

Abstract
Systemic sclerosis (SSc) is an autoimmune disease characterized by vasculopathy, which may be the consequence of inflammation and oxidative stress that ultimately leads to a reduced nitric oxide (NO) bioavailability. Passive leg movement (PLM) is a novel methodology for assessing lower limb vascular function that is predominantly NO-dependent. We combined this vascular assessment with a comprehensive panel of plasma biomarkers to assess the axis of inflammation, oxidative stress, and NO in SSc patients (n = 12, 62±11 yrs) compared to healthy controls (n = 17, 60±16 yrs). PLM-induced changes in leg blood flow (LBF) (191±104 vs 327±217 ml/min) and LBF area under the curve (AUC) (39±104 vs 125±131 ml) were reduced in SSc compared to controls. Stratification of patients according to history of digital ulcer (DU) formation revealed a further reduction in LBF AUC in DU (−13±83) vs non-DU (91±102) patients. Biomarkers of inflammation (C-reactive protein, CRP) and oxidative stress (malondialdehyde, MDA and protein carbonyl, PC) were all elevated in SSc (CRP: 3299±2372 vs 984±565 ng/ml; MDA: 3.2±1.1 vs 1.1±0.7 uM; PC: 0.15±0.05 vs 0.12±0.03 nmol/mg) and CRP was further elevated in patients with a history of DU (4551±2752 vs 2047±1019 ng/ml) compared to non-DU, although these were not individually correlated to changes in LBF. These findings of impaired NO-mediated vascular function, linked to DU, and a milieu of inflammation and oxidative stress, suggest that redox balance plays an important, but not necessarily deterministic, role in the vascular pathophysiology of SSc.
Keywords: Vasculopathy, nitric oxide, oxidative stress, inflammation, digital ulcers
INTRODUCTION
Systemic sclerosis (SSc) is a multisystem disorder characterized by autoimmunity, tissue fibrosis, and pronounced vasculopathy, and is often associated with increased inflammation and oxidative stress. Vasculopathy is an early and important component of SSc and has clinical presentations that include Raynaud’s phenomenon (RP), which is often the initial indicator of the disease, and digital ulcers (DU), pulmonary arterial hypertension (PAH), and scleroderma renal crisis (SRC), which are severe end-stage vascular complications (Matucci-Cerinic, Kahaleh, & Wigley, 2013; Strange & Nash, 2009). In SSc, an initial vascular insult leads to endothelial cell activation and the initiation of pro-inflammatory cascades, as well as a vicious cycle of chronic ischemia and intermittent reperfusion that results in deleterious histological changes and the generation of an excess of free radicals and reactive oxygen species (ROS) (Fuschiotti, 2016; Gabrielli, Svegliati, Moroncini, & Amico, 2012). When the generation of ROS outstrips the body’s antioxidant capacity, this leads to a state of “oxidative stress” and can cause oxidative damage to cells, as well as to lipids, carbohydrates, and proteins (Incalza et al., 2017). ROS also attenuate the bioavailability of nitric oxide (NO), a potent endogenous vasodilator, thus, potentially, further impacting vascular function in this patient group.
Our group (Frech et al., 2015) and others (Bartoli et al., 2007; Cypiene et al., 2008; Rollando et al., 2010; Rossi, Granel, Marziale, Le Mee, & Frances, 2010; Szucs et al., 2007; Takahashi et al., 2013) have investigated the degree to which vascular function is impaired in SSc by means of flow-mediated dilation (FMD), and have reported that FMD is attenuated in SSc patients compared to healthy controls. Moreover, within the SSc population, FMD has been documented to be further attenuated with disease duration (Takahashi et al., 2013), increasing severity of microvascular changes (Rollando et al., 2010), and in those with a history of DU (Frech et al., 2015). FMD has thus been well established as a method for assessing vascular health. However, recent studies utilizing the endothelial nitric oxide synthase (eNOS) inhibitor N(G)-monomethyl-L-arginine (L-NMMA) have challenged FMD as a bioassay for NO, finding modest (Wray et al., 2013) to no difference (Pyke et al., 2010) in the post-occlusion vasodilatory response in the presence of this inhibitor, thus questioning the magnitude of the NO contribution to the FMD response. In light of this, our group developed the passive leg movement (PLM) test as an alternative, non-invasive, method for assessing NO-mediated vascular function. The hyperemic response to PLM has been documented to be predominantly NO-mediated (Groot, Trinity, et al., 2015; Mortensen, Askew, Walker, Nyberg, & Hellsten, 2012; Trinity et al., 2012), and while this technique has been used to describe decrements in NO-mediated vascular function with aging (Groot, Rossman, et al., 2015; Groot, Trinity, et al., 2015; Groot et al., 2013; Mortensen et al., 2012) and disease states such as peripheral artery disease (Mortensen et al., 2012), heart failure (Witman et al., 2015), and sepsis (Nelson et al., 2016), it has not yet been used to characterize the extent to which NO-mediated vascular function may be impaired in SSc. In addition to being NO-dependent, the PLM response reflects vasodilation of the feed arteries and microvascular beds, further distinguishing it from FMD which assesses conduit artery dilation. Moreover, this test specifically interrogates vascular function in the leg. To date, vascular function assessments in SSc have been performed largely in the arm, despite clinical vascular complications being reported in both the upper and lower limbs. PLM testing thus provides the opportunity to extend previous studies in SSc (Bartoli et al., 2007; Cypiene et al., 2008; Frech et al., 2015; Luders et al., 2017; Rollando et al., 2010; Rossi et al., 2010; Szucs et al., 2007; Takahashi et al., 2013) and offer new insight into the nature and degree of lower limb vascular dysfunction in this unique patient population.
Therefore, the purpose of this investigation was to combine plasma biomarkers of inflammation, antioxidant capacity, and oxidative stress with a novel assessment of NO bioavailability to evaluate the mechanisms contributing to the vascular pathophysiology in SSc patients. We hypothesized that the PLM-induced hyperemic response would be attenuated in SSc compared to that of healthy controls, and that this would be accompanied by elevated systemic inflammation, impaired antioxidant capacity and greater oxidative stress in these patients. An additional aim of this study was to evaluate the relationship between the PLM response in SSc and the incidence of DU, an important clinical cardiovascular end point, with the hypothesis that patients with a history of DU would demonstrate a greater impairment in vascular function than those without DU.
METHODS
Ethical approval
The experimental protocol was approved by the University of Utah and Salt Lake City VA Medical Center Institutional Review Board (IRB_00040212) and was in compliance with clause 35 of the Declaration of Helsinki, except for registration in a database. All participants gave written informed consent.
Participants
12 SSc patients and 17 healthy, age-matched controls were recruited through the University of Utah SSc Clinic or the Utah Vascular Research Laboratory (UVRL). All participants were non-smokers and were normally active. Patients met classification criteria for a diagnosis of SSc (van den Hoogen et al., 2013). Female participants who were premenopausal were studied during the follicular phase of the menstrual cycle. All participants abstained from exercise, alcohol, and caffeine for 24 hours prior to the study visit and were at least 5 hours post-prandial. SSc patients abstained from any vasoactive medications for 12 hours prior to the study visit, as directed by their physician. All data collection took place at the UVRL located in the Salt Lake City VA Medical Center’s Geriatric Research, Education, and Clinical Center.
Passive leg movement protocol and measurements
Patients were instrumented and rested quietly in a thermoneutral room in the supine position for at least 15 minutes prior to the start of PLM. After 1 minute of baseline measures, PLM was performed by a trained investigator who moved the patient’s knee joint from 180˚ (full extension) to 90˚ at a rate of 1 Hz continuously for 2 minutes. Patients were instructed to remain passive and not resist or assist with the movement. To avoid a startle reflex, patients were made aware that PLM would start in 5-10 seconds, but to avoid an anticipatory response, they were not made aware of exactly when the movement would begin.
Simultaneous measurements of femoral artery blood velocity and common femoral artery diameter were performed using a linear array transducer operating in duplex mode, with imaging frequency of 10 MHz and Doppler frequency of 5 MHz (Logiq 7; GE Medical Systems, Milwaukee, WI). The femoral artery was insonated distal to the inguinal ligament and proximal to the deep and superficial femoral bifurcation. Vessel diameter was determined at a perpendicular angle along the central axis of the scanned area. All blood velocity measurements were obtained with the probe appropriately positioned to maintain an insonation angle of 60° or less. The sample volume was maximized according to vessel size and was centered within the vessel on the basis of real-time ultrasound visualization. Mean velocity (Vmean) values (angle-corrected and intensity-weighted AUC) were automatically calculated using commercially available software (Logiq 7). Using arterial diameter and Vmean, leg blood flow was calculated as: LBF = Vel × π(vessel diameter/2)2 × 60, expressed in milliliters per minute. Anterograde (Ant) and retrograde (Ret) leg blood flow (LBF) were calculated from the Vmean above and below the zero line, respectively. LBF was assessed second-by-second and then reported as a 3 second rolling average. LBF responses to PLM are reported as peak change from baseline and the area under the curve (AUC) normalized for baseline during the first minute of movement. Supine resting systolic (SBP) and diastolic blood pressures (DBP) were obtained using an automated blood pressure monitor (Tango+, Suntech, Morrisville, NC, USA) and resting heart rate (HR) was determined from a three-lead ECG interfaced with the automated blood pressure monitor. Mean arterial pressure (MAP) was calculated as MAP=DBP+[(SBP-DBP)/3].
Inflammation, antioxidant capacity, and oxidative stress
Venous blood samples were obtained at the beginning of the study visit and were centrifuged to collect plasma. These samples were then stored at −80C until analysis. Plasma levels of tumor necrosis factor alpha (TNFα), interleukin 6 (IL-6), and C-reactive protein (CRP) were determined using high sensitivity ELISA (R&D Systems, Minneapolis, MN, USA). Plasma free radical levels were directly assessed by spin trapping and electron paramagnetic resonance (EPR) spectroscopy. Briefly, 3.0 ml of venous blood was collected into a vacutainer containing 1.0 ml α-phenyl-tert-butylnitrone (PBN; 0.14 M), then the PBN adduct was extracted under toluene and pipetted into a precision-bore quartz EPR spectroscopy sample tube (Wilmad-LabGlass, Vineland, NJ) that had been flushing with compressed N2. EPR spectroscopy was performed at 21°C with an EMX X-band spectrometer (Bruker, Billerica, MA) and analyzed with commercially available software (Bruker Xenon, version 1.1b.51) with the analyst blinded to experimental condition and reported as PBN adduct in arbitrary units (AU). Total antioxidant capacity was determined by the ferric reducing ability of plasma (FRAP) using the method previously described by Benzie and Strain (Benzie & Strain, 1996). The activity of superoxide dismutase (SOD), an important endogenous antioxidant that scavenges superoxide and spares NO, was assessed by colorimetry (Cayman Chemical Company, Ann Arbor, MI, USA). Lipid peroxidation was assessed by malondialdehyde (MDA) levels using colorimetry (Bioxytech MDA-586 assay, OxisResearch, Burlingame, CA, USA), and protein oxidation was assessed by plasma protein carbonyl (PC) levels using colorimetry (PC test kit, Biocell Corporation LTD, Papatoetoe, NZ).
Statistics
Physical characteristics, resting cardiovascular measures, PLM-induced blood flow responses, and plasma biomarkers were compared between groups (CON vs SSc and DU vs non-DU) using unpaired Student’s t-tests to identify any group differences. When comparing DU and non-DU groups to CON, analysis of variance (ANOVA) was used, and post hoc testing was performed using the Tukey test when main effects were found. The Pearson Correlation Coefficient test was used to assess relationships between plasma biomarkers and PLM responses as well as between baseline LBF and PLM responses. Significance was set at P < 0.05, and data are expressed as mean ± SD.
RESULTS
Participants
SSc patients and controls were well-matched for gender, age, height, weight, and BMI (Table 1). Disease-specific characteristics as well as pharmacological therapeutic information for the SSc patients are documented in Table 2. Among the patient population, SSc disease duration and duration since onset of RP symptoms both ranged from 1 to 36 years, with a mean of 9.6 and 10.0 years, respectively (Table 2). Of the 12 patients, 3 had prescriptions for cardiovascular medications at the time of the study, which included calcium channel blockers (CCB), angiotensin receptor blockers (ARB), and diuretics. All SSc patients were positive for antinuclear antibodies (ANA) and a slight majority (58%) were positive for anti-centromeric antibodies. Two-thirds of the patients were identified as having limited SSc, while the remaining one-third was identified as having the diffuse phenotype. Half of the SSc patients had a history of DU.
Table 1.
Characteristics of SSc patients and healthy controls
| Control | SSc | |
|---|---|---|
| n | 17 | 12 |
| Female, n (%) | 12 (71%) | 9 (75%) |
| Age, years | 60±16 | 62±11 |
| Height, cm | 167±9 | 169±9 |
| Weight, kg | 72.7±12.2 | 71.4±11.6 |
| BMI, kg/m2 | 26±4 | 25±4 |
Values are mean ± SD. n, number of participants; SSc, systemic sclerosis; BMI, body mass index. p<0.05; there were no significant differences.
Table 2.
SSc patient health history and medication use.
| SSc disease duration, years | 9.6±9.9 |
| RP duration, years | 10.0±9.7 |
| SSc subtype, n (%) | |
| Limited | 8 (67%) |
| Diffuse | 4 (33%) |
| mRSS | 6.3±6.4 |
| End-stage vasculopathy, n (%) | |
| DU | 6 (50%) |
| PAH | 1 (8%) |
| SRC | 1 (8%) |
| Antibody presence, n (%) | |
| ANA | 12 (100%) |
| SCL70 | 1 (8%) |
| Anti-centromeric | 7 (58%) |
| RNA polymerase III | 0 |
| CVD risk factor, n (%) | 7 (58%) |
| Medication use, n (%) | |
| CCB | 2 (17%) |
| PDE5I | 0 |
| ERA | 0 |
| PCA | 0 |
| ARB | 2 (17%) |
| Diuretic | 2 (17%) |
| Immunosuppression | 2 (17%) |
Values are mean ± SD. RP, Raynaud’s phenomenon; mRSS, modified Rodnan skin score; DU, digital ulcer; PAH, pulmonary arterial hypertension; SRC, scleroderma renal crisis; ANA, antinuclear antibody; SCL70, anti-Scl-70 antibody; CVD, cardiovascular disease; CCB, calcium channel blocker; PDE5I, phosphodiesterase 5 inhibitor; ERA, endothelin receptor antagonist; PCA, prostacyclin analog; ARB, angiotensin receptor blocker.
Baseline measurements
At rest, HR, MAP, and femoral artery diameter were not different between controls and patients, nor were there any differences between patients with a history of DU and those without (Table 3). As a group, SSc patients had higher Vmean, LBF, and anterograde LBF than controls. Correlation analysis failed to identify a significant relationship between baseline LBF and peak change in LBF (r2=0.031, p=0.363) or LBF AUC (r2=0.007, p=0.675). There were no differences in any baseline measurements between patients with and without DU.
Table 3.
Resting cardiovascular measurements.
| Control | SSc | |||
|---|---|---|---|---|
| (n=17) |
All (n=12) |
No DU (n=6) |
DU (n=6) |
|
| HR (bpm) | 61±11 | 67±10 | 66±11 | 68±10 |
| MAP (mmHg) | 92±13 | 84±7 | 82±8 | 86±6 |
| FA Diam (cm) | 0.86±0.15 | 0.80±0.12 | 0.74±0.10 | 0.87±0.10 |
| Vel (cm/sec) | 8.9±3.4 | 14.8±7.9 * | 16.9±9.6 * | 12.7±6.1 |
| LBF (ml/min) | 299±94 | 418±140 * | 412±182 | 424±100 |
| Ant LBF (ml/min) | 402±81 | 516±143 * | 502±180 | 531±109 |
| Ret LBF (ml/min) | 104±49 | 99±63 | 90±57 | 107±72 |
Values are mean ± SD. SSc, systemic sclerosis; No DU, no history of digital ulcers; DU, history of digital ulcers; HR, heart rate; MAP, mean arterial pressure; FA diam, femoral artery diameter; Vel, blood velocity; LBF, leg blood flow; Ant, anterograde; Ret, retrograde.
Significantly different from control, p<0.05.
Response to passive leg movement
PLM elicited a hyperemic response in both controls and SSc patients (Figure 1A). The AUC for this response was ≈69% lower in SSc (Figure 1B) and this was largely due to a ≈51% attenuation in Ant LBF AUC (Figure 1D). Ret LBF AUC differences were not significant (Figure 1F). When assessed as the change in LBF, the SSc PLM response was notably different from controls, and the peak change in LBF was ≈42% lower than controls (Figure 2).
Figure 1.
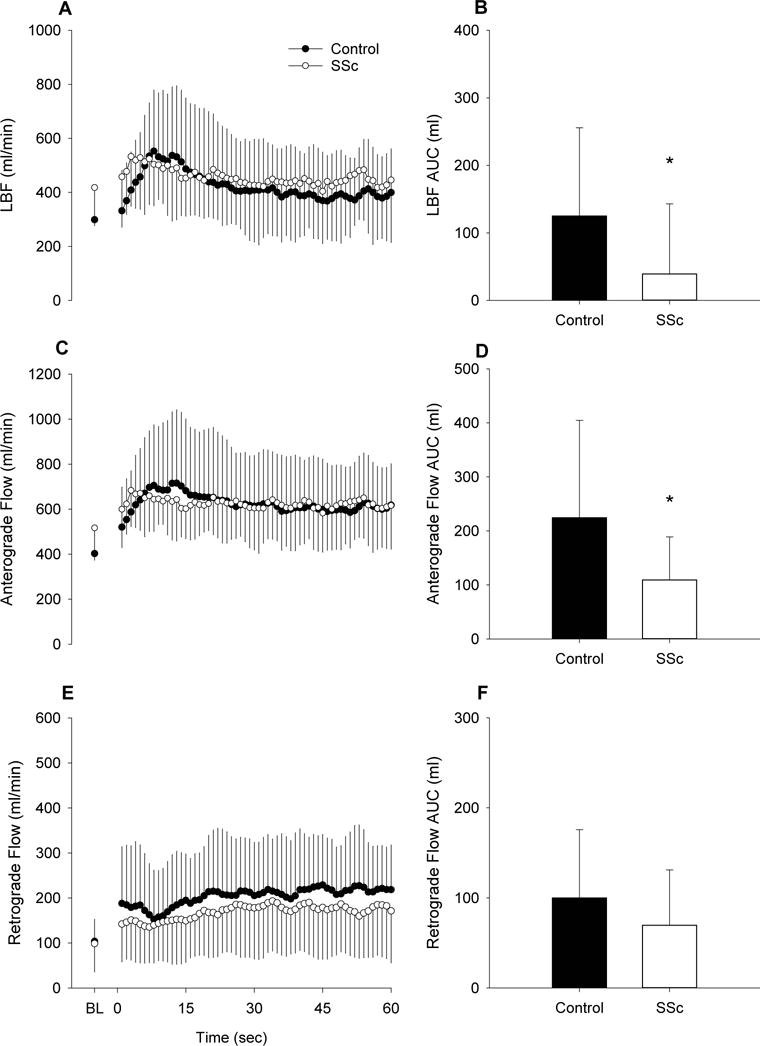
Leg blood flow (LBF) responses to passive leg movement (PLM) in patients with systemic sclerosis (SSc) and healthy controls. A, second-by-second tracing of LBF (ml/min) at baseline (BL) and for 1 min of PLM. B, LBF area under the curve (AUC) (ml) for 1 min. C, second-by-second tracing of anterograde (Ant) LBF (ml/min) at BL and for 1 min of PLM. D, Ant LBF AUC (ml) for 1 min. E, second-by-second tracing of retrograde (Ret) LBF (ml/min) at BL and for 1 min of PLM. F, Ret LBF AUC (ml) for 1 min. Control, n=17; SSc, n=12. * Significantly different from control, p<0.05. Values are mean ± SD.
Figure 2.
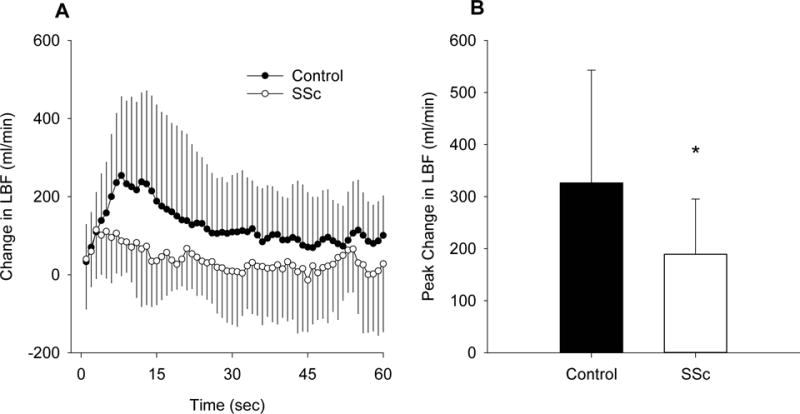
Change in LBF in response to PLM in controls and SSc. A, second-by second tracing of the change in LBF (ml/min). B, Peak change in LBF (ml/min). Control, n=17; SSc, n=12. * Significantly different from control, p<0.05. Values are mean ± SD.
Inflammation, antioxidant capacity, and oxidative stress
There were no differences between groups in the pro-inflammatory cytokines TNFα (1.52±0.72 vs 1.12±0.59 pg/ml, controls vs SSc) or IL-6 (1.55±1.71 vs 2.14± pg/ml, controls vs SSc), but systemic inflammation, as assessed by CRP, was greater in the SSc group (984±565 vs 3299±2372 ng/ml, controls vs SSc, Figure 3). Plasma free radical levels, as assessed by EPR spectroscopy, were not different between controls and patients (6.9±7.4 vs 10.4±9.7 AU, controls vs SSc, Figure 4). FRAP, a measure of total antioxidant capacity, was elevated in SSc (1.2±0.46 vs 2.2±1.0 mM, controls vs SSc) while SOD, a measure of endogenous antioxidant capacity, was lower (12.1±4.2 vs 8.8±4.1 U/ml, controls vs SSc, Figure 5). Lipid peroxidation, as determined by plasma MDA, was increased in SSc patients (1.1±0.7 vs 3.2±1.1 uM, controls vs SSc). Likewise, protein oxidation, as assessed by plasma PC, was also elevated in the SSc group (0.12±0.03 vs 0.15±0.05 nmol/mg, controls vs SSc, Figure 6). There were no significant correlations between any biomarkers and either LBF AUC or peak change in LBF, although there was a trend towards significance for CRP and LBF AUC (r2=0.130, p=0.06).
Figure 3.
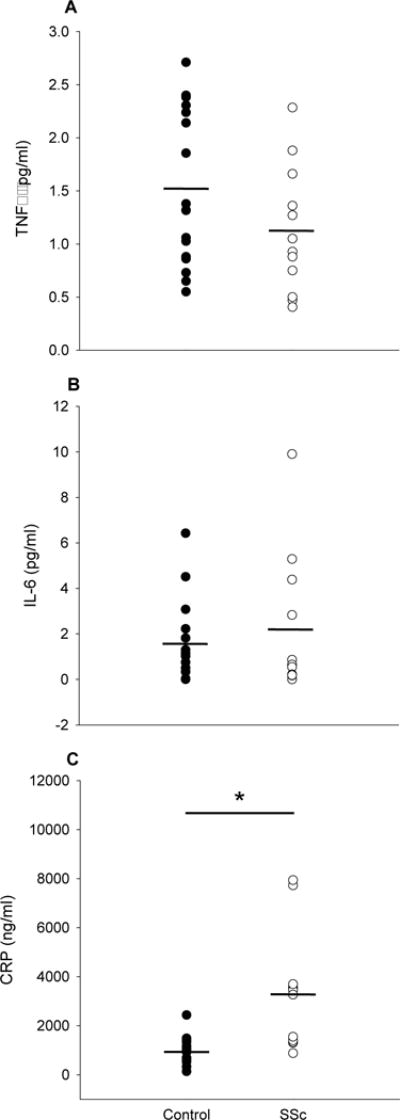
Markers of inflammation in controls and SSc. A, Tumor necrosis factor alpha (TNFα) (pg/ml). B, Interleukin 6 (IL-6) (pg/ml). C, C-reactive protein (CRP) (ng/ml). * Significantly different from control, p<0.05. Mean indicated by short horizontal bar.
Figure 4.
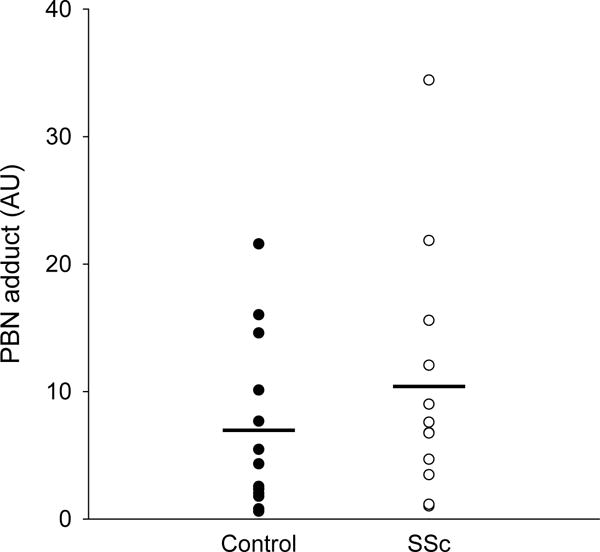
Plasma free radical levels as measured by spin trapping by α-phenyl-tert-butylnitrone (PBN) and electron paramagnetic resonance (EPR) spectroscopy in controls and SSc (arbitrary units, AU). Mean indicated by horizontal bar.
Figure 5.
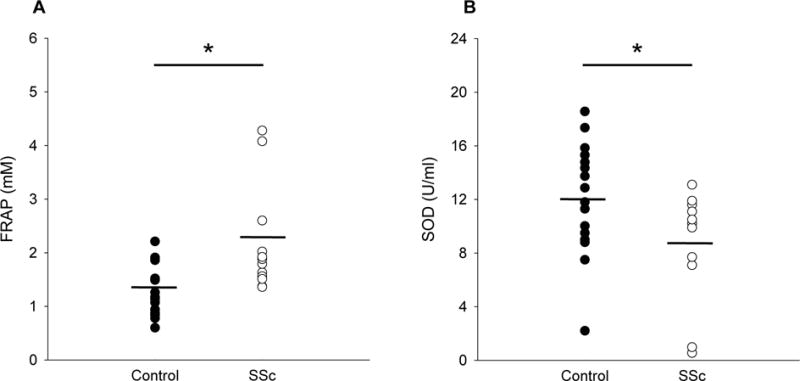
Total and endogenous antioxidant capacity in controls and SSc. A, Ferric reducing ability of plasma (FRAP) (mM). B, Superoxide dismutase (SOD) (U/ml). * Significantly different from control, p<0.05. Mean indicated by short horizontal bar.
Figure 6.

Markers of oxidative damage in controls and SSc. A, Malondiadlehyde (MDA) (uM). B, Protein carbonyls (PC) (nmol/mg). * Significantly different from control, p<0.05. Mean indicated by short horizontal bar.
Digital ulcers, PLM responses, and inflammation markers
SSc patients with DU history demonstrated markedly attenuated LBF AUC in response to PLM compared to patients with no incidence of DU (Figure 7A), which may be attributed in large part to a ≈155% greater retrograde blood flow in the DU group (Figure 7D). There were no significant differences in inflammation, antioxidant capacity, and oxidative stress biomarkers between patients with and without DU history, with the exception of CRP, which was ≈122% greater in patients with DU (Table 4). However, CRP levels were not predictive of peak change in LBF (r2=0.06, p=0.43) or LBF AUC (r2=0.08, p=0.39) in SSc patients.
FIGURE 7.
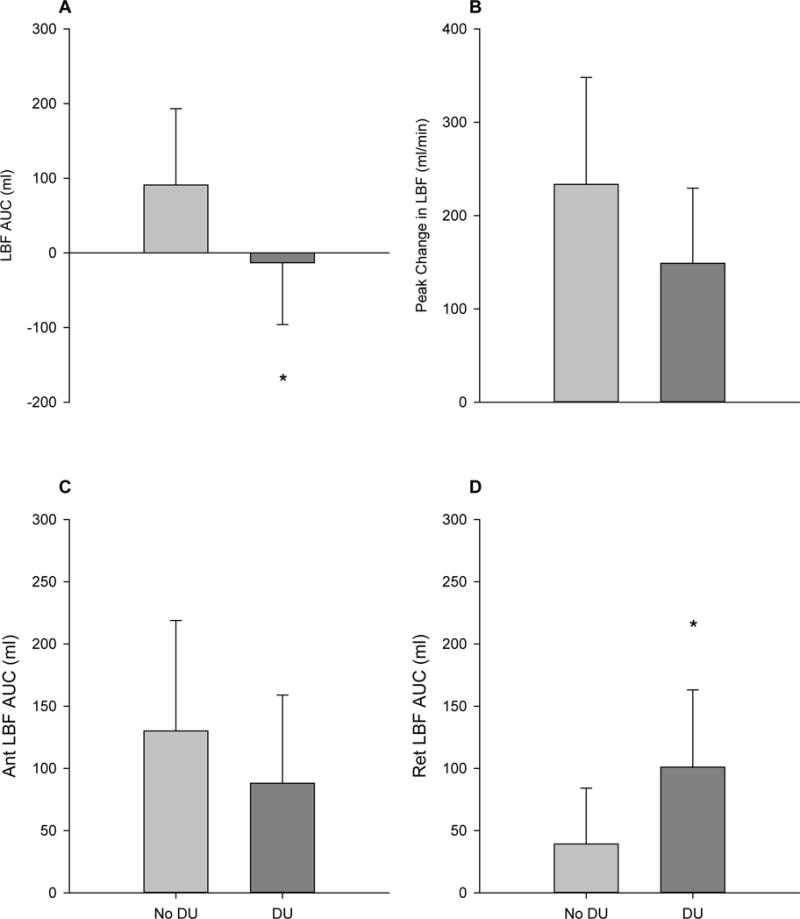
PLM-induced LBF responses in SSc patients without history of digital ulcers (No DU) and with history of digital ulcers (DU). A, Net LBF AUC (ml) for No DU and DU. B, Peak change in LBF for No DU and DU. C, Anterograde (Ant) LBF AUC (ml) for No DU and DU. D, Retrograde (Ret) LBF AUC (ml) for No DU and DU. No DU, n=6; DU, n=6. * Significantly different from No DU, p<0.05. Values are mean ± SD.
Table 4.
Markers of inflammation, oxidative capacity, and oxidative stress in SSc patients with and without a history of DU.
| No DU (n=6) |
DU (n=6) |
|
|---|---|---|
| TNFα (pg/ml) | 0.9±0.5 | 1.4±0.6 |
| IL-6 (pg/ml) | 1.3±2.0 | 3.0±3.8 |
| CRP (ng/ml) | 2047±1019 | 4551±2752* |
| PBN adduct (AU) | 13.5±12.3 | 7.3±5.5 |
| FRAP (mM) | 2.1±1.1 | 2.3±1.0 |
| SOD (U/ml) | 7.7±3.8 | 9.9±4.4 |
| MDA (uM) | 3.1±1.1 | 3.4±1.2 |
| PC (nmol/mg) | 0.14±0.04 | 0.16±0.05 |
Values are mean ± SD. No DU, no history of digital ulcers; DU, history of digital ulcers; TNFα, tumor necrosis factor alpha; IL-6, Interleukin 6; CRP, C-reactive protein; PBN, α-phenyl-tert-butylnitrone; FRAP, ferric reducing ability of plasma; SOD, superoxide dismutase; MDA, malondiadlehyde; PC, protein carbonyls.
Significantly different from No DU, p<0.05.
DISCUSSION
This study combined PLM, a novel method for determining NO-mediated vascular function, with a comprehensive panel of plasma biomarkers to assess disease-related changes in the axis of inflammation, oxidative stress, and NO in SSc. The PLM-induced hyperemic response, whether expressed as the LBF AUC or the peak change in LBF, was significantly attenuated in SSc patients, suggesting a decrement in NO bioavailability that may contribute to the impaired vascular function in this patient group. When SSc patients were stratified based on the presence or absence of DU, LBF AUC was markedly attenuated in patients with a history of DU compared to their non-DU counterparts, highlighting the link between vascular dysfunction and disease severity. SSc patients also exhibited a clear elevation in several inflammatory and oxidative stress plasma biomarkers and a reduction in endogenous antioxidant capacity, with additional evidence of a greater degree of inflammation in those patients with DU. However, there were no significant correlations between these plasma biomarkers and the blood flow responses. Taken together, these findings demonstrate an impairment in NO-mediated vascular function that appears to reflect the degree of vasculopathy in SSc, and suggests that the presence of a vascular milieu characterized by inflammation and oxidative stress may play an important, although not necessarily deterministic, role in the vascular pathophysiology of this patient group.
PLM Response in SSc
To better characterize vascular dysfunction in SSc, we assessed PLM-induced hyperemia. Decrements in vascular function in SSc have been previously reported using FMD (Bartoli et al., 2007; Cypiene et al., 2008; Frech et al., 2015; Rollando et al., 2010; Rossi et al., 2010; Szucs et al., 2007; Takahashi et al., 2013), however, there are several key distinctions between FMD and PLM. While recent studies have identified additional uncertainty regarding the ability of FMD testing to assess NO bioavailability (Pyke et al., 2010; Wray et al., 2013), PLM has emerged as a vascular function test that is largely NO-dependent (Groot, Trinity, et al., 2015; Mortensen et al., 2012; Trinity et al., 2012). Furthermore, while FMD provokes vasodilation of a conduit artery, PLM elicits a hyperemic response through dilation of the smaller feed arteries and microvasculature distal to the measurement site (the femoral artery). And lastly, unlike FMD, which is traditionally performed in the arm, PLM interrogates the legs, and thus is unique in that this method evaluates the vasodilatory response in a major locomotory muscle group. As it has been demonstrated that upper limb vascular function measures do not directly correlate with those of the lower limb (Thijssen et al., 2011), independently examining vascular function in the lower limb is not only necessary for a more comprehensive assessment of vascular health, it may provide specific insights into the mechanisms contributing to attenuated skeletal muscle blood flow and impaired exercise capacity during locomotor activities that are not evident in the arm. To date, NO-mediated microvascular function of the lower limbs has not been examined in SSc, and thus the present study provided an opportunity to better define the degree of vascular dysfunction in this patient population.
SSc patients demonstrated an increase in net femoral artery blood flow with PLM, as well as an increase in anterograde and retrograde blood flow, that had temporal characteristics similar to those of the controls (Figures 1A, 1C, 1E). However, the magnitude of both the peak change in LBF (Figure 2) and the AUC for net and anterograde blood flow (Figures 1B, 1D) were significantly attenuated. These findings are consistent with those documented in other disease populations using PLM, where a hyperemic response is still present and follows a similar timeline to that of healthy controls, but with a magnitude that is severely blunted (Nelson et al., 2016; Witman et al., 2015). Indeed, our group recently reported strikingly similar results to those seen here in SSc in a group of patients with severe sepsis (Nelson et al., 2016). In this previous study, we speculated that the attenuated PLM response was likely the result of a combination of vascular insensitivity to shear stress from eNOS downregulation and NO saturation from inducible NOS (iNOS) upregulation. In SSc, there is also evidence of a downregulation of eNOS and upregulation of iNOS (Cotton, Herrick, Jayson, & Freemont, 1999; Yamamoto, Katayama, & Nishioka, 1998) as well as an overproduction of NO (Devrim et al., 2008; Luo, Liu, Jiang, Zhao, & Zhao, 2016; Mok et al., 2008; Yamamoto et al., 1998), making the dysregulation of NO signaling from eNOS and iNOS a likely culprit for the attenuated vascular responsiveness in this patient group.
Inflammation, Oxidative Stress, and Antioxidant Capacity in SSc
The PLM response is largely mediated by NO bioavailability (Groot, Trinity, et al., 2015; Mortensen et al., 2012; Trinity et al., 2012), which in turn is strongly influenced by the presence of oxidative stress. Indeed, oxidative stress attenuates NO bioavailability by both decreasing NO production, predominantly by uncoupling eNOS (Landmesser et al., 2003), and increasing NO degradation, namely through its conversion to peroxynitrite by superoxide (Ferrer-Sueta & Radi, 2009). Moreover, uncoupled eNOS will produce superoxide rather than NO, and the elevated levels of superoxide will then go on to degrade existing pools of NO, further attenuating NO bioavailability. Indeed, elevated levels of oxidative stress are often associated with impairments in NO signaling and endothelial function, and studies aimed at lowering oxidative stress through antioxidant supplementation have reported improvements in endothelial function (Ives et al., 2014; Witman et al., 2012; Wray et al., 2012). Thus, to explore potential mechanisms responsible for the observed decrement in NO-mediated PLM responses in SSc patients, we assessed an extensive panel of biomarkers specific for systemic inflammation, oxidative stress, and total/endogenous antioxidant capacity. The rationale for evaluating these biomarkers is related to the pathogenesis of SSc, as it has been proposed that following an initial insult or vascular injury, endothelial activation and pro-inflammatory signaling occur early in the disease, and lead to inflammation, autoimmunity, and vascular changes, and eventually to vasculopathy, tissue hypoxia, and oxidative stress. It is important to note, however, that oxidative stress has been postulated as a potential culprit responsible for instigating endothelial activation. Thus, in the case of SSc, it remains unclear if oxidative stress leads to inflammation, or if inflammation leads to oxidative stress (Fuschiotti, 2016; Gabrielli et al., 2012; Incalza et al., 2017; Matucci-Cerinic et al., 2013). However, while the seminal event remains elusive, it is evident that both inflammation and oxidative stress contribute significantly to the progression of the disease.
Surprisingly, the two pro-inflammatory cytokines measured in this study, TNF-α and IL-6, were not significantly different between SSc and the healthy controls (Figures 3A and 3B). While this differs from some previously reported findings (Hussein et al., 2005; Pehlivan et al., 2012), these former studies examined individuals who were notably younger, with mean ages in the 30’s and 40’s, compared to the current study with a mean age in the 60’s. As aging per se is associated with increased expression of both TNF-α and IL-6 (Donato, Black, Jablonski, Gano, & Seals, 2008), the advanced age of both the SSc and control groups here may have diminished the ability to detect disease-specific differences in these biomarkers. Additionally, these previous studies also failed to report medication use, which is important given the known ability of immunosuppressive and antihypertensive pharmacotherapy to reduce circulating levels of TNF-α and IL-6 (Toledo et al., 2015). In the current study, several SSc patients were on immunosuppressive or antihypertensive medications which may have exerted anti-inflammatory effects. However, despite the lack of evidence for an elevation in the pro-inflammatory cytokines TNF-α and IL-6, CRP was elevated in SSc patients compared to controls (Figure 3C), which is consistent with previous studies in this patient group (Cypiene et al., 2008). While these biomarkers represent major players in the inflammatory cascade, inflammation is the result of a shift in the complex balance of both pro- and anti-inflammatory cytokines. Together, these findings suggest that there is some degree of derangement in the inflammatory profile of patients with SSc compared to their age match controls.
Perhaps related to this increase in inflammation, we observed a markedly higher resting leg blood flow in SSc patients compared to their age-matched counterparts (Table 3). This seems contrary to what would be expected in SSc, as these patients are typically characterized by severely compromised blood flow in the peripheral circulation, particularly the digital arteries. Our group has previously reported that resting blood flow through the brachial artery is preserved in SSc (Frech et al., 2015), but resting blood flow in the femoral artery has not been well described. Interestingly, we also observed an elevation in resting LBF in sepsis patients (Nelson et al., 2016) that was no longer present at a 3 day follow-up, likely the result of a subsidence of the initial inflammatory response. Given that inflammation is considered a key component in the pathophysiology of SSc (Fuschiotti, 2016), it is conceivable that aspects of the inflammatory cascade could be contributing to this paradoxical increase in resting LBF in the present study.
Although the plasma free radical levels (as assessed by EPR spectroscopy) were not different between groups (Figure 4), FRAP, a measure of the total antioxidant capacity (TAC) and marker of antioxidant system activation, was greater in SSc patients (Figure 5A). Previous studies that employed this same assay found TAC to be slightly, but not significantly, higher in patients with SSc compared to healthy controls. This observation was contrary to the expected diminution in TAC in these patients, and the authors speculated that these findings may be explained by enhanced antioxidant production in response to the elevated levels of ROS (Firuzi et al., 2006; Riccieri et al., 2008). Similarly, a study using the copper reducing ability of serum to assess total antioxidant power found this to be significantly greater in SSc, and, like the aforementioned studies, suggested this was an indicator of a global response to the oxidative stress associated with SSc (Ogawa, Shimizu, Muroi, Hara, & Sato, 2011). While our findings of elevated FRAP also suggest a generalized response to oxidative challenges in this patient group, a specific endogenous antioxidant enzyme, SOD, was significantly lower in SSc (Figure 5B), which is consistent with other findings (Luo et al., 2016). Taken together, these data suggest that although the global response to oxidative stress may be elevated in patients with SSc, some of their endogenous capacity to deal with oxidative stress may be compromised.
Markers of oxidative damage were greater in the SSc patients. MDA, a by-product of lipid peroxidation, was elevated in SSc patients (Figure 6A), which is in agreement with several studies reporting evidence of lipid peroxidation through MDA (Devrim et al., 2008; Luo et al., 2016) as well peroxide levels (Firuzi et al., 2006; Luo et al., 2016; Riccieri et al., 2008) in this population. We also observed that protein oxidation, indicated by PC, was elevated in SSc (Figure 6B), although some earlier studies did not detect any differences (Firuzi et al., 2006; Riccieri et al., 2008). While direct assessment of plasma free radical concentration (EPR spectroscopy) did not reveal differences between groups, elevated MDA and PC suggest a cumulative effect of intermittent bouts of oxidative stress that produce a footprint of oxidative damage in these patients, in spite of greater total antioxidant capacity (as measured by FRAP).
PLM responses in DU vs non-DU patients
DUs are a debilitating vascular complication in SSc, and a history of DU is associated with more rapid disease progression and increased risk of cardiovascular events and mortality (Mihai et al., 2016). Though the etiology of DU formation remains incompletely understood, it is likely that disease-related alterations in vascular function may play a critical role. Indeed, our group has previously reported that SSc patients with a history of DU demonstrate impaired endothelium-dependent vasodilation, as assessed by FMD, compared to their non-DU SSc counterparts (Frech et al., 2015). During PLM, LBF AUC was significantly attenuated in patients with a history of DU (Figure 7A). As PLM responses are governed by the microvasculature, and DU are associated with structural and functional derangements of these vascular beds, it is perhaps not surprising that the hyperemic response to PLM is markedly attenuated in the DU group. Interestingly, the reduction in LBF AUC appears to be largely attributable to an increase in the retrograde component of net blood flow in the DU group (Figure 7D). While greater anterograde blood flow and shear stress have been associated with improved endothelial function (Naylor et al., 2011; Tinken et al., 2009), greater retrograde or oscillatory blood flow and shear stress have been documented to impair endothelial function and NO bioavailability (Hwang et al., 2003; Schreuder, Green, Hopman, & Thijssen, 2014; Ziegler, Bouzourene, Harrison, Brunner, & Hayoz, 1998). However, in this case we are unable to determine if having a more severe manifestation of the disease is causing the greater retrograde blood flow, or if greater retrograde blood flow in these patients results in the disease following a more deleterious path. Interestingly, plasma markers of inflammation were >2-fold higher in DU vs non-DU patients (Table 4), providing further support that this group has a more severe manifestation of the disease. Given the clinical importance of DU, further studies to determine whether PLM testing may be of prognostic relevance in predicting the development of DUs, as previously demonstrated for FMD (Frech et al., 2015), are warranted.
Limitations
The PLM response is reported as the peak change in LBF from baseline and the LBF AUC normalized to baseline, which accounts for differences in baseline femoral artery blood flow across different populations. While we cannot exclude the possibility that differences in resting LBF in the present study may have influenced PLM responses, this concern is mitigated by the lack of correlation between baseline LBF and both peak change in LBF and LBF AUC. Our group and others have demonstrated that NOS inhibition by L-NMMA administration reduces, but does not completely abolish, the PLM response (Groot, Trinity, et al., 2015; Mortensen et al., 2012; Trinity et al., 2012), suggesting that other vasodilators may contribute to the hyperemic response. We therefore cannot exclude the possibility that the reduced PLM response observed in SSc patients may also be attributable to disease-related changes in non-NO vasodilator pathways. Additionally, it is noteworthy that SSc patients demonstrate structural remodeling of the vasculature, including narrowing or occlusion of small arteries (Luders et al., 2017) and changes in capillary morphology and density (Pizzorni et al., 2016). Given that PLM elicits vasodilation of these smaller vessels and capillary beds, it is possible that these structural deformations may be limiting, at least in part, the ability of SSc patients to augment blood flow in response to passive movement. Further study is needed to determine if the attenuated PLM response in SSc is a functional limitation arising from maladaptations in NO signaling, changes in other vasodilatory signaling mechanisms, or is the result of structural limitations caused by luminal narrowing and capillary rarefication.
Conclusions
The hyperemic response to PLM is significantly attenuated in SSc, supporting the presence of impaired NO-mediated vascular function in these patients, with a more severe decrement in patients with a history of DU. Moreover, the identification of disease-specific changes in several key indicators of systemic inflammation, antioxidant capacity, and oxidative stress, provide evidence of a deleterious vascular milieu that likely plays an important role in the vascular pathophysiology of this patient group. However, the absence of significant correlations between individual biomarkers and decrements in the blood flow responses suggests that the vascular dysfunction observed in SSc may not be solely the result of derangements in the redox balance or inflammatory signaling.
NEW FINDINGS.
What is the central question of this study?
Do systemic sclerosis (SSc) patients exhibit impaired nitric oxide (NO)-mediated vascular function of the lower limb, and are these decrements correlated with plasma biomarkers for inflammation and oxidative stress?
What is the main finding and its importance?
Findings indicate impaired NO-mediated vascular function, linked to the incidence of digital ulcers, and a milieu of inflammation and oxidative stress. However, the absence of significant correlations between individual biomarkers and blood flow responses suggests the vasculopathy observed in SSc may not be solely the result of derangements in the redox balance or inflammatory signaling.
Acknowledgments
Funding.
Funded in part by grant from the National Institutes of Health (HL118313, K23AR067889, R01AG050238, K02AG045339) and the U.S. Department of Veterans Affairs (RX001311, RX001697, CX001183).
Footnotes
Competing Interests.
None declared.
Author contributions.
All data acquisition took place in the Utah Vascular Research Laboratory located in the Salt Lake City Department of Veterans Affairs Medical Center. HLC, DRM, TMF, AJD, RSR, and DWW contributed to the concept or design of the work; HLC, DRM, HJG, TMF, AJD, and DWW contributed to the acquisition, analysis, or interpretation of data; and HLC, DRM, HJG, TMF, AJD, RSR, and DWW contributed to drafting the work or revising it critically for important intellectual content. All authors approved of the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as coauthors qualify for authorship and all those who qualify for authorship are listed as coauthors.
References
- Bartoli F, Blagojevic J, Bacci M, Fiori G, Tempestini A, Conforti ML, Cerinic MM. Flow-mediated vasodilation and carotid intima-media thickness in systemic sclerosis. Ann N Y Acad Sci. 2007;1108:283–290. doi: 10.1196/annals.1422.030. [DOI] [PubMed] [Google Scholar]
- Benzie IF, Strain JJ. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Anal Biochem. 1996;239(1):70–76. doi: 10.1006/abio.1996.0292. [DOI] [PubMed] [Google Scholar]
- Cotton SA, Herrick AL, Jayson MI, Freemont AJ. Endothelial expression of nitric oxide synthases and nitrotyrosine in systemic sclerosis skin. J Pathol. 1999;189(2):273–278. doi: 10.1002/(SICI)1096-9896(199910)189:2<273::AID-PATH413>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Cypiene A, Laucevicius A, Venalis A, Dadoniene J, Ryliskyte L, Petrulioniene Z, Gintautas J. The impact of systemic sclerosis on arterial wall stiffness parameters and endothelial function. Clin Rheumatol. 2008;27(12):1517–1522. doi: 10.1007/s10067-008-0958-1. [DOI] [PubMed] [Google Scholar]
- Devrim E, Erten S, Erguder IB, Namuslu M, Turgay M, Durak I. Malondialdehyde and nitric oxide levels in erythrocytes from patients with systemic sclerosis. Med Princ Pract. 2008;17(4):349–350. doi: 10.1159/000129620. [DOI] [PubMed] [Google Scholar]
- Donato AJ, Black AD, Jablonski KL, Gano LB, Seals DR. Aging is associated with greater nuclear NF kappa B, reduced I kappa B alpha, and increased expression of proinflammatory cytokines in vascular endothelial cells of healthy humans. Aging Cell. 2008;7(6):805–812. doi: 10.1111/j.1474-9726.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Sueta G, Radi R. Chemical biology of peroxynitrite: kinetics, diffusion, and radicals. ACS Chem Biol. 2009;4(3):161–177. doi: 10.1021/cb800279q. [DOI] [PubMed] [Google Scholar]
- Firuzi O, Fuksa L, Spadaro C, Bousova I, Riccieri V, Spadaro A, Saso L. Oxidative stress parameters in different systemic rheumatic diseases. J Pharm Pharmacol. 2006;58(7):951–957. doi: 10.1211/jpp.58.7.0010. [DOI] [PubMed] [Google Scholar]
- Frech T, Walker AE, Barrett-O’Keefe Z, Hopkins PN, Richardson RS, Wray DW, Donato AJ. Systemic sclerosis induces pronounced peripheral vascular dysfunction characterized by blunted peripheral vasoreactivity and endothelial dysfunction. Clin Rheumatol. 2015;34(5):905–913. doi: 10.1007/s10067-014-2834-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuschiotti P. Current perspectives on the immunopathogenesis of systemic sclerosis. Immunotargets Ther. 2016;5:21–35. doi: 10.2147/ITT.S82037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielli A, Svegliati S, Moroncini G, Amico D. New insights into the role of oxidative stress in scleroderma fibrosis. Open Rheumatol J. 2012;6:87–95. doi: 10.2174/1874312901206010087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groot HJ, Rossman MJ, Trinity JD, Layec G, Ives SJ, Richardson RS. Passive leg movement-induced vasodilation in women: the impact of age. Am J Physiol Heart Circ Physiol. 2015;309(5):H995–H1002. doi: 10.1152/ajpheart.00422.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groot HJ, Trinity JD, Layec G, Rossman MJ, Ives SJ, Morgan DE, Richardson RS. The role of nitric oxide in passive leg movement-induced vasodilatation with age: insight from alterations in femoral perfusion pressure. J Physiol. 2015;593(17):3917–3928. doi: 10.1113/JP270195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groot HJ, Trinity JD, Layec G, Rossman MJ, Ives SJ, Richardson RS. Perfusion pressure and movement-induced hyperemia: evidence of limited vascular function and vasodilatory reserve with age. Am J Physiol Heart Circ Physiol. 2013;304(4):H610–619. doi: 10.1152/ajpheart.00656.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein MR, Hassan HI, Hofny ER, Elkholy M, Fatehy NA, Abd Elmoniem AE, Rashed HG. Alterations of mononuclear inflammatory cells, CD4/CD8+ T cells, interleukin 1beta, and tumour necrosis factor alpha in the bronchoalveolar lavage fluid, peripheral blood, and skin of patients with systemic sclerosis. J Clin Pathol. 2005;58(2):178–184. doi: 10.1136/jcp.2004.019224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J, Saha A, Boo YC, Sorescu GP, McNally JS, Holland SM, Jo H. Oscillatory shear stress stimulates endothelial production of O2- from p47phox-dependent NAD(P)H oxidases, leading to monocyte adhesion. J Biol Chem. 2003;278(47):47291–47298. doi: 10.1074/jbc.M305150200. [DOI] [PubMed] [Google Scholar]
- Incalza MA, D’Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol. 2017 doi: 10.1016/j.vph.2017.05.005. [DOI] [PubMed] [Google Scholar]
- Ives SJ, Harris RA, Witman MA, Fjeldstad AS, Garten RS, McDaniel J, Richardson RS. Vascular dysfunction and chronic obstructive pulmonary disease: the role of redox balance. Hypertension. 2014;63(3):459–467. doi: 10.1161/HYPERTENSIONAHA.113.02255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111(8):1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luders S, Friedrich S, Ohrndorf S, Glimm AM, Burmester GR, Riemekasten G, Backhaus M. Detection of severe digital vasculopathy in systemic sclerosis by colour Doppler sonography is associated with digital ulcers. Rheumatology (Oxford) 2017;56(11):1865–1873. doi: 10.1093/rheumatology/kex045. [DOI] [PubMed] [Google Scholar]
- Luo JY, Liu X, Jiang M, Zhao HP, Zhao JJ. Oxidative stress markers in blood in systemic sclerosis: A meta-analysis. Mod Rheumatol. 2016:1–9. doi: 10.1080/14397595.2016.1206510. [DOI] [PubMed] [Google Scholar]
- Matucci-Cerinic M, Kahaleh B, Wigley FM. Review: evidence that systemic sclerosis is a vascular disease. Arthritis Rheum. 2013;65(8):1953–1962. doi: 10.1002/art.37988. [DOI] [PubMed] [Google Scholar]
- Mihai C, Landewe R, van der Heijde D, Walker UA, Constantin PI, Gherghe AM, co-authors, E Digital ulcers predict a worse disease course in patients with systemic sclerosis. Ann Rheum Dis. 2016;75(4):681–686. doi: 10.1136/annrheumdis-2014-205897. [DOI] [PubMed] [Google Scholar]
- Mok MY, Fung PC, Ooi C, Tse HF, Wong Y, Lam YM, Lau CS. Serum nitric oxide metabolites and disease activity in patients with systemic sclerosis. Clin Rheumatol. 2008;27(3):315–322. doi: 10.1007/s10067-007-0708-9. [DOI] [PubMed] [Google Scholar]
- Mortensen SP, Askew CD, Walker M, Nyberg M, Hellsten Y. The hyperaemic response to passive leg movement is dependent on nitric oxide: a new tool to evaluate endothelial nitric oxide function. J Physiol. 2012;590(17):4391–4400. doi: 10.1113/jphysiol.2012.235952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor LH, Carter H, FitzSimons MG, Cable NT, Thijssen DH, Green DJ. Repeated increases in blood flow, independent of exercise, enhance conduit artery vasodilator function in humans. Am J Physiol Heart Circ Physiol. 2011;300(2):H664–669. doi: 10.1152/ajpheart.00985.2010. [DOI] [PubMed] [Google Scholar]
- Nelson AD, Rossman MJ, Witman MA, Barrett-O’Keefe Z, Groot HJ, Garten RS, Richardson RS. Nitric oxide-mediated vascular function in sepsis using passive leg movement as a novel assessment: a cross-sectional study. J Appl Physiol (1985) 2016;120(9):991–999. doi: 10.1152/japplphysiol.00961.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa F, Shimizu K, Muroi E, Hara T, Sato S. Increasing levels of serum antioxidant status, total antioxidant power, in systemic sclerosis. Clin Rheumatol. 2011;30(7):921–925. doi: 10.1007/s10067-011-1695-4. [DOI] [PubMed] [Google Scholar]
- Pehlivan Y, Onat AM, Ceylan N, Turkbeyler IH, Buyukhatipoglu H, Comez G, Tarakcioglu M. Serum leptin, resistin and TNF-alpha levels in patients with systemic sclerosis: the role of adipokines in scleroderma. Int J Rheum Dis. 2012;15(4):374–379. doi: 10.1111/j.1756-185X.2012.01755.x. [DOI] [PubMed] [Google Scholar]
- Pizzorni C, Sulli A, Smith V, Llado A, Paolino S, Cutolo M, Ruaro B. Capillaroscopy 2016: new perspectives in systemic sclerosis. Acta Reumatol Port. 2016;41(1):8–14. [PubMed] [Google Scholar]
- Pyke K, Green DJ, Weisbrod C, Best M, Dembo L, O’Driscoll G, Tschakovsky M. Nitric oxide is not obligatory for radial artery flow-mediated dilation following release of 5 or 10 min distal occlusion. Am J Physiol Heart Circ Physiol. 2010;298(1):H119–126. doi: 10.1152/ajpheart.00571.2009. [DOI] [PubMed] [Google Scholar]
- Riccieri V, Spadaro A, Fuksa L, Firuzi O, Saso L, Valesini G. Specific oxidative stress parameters differently correlate with nailfold capillaroscopy changes and organ involvement in systemic sclerosis. Clin Rheumatol. 2008;27(2):225–230. doi: 10.1007/s10067-007-0769-9. [DOI] [PubMed] [Google Scholar]
- Rollando D, Bezante GP, Sulli A, Balbi M, Panico N, Pizzorni C, Ghio M. Brachial artery endothelial-dependent flow-mediated dilation identifies early-stage endothelial dysfunction in systemic sclerosis and correlates with nailfold microvascular impairment. J Rheumatol. 2010;37(6):1168–1173. doi: 10.3899/jrheum.091116. [DOI] [PubMed] [Google Scholar]
- Rossi P, Granel B, Marziale D, Le Mee F, Frances Y. Endothelial function and hemodynamics in systemic sclerosis. Clin Physiol Funct Imaging. 2010;30(6):453–459. doi: 10.1111/j.1475-097X.2010.00965.x. [DOI] [PubMed] [Google Scholar]
- Schreuder TH, Green DJ, Hopman MT, Thijssen DH. Acute impact of retrograde shear rate on brachial and superficial femoral artery flow-mediated dilation in humans. Physiol Rep. 2014;2(1):e00193. doi: 10.1002/phy2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange G, Nash P. The manifestations of vasculopathy in systemic sclerosis and its evidence-based therapy. Int J Rheum Dis. 2009;12(3):192–206. doi: 10.1111/j.1756-185X.2009.01410.x. [DOI] [PubMed] [Google Scholar]
- Szucs G, Timar O, Szekanecz Z, Der H, Kerekes G, Szamosi S, Soltesz P. Endothelial dysfunction precedes atherosclerosis in systemic sclerosis–relevance for prevention of vascular complications. Rheumatology (Oxford) 2007;46(5):759–762. doi: 10.1093/rheumatology/kel426. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Asano Y, Amiya E, Hatano M, Tamaki Z, Takata M, Sato S. Clinical correlation of brachial artery flow-mediated dilation in patients with systemic sclerosis. Mod Rheumatol. 2013 doi: 10.1007/s10165-013-0867-2. [DOI] [PubMed] [Google Scholar]
- Thijssen DH, Rowley N, Padilla J, Simmons GH, Laughlin MH, Whyte G, Green DJ. Relationship between upper and lower limb conduit artery vasodilator function in humans. J Appl Physiol (1985) 2011;111(1):244–250. doi: 10.1152/japplphysiol.00290.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinken TM, Thijssen DH, Hopkins N, Black MA, Dawson EA, Minson CT, Green DJ. Impact of shear rate modulation on vascular function in humans. Hypertension. 2009;54(2):278–285. doi: 10.1161/HYPERTENSIONAHA.109.134361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo JO, Moraes CF, Souza VC, Tonet-Furioso AC, Afonso LC, Cordova C, Nobrega OT. Tailored antihypertensive drug therapy prescribed to older women attenuates circulating levels of interleukin-6 and tumor necrosis factor-alpha. Clin Interv Aging. 2015;10:209–215. doi: 10.2147/CIA.S74790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinity JD, Groot HJ, Layec G, Rossman MJ, Ives SJ, Runnels S, Richardson RS. Nitric oxide and passive limb movement: a new approach to assess vascular function. J Physiol. 2012;590(6):1413–1425. doi: 10.1113/jphysiol.2011.224741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, Pope JE. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013;65(11):2737–2747. doi: 10.1002/art.38098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witman MA, Fjeldstad AS, McDaniel J, Ives SJ, Zhao J, Barrett-O’Keefe Z, Richardson RS. Vascular function and the role of oxidative stress in heart failure, heart transplant, and beyond. Hypertension. 2012;60(3):659–668. doi: 10.1161/HYPERTENSIONAHA.112.193318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witman MA, Ives SJ, Trinity JD, Groot HJ, Stehlik J, Richardson RS. Heart failure and movement-induced hemodynamics: partitioning the impact of central and peripheral dysfunction. Int J Cardiol. 2015;178:232–238. doi: 10.1016/j.ijcard.2014.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray DW, Nishiyama SK, Harris RA, Zhao J, McDaniel J, Fjeldstad AS, Richardson RS. Acute reversal of endothelial dysfunction in the elderly after antioxidant consumption. Hypertension. 2012;59(4):818–824. doi: 10.1161/HYPERTENSIONAHA.111.189456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray DW, Witman MA, Ives SJ, McDaniel J, Trinity JD, Conklin JD, Richardson RS. Does brachial artery flow-mediated vasodilation provide a bioassay for NO? Hypertension. 2013;62(2):345–351. doi: 10.1161/HYPERTENSIONAHA.113.01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Katayama I, Nishioka K. Nitric oxide production and inducible nitric oxide synthase expression in systemic sclerosis. J Rheumatol. 1998;25(2):314–317. [PubMed] [Google Scholar]
- Ziegler T, Bouzourene K, Harrison VJ, Brunner HR, Hayoz D. Influence of oscillatory and unidirectional flow environments on the expression of endothelin and nitric oxide synthase in cultured endothelial cells. Arterioscler Thromb Vasc Biol. 1998;18(5):686–692. doi: 10.1161/01.atv.18.5.686. [DOI] [PubMed] [Google Scholar]