

Abstract
This study evaluated the effects of elevated homocysteine (Hcy) on the oxidative stress response in retinal Müller glial cells. Elevated Hcy has been implicated in retinal diseases including glaucoma and optic neuropathy, which are characterized by retinal ganglion cell (RGC) loss. To understand the mechanisms of Hcy-induced RGC loss, in vitro and in vivo models have been utilized. In vitro isolated RGCs are quite sensitive to elevated Hcy levels, while in vivo murine models of hyperhomocysteinemia (HHcy) demonstrate a more modest RGC loss (~20%) over a period of many months. This differential response to Hcy between isolated cells and the intact retina suggests that the retinal milieu invokes mechanisms that buffer excess Hcy. Oxidative stress has been implicated as a mechanism of Hcy-induced neuron loss and NRF2 is a transcription factor that plays a major role in regulating cytoprotective responses to oxidative stress. In the present study we investigated whether HHcy upregulates NRF2-mediated stress responses in Müller cells, the chief retinal glial cell responsible for providing trophic support to retinal neurons. Primary Müller cells were exposed to L-Hcy-thiolactone [50μM–10mM] and assessed for viability, reactive oxygen species (ROS), and glutathione (GSH) levels. Gene/protein levels of Nrf2 and levels of NRF2-regulated antioxidants (NQO1, CAT, SOD2, HMOX1, GPX1) were assessed in Hcy-exposed Müller cells. Unlike isolated RGCs, isolated Müller cells are viable over a wide range of Hcy concentrations [50μM – 1mM]. Moreover, when exposed to elevated Hcy, Müller cells demonstrate decreased oxidative stress and decreased ROS levels. GSH levels increased by ~20% within 24h exposure to Hcy. Molecular analyses revealed 2-fold increase in Nrf2 expression. Expression of antioxidant genes Nqo1, Cat, Sod2, Hmox1, Gpx1 increased significantly. The consequences of Hcy exposure were evaluated also in Müller cells harvested from Nrf2−/− mice. In contrast to WT Müller cells, in which oxidative stress decreased upon exposure to Hcy, the Nrf2−/− Müller cells showed a significant increase in oxidative stress. Our data suggest that at least during early stages of Hhcy, a cytoprotective response may be in place, mediated in part by NRF2 in Müller cells.
Keywords: retinal glial cells, exfoliative glaucoma, optic neuropathy, oxidative stress, mouse, hyperhomocysteinemia
Graphical Abstract
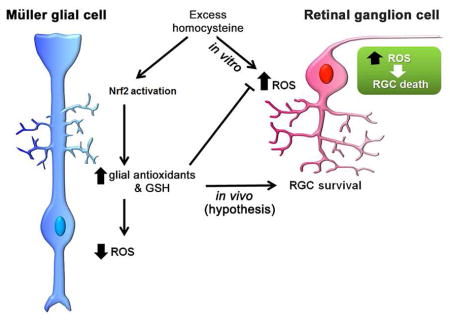
Introduction
This study addresses the consequences in retinal Müller glial cells of exposure to excess homocysteine (Hcy). Hcy is an excitatory non-proteinogenic amino acid formed by the demethylation of methionine. It sits at the intersection of the remethylation and transsulfuration pathways (Ubbink et al, 1996; Selhub, 1999). Enzymes involved in the remethylation pathway include methylene tetrahydrofolate reductase (MTHFR) and methionine synthase, which in the presence of folate and vitamin B12, convert Hcy to methionine used in DNA methyltransfer reactions. Enzymes involved in the transsulfuration pathway include cystathionine-β-synthase (CBS) and cystathionine-γ-lyase, which in the presence of vitamin B6 form cysteine used to produce glutathione (GSH), hydrogen sulfide and taurine. Dietary deficiency of folate or B vitamins can lead to elevated plasma Hcy, a condition termed hyperhomocysteinemia (Hhcy) (Clarke et al, 1991; Jacques et al, 1996; Cantu et al, 2004). Deficiency of the aforementioned enzymes is a common cause of Hhcy, with the most frequent mutation involving MTHFR (677 C→T) (Leclerc et al, 2005). There is a significant association between Hhcy, neurodegenerative and cardiovascular diseases (Clarke et al, 1991; Boushey et al, 1995; Duan et al, 2002; Seshadri et al, 2002; Obeid and Herrmann, 2006; Religa et al, 2006).
Retina is a neurovascular tissue prompting interest in the role of Hhcy in retinal disease (Ajith and Ranimenon, 2015). Several studies implicate Hhcy in the pathogenesis of retinopathies involving retinal ganglion cells (RGCs) such as exfoliation glaucoma (Leibovitch et al, 2003; Bleich et al, 2004; Puustjarvi, et al, 2004; Roedl et al, 2007), which is the most common secondary form of glaucoma worldwide (Ritch, 1994). The exact role of Hhcy in this disease, remains to be determined (Xu et al, 2012; Li et al, 2016; Pasquale et al, 2016). In at least two murine models of Hhcy, there is significant reduction of RGCs and compromised visual function. In mice with deficiency of Mthfr, there is ~20% loss of RGCs in mice at 24 wks, decreased NFL thickness and reduced positive scotopic threshold response (Markand et al, 2015). In mice with deficiency of Cbs, there is also ~20% loss of RGCs and delayed visual evoked potentials in mice by 30 wks (Ganapathy et al, 2009; Yu et al, 2012). Administration of Hcy intravitreally in rodents induces RGC loss, the severity of which is dose-dependent (Moore et al, 2001; Chang et al, 2011).
Efforts to determine the mechanism by which Hhcy preferentially induces RGC loss have utilized primary RGC culture as a model system (Ganapathy et al, 2011a; Ganapathy et al, 2011b). ~50–60% of RGCs, purified by immunopanning, die within 18 h exposure to 50μM Hcythiolactone (Dun et al, 2007), whereas in the endogenous (in vivo) models, the loss of RGCs is more modest (~20%). This observation suggests that in vivo there are cellular and molecular mechanisms that buffer excess Hcy and dampen the deleterious consequences of moderate Hhcy to RGCs. We hypothesize that retinal Müller cells play a role in this process. Müller cells are the principal retinal glial cells; they maintain homeostasis by providing trophic support to retinal neurons including RGCs (Bringmann et al, 2006; Bringmann et al, 2009).
Evidence from studies of several cell types, including neurons, suggests that oxidative stress is a major mechanism by which Hhcy induces cellular damage (Kruman et al, 2000; Ho et al, 2001; Bhattacharjee et al, 2016). In response to oxidative stress, Müller cells upregulate the gene encoding nuclear factor erythroid 2-related factor 2 (NRF2), which is an important antioxidant molecule that regulates transcription of more than 500 antioxidant/cytoprotective genes (Sporn and Liby, 2012; Gorrini et al, 2013). There have been no studies of the effects of Hhcy on Müller cells. Here we evaluated the effects of Hhcy on the viability of primary retinal Müller cells and analyzed the oxidative stress response of Müller cells to excess Hcy, specifically focusing on its effects related to NRF2.
Materials and methods
Animals, cell culture and Hcy treatment
C57Bl/6J mice were the source of retinal cells used in this study. The mice were the offspring of our breeding colony. Original breeding pairs, obtained from the Jackson Laboratories (Bar Harbor, ME), were maintained and their offspring utilized according to our IACUC approved protocol, which is consistent with the NIH guide for care and use of laboratory animals and complies with ARRIVE guidelines. Müller cells were also isolated from Nrf2−/− mice, generated on the C57BL/6 background. Details about Nrf2−/− mice have been described (Kaidery et al, 2013).
Isolation of Müller cells from P6-7 day old pups followed our published protocol (Jiang et al, 2006; Mysona et al, 2009; Wang et al, 2015) based on the method of Hicks and Cortois (1990). The decision to isolate cells from mice at this age was based upon 3H-thymidine labeling studies performed in mouse retina demonstrating the significant number of Müller cells present at this time period (Young, 1985). Cells were used at passage 4–5. Earlier studies from our lab confirmed purity of the cells using this isolation method (Jiang et al, 2006; Mysona et al, 2009; Wang et al, 2015) and were repeated in the current study. Regarding primary RGCs, the isolation of cells was performed using retinas of mouse pups (P1–P5) following our published protocol (Dun et al, 2006; Dun et al, 2007; Dun et al, 2008; Ganapathy et al, 2011a; Ganapathy et al, 2011b) that is based upon a two-step immunopanning procedure (Barres et al, 1998). The experiments were conducted on day 3 following isolation.
Treatment of cells with Hcy used L-homocysteine thiolactone hydrochloride. Hcy treatment stocks and dilutions were made fresh for each experiment. Hcy and all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise specified.
Assessment of cell purity
Cells were seeded on coverslips and grown overnight. They were fixed with 4% paraformaldehyde, washed with PBS-Triton X-100, and incubated with Power Block (BioGenex, Fremont, CA). Cells were incubated overnight at 4°C with primary antibodies to validate the identity of the primary Müller cell cultures (Hu et al, 2014) including vimentin, glutamine synthetase (GS), cellular retinaldehyde binding protein (CRALBP) (Table 1). Additionally, they were incubated with primary antibodies that are specific for neurons (neuronal nuclei, NeuN) and retinal pigment epithelial cells (RPE65). RGC purity was confirmed using the neuronal marker Brn3a (Sajgo et al, 2017) (Table 1). Cells were washed with PBS-Triton X-100 and incubated with the appropriate secondary antibodies (Table 1) for 1 h at 37°C. Cells were washed with PBS-Triton X-100 and mounted on slides using Fluoroshield with DAPI (4,6-diamidino-2-phenylindole, dihydrochloride) (Sigma-Aldrich) to label nuclei. Immunofluorescence images were obtained using either an Axioplan-2 fluorescent microscope (Carl Zeiss, Göttingen, Germany) equipped with an HRM camera or a Zeiss LSM 780 upright confocal microscope. Immunofluorescent images were processed using Zeiss Axiovision digital image processing software provided with the microscopes. Fluorescence intensity was quantified using ImageJ software (version 1.48, http://imagej.nih.gov/ij/, NIH).
Table 1.
Antibodies used in immunoblotting and immunocytochemical studies.
| Antibody | Supplier | Dilution |
|---|---|---|
| Primary antibodies | ||
| Rabbit anti-GFAP (Z0334) | Dako, Carpentaria, CA | 1:500 |
| Rabbit anti-NRF2 (SC722) | Santa Cruz Corp., Santa Cruz CA | 1:500 |
| Rabbit anti-NRF2 (ab31163) | Abcam, Cambridge, MA | 1:500 |
| Rabbit anti-HMOX1 (SC-10789) | Santa Cruz Corp., Santa Cruz CA | 1:500 |
| Rabbit anti-Catalase (SC-50508) | Santa Cruz Corp., Santa Cruz CA | 1:1000 |
| Rabbit anti-HDAC1 (SC-7872) | Santa Cruz Corp., Santa Cruz CA | 1:1000 |
| Mouse anti-GAPDH (MAB374) | EMD Millipore, Temecula, CA | 1:5000 |
| Goat anti-Vimentin (AB1620) | Chemicon Intl., Temecula, CA | 1:100 |
| Mouse anti-RPE65 (ab13826) | Abcam, Cambridge, MA | 1:500 |
| Goat anti-Brn3a C20 (SC31984) | Santa Cruz Corp., Santa Cruz CA | 1:200 |
| Mouse anti-NeuN (MAB377) | Chemicon Intl., Temecula, CA | 1:100 |
| Mouse anti-CRALBP (NB100-74392) | Novus Biologicals, Littleton, CO | 1:50 |
| Rabbit anti Glutamine synthetase (FL-373) | Santa Cruz Corp., Santa Cruz CA | 1:50 |
| Secondary Antibodies | ||
| Goat anti-Rabbit IgG-HRP (SC-2004) | Santa Cruz Corp., Santa Cruz CA | 1:10000 |
| Goat anti-Mouse IgG-HRP (SC-2005) | Santa Cruz Corp., Santa Cruz CA | 1:10000 |
| Alexa Fluor 488 anti-Rabbit IgG (H+L) | Invitrogen Molecular Probes, NY | 1:1000 |
| Alexa Fluor 555 anti-Mouse IgG (H+L | Invitrogen Molecular Probes, NY | 1:1000 |
| Alexa Fluor 488 anti-Goat IgG (H+L) | Invitrogen Molecular Probes, NY | 1:1000 |
| Alexa Fluor 555 anti-Goat IgG (H+L) | Invitrogen Molecular Probes, NY | 1:1000 |
| Alexa Flour 488 anti-Mouse IgG(H+L) | Invitrogen Molecular Probes, NY | 1:1000 |
| Alexa Fluor 555 anti-Rabbit IgG (H+L) | Invitrogen Molecular Probes, NY | 1:1000 |
Assessment of cellular viability
The effects of Hcy on cell viability were assessed using PrestoBlue Viability reagent (Invitrogen, Carlsbad, CA). Cells were incubated 24 h in media containing [12.5-50μM] Hcy for primary RGCs; [50μM-10mM] Hcy for primary Müller cells. Cells were then incubated 30 min in PrestoBlue reagent. The resazurin-based cell permeant dye penetrates viable cells and the reducing environment of these cells modifies the dye to yield a highly fluorescent red color. The fluorescence was detected using a plate reader (Molecular Devices, Sunnyvale, CA) at 570nm excitation/590nm emission.
Assessment of oxidative stress
Oxidative stress was detected in primary Müller cells using 2’,7’ –dichlorofluorescein diacetate (DCFDA) (Abcam, Cambridge, MA). The cells were grown in black-sided, clear-bottom 96-well plates and incubated with DCFDA reagent [40μM]. DCFDA is a cell permeant fluorogenic dye, which can detect hydroxyl, peroxyl and other reactive oxygen species (ROS) activity within cells. After 45 min incubation in DCFDA, cells were washed and treated with Hcy [50μM – 10mM] for 3 h or 24 h. Cells were then subjected to fluorescence detection using a plate reader at 485nm excitation/535nm emission. Tertiary-butyl hydroperoxide (tBHP) [55μM] treatment for 3h was used as the positive control.
We confirmed these results using CellROX Green (Thermo Fisher Scientific, Waltham, MA), a cell permeant dye, which upon oxidation binds to DNA yielding a green fluorescent image; the signal localizes mainly to mitochondria and nuclei. Primary Müller cells were grown overnight on coverslips in 24 well plates. After Hcy treatment [50μM – 10 mM] for 3h or 24h, the medium was aspirated and cells were incubated 30 min with the CellROX reagent [5μM], after which the dye was removed by aspiration, cells were washed with PBS and fixed with 4% paraformaldehyde (PFA, Electron Microscopy Science, Hatfield, PA). The coverslips were mounted on microscope slides using DAPI mounting medium and images were captured using the Zeiss LSM 780 upright confocal microscope. Fluorescence intensity was quantified using ImageJ software (NIH). As a positive control, Müller cells were exposed 30 min to 100μM menadione, which increases ROS and reduces mitochondrial NADPH levels (Criddle et al, 2006).
Assessment of intracellular GSH levels
We used the Glutathione (GSSG/GSH) Detection Kit (ADI-900–160, Enzo Life Sciences, Farmingdale, NY) to detect total, oxidized and reduced GSH in Hcy-treated primary Müller cells. Cells were grown in 6 well plates. After Hcy treatments [50μm, 1mM] for 24 h, cells were trypsinized, washed with PBS, and suspended in 5% (w/v) Metaphosphoric acid. After sonication, cell lysates were centrifuged. To the supernatant, glutathione reductase, provided in the kit, was added to reduce GSSG to GSH. The sulfhydryl group of GSH, thus formed, reacts with 5, 5’-dithiobis-2-nitrobenzoic acid (Ellman’s reagent) to produce a yellow colored 5-thio-2-nitrobenzoic acid (TNB). The reaction kinetics was determined by measuring absorbance at 405nm and readings were taken at 1 min intervals for 10 min. Serially diluted GSSG or oxidized glutathione was used to prepare a standard curve. Similarly, oxidized GSH or GSSG in the samples were detected by adding 4-vinyl pyridine, which blocks any free thiols present in the reaction. The absorbance readings were taken at 405nm using a plate reader.
Assessment of gene expression
Müller cells were incubated with Hcy [50μM, 1mM] 3-20 h and were used to analyze genes involved in the Nrf2 antioxidant pathway (Nrf2, Hmox1, Nqo1, Cat, Sod2). RNA was prepared using Trizol (Invitrogen). 2 μg RNA was converted to cDNA using iScript Reverse Transcription Supermix (Bio-Rad Laboratories, Hercules, CA). Primer pairs used for the genes analyzed were obtained from Integrated DNA Technologies (Coralville, IA) and are listed in Table 2. qRT PCR was performed using SsoAdvanced SYBR Green Supermix from Bio-Rad (Hercules, CA) and the Bio-Rad CFX96-Real Time System Thermal Cycler (Hercules, CA). PCR was performed at 95°C for 10s and 56–57°C for 30s; melt curve analysis confirmed the purity of the end products. Relative mRNA levels were normalized to that of 18S. Expression levels were detected by comparing the Ct values (ΔΔCt) (Livak and Schmittgen, 2001). The analyses were performed in triplicate.
Table 2.
List of primers used for qRT-PCR analysis.
| Accession | Species | Gene | Primer | Sequence |
|---|---|---|---|---|
| NM_010902.3 | Mouse | Nrf2 | Forward | 5’AGCCCCATTCACAAAAGACA-3’ |
| Reverse | 5’-GAAGTCATCAACAGGGAGGTTA-3’ | |||
| NM_010442.2 | Mouse | Hmox1 | Forward | 5’-GTCAAGCACAGGGTGACAGA-3’ |
| Reverse | 5’-ATCACCTGCAGCTCCTCAAA-3’ | |||
| NM_008706.5 | Mouse | Nqo1 | Forward | 5’-AGCGTTCGGTATTACGATCC-3’ |
| Reverse | 5’-AGTACAATCAGGGCTCTTCTCG-3’ | |||
| NM_013671.3 | Mouse | Sod2 | Forward | 5’-GACCCATTGCAAGGAACAA-3’ |
| Reverse | 5’-GTAGTAAGCGTGCTCCCACAC-3’ | |||
| NR_003278.1 | Mouse | 18S rRNA | Forward | 5’-GCAATTATTCCCCATGAACG-3’ |
| Reverse | 5’-GGGACTTAATCAACGCAAGC-3’ | |||
| NM_008160.6 | Mouse | Gpx1 | Forward | 5’-TGCAATCAGTTCGGACACCA-3’ |
| Reverse | 5’-AAGGTAAAGAGCGGGTGAGC-3’ | |||
| NM_009804.2 | Mouse | Catalase | Forward | 5’-CCAGATACTCCAAGGCAAAGGT-3’ |
| Reverse | 5’-CGAGGGTCACGAACTGTGTC-3’ |
Assessment of protein levels
Cells were incubated with Hcy [50μM, 1mM] 3-20 h, after which they were lysed in a buffer containing 50mM Tris HCl (pH 7.4), 5mM EDTA, 150mM NaCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1% SDS and 1x protease-phosphatase inhibitor cocktail (Thermo Fisher Scientific). 50–60 μg of protein were subjected to SDS polyacrylamide gel electrophoresis at 100 volts and transferred to nitrocellulose membranes at 90 volts for 1.5 h. Membranes were blocked in 5% milk in TBST (1h RT) followed by incubation with primary antibodies (Table 1) in 5% milk in TBST buffer at 4°C overnight. Membranes were washed three times for 5 min in TBST and incubated with HRP-conjugated goat anti-rabbit or anti-mouse IgG antibody (Table 1) for 1 h at RT. The ECL Western blot detection system (Thermo Fisher Scientific) was used to visualize protein bands. Membranes were reprobed with anti-GAPDH (Millipore, Temecula, CA), which served as the loading control. To analyze nuclear versus cytoplasmic proteins, Müller cells were exposed to Hcy [50μM, 1mM] for 4 h. Proteins were extracted using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific) per the manufacturer’s protocol. Fraction purity was confirmed using anti-HDAC1 and anti-GAPDH for the nuclear and cytoplasmic fractions, respectively (Table 1). Protein band densities were quantified using ImageJ software and confirmed using Quantity One Software (Bio-Rad Laboratories). All immunoblot images represent three or more independent experiments.
Data analysis
The data were subjected to statistical analysis using Prism 6 for Windows Version 6.01 statistical analysis software (GraphPad, La Jolla, CA). All values are reported as mean ± SEM. Data were either analyzed by Kruskal-Wallis non-parametric test followed by Dunn’s multiple comparison as the post-hoc test or were subjected to D’Agostino and Pearson Omnibus Normality test and then, as appropriate, either one-way ANOVA followed by Dunnett’s test or two-way ANOVA (to compare multiple parameters) followed by Sidak’s post-hoc comparison test. For all analyses p < .05 was considered statistically significant.
Results
Confirmation that primary RGCs are sensitive to Hhcy
Prior to investigating effects of excess Hcy on Müller glial cells, we confirmed the sensitivity of primary RGCs to Hhcy. Fig. 1A-C shows isolated RGCs, the cells are positive for the RGC marker Brn3 (Fig. 1A) and they are negative for the glial cell marker GFAP (Fig. 1B). The neurite processes of the cells are visible by DIC microscopy (Fig. 1C). RGCs were incubated with Hcy over a concentration range [12.5 – 50μM] and viability was detected using PrestoBlue. Primary RGCs showed ~40% cell death within 24h exposure to 50μM (Fig. 1D). We then examined a shorter time period of Hcy exposure to determine whether there was a detectable increase in oxidative stress in RGCs incubated for only three hours with increasing Hcy concentrations [25, 50, 100μM]. Within 3h there was a significant increase in DCFDA fluorescence in cells exposed to 100μM Hcy (Fig. 1E). The data confirm our previously published findings that primary RGCs are very sensitive to Hcy exposure leading to decreased viability and increased oxidative stress (Ganapathy et al, 2011a; Ganapathy et al, 2011b; Dun et al, 2007).
Fig 1. Confirmation of the sensitivity of primary retinal ganglion cells (RGCs) to Hhcy.

RGCs were isolated from mouse retina by immunopanning. Cells were positive for (A) Brn3, a neuronal marker and were negative for (B) GFAP, a marker of glial cells (calibration bar 50μm.) Nuclei were labeled with 4,6-diamidino-2-phenylindole, dihydrochloride (DAPI, blue fluorescence). (C) Differential interference contrast (DIC) microscopic image of primary RGCs demonstrating extension of neurite processes (calibration bar 20μm). (D) Assessment of cell viability after 24h exposure to increasing concentrations of Hcy; data are presented as a percentage of control cells (cells incubated in buffer containing no Hcy). n=3, *p<0.05 (D’Agostino and Pearson Omnibus Normality test; data were normally distributed and were analyzed by one-way ANOVA and Dunnett’s post-hoc test). (E) Detection of oxidative stress using DCFDA in primary RGCs exposed to 25μM, 50μM or100μM Hcy for 3h. Cells were exposed to t-BHP [55μM] for 3 h as a positive inducer of oxidative stress; n=3, *p<0.05 ****p<0.0001 (Kruskal-Wallis and Dunn’s multiple comparison tests).
Assessment of effects of Hhcy on primary Müller cell viability and oxidative stress
We followed our published procedure for isolating Müller cells from mouse retina (Jiang et al, 2006; Mysona et al, 2009; Wang et al, 2015) and confirmed the presence of known Müller cell markers glutamine synthetase (GS) (Fig. 2A), cellular retinaldehyde binding protein (CRALBP) (Fig. 2B), and vimentin (Fig. 2C) in the cells. The isolated cells were negative for proteins not typically found in Müller cells (NeuN, a marker for neurons, Fig. 2D; RPE65, a marker for retinal pigment epithelial cells Fig. 2E). While the vulnerability of CNS neurons to high levels of Hcy is established (Mattson and Shea, 2003), much less is known about how glial cells react to this excitotoxic amino acid.
Fig 2. Assessment of viability and oxidative stress in Müller cells exposed to Hhcy.

Primary Müller cells were isolated from mouse (P6-7) retinas as described in the text. Immunofluorescent detection experiments were performed using markers for Müller glial cells including (A) glutamine synthetase (GS, red fluorescence), (B) cellular retinaldehyde binding protein (CRALBP, green fluorescence), (C) vimentin (green fluorescence). The cells were negative for markers of neurons (D) neuronal nuclei (NeuN, red fluorescence) and RPE cells (E) retinal pigment epithelium 65 (RPE65, red fluorescence). DAPI was used to label nuclei. (F) Viability of Müller cells exposed 24h to increasing concentrations of Hcy. Data are presented as a percentage of control cells (cells incubated in buffer containing no Hcy). n=3, ****p<0.0001 (D’Agostino and Pearson Omnibus Normality test; data were normally distributed and were analyzed by one-way ANOVA and Dunnett’s post-hoc test). (G) Detection of oxidative stress using DCFDA in primary Müller cells exposed to 50μM, 100μM, 500μM or 1mM Hcy for 3h. Cells were exposed to t-BHP [55μM] for 3 h as a positive inducer of oxidative stress; n=3, *p<0.05, **p<0.01, ****p<0.0001 (Kruskal-Wallis and Dunn’s multiple comparison tests). (H and I) Confocal images of primary Müller cells stained with CellROX green reagent after 3h and 24h exposures, respectively, to 50μM and 1mM Hcy. CellROX green fluorescence from primary Müller cells exposed with 100μM Menadione (MEN), an inducer of oxidative stress, was used as the positive control. (Calibration bar is 50μm.) Quantification of CellROX green fluorescence intensities from the cells treated with 50μM and 1mM Hcy for 3h (J) or 24h (K). n=3, *p<0.05, (Kruskal-Wallis and Dunn’s multiple comparison tests).
We evaluated viability in Müller cells exposed to increasing concentrations of Hcy. Unlike the primary RGCs, which show a significant decrease in viability when exposed to 50μM Hcy (Fig. 1D), the primary Müller cells showed no change in viability following 24h exposure to Hcy at a concentration range of 50μM through 1mM (Fig. 2F). Only after the concentration of Hcy was extremely high, was there a significant effect on viability (e.g. 4mM and 10mM induced ~40% and 90% cell death of Müller cells, respectively). These higher dosages [4mM, 10mM] far exceed physiological levels of Hcy, but the findings underscore the remarkable resilience of these retinal glial cells.
Oxidative stress is implicated as a mechanism by which Hhcy induces neuronal death (Lipton et al, 1997; Kruman et al, 2000; Mattson and Shea, 2003; Bhattacharjee et al, 2016). We were interested in the role of Hcy in modulating oxidative stress in Müller glia. We used DCFDA to quantify the level of oxidative stress in the cells. Interestingly, in Müller cells we did not detect an increase in fluorescence in cells following 3h exposure to Hcy, rather we observed a significant decrease in fluorescence in cells exposed to 50μM Hcy, which decreased further in cells exposed to 100μM and 500μM Hcy (Fig. 2G). We next exposed primary Müller cells for 3h or 24h to two concentrations of Hcy [50μM or 1mM] and used a second assay to detect oxidative stress, the CellROX green reagent. The reagent exhibits minimal fluorescence in the reduced state, but strong fluorescence in the oxidized state. We detected minimal fluorescence in cells exposed to Hcy [50 μM, or 1mM] for 3h (Fig. 2H) or 24h (Fig. 2I), whereas cells exposed to menadione (a known oxidative stress inducer) showed robust fluorescence (Fig. 2H, 2I). The quantitative data from replications of the experiment are shown in Fig. 2J and 2K and indicate that at neither time point (3 or 24 h) nor concentration [50μM or 1mM] was there a significant increase in oxidative stress due to Hcy exposure.
Hhcy is associated with a robust antioxidant response in Müller glial cells
The finding that oxidative stress was actually reduced, rather than increased, when Müller cells were exposed to Hhcy prompted investigation of several antioxidants. A key cellular antioxidant in retina is glutathione and the reduced form (GSH) attenuates ROS-induced cellular damage by reducing disulfide bonds. We used a commercially available kit to analyze levels of GSH in Müller cells exposed to 50μM or 1mM Hhcy for 24 h. The levels of total GSH increased in Hhcy-exposed Müller cells (Fig. 3A) and importantly the level of reduced GSH increased (Fig. 3B). Whether expression of other antioxidant genes would be altered in Müller glia in the presence of excess Hcy was not known. To address this, we evaluated the consequences of Hhcy on expression of genes encoding heme oxygenase-1 (Hmox1), superoxide dismutase 2 (Sod2), catalase (Cat), glutathione peroxidase 1 (Gpx1) and NAD(P)H quinone dehydrogenase 1 (Nqo1) (Fig. 3C). Cells were exposed to 50μM or 1mM Hcy for 3h or 8h, RNA was isolated and qRT-PCR performed with gene-specific primers (Table 2). There was an increase in expression of all of these genes, although the extent varied depending upon dosage and duration of exposure to Hcy. For example, levels of Hmox1 increased dramatically within 3h exposure to 50μM Hcy, but returned to baseline within 8h exposure, whereas Sod2 expression did not differ significantly from control at 3h, but was significantly increased at 8h.
Fig 3. Assessment of glutathione and antioxidant genes in Müller cells exposed to Hhcy.

Glutathione levels were detected in primary Müller cells exposed to 50μM or1mM Hcy for 24h as described in the text. Levels are shown for (A) total GSH, and (B) reduced GSH. n=3, ***p<0.001, ****p<0.0001. (C) Expression of genes encoding several antioxidant proteins was analyzed by qRT-PCR in primary Müller cells exposed to 50μM or 1mM Hcy for 3h or 8h. n=3, *p<0.05, **p<0.01, ***p<0.001. (D’Agostino and Pearson Omnibus Normality test; data were normally distributed and were analyzed by one-way ANOVA and Dunnett’s post-hoc test).
Hhcy upregulates Nrf2 in Müller glial cells
NRF2 is a transcription factor that regulates expression of numerous antioxidant genes including those analyzed in Fig. 3C. Given the increase in expression of these genes, we examined Nrf2 expression in Müller cells exposed to Hhcy. We used primers specific for Nrf2 in qRT-PCR analysis and quantified Nrf2 expression as a consequence of two Hcy concentrations and subsequently over a time course for one of these concentrations. The Hcy concentrations utilized were 50μM and 1mM; we observed upregulation of Nrf2 in Müller cells exposed 3 h to 50μM Hcy and 1mM Hcy (Fig. 4A). We then explored the temporal changes in Nrf2 gene expression by exposing Müller cells to 50μM Hcy for 1h, 3h, 6h, 8h and 18h (Fig. 4B). The most significant increase in Nrf2 levels occurred between 1 and 3h exposure to Hcy and the peak increase in Nrf2 expression was at 3h followed by a gradual return to baseline over the next 15h. Interestingly, the increase in Nrf2 gene levels in Müller cells was not apparent at the protein level when we analyzed protein by western blotting in whole cell lysates. The immunodetection of bands for control cells (no Hcy exposure) and cells exposed to 50μM Hcy for 3h or 20h is shown in (Fig. 4C). Using anti-NRF2 antibodies, we detected two bands from the whole cell lysate, which is characteristic for detection of NRF2 as described by others (Macari and Lowrey, 2011; Ahuja et al, 2016). Both bands were utilized to quantify the level of NRF2 as a ratio to GAPDH. We did not observe a statistically significant difference NRF2 levels in Hcy-treated versus non-treated cells (Fig. 4D). This observation prompted analysis of NRF2 in nuclear versus cytosolic fractions. Typically cells maintain NRF2 at a basal level in the cytosol and excess NRF2 is degraded by the proteasome. Under stress however, increased NRF2 translocates from the cytosol to the nucleus. We isolated the nuclear and cytosolic fractions of Müller cells exposed to 50μM or 1mM Hcy. In the nuclear and cytosolic fractions, only one NRF2 band is typically observed for each fraction (Ahuja et al, 2016). Nuclear and cytosolic NRF2 levels were quantified as a ratio to HDAC1 or GAPDH, respectively. There was a discernable increase in NRF2 in the nuclear fraction of cells exposed to 50μM (~59% increase) and 1mM Hcy (65% increase) compared with the control cells, whereas NRF2 levels in the cytosol of Hcy-exposed Müller cells were decreased (50μM: 17% decrease; 1mM: 33% decrease) compared to controls (Fig. 4E). Taken collectively, the data indicate that there is an increase in Nrf2 at the gene and protein level in Müller cells exposed to Hhcy, which may reflect an important cytoprotective response mechanism in these cells.
Fig 4. Assessment of Nrf2 gene and protein in Müller cells exposed to Hhcy.

Relative Nrf2 mRNA expression levels in Müller cells (A) exposed to 50μM and 1mM Hcy for 3h and 8h; (B) Müller cells exposed to 50 μM Hcy for 1h, 3h, 6h, 8h and 18h, n=3, *p<0.05, **p<0.01 (Kruskal-Wallis and Dunn’s multiple comparison tests). (C) Western blot analysis of NRF2 protein levels in Müller cells exposed to 50μM Hcy for 3h and 20h, TBHQ is a positive inducer of Nrf2. (D) Quantification of data shown in panel F (ns = not significant, Krusal-Wallis and Dunn’s). (E) Detection of NRF2 in the nuclear and cytoplasmic fractions of Müller cells exposed 4h to 50 μM or 1 mM Hcy; HDAC1 was used as a marker for the nuclear fraction and GAPDH for the cytosolic fraction.
In the absence of NRF2, the antioxidant response to Hhcy is attenuated
Given that exposure of Müller cells to Hhcy triggers a robust antioxidant response characterized by increased expression of Nrf2, increased levels of NRF2 in the nuclear fraction, and upregulation of several antioxidant genes, we investigated the consequences of Hcy exposure in Müller cells lacking Nrf2. Müller cells harvested from Nrf2−/− and WT mice were exposed to Hcy over a concentration range of 50μM, 1mM, 4mM and 10mM. WT cells showed minimal alteration in viability at 50μM or 1mM Hcy and a decrease in viability at 4mM and 10mM, similar to the data shown in Fig. 2. In the Nrf2−/− Müller cells viability in general was diminished compared to WT. For example, Nrf2−/− Müller cells that received no exposure to Hcy showed compromised viability compared to WT cells (Fig. 5A). Exposure of Nrf2−/− Müller cells to Hcy (50μM or 1mM) did not alter the viability significantly beyond the non-Hcy-treated Nrf2−/− cells, though exposure to higher concentrations of Hcy (4mM) compromised viability markedly (Fig. 5A). We asked whether levels of oxidative stress differed between WT versus Nrf2−/− Müller cells when exposed to Hhcy. As we observed in Fig. 2G, WT cells showed a decrease in DCFDA fluorescence when exposed to 50μM and 1mM Hcy compared to no Hcy treatment (control), however Nrf2−/− Müller cells showed a significant increase in oxidative stress levels compared to WT (Fig. 5B). There was a significant (~50%) increase in DCFDA in non-Hcy-treated Nrf2−/- Müller cells versus WT cells (Fig. 5B). In the Nrf2−/− Müller cells exposed to Hhcy, we observed a higher level of oxidative stress compared to WT cells. We evaluated the levels of several antioxidant genes in the Nrf2−/− Müller cells in the presence or absence of Hhcy exposure. As expected, there was no expression of Nrf2 in Nrf2−/− Müller cells. There was a dramatic decrease in levels of Nqo1, Cat, and Gpx1 in Nrf2−/− Müller cells regardless of Hcy exposure (Fig. 5C). Interestingly, levels of Hmox1 increased slightly in the Nrf2−/− Müller cells (without Hcy exposure) possibly reflecting other transcription factors (besides NRF2) that can regulate Hmox1 expression. Taken collectively, Müller cells lacking Nrf2 are less well equipped to mount a significant antioxidant response to excess Hcy compared with WT cells.
Fig 5. Analysis of the antioxidant response to Hhcy in Nrf2−/− Müller cells.

(A) Data from the PrestoBlue viability assay in primary Müller cells isolated from WT and Nrf2−/− mice exposed 24 h to several concentrations of Hcy. (n=3) (B) Fluorescence levels of DCFDA as an indicator of oxidative stress in primary Müller cells isolated from WT and Nrf2−/− mice exposed 3h to 50μm or 1mM Hcy; 55μM TBHP treatment for 3h was the positive control. (C) Expression of antioxidant genes in primary Müller cells isolated from WT and Nrf2−/− mice exposed 3h to 50μm or 1mM Hcy. The relative mRNA levels in Hcy treated Nrf2−/− cells were compared with corresponding relative mRNA levels in WT and Nrf2−/− untreated control cells. n=3, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 (D’Agostino and Pearson Omnibus Normality test; data were normally distributed and were analyzed by two-way ANOVA and Sidak’s post-hoc test.)
Discussion
This study was undertaken to investigate the effects of excess Hcy on retinal Müller glial cells. Accumulation of Hcy, a highly reactive amino acid, is implicated in retinal diseases (Ajith and Ranimenon, 2015), notably those affecting RGCs in optic neuropathies such as exfoliation glaucoma (Ritch, 1994; Roedl et al, 2007). Müller cells play a key role in maintaining RGCs, thus in the current study, we evaluated effects of Hcy in this glial cell type, specifically evaluating viability and response to oxidative stress.
We were particularly interested in the Müller cell response to Hhcy because of a conundrum observed in in vivo versus in vitro models of excess Hcy. Studies from two in vivo murine models of Hhcy, the Mthfr+/− mouse and the Cbs+/− mouse, both demonstrate ~20% loss of RGCs over a 30-week period (Ganapathy et al, 2009; Markand et al, 2015). While this loss is sufficient to alter retinal function in the mice, it is not blinding. In contrast, isolated (primary) RGCs are quite sensitive to Hcy such that ~50–60% of the cells die when exposed 18h to 50μM Hcy (Dun et al, 2007; Ganapathy et al, 2011a; Ganapathy et al, 2011b). An obvious and critical difference in the experimental systems is the in vivo models reflect the collective and potentially adaptive response of neurons and glia to Hcy-induced excitotoxicity, whereas in the in vitro studies, the primary RGCs are a purified neuronal population existing in a nutrient-supplemented, but nevertheless artificial milieu. We were interested in whether isolated Müller cells would also manifest a heightened sensitivity to Hcy when they were removed from their typical neuronal-vascular-glial environment in a manner similar to isolated RGCs.
The Hhcy-exposed primary Müller cells did not demonstrate heightened sensitivity to excess Hcy in vitro. They were viable over a broad range of Hcy exposure levels [50μM – 1 mM]. Müller cells demonstrated far more resilience to Hhcy exposure than primary RGCs. In addition, exposure of Müller cells to moderately elevated Hcy did not increase oxidative stress; rather oxidative stress actually decreased under Hhcy conditions. This result was confirmed using two different assays. The levels of oxidative stress decreased over a Hcy concentration range of [50μM – 1.0 mM], which was in contrast to primary RGCs in which oxidative stress increased markedly within 3 h exposure to 100μM Hcy. Previous studies in Hhcy-exposed primary RGCs using electron paramagnetic resonance spectroscopy also detected marked oxidative stress including induction of reactive species superoxide, nitric oxide and peroxynitrite (Ganapathy et al, 2011a). We conclude that while Hhcy increases oxidative stress dramatically in neurons such as RGCs, this is not the case in Müller cells.
The decreased oxidative stress observed in Müller cells prompted analyses of GSH levels and expression of several antioxidant genes. We detected a robust antioxidant response by Müller cells exposed to Hhcy. Reduced GSH, one of the most important scavengers of ROS, increased markedly in Müller cells exposed to Hhcy. This may represent a higher capacity to metabolize Hcy and is consistent with reports using HepG2 cells, which demonstrated an Hcy-induced dose-dependent increase in GSH (Zhang et al, 2017). Expression of a number of antioxidant genes increased also in Müller cells exposed to Hcy compared to non-treated cells, including Hmox1, Sod2, Cat, Gpx1, Nqo1. NRF2, a redox-sensitive transcription factor that binds to antioxidant response elements located in the promoter region of these (and many other) genes, was elevated at the mRNA level. The analysis of the temporal pattern of Nrf2 expression showed a significant upregulation by 3h. The NRF2 protein level detected in the whole cell lysate did not differ between control and Hcy-treated cells, however there was an increase in NRF2 in the nuclear fraction of the cells when treated with Hcy. Translocation of NRF2 to the nucleus is required for the activation of downstream antioxidant genes. Our data show that Müller cells respond to Hhcy by translocating NRF2 to the nucleus Taken collectively the data provide compelling evidence that exposure of isolated Müller cells to Hhcy activates NRF2 enhancing the capacity of the cells to manage excitotoxic insult.
We investigated the extent to which Hcy would yield an oxidative response in the absence of NRF2 using Müller cells harvested from mice that lack this gene (Nrf2−/ − mice). There was a significant decrease (~40%) in viability of Nrf2−/ − Müller cells compared to WT cells; this was observed not only in cells exposed to Hcy, but also in cells that received no Hcy. The Nrf2−/ − Müller cells demonstrated increased oxidative stress as measured by DCFDA fluorescence, whether exposed to Hcy or not. We note that most of the antioxidant genes that are regulated primarily by NRF2 (e.g. Nqo1, Cat, Gpx1) were reduced markedly when Nrf2−/ − cells were exposed to Hhcy. We did observe a slight increase in Hmox1 expression in Nrf2−/ − Müller cells compared to WT, but it was not altered by Hcy treatment. The increase in Hmox1 expression may reflect influence of transcription factors such as HIF1α and PGC-1α, which, in addition to Nrf2, are known to regulate Hmox1 expression (Kang et al, 2005; Hosick and Stec, 2012).
A number of recent studies have demonstrated that RGC loss can be attenuated by activating NRF2 (Himori et al, 2013; Cho et al, 2015; Xu et al, 2015; Cheng et al, 2017; Wan et al, 2017). It is recognized that Müller cells have a robust and remarkable capacity to buffer cellular stress (Bringmann et al, 2006), although their activation in response may vary depending upon the disease (Hippert et al, 2015). Some of the mechanisms by which Müller cells can attenuate oxidative stress (besides activation of Nrf2) or attenuate other forms of cellular stress include release of vascular endothelial growth factor (Yamada et al, 1999), uptake of excess glutamate within the retina (Kawasaki et al, 2000), and production of neurotrophic factors and growth factors (Garcia and Vecino, 2003). These were not examined in the present study, but could be fruitful areas of further investigation. In RGCs, the effects of homocysteine have been reported to be mediated by over activation of the N-methyl-D-aspartate receptor (NMDA) subtype of glutamate receptor (Lipton et al, 1997; Martin et al, 2004; Ganapathy et al, 2011b), which can in turn lead to increased oxidative stress. There is considerable evidence that this is true for other non-retinal neuronal cell types as well (Bhatia and Singh, 2015 Sibarov et al, 2016). Cortical glial cells are also known to express NMDA receptors (Dzamba et al, 2015), however much less is known about the presence of NMDA receptors in mammalian retinal Müller glial cells, though they have been reported in Müller glia derived from chicken retina (Lamas et al, 2005). Whether the differential sensitivity of the primary Müller cells versus primary RGCs to Hhcy observed in the present study reflects a difference in NMDA receptor activation remains to be investigated.
Our findings are relevant to in vivo models of Hcy in retinal disease. Earlier studies of endogenous murine models of Hcy (Mthfr+/− and Cbs+/− mice) showed clear evidence of RGC loss and diminished RGC function. However, compared to the effects of Hhcy on isolated RGCs, observed in this and other studies (Dun et al, 2007; Ganapathy et al, 2011a, Ganapathy et al, 2011b;), the severity of the neuronal loss in the endogenous models was mild-moderate. This is intriguing. We speculate that Müller cells in these mouse models may be mounting a significant neuroprotective response to Hhcy. The present study, using the isolated Müller cell, supports this theory, i.e. Müller cells buffer effects of Hhcy, which may protect retinal neurons – at least for some time period. Our data suggest that the mechanism involves a robust antioxidant response. It is noteworthy that earlier studies performed in neural retinas of Mthfr+/− and Cbs+/− mice showed that, rather than being decreased, levels of GSH were similar to WT mice as were levels of xCT, a protein essential for the function of the cystine-glutamate exchanger involved in GSH synthesis (Cui et al, 2017). Levels of antioxidants and NRF2 have not been investigated in Mthfr+/− and Cbs+/− mice, but this would also be an area of fruitful investigation in the future.
The data obtained from the present study demonstrate a differential response to excess Hcy by glial cells versus neurons, one that is protective rather than deleterious – at least under acute conditions. The studies reflect a response to an insult spanning 24 h or less. In the rodent models, the exposure to the modest elevation of Hcy spans several weeks and the loss of the ganglion cells is gradual. In Mthfr−/ − mice for example, RGC loss is detectable by 24 weeks, but not at 12 weeks. It may be that the capacity of Müller cells to buffer excess Hcy within the intact retina decreases with time. Long-term studies of these murine models at ages exceeding 1 year may disclose more advanced RGC loss. The observations of the current study are relevant to humans, especially patients with exfoliation glaucoma. Hcy is modestly, but consistently higher in serum, aqueous humor and tears in patients with this disease (Roedl et al, 2007; Xu et al, 2012). It is still unclear, however, whether Hcy is ‘a disease driver, disease biomarker or an innocent bystander to some biochemical process related to Hcy metabolism’ (Pasquale et al, 2016). The present study does not answer this question, but it does provide insights that retinal cells respond differently to excess Hcy – neurons in isolation demonstrate marked vulnerability and decreased viability, whereas glial cells in isolation demonstrate a robust antioxidant response coupled with sustained viability over a broad Hcy concentration range. Some of the confusion as to whether Hhcy is a marker for human retinal disease or is actually pathogenic may reflect varied responses by key supportive cells such as Müller cells. The work that we and others have performed in laboratory studies using in vitro and in vivo models provides compelling evidence that Hhcy can kill neurons and that glial cells can buffer the effects of Hhcy, at least for a period of time. If more clinical evidence continues to emerge that implicates Hhcy as a factor in RGC-related retinal diseases, there may be a benefit in modulating diet since increased folate consumption can decrease Hcy levels. It remains to be determined in controlled in vivo studies whether dietary modification would attenuate RGC loss, but such studies would be extremely informative as patient compliance with vitamin supplementation is certainly feasible.
Highlights.
Homocysteine (Hcy) is an excitatory amino acid that has been implicated in loss of retinal ganglion cells (RGCs) in conditions such as exfoliative glaucoma.
While several studies report vulnerability of isolated RGCs to elevated Hcy, there have been few studies of the effects of excess Hcy on isolated Müller cells.
Primary Müller cells exposed to excess Hcy did not demonstrate altered viability nor increased oxidative stress, which is observed in Hcy-exposed RGCs. Instead, Müller cells demonstrated a cytoprotective response characterized by increased levels of antioxidants and increased expression of Nrf2.
Müller cells isolated from Nrf2−/− mice show decreased viability and increased levels of oxidative stress compared with wildtype Müller cells, underscoring the importance of Nrf2 as an important mediator of oxidative stress in Müller cells.
Acknowledgments
This work was supported by the National Institutes of Health (National Eye Institute, R01 EY012830 and R01 NS101967), and the James and Jean Culver Vision Discovery Institute of Augusta University. We acknowledge the Cell Imaging Core (Department of Cellular Biology and Anatomy, Medical College of Georgia at Augusta University) for the excellent instrumentation and technical assistance in image acquisition. We thank Dr. Jennifer Waller, Professor of Population Health Science, Medical College of Georgia at Augusta University for excellent advice and guidance on the statistical analysis of the data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ajith TA, Ranimenon Homocysteine in ocular diseases. Clinica Chimica Acta. 2015;450:316–321. doi: 10.1016/j.cca.2015.09.007. [DOI] [PubMed] [Google Scholar]
- Ahuja M, Ammal Kaidery N, Yang L, Calingasan N, Smirnova N, Gaisin A, Gaisina IN, Gazaryan I, Hushpulian DM, Kaddour-Djebbar I, Bollag WB, Morgan JC, Ratan RR, Starkov AA, Beal MF, Thomas B. Distinct Nrf2 Signaling Mechanisms of Fumaric Acid Esters and Their Role in Neuroprotection against 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Experimental Parkinson's-Like Disease. J Neurosci. 2016;36:6332–6351. doi: 10.1523/JNEUROSCI.0426-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA, Silverstein BE, Corey DP, Chun LL. Immunological, morphological, and electrophysiological variation among retinal ganglion cells purified by panning. Neuron. 1998;1:791–803. doi: 10.1016/0896-6273(88)90127-4. [DOI] [PubMed] [Google Scholar]
- Bhatia P, Singh N. Homocysteine excess: delineating the possible mechanism of neurotoxicity and depression. Fundam Clin Pharmacol. 2015 Dec;29(6):522–8. doi: 10.1111/fcp.12145. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee N, Paul R, Giri A, Borah A. Chronic exposure of homocysteine in mice contributes to dopamine loss by enhancing oxidative stress in nigrostriatum and produces behavioral phenotypes of Parkinson’s disease. Biochem Biophys Rep. 2016;6:47–53. doi: 10.1016/j.bbrep.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleich S, Roedl J, Von Ahsen N, Schlötzer-Schrehardt U, Reulbach U, Beck G, Kruse FE, Naumann GO, Kornhuber J, Jünemann AG. Elevated homocysteine levels in aqueous humor of patients with pseudoexfoliation glaucoma. Amer J Ophthalmol. 2004;138:162–164. doi: 10.1016/j.ajo.2004.02.027. [DOI] [PubMed] [Google Scholar]
- Boushey CJ, Beresford SA, Omenn GS, Motulsky AG. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. J Amer Med Assoc. 1995;274:1049–1057. doi: 10.1001/jama.1995.03530130055028. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Iandiev I, Pannicke T, Wurm A, Hollborn M, Wiedemann P, Osborne NN, Reichenbach A. Cellular signaling and factors involved in Müller cell gliosis: neuroprotective and detrimental effects. Prog Ret Eye Res. 2009;28:423–451. doi: 10.1016/j.preteyeres.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A. Müller cells in the healthy and diseased retina. Prog Ret Eye Res. 2006;25:397–424. doi: 10.1016/j.preteyeres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Cantu C, Alonso E, Jara A, Martínez L, Ríos C, de Fernández ML, Garcia I, Barinagarrementeria F. Hyperhomocysteinemia, low folate and vitamin B12 concentrations, and methylene tetrahydrofolate reductase mutation in cerebral venous thrombosis. Stroke. 2004;35:1790–1794. doi: 10.1161/01.STR.0000132570.24618.78. [DOI] [PubMed] [Google Scholar]
- Chang HH, Lin DP, Chen YS, Liu HJ, Lin W, Tsao ZJ, Teng MC, Chen BY. Intravitreal homocysteinethiolactone injection leads to the degeneration of multiple retinal cells, including photoreceptors. Mol Vis. 2011;17:1946–1956. [PMC free article] [PubMed] [Google Scholar]
- Cheng LB, Li KR, Yi N, Li XM, Wang F, Xue B, Pan YS, Yao J, Jiang Q, Wu ZF. miRNA-141 attenuates UV-induced oxidative stress via activating Keap1-Nrf2 signaling in human retinal pigment epithelium cells and retinal ganglion cells. Oncotarget. 2017;8:13186–13194. doi: 10.18632/oncotarget.14489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Hartsock MJ, Xu Z, He M, Duh EJ. Monomethyl fumarate promotes Nrf2-dependent neuroprotection in retinal ischemia-reperfusion. J Neuroinflam. 2015;21(12):239. doi: 10.1186/s12974-015-0452-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke R, Daly L, Robinson K, Naughten E, Cahalane S, Fowler B, Graham I. Hyperhomocysteinemia: an independent risk factor for vascular disease. New Engl J Med. 1991;324:1149–1155. doi: 10.1056/NEJM199104253241701. [DOI] [PubMed] [Google Scholar]
- Criddle DN, Gillies S, Baumgartner-Wilson HK, Jaffar M, Chinje EC, Passmore S, Chvanov M, Barrow S, Gerasimenko OV, Tepikin AV, Sutton R, Petersen OH. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J Biol Chem. 2006;281:40485–40492. doi: 10.1074/jbc.M607704200. [DOI] [PubMed] [Google Scholar]
- Cui X, Navneet S, Wang J, Roon P, Chen W, Xian M, Smith SB. Analysis of MTHFR, CBS, Glutathione, Taurine, and Hydrogen Sulfide Levels in Retinas of Hyperhomocysteinemic Mice. Invest Ophthalmol Vis Sci. 2017;58:1954–1963. doi: 10.1167/iovs.16-21247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzamba D, Honsa P, Valny M, Kriska J, Valihrach L, Novosadova V, Kubista M, Anderova M. Quantitative Analysis of Glutamate Receptors in Glial Cells from the Cortex of GFAP/EGFP Mice Following Ischemic Injury: Focus on NMDA Receptors. Cell Mol Neurobiol. 2015;35:1187–2202. doi: 10.1007/s10571-015-0212-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Ladenheim B, Cutler RG, Kruman II, Cadet JL, Mattson MP. Dietary folate deficiency and elevated homocysteine levels endanger dopaminergic neurons in models of Parkinson's disease. J Neurochem. 2002;80:101–110. doi: 10.1046/j.0022-3042.2001.00676.x. [DOI] [PubMed] [Google Scholar]
- Dun Y, Duplantier J, Roon P, Martin PM, Ganapathy V, Smith SB. Serine racemase expression and D-serine content are developmentally regulated in neuronal ganglion cells of the retina. J Neurochem. 2008;104:970–8. doi: 10.1111/j.1471-4159.2007.05015.x. [DOI] [PubMed] [Google Scholar]
- Dun Y, Mysona B, Van Ells T, Amarnath L, Ola MS, Ganapathy V, Smith SB. Expression of the cystine-glutamate exchanger (xc-) in retinal ganglion cells and regulation by nitric oxide and oxidative stress. Cell Tissue Res. 2006;324:189–202. doi: 10.1007/s00441-005-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dun Y, Thangaraju M, Prasad P, Ganapathy V, Smith SB. Prevention of excitotoxicity in primary retinal ganglion cells by (+)-pentazocine, a sigma receptor-1 specific ligand. Invest Ophthalmol Vis Sci. 2007;48:4785–4794. doi: 10.1167/iovs.07-0343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy PS, Moister B, Roon P, Mysona BA, Duplantier J, Dun Y, Moister TK, Farley MJ, Prasad PD, Liu K, Smith SB. Endogenous elevation of homocysteine induces retinal neuron death in the cystathionine-beta-synthase mutant mouse. Invest Ophthalmol Vis Sci. 2009;50:4460–4470. doi: 10.1167/iovs.09-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy PS, Perry RL, Tawfik A, Smith RM, Perry E, Roon P, Bozard BR, Ha Y, Smith SB. Homocysteine-mediated modulation of mitochondrial dynamics in retinal ganglion cells. Invest Ophthalmol Vis Sci. 2011a;52:5551–5558. doi: 10.1167/iovs.11-7256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy PS, White RE, Ha Y, Bozard BR, McNeil PL, Caldwell RW, Kumar S, Black SM, Smith SB. The role of N-methyl-D-aspartate receptor activation in homocysteine-induced death of retinal ganglion cells. Invest Ophthalmol Vis Sci. 2011b;52:5515–5524. doi: 10.1167/iovs.10-6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García M, Vecino E. Role of Müller glia in neuroprotection and regeneration in the retina. Histol Histopathol. 2003;18:1205–1218. doi: 10.14670/HH-18.1205. [DOI] [PubMed] [Google Scholar]
- Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Dis. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- Hicks D, Courtois Y. The growth and behaviour of rat retinal Müller cells in vitro. 1. An improved method for isolation and culture. Exp Eye Res. 1990;51:119–129. doi: 10.1016/0014-4835(90)90063-z. [DOI] [PubMed] [Google Scholar]
- Himori N, Yamamoto K, Maruyama K, Ryu M, Taguchi K, Yamamoto M, Nakazawa T. Critical role of Nrf2 in oxidative stress-induced retinal ganglion cell death. J Neurochem. 2013;127:669–680. doi: 10.1111/jnc.12325. [DOI] [PubMed] [Google Scholar]
- Hippert C, Graca AB, Barber AC, West EL, Smith AJ, Ali RR, Pearson RA. Müller glia activation in response to inherited retinal degeneration is highly varied and disease-specific. PLoS One. 2015 Mar 20;10(3):e0120415. doi: 10.1371/journal.pone.0120415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PI, Collins SC, Dhitavat S, Ortiz D, Ashline D, Rogers E, Shea TB. Homocysteine potentiates beta-amyloid neurotoxicity: role of oxidative stress. J Neurochem. 2001;78:249–253. doi: 10.1046/j.1471-4159.2001.00384.x. [DOI] [PubMed] [Google Scholar]
- Hosick PA, Stec DE. Heme oxygenase, a novel target for the treatment of hypertension and obesity? Amer J Physiology: Reg, Integ, Comp Phys. 2012;302:R207–214. doi: 10.1152/ajpregu.00517.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Popp R, Frömel T, Ehling M, Awwad K, Adams RH, Hammes HP, Fleming I. Müller glia cells regulate Notch signaling and retinal angiogenesis via the generation of 19,20-dihydroxydocosapentaenoic acid. J Exp Med. 2014;211:281–295. doi: 10.1084/jem.20131494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques PF, Bostom AG, Williams RR, Ellison RC, Eckfeldt JH, Rosenberg IH, Selhub J, Rozen R. Relation between folate status, a common mutation in methylenetetrahydrofolate reductase, and plasma homocysteine concentrations. Circulation. 1996;93:7–9. doi: 10.1161/01.cir.93.1.7. [DOI] [PubMed] [Google Scholar]
- Jiang G, Mysona B, Dun Y, Gnana-Prakasam JP, Pabla N, Li W, Dong Z, Ganapathy V, Smith SB. Expression, subcellular localization, and regulation of sigma receptor in retinal Müller cells. Invest Ophthalmol Vis Sci. 2006;47:5576–5582. doi: 10.1167/iovs.06-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidery NA, Banerjee R, Yang L, Smirnova NA, Hushpulian DM, Liby KT, Williams CR, Yamamoto M, Kensler TW, Ratan RR, Sporn MB, Beal MF, Gazaryan IG, Thomas B. Targeting Nrf2-mediated gene transcription by extremely potent synthetic triterpenoids attenuate dopaminergic neurotoxicity in the MPTP mouse model of Parkinson's disease. Antiox Redox Sig. 2013;18:139–157. doi: 10.1089/ars.2011.4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MJ, Kim HJ, Kim HK, Lee JY, Kim DH, Jung KJ, Kim KW, Baik HS, Yoo MA, Yu BP, Chung HY. The effect of age and calorie restriction on HIF-1-responsive genes in aged liver. Biogerontology. 2005;6:27–37. doi: 10.1007/s10522-004-7381-z. [DOI] [PubMed] [Google Scholar]
- Kawasaki A, Otori Y, Barnstable CJ. Müller cell protection of rat retinal ganglion cells from glutamate and nitric oxide neurotoxicity. Invest Ophthalmol Vis Sci. 2000;41:3444–3450. [PubMed] [Google Scholar]
- Kruman II, Culmsee C, Chan SL, Kruman Y, Guo Z, Penix L, Mattson MP. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J Neurosci. 2000;20:6920–6926. doi: 10.1523/JNEUROSCI.20-18-06920.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamas M, Lee-Rivera I, López-Colomé AM. Cell-specific expression of N-methyl-D-aspartate receptor subunits in Müller glia and neurons from the chick retina. Invest Ophthalmol Vis Sci. 2005;46:3570–3577. doi: 10.1167/iovs.04-1398. [DOI] [PubMed] [Google Scholar]
- Leclerc D, Sibani S, Rozen R. Molecular Biology of Methylenetetrahydrofolate Reductase (Mthfr) and Overview of Mutations/Polymorphisms. In: Ueland PM, Rozen R, editors. Mthfr Polymorphisms and Disease. Georgetown, TX: Landes Bioscience; 2005. pp. 1–20. [Google Scholar]
- Leibovitch I, Kurtz S, Shemesh G, Goldstein M, Sela BA, Lazar M, Loewenstein A. Hyperhomocystinemia in pseudoexfoliation glaucoma. J Glaucoma. 2003 Feb;12(1):36–9. doi: 10.1097/00061198-200302000-00007. [DOI] [PubMed] [Google Scholar]
- Li J, Xu F, Zeng R, Gong H, Lan Y. Plasma Homocysteine, Serum Folic Acid, Serum Vitamin B12, Serum Vitamin B6, MTHFR, and Risk of Normal-Tension Glaucoma. J Glaucoma. 2016;25:e94–98. doi: 10.1097/IJG.0000000000000269. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Kim WK, Choi YB, Kumar S, D'Emilia DM, Rayudu PV, Arnelle DR, Stamler JS. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc Natl Acad Sci USA. 1997;94:5923–5928. doi: 10.1073/pnas.94.11.5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Macari ER, Lowrey CH. Induction of human fetal hemoglobin via the NRF2 antioxidant response signaling pathway. Blood. 2011;117:5987–5997. doi: 10.1182/blood-2010-10-314096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markand S, Saul A, Roon P, Prasad P, Martin P, Rozen R, Ganapathy V, Smith SB. Retinal Ganglion Cell Loss and Mild Vasculopathy in Methylene Tetrahydrofolate Reductase (Mthfr)-Deficient Mice: A Model of Mild Hyperhomocysteinemia. Invest Ophthalmol Vis Sci. 2015;56:2684–2695. doi: 10.1167/iovs.14-16190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PM, Ola MS, Agarwal N, Ganapathy V, Smith SB. The sigma receptor ligand (+)-pentazocine prevents apoptotic retinal ganglion cell death induced in vitro by homocysteine and glutamate. Brain Res Mol Brain Res. 2004;123:66–75. doi: 10.1016/j.molbrainres.2003.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Shea TB. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci. 2003;26:137–146. doi: 10.1016/S0166-2236(03)00032-8. [DOI] [PubMed] [Google Scholar]
- Moore P, El-sherbeny A, Roon P, Schoenlein PV, Ganapathy V, Smith SB. Apoptotic cell death in the mouse retinal ganglion cell layer is induced in vivo by the excitatory amino acid homocysteine. Exp Eye Res. 2001;73:45–57. doi: 10.1006/exer.2001.1009. [DOI] [PubMed] [Google Scholar]
- Mysona B, Dun Y, Duplantier J, Ganapathy V, Smith SB. Effects of hyperglycemia and oxidative stress on the glutamate transporters GLAST and system xc- in mouse retinal Müller glial cells. Cell Tiss Res. 2009;335:477–488. doi: 10.1007/s00441-008-0742-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeid R, Herrmann W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Letters. 2006;580:2994–3005. doi: 10.1016/j.febslet.2006.04.088. [DOI] [PubMed] [Google Scholar]
- Pasquale LR, Borras T, Fingert JH, Wiggs JL, Ritch R. Exfoliation syndrome: assembling the puzzle pieces. Acta Ophthalmol. 2016;94:e505–512. doi: 10.1111/aos.12918. [DOI] [PubMed] [Google Scholar]
- Puustjärvi T, Blomster H, Kontkanen M, Punnonen K, Teräsvirta M. Plasma and aqueous humour levels of homocysteine in exfoliation syndrome. Graefes Arch Clin Exp Ophthalmol. 2004 Sep;242(9):749–54. doi: 10.1007/s00417-004-0918-7. [DOI] [PubMed] [Google Scholar]
- Religa D, Czyzewski K, Styczynska M, Peplonska B, Lokk J, Chodakowska-Zebrowska M, Stepien K, Winblad B, Barcikowska M. Hyperhomocysteinemia and methylenetetrahydrofolate reductase polymorphism in patients with Parkinson's disease. Neurosci Letters. 2006;404:56–60. doi: 10.1016/j.neulet.2006.05.040. [DOI] [PubMed] [Google Scholar]
- Ritch R. Exfoliation syndrome-the most common identifiable cause of open-angle glaucoma. J Glaucoma. 1994;3(2):176–7. [PubMed] [Google Scholar]
- Roedl JB, Bleich S, Reulbach U, Rejdak R, Naumann GO, Kruse FE, Schlötzer-Schrehardt U, Kornhuber J, Jünemann AG. Vitamin deficiency and hyperhomocysteinemia in pseudoexfoliation glaucoma. J Neural Transm (Vienna) 2007;114(5):571–5. doi: 10.1007/s00702-006-0598-z. [DOI] [PubMed] [Google Scholar]
- Sajgo S, Ghinia MG, Brooks M, Kretschmer F, Chuang K, Hiriyanna S, Wu Z, Popescu O, Badea TC. Molecular codes for cell type specification in Brn3 retinal ganglion cells. Proc Natl Acad Sci U S A. 2017;114:E3974–E3983. doi: 10.1073/pnas.1618551114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selhub J. Homocysteine metabolism. Ann Rev Nutrition. 1999;19:217–246. doi: 10.1146/annurev.nutr.19.1.217. [DOI] [PubMed] [Google Scholar]
- Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D'Agostino RB, Wilson PW, Wolf PA. Plasma homocysteine as a risk factor for dementia and Alzheimer's disease. New Engl J Med. 2002;346:476–483. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- Sibarov DA, Abushik PA, Giniatullin R, Antonov SM. GluN2A Subunit-Containing NMDA Receptors Are the Preferential Neuronal Targets of Homocysteine. Front Cell Neurosci. 2016;10:246. doi: 10.3389/fncel.2016.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564–571. doi: 10.1038/nrc3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubbink JB, van der Merwe A, Delport R, Allen RH, Stabler SP, Riezler R, Vermaak WJ. The effect of a subnormal vitamin B-6 status on homocysteine metabolism. J Clin Invest. 1996;98:177–184. doi: 10.1172/JCI118763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan P, Su W, Zhang Y, Li Z, Deng C, Zhuo Y. Trimetazidine protects retinal ganglion cells from acute glaucoma via the Nrf2/Ho-1 pathway. Clin Sci (Lond) 2017;131:2363–2375. doi: 10.1042/CS20171182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Shanmugam A, Markand S, Zorrilla E, Ganapathy V, Smith SB. Sigma 1 receptor regulates the oxidative stress response in primary retinal Müller glial cells via NRF2 signaling and system xc(-), the Na(+)-independent glutamate-cystine exchanger. Free Rad Biol Med. 2015;86:25–36. doi: 10.1016/j.freeradbiomed.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Zhang L, Li M. Plasma homocysteine, serum folic acid, serum vitamin B12, serum vitamin B6, MTHFR and risk of pseudoexfoliation glaucoma: a meta-analysis. Graefes Arch Clin Exp Ophthalmol. 2012;250:1067–74. doi: 10.1007/s00417-011-1877-4. [DOI] [PubMed] [Google Scholar]
- Xu Z, Cho H, Hartsock MJ, Mitchell KL, Gong J, Wu L, Wei Y, Wang S, Thimmulappa RK, Sporn MB, Biswal S, Welsbie DS, Duh EJ. Neuroprotective role of Nrf2 for retinal ganglion cells in ischemia-reperfusion. J Neurochem. 2015;133:233–241. doi: 10.1111/jnc.13064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Yamada E, Hackett SF, Ozaki H, Okamoto N, Campochiaro PA. Hyperoxia causes decreased expression of vascular endothelial growth factor and endothelial cell apoptosis in adult retina. J Cell Physiol. 1999;179:149–156. doi: 10.1002/(SICI)1097-4652(199905)179:2<149::AID-JCP5>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Young RW. Cell differentiation in the retina of the mouse. Anat Rec. 1985;212:199–205. doi: 10.1002/ar.1092120215. [DOI] [PubMed] [Google Scholar]
- Yu M, Sturgill-Short G, Ganapathy P, Tawfik A, Peachey NS, Smith SB. Age-related changes in visual function in cystathionine-beta-synthase mutant mice, a model of hyperhomocysteinemia. Exp Eye Res. 2012;96:124–131. doi: 10.1016/j.exer.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Dong JL, Chen YL, Liu Y, Huang SS, Zhong XL, Cheng YH, Wang ZG. Nrf2 mediates the protective effects of homocysteine by increasing the levels of GSH content in HepG2 cells. Mol Med Rep. 2017;16:597–602. doi: 10.3892/mmr.2017.6633. [DOI] [PMC free article] [PubMed] [Google Scholar]