
Figure 1: The unique biochemical activities of LMW-E versus FL cyclin E and their consequences.
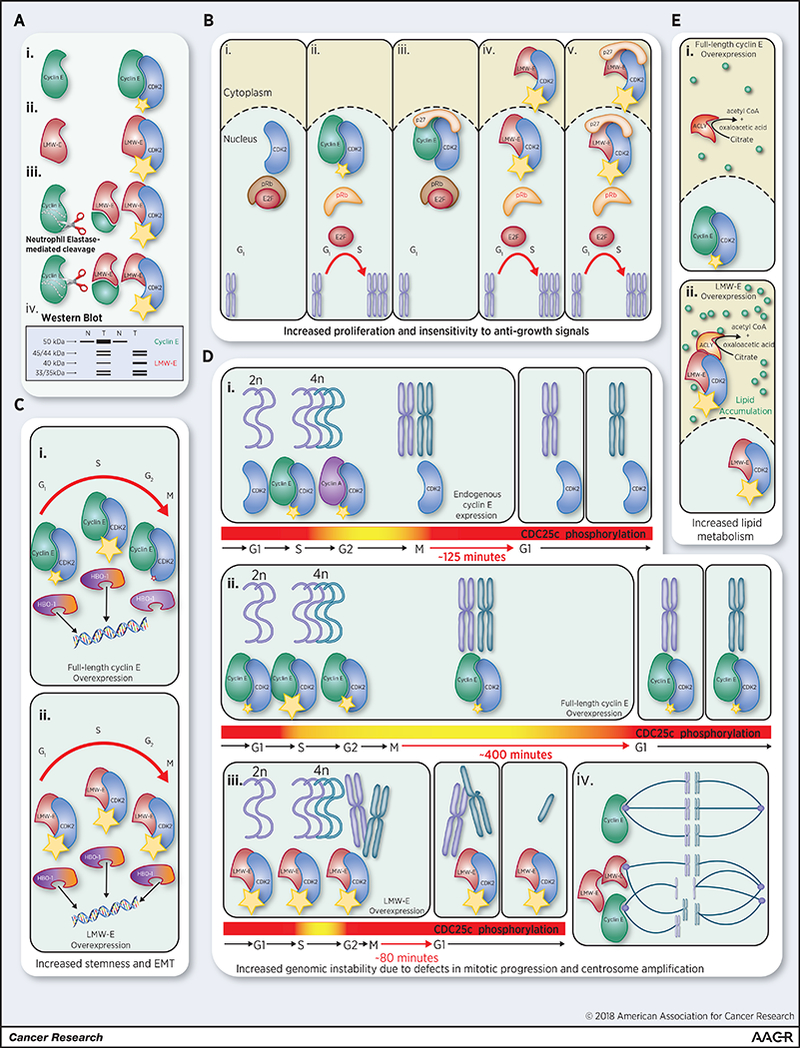
(A) (i) Cyclin E (FL 50 kDa) is an activating subunit of CDK2 that promotes kinase activity (small yellow star) during the G1-S-phase transition. Tumor-specific LMW-E isoforms are generated by (ii) alternative translation from methionine 46 (40 kDa) and (iii) NE-mediated cleavage of full-length cyclin E at two N-terminal sites (45/44 kDa and 35/33 kDa; doublets due to phosphorylation events) (66,105). LMW-E isoforms demonstrate higher binding affinity for CDK2, promoting hyperactivation of the kinase (large yellow star). (iv) In western blot analysis of normal (N) and tumor (T) tissue (using a C-terminally-directed antibody), LMW-E isoforms characteristically resolve as five distinct bands beneath FL cyclin E. In half of LMW-E-expressing breast tumors, cyclin E is also overexpressed; in the other half, however, LMW-E is expressed in the absence of full-length cyclin E (B). (i) When the pRb pathway is unaltered by oncogenic events, hypophosphorylated pRb binds to and sequesters E2F family transcription factors in G0-phase. (ii) FL cyclin E activates CDK2, leading to the hyperphosphorylation and inactivation of pRb, thereby releasing E2Fs to activate S-phase gene expression and progression. (iii) However, CDK inhibitors (e.g. p27) can inhibit the FL cyclin E-CDK2 complex (even if FL cyclin E is overexpressed) and prevent S-phase progression. (iv) Hyperactive LMW-E-CDK2 complexes can localize to the nucleus and hyperphosphorylate pRB; (v) even in the presence of CDK inhibitors, thereby promoting insensitivity to negative growth signals. (C) Hyperactive LMW-E-CDK2 complexes have other consequences. (i) FL Cyclin E-CDK2 can phosphorylate the substrate HBO-1; however, (ii) only constitutive hyperphosphorylation by LMW-E-CDK2 can promote HBO-1-dependant EMT and stemness properties, suggesting that cell cycle context-independent phosphorylation of HBO-1 alters its histone acetyltransferase activity in a pro-tumorigenic manner. (D) The proper timing of DNA replication and mitosis is essential to genome integrity. (i) One mechanism to ensure the fidelity of this process is feedback control through CDC25c, which promotes the proper timing of mitotic entry and exit through the activation of cyclin B-CDK1 and PLK1. (ii) Overexpression of FL cyclin E results in the improper phosphorylation of CDC25c and premature mitotic entry, but maintains the phosphorylation of CDC25c, delaying mitotic progression to cytokinesis and thereby largely preventing genomic instability. (iii) LMW-E overexpression also initiates premature mitotic entry; however unlike FL cyclin E, LMW-E cannot sustain CDC25c phosphorylation, resulting in faster mitotic exit and genomic instability. (iv) Genomic instability is further promoted by centrosome amplification induced by both FL cyclin E and LMW-E overexpression. (E) (i) FL cyclin E is largely restricted to the nucleus and therefore has limited opportunities to interact with cytoplasmic proteins. (ii) In contrast, LMW-E lacks an N-terminal nuclear localization signal promoting its accumulation in the cytoplasm where it can interact with novel binding partners including ACLY. LMW-E-CDK2 enhances ACLY activity (independent of phosphorylation), thereby promoting intracellular lipid accumulation and pro-tumorigenic phenotypes, including migration and invasion.