

Abstract
The population of the Mediterranean island of Sardinia has made important contributions to genome-wide association studies of complex disease traits and, based on ancient DNA (aDNA) studies of mainland Europe, Sardinia is hypothesized to be a unique refuge for early Neolithic ancestry. To provide new insights on the genetic history of this flagship population, we analyzed 3,514 whole-genome sequenced individuals from Sardinia. We find Sardinian samples show elevated levels of shared ancestry with Basque individuals, especially samples from the more historically isolated regions of Sardinia. Our analysis also uniquely illuminates how levels of genetic similarity with mainland aDNA samples varies subtly across the island. Together, our results indicate within-island sub-structure and sex-biased processes have substantially impacted the genetic history of Sardinia. These results give new insight to the demography of ancestral Sardinians and help further the understanding of sharing of disease risk alleles between Sardinia and mainland populations.
How complex traits change through time is a central question in evolutionary biology and genetics. Human genetics provides a compelling context for studying this process but requires populations where it is possible to integrate trait mapping with a detailed knowledge of population history. The people of the Mediterranean island of Sardinia are particularly well suited for genetic studies as evident from a number of successes in complex trait and disease mapping1. For example, early studies illuminated the genetic basis of thalassemia and more recent studies have mapped novel quantitative trait loci for traits such as hemoglobin levels2, inflammation levels2,3, height4, and diseases such as multiple sclerosis5 and type 1 diabetes6. These autoimmune diseases and hematological diseases like beta thalassemia show unique incidences in Sardinia (e.g. ref. 7–9). Understanding how and why these conditions reached their frequencies in Sardinia would provide valuable insights to the dynamics of complex trait evolution. Yet to empower such studies, a detailed background population history of Sardinia is needed.
One key characteristic of Sardinia is its differentiation from mainland populations, as evidenced by a distinctive cultural, linguistic, and archaeological legacy10. Early genetic studies made clear that Sardinia has also been a genetically isolated population on the basis of classical autosomal markers, uniparental markers, and elevated linkage disequilibrium11–16. Partly on this basis, Sardinia was included in the Human Genome Diversity Panel (HGDP; ref. 17), which has been used as a reference sample in many studies, including recent ancient DNA (“aDNA”) studies of Europe18–22. Despite substantial research and interest in Sardinia, genetic studies of its demographic history are still incomplete.
One of the most remarkable findings to date regarding Sardinia’s demographic history is that it has the highest detected levels of genetic similarity to ancient Neolithic farming peoples of Europe18–20,22–26. This result is currently interpreted in a model with three ancestral populations that contribute ancestry to modern European populations18,20,23,25. The model postulates that early Neolithic farmers (“EF”) from the Near East and Anatolia expanded into Europe ~7,500 to ~8,000 years ago and mixed in varying proportions with the existing, pre-Neolithic, hunter-gatherers (“HG”) in Europe. Then a substantial post-Neolithic expansion of Steppe pastoralists (“SP”, associated with the Yamnaya culture) in the Bronze Age ~4,500 to ~5,000 years ago introduced a third major component of ancestry across Europe. This model has been useful in explaining patterns observed in ancient and modern DNA data throughout Europe; here we look closely at how well it explains Sardinian population history.
In this model, Sardinia is effectively colonized by the EF during the European Neolithic, with minor contributions from pre-Neolithic HG groups. Sardinia then remained largely isolated from subsequent migrations on the mainland26, including the Bronze Age expansions of the SP18,20,23. Support for this model is based on SNP array data, and has additional support from a ancient mtDNA study in Sardinia, which showed relative isolation from mainland Europe since the Bronze Age, particularly for the Ogliastra region27. There is also support for this model in the relatively low frequency in Sardinia of U haplogroups that are markers of hunter-gather ancestry18,28–31. The archaeological record in Sardinia is also broadly supportive of such a model – there are few notable sites from the pre-Neolithic, followed by an expansion of sites in the Neolithic and subsequent development of a unique local cultural assemblage (Nuragic culture) by the Bronze Age in Sardinia (see ref. 32,33).
The conclusion that Sardinia was effectively descended from early Neolithic farmers is not without question though. The first human remains on Sardinia date to the Upper Paleolithic time and flint-stone instruments are found in the Lower Paleolithic period12,34, so the potential earliest residents arrived in Sardinia ~14,000–18,000 years ago, much earlier than the Neolithic period. Studies using Y-chromosome haplotypes (particularly haplotype I2a1a1) have found Mesolithic or Paleolithic dates for the common ancestor of Sardinian-specific haplotypes and have interpreted these results as evidence for a strong pre-Neolithic component of Sardinian ancestry12,35–38, although this interpretation is controversial27,28,33,39,40. Moreover, the relatively high prevalence of R haplogroup R1b1a2 (R-M269) haplogroup in Sardinia (~18%) has been interpreted to reflect a large component of pre-Neolithic HG ancestry in Sardinia (Contu et al. 2008, Morelli et al. 2010), although recent modern and aDNA studies support a recent coalescent time for the haplogroup throughout Europe, in line with an expansion during the late Neolithic and Bronze Age18,41,42. Sex-biased migration processes43,44 are a common occurrence in humans, and such processes can give rise to differing patterns on autosomal versus uniparental markers.
Here, to provide novel insight to the peopling of Sardinia and its relationship to mainland populations, we analyze a collection of 3,514 individual whole-genome sequences sampled as part of the SardiNIA project2. We specifically assess whether the isolation of Sardinia is consistent with a dominantly EF or HG peopling and whether there is evidence for sex-biased processes. As part of our analysis we address the hypothesis of an ancestral connection between Sardinia and the Basque populations of Spain and France (the Basque also show high affinity with early Neolithic farmer aDNA20,45). We also address whether gene flow from North African populations to Sardinia32,46–48 has been substantial, as Sardinia may be the result of a recent multiway admixture involving sources from around the Mediterranean46.
A key factor in our analysis is the evaluation of internal sub-structure within Sardinia. Numerous studies have noted relative heterogeneity of small sub-populations within Sardinian27–29,33,49,50. Our dataset including broadly dispersed samples from across Sardinia51,52, as well as a deep sampling of several villages from the Lanusei Valley region of the Ogliastra province2,53 is however, especially well suited to systematically assess the extent and source of such heterogeneity (Figure 1). We show samples from the Ogliastra province and the broader, mountainous Gennargentu region have signs of elevated isolation. Thus, we frame our analyses of the demographic history of Sardinia by contrasting results for sampled individuals from the Gennargentu region with those outside of the region.
Figure 1: Geographical map of Sardinia.
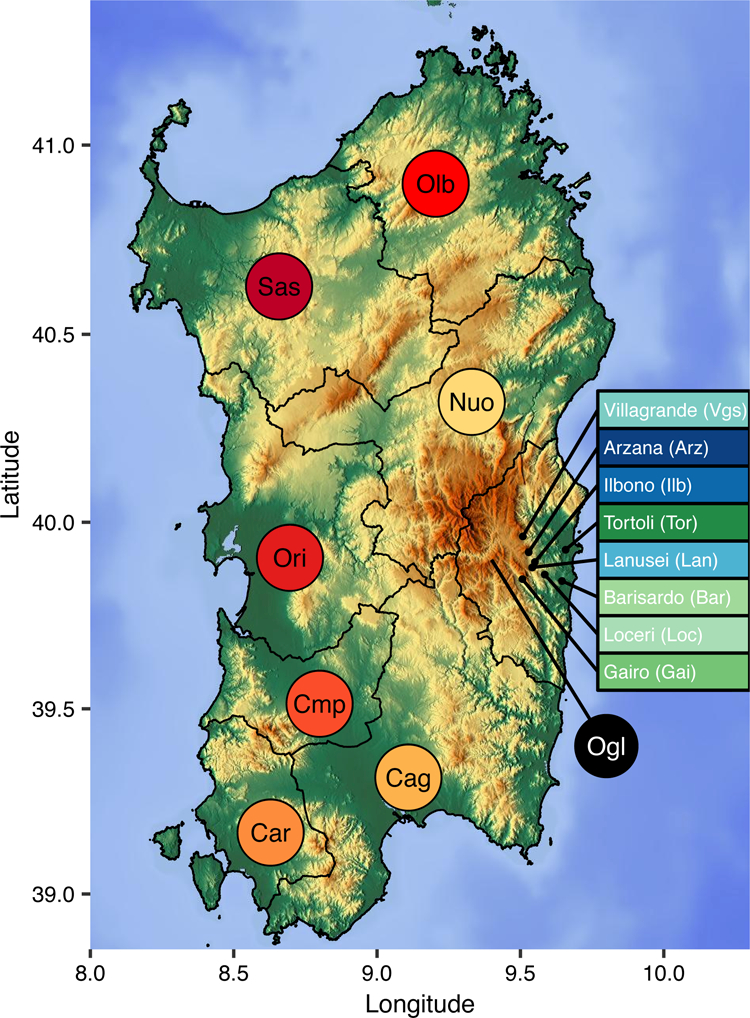
The provincial boundaries are given as black lines. The provinces are abbreviated as Cag (Cagliari), Cmp (Campidano), Car (Carbonia), Ori (Oristano), Sas (Sassari), Olb (Olbia-tempio), Nuo (Nuoro), and Ogl (Ogliastra). For sampled villages within Ogliastra, the names and abbreviations are indicated in colored boxes. Color corresponds to the color used in the PCA plot (Figure 2). The Gennargentu region referred to in the main text is the mountainous area shown in brown that is centered in western Ogliastra (Ogl) and southeastern Nuoro.
RESULTS
Before addressing broader-scale questions regarding Sardinian demographic history, we first examine population structure within Sardinia. We focus on a subset (N=1,577) of unrelated individuals with at least three grandparents originating from the same geographical location to lessen the confounding of recent internal migrations.
We find the strongest axis of genetic variation is between individuals from Ogliastra (Ogl) and those outside of Ogliastra (Figure 2A-B). A notable exception is the sub-population from Tortoli, a recently developed coastal city and the main seaport of the Ogliastra region. Samples from Tortoli show closer affinity in the PCA to samples from the western part of the island. Samples from outside Ogliastra show lower levels of differentiation by FST (Figure 2C) and higher levels of allele sharing amongst themselves (Figure 3), despite the greater geographical distance between populations. When we use a spatially explicit statistical method (EEMS, ref 54) for visualizing genetic diversity patterns, the resulting effective migration surface (Figure 2D) is consistent with high effective migration in western regions of Sardinia connecting the major populations centers of Cagliari (Cag), Oristano (Ori), and Sassari (Sas). Low effective migration rates separate these provinces from a broad area that extends to the mountainous Gennargentu Massif region, including inland Ogliastra to the west. The Gennargentu region is also where some of the Sardinian individuals in the Human Genome Diversity Project (HGDP) originate (A. Piazza, personal communication). We find the HGDP Sardinia individuals partially overlap with our dataset and include a subset that clusters near the Ogliastra sub-population (Figure S1, S2, Table S1, S2). Thus, we use the term “Gennargentu-region” to describe this ancestry component (red component in Figure 2B). Based on these results, and to simplify analyses going forward, we use individuals from the town of Arzana as a representative of the Gennargentu-region ancestry component and Cagliari as a representative of ancestry outside of the Gennargentu region.
Figure 2: Within-island population structure.
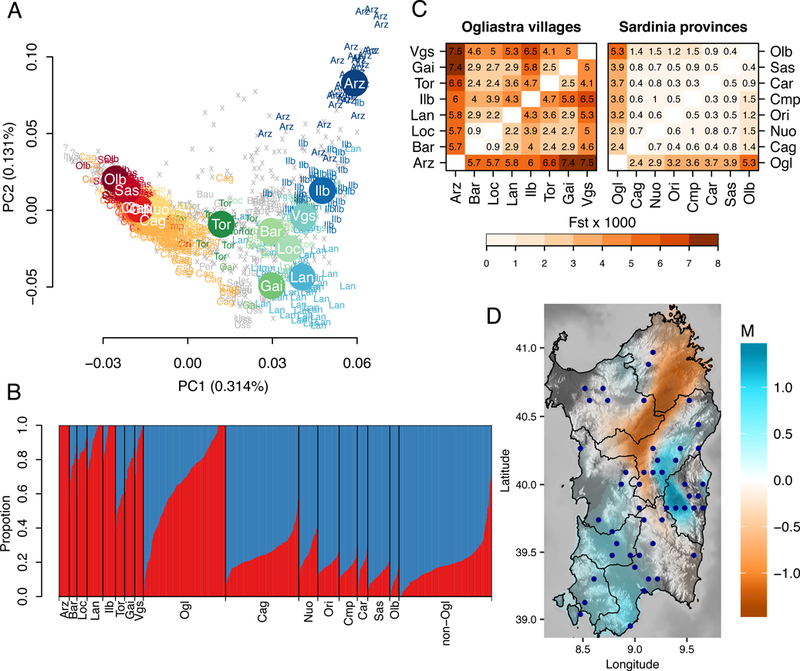
(A) Top two principal components of PCA based on 1,577 unrelated Sardinians. Each individual is labeled by geographical origin as defined by grandparental birthplaces (Methods); otherwise they are labeled with x or with ? for missing information. Subpopulations with less than 8 individuals are displayed in the background in grey color. (B) Admixture result at K = 2, which had the lowest cross-validation errors from K = 2 to K = 7 (not shown). Individuals from Ogliastra and outside of Ogliastra that were not assigned to a major location or had mixed grandparental origins are grouped under Ogl and non-Ogl, respectively. Bars for locations with individuals fewer than 40 (Bar, Gai, Loc, Olb, Tor, Vgs) were expanded to a fixed minimum width to aid visualization. (C) Genetic differentiation among Ogliastra villages (left) or among Sardinia provinces (right) as measured by Weir and Cockerham’s unbiased estimator of Fst. Ogliastra appears to be the most differentiated from other provinces and within Ogliastra the level of differentiation between villages is substantial (reaching as high as 0.0075 between Villagrande and Arzana), with Arzana being consistently well differentiated from other villages. (D) Estimated effective migration surface plot within Sardinia based on 181 Sardinians with all four grandparents born in the same location. Refer to Figure 1 for abbreviations of subpopulations.
Figure 3: Allele-sharing across the island and within Ogliastra.
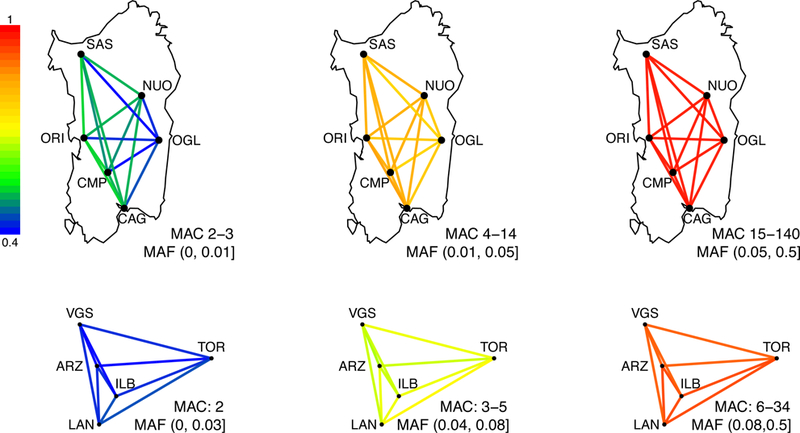
As a function of allele frequencies, allele-sharing across the island (top) and within Ogliastra (bottom) are shown. Allele-Sharing between a pair of population is defined as the ratios of the probability that two randomly drawn carriers of the allele of a given minor allele frequency are from different populations, normalized by the panmictic expectation, and visualized here by the color of the lines connecting two populations. Island-wide analysis used subpopulations with at least 70 individuals, and the minor allele counts of each of 1 million randomly selected variants were down-sampled to 140 chromosomes. Within Ogliastra analysis used subpopulations with at least 17 individuals, and the minor allele counts were down-sampled to 34 chromosomes.
Sardinia as an isolated Mediterranean population
To assess Sardinian variation in a regional context, we created a merged dataset of Sardinian, with Mediterranean populations from the Human Origins Array (HOA)20. PCA of this shows a one-dimensional isolation-by-distance pattern around the Mediterranean, from North Africa through the Near East and then towards Iberia55–58, with Sardinian samples clustering offset from southern European samples (Figure 4A). The effective migration surface shows the Mediterranean Sea isolating Sardinia from neighboring mainland populations, with stronger isolation between Sardinia and North Africa than Sardinia and mainland Europe (Figure 4B). An analysis with ADMIXTURE further supports this isolation of Sardinian populations (Figure 4C, Figure S3). Across analyses of varying number of population clusters, Sardinians tend to form a distinct cluster with all individuals near 100% ancestry (Figure S3); this is consistent with relatively high levels of differentiation (FST ~ 0.023–0.037 between the “blue” component and other ancestral components in Figure S3), which may results from extended divergence and/or elevated rates of drift.
Figure 4: Population structure relative to mainland Europeans.
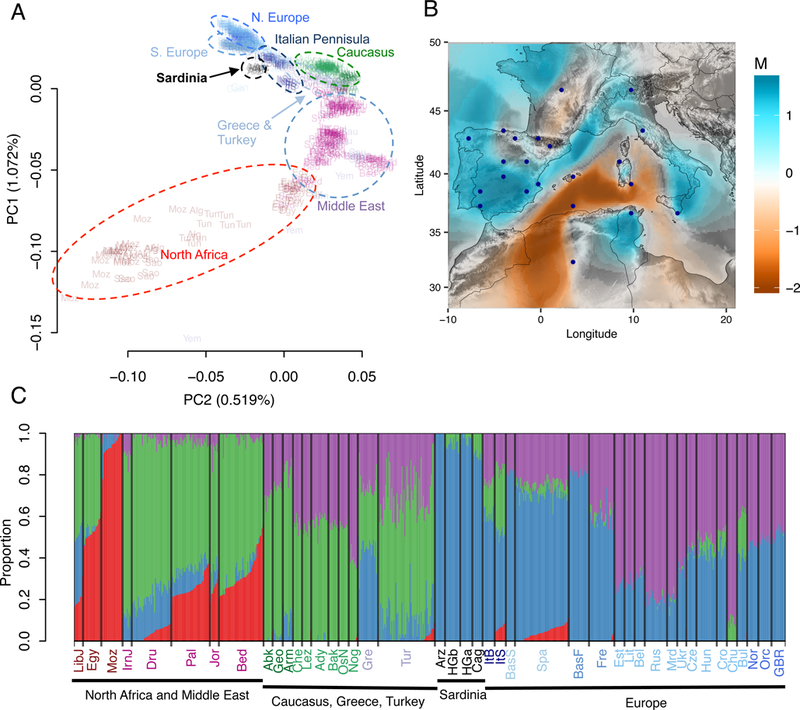
(A) Top two principal components of PCA of the merged dataset of Sardinia and Human Origins Array data. Populations are enclosed in dashed ellipses by major subcontinents. (B) Estimated effective migration surface result for the pan-Mediterranean analysis. (C) Admixture results at K = 4, which has the lowest cross-validation error in analysis from K = 2 to K = 15. For clarity, only populations with sample size > 8 are visualized. Arzana and Cagliari contained 100% and 89% of the European-dominant, “blue”, ancestry. Populations are ordered by sub-continental regions and then by population median values in PC1. See Figure S3 for the full result. Populations labels are color coded by major sub-continental regions. Abbreviations are: (North Africa) LibJ, Jewish in Libya; Egy, Egyptian; Moz, Mozabite; (Middle East) IrnJ, Jewish in Iran; Dru, Druze; Pal, Palestinian; Jor, Jordanian; Bed, Bedouin; (Caucasus) Abk, Abkhasian; Geo, Georgian; Arm, Armenian; Che, Chechen; Lez, Lezgin; Ady, Adygei; Bak, Balkar; OsN, North Ossetian; Nog, Nogai; (Turkey and Greece) Gre, Greece; Tur, Turkey; (Europe) Arz, Arzana; HGb, HGDP Sardinian; HGa, HGDP Sardinian; Cag, Cagliari; ItB, Bergamo; ItS, Sicilians; BasS, Spanish Basque; Spa, Spanish; BasF, French Basque; Fre, French; Est, Estonian; Lit, Lithuanian; Bel, Belarusian; Rus, Russian; Mrd, Mordovian; Ukr, Ukranian; Cze, Czech Republican; Hun, Hungarian; Cro, Croatian; Chu, Chuvash; Bul, Buglarian; Nor, Norwegian; Orc, Orcadian; GBR, British Great Britain.
Time-scale of divergence and population size history
While the relative isolation of Sardinia is apparent, the time-scale of the divergence is unclear. We used a recent approach that leverages information from both sequential Markovian coalescent and site-frequency-spectra based frameworks (SMC++, ref 59) to infer an approximate divergence time and population size history. SMC++ infers a divergence time reflecting the time point in an idealized two-population split model after which effective migration between populations becomes negligible, and thus it should be expected to underestimate divergence times when post-divergence gene flow has taken place. For the mainland European ancestry CEU and TSI populations, SMC++ infers a divergence time of 14.4 +/− 3.5 generations (or ~430 years ago). Sardinia is estimated as having a deeper divergence time with each of these populations with estimated divergence time of 143.3 +/− 1.3 gen (~4,300 years ago) between Sardinia and TSI, and 231.7 +/− 12.9 gen (~7,000 years ago) between Sardinia and CEU (Figure 5A). We complemented this approach with another commonly used method (MSMC, ref 60). Consistently, MSMC estimates Sardinia as more deeply diverged from the CEU and TSI populations, than CEU and TSI are to each other (Figure S4A). Both methods also show that Sardinia has had lower long-term effective population sizes, and lacks the signature of strong population growth typical of mainland European populations (Figure 5B, S4B). Sardinian populations from the Ogliastra province (Arzana, Lanusei, and Ilbono) showed consistently lower population size, while Sardinian populations from outside of Ogliastra (Cagliari) showed a pattern of growth more similar to that observed in CEU and TSI (Figure S5A, S5B). Sardinian populations from Ogliastra province showed a more ancient split time with mainland European populations, while Cagliari showed a more recent split time (Figure S5C).
Figure 5: Coalescent-based inference of demographic history using SMC++.
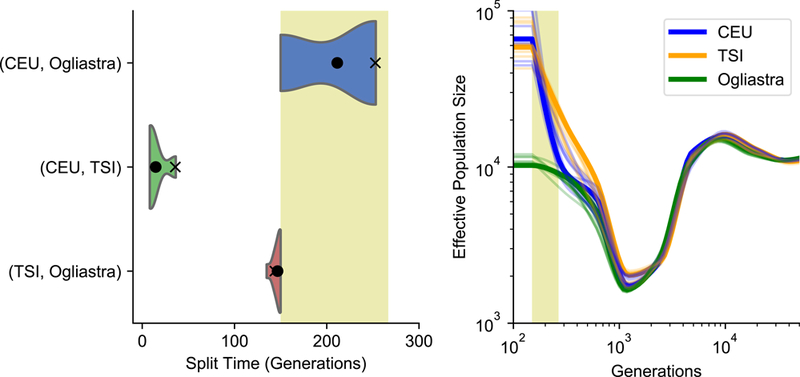
(A) Inference of population divergence times and (B) population size history. Analysis based on 4 focal individuals and 90 low-coverage samples from the combined dataset of Lanusei and Arzana individuals (Ogliastra) and 1000 Genomes CEU and TSI. The uncertainty and mean point estimates of population divergence time are shown using 10 bootstrap samples (the violin plot and the black dot, respectively). We also show the point estimate using all of the data by the black cross. For population size trajectories, we estimated the size until 150 generations in the past and use 10 internal spline knots when running SMC++. Uncertainty reflected through 10 bootstrap samples are also shown in the same but lighter colors. The orange shaded box denotes the Neolithic period, approximately 4500 to 8000 years ago, converted to units of generations assuming 30 years per generation.
Sardinia in relation to other Mediterranean populations
Due to its smaller long-term effective population size (Figure 5B), Sardinia is expected to have undergone accelerated genetic drift. To correct for this when measuring similarity to other mainland populations, we used “shared drift” outgroup-f3 statistics61, which measure the length of shared branch length between two populations relative to an outgroup. Using this metric, we find the Basque are the most similar to Sardinia, even more so than mainland Italian populations such as Tuscany and Bergamo (Figure S6A, S6B). We also tested the affinity between Sardinians and Basque with the D-statistics of the form D(Outgroup, Sardinia; Bergamo or Tuscan, Basque). In this formulation, significant allele sharing between Sardinia and Basque, relative to sharing between Sardinia and Italian populations, will result in positive values for the D-statistic. We find that Sardinia consistently showed increased sharing with the Basque populations compared to mainland Italians (|Z|> 4; Figure S6C), and the result was stronger when using the Arzana than Cagliari sample (D(Outgroup, Basque, CAG, ARZ) = 0.0020 and 0.0021 for French Basque and Spanish Basque, respectively; |Z| > 3.2). In contrast, sharing with other Spanish samples was generally weaker and not significant (Figure S6C), suggesting the shared drift with the Basque is not mediated through modern Spanish ancestry.
The ADMIXTURE and PCA analyses above (Figure 4) suggest that Sardinian samples, particularly outside of Ogliastra, may be admixed with mainland sources, as suggested previously46–48. For example, Cagliari individuals demonstrated ~10% of a non-Sardinian component (“green” in Figure 4C) that is found among extant individuals from Southern Europe, Middle East, Caucasus, and North Africa. To assess this further we used the f3-test for admixture62 and contrary to mainland Europeans, we found none of the Sardinian populations showed evidence of admixture (Figure S7). Because f3-based tests may lose power when applied to populations that have experienced extensive drift post-admixture62, we also tested for admixture using a complementary LD-based approach (ALDER, ref 47). Using this approach, a number of Sardinian populations outside of Ogliastra are inferred to be admixed (Table 1, Table S3). The inferred source populations are typically a mainland Eurasian population and a sub-Saharan African population. The admixture proportions range from 0.9% to 5% of sub-Saharan ancestry by the f4-ratio estimator62 with estimated admixture dates of approximately 62–101 generations (Table 1, Table S3).
Table 1: Evidence of admixture as inferred by ALDER.
P-value has been corrected for multiple hypothesis testing (both number of pairs of source populations and analysis-wide number of test populations). Admixture proportions of the sub-Saharan ancestry are estimated by the f4 ratio test, using Finnish and Chimp as the outgroups.
| Test Population |
Source 1 | Source 2 | Admixture Date (gen) | Fitted amplitude (x10−5) | P-value | Admixture Proportion | |||
|---|---|---|---|---|---|---|---|---|---|
| mean | s.e. | mean | s.e. | mean | s.e. | ||||
|
Outside of Ogliastra |
|||||||||
| Cagliari | Wambo | Spanish (Castilla y Leon) |
62.57 | 6.61 | 2.70 | 0.310 | 1.28×10−13 | 0.0039 | 0.0036 |
| Campidano | Luhya | Tuscan | 63.56 | 12.80 | 2.59 | 0.506 | 0.0285 | 0.0086 | 0.0030 |
| Carbonia | No successful fit | ||||||||
| Nuoro | No successful fit | ||||||||
| Olbiatempio | No successful fit | ||||||||
| Oristano | Wambo | Estonian | 101.05 | 20.08 | 3.51 | 0.638 | 0.0195 | 0.049 | 0.0029 |
| Sassari | Khwe | Lithuanian | 82.21 | 15.92 | 2.53 | 0.430 | 2.25×10−8 | 0.039 | 0.0027 |
|
Ogliastra |
|||||||||
| Arzana | No successful fit | ||||||||
| Barisardo | No successful fit | ||||||||
| Gairo | No successful fit | ||||||||
| Ilbono | No successful fit | ||||||||
| Lanusei | No successful fit | ||||||||
| Loceri | No successful fit | ||||||||
| Tortoli | No successful fit | ||||||||
| Villagrande | No successful fit | ||||||||
|
HGDP Sardinians |
|||||||||
| SarHGDPa | No successful fit | ||||||||
| SarHGDPb | No successful fit | ||||||||
Elevated Neolithic and pre-Neolithic ancestry
Ancient DNA studies have shown that across the autosome, Sardinians exhibit higher levels of Neolithic Farmer ancestry compared to mainland Europeans18,20. However, because previous samples from Sardinia have been limited in sample size, we revisit the question using our dataset and addressing within-island variation.
We confirm that Sardinians have the highest observed levels of shared drift with early Neolithic farming cultures (represented by the LBK380 sample from Stuttgart, Germany20; hereafter referred to as “Stuttgart”) and relatively low levels of shared drift with earlier hunter-gather cultures (represented by an aDNA sample from Loschbour rock shelter in Luxembourg20; hereafter referred to as “Loschbour”) (Figure 6A, Figure S8). As expected, the Neolithic farmer ancestry component is more abundant than the hunter-gatherer ancestry component across all Sardinian populations (Table S4). Surprisingly though, using supervised estimation of ancestry proportions18 based on aDNA, we found an indication of higher levels of Neolithic and pre-Neolithic ancestries in the Gennargentu-region, and higher levels of Steppe Pastoralist ancestry outside the region (Figure S9, Table S5). Investigating this further, we find that shared drift with Neolithic farmers and with pre-Neolithic hunter-gatherers are significantly correlated with the proportion of “Gennargentu-region” ancestral component estimated from ADMIXTURE analysis, while that with Steppe pastoralists is weakly negative and non-significantly correlated with Gennargentu-region ancestry ( |Z| > 6 for Neolithic farmers and pre-Neolithic hunter-gatherers, |Z| < 2 for Steppe pastoralists; Figure 6B, Table S6). Moreover, D-statistics of the form D(Outgroup, Ancient, Ogliastra, Non-Ogliastra) also support increased sharing with Neolithic and pre-Neolithic individuals, but not post-Neolithic individuals from the Steppe, in the Ogliastra samples (D = −0.0029 and −0.0035, |Z| = 6.1 and 6.8 when aDNA sample = Stuttgart and Loschbour, respectively; D = −0.0002, |Z| = 0.7, when aDNA sample = Yamnaya).
Figure 6: Similarity of ancient samples to populations across Europe and within Sardinia.
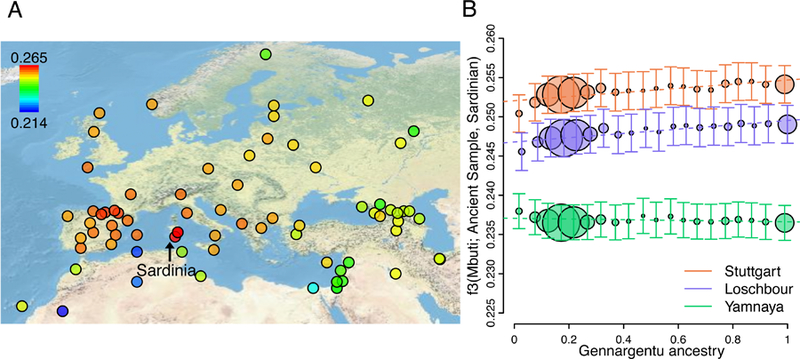
(A) Outgroup f3 statistics of the form f3(Mbuti; Stuttgart, X), where X is a population across the merged dataset of Sardinia and Human Origins Array data. Higher f3 values suggest larger shared drift between a pair of populations. Arrow indicates Sardinia populations. (B) Outgroup f3 statistics of the form f3(Mbuti; Ancient, Sardinian) across Sardinian samples binned in steps of 5% of Gennargentu ancestry estimated in Figure 2B. The increase of outgroup f3 statistics as function of ancestry is positive for Stuttgart and Loschbour (0.00263 and 0.00274, respectively), and slightly negative for Yamnaya (−4.4×10−4). Ancient samples used include a reference Neolithic farmer individual (Stuttgart, orange), a reference pre-Neolithic hunter-gather individual, (Loschbour, blue), and a reference Steppe population(Yamnaya, green) from the merged dataset with Haak et al. (Methods). Error bars represent the s.e. of the estimated f3 values from blocked jackknife procedure. Sizes of the circle are proportional to the number of samples per bin (max N = 281 per bin).
Together, these results confirm that relative to the mainland, Sardinia appears to harbor the highest amounts of Neolithic farmer ancestry and very little of the pre-Neolithic hunter-gatherer or Bronze Age pastoralists ancestries. We further find within-island variation of ancestry. Specifically, we find that with increasing level of isolation (represented by increasing level of the Gennargentu ancestry), there is greater Neolithic farmer and pre-Neolithic hunter-gatherer ancestry, while the Steppe ancestry generally showed no significant correlation.
Sex-biased demography in prehistoric Sardinia
The relatively high frequencies and low divergences within two particular Y-chromosome haplogroups32,35–38 (I2a1a1 at ~39% and R1b1a2 at ~18%) in Sardinia are a notable feature of Sardinian genetic variation. Neither haplogroup is typically affiliated with Neolithic ancestry in ancient DNA data, raising the potential of sex-biased processes in the history of Sardinia.
To investigate further, we first use ADMIXTURE and contrast the inferred Gennargentu-region ancestry on the X chromosome versus the autosome. Intriguingly, on average, we find a higher proportion of the Gennargentu-region ancestry (“red” component in Figure S10) on the X-chromosome (37%) than on the autosome (30%, P < 1×10−6 by permutation). The Gennargentu-region ancestry is correlated with Neolithic or pre-Neolithic ancestries rather than more recent Bronze Age Steppe ancestry (Figure 6B), suggesting this result may be due to sex-biased processes in which more females than males carried the non-Steppe ancestries. We also examined relative levels of nucleotide diversity on the X-chromosome versus the autosome. Doing so, we find that Sardinia shows a high ratio of X-to-A diversity, particularly comparing to most mainland European populations (Figure S11), suggesting Sardinian demographic history has had a relatively low male effective size.
DISCUSSION
We investigated the fine-scale population structure and demography of the people of Sardinia using a whole-genome sequences of 3,514 Sardinians with detailed self-reported ancestry that goes back two generations. The genotype calling leveraged extensive haplotype sharing to produce a high quality call set2, and we integrated the data with the 1000 Genomes, HGDP, and Human Origin Array reference sets. From our analyses, we were able to confirm a number of major features of previous analyses and provide more detail regarding the isolation between Sardinia and the mainland.
Our analysis of divergence times suggests the population lineage ancestral to modern-day Sardinia was effectively isolated from the mainland European populations approximately 140–250 generations ago, corresponding to approximately 4,300 to 7,000 years ago assuming a generation time of 30 years and mutation rate of 1.25×10−8 per basepair per generation. However, these quantitative estimates should be treated with caution, as the SMC++ model assumes an idealize model of homogeneous ancestries with no post-divergence gene-flow. Nevertheless, in terms of relative values, the divergence time between Northern and Southern Europeans are much more recent than either is to Sardinia, signaling the relative isolation of Sardinia from mainland Europe.
We documented fine-scale variation in the ancient population ancestry proportions across the island. The most remote and interior areas of Sardinia – the Gennargentu Massif covering the central and eastern regions, including the present day province of Ogliastra, are thought to have been the least exposed to contact with outside populations27,29,50. We find pre-Neolithic hunter-gatherer and Neolithic farmer ancestry are enriched in this region of isolation. Under the premise that Ogliastra has been more buffered from recent immigration to the island, one interpretation of the result is that the early populations of Sardinia were an admixture of the two ancestries, rather than the pre-Neolithic ancestry arriving via later migrations from the mainland. Such admixture could have occurred principally on the island or on the mainland prior to the hypothesized Neolithic era influx to the island. Under the alternative premise that Ogliastra is simply a highly isolated region that has differentiated within Sardinia due to genetic drift, the result would be interpreted as genetic drift leading to a structured pattern of pre-Neolithic ancestry across the island, in an overall background of high Neolithic ancestry.
We found Sardinians show a signal of shared ancestry with the Basque, in terms of the outgroup f3 shared-drift metric. This consistent with long-held arguments of a connection between the two populations, including claims of Basque-like non-Indo-European language words among Sardinian placenames63. More recently the Basque have been shown to be enriched for Neolithic farmer ancestry20,45 and Indo-European languages have been associated with Steppe population expansions in the post-Neolithic Bronze Age18,23. These results support a model in which Sardinians and the Basque may both retain a legacy of pre-Indo-European, Neolithic ancestry45. To be cautious, while it seems unlikely, we cannot exclude that the genetic similarity between the Basque and Sardinians is due to an un-sampled pre-Neolithic population that has affinities with the Neolithic representatives analyzed here.
We also examined possible sources of African admixture to Sardinia. Prior to our studies, there have been reports of a minor proportion (0.6% to 2.9%) of sub-Saharan admixture47,48 and a multi-way admixture involving an African source46 in the HGDP Sardinians. In light of the close geographical proximity of Sardinia and North Africa, as well as the substantial admixture proportion from North Africa in Southern Europe56, we tested for admixture using modern North African reference populations included in the Human Origins Array data (Tunisia, Algeria, Mozabite, Egypt, and Saharawi). We found the best proxy for African admixture is sub-Saharan African populations, rather than Mediterranean North African populations, and we inferred the date of admixture as approximately 1,800–3,000 years ago (assuming 30 years per generation). The lack of a strong signal of North African autosomal admixture may be due to inadequate coverage of modern North African diversity in our reference sample, such that the sub-Saharan component of admixture we detect may be an indirect reflection of recent North African admixture (particularly if the North African source was admixed with sub-Saharan Africans; for example, see ref. 64). Alternatively, it may be due to a poor representation of ancestral North Africans. Present-day North African ancestry reflects large-scale recent gene flow during the Arab expansion (~1,400 years ago57). The sub-Saharan African admixture observed in the non-Ogliastra samples could be mediated through an influx of migrants from North Africa prior to the Arab expansion, for example during the eras of trade relations and occupations from the Phoenicians, Carthaginians, and Romans (~700 B.C.- ~200 B.C.; ref. 10).
While we confirm the Sardinians principally have Neolithic ancestry on the autosomes, the high frequency of two Y-chromosome haplogroups32,35–38 (I2a1a1 at ~39% and R1b1a2 at ~18%) that are not typically affiliated with Neolithic ancestry is one challenge to this model. Whether these haplogroups rose in frequency due to extensive genetic drift and/or reflect sex-biased demographic processes has been an open question. Our analysis of X vs autosome diversity suggests a smaller effective size for males, which can arise due to multiple processes, including polygyny, patrilineal inheritance rules, or transmission of reproductive success65. We also find the genetic ancestry enriched in Sardinia is more prevalent on the X chromosome than the autosome, suggesting that male lineages may more rapidly trace back to the mainland. Considering that the R1b1a2 haplogroup may be associated with post-Neolithic Steppe ancestry expansions in Europe18, and the recent timeframe when the R1b1a2 lineages expanded in Sardinia32, the patterns raise the possibility of recent male-biased Steppe ancestry migration to Sardinia, as has been reported among mainland Europeans at large44 (though see 66,67). Such a recent influx is difficult to square with the overall divergence of Sardinian populations observed here. Thus, our results make clear that future studies aimed to understand sex-biased processes in the history of Sardinia and European populations in general will be illuminating, especially as systems of mating and dispersal may have shifted alongside modes of subsistence68.
For the purposes of understanding complex trait evolution in Sardinian history, the results suggest that while Sardinia has clearly had influence from pre-Neolithic sources and contact with Steppe ancestry populations, the demographic history is one of substantial isolation and abundant Neolithic ancestry relative to the mainland. For traits with a strong sex-linked component our results encourage accounting for the sex-biased processes detected here. The relatively constant size of Sardinian populations predicts there has been less of an influx of rare variants69, as well as an increase of homozygosity, relative to other expanding populations. These two factors may increase the impact of dominance components of variation70, and reduce the allelic heterogeneity of complex traits71–73. Armed with a better understanding of Sardinian prehistory and demographic events, we anticipate a more nuanced understanding of complex trait variation and disease incidences in Sardinia. The affinity to Neolithic farmer populations (and to a lesser extent, pre-Neolithic hunter-gatherer populations) also means Sardinia is a potential reservoir for variants that may have been lost in mainland Europeans.
ONLINE METHODS
Cohort description.
We included in this study individuals from the SardiNIA/Progenia longitudinal study of aging2,53 based in the Ogliastra region and from the case-control studies of Multiple Sclerosis51 and Type 1 Diabetes52 across the general population of Sardinia. For the case-control study cohort, we required individuals to have at least three Sardinian grandparents. All participants gave informed consent, with protocols approved by institutional review boards for the University of Cagliari, the National Institute on Aging, and the University of Michigan.
Whole-genome sequenced Sardinian dataset.
The dataset includes 3,514 individuals sequenced at low-coverage (average coverage 4.2x) and 131 individuals sequenced at high-coverage (average coverage 36.7x). 2,090 individuals belong to the SardiNIA cohort, the remaining 1,424 are derived from the case-control study. A subset of 2,120 low-coverage individuals was previously described2. The additional 1,394 individuals and the 131 high coverage individuals were aligned, recalibrated and quality checked using the same criteria2 to guarantee sample uniformity. Variant calling was performed using GotCloud as described in ref. 2, which also include a step of genotyping refinement with Beagle74 to increase genotype accuracy in the low-coverage individuals. For chromosome X we performed standard variant calling to generate genotype likelihoods for each individual genotype. We then set heterozygotes GL to 500 among males and ran genotype refinement as described for autosomal markers. The most likely homozygous genotypes for males generated by Beagle are then converted to haploid genotypes.
To process the high-coverage data, we created a pileup of raw sequence reads using samtools v0.2 (“samtools mpileup”), filtering out bases with a base quality score < 20 and reads with a map quality score < 20. We then used the bcftools (v1.2) variant caller (“bcftools call”) in conjunction with custom scripts (see URLs) to call single nucleotide polymorphisms.
Filtering individual samples by poor sequencing quality and relatedness.
For each individual we examined the proportion of imputed genotypes with highest posterior genotype probability less than 0.9 and removed 8 outlier individuals with an excessive proportion (> 0.008), likely due to an overall low coverage of these samples.
To prune the dataset for related individuals, we first extracted a subset of 153.7K SNPs with maximum pairwise r2 of 0.2 (pruned from 1.21M SNPs overlapping between the Sardinian whole genome sequence data and HapMap 3), and then computed the genome-wide proportion of pairwise identity by descent (pihat) using PLINK v1.08. The distribution of pihat showed distinct modes corresponding to different degrees of relatedness, as well as extensive low level sharing (pihat < 0.1) between individuals, consistent with long-term isolation. We removed one individual from each pair of individuals with pihat > 0.07 to retain 1,577 approximately unrelated individuals, including 615 individuals from the SardiNIA sample and 964 individuals from the case-control cohort.
Defining sample origin based on self-reported grandparental ancestry.
We assigned a 4-part ancestral origin to each study participants based on self-reported geographical birth locations of each of their parents and grandparents. We first categorize each location by three levels of resolutions: (1) macro-regions (e.g. Sardinia, South Italy, France, Tunisia), (2) provinces within Sardinia (e.g. Cagliari, Sassari, Ogliastra), (3) town level (e.g. Arzana, Lanusei, Tortoli). For each parental lineage, we preferentially used grandparental origin if available, or parental information to represent the ancestry from both grandparents if grandparental information is missing. Given the more detailed information provided by the participants of the SardiNIA project, we defined SardiNIA samples down to town resolution, but only define the case-control samples down to the province resolution unless noted otherwise. In initial PCA and ADMIXTURE analysis stratified by these labels, we find that the genetic ancestry did not significantly differ for individuals having 4- or 3- parts of their ancestry coming from a particular location, nor did it differ by whether the 4-part origin came from self-reported grandparental origin or self-reported parental origin (data not shown). However, we did observe more heterogeneity among individuals with 2-part origin. Thus unless noted otherwise, for any analysis where discrete geographical labeling is used we restrict to individuals having at least 3 out of the 4-part origin from the same geographical location.
Merging with other datasets.
To merge the Sardinia sequenced data with the Human Origin Dataset20 we repeated the variant calling pipeline in the Sardinian dataset specifically at the 600,841 variable sites released with the Human Origin Dataset. Comparisons of this callset with the array genotypes on the same individuals suggest that the resulting genotype calls are of high quality (genotype discordant rate = 0.43% and 0.24% at heterozygous sites and all call sites, respectively). We then merged this callset with the Human Origins dataset across the autosome (594,924 SNPs), of which 95,853 are monomorphic in Sardinia. Some basic information and summary statistics of the reference panels and populations used in the merge can be found in Table S7. Unless denoted specifically, the Human Origins merge is with the Lazaridis et al data20, which also provided ancient DNA samples for the Neolithic Farmer (LBK380, or “Stuttgart”), and the pre-Neolithic Hunter-gatherer (“Loschbour”). For estimating mixture proportions in a 3-way model of European admixture, we merged our dataset with the version of Human Origin Array data published by ref. 18, which contains additional ancient samples (particularly additional Early Neolithic farmers, “LBK_EN”, and post-Neolithic Steppe pastoralists, “Yamnaya”) but fewer SNPs (354,212 SNPs).
PCA.
Sardinia-specific PCA was conducted using all unrelated individuals genotyped at SNPs found in HapMap 3 (ref. 75). For regional PCA, since a significant imbalance of sample sizes across populations may distort the PCA, a random subset of 10 unrelated Sardinians from Arzana and Cagliari were chosen to represent Sardinia and merged with the Human Origin dataset. Only populations from North Africa, Middle East, Caucasus, and Europe from the Human Origins Array data were included. PCA analysis was performed using EIGENSTRAT v5.0 after removing one SNP of each pair of SNPs with r2 ≥0.8 (in windows of 50 SNPs and steps of 5 SNPs) as well as SNPs in regions known to exhibit extended long-range LD (ref. 76).
ADMIXTURE.
Similar to the PCA analyses, Sardinia-specific Admixture analysis was conducted using all unrelated individuals genotyped at SNPs found in HapMap 3. The regional analysis was conducted with sub-sampling of 10 Sardinians each with self-reported Cagliari and Arzana ancestries, merged with individuals from relevant populations from the Human Origins Array. Analysis was performed using Admixture v1.22, following the recommended practice in the manual for LD filtering (removing one SNP of each pair of SNPs with r2 ≥0.1 in windows of 50 SNPs and steps of 5 SNPs). Ten independent unsupervised runs for K = 2 to 15 were performed, and for each value of K the run with maximum likelihood as estimated by the program is retained.
EEMS.
The EEMS analysis was conducted using the same set of SNPs as the PCA. As EEMS requires fine-scale geographically indexed samples, we only used individuals whose four grandparents were all born in the same location at the town level. This resulted in 181 individuals across the island for analysis in the Sardinia-only analysis. For the Mediterranean region analysis, the merged dataset with Human Origins Array was used. Because of the scale of the Mediterranean region, we only used the two Sardinian populations of Cagliari and Sassari, the two Sardinian populations with the largest sample sizes that are geographically sufficiently distant to not be merged by EEMS. Populations from Human Origins Array data used in this analysis are: Spanish (Castilla y Leon, Castilla la Mancha, Extremadura, Cantabria, Cataluna, Valencia, Murcia, Andalucia, Baleares, Aragon, Galicia), Spanish_North, French_South, French, Bergamo, Italian_South, Tuscan, Sicilian, Mozabite, Algeria, Tunisian, and Spanish_Basque. We used the default settings for the EEMS hyper-parameters. For each run, we ran a burn-in of 1 million iterations followed by an additional 1 million iterations with posterior samples taken every 1000 iterations. We assessed the convergence of the MCMC chain by the posterior probability trace plot. We further assessed model fit by comparing the expected distance fitted by EEMS to the raw observed distances. Repeating the analysis with different combinations of grid sizes and random seeds produced qualitatively similar results. Results were displayed geographically using the mapdata package and ggplot2 in R.
f3/D.
For admixture f3 analyses, aimed to test for evidence of admixture in a target population, we computed the f3 statistic using all pairs of population from Europe (including Turkey/Greece, Italian peninsula and Iberian peninsula), Caucasus, Middle East, North Africa, and Subsaharan Africa (Table S7). For the outgroup f3 analyses, aimed to estimate the amount of shared drift between a pair of populations, we computed the f3 statistics between a Sardinian (Arzana or Cagliari) and another mainland population, while using the Mbuti individuals as the outgroup. Both f3 and D statistics were calculated using Admixtools v3.0 (ref. 62). Statistical significance was assessed using the default blocked jackknife implementation in Admixtools. Results were displayed geographically using ‘nps’ set of maps available through the OpenStreetMap package in R.
ALDER.
We used the full set of Human Origin Array SNPs, except for SNPs lacking recombination map information (based on sex-averaged deCODE map77), or found in regions of long range LD (ref. 76). For each Sardinian test populations, we tested all pairwise combinations of mainland populations as in the f3 admixture analysis using ALDER v1.03.
In contrast to the approach taken by Loh et al.47, we opted to be conservative with interpreting ALDER results where the 2-reference LD decay fit is inconsistent with 1-reference LD decay fit from the program (i.e. a “successful fit” with warnings). We find that in general, successful fits where the 2-reference decay curve and the 1-reference decay curve agree with each other, the amplitude of the fit tend to be negatively correlated with the f3 statistics of the same source/target population triplets, even if the f3 statistics may be positive (i.e. suggesting no evidence of admixture). However, when the decay curve under the two scenarios do not agree with each other, the correlation with f3 statistics is also poorer and/or becomes positive. These results suggest complications from the shared past demography between source and target populations could have influenced the LD decay curve fitting47. Thus we reported only successful fits for up to 5 pairs of populations with the highest estimated amplitude among those with significant evidence of admixture (P < 0.05 after multiple-testing correction by ALDER and Bonferroni correction for testing 15 Sardinian subpopulations), if available (Table S3). The pairs of source populations with the highest estimated amplitude of LD decay are the populations closest to the true ancestral populations47 among those available in our analyses. We thus estimate the admixture proportion using these pairs of source population via f4 ratio test62 (Table S3).
Estimating ancient population mixture proportions.
Following Haak et al18, we estimate mixture proportion with respect to the early European farmers (LBK_EN), western hunter-gatherers (Loschbour), and the Yamnaya steppe pastoralists (Yamnaya) using the command lsqlin in matlab, based on a matrix of relationships between the test sample, the three ancient reference samples and a set of 15 worldwide outgroups (Ami, Biaka, Bougainville, Chukchi, Eskimo, Han, Ju_hoan_North, Karitiana, Kharia, Mbuti, Onge, Papuan, She, Ulchi, and Yoruba)18,20. We assessed the uncertainty of these estimate with a blocked jackknife with 10 Mb blocks.
SMC++.
Our main SMC++ analysis is based on 4 high-coverage unrelated individuals and 90 low-coverage unrelated individuals. The CEU and TSI high-coverage individuals were sequenced by Complete Genomics (see URLs), and variants were called using the same pipeline that was applied to the 131 high-coverage Sardinian samples described above and merged with 90 randomly selected individuals from 1000 Genomes. For Sardinia, we selected 2 high-coverage individuals each from Lanusei and Arzana (all with 4 grandparents from each village), and 50 and 40 low-coverage individuals each from Lanusei and Arzana, to match 90 samples in 1000 Genomes. Only bi-allelic sites were used. Moreover, we kept only regions where reads can be uniquely mapped (see URLs). For our supplementary analysis to explore finer-scale differentiation among Sardinian populations, we selected 2 high-coverage individuals each from CEU, TSI, Arzana, Lanusei, Ilbono, and Cagliari, merged with 40 low-coverage individuals from each population. The high-coverage Sardinian individuals all have 4 grandparents from the same population, with the exception of one Cagliari individual who has 3 grandparents from Cagliari and one grandparent with no information.
We used SMC++ v1.9.3 to estimate population size trajectories and divergence time between populations. We used default parameters except that we set t1, the most recent time point for population size history inference, to 150 generations and the number of spline knots used to anchor the size history to 10. We had simulated a plausible European population growth model78 and found that this combination of t1 and number of knots produced the best fitted population size trajectory from the simulated demography (data not shown). We evaluated the variability in the estimated size trajectory and divergence times by resampling 10 replicates of the genome in blocks of 10 Mb. We note that in bootstrap samples we sometimes observe bimodally distributed estimates of divergence times. Therefore we report both the point estimates for divergence times from both the whole dataset and the average of the 10 bootstrap replicates, as reflected in Figure 5A and Figure S5C. Importantly, the order of divergence time estimates among the pairs we examined remain unchanged. An alternative combination that could also recapitulate the simulated demography is t1 = 100 and knots = 12, however we find this combination of parameters produces less stable population size trajectories for the recent past based on the bootstrapping results. Following the practice in Terhorst et al. (ref 59) we used a mutation rate of 1.25×10−8 per basepair per generation to scale time.
MSMC.
We additionally phased the high-coverage variant callset from SMC++ analysis with SHAPEIT2 (v2, r790) using the 1000 Genomes Phase 3 reference panel (see URLs). Input files for msmc were then generated using custom scripts from the msmc github repository (see URLs). We used msmc v0.1.0 to estimate effective population size and cross-coalescent rates. For effective population size inference, we used 4 individuals (8 phased haplotypes) from each of CEU, TSI, Arzana, and Lanusei; for cross-coalescent rate inference we used pairs of two individuals from each population. We define the estimated divergence time between a pair of populations as first time point at which the cross-coalescent rate is at or above 0.5. The mutation rate used to scale time is 1.25×10−8 per basepair per generation.
Allele-sharing.
We computed the allele-sharing ratios between pairs of populations as the probability that two randomly drawn carriers of the allele of a given minor allele frequency are from different populations, normalized by the panmictic expectation79,80. Specifically, we defined and as the relative fraction of sample sizes and and j as the frequency of the minor allele in populations and . Then the probability of two randomly drawn carriers are from different populations is the probability of sampling two carriers from different populations over the total probability of sampling two carriers. In terms of the variables defined above, this is . This quantity is normalized by the panmictic expectation, which is .
X vs autosome analysis.
Both autosomal and chromosome X data were first filtered to retain only SNPs with minor allele frequencies > 0.02 in Europeans (1000 Genomes Europeans + 1,577 unrelated Sardinians). This left 6,740,788 SNPs on the autosome and 221,434 SNPs on the X chromosome. SNPs were then pruned by LD using 868 unrelated Sardinian females by removing one SNP of each pair of SNPs with r2 ≥0.1 (in windows of 50 SNPs and steps of 5 SNPs), leaving 433,704 autosomal SNPs and 18,918 X chromosome SNPs. We ran ADMIXTURE with K = 3, using all of unrelated Sardinians and TSI from 1000 Genomes. In general, TSI individuals form the first ancestry component, while the Sardinians are distributed in two different components as was observed in Figure 1B. We then compared between the autosome and chromosome X the distribution of the component showing the largest FST from the TSI-dominated component to evaluate excess of the “Sardinian-specific” ancestry on chromosome X. Significance was assessed by permuting individual ancestries 1 million times, as well as by bootstrapping individuals. Analysis was done on both male and females using Admixture v. 1.3 (ref. 81), and confirmed by rerunning the analysis in the subset of 868 unrelated Sardinian females and in 839 non-Arzana Sardinian females. We also compared chr X to chr 7 only. SNPs were filtered similarly as above; in total we compared 26,164 SNPs on chr 7 to 18,918 SNPs on chromosome X.
To compare heterozygosity of chromosome X versus autosome, we utilized the whole-genome sequencing data released by the Simons Genome Diversity Project82 (see URLs; accessed March 2015). We restricted analysis to only the 21 female Europeans in SGDP, and only using data from the presumed neutral regions of the autosome and the X-chromosome (A.E. Woerner, personal communication; see URLs) consisting of 3,606 and 787 10kb-windows, respectively. Heterozygosity is computed as the number of non-missing heterozygous site of an individual normalized by the total genomic span in the neutral region. We then compared the ratio of the heterozygosity across populations.
Data Availability.
Allele frequency summary data analyzed in the study will be deposited to EGA under accession number EGAS00001002212. The disaggregated individual-level sequence data for 2105 samples (adult volunteers of the SardiNIA cohort longitudinal study) analyzed in this study are from Sidore et al (2015) and are available from dbGAP under project identifier phs000313 (v4.p2). The remaining individual-level sequence data are from a case-control study of autoimmunity from across Sardinia, and per the obtained consent and local IRB, these data are only available for collaboration by request from the project leader (Francesco Cucca, Consiglio Nazionale delle Ricerche, Italy).
URLs
MSMC and related utilities repository: https://github.com/stschiff/msmc-tools
1000 Genomes phasing reference panel: https://mathgen.stats.ox.ac.uk/impute/impute_v2.html#reference
Complete Genomics Public Data: http://www.completegenomics.com/public-data/69-genomes/
Uniquely mapped reads: https://oc.gnz.mpg.de/owncloud/index.php/s/ RNQAkHcNiXZz2fd
Simons Genome Diversity Project: https://www.simonsfoundation.org/life-sciences/simons-genome-diversity-project-dataset/
Neutral regions of the autosome and chromosome X: http://hammerlab.biosci.arizona.edu/Neutralome/Neutralome.bed
Supplementary Material
ACKNOWLEDGEMENTS
This study was funded in part by the National Institutes of Health, including support by National Human Genome Research Institute grants HG005581, HG005552, HG006513, HG007022 to G.R.A., and HG007089 to J.N.; by National Heart Lung and Blood Institute grant HL117626 to G.R.A.; by National Institute of General Medical Sciences grant GM108805 to J.N., F32GM106656 to C.W.K.C., and T32GM007197 to J.H.M. and A.B.; by National Institute of Neurological Disorders and Stroke T32NS048004 to C.W.K.C.; by the Intramural Research Program of the NIH, National Institute on Aging, with contracts N01-AG-1–2109 and HHSN271201100005C to the Italian National Research Council; and by National Science Foundation fellowship DGE-1746045 to J.H.M. and H.A.. This research was also supported by Sardinian Autonomous Region (L.R. no. 7/2009) grant cRP3–154, PB05 InterOmics MIUR Flagship Project, and grant FaReBio2011 “Farmaci e Reti Biotecnologiche di Qualità” to F.C.. We also would like to thank Iosif Lazaridis, Pontus Skoglund, Nick Patterson, Sohini Ramanchandran, Alan Rogers, and Robert Brown for discussion and technical assistance, as well as members of the Novembre and Lohmueller labs for constructive comments regarding this research.
Footnotes
COMPETING FINANCIAL INTEREST
The authors declared no competing interests
REFERENCES
- 1.Lettre G & Hirschhorn JN Small island, big genetic discoveries. Nat Genet 47, 1224–5 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Sidore C et al. Genome sequencing elucidates Sardinian genetic architecture and augments association analyses for lipid and blood inflammatory markers. Nat Genet 47, 1272–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Naitza S et al. A genome-wide association scan on the levels of markers of inflammation in Sardinians reveals associations that underpin its complex regulation. PLoS Genet 8, e1002480 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zoledziewska M et al. Height-reducing variants and selection for short stature in Sardinia. Nat Genet 47, 1352–6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steri M et al. Overexpression of the Cytokine BAFF and Autoimmunity Risk. N Engl J Med 376, 1615–1626 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cucca F et al. The distribution of DR4 haplotypes in Sardinia suggests a primary association of type I diabetes with DRB1 and DQB1 loci. Hum Immunol 43, 301–8 (1995). [DOI] [PubMed] [Google Scholar]
- 7.Marrosu MG et al. The co-inheritance of type 1 diabetes and multiple sclerosis in Sardinia cannot be explained by genotype variation in the HLA region alone. Hum Mol Genet 13, 2919–24 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Pugliatti M et al. The epidemiology of multiple sclerosis in Europe. Eur J Neurol 13, 700–22 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Cao A & Galanello R Beta-thalassemia. Genet Med 12, 61–76 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Dyson SL & Rowland RJ Archaeology and History in Sardinia from the Stone Age to the Middle Ages: Shepherds, Sailors, & Conquerors, 240 (University of Pennsylvania Museum of Archaeology and Anthropology, Philadelphia, PA, 2007). [Google Scholar]
- 11.Eaves IA et al. The genetically isolated populations of Finland and sardinia may not be a panacea for linkage disequilibrium mapping of common disease genes. Nat Genet 25, 320–3 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Calo CM, Melis A, Vona G & Piras IS Sardinian Population (Italy): a Genetic Review. International Journal of Modern Anthropology 1, 39–65 (2008). [Google Scholar]
- 13.Cavalli-Sforza LL & Piazza A Human genomic diversity in Europe: a summary of recent research and prospects for the future. Eur J Hum Genet 1, 3–18 (1993). [DOI] [PubMed] [Google Scholar]
- 14.Barbujani G & Sokal RR Zones of sharp genetic change in Europe are also linguistic boundaries. Proc Natl Acad Sci U S A 87, 1816–9 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zavattari P et al. Major factors influencing linkage disequilibrium by analysis of different chromosome regions in distinct populations: demography, chromosome recombination frequency and selection. Hum Mol Genet 9, 2947–57 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Elhaik E et al. Geographic population structure analysis of worldwide human populations infers their biogeographical origins. Nat Commun 5, 3513 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cann HM Human genome diversity. C R Acad Sci III 321, 443–6 (1998). [DOI] [PubMed] [Google Scholar]
- 18.Haak W et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 522, 207–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keller A et al. New insights into the Tyrolean Iceman’s origin and phenotype as inferred by whole-genome sequencing. Nat Commun 3, 698 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Lazaridis I et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature 513, 409–13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li JZ et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science 319, 1100–4 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Skoglund P et al. Origins and genetic legacy of Neolithic farmers and hunter-gatherers in Europe. Science 336, 466–9 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Allentoft ME et al. Population genomics of Bronze Age Eurasia. Nature 522, 167–72 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Hofmanova Z et al. Early farmers from across Europe directly descended from Neolithic Aegeans. Proc Natl Acad Sci U S A 113, 6886–91 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mathieson I et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature 528, 499–503 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sikora M et al. Population genomic analysis of ancient and modern genomes yields new insights into the genetic ancestry of the Tyrolean Iceman and the genetic structure of Europe. PLoS Genet 10, e1004353 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghirotto S et al. Inferring genealogical processes from patterns of Bronze-Age and modern DNA variation in Sardinia. Mol Biol Evol 27, 875–86 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Fraumene C, Petretto E, Angius A & Pirastu M Striking differentiation of sub-populations within a genetically homogeneous isolate (Ogliastra) in Sardinia as revealed by mtDNA analysis. Hum Genet 114, 1–10 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Morelli L et al. Frequency distribution of mitochondrial DNA haplogroups in Corsica and Sardinia. Hum Biol 72, 585–95 (2000). [PubMed] [Google Scholar]
- 30.Pala M et al. Mitochondrial haplogroup U5b3: a distant echo of the epipaleolithic in Italy and the legacy of the early Sardinians. Am J Hum Genet 84, 814–21 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olivieri A et al. Mitogenome Diversity in Sardinians: A Genetic Window onto an Island’s Past. Mol Biol Evol 34, 1230–1239 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Francalacci P et al. Low-pass DNA sequencing of 1200 Sardinians reconstructs European Y-chromosome phylogeny. Science 341, 565–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caramelli D et al. Genetic variation in prehistoric Sardinia. Hum Genet 122, 327–36 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Vona G The peopling of Sardinia (Italy): history and effects. International Journal of Anthropology 12, 71–87 (1997). [Google Scholar]
- 35.Contu D et al. Y-chromosome based evidence for pre-neolithic origin of the genetically homogeneous but diverse Sardinian population: inference for association scans. PLoS One 3, e1430 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morelli L et al. A comparison of Y-chromosome variation in Sardinia and Anatolia is more consistent with cultural rather than demic diffusion of agriculture. PLoS One 5, e10419 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Semino O et al. The genetic legacy of Paleolithic Homo sapiens sapiens in extant Europeans: a Y chromosome perspective. Science 290, 1155–9 (2000). [DOI] [PubMed] [Google Scholar]
- 38.Rootsi S et al. Phylogeography of Y-chromosome haplogroup I reveals distinct domains of prehistoric gene flow in europe. Am J Hum Genet 75, 128–37 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chikhi L, Nichols RA, Barbujani G & Beaumont MA Y genetic data support the Neolithic demic diffusion model. Proc Natl Acad Sci U S A 99, 11008–13 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Passarino G et al. Y chromosome binary markers to study the high prevalence of males in Sardinian centenarians and the genetic structure of the Sardinian population. Hum Hered 52, 136–9 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Olalde I et al. The Beaker phenomenon and the genomic transformation of northwest Europe. Nature 555, 190–196 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kivisild T The study of human Y chromosome variation through ancient DNA. Hum Genet 136, 529–546 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skoglund P et al. Genomic insights into the peopling of the Southwest Pacific. Nature 538, 510–513 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goldberg A, Gunther T, Rosenberg NA & Jakobsson M Ancient X chromosomes reveal contrasting sex bias in Neolithic and Bronze Age Eurasian migrations. Proc Natl Acad Sci U S A 114, 2657–2662 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gunther T et al. Ancient genomes link early farmers from Atapuerca in Spain to modern-day Basques. Proc Natl Acad Sci U S A 112, 11917–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hellenthal G et al. A genetic atlas of human admixture history. Science 343, 747–51 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loh PR et al. Inferring admixture histories of human populations using linkage disequilibrium. Genetics 193, 1233–54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moorjani P et al. The history of African gene flow into Southern Europeans, Levantines, and Jews. PLoS Genet 7, e1001373 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barbujani G, Bertorelle G, Capitani G & Scozzari R Geographical structuring in the mtDNA of Italians. Proc Natl Acad Sci U S A 92, 9171–5 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pistis G et al. High differentiation among eight villages in a secluded area of Sardinia revealed by genome-wide high density SNPs analysis. PLoS One 4, e4654 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanna S et al. Variants within the immunoregulatory CBLB gene are associated with multiple sclerosis. Nat Genet 42, 495–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zoledziewska M et al. Variation within the CLEC16A gene shows consistent disease association with both multiple sclerosis and type 1 diabetes in Sardinia. Genes Immun 10, 15–7 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Pilia G et al. Heritability of cardiovascular and personality traits in 6,148 Sardinians. PLoS Genet 2, e132 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Petkova D, Novembre J & Stephens M Visualizing spatial population structure with estimated effective migration surfaces. Nat Genet 48, 94–100 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Novembre J & Stephens M Interpreting principal component analyses of spatial population genetic variation. Nat Genet 40, 646–9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Botigue LR et al. Gene flow from North Africa contributes to differential human genetic diversity in southern Europe. Proc Natl Acad Sci U S A 110, 11791–6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Henn BM et al. Genomic ancestry of North Africans supports back-to-Africa migrations. PLoS Genet 8, e1002397 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paschou P et al. Maritime route of colonization of Europe. Proc Natl Acad Sci U S A 111, 9211–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Terhorst J, Kamm JA & Song YS Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat Genet 49, 303–309 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schiffels S & Durbin R Inferring human population size and separation history from multiple genome sequences. Nat Genet 46, 919–25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raghavan M et al. Upper Palaeolithic Siberian genome reveals dual ancestry of Native Americans. Nature 505, 87–91 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Patterson N et al. Ancient admixture in human history. Genetics 192, 1065–93 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blasco Ferrer E Paleosardo: Le Radici Linguistiche Della Sardegna Neolitica, (Walter De Gruyter Inc, 2010). [Google Scholar]
- 64.Pickrell JK et al. Ancient west Eurasian ancestry in southern and eastern Africa. Proc Natl Acad Sci U S A 111, 2632–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heyer E, Chaix R, Pavard S & Austerlitz F Sex-specific demographic behaviours that shape human genomic variation. Mol Ecol 21, 597–612 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Goldberg A, Gunther T, Rosenberg NA & Jakobsson M Reply to Lazaridis and Reich: Robust model-based inference of male-biased admixture during Bronze Age migration from the Pontic-Caspian Steppe. Proc Natl Acad Sci U S A 114, E3875–E3877 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lazaridis I & Reich D Failure to replicate a genetic signal for sex bias in the steppe migration into central Europe. Proc Natl Acad Sci U S A 114, E3873–E3874 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wilkins JF & Marlowe FW Sex-biased migration in humans: what should we expect from genetic data? Bioessays 28, 290–300 (2006). [DOI] [PubMed] [Google Scholar]
- 69.Keinan A & Clark AG Recent explosive human population growth has resulted in an excess of rare genetic variants. Science 336, 740–3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Joshi PK et al. Directional dominance on stature and cognition in diverse human populations. Nature 523, 459–62 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lohmueller KE The impact of population demography and selection on the genetic architecture of complex traits. PLoS Genet 10, e1004379 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Simons YB, Turchin MC, Pritchard JK & Sella G The deleterious mutation load is insensitive to recent population history. Nat Genet 46, 220–4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Uricchio LH, Zaitlen NA, Ye CJ, Witte JS & Hernandez RD Selection and explosive growth alter genetic architecture and hamper the detection of causal rare variants. Genome Res 26, 863–73 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Browning BL & Browning SR A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet 84, 210–23 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.International HapMap C et al. Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Price AL et al. Long-range LD can confound genome scans in admixed populations. Am J Hum Genet 83, 132–5; author reply 135–9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kong A et al. Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467, 1099–103 (2010). [DOI] [PubMed] [Google Scholar]
- 78.Tennessen JA et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337, 64–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gravel S et al. Demographic history and rare allele sharing among human populations. Proc Natl Acad Sci U S A 108, 11983–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nelson MR et al. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science 337, 100–4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shringarpure SS, Bustamante CD, Lange K & Alexander DH Efficient analysis of large datasets and sex bias with ADMIXTURE. BMC Bioinformatics 17, 218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mallick S et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538, 201–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Allele frequency summary data analyzed in the study will be deposited to EGA under accession number EGAS00001002212. The disaggregated individual-level sequence data for 2105 samples (adult volunteers of the SardiNIA cohort longitudinal study) analyzed in this study are from Sidore et al (2015) and are available from dbGAP under project identifier phs000313 (v4.p2). The remaining individual-level sequence data are from a case-control study of autoimmunity from across Sardinia, and per the obtained consent and local IRB, these data are only available for collaboration by request from the project leader (Francesco Cucca, Consiglio Nazionale delle Ricerche, Italy).