.
- SiglecH expression was analyzed on pDC isolated from the spleen of indicated mice and gated as in (A).
- Dot plots represent IFN‐I versus CD317 (left panels) or versus SiglecH (right panels) stainings in pDC isolated from indicated uninfected or MCMV‐infected mice. Data are representative of two animals for uninfected mice and six animals for MCMV‐infected mice from two independent experiments.
Figure EV2. Comparative gene expression profiling of sorted pDC with other cells sorted from the same mice and with an ImmGen compendium confirms specificity of gating strategy.
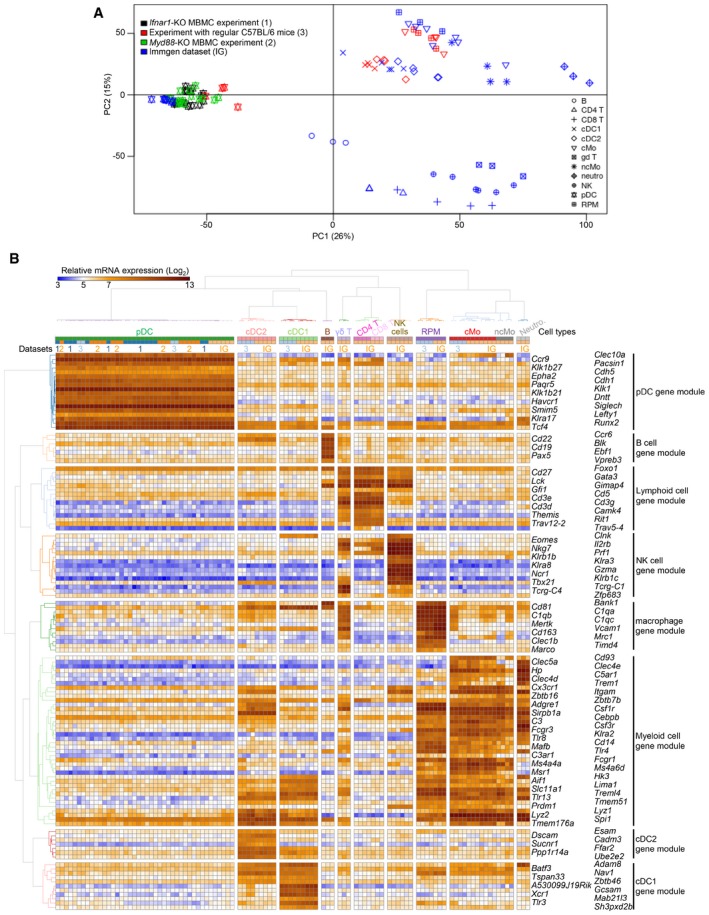
- Principal component analysis demonstrating overall close transcriptomic proximity between splenic pDC sorted from MBMC mice or from C57BL6 mice under steady state or MCMV infection conditions and splenic pDC sorted from uninfected C57BL/6 mice by the ImmGen consortium. The data shown were batch corrected using ComBat with a model considering each independent dataset as one batch and identity between cDC1, cDC2, RPM, and cMo across batches, but without any inference on the relationships between other cell types including pDC across batches (see Appendix Fig S1 for uncorrected data).
- Heatmap showing the expression patterns of well‐known cell type‐specific genes across all samples, with hierarchical clustering of cell samples (columns) and genes (rows). Note that all pDC cluster together in (A) and (B) and that each of the individual pDC samples analyzed strongly expresses the pDC gene module but none of the genes from the other cell type gene modules. Dataset numbers correspond to those indicated in the legend of panel (A).
In hierarchical clustering analysis, Ifnar1‐KO pDC isolated from infected Ifnar1‐TST MBMC segregated with the pDC isolated from uninfected MBMC and C57BL/6 mice (Fig 1C). In contrast, MyD88‐KO pDC isolated from infected Myd88‐TST MBMC regrouped with all the other pDC populations isolated from infected animals in the second major branch of the tree. Consistently, in principal component analysis (PCA), the first axis (PC1) accounting for most of the variability of the dataset (26%) separated pDC samples according to the infection status of the mice, except for Ifnar1‐KO pDC from infected MBMC which regrouped with pDC from uninfected animals (Fig 1D). The second axis (PC2, 17%) separated pDC populations according to each experiment, with a stronger difference between the Myd88‐KO and Ifnar1‐KO MBMC experiments than between the latter and non‐irradiated C57BL/6 animals, consistent with hierarchical clustering. Hence, MCMV infection had a strong impact on pDC transcriptional reprogramming, which was largely unaffected by the BM engraftment protocol and much more profoundly driven by IFN‐I‐dependent signaling pathways than by MyD88.
To precisely identify how MCMV infection affected the gene expression program of pDC, and the contribution of cell‐intrinsic IFN‐I or MyD88 signaling to this response, we identified the genes that were (i) differentially expressed in WT pDC between uninfected versus MCMV‐infected mice (Fig 1E, gray circle), (ii) expressed to lower levels in Ifnar1‐KO pDC from infected mice as compared to their WT counterparts (pDC IFN‐I‐stimulated genes, ISG, Fig 1E, red circle), or (iii) expressed to lower levels in Myd88‐KO pDC from infected mice as compared to their WT counterparts (pDC MyD88‐stimulated genes, MSG, Fig 1E, green circle) (Table EV1). We generated a Venn diagram for visual analysis of the relative size of these three gene lists and of their overlap (Fig 1E). pDC ISG were much more numerous that pDC MSG. Indeed, 56% of all the DEGs upregulated in pDC during MCMV infection (n = 292) were ISG (n = 164), whereas only 8% were MSG (n = 23). Apparently, 37% (n = 109) of these DEGs required neither cell‐intrinsic IFN‐I nor MyD88 signaling for their induction. Hence, this analysis confirmed that pDC gene expression reprograming during MCMV infection is much more profoundly driven by IFN‐I‐dependent than by MyD88‐dependent signaling. Representative genes were selected to generate a heatmap illustrating these different expression patterns (Fig 2).
Figure 2. Heatmap showing the expression profiles of selected genes representative of distinct patterns of molecular control in pDC.
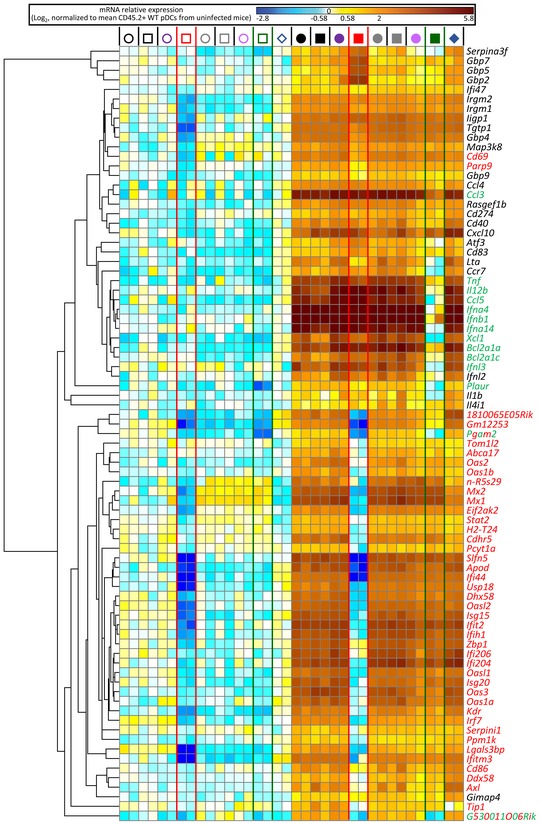
Representative genes were selected for modules identified on Venn diagram in Fig 1E and their expression pattern across all samples shown as a heatmap. Red font, pDC ISG (module A); green font, pDC MSG (module D); mixed red and green font, pDC ISG and MSG (module E); black font, non‐ISG and non‐MSG (module B). The relative mRNA expression levels for each gene (lanes) across biological samples (rows) are represented by colors according to the scale shown above the heatmap. For the legend of the symbols depicting the classes of biological samples above each column please refer to the symbol key in Fig 1D.
We mined the different gene modules established through the Venn diagram analysis for enrichment in annotations linked to specific signaling pathways, biological functions, diseases, or upstream regulators, using ingenuity pathway analysis (IPA) (Fig 1F).
Both pDC gene modules (A) and (B) were enriched for IFNB1‐, IRF7‐, and IRF3‐regulated genes as well as for ISG induced in vivo in splenic CD11c+ MHC‐II+ Flt3+ cells 2 h after subcutaneous IFN‐α administration in mice (“ImmGen DC ISG”) (Mostafavi et al, 2016). However, only the pDC ISG gene module (A) was significantly enriched in targets of the canonical transcription factor complex downstream of IFNAR1 triggering, ISGF3, consistent with the exclusive dependency on cell‐intrinsic IFN‐I signaling of its induction in pDC from infected mice, as exemplified by the OAS gene family (Fig 2). Of note, among the 164 genes whose induction in pDC was dependent on cell‐intrinsic IFN‐I signaling (Table EV1), over 50 had not been identified as ISG in other mononuclear phagocytes or in lymphocytes (Mostafavi et al, 2016), further emphasizing cell type‐dependency of IFN‐I responses. The gene module (B) was more significantly enriched in IFN‐γ target genes. In the ImmGen experiment, many genes were induced in DC by subcutaneous injection of either IFN‐I or IFN‐γ. Since no MBMC experiments were performed, the apparent redundancy between IFN‐I and IFN‐γ effects on DC might have been indirect, consecutive to IFN‐γ induction upon IFN‐I injection, and reciprocally. In our experiments, Irgm1/2 induction in pDC must have been driven by IFN‐γ, since their expression was not affected in Ifnar1‐KO pDC (Fig 2) and these genes are considered to be archetypical IFN‐γ targets. Unexpectedly, although Cxcl10 is considered to be selectively induced by IFN‐I, it belonged to module (B) suggesting redundancy between IFN‐I and IFN‐γ signaling for its induction in pDC. The expression of certain known IFN‐γ‐stimulated genes, such as Gbp family members (Fig 2), was higher in Ifnar1‐KO pDC as compared to their WT counterparts, consistent with the known antagonistic effects of IFN‐I signaling on IFN‐γ responses. Whereas IPA highlighted MyD88 and IFNAR1 as potential upstream regulators of both A and D gene modules, our data show that the former requires IFN‐I but not MyD88 cell‐intrinsic signaling for its induction in pDC, and reciprocally for the latter. Hence, we rigorously determined which gene modules require IFN‐I versus MyD88 cell‐intrinsic signaling for their induction in pDC during a physiological viral infection in vivo, and delineated non‐redundant versus shared versus antagonistic pDC responses to IFN‐I and IFN‐γ.
The pDC ISG gene module (A) was the most significantly enriched in genes upregulated during DC maturation (“DC maturation core up”) (Vu Manh et al, 2013) or associated with annotations pertaining to antiviral/antibacterial responses or psoriasis. Both gene modules A and B were highly enriched for genes associated with insulin‐dependent diabetes mellitus or systemic autoimmune syndrome.
The pDC MSG gene module (D) encompassed mainly genes encoding cytokines and chemokines, including all the genes for IFN‐I/III (Fig 2), and was enriched for TLR‐, MyD88‐, CD40‐, IRAK3‐, NFKB1‐, IKBKB‐, IRF1‐, IRF3‐, IRF7‐, and IRF8‐regulated genes (Fig 1F). It was significantly enriched in genes associated with annotations pertaining to antiviral/antibacterial responses, inflammation, dendritic cell maturation, and the roles of cytokines in the communication between immune cells.
Altogether, these results indicate that, in vivo during MCMV infection, pDC undergo a major transcriptional reprogramming, under combined instruction of IFN‐I, IFN‐γ, and direct TLR triggering. However, these different stimuli drive specific, largely distinct, gene expression programs. In particular, during MCMV infection, contrary to MyD88 cell‐intrinsic signaling, IFN‐I responsiveness is dispensable in pDC for their induction of the expression of many cytokine genes, including for all IFN‐I/III.
IFN‐I production by pDC requires neither IFN‐I positive feedback nor AP3‐driven TLR endosomal routing during MCMV infection as opposed to CpG injection
In contrast to what our microarrays showed for pDC transcriptional responses to MCMV infection, IFN‐I positive feedback is necessary for high‐level IFN‐I production by pDC upon stimulation by CpG (Kerkmann et al, 2003; Asselin‐Paturel et al, 2005; Kim et al, 2014; Wu et al, 2016). Hence, we next examined at the single‐cell protein level whether the molecular requirements for in vivo pDC IFN‐I production and phenotypic maturation differed between MCMV infection and CpG injection. We examined responses 36 h after MCMV infection versus 7 h after CpG injection, because these time points correspond to the peak of pDC IFN‐I production for these two stimuli, respectively (Dalod et al, 2002; Krug et al, 2004; Asselin‐Paturel et al, 2005; Zucchini et al, 2008a). MCMV is slowly replicating, needing 24–30 h to complete its first cycle. Hence, during MCMV infection in vivo, the peak of pDC IFN‐I production occurs 6–12 h after the release of the first wave of endogenously synthesized viral CpG DNA sequences, consistent with the kinetics of the responses to high‐dose injection of synthetic CpG. Indeed, reanalysis of public microarray data showed that all the genes found here to be significantly modulated in pDC at 36 h after MCMV infection were similarly modulated upon pDC stimulation with CpG in vitro for 4–12 h (Appendix Fig S2). We isolated splenic pDC from MCMV‐infected CTR and Ifnar1‐KO MBMC and analyzed by flow cytometry their expression of CD86 and IFN‐I. MCMV infection leads to a decrease in SiglecH expression on pDC (Fig EV1; Puttur et al, 2013). Moreover, IFN‐I production occurred specifically in pDC expressing higher CD317 but lower SiglecH levels than the bulk of the pDC population (Fig EV1; Zucchini et al, 2008a). Hence, in order to ensure the best detection of all IFN‐I‐producing pDC, we did not add SiglecH in our pDC gating strategy. Consistent with our microarray data, during MCMV infection CD86 upregulation but not IFN‐I production was significantly impaired in Ifnar1‐KO pDC (Figs 3A–C and EV1). In contrast, upon in vivo injection of CpG both responses were abrogated in Ifnar1‐KO pDC (Fig 3D). Hence, pDC IFN‐I production does not require IFN‐I positive feedback in vivo during MCMV infection, contrary to CpG injection. IFN‐γ is one of the upstream regulators of genes induced in pDC during MCMV infection, including for the Myd88‐dependent gene module encompassing all IFN‐I/III genes (Fig 1F). During MCMV infection, IFN‐γ and IFN‐III are induced to high levels at the same time as IFN‐I. Hence, we wondered whether different IFN types could play redundant roles in promoting pDC cytokine production during MCMV infection. Signal transducer and activator of transcription 1 (STAT‐1) is a key element of the signaling cascades downstream of the receptors to all the three types of IFN. Hence, we compared IFN‐I production in pDC isolated from MCMV‐infected CTR versus Stat1‐KO MBMC. No major defect was detected in Stat1‐KO pDC (Fig EV3A), thus excluding a critical contribution of any of the three types of IFN in promoting IFN‐I production by pDC during MCMV infection.
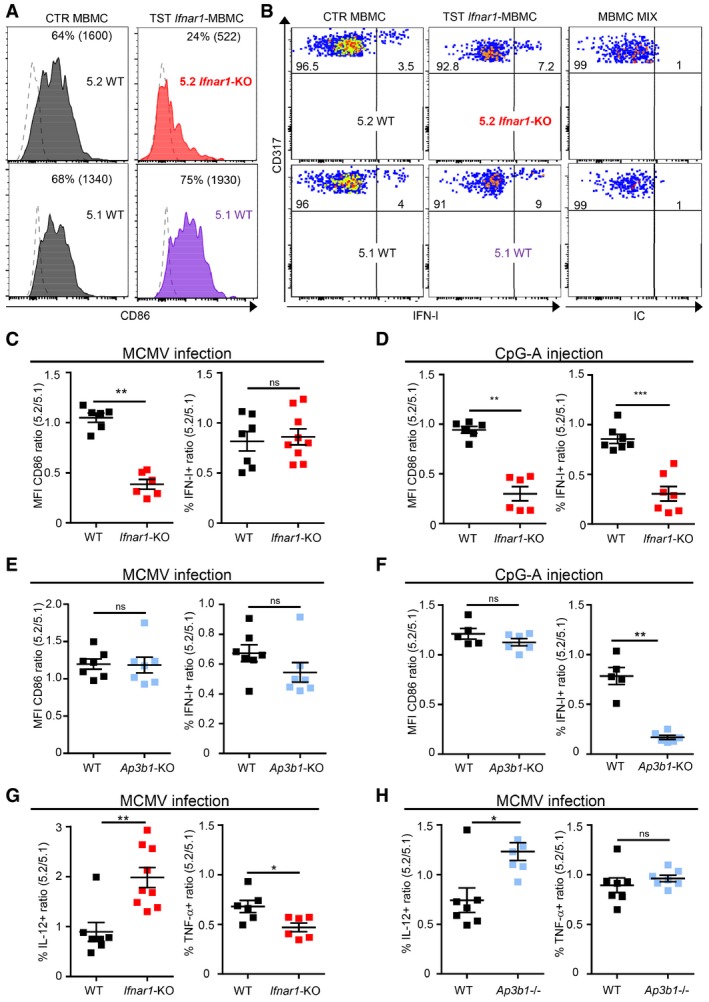
Figure 3. IFN‐I‐responsiveness and AP3 are dispensable for in vivo IFN‐I production in pDC during MCMV infection, but required upon stimulation with CpG.
-
AHistogram representing CD86 expression in 5.2+ versus 5.1+ pDC isolated from indicated MCMV‐infected MBMC; gray, 5.2+ and 5.1+ pDC from CTR MBMC; red, 5.2+ Ifnar1 −/− pDC isolated from Ifnar1‐TST MBMC; purple, 5.1+ WT pDC isolated from Ifnar1‐TST MBMC. Dotted line shows isotype control staining. One histogram representative of six animals from three independent experiments is shown.
-
BDot plots represent intracellular IFN‐I staining in 5.2+ versus 5.1+ pDC isolated from indicated MCMV‐infected MBMC. Isotype control (IC) staining was performed on a mixture of cells isolated from both types of MCMV‐infected MBMC.
-
C–HThe expression of indicated markers was analyzed in 5.2+ versus 5.1+ pDC of each MBMC type. (C–F) Results were expressed as 5.2/5.1 ratio of mean fluorescence intensity (MFI) values for CD86 or of the percentages of IFN‐I‐producing cells obtained from MCMV‐infected (C, E) or CpG‐stimulated (D, F) mice. (G, H) Ratio of IL‐12‐ and TNF‐producing cells in pDC isolated from indicated MCMV‐infected MBMC. Black, CTR MBMC; red, Ifnar1‐TST MBMC (red); pale blue, Ap3b1‐TST MBMC. Data shown (mean ± SEM) are from two pooled independent experiments each with at least three mice per group. ns, not significant (P > 0.05); *P < 0.05; **P < 0.01; ***P < 0.001; (nonparametric Mann–Whitney test) for panels (C–H).
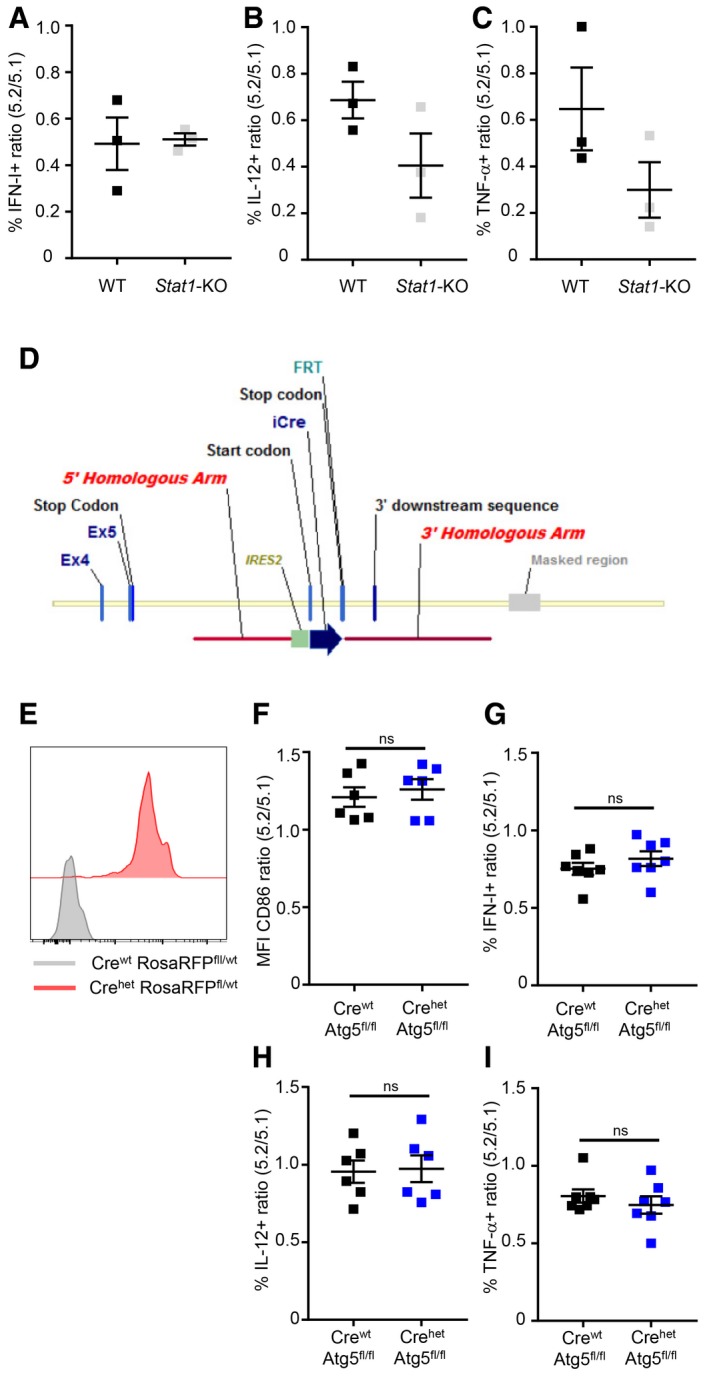
Figure EV3. STAT1 and ATG5 are dispensable for pDC cytokine production in vivo during MCMV infection.
-
A–C5.2/5.1 ratio (mean ± SEM) of indicated cytokine‐producing pDC in CTR (black) versus Stat1‐KO (gray) MBMC mice (n = 3).
-
DSchematic representation of the construct used for targeting the mouse Siglech gene to generate the Siglech‐iCre KI mouse strain. The cassette encoding improved Cre (iCre) gene was inserted in the 3′ non‐coding region of Siglech gene. The initiation of iCre gene translation was under the control of internal ribosome entry site (IRES) sequences. FRT is a residual recombination site left after mouse breeding with Flipper mice expressing Flippase O in the germline, a step that was necessary to remove the neo selection cassette.
-
ESiglech‐iCre mice were bred with Rosa26‐LSL‐RFP mice to fate‐map cells in which Siglech‐driven Cre recombination had been effective. RFP expression in splenic pDC isolated from Siglech‐iCrewt Rosa26‐LSL‐RFPfl/wt (gray) versus Siglech‐iCrehet Rosa26‐LSL‐RFPfl/wt (red) is depicted. Histograms are representative of one out of ten mice tested.
-
F–I5.2/5.1 ratio of CD86 MFI or of the percentages of cytokine‐producing cells in pDC from Siglech‐iCrewt Atg5 fl/fl (Crewt Atg5 fl/fl, black) or Siglech‐iCrehet Atg5 fl/fl (Crehet Atg5 fl/fl, dark blue) MBMC. Data shown (mean ± SEM) are from two pooled independent experiments each with at least three mice per group. ns, not significant (P > 0.05); (nonparametric Mann–Whitney test).
The AP3 adaptor complex is critical for cargo‐selective transport of CpG DNA/TLR/MyD88 complexes to specialized endosomes where they can recruit IRF7 for IFN‐I production (Blasius et al, 2010; Sasai et al, 2010; Prandini et al, 2016). To our knowledge, the role of AP3 in pDC response to stimulation with viruses was assessed exclusively in vitro (Blasius et al, 2010; Prandini et al, 2016). Therefore, we compared the cell‐intrinsic impact of AP3 loss on in vivo pDC responses to MCMV infection versus CpG injection (Fig 3E and F). Neither CD86 upregulation nor IFN‐I production was impaired in Ap3b1‐KO pDC during MCMV infection (Fig 3E). In contrast, during CpG injection, IFN‐I production but not CD86 upregulation was significantly decreased in Ap3b1‐KO pDC (Fig 3F), consistent with previous reports (Blasius et al, 2010; Sasai et al, 2010). During MCMV infection, IL‐12 production was enhanced in both Ifnar1‐KO and Ap3b1‐KO pDC (Fig 3G and H), while slightly decreased in Stat1‐KO pDC (Fig EV3B), consistent with the previous observations (Dalod et al, 2002; Krug et al, 2004; Sasai et al, 2010). TNF production was slightly decreased in Ifnar1‐KO and in Stat1‐KO but not Ap3b1‐KO pDC (Figs 3G and H, and EV3C). Thus, we showed that AP3 is dispensable for IFN‐I production by pDC during MCMV infection, whereas we confirmed that it is necessary for this function upon CpG stimulation.
pDC IFN‐I production can be promoted via an AP3‐independent but autophagy‐related 5 (Atg5)‐dependent LC3‐associated phagocytosis (LAP) (Henault et al, 2012; Hayashi et al, 2018). Hence, we investigated whether Atg5 deletion in pDC affected their IFN‐I production during MCMV infection. We generated Siglech‐iCre‐knock‐in mice in which the improved Cre (iCre) recombinase is expressed under the control of the Siglech promoter (Fig EV3D). When Siglech‐iCre mice were bred with Rosa26‐LSL‐RFP mice, RFP fluorescent protein was expressed in all pDC, thus confirming that this new mouse model allows selective and efficient gene targeting in pDC (Fig EV3E). Hence, Atg5 was inactivated in pDC by crossing Siglech‐iCre with Atg5‐floxed (fl) mice. Siglech‐iCrewt;Atg5 fl/fl and Siglech‐iCrehet;Atg5 fl/fl mice were used to generate CTR and Atg5 fl/fl MBMC, respectively. Cytokine production and CD86 induction were not compromised in pDC defective for Atg5 expression (Fig EV3F–I). Hence, Atg5‐dependent LAP is not required for pDC IFN‐I production during MCMV infection.
In summary, our results show different molecular requirements for pDC production of IFN‐I in vivo during CpG injection versus MCMV infection, with a strict requirement for IFN‐I responsiveness and AP3 activity only in the former context.
IFN‐I production by pDC during MCMV infection requires TLR9‐to‐MyD88 signaling, irrespective of their responsiveness to IFN‐I
Upon stimulation with synthetic TLR ligand or in vitro exposure to viruses, IFN‐I positive feedback signaling and AP3 activity are proposed to promote IFN‐I production by pDC by enforcing IRF7 activity. They, respectively, amplify its transcription (Kerkmann et al, 2003; Honda et al, 2005b; Prakash et al, 2005; O'Brien et al, 2011; Kim et al, 2014; Swiecki & Colonna, 2015) or favor its association with TLR7/9 and MyD88 in dedicated endosomes (Blasius et al, 2010; Sasai et al, 2010). Hence, the lack of requirement of IFN‐I positive feedback and AP3 activity in pDC for IFN‐I production during MCMV infection calls into question this prevailing model of the molecular mechanisms controlling IFN‐I production by pDC. In mice infected with the Newcastle disease virus (NDV), loss of IFN‐I responses led to pDC infection and shifted their mechanism of IFN‐I induction from TLR7/MyD88‐dependent to viral replication/cytosolic sensor‐dependent (Kumagai et al, 2009). Thus, we wondered whether the differences observed in the molecular requirement for pDC IFN‐I production between MCMV infection and CpG injection could result from a similar shift in how Ifnar1‐KO, Stat1‐KO, or Ap3b1‐KO pDC sense MCMV. Indeed, a higher rate of infection of mutant pDC might allow them compensating impaired TLR7/9‐to‐IRF7‐to‐IFN‐I signaling by enhanced triggering of cytosolic sensors of viral nucleic acids. To assess in vivo the frequency of infected cells within IFN‐producing pDC, we used an MCMV strain engineered to express the fluorescent reporter GFP under control of an immediate early gene (Henry et al, 2000; Dalod et al, 2003). MCMV infection was only slightly increased in Ifnar1 −/− pDC (Fig 4A and B). Moreover, in both WT and Ifnar1 −/− pDC, most IFN‐I‐producing cells were not infected since they did not express GFP (Fig 4A and C). Thus, IFN‐I production in Ifnar1 −/− pDC was unlikely to result from cytosolic virus sensing, at least not from endogenous MCMV replication. To further confirm that, during MCMV infection, even in the absence of IFN‐I responsiveness, IFN‐I production by pDC was driven by their endosomal recognition of engulfed viral material through a TLR7/9‐to‐MyD88‐to‐IRF7 signaling pathway, we analyzed infected Myd88‐KO and CTR MBMC treated with blocking anti‐IFNAR1 antibodies. In both MBMC types, IFNAR1 blockade strongly decreased ISG transcription (Fig 4D) and CD86 upregulation (Fig 4E), thus validating treatment efficacy for abrogating IFN‐I responsiveness. If IFNAR1 blockade was shifting the molecular mechanisms promoting pDC IFN‐I production from TLR9/MyD88‐dependent to viral replication/cytosolic sensor‐dependent, it would rescue this function at least partially in Myd88 −/− pDC. This did not occur. The production of both IFN‐I (Fig 4F) and IL‐12 (Fig 4G) remained defective in Myd88 −/− pDC despite IFNAR1 blockade. This was also the case in MCMV‐infected Ifnar1‐KO × Myd88‐KO mice (Fig 4H–J) and in Tlr9‐KO animals (Figs 4K and L, and EV4). Thus, pDC IFN‐I production during MCMV infection strictly requires TLR9‐to‐MyD88 signaling and occurs mostly in uninfected cells, irrespective of their responsiveness to IFN‐I.
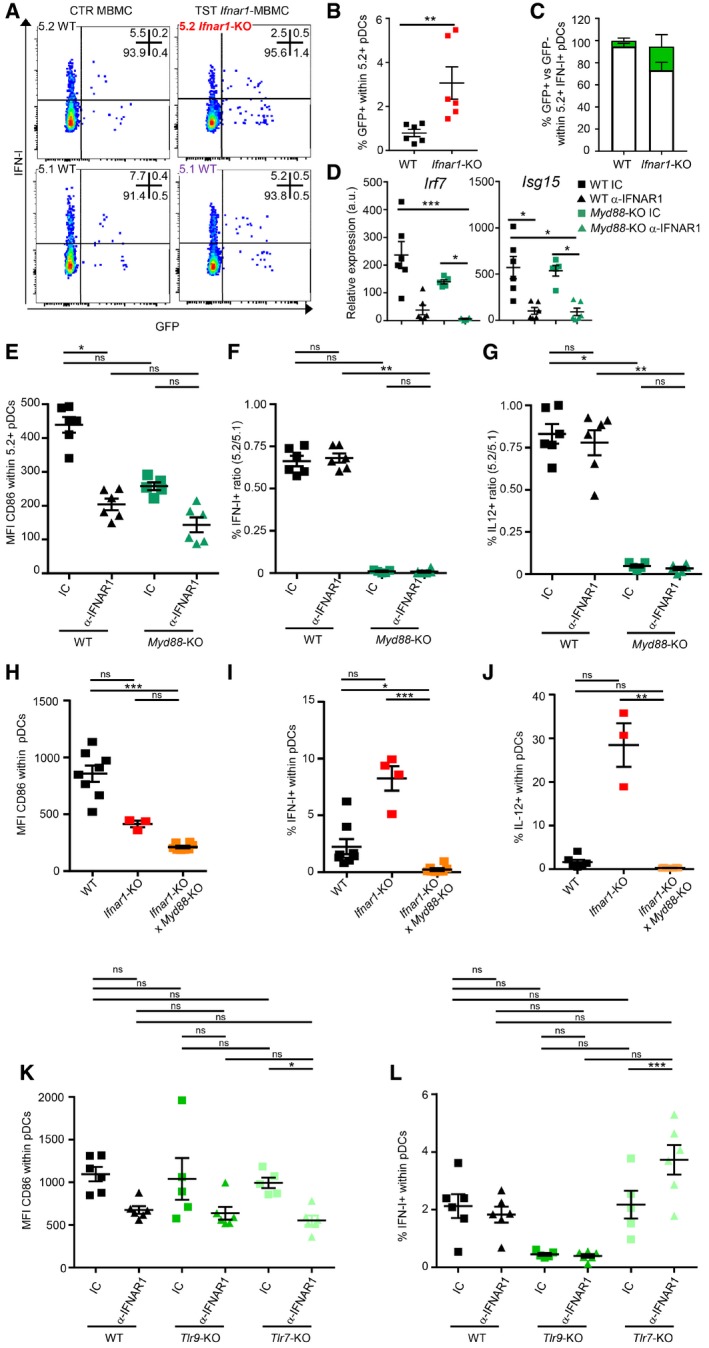
Figure 4. During MCMV infection, the TLR9/Myd88‐dependent pathway is necessary for pDC cytokine production, irrespective of their IFN‐I responsiveness.
-
ADot plots represent IFN‐I versus GFP staining in 5.2+ versus 5.1+ pDC isolated from indicated MCMV‐GFP‐infected MBMC. One staining representative of six animals from two independent experiments is shown.
-
B, CPercentages of GFP+ cells within 5.2+ pDC (B) or of GFP+ (green) versus GFP− (white) cells within 5.2+ IFN‐I‐producing pDC (C) isolated from indicated MCMV‐GFP‐infected MBMC. Data shown (mean ± SEM) are from two pooled independent experiments each with three mice per group.
-
DqPCR for the expression of the indicated genes was performed on whole mRNA isolated from the spleen of indicated MCMV‐infected MBMC. CTR MBMC (black) and Myd88 −/− TST MBMC (dark green) were treated with isotype control (IC, square) or blocking α‐IFNAR1 mAbs (triangle). Data shown (mean ± SEM) are from two pooled independent experiments each with two to three mice per group.
-
E–GCD86 MFI in 5.2+ pDC of each indicated MBMC type (E) and 5.2/5.1 ratio of cytokine‐producing pDC (F, G). Data (mean ± SEM) are representative of two pooled independent experiments each with two to three mice per group.
-
H–LCD86 MFI (H, K) and percentages of cytokine‐producing cells (I, J and L) within pDC of each indicated mouse strain. Black, C57BL6; red, Ifnar1 −/−; orange, Ifnar1 −/− × Myd88 −/−; green, Tlr9 −/−; and pale green, Tlr7 −/−. In (K–L), indicated mice were treated with IC (square) or blocking α‐IFNAR1 mAbs (triangle). Data depicted (mean ± SEM) are from two pooled independent experiments each with two to three mice per group.
Figure EV4. During MCMV infection, the TLR9/Myd88‐dependent pathway is necessary for pDC IL‐12 production, irrespective of their IFN‐I responsiveness.
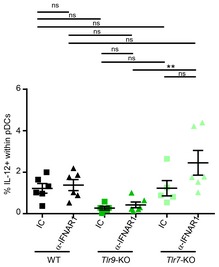
Percentages of IL‐12‐producing cells within pDC of each indicated mouse strain. Black, C57BL6; green, Tlr9 −/− and pale green, Tlr7 −/−. Mice were treated with IC (square) or blocking α‐IFNAR1 mAbs (triangle). Data depicted (mean ± SEM) are from two pooled independent experiments each with two to three mice per group. ns, not significant (P > 0.05); **P < 0.01; (nonparametric Mann–Whitney test).
IRF7 is necessary for IFN‐I production by pDC during MCMV infection, but low levels of this protein are sufficient to promote this function
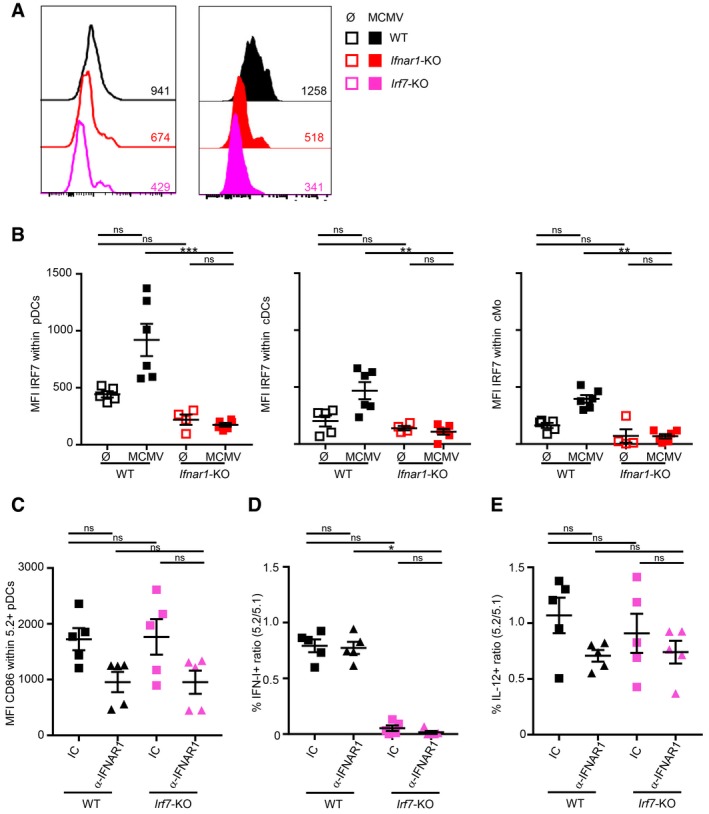
In Ifnar1‐KO pDC, Irf7 expression was significantly diminished at steady state and failed to increase upon MCMV infection (Fig 2). Despite this dramatic reduction in their Irf7 expression, Ifnar1‐KO pDC were not impaired for IFN‐I production (Figs 2 and 3B and C). This suggested that high‐level expression of IRF7 is not necessary to endow pDC with their unique function of professional IFN‐I producers, contrary to current dogma (Kerkmann et al, 2003; Honda et al, 2005b; Prakash et al, 2005; O'Brien et al, 2011; Kim et al, 2014; Swiecki & Colonna, 2015). However, as IRF7 protein stability can vary depending on cell activation states (Prakash & Levy, 2006), mRNA levels might not mirror IRF7 protein quantity. We thus analyzed IRF7 protein expression in individual pDC by flow cytometry. The specificity of the anti‐IRF7 mAb used was validated by using Irf7‐KO pDC (Fig 5A, pink and Fig EV5A, left panel). In WT pDC, IRF7 protein was clearly detectable at steady state (Fig 5A, left panel, black and Fig EV5A, middle panel) and significantly increased upon MCMV infection (Fig 5A, right panel and Fig EV5A, middle panel, black). In WT mice, IRF7 expression was significantly higher in pDC than in other cells, including conventional DC (cDC) and classical monocytes (cMo), although MCMV infection enhanced expression in all cell types (Fig 5B). In steady‐state Ifnar1‐KO pDC, IRF7 was still detectable, but to very low levels as compared to their WT counterparts (Fig 5A, left panel, red and Fig 5B, left panel, Fig EV5A, right panel). No increase in IRF7 expression occurred in Ifnar1‐KO pDC upon MCMV infection (Fig 5A, right panel, red and Fig EV5A, right panel). IRF7 expression was thus very low in Ifnar1‐KO pDC irrespective of MCMV infection and comparable to that observed in steady‐state WT cDC or cMo (Fig 5B). The analysis of microarray data highlighted IRF1 as a potential upstream regulator of genes induced in pDC upon MCMV infection (Fig 1F). Thus, in the absence of positive IFN‐I feedback loop, decreased pDC IRF7 activity might be compensated by IRF1 upregulation. Indeed, IRF1 can drive TLR9/MyD88‐dependent IFN‐β production by CpG‐B‐stimulated monocyte‐derived DC and macrophages, independently of IRF3 and IRF7 (Schmitz et al, 2007). However, IFNAR1 blockade did not rescue IFN‐I production by Irf7‐KO pDC during MCMV infection (Fig 5C and D), showing that IRF1 upregulation did not functionally compensate IRF7 loss. IL‐12 production and TNF production were not affected in Irf7‐KO pDC (Figs 5E and EV5B), consistent with the specific role of IRF7 in the regulation of IFN‐I production. To more specifically test the role of IRF1, CTR and Irf1‐KO MBMC were infected with MCMV and treated with blocking a‐IFNAR1 antibodies. IFN‐I production was not affected in Irf1‐KO pDC, even upon a‐IFNAR1 antibody treatment (Fig EV5C). Thus, low IRF7 expression did not require compensation by IRF1 for IFN‐I production by IFNAR1‐unresponsive pDC. IL‐12 and TNF‐α were also normal in Irf1‐KO pDC and this independently of a‐IFNAR1 antibody treatment (Fig EV5D and E). Thus, during MCMV infection, IRF7 is necessary for IFN‐I production by pDC irrespective of their responsiveness to IFN‐I, and very low levels of this transcription factor are sufficient to promote this function.
Figure 5. IRF7 is necessary for IFN‐I production in pDC, but low protein levels are sufficient to promote this function during MCMV infection.
-
AIRF7 protein expression in pDC isolated from Irf7 −/− (pink, bottom), C57BL/6 (WT, black, top), and Ifnar1 −/− (red, middle) uninfected (empty histograms, Ø, left) versus MCMV‐infected mice (filled histograms, MCMV, right). For each condition, histograms are representative of one out of four to six mice from two independent experiments.
-
BIRF7 protein expression in pDC (left), cDC (middle), and conventional monocytes (cMo, right) isolated from uninfected (Ø, empty) or MCMV‐infected (MCMV, filled) C57BL/6 (black) or Ifnar1 −/− (red) mice. Data are shown as net IRF7 MFI, after subtraction of the MFI obtained using isotype control mAbs. Data shown (mean ± SEM) are pooled from two independent experiments with five to six mice for each group.
-
C–EThe expression of CD86 (C) or of indicated cytokines (D, E) was analyzed, as described in Fig 2. Black, CTR MBMC; pink, Irf7‐TST MBMC. Mice were treated with isotype control (IC, square) or blocking α‐IFNAR1 mAbs (triangle). Data shown (mean ± SEM) are from two pooled independent experiments each with two to three mice per group.
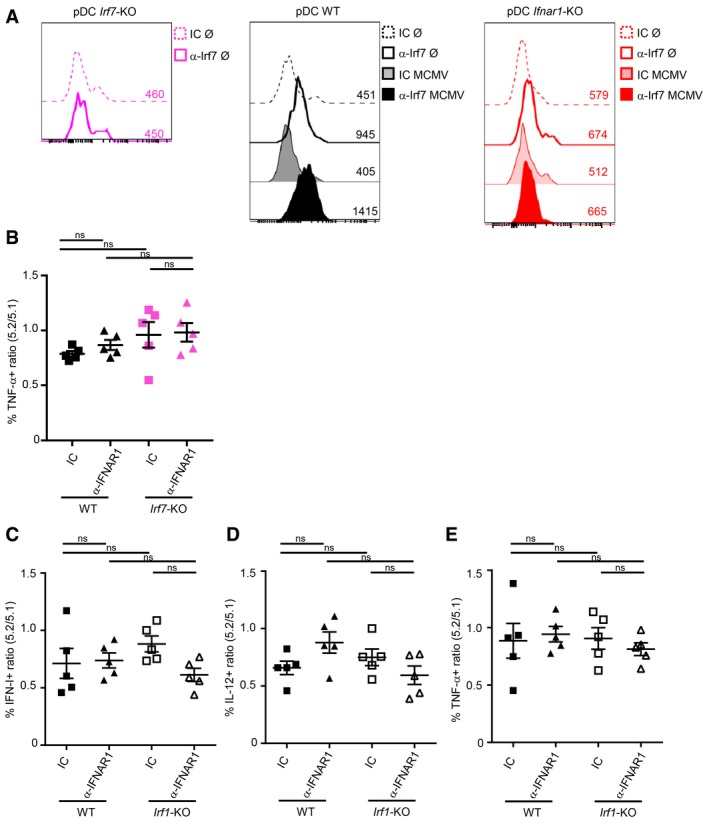
Figure EV5. IRF1 is not necessary for IFN‐I production in pDC during MCMV infection, even in the absence of IFN‐I‐responsiveness.
-
AIRF7 protein expression in pDC isolated from Irf7 −/− (pink, left), C57BL/6 (WT, black, middle), and Ifnar1 −/− (red, right) mice. Empty histograms, uninfected mice (Ø); with bold line, IRF7 staining (a‐IRF7) and dotted line, isotype control (IC) staining. Filled histograms, infected mice (MCMV); with dark color, IRF7 staining, and pale color, IC staining. For each condition, histograms are representative of one out of four to six mice from two independent experiments.
-
BRatio of TNF‐producing cells in 5.2+ versus 5.1+ pDC of each indicated MBMC. Black, CTR MBMC; pink, Irf7 −/− TST MBMC. Mice were treated with isotype control (IC, square) or blocking α‐IFNAR1 mAbs (triangle). Data shown (mean ± SEM) are from two pooled independent experiments each with two to three mice per group.
-
C–ERatio of cytokine‐producing cells in 5.2+ versus 5.1+ pDC of each indicated MBMC. Black, CTR MBMC; white, Irf1 −/− TST MBMC. Mice were treated with isotype control (IC, square) or blocking α‐IFNAR1 mAbs (triangle). Data shown (mean ± SEM) are from two pooled independent experiments each with two to three mice per group. ns, not significant; (nonparametric Kruskal–Wallis test combined with Dunn's multiple correction test) for panels (B–E).
IKKβ, but not IKKα kinase activity, is required for IFN‐I production by pDC
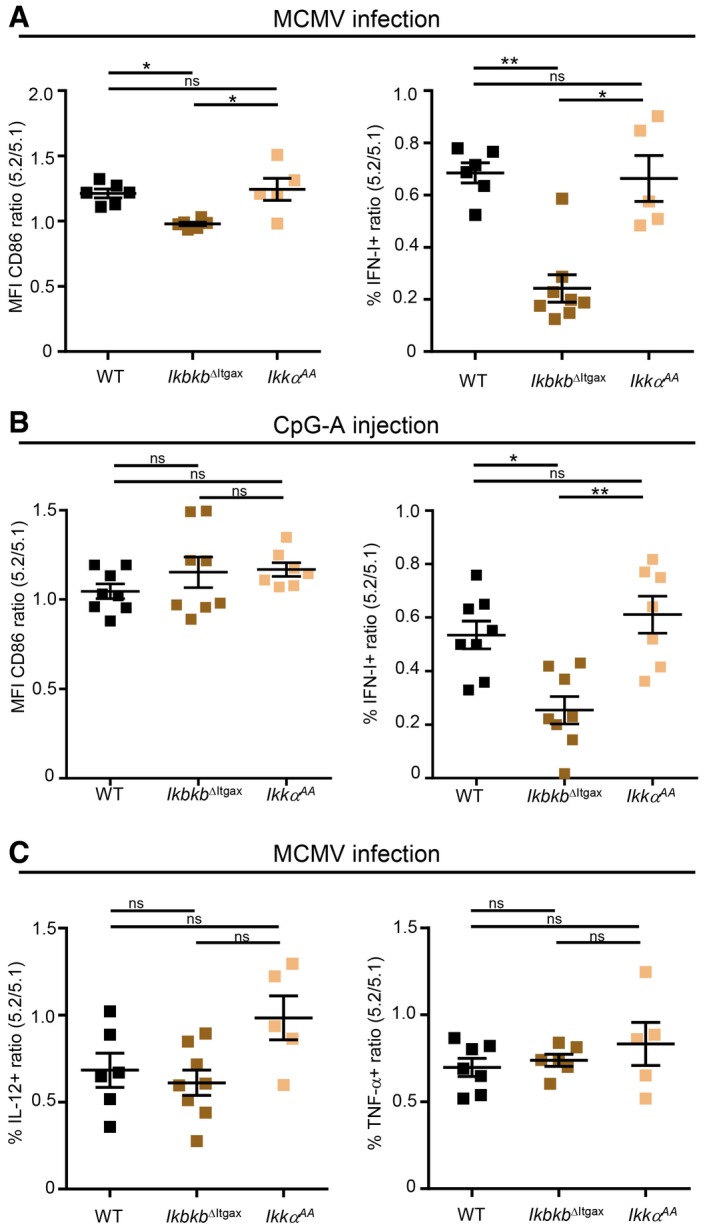
Our gene expression profiling of pDC identified the kinase IKKβ and the NF‐κB complex among the candidate upstream regulators inducing cytokine transcription in vivo during MCMV infection (Fig 1F). Both IKKα and IKKβ were reported to promote IRF7‐dependent IFN‐I production by pDC in response to stimulation with synthetic TLR ligands (Hoshino et al, 2006; Pauls et al, 2012). Hence, we compared their role in the promotion of pDC IFN‐I production during MCMV infection or CpG injection. We generated three types of MBMC: CTR MBMC, Ikbkb ΔItgax TST MBMC in which IKKβ is inactivated in all CD45.2+ CD11c+ cells, including pDC (Baratin et al, 2015), and Ikkα AA TST MBMC where all CD45.2+ cells express a mutant IKKα bearing alanine substitutions for serines in the kinase activation loop (Cao et al, 2001). The upregulation of CD86 was slightly reduced specifically in Ikbkb ΔItgax pDC during MCMV infection (Fig 6A). IFN‐I production was nearly abrogated specifically in Ikbkb ΔItgax pDC both during MCMV infection (Fig 6A) and CpG injection (Fig 6B). pDC production of IL‐12 and TNF was not impaired in Ikbkb ΔItgax pDC, while IL‐12 produced appeared slightly enhanced in Ikkα AA pDC (Fig 6C). Thus, IKKβ, but not IKKα kinase activity, is critical for IFN‐I production by mouse pDC in vivo during both MCMV infection and CpG injection.
Figure 6. IKKβ is required for pDC IFN‐I production.
-
A, BRatio of CD86 expression or IFN‐I production in 5.2+ versus 5.1+ pDC isolated from MCMV‐infected (A) or CpG‐stimulated (B) MBMC of each indicated type.
-
CRatio of IL‐12‐ or TNF‐producing cells in 5.2+ versus 5.1+ pDC of indicated MCMV‐infected MBMC mice.
LFA‐1 expression on pDC promotes their production of IFN‐I
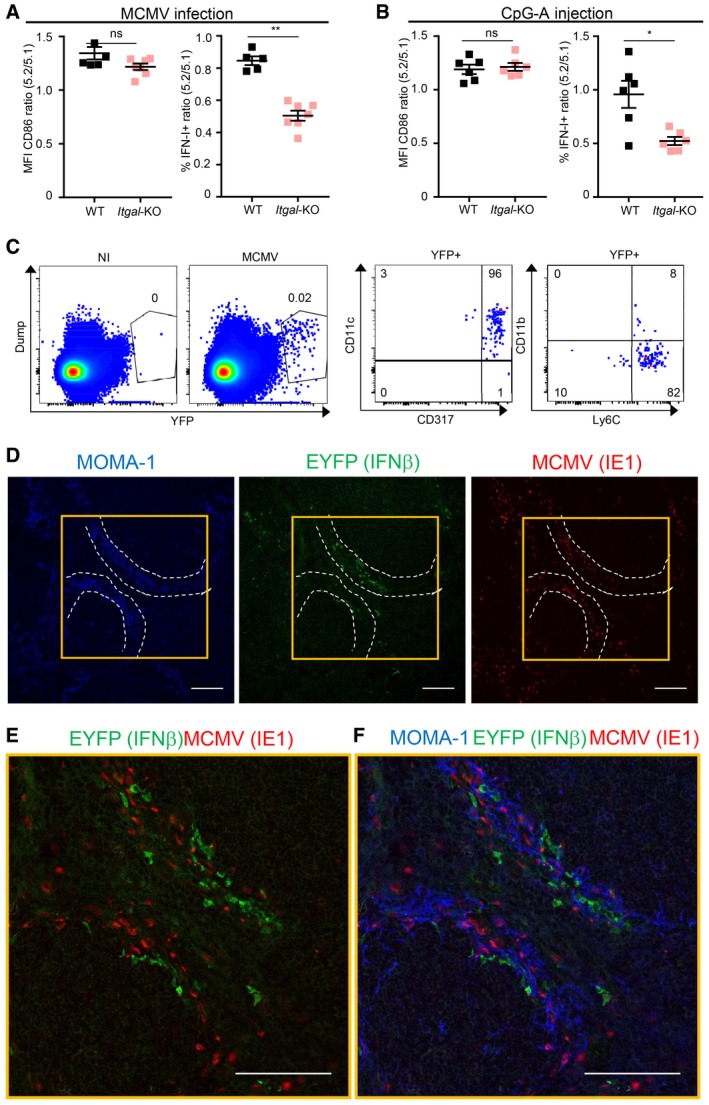
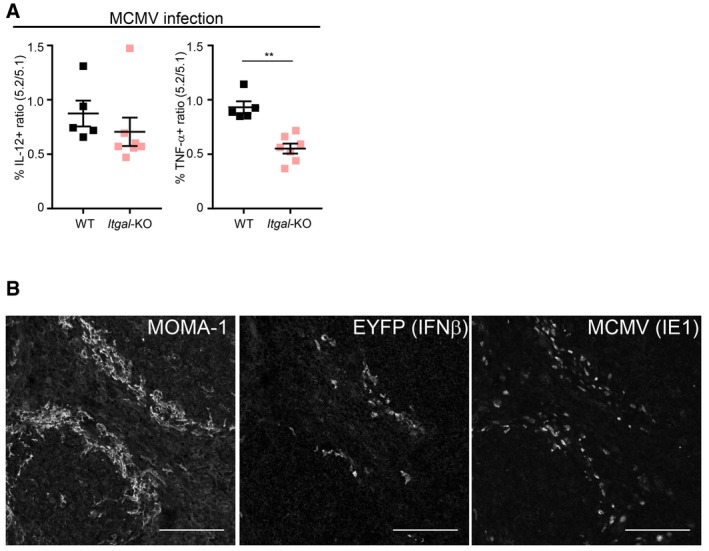
The integrin molecule LFA‐1 (alias CD11a) promotes IFN‐I production by human (Hagberg et al, 2011) and mouse (Saitoh et al, 2017) pDC activated by immunocomplexes or by synthetic TLR7 ligands, respectively. Whether this concept can be extended to in vivo antiviral responses has not been yet investigated. Hence, we examined whether loss of the Itgal gene encoding LFA‐1 perturbed pDC activation in vivo during MCMV infection (Fig 7A) or CpG injection (Fig 7B). Itgal‐KO pDC were not affected in their upregulation of CD86 but significantly impaired in their production of IFN‐I under both stimuli. During MCMV infection, Itgal‐KO pDC were also impaired for TNF, but not for IL‐12 production (Fig EV6A). Hence, LFA‐1 expression on pDC promotes their ability to produce IFN‐I in vivo during MCMV infection or upon CpG injection. This suggested that pDC IFN‐I production may be promoted by the establishment of LFA1‐dependent cell–cell interactions whose precise nature might differ between viral infection versus synthetic CpG administration. Although this hypothesis will definitely require dedicated experimental testing, it is consistent with several previously published reports. First, in vitro, interactions between pDC themselves promote their activation in response to CpG stimulation (Kim et al, 2014; Saitoh et al, 2017). Second, co‐culture with infected cells boosts pDC antiviral responses as compared with exposure to free virus, mainly in a cell contact‐dependent manner (Megjugorac et al, 2007; Takahashi et al, 2010; Dreux et al, 2012; Decembre et al, 2014; Frenz et al, 2014; Wieland et al, 2014; Garcia‐Nicolas et al, 2016). Third, pDC IFN‐I production during MCMV infection requires completion of at least one cycle of viral replication and is more strongly dependent on in vivo viral replication than on the initial viral inoculum dose, suggesting that pDC recognize MCMV‐infected cells rather than viral particles (Robbins et al, 2007). Therefore, we sought to investigate whether IFN‐I production by pDC in vivo during MCMV infection occurs in close proximity to infected cells. We used knocked‐in animals expressing the fluorescent reporter protein EYFP in the 3′ UTR of the Ifnb1 gene (Ifnb1 EYFP mice), leading to induction of yellow fluorescence during MCMV infection mainly in CD11cdim CD317high CD11b− Ly6C+ cells corresponding to pDC (Fig 1B; Scheu et al, 2008; Fig 7C). In spleen sections of MCMV‐infected Ifnb1 EYFP mice, EYFP+ (IFNβ+) cells were mainly found in the marginal zone, often in close proximity to MCMV‐infected (IE‐1+) cells (Figs 7D and EV6B). Despite this co‐localization, we did not detect any double IFNβ+ (EYFP+) infected (IE‐1+) cells (Fig 7E and F), consistent with our FACS data showing that the vast majority of IFN‐I‐producing pDC are not productively infected (Fig 4A–C). Some of the infected (IE‐1+) cells were also MOMA‐1+ (Fig 7F), indicating that metallophilic marginal zone macrophages were targets of MCMV infection. Altogether, these results demonstrate that optimal activation of IFN‐I production by pDC in vivo requires LFA‐1 expression and that, during MCMV infection, IFN‐I‐producing cells co‐localize with MCMV‐infected cells in the marginal zone of the spleen, thus suggesting their potential interaction in a LFA1‐dependent manner.
Figure 7. pDC IFN‐I production is promoted by LFA1 engagement and occurs in the vicinity of infected cells during MCMV infection.
-
A, BRatio of CD86 expression or IFN‐I production in 5.2+ versus 5.1+ pDC isolated from MCMV‐infected (A) or CpG‐stimulated (B) MBMC of each indicated type. Black, CTR MBMC; pale pink, Itgal‐TST MBMC. Data shown (mean ± SEM) are from two pooled independent experiments each with three mice per group. ns, not significant (P > 0.05); *P < 0.05; **P < 0.01; nonparametric Mann–Whitney test.
-
CEYFP expression in splenocytes isolated from uninfected (NI) versus MCMV‐infected (MCMV) IFNb tm1(EYFP) reporter mice. The expression of indicated markers was analyzed on YFP+ cells. Plots are representative of one out of six mice from two independent experiments.
-
D–FImmunohistological analysis of splenic sections stained with mAb against MOMA1/CD169 (blue), GFP/EYFP (green), whose expression correlates with that of IFN‐β, and MCMV IE‐1 (red). Scale bars, 100 μm. The image shown is one representative of 15 obtained from five different mice. Optical magnification was 20×, and images were scanned on the microscope at zoom x2 for (E, F). White dotted lines delimit marginal zone. Yellow squares in (D) defines zoomed region in (E, F).
Figure EV6. LFA‐1 is necessary for TNF, but not for IL‐12 production in pDC during MCMV infection.
- Ratio of TNF and IL‐12 production in 5.2+ versus 5.1+ pDC isolated from MCMV‐infected MBMC of each indicated type. Black, CTR MBMC; pale pink, Cd11a −/− TST MBMC. Data shown (mean ± SEM) are from two pooled independent experiments each with three mice per group. **P < 0.01; (nonparametric Mann–Whitney test).
- Individual color images of Fig 7D have been represented as black and white. Scale bars, 100 μm.
Discussion
pDC are professional producers of IFN‐I/III, able to rapidly produce very high levels of all these cytokines upon exposure to viruses, without being themselves infected (Cella et al, 1999; Siegal et al, 1999; Asselin‐Paturel et al, 2001; Dalod et al, 2003; Ito et al, 2006; Zucchini et al, 2008a). Indeed, pDC sense and engulf viral particles or material from infected cells and route them to dedicated endosomes for triggering of a TLR7/9‐to‐MyD88‐to‐IRF7 signaling pathway (Swiecki & Colonna, 2015). Both in human and in mice, other cell types can sense and engulf viral material. They co‐express endosomal TLR recognizing these molecules, together with their signaling adaptors. Type 2 cDC and cells of the monocyte/macrophage lineages express TLR7 or TLR8 with MyD88, and type 1 cDC TLR8/MyD88 and TLR3/TRIF (Hemont et al, 2013; Dalod et al, 2014). Type 1 cDC are professional producers of IFN‐III but not of IFN‐I in response to TLR3 triggering (Tomasello et al, 2014), whereas uninfected type 2 cDC and monocytes/macrophages do not produce high levels of IFN‐I/III upon stimulations with viruses or synthetic TLR ligands (Diebold et al, 2003; Ito et al, 2006; Zucchini et al, 2008a). Thus, the professional IFN‐I/III production by pDC must result from molecular and cellular mechanisms selectively present in these cells and not in cDC or monocytes/macrophages. The currently prevailing model is that pDC functional specialization depends on their high constitutive IRF7 expression combined with their ability to use AP3 for sorting TLRs and engulfed material into dedicated endosomes (Kerkmann et al, 2003; Honda et al, 2005b; Prakash et al, 2005; Blasius et al, 2010; Sasai et al, 2010; O'Brien et al, 2011; Kim et al, 2014; Swiecki & Colonna, 2015). However, to our knowledge, these mechanisms have not been investigated in vivo during a physiological viral infection.
We compared the cell‐intrinsic impact of IFN‐I, MyD88, IRF7, and AP3 activities on pDC IFN‐I production in vivo during MCMV infection versus CpG injection. We confirmed that in both activation conditions pDC IFN‐I production was triggered downstream of a cell‐intrinsic TLR7/9‐to‐MyD88‐to‐IRF7/NFKB signaling pathway. However, AP3 and high‐level expression of IRF7 were not necessary to endow pDC with their professional IFN‐I production during MCMV infection as opposed to CpG administration. Hence, for the first time to the best of our knowledge, we show that the dependency of in vivo pDC IFN‐I production on IFN‐I positive feedback and AP3 activity depends on the stimulus encountered. Further studies will be necessary to better understand what confers to pDC their unique ability for rapid and high‐level IFN‐I production in vivo during viral infections.
Global gene expression profiling of pDC isolated from MBMC demonstrated a much broader impact of cell‐intrinsic IFN‐I responses than of MyD88 signaling on the transcriptional reprogramming of pDC during MCMV infection. Whereas cytokine production is preserved in pDC lacking IFN‐I responsiveness, these cells fail to upregulate molecules involved in the cross‐talk with T lymphocytes and must more generally be strongly affected for other processes. Hence, even though loss of IFN‐I responsiveness by pDC during MCMV infection does not compromise their cytokine production, it is likely to affect their contribution to the orchestration of innate and adaptive immune responses, as previously shown for cDCs (Baranek et al, 2012). Studies addressing this issue are beyond the scope of the current study and will be pursued independently.
In our experimental settings, IKKα kinase activity is dispensable for pDC IFN‐I production. This seems contradictory with the major defect of IFN‐I production reported in mouse or human pDC lacking IKKα (Hoshino et al, 2006; Pauls et al, 2012). However, explanations can be proposed to reconcile these observations. Indeed, here we did not use Ikka −/− animals, but a Ikka A/A knock‐in mouse strain expressing a mutated protein devoid of kinase activity (Cao et al, 2001). Hence, for pDC IFN‐I production, IKKα might mostly function as a docking partner within the IKKα/IKKβ/NEMO complex and not as a kinase. Hence, loss of IKKα might impact pDC IFN‐I production indirectly, by affecting the availability, stoichiometry, and function of the other molecules composing the complex, including IKKβ. Indeed, we showed that IKKβ deficiency in pDC mainly impaired IFN‐I production without affecting the secretion of other pro‐inflammatory cytokines, such as IL‐12 and TNF, thus reproducing results obtained by using mouse Ikka −/− pDC (Hoshino et al, 2006). Alternatively, both IKKα and IKKβ could directly signal to promote IRF7‐dependent IFN‐I production in pDC, but by using distinct and complementary mechanisms not requiring Ikkα canonical kinase activity (Pauls et al, 2012).
The lack of AP3 requirement for pDC IFN‐I production during MCMV infection is consistent with the normal levels of circulating IFN‐I in the sera of MCMV‐infected Ap3b1 −/− mice (Del Prete et al, 2015). However, our conclusions drastically differ from those of that previous report. In that study, pDC from Ap3b1 −/− mice showed an impaired IFN‐α and TNF production in vitro in response to free RNA and DNA viruses, including MCMV. Based on this observation, the authors inferred that pDC were unable to produce high levels of IFN‐I in MCMV‐infected Ap3b1 −/− mice but that this did not lead to a decrease in the peak serum levels of these cytokines. Thus, without measuring cytokine production in pDC in vivo, they concluded that pDC are dispensable for high systemic IFN‐I production during MCMV infection (Del Prete et al, 2015), which is in contradiction with previous reports (Dalod et al, 2002, 2003; Krug et al, 2004; Swiecki et al, 2010; Cocita et al, 2015). Here, we showed that Ap3b1 −/− pDC mount normal cytokine responses in vivo during MCMV infection. Altogether, these results show that the molecular requirements for pDC IFN‐I production upon stimulation with viruses differ between artificial in vitro settings and a physiological infection in vivo. Hence, caution must be exerted for extrapolation to in vivo conditions of molecular mechanisms described to control pDC activation in vitro.
An outstanding question opened by our study is how MCMV infection allows pDC IFN‐I production in vivo to overcome the need for IFN‐I positive feedback and for AP3 that is required for responses to CpG. We confirmed that cell‐intrinsic TLR9 signaling is critical for promoting pDC cytokine production during MCMV infection, independently of IFN‐I responsiveness. However, we speculate that MCMV infection delivers additional signals to pDC, overlapping with IFN‐I positive feedback or bypassing AP3 function. Since Stat1‐KO pDC were not affected for IFN‐I production during MCMV infection, we can rule out a redundancy between different IFN types as the mechanism underlying independency of pDC activation from IFN‐I responsiveness during MCMV infection. We also ruled out triggering of LC3‐ and Atg5‐dependent phagocytosis (LAP) in pDC as the mechanism underlying their lack of AP3 requirement for IFN‐I production during MCMV infection. TLR7 and TLR9 can exert overlapping functions during MCMV infection (Zucchini et al, 2008b). If this cooperation can override the need for IFN‐I positive feedback, we would expect a strong impairment of pDC IFN‐I production in TLR7‐KO mice treated with a‐IFNAR1 antibodies, which is not the case. However, we cannot exclude the involvement of other TLR or pathogen sensors.
Exposure of splenocytes to viruses significantly increases the stability of IRF7 protein, at least in part through STAT‐1‐independent mechanisms (Prakash & Levy, 2006), consistent with our observation that IFN‐I production by Stat1‐KO pDC is not affected during MCMV infection. Hence, differences in the signals received by pDC upon different contexts might lead to differential stability and/or nuclear translocation efficacy of IRF7, explaining the requirements for different absolute levels of IRF7. In any case, how pDC can capture and sense viruses without being infected themselves remains one of the most puzzling question regarding the biology of these cells, because the exact nature of the viral material engulfed by pDC and the endocytic receptors used for this process are still unknown (Puttur et al, 2013; Alexandre et al, 2014). Physical proximity and eventually cell contact‐dependent interaction with virally infected cells are necessary to promote maximal IFN‐I production in vitro (Takahashi et al, 2010; Decembre et al, 2014; Wieland et al, 2014; Garcia‐Nicolas et al, 2016). This interaction allows transfer of viral material to pDC (Dreux et al, 2012; Wieland et al, 2014). However, whether similar contacts are required in vivo is unknown and the underlying molecular mechanisms remain a mystery. Our data suggest that, during MCMV infection, in the marginal zone of the spleen, pDC might interact with infected cells through LFA1‐dependent contacts which would promote their IFN‐I production. Additional studies will be required to rigorously test this hypothesis and to further decipher the nature of the signals delivered by infected cells, including how they might compensate the loss of IFNAR1 and AP3 functions for promoting pDC IFN‐I production. However, these studies are currently hampered by the lack of proper mutant mouse models to specifically track and genetically manipulate pDC in vivo. This difficulty stems from the fact that no cell surface marker, and not even a single gene, has been found to be specifically expressed on mouse pDC. This includes CD317, which is induced on other cell types in response to IFN‐I, although, on other CD11c‐expressing cells, to lower levels than those observed on pDC, as well as SiglecH which is also expressed on DC progenitors and on specific subsets of macrophages (Swiecki et al, 2014; Schlitzer et al, 2015). Consequently, only complex strategies relying on the use of several positive and negative markers allow unequivocal phenotypic identification of bona fide pDC, as performed here for flow cytometry by defining them as lineage− CD11b− CD11clow/int CD317high cells. However, similar types of logic gating strategies are much more difficult to apply for immunohistofluorescence studies and for the engineering of mutant mice. Several teams are currently putting a major effort to overcome this roadblock; once it is achieved, our understanding of pDC functions and of their molecular regulation under physiological conditions should advance tremendously.
Materials and Methods
Mice
C57BL/6 mice were purchased from Janvier, France. Congenic CD45.1 C57BL/6 mice were purchased from Charles River, Italy. All other mouse strains were bred under specific pathogen‐free conditions at the Centre d'ImmunoPhénomique (CIPHE) or the Centre d'Immunologie de Marseille‐Luminy, Marseille, France. Ap3b1‐KO (B6Pin.C3‐Ap3b1 pe/J), IFNb EYFP (B6.129‐Ifnb1 tm1Lky) reporter mice and Itgal‐KO (B6.129S7‐Itgal tm1Bll) mice were purchased from Jackson Laboratories, USA. Ifnar1‐KO (B6.129S2‐Ifnar1 tm1agt ), Myd88‐KO (B6.129P2‐Myd88 tm1Aki), Tlr9‐KO (129P2‐Tlr9 tm1Aki ), Tlr7‐KO (B6.129P2‐Tlr7 tm1Aki), Irf7‐KO (B6.129P2‐Irf7 tm1Ttg) (Honda et al, 2005b), Ikka A/A (B6.129P2‐Chuk tm2Mka) (Cao et al, 2001), Ikbkb ΔItgax (Baratin et al, 2015), and Atg5 fl/fl (B6.129S‐Atg5 tm1Myok) (Hara et al, 2006; Terawaki et al, 2015) mice were backcrossed on C57BL/6 background for over eight generations. Rosa26‐LoxP_STOP_LoxP(LSL)‐Red Fluorescent protein, Rosa26‐LSL‐RFP (B6‐Gt(ROSA)26Sor tm1Hjf) mice (Luche et al, 2007) were kindly provided by B. Malissen (CIML). Ifnar1‐KO × Myd88‐KO mice were generated by interbreeding single knock‐out mice. Stat1‐KO (B6‐Stat1 tm1d(EUCOMM)Ciphe), Irf1‐KO (B6‐Irf1 tm1a(EUCOMM)Wtsi), and Siglech‐iCre (B6‐Siglech tm1(iCre)Ciphe) mice were generated at CIPHE, Marseille, France. All animals used were sex‐ and age‐matched. The animal care and use protocols were designed in accordance with national and international laws for laboratory animal welfare and experimentation (EEC Council Directive 2010/63/EU, September 2010). They were approved by the Marseille Ethical Committee for Animal Experimentation (registered by the Comité National de Réflexion Ethique sur l'Expérimentation Animale under no. 14; authorization #11–09/09/2011 and APAFIS#1212‐2015072117438525 v5).
Viruses and viral titers
Virus stocks were prepared from salivary gland extracts of 3‐week‐old MCMV‐infected BALB/c mice. Mice were infected i.p. with 1 × 105 pfu Smith MCMV or 5 × 104 pfu MCMV‐GFP (Henry et al, 2000) and sacrificed 36 h after infection. Viral titers were measured by real‐time quantitative RT–PCR as absolute levels of expression of the Ie1 gene as described (Baranek et al, 2012).
In vivo treatment with synthetic TLR ligands or with blocking antibodies
Mice were injected i.v. with 10 μg/mouse of the synthetic TLR9 ligand CpGA 2216 (Invivogen, France), complexed with DOTAP liposomal transfection reagent (Roche Applied Sciences, France), and sacrificed 7 h after injection. To block IFNAR1, mice were treated 24 h before sacrifice with 1 mg of isotype control or anti‐IFNAR1 MAR‐1 mAbs (BioXcell, USA).
Generation of MBMC
Congenic CD45.1 mice were 8 Gy irradiated, and then reconstituted with equal proportions of BM cells derived from CD45.1 animals and from CD45.2 mice either wild type (WT) or deficient for indicated genes. Mice were used at least 8 weeks after BM reconstitution.
Cell preparation, flow cytometry analysis, and cell sorting
Spleen was harvested and submitted to enzymatic digestion for 25 min at 37°C with collagenase IV (Worthington biochemicals), DNAse (Roche Diagnostics), and Brefeldin A (Sigma‐Aldrich). Red blood cells were then lysed by using RBC lysis buffer (Affymetrix eBiosciences, France), and pDC were enriched with the mouse plasmacytoid dendritic cell isolation kit (Miltenyi Biotec, France). Cell suspensions were incubated with 2.4G2 (anti‐CD16/CD32) mAbs for 10 min, and then, extracellular staining was performed at 4°C in 1× PBS supplemented with 2 mM EDTA (Sigma‐Aldrich) and 0.5% bovine serum albumin (BSA, H2B, Limoges, France). Intracellular staining was performed after fixation with Cytofix/Cytoperm (BD Biosciences), by incubation with specific anti‐cytokine mAbs diluted in PermWash (BD Biosciences). mAbs recognizing mouse CD3e (2C11), CD19 (1D3), Ly6G (1A8), NK1.1 (PK136), CD11b (M1/70), CD11c (HL3), CD86 (GL3), TNF (MP6‐XT22), and IL‐12 (C15‐6), as well as their respective isotype controls, were purchased from BD Biosciences. Purified anti‐mouse IFNα (RMMA1) and IFNβ (RMMB1) were purchased from PBL Interferon Source (Tebu, France); purified rat IgG was used as isotype control (Jackson Immuno Research); these mAbs were coupled to Alexa647 by using a monoclonal labeling kit (Southern Biotech, France). Anti‐mouse Bst2/CD317/PDCA1 (eBio927), Irf7 (MNGPKL), and its isotype control were purchased from Affymetrix eBiosciences, France. Irf7 intracellular staining was performed by using a FoxP3 staining kit (Affymetrix eBiosciences, France). Samples were acquired on a FACS LSR II (BD Biosciences, France) and analyzed with FlowJo software (FlowJo, LLC, USA). For microarray studies, pDC were sorted to high purity by flow cytometry on a FACS Aria II cell sorter (BD Biosciences, France) and directly collected in RLT buffer (Qiagen, France) supplemented with 10% β‐mercaptoethanol.
Immunohistological analysis
Spleen fragments isolated from uninfected or MCMV‐infected IFNb EYFP reporter mice were fixed for 2 h with Antigenfix (DiaPath), then washed in phosphate buffer (PB1X: 0.025 M NaH2PO4 and 0.1 M Na2HPO4) for 1 h, dehydrated in 30% sucrose overnight at 4°C, and embedded in OCT freezing media (Sakura Finetek). Eight‐micrometer‐thick tissue sections were blocked in PB1X containing 0.1% saponin, 2% BSA, 2% 2.4G2 supernatant, and streptavidin/biotin blocking kit (Vector Laboratories) and stained in PB1X with the following antibodies: Alexa488‐conjugated rabbit anti‐GFP (Invitrogen), mouse Croma anti‐MCMV IE‐1 mAb, followed by Alexa633‐conjugated goat anti‐mouse IgG2a mAb (Molecular Probes) and biotin rat anti‐mouse CD169 (MOMA1, Abcam), followed by Alexa546‐conjugated streptavidin (Thermo Fisher). Stained sections were mounted in ProLong Gold Antifade reagent (Invitrogen), acquired on a ZEISS LSM780 confocal microscope (Zeiss), and analyzed with ImageJ software.
Microarray data generation and analysis
Independent duplicate samples were generated by using pooled spleens of uninfected or MCMV‐infected indicated mice. Total RNA was prepared and converted to biotinylated double‐strand cDNA targets which were hybridized on GeneChip Mouse Gene 1.0 ST arrays (Affymetrix) that were scanned to extract raw data (.CEL Intensity files) and perform microarray bioinformatics analyses, as previously described (Baranek et al, 2012; Tamoutounour et al, 2013) and specified in Figure legends. List of genes showing differential expression between two different conditions were generated by statistical analysis using Limma. All WT pDC samples from infected mice were compared with those from uninfected animals (n = 14 in each group). Duplicates from Ifnar1‐KO or Myd88‐KO pDC from infected MBMC were compared with WT pDC from the same experiment (n = 2 for mutant pDC and n = 6 for WT control pDC). Heatmaps were performed using Gene‐E (http://www.broadinstitute.org/cancer/software/GENE-E/). The microarray data have been deposited in the GEO database (http://www.ncbi.nlm.nih.gov/geo/) under the series accession number GEO: GSE115450.
qPCR
Total mRNA was converted into cDNA by using QuantiTect Reverse Transcription kit (Qiagen). qPCR was performed by using a SybrGreen kit (Takara) and run on a 7500 Real Time PCR System apparatus (Applied Biosystems). We performed two independent replicates for each indicated condition by using 2 ng of cDNA/sample. Relative gene expression was calculated with the ΔΔC t method using Hprt as housekeeping gene for normalization. Primers used were the following: Hprt Forward 5′‐GGCCCTCTGTGTGCTCAAG‐3′; Hprt Reverse 5′‐CTGATAAAATCTACAGTCATAGGAATGGA‐3′; Irf7 Forward 5′‐TCCAGTTGATCCGCATAAGGT‐3′; Irf7 Reverse 5′‐CTTCCCTATTTTCCGTGGCTG‐3′; Isg15 Forward 5′‐GGTGTCCGTGACTAACTCCAT‐3′; Isg15 Reverse 5′‐TGGAAAGGGTAAGACCGTCCT‐3′.
Statistical analyses
Statistical analyses were performed using a nonparametric Mann–Whitney test for comparisons between two groups. For comparisons among three or more groups, we used a nonparametric Kruskal–Wallis test combined with Dunn's multiple correction test. All the analyses were performed with GraphPad Prism 6 software. ns, not significant (P > 0.05); *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Mean values are indicated as horizontal black bars.
Author contributions
ET performed most of the experiments; KN bred mice and performed histology experiments; GB bred mice; RC, EP, and T‐PVM performed bioinformatics analysis; AF contributed to the generation of microarray samples and to the setting of functional assays; EG performed qPCR experiments; AA helped to perform some experiments; PP and TL contributed to the editing of the manuscript and, respectively, provided Atg5 fl/fl versus Irf1‐KO, Ikkβ, and Ikkα mutant mice; MD and ET directed the project, designed the experiments, analyzed data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Review Process File
Acknowledgements
We thank all the staff of the CIML and CIPHE mouse houses, Atika Zouine, Marc Barad, and Sylvain Bigot of the CIML flow cytometry facility, Mathieu Fallet of the CIML microscopy facility, and the France‐BioImaging infrastructure supported by the Agence Nationale de la Recherche (ANR‐10‐INSB‐04‐01, call “Investissements d'Avenir”). We thank Violaine Alunni and Christelle Thibault‐Carpentier from Plate‐forme Genomeast (Strasbourg, France) for microarray experiments (http://www.igbmc.fr/technologies/5/team/54/), and Pr. Stipan Jonjic for the anti‐MCMV Croma antibody. We thank Frédéric Fiore, Bernard Malissen, and all the staff of Centre d'Immunophénomique [CIPHE] (UM2 Aix‐Marseille Université, Institut National de la Santé et de la Recherche Médicale US012, Centre National de la Recherche Scientifique UMS3367, Marseille, France) for generating mutant mice. We acknowledge Pr. Tadatsugu Taniguchi (University of Tokyo, Japan), Dr. Claude Leclerc, and Dr. Molly Ingersoll (Institut Pasteur, Paris) for generous gift of mutant mice and/or critical reading of the manuscript. This work benefited from data assembled by the ImmGen consortium. This research was funded by grants from the European Research Council under the European Community's Seventh Framework Programme (FP7/2007‐2013 Grant Agreement 281225, “SystemsDendritic”), the Fondation pour la Recherche Médicale (FRM, reference DEQ20110421284), and the Agence Nationale de la Recherche (ANR) (SCAPIN, ANR‐15‐CE15‐0006‐01). We also acknowledge support from the DCBIOL Labex (ANR‐11‐LABEX‐0043, grant ANR‐10‐IDEX‐0001‐02 PSL*), the A*MIDEX project (ANR‐11‐IDEX‐0001‐02) funded by the French Government's “Investissements d'Avenir” program managed by the ANR, and institutional support from CNRS, Inserm, Aix Marseille Université, and Marseille Immunopole.
The EMBO Journal (2018) 37: e98836
Contributor Information
Elena Tomasello, Email: tomasell@ciml.univ-mrs.fr.
Marc Dalod, Email: dalod@ciml.univ-mrs.fr.
References
- Alexandre YO, Cocita CD, Ghilas S, Dalod M (2014) Deciphering the role of DC subsets in MCMV infection to better understand immune protection against viral infections. Front Microbiol 5: 378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asselin‐Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter‐Dambuyant C, Vicari A, O'Garra A, Biron C, Briere F, Trinchieri G (2001) Mouse type I IFN‐producing cells are immature APCs with plasmacytoid morphology. Nat Immunol 2: 1144–1150 [DOI] [PubMed] [Google Scholar]
- Asselin‐Paturel C, Brizard G, Chemin K, Boonstra A, O'Garra A, Vicari A, Trinchieri G (2005) Type I interferon dependence of plasmacytoid dendritic cell activation and migration. J Exp Med 201: 1157–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranek T, Manh TP, Alexandre Y, Maqbool MA, Cabeza JZ, Tomasello E, Crozat K, Bessou G, Zucchini N, Robbins SH, Vivier E, Kalinke U, Ferrier P, Dalod M (2012) Differential responses of immune cells to type I interferon contribute to host resistance to viral infection. Cell Host Microbe 12: 571–584 [DOI] [PubMed] [Google Scholar]
- Baratin M, Foray C, Demaria O, Habbeddine M, Pollet E, Maurizio J, Verthuy C, Davanture S, Azukizawa H, Flores‐Langarica A, Dalod M, Lawrence T (2015) Homeostatic NF‐kappaB signaling in steady‐state migratory dendritic cells regulates immune homeostasis and tolerance. Immunity 42: 627–639 [DOI] [PubMed] [Google Scholar]
- Barchet W, Cella M, Odermatt B, Asselin‐Paturel C, Colonna M, Kalinke U (2002) Virus‐induced interferon alpha production by a dendritic cell subset in the absence of feedback signaling in vivo . J Exp Med 195: 507–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasius AL, Arnold CN, Georgel P, Rutschmann S, Xia Y, Lin P, Ross C, Li X, Smart NG, Beutler B (2010) Slc15a4, AP‐3, and Hermansky‐Pudlak syndrome proteins are required for Toll‐like receptor signaling in plasmacytoid dendritic cells. Proc Natl Acad Sci USA 107: 19973–19978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewitz A, Eickhoff S, Dahling S, Quast T, Bedoui S, Kroczek RA, Kurts C, Garbi N, Barchet W, Iannacone M, Klauschen F, Kolanus W, Kaisho T, Colonna M, Germain RN, Kastenmuller W (2017) CD8+ T cells orchestrate pDC‐XCR1+ dendritic cell spatial and functional cooperativity to optimize priming. Immunity 46: 205–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Bonizzi G, Seagroves TN, Greten FR, Johnson R, Schmidt EV, Karin M (2001) IKKalpha provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell 107: 763–775 [DOI] [PubMed] [Google Scholar]
- Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, Colonna M (1999) Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med 5: 919–923 [DOI] [PubMed] [Google Scholar]
- Cocita C, Guiton R, Bessou G, Chasson L, Boyron M, Crozat K, Dalod M (2015) Natural killer cell sensing of infected cells compensates for MyD88 deficiency but not IFN‐I activity in resistance to mouse cytomegalovirus. PLoS Pathog 11: e1004897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalod M, Salazar‐Mather TP, Malmgaard L, Lewis C, Asselin‐Paturel C, Briere F, Trinchieri G, Biron CA (2002) Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo . J Exp Med 195: 517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalod M, Hamilton T, Salomon R, Salazar‐Mather TP, Henry SC, Hamilton JD, Biron CA (2003) Dendritic cell responses to early murine cytomegalovirus infection: subset functional specialization and differential regulation by interferon alpha/beta. J Exp Med 197: 885–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalod M, Chelbi R, Malissen B, Lawrence T (2014) Dendritic cell maturation: functional specialization through signaling specificity and transcriptional programming. EMBO J 33: 1104–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decembre E, Assil S, Hillaire ML, Dejnirattisai W, Mongkolsapaya J, Screaton GR, Davidson AD, Dreux M (2014) Sensing of immature particles produced by dengue virus infected cells induces an antiviral response by plasmacytoid dendritic cells. PLoS Pathog 10: e1004434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Prete A, Luganini A, Scutera S, Rossi S, Anselmo A, Greco D, Landolfo S, Badolato R, Gribaudo G, Sozzani S, Musso T (2015) Interferon‐alpha production by plasmacytoid dendritic cells is dispensable for an effective anti‐cytomegalovirus response in adaptor protein‐3‐deficient mice. J Interferon Cytokine Res 35: 232–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delale T, Paquin A, Asselin‐Paturel C, Dalod M, Brizard G, Bates EE, Kastner P, Chan S, Akira S, Vicari A, Biron CA, Trinchieri G, Briere F (2005) MyD88‐dependent and ‐independent murine cytomegalovirus sensing for IFN‐alpha release and initiation of immune responses in vivo . J Immunol 175: 6723–6732 [DOI] [PubMed] [Google Scholar]
- Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, Al‐Shamkhani A, Flavell R, Borrow P, Reis e Sousa C (2003) Viral infection switches non‐plasmacytoid dendritic cells into high interferon producers. Nature 424: 324–328 [DOI] [PubMed] [Google Scholar]
- Dreux M, Garaigorta U, Boyd B, Decembre E, Chung J, Whitten‐Bauer C, Wieland S, Chisari FV (2012) Short‐range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 12: 558–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonteneau JF, Guillerme JB, Tangy F, Gregoire M (2013) Attenuated measles virus used as an oncolytic virus activates myeloid and plasmacytoid dendritic cells. Oncoimmunology 2: e24212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenz T, Graalmann L, Detje CN, Doring M, Grabski E, Scheu S, Kalinke U (2014) Independent of plasmacytoid dendritic cell (pDC) infection, pDC triggered by virus‐infected cells mount enhanced type I IFN responses of different composition as opposed to pDC stimulated with free virus. J Immunol 193: 2496–2503 [DOI] [PubMed] [Google Scholar]
- Garcia‐Nicolas O, Auray G, Sautter CA, Rappe JC, McCullough KC, Ruggli N, Summerfield A (2016) Sensing of porcine reproductive and respiratory syndrome virus‐infected macrophages by plasmacytoid dendritic cells. Front Microbiol 7: 771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg N, Berggren O, Leonard D, Weber G, Bryceson YT, Alm GV, Eloranta ML, Ronnblom L (2011) IFN‐alpha production by plasmacytoid dendritic cells stimulated with RNA‐containing immune complexes is promoted by NK cells via MIP‐1beta and LFA‐1. J Immunol 186: 5085–5094 [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki‐Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441: 885–889 [DOI] [PubMed] [Google Scholar]
- Hayashi K, Taura M, Iwasaki A (2018) The interaction between IKKalpha and LC3 promotes type I interferon production through the TLR9‐containing LAPosome. Sci Signal 11: eaan4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemont C, Neel A, Heslan M, Braudeau C, Josien R (2013) Human blood mDC subsets exhibit distinct TLR repertoire and responsiveness. J Leukoc Biol 93: 599–609 [DOI] [PubMed] [Google Scholar]
- Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L, Sasai M, Latz E, Brinkmann MM, Iwasaki A, Coyle AJ, Kolbeck R, Green DR, Sanjuan MA (2012) Noncanonical autophagy is required for type I interferon secretion in response to DNA‐immune complexes. Immunity 37: 986–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry SC, Schmader K, Brown TT, Miller SE, Howell DN, Daley GG, Hamilton JD (2000) Enhanced green fluorescent protein as a marker for localizing murine cytomegalovirus in acute and latent infection. J Virol Methods 89: 61–73 [DOI] [PubMed] [Google Scholar]
- Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, Taya C, Taniguchi T (2005a) Spatiotemporal regulation of MyD88‐IRF‐7 signalling for robust type‐I interferon induction. Nature 434: 1035–1040 [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T (2005b) IRF‐7 is the master regulator of type‐I interferon‐dependent immune responses. Nature 434: 772–777 [DOI] [PubMed] [Google Scholar]
- Hoshino K, Sugiyama T, Matsumoto M, Tanaka T, Saito M, Hemmi H, Ohara O, Akira S, Kaisho T (2006) IkappaB kinase‐alpha is critical for interferon‐alpha production induced by Toll‐like receptors 7 and 9. Nature 440: 949–953 [DOI] [PubMed] [Google Scholar]
- Ito T, Kanzler H, Duramad O, Cao W, Liu YJ (2006) Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood 107: 2423–2431 [DOI] [PubMed] [Google Scholar]
- Kadowaki N, Antonenko S, Liu YJ (2001) Distinct CpG DNA and polyinosinic‐polycytidylic acid double‐stranded RNA, respectively, stimulate CD11c‐ type 2 dendritic cell precursors and CD11c+ dendritic cells to produce type I IFN. J Immunol 166: 2291–2295 [DOI] [PubMed] [Google Scholar]
- Kerkmann M, Rothenfusser S, Hornung V, Towarowski A, Wagner M, Sarris A, Giese T, Endres S, Hartmann G (2003) Activation with CpG‐A and CpG‐B oligonucleotides reveals two distinct regulatory pathways of type I IFN synthesis in human plasmacytoid dendritic cells. J Immunol 170: 4465–4474 [DOI] [PubMed] [Google Scholar]
- Kim S, Kaiser V, Beier E, Bechheim M, Guenthner‐Biller M, Ablasser A, Berger M, Endres S, Hartmann G, Hornung V (2014) Self‐priming determines high type I IFN production by plasmacytoid dendritic cells. Eur J Immunol 44: 807–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M (2004) TLR9‐dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 21: 107–119 [DOI] [PubMed] [Google Scholar]
- Kumagai Y, Kumar H, Koyama S, Kawai T, Takeuchi O, Akira S (2009) Cutting Edge: TLR‐Dependent viral recognition along with type I IFN positive feedback signaling masks the requirement of viral replication for IFN‐{alpha} production in plasmacytoid dendritic cells. J Immunol 182: 3960–3964 [DOI] [PubMed] [Google Scholar]
- Le Mercier I, Poujol D, Sanlaville A, Sisirak V, Gobert M, Durand I, Dubois B, Treilleux I, Marvel J, Vlach J, Blay JY, Bendriss‐Vermare N, Caux C, Puisieux I, Goutagny N (2013) Tumor promotion by intratumoral plasmacytoid dendritic cells is reversed by TLR7 ligand treatment. Cancer Res 73: 4629–4640 [DOI] [PubMed] [Google Scholar]
- Luche H, Weber O, Nageswara Rao T, Blum C, Fehling HJ (2007) Faithful activation of an extra‐bright red fluorescent protein in “knock‐in” Cre‐reporter mice ideally suited for lineage tracing studies. Eur J Immunol 37: 43–53 [DOI] [PubMed] [Google Scholar]
- Madera S, Sun JC (2015) Cutting edge: stage‐specific requirement of IL‐18 for antiviral NK cell expansion. J Immunol 194: 1408–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malleret B, Maneglier B, Karlsson I, Lebon P, Nascimbeni M, Perie L, Brochard P, Delache B, Calvo J, Andrieu T, Spreux‐Varoquaux O, Hosmalin A, Le Grand R, Vaslin B (2008) Primary infection with simian immunodeficiency virus: plasmacytoid dendritic cell homing to lymph nodes, type I interferon, and immune suppression. Blood 112: 4598–4608 [DOI] [PubMed] [Google Scholar]
- Marie I, Durbin JE, Levy DE (1998) Differential viral induction of distinct interferon‐alpha genes by positive feedback through interferon regulatory factor‐7. EMBO J 17: 6660–6669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megjugorac NJ, Jacobs ES, Izaguirre AG, George TC, Gupta G, Fitzgerald‐Bocarsly P (2007) Image‐based study of interferongenic interactions between plasmacytoid dendritic cells and HSV‐infected monocyte‐derived dendritic cells. Immunol Invest 36: 739–761 [DOI] [PubMed] [Google Scholar]
- Mostafavi S, Yoshida H, Moodley D, LeBoite H, Rothamel K, Raj T, Ye CJ, Chevrier N, Zhang SY, Feng T, Lee M, Casanova JL, Clark JD, Hegen M, Telliez JB, Hacohen N, De Jager PL, Regev A, Mathis D, Benoist C et al (2016) Parsing the interferon transcriptional network and its disease associations. Cell 164: 564–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien M, Manches O, Sabado RL, Baranda SJ, Wang Y, Marie I, Rolnitzky L, Markowitz M, Margolis DM, Levy D, Bhardwaj N (2011) Spatiotemporal trafficking of HIV in human plasmacytoid dendritic cells defines a persistently IFN‐alpha‐producing and partially matured phenotype. J Clin Invest 121: 1088–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohto U, Ishida H, Shibata T, Sato R, Miyake K, Shimizu T (2018) Toll‐like receptor 9 contains two DNA binding sites that function cooperatively to promote receptor dimerization and activation. Immunity 48: 649–658 e4 [DOI] [PubMed] [Google Scholar]
- Pauls E, Shpiro N, Peggie M, Young ER, Sorcek RJ, Tan L, Choi HG, Cohen P (2012) Essential role for IKKbeta in production of type 1 interferons by plasmacytoid dendritic cells. J Biol Chem 287: 19216–19228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash A, Smith E, Lee CK, Levy DE (2005) Tissue‐specific positive feedback requirements for production of type I interferon following virus infection. J Biol Chem 280: 18651–18657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash A, Levy DE (2006) Regulation of IRF7 through cell type‐specific protein stability. Biochem Biophys Res Commun 342: 50–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prandini A, Salvi V, Colombo F, Moratto D, Lorenzi L, Vermi W, De Francesco MA, Notarangelo LD, Porta F, Plebani A, Facchetti F, Sozzani S, Badolato R (2016) Impairment of dendritic cell functions in patients with adaptor protein‐3 complex deficiency. Blood 127: 3382–3386 [DOI] [PubMed] [Google Scholar]
- Puttur F, Arnold‐Schrauf C, Lahl K, Solmaz G, Lindenberg M, Mayer CT, Gohmert M, Swallow M, van Helt C, Schmitt H, Nitschke L, Lambrecht BN, Lang R, Messerle M, Sparwasser T (2013) Absence of Siglec‐H in MCMV infection elevates interferon alpha production but does not enhance viral clearance. PLoS Pathog 9: e1003648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puttur F, Francozo M, Solmaz G, Bueno C, Lindenberg M, Gohmert M, Swallow M, Tufa D, Jacobs R, Lienenklaus S, Kuhl AA, Borkner L, Cicin‐Sain L, Holzmann B, Wagner H, Berod L, Sparwasser T (2016) Conventional dendritic cells confer protection against mouse cytomegalovirus infection via TLR9 and MyD88 signaling. Cell Rep 17: 1113–1127 [DOI] [PubMed] [Google Scholar]
- Robbins SH, Bessou G, Cornillon A, Zucchini N, Rupp B, Ruzsics Z, Sacher T, Tomasello E, Vivier E, Koszinowski UH, Dalod M (2007) Natural killer cells promote early CD8 T cell responses against cytomegalovirus. PLoS Pathog 3: e123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins SH, Walzer T, Dembele D, Thibault C, Defays A, Bessou G, Xu H, Vivier E, Sellars M, Pierre P, Sharp FR, Chan S, Kastner P, Dalod M (2008) Novel insights into the relationships between dendritic cell subsets in human and mouse revealed by genome‐wide expression profiling. Genome Biol 9: R17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh SI, Abe F, Kanno A, Tanimura N, Mori Saitoh Y, Fukui R, Shibata T, Sato K, Ichinohe T, Hayashi M, Kubota K, Kozuka‐Hata H, Oyama M, Kikko Y, Katada T, Kontani K, Miyake K (2017) TLR7 mediated viral recognition results in focal type I interferon secretion by dendritic cells. Nat Commun 8: 1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai M, Linehan MM, Iwasaki A (2010) Bifurcation of Toll‐like receptor 9 signaling by adaptor protein 3. Science 329: 1530–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N (1998) Positive feedback regulation of type I IFN genes by the IFN‐inducible transcription factor IRF‐7. FEBS Lett 441: 106–110 [DOI] [PubMed] [Google Scholar]
- Scheu S, Dresing P, Locksley RM (2008) Visualization of IFNbeta production by plasmacytoid versus conventional dendritic cells under specific stimulation conditions in vivo . Proc Natl Acad Sci USA 105: 20416–20421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlitzer A, Sivakamasundari V, Chen J, Sumatoh HR, Schreuder J, Lum J, Malleret B, Zhang S, Larbi A, Zolezzi F, Renia L, Poidinger M, Naik S, Newell EW, Robson P, Ginhoux F (2015) Identification of cDC1‐ and cDC2‐committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat Immunol 16: 718–728 [DOI] [PubMed] [Google Scholar]
- Schmitz F, Heit A, Guggemoos S, Krug A, Mages J, Schiemann M, Adler H, Drexler I, Haas T, Lang R, Wagner H (2007) Interferon‐regulatory‐factor 1 controls Toll‐like receptor 9‐mediated IFN‐beta production in myeloid dendritic cells. Eur J Immunol 37: 315–327 [DOI] [PubMed] [Google Scholar]
- Shibata T, Ohto U, Nomura S, Kibata K, Motoi Y, Zhang Y, Murakami Y, Fukui R, Ishimoto T, Sano S, Ito T, Shimizu T, Miyake K (2016) Guanosine and its modified derivatives are endogenous ligands for TLR7. Int Immunol 28: 211–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegal FP, Kadowaki N, Shodell M, Fitzgerald‐Bocarsly PA, Shah K, Ho S, Antonenko S, Liu YJ (1999) The nature of the principal type 1 interferon‐producing cells in human blood. Science 284: 1835–1837 [DOI] [PubMed] [Google Scholar]
- Strobl B, Bubic I, Bruns U, Steinborn R, Lajko R, Kolbe T, Karaghiosoff M, Kalinke U, Jonjic S, Muller M (2005) Novel functions of tyrosine kinase 2 in the antiviral defense against murine cytomegalovirus. J Immunol 175: 4000–4008 [DOI] [PubMed] [Google Scholar]
- Swiecki M, Gilfillan S, Vermi W, Wang Y, Colonna M (2010) Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity 33: 955–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecki M, Wang Y, Gilfillan S, Colonna M (2013) Plasmacytoid dendritic cells contribute to systemic but not local antiviral responses to HSV infections. PLoS Pathog 9: e1003728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecki M, Wang Y, Riboldi E, Kim AH, Dzutsev A, Gilfillan S, Vermi W, Ruedl C, Trinchieri G, Colonna M (2014) Cell depletion in mice that express diphtheria toxin receptor under the control of SiglecH encompasses more than plasmacytoid dendritic cells. J Immunol 192: 4409–4416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecki M, Colonna M (2015) The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol 15: 471–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Asabe S, Wieland S, Garaigorta U, Gastaminza P, Isogawa M, Chisari FV (2010) Plasmacytoid dendritic cells sense hepatitis C virus‐infected cells, produce interferon, and inhibit infection. Proc Natl Acad Sci USA 107: 7431–7436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, Kano S, Honda K, Ohba Y, Mak TW, Taniguchi T (2005) Integral role of IRF‐5 in the gene induction programme activated by Toll‐like receptors. Nature 434: 243–249 [DOI] [PubMed] [Google Scholar]
- Tamoutounour S, Guilliams M, Montanana Sanchis F, Liu H, Terhorst D, Malosse C, Pollet E, Ardouin L, Luche H, Sanchez C, Dalod M, Malissen B, Henri S (2013) Origins and functional specialization of macrophages and of conventional and monocyte‐derived dendritic cells in mouse skin. Immunity 39: 925–938 [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Takaoka A (2001) A weak signal for strong responses: interferon‐alpha/beta revisited. Nat Rev Mol Cell Biol 2: 378–386 [DOI] [PubMed] [Google Scholar]
- Terawaki S, Camosseto V, Prete F, Wenger T, Papadopoulos A, Rondeau C, Combes A, Rodriguez Rodrigues C, Vu Manh TP, Fallet M, English L, Santamaria R, Soares AR, Weil T, Hammad H, Desjardins M, Gorvel JP, Santos MA, Gatti E, Pierre P (2015) RUN and FYVE domain‐containing protein 4 enhances autophagy and lysosome tethering in response to interleukin‐4. J Cell Biol 210: 1133–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasello E, Pollet E, Vu Manh TP, Uze G, Dalod M (2014) Harnessing mechanistic knowledge on beneficial versus deleterious IFN‐I effects to design innovative immunotherapies targeting cytokine activity to specific cell types. Front Immunol 5: 526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu Manh TP, Alexandre Y, Baranek T, Crozat K, Dalod M (2013) Plasmacytoid, conventional, and monocyte‐derived dendritic cells undergo a profound and convergent genetic reprogramming during their maturation. Eur J Immunol 43: 1706–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu Manh TP, Elhmouzi‐Younes J, Urien C, Ruscanu S, Jouneau L, Bourge M, Moroldo M, Foucras G, Salmon H, Marty H, Quere P, Bertho N, Boudinot P, Dalod M, Schwartz‐Cornil I (2015) Defining mononuclear phagocyte subset homology across several distant warm‐blooded vertebrates through comparative transcriptomics. Front Immunol 6: 299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterstrat A, Liang Y, Swiderski CF, Shelton BJ, Van Zant G (2010) Congenic interval of CD45/Ly‐5 congenic mice contains multiple genes that may influence hematopoietic stem cell engraftment. Blood 115: 408–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland SF, Takahashi K, Boyd B, Whitten‐Bauer C, Ngo N, de la Torre JC, Chisari FV (2014) Human plasmacytoid dendritic cells sense lymphocytic choriomeningitis virus‐infected cells in vitro . J Virol 88: 752–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Sanin DE, Everts B, Chen Q, Qiu J, Buck MD, Patterson A, Smith AM, Chang CH, Liu Z, Artyomov MN, Pearce EL, Cella M, Pearce EJ (2016) Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity 44: 1325–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Dai J, Deng J, Sheikh F, Natalia M, Shih T, Lewis‐Antes A, Amrute SB, Garrigues U, Doyle S, Donnelly RP, Kotenko SV, Fitzgerald‐Bocarsly P (2012) Type III IFNs are produced by and stimulate human plasmacytoid dendritic cells. J Immunol 189: 2735–2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Ohto U, Shibata T, Krayukhina E, Taoka M, Yamauchi Y, Tanji H, Isobe T, Uchiyama S, Miyake K, Shimizu T (2016) Structural analysis reveals that toll‐like receptor 7 is a dual receptor for guanosine and single‐stranded RNA. Immunity 45: 737–748 [DOI] [PubMed] [Google Scholar]
- Zucchini N, Bessou G, Robbins SH, Chasson L, Raper A, Crocker PR, Dalod M (2008a) Individual plasmacytoid dendritic cells are major contributors to the production of multiple innate cytokines in an organ‐specific manner during viral infection. Int Immunol 20: 45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucchini N, Bessou G, Traub S, Robbins SH, Uematsu S, Akira S, Alexopoulou L, Dalod M (2008b) Cutting edge: overlapping functions of TLR7 and TLR9 for innate defense against a herpesvirus infection. J Immunol 180: 5799–5803 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Review Process File