

Abstract
Purpose
Desmoplastic small round cell tumor (DSRCT), which harbors EWSR1-WT1 t(11;22)(p13:q12) chromosomal translocation, is an aggressive malignancy that typically presents as intra-abdominal sarcomatosis in young males. Given its rarity, optimal treatment has not been defined.
Experimental Design
We conducted a retrospective study of 187 DSRCT patients treated at MD Anderson Cancer Center over two decades. Univariate and multivariate regression analyses were performed. We determined whether chemotherapy, complete cytoreductive surgery (CCS), hyperthermic intraperitoneal cisplatin (HIPEC), and/or whole abdominal radiation (WART) improve overall survival in DSRCT patients. Critically, since our institutional practice limits HIPEC and WART to patients with less extensive, potentially resectable disease that had benefited from neoadjuvant chemotherapy, a time-variant analysis was performed to evaluate those adjunct treatment modalities.
RESULTS
The pre-2003 5-year overall survival (OS) rate of 5% has substantially improved to 25% with the advent of newer chemotherapies and better surgical and radiotherapy techniques (HR 0.47, 95% CI 0.29–0.75). Chemotherapy response (log rank p=0.004) and CCS (log rank p<0.0001) were associated with improved survival. Though WART and HIPEC lacked statistical significance, our study was not powered to detect their potential impact upon OS.
CONCLUSIONS
Improved 3- and 5-year overall survival were observed following multidisciplinary treatment that includes Ewing sarcoma (ES)-based chemotherapy and complete tumor cytoreductive surgery, but few if any patients are cured. Prospective randomized studies will be required to prove whether HIPEC or WART are important. In the meantime, chemotherapy and CCS remain the cornerstone of treatment and provide a solid foundation to evaluate new biologically targeted therapies.
Keywords: DSRCT, EWS-WT1, Ewing sarcoma, sarcoma, therapy and round cell sarcoma
INTRODUCTION
Desmoplastic small round cell tumor (DSRCT) is an aggressive malignancy that occurs predominantly in young adult males and is characterized by abdominopelvic sarcomatosis exhibiting multi-lineage cellular nests of epithelial, muscular, mesenchymal, and neural differentiation admixed with desmoplastic stroma.(1–6) Owing to the disease’s low age-adjusted incidence and peak incidence of 0.3 and 0.74 cases per million, respectively,(7) it wasn’t until 1989 that Gerald and Rosai recognized a pathognomonic EWSR1-WT1 t(11;22)(p13:q12) chromosomal translocation that pairs the Ewing sarcoma (ES) gene (EWSR1) with the Wilm’s tumor suppressor gene (WT1).(3) The functional loss of the WT1 tumor suppressor protein and the oncogenic effects caused by the aberrant 59 kDa fusion protein results in hundreds to thousands of nodules coating the intraabdominal serosal and subdiaphragmatic surfaces. Most DSRCT patients present with advanced disease and symptoms of abdominal pain or distention, nausea, constipation, and/or weight loss.(8)
Prior to the recognition of the disease as a distinct clinical entity, DSRCT was invariably misclassified as poorly differentiated atypical cancer of the testes, ovary, mesentery, or gastrointestinal tract, and the chemotherapies used for those malignancies elicited poor clinical response. As previously reported,(5,9) a tectonic shift in the treatment of these patients occurred after researchers made two astute observations: 1) DSRCT microscopically resembles other small round “blue cell” sarcoma subtypes (e.g., ES, rhabdomyosarcoma, synovial sarcoma), and 2) DSRCT and ES have the same N-terminal EWSR1 fusion partner.(10,11) A fuller historical context of these discoveries is summarized in a recent review by Mora and Rosai.(12)
The morphological and genomic characterization of DSRCT as distinct sarcoma subtype has provided a relatively recent opportunity to systematically evaluate which treatment modalities yield clinical benefits in DSRCT. Yet, the relative rarity of this malignancy has contributed to the lack of standardization, and most literature to date is in the format of anecdotal case reports. As a tertiary cancer hospital that cares for one of the largest groups of DSRCT patients in the world, our multidisciplinary team of medical, pediatric, surgical, and radiation oncologists have reviewed our entire clinical experience treating DSRCT since 1990 and, herein, share our seminal findings. As will be discussed, though carefully sequenced trimodality therapy reliant upon chemotherapy, complete cytoreductive surgery (CCS), and WART represents the standard DSRCT treatment approach used at our institution and others, our analysis suggests the current approach should undergo major revision, or at least prompt prospective clinical investigation.
PATIENTS AND METHODS
Demographics and data
The patient treatments and study analysis were reviewed and approved by the Institutional Review Board of MDACC and conducted in accordance with the principals of Declaration of Helsinki. Written informed consent was obtained from all patients for treatments, but it was not required for this retrospective review, and all the patient records were de-identified prior to analysis.
We identified 187 DSRCT patients treated at MDACC from 1990 to 2016 and collected data in accordance with the guidelines of MD Anderson’s Institutional Review Board. Specialist pathologists used clinical information, immunohistochemistry, and cytogenetic analysis for the EWSR1-WT1 fusion to confirm the DSRCT diagnoses. The date of original biopsy was used to calculate each patient’s age at initial diagnosis. In the comprehensive analysis of prognostic factors, patients were used to assess the association between prognostic factors and overall survival time, i.e., from the time of diagnosis until the time of death or last follow-up. More comprehensive clinical information was available which allowed for a thorough statistical analysis of a number of factors, including: (a) the era in which a patient was treated, taking into account the advent of the ifosfamide/etoposide combination used more frequently after 2003, (b) the ability to achieve an optimal surgical cytoreduction, and (c) the tumor response to neoadjuvant chemotherapy. A complete tumor surgical debulking was defined as removal of the primary dominant tumor mass and all visualized metastatic nodules if present. Given the difficulty in accurately measuring the extent of abdominal sarcomatosis, a positive neoadjuvant chemotherapy response was defined as any reduction in tumor size, not necessarily a clinical response as would otherwise be reported using WHO or RECIST criteria that are better suited to assess well-demarcated tumors. Major response to chemotherapy was defined using RECIST criteria (greater than or equal to 30% reduction in one-dimensional measurement in the predominant mass) to any course of chemotherapy whether provide in the neoadjuvant, adjuvant, or relapsed setting. As a tertiary referral center, many patients had received chemotherapy and exhibited chemoresistance prior to referral. In those circumstances, external medical records were reviewed to determine the type, length, and dose of chemotherapy. All but four patients received chemotherapy and despite vast treatment heterogeneity most patients received ES-based chemotherapy regimens that were the standard at their respective times of diagnosis.
Chemotherapy
Most regimens included combinations of vincristine, dactinomycin, cyclophosphamide, doxorubicin, ifosfamide, and etoposide. Temozolomide/irinotecan, cyclophosphamide/topotecan, and high-dose ifosfamide, which have proven clinical activity in chemotherapy-resistant ES, were common second-line regimens. Less common salvage regimens included cyclophosphamide/vinorelbine, gemcitabine/docetaxel, and dacarbazine. A small minority of DSRCT patients treated since 2007 received insulin-like growth factor 1– targeted monoclonal antibodies alone or in combination with mammalian target of rapamycin (mTOR) inhibitors, which was in keeping with early-phase clinical trials that revealed that these therapies had striking, albeit short-lived, clinical activity in ES patients. Other biologically targeted therapies, such as polyethylene glycol–conjugated interferon and pazopanib, were used infrequently. Finally, as an induction regimen, busulphan was given to a handful of patients who underwent bone marrow transplantation.
Clinical staging
Clinical data incorporated information from newer ifosfamide/etoposide-containing regimens (which were used more frequently after 2003), the ability to achieve a complete surgical cytoreduction, and response to neoadjuvant chemotherapy. An extensive discussion about DSRCT staging is beyond the scope of this publication. No prospectively validated staging system for DSRCT exists, and those for soft tissue sarcomas have the unintended effect of identifying most DSRCT as stage 4 disease. Similar to the American Joint Committee on Cancer’s staging system, that proposed by Hayes-Jordan et al. is based on tumor burden (peritoneal cancer index as defined by Sugerbaker et al.,39) regional spread (liver, rather than nodal, metastases), and the presence or absence of extra-abdominopelvic metastases.40 While the latter two metrics of extra-abdominopelvic metastases and liver spread were readily assessed, our experience suggests that surgeons, radiologists, and pathologists do not consistently document the peritoneal cancer index score, whose calculation requires that each of 13 anatomical regions be scored from 1 to 3 based upon maximal lesion size, and in our opinion, this is too time-consuming to gain widespread traction in clinical practice. We suggest a pragmatic staging alternative that replaces the peritoneal cancer index with a simpler method based on the American Joint Committee on Cancer’s practice of recording the maximal tumor diameter of the largest mass. In addition, given its clear utility, fluorodeoxyglucose positron emission tomography/CT should be part of DSRCT staging.
Statistical Analysis
Continuous variables were summarized by their means, standard deviations, and ranges, whereas categorical variables were summarized by their frequencies or percentages. A univariate analysis included demographic information (age and gender), treatment type (radiotherapy, surgery, and stem cell transplantation [SCT]) and treatment era (two analyses of patients diagnosed before 2003 or after 2003). The association between diagnosis time (≤2003 vs. >2003, when more effective chemotherapy regimens became available for pediatric sarcomas) and other relevant clinical variables were assessed using the Fisher exact test. Kaplan-Meier curves were calculated to estimate unadjusted OS. A Cox proportional hazards model was used to evaluate the ability of the covariates to predict OS. Within the multivariate Cox regression model, non-significant variables were eliminated in a step-down fashion with a p-value cut-off of 0.05. All computations were carried out in SAS 9.4 and S-Plus version 8 software.
RESULTS
Patient Demographics and Prognostic Factors
The demographic and treatment information of 187 evaluable DSRCT patients is summarized in Table 1. Consistent with prior reports, an overwhelming majority of patients were male (82.9%). The patients’ mean age ± standard deviation was 22.6 ± 10.6 years (range, 0.5–52.9 years), a relatively broad spectrum compared to earlier reports from children’s hospitals, which naturally have a younger referral base. Local control modalities included cytoreductive surgery (106 patients; 57%) and whole abdominal radiotherapy (WART, 92 patients; 49.2%). A subset of patients had either hyperthermic intra-peritoneal perfusion of chemotherapy (HIPEC) with cisplatin (200 mg/m2) following complete cytoreductive surgery (CCS, 82 patients; 43.9%) or SCT (12 patients; 6.4%). Treatment evolved as we gained more experience using HIPEC, radiation, and SCT in patients with DSRCT, and each of these modalities were more commonly used after 2003.
Table 1.
Desmoplastic Small Round Cell Tumor Clinical Trials
| Cancer Center | |||||
|---|---|---|---|---|---|
| Published Clinical Trials | Current Study |
MSKCC | MDACC | UK Centers |
France GR Center |
| Study dates | 1990–2016 | 1972–2003 | Until 1998 | 1991–2012 | 1991–2013 |
| Patient Demographics | |||||
| No. of patients | 187 | 66 | 39 | 41 | 38 |
| Median age, y | 22.6 | 19 | 25 | 27 | 27 |
| Male:Female ratio | 4.8:1 | 10:1 | 4.5:1 | 3.1:1 | 3.5:1 |
| Caucasian race | 75.4% | 75% | N/A | N/A | N/A |
| Treatment modality (% of patients) | |||||
| Chemotherapy | 98% | >92% | 92% | 93% | 100% |
| Surgery | 61% | 71% | 92% | 20% | 60.5% |
| Radiotherapy | 49% | >44% | N/A | 15% | 30% |
| HIPEC1 | 72% | N/A | N/A | N/A | 5% |
| SCT | 6.4% | 0% | 0% | 0% | N/A |
| Survival | |||||
| Median survival (months) | 35 | 28 | N/A | 16 | 25.7 |
| 3-year OS rate | 48.2% | 44% | N/A | N/A | 31.6% |
| 5-year OS rate | 21.6% | 15% | N/A | N/A | 8.3% |
| Improved by surgery | Yes (p=0.04) | Yes | Yes (p=0.024) | Yes (p=0.026) | |
| Improved by radiation | Yes (p<0.01) | Yes | Yes (p=0.015) | Yes (p=0.022) | |
| Improved by HIPEC | No (p=0.26) | N/A | N/A | N/A | N/A |
Abbreviations: MSKCC, Memorial Sloan Kettering Cancer Center; MDACC, The University of Texas MD Anderson Cancer Center; UK, United Kingdom; GR Center, Gustave Roussy; SCT, stem cell transplantation; OS, overall survival.
NOTE:
HIPEC was only offered to patients that had undergone a complete cytoreductive surgery (n=114).
When assessing the effect of treatment modality upon overall survival, chemotherapy (p<0.01; hazard ratio [HR]=0.12), radiotherapy (p<0.01; HR=0.44) and CCS (p=0.006; HR=0.59) were statistically significant in the univariate analysis (UVA; Supplemental Table 1). In the subsequent multivariate analysis (MVA), chemotherapy (p<0.01; HR=0.17) and radiation (p<0.01; HR=0.46) retained statistical significance (Supplemental Table 2). Notably, WART—the predominant form of XRT provided—was not statistically significant when CCS was used as the reference timepoint (Supplemental Table 2). Our results indicate that patients diagnosed after 2003 (p<0.01; HR=0.47) had improved survival (Fig. 1A). Most patients (88.2%) were treated after 2003, when results from INT-0091 demonstrated that ifosfamide/etoposide, added to a VAC (Vincristine, Adriamycin, Cyclophosphamide) backbone, improved clinical outcomes for those with localized ES.(13) Hearteningly, we observed a striking time-dependent improvement on 3- and 5-year OS rates that had risen, respectively, to 50.9% and 25.1% after 2003 compared to earlier OS rates of just 31.8% (3-year) and 4.6% (5-year).
Figure 1. Chemotherapy impact upon OS in DSRCT patients.
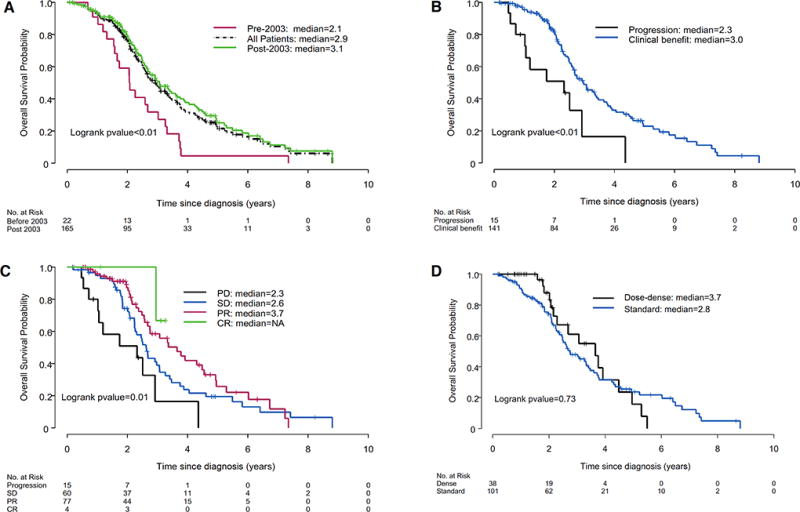
(A) Overall survival for all patients according to treatment era (before 2003, after 2003, and both). (B) Overall survival in patients who achieved clinical benefit or had progressive disease to their initial chemotherapy regimen. (C) Stratification of overall survival by RECIST response to the first chemotherapy regimen each patient received, and (D) Survival effect resulting from standard 3-week chemotherapy treatment vs. dose-dense treatment, when the traditional neoadjuvant VAC/IE regimen was provided. PD = partial disease; SD = stable disease; PR = partial response; CR = complete response.
Individual Treatment Effects
Chemotherapy
As DSRCT almost universally presents with advanced-stage disease and is remarkably chemosensitive, all but four patients in the present study received systemic chemotherapy. Since nearly all patients received chemotherapy, it was not possible to prove that chemotherapy was superior to observation. Instead, as an indirect measure of chemotherapy effect, we assessed whether chemotherapy response prolonged patient survival, recognizing that most DSRCT patients eventually die from their malignancy. Of the 156 patients that had computed tomography (CT) and/or fluorodeoxyglucose (FDG)-positron emission tomography (PET)/CT imaging available for review, 90.4% achieved clinical benefit (SD/PR/CR) and just over half the patients (51.9%) had a pre-surgical response (CR/PR) according to Response Evaluation Criteria in Solid Tumors (RECIST) at some point during treatment. OS was significantly better for those who achieved clinical benefit vs. no benefit from their initial chemotherapy regimen (Fig 1B), as could be further stratified by clinical response (Fig 1C). Consistent with the findings of Schwarz et al., our univariate analysis revealed that patients whose disease responded to chemotherapy in the neoadjuvant setting (p = 0.03; HR=0.63; Supplemental Table 1) had better OS than patients whose disease did not respond to chemotherapy.(14)
Though not previously assessed in DSRCT, our univariate data suggests that dose-dense VAC/IE-based therapy could be superior to 3-week dosing (68.4% vs. 48.2% response rate; p=0.05). However, our study was not powered to detect a difference in dosing schedules and the Kaplan-Meier curves failed to reach significance (Fig 1D). Almost half the patients received variants of the Children Oncology Group’s alternating VAC/IE (VAC/Ifosfamide and Etoposide) protocol (AEWS-0031), VAI, Europe’s VIDE (EURO-EWING99) protocol, or less frequently the P6 protocol when the patients were referred from other cancer centers (Fig 2A), and no regimen proved superior to another (Supplemental Figure S1).(9) Nearly nine in ten patients received neoadjuvant chemotherapy, the standard of care at MDACC, and a small minority first underwent chemotherapy as part of an adjuvant or salvage approach when the initial surgery was performed prior to their arrival at MDACC (Fig 2B). Most received standard interval therapy every 3 weeks (72.7%) and roughly one-quarter of the patients received dose-dense therapy every 2 weeks (Fig 2C). The second-line regimens used for salvage therapy is shown in Fig. 2D. Our analysis was underpowered to detect any difference in the response rates to second-line chemotherapies (data not shown). However, salvage therapy was extensively used since 61% of the patients (114 of 187) had metastatic disease at the time of diagnosis (Fig 2E). A univariate analysis of the number and location of metastatic sites is included in Supplemental Table 3.
Figure 2. Chemotherapy patterns in DSRCT patients.

(A) The initial chemotherapy regimens used to treat DSRCT patients. (B) Chemotherapy administration in relation to surgery: included neoadjuvant (87.3%), adjuvant (6.6%), or salvage (6.1%) treatments. (C) Dosing interval in DSRCT patients. 72.7% of the patients received standard therapy every 3 weeks and 27.3% received dense therapy every two weeks. (D) Second-line chemotherapy treatment regimens used in DSRCT patients. (E) Organ sites involved among the 114 patients who presented with metastases at the time of diagnosis. P6 = 7 cycles of IE/VDC (Ifosfamide, Etoposide, Vincristine, Adriamycin, and Cytoxan); BEP = bleomycin, etoposide, and cisplatin; VAI = Vincristine, actinomycin & ifosfamide; VAC/IE = Vincristine, Adriamycin, Cyclophosphamide, Ifosfamide and Etoposide; Cy = cyclophosphamide; Topo = topotecan; HD ifos = high dose ifosfamide; Tem = temozolomide; Irino = irinotecan; Vcr = vincristine; N = number of metastatic sites.
Surgery and HIPEC
Fifty-seven percent of the patients underwent CCS when clinically appropriate, and this group had substantially better OS (p = 0.006; HR=0.59) than those who were gauged to be inappropriate for CCS (Fig 3A & Supplemental Table 1). Our data suggests the pre-operative imaging is reasonably accurate assessing a patient’s suitability for CCS, as 75.5% of the patients achieved the expected surgical outcome by eliminating all macroscopically evident disease (i.e. a completeness of cytoreduction score [CCR] score of 0; 61.3% of CCS patients) or leaving behind measurable tumor implants less than 2.5 mm in diameter (14.2% of patients). Care should be taken in extrapolating this result, since CCS was performed by only a handful of surgeons at our center.
Figure 3. Impact upon OS by surgery, HIPEC, radiation, and WART.

(A) Overall survival in patients who underwent complete cytoreductive surgery (CCS). (B) Overall survival benefit of CCS in patients who achieved clinical benefit from their first neoadjuvant chemotherapy regimen. (C) Overall survival in patients that underwent CCS +/− HIPEC (D) Overall survival in patients who received radiation at some time during their clinical care. (E) The overall survival effect of whole abdominal radiotherapy (WART), which was almost always provided 6–8 weeks following CCS.
Importantly, our institutional practice has been to offer patients CCS only if they achieved clinical benefit from neoadjuvant chemotherapy since most chemo-resistant tumors quickly recur. As this approach naturally biased the CCS-selected patients towards an improved survival outcome, we performed a secondary analysis limited to those who achieved clinical benefit from their initial neoadjuvant chemotherapy regimen. Shown in Figure 3B, the benefit of CCS persisted in those who had responded to neoadjuvant chemotherapy treatment (p<0.01). An identical subset analysis was conducted to determine whether CCS improved survival in those whose tumors progressed to their first neoadjuvant chemotherapy regimen, however only eight patients met that criteria (data not shown). Of those, four patients obtained clinical benefit from a second neoadjuvant regimen and one achieved clinical benefit from a third neoadjuvant regimen, rendering them operable candidates. Without more evidence, we cannot routinely advocate for CCS in patients that progress on neoadjuvant chemotherapy.
Seventy-two percent of the patients also underwent HIPEC with cisplatin after CCS in an attempt to eliminate micrometastatic cells inevitably left behind at the time of surgery. A subtle, statistically insignificant trend towards improved survival was observed in the first two years following CCS, however this effect was transitory and did not alter 3- or 5-year OS (Fig 3C, p=0.16). The results were unchanged when CCS and HIPEC were secondarily evaluated in patients with abdomen-only disease devoid of hepatic surface implants (Supplemental Figure S2, panels A-C) or within an even more stringent subset of patients that achieved complete cytoreductive resection (CCR0) status from CCS (Supplemental Figure S3). As the current study was underpowered to detect a subtle benefit from HIPEC, if one exists, more definitive results are anticipated later this year from a HIPEC-specific trial that continues to enroll at our institution.
Radiation and Other Therapies
Radiation was commonly used for palliation at the time of tumor recurrence. However, our clinical practice evolved over time, and we increasingly used WART as an adjuvant therapy before consolidative chemotherapy after several publications seemed to indicate that trimodality therapy (i.e. chemotherapy, radiation, and surgery) led to better clinical outcomes than bimodality treatment (i.e. chemotherapy and surgery).(15) Our results supported this trimodality approach when the time of diagnosis was used as the reference timepoint to gauge OS (Supplemental Figure S4). However, since WART cannot contribute to improved OS before it is provided, we conducted a time-variant analysis of the effect of WART vs. no WART, this time using the date each patient underwent their respective CCS as the reference point. Surprisingly, in this more homogeneous subset of patients who had undergone WART shortly after recovering from CCS (n=69), no benefit was observed (Fig 3E). Subsequent analysis in patients with abdomen-only disease yielded identical results (Supplemental Figure S2D). This unexpected result conflicts with our current treatment paradigm and prompted an updated treatment recommendation to consider WART only in highly-selected patients that are prospectively monitored in clinical trials specifically designed to quantify the post-WART toxicity and in-field recurrence rate.
In an era where immunotherapy plays a prominent role in treatment for many different cancer types, the use immunotherapies, check-point inhibitors, or SCT and other cellular therapies can also be studied in DSRCT. At the time of this writing, we are unaware of any DSRCT patients who have responded to immunotherapy, and to the extent one can draw from the sarcoma community’s experience with ES and other translocation-positive sarcomas that have otherwise relatively quiescent genomes, current evidence suggests limited clinical effect.(16–18) SCT is not standard-of-care at MD Anderson and only 12 of 187 DSRCT patients underwent this treatment modality throughout the almost two-decade long experience presented in this work. Nine of the twelve DSRCT patients who underwent SCT received this before 2006 when this treatment modality was more commonly used and all but one of the patients had an autologous rather than allogeneic SCT, so we have insufficient data to evaluate the effect of SCT in the present era. Nevertheless, rapid advancements in the immunotherapy space provide a tantalizing opportunity to study them in patients combating DSRCT.
DISCUSSION
Our retrospective study of DSRCT patients is the largest conducted to date at a single-institution and unique for its inclusion of children and adults treated throughout a two-decade long period. The treatment approach taken by our multidisciplinary team has steadily improved (Fig 4), and significant strides in surgical technique, radiation delivery, supportive care, and novel chemotherapies led to improved time-dependent patient survival. We report the 5-year OS rate of DSRCT patients is considerably higher than reported in recent case series and now approaches 25.1% when patients receive trimodality care from skilled experts.(19–21) Nonetheless, despite clear clinical advancements, most survivors harbor residual disease that requires near-perpetual systemic chemotherapy to control.
Figure 4. Revised DSRCT Treatment algorithm adapted by MDACC.

The diagnosis of DSRCT is confirmed using immunohistochemistry and/or cytology, and the staging workup is completed. All patients receive neoadjuvant chemotherapy, usually six cycles of the current standard-of-care for ES. Clinical response is assessed with PET/CT every two cycles and the small minority that fail to respond to chemotherapy are transitioned to second-line regimens. Patients that harbor extra-peritoneal disease (EPD) after 6 neoadjuvant chemotherapy cycles are considered poor surgical candidates and continue with chemotherapy. Those lacking EPD proceed to complete cytoreductive surgery (CCS), with the aim of removing all measurable disease. In light the current study findings, we recommend HIPEC only in conjunction with an ongoing clinical study. Whole abdominopelvic intensity-modulated radiotherapy (WART), if provided, is also given as part of a prospective clinical trial designed to assess disease-free survival and intra-abdominal recurrence-free survival. Adjuvant chemotherapy with temozolomide/irinotecan or other agents are provided and oligo-metastatic sites are treated, when needed, using radiofrequency ablation or stereotactic radiation. VAC/IE = Vincristine, Adriamycin, Cyclophosphamide, Ifosfamide and Etoposide; VAI = Vincristine, actinomycin & ifosfamide; VIDE = Vincristine, Adriamycin, Ifosfamide and Etoposide; P6 = 7 cycles of IE/VDC (Ifosfamide, Etoposide, Vincristine, Adriamycin, and Cytoxan); LN = lymph nodes; HIPEC = hyperthermic peritoneal perfusion of chemotherapy; IGF-1R = insulin-like growth factor receptor 1; mTOR = mammalian target of Rapamycin.
Our study was not adequately powered to discern regimen-specific differences in chemotherapy response. The majority of patients received the P6 protocol or a similar ES-oriented regimen (e.g. VID or VAC/IE) as first-line neoadjuvant treatment, and we favor this approach because it typically leads to rapid tumor control and selects chemosensitive patients likely to benefit from CCS.(22) High-dose ifosfamide and topoisomerase-containing regimens such as temozolomide/irinotecan or cyclophosphamide/topotecan have proven activity as second- or third-line options, and trabectedin (recently approved for soft-tissue sarcoma) has demonstrated clinical responses in several case reports.(23–25) A substantial number of DSRCTs overexpress vascular endothelial growth factor receptor 2, and a handful of patients have had clinical responses to sunitinib,(26) sorafenib, or pazopanib.(27,28) Other biological therapies, including anti-ganglioside GD2 antibodies,(29,30) imatinib,(31) mammalian target of rapamycin (mTOR) inhibitors,(32,33) and a combination of insulin-like growth factor 1 and mTOR inhibitors,(34) have shown limited success. As reported by Fine et al., DSRCT highly expresses the androgen receptor (AR), which remains to be investigated as a cancer target.(35)
Expectedly, chemotherapy responders exhibited a better OS than non-responders. To tease apart the confounding effect between chemotherapy response and surgery, we performed a secondary Kaplan-Meier analysis limited to 141 patients who derived preoperative clinical benefit from their first chemotherapy regimen. As hypothesized, CCS performed by surgeons that routinely care for DSRCT patients was associated with improved the OS of patients whose disease responded to neoadjuvant chemotherapy, solidifying this clinical practice recommendation. Insufficient data existed to determine whether CCS improves the OS of chemotherapy non-responders and further pursuit of this question would likely require international collaboration by the handful of cancer centers that have surgeons experienced in CCS for DSRCT.
At MDACC, surgeons routinely performed CCS when possible since incomplete resections lead to inferior outcomes and the current study validates this approach. HIPEC has been used extensively at MDACC, with the goal of eradicating micrometastatic disease, however our study was underpowered to detect a statistically significant survival benefit. We eagerly await the results from a prospective clinical trial studying this treatment modality.(36)
Given the exquisite sensitivity of ES to radiation and the molecular and morphological similarity of ES and DSRCT, radiotherapy for DSRCT patients has been evaluated and is most often applied as an adjunct to chemotherapy and surgery. First described by Kushner et al. in 1996, the treatment approach at MSKCC relied on conventional two-dimensional WART (30 Gy plus boosts to sites of residual disease).(9) In that initial series,(37) 86% of tumors responded at the expense of serious gastrointestinal and hematological complications.
Recent data using intensity-modulated radiotherapy is associated with considerably less toxicity than conventional WART.(38) Nevertheless, despite safer methods to deliver radiation, we were concerned that the toxicity of WART would outweigh its putative benefits. At first pass, our univariate analysis indicated that radiotherapy conferred a survival benefit (p=0.01; HR=0.44) when analyzed from time of diagnosis. Because radiotherapy was given almost exclusively to patients whose disease responded to chemotherapy and who later underwent successful CCS (two variables independently linked to improved outcomes), we sought to remove these confounding effects using a planned subset analysis that compared trimodality therapy (chemotherapy/CCS/WART) with bimodality therapy (chemotherapy/CCS). Unlike prior studies that assessed the effects of WART from the date of diagnosis, our analysis used CCS as the study start date, which enabled an unbiased measure of WART’s impact in the most common clinical scenario (chemosensitive patients who underwent CCS).
Remarkably, in this more uniform patient population, WART did not improve OS. Reconciling the potential benefit of focal radiation but not WART, one explanation is that radiation was usually provided in an attempt to control solitary extraperitoneal disease in patients that harbored less extensive tumor spread as an adjunct to CCS. Thus, in many instances radiation and CCS were carefully coordinated in an attempt to control all known detectable sites of disease. Compared to historical controls, WART appears to delay extrahepatic intra-abdominal tumor recurrence and may still provide value by controlling symptoms and preserving quality of life.(39) However, those end points must be balanced by the potential side effects, cost, and time required for radiation administration, and should be preferably assessed in any future trial implementing WART for DSRCT.
Among our key findings, multimodality care by experts well-versed in DSRCT treatment can achieve 5-year overall survival (OS) rates of 25.1%, which exceeds the 15% survival rate cited historically(7) and is considerably better than the pre-2003 OS survival rate (4.6%). Nevertheless, few patients are cured, and most have rapid tumor recurrence unless they perpetually receive systemic chemotherapy. Importantly, our data indicate that complete cytoreductive surgery (CCS) and chemotherapy are essential modalities of proven clinical benefit, whereas HIPEC and WART warrant further study in randomized prospective clinical trials.
CONCLUSION
Persistent mesenteric implants and common extra-abdominal metastases continue to pose formidable clinical challenges for patients diagnosed with DSRCT and durable disease-free intervals are brief when chemotherapy is withheld for any significant period. In this sense, the treatment of DSRCT more closely resembles that of metastatic ES, in which traditional cytotoxic chemotherapies and nascent biologically targeted therapies are used to halt or reverse tumor growth in an effort to alleviate cancer-related symptoms and extend patient survival. Though chemotherapy and CCS were associated with improved OS, our research calls into question the role of HIPEC and WART. Prospective, randomized, multi-center cooperative trials will be required to evaluate those local-control modalities and to advance novel biologically-targeted therapies.
Supplementary Material
Translational Relevance.
Desmoplastic small round cell tumor (DSRCT) is an aggressive malignancy harboring an EWSR1-WT1 t(11;22)(p13:q12) chromosomal translocation that typically presents as intra-abdominal sarcomatosis in young males. In the largest retrospective review conducted to date in patients with DSRCT, chemotherapy and complete cytoreductive surgery (CCS) were associated with improved overall survival and remain the cornerstone of our institutional treatment approach. Hyperthermic intraperitoneal cisplatin (HIPEC) and whole abdominal radiation (WART) are investigational options and further prospective study is warranted. New drugs are needed for an orphan tumor type like DSRCT that lacks a curative therapy.
Acknowledgments
Research Support: The University of Texas MD Anderson Cancer Center is supported by the National Institutes of Health through Cancer Center Support Grant CA016672. JAL is supported by R01-CA180279-01A1.
Abbreviations
- VAC/IE
Vincristine, Adriamycin, Cyclophosphamide, Ifosfamide and Etoposide
- VACD
Vincristine, Adriamycin, Cyclophosphamide, Dacarbazine
- VID
Vincristine, Ifosfamide, Doxorubicin (Adriamycin)
- VIDE
Vincristine, Adriamycin, Ifosfamide and Etoposide
- MSKCC
Memorial Sloan Kettering Cancer Center
- MDACC
The University of Texas MD Anderson Cancer Center
- UK
United Kingdom
- SCT
stem cell transplantation
- OS
overall survival
- HIPEC
hyperthermic peritoneal perfusion of chemotherapy
- RECIST
Response Evaluation Criteria in Solid Tumors
Footnotes
Financial Disclosures: No financial disclosures are reported.
References
- 1.Chang F. Desmoplastic small round cell tumors: cytologic, histologic, and immunohistochemical features. Archives of pathology & laboratory medicine. 2006;130(5):728–32. doi: 10.1043/1543-2165(2006)130[728:DSRCTC]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 2.Benjamin LE, Fredericks WJ, Barr FG, Rauscher FJ., 3rd Fusion of the EWS1 and WT1 genes as a result of the t(11;22)(p13;q12) translocation in desmoplastic small round cell tumors. Medical and pediatric oncology. 1996;27(5):434–9. doi: 10.1002/(SICI)1096-911X(199611)27:5<434::AID-MPO8>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 3.Gerald WL, Rosai J. Case 2. Desmoplastic small cell tumor with divergent differentiation. Pediatric pathology / affiliated with the International Paediatric Pathology Association. 1989;9(2):177–83. doi: 10.3109/15513818909022347. [DOI] [PubMed] [Google Scholar]
- 4.Gerald WL, Rosai J, Ladanyi M. Characterization of the genomic breakpoint and chimeric transcripts in the EWS-WT1 gene fusion of desmoplastic small round cell tumor. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(4):1028–32. doi: 10.1073/pnas.92.4.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerald WL, Ladanyi M, de Alava E, Cuatrecasas M, Kushner BH, LaQuaglia MP, et al. Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): desmoplastic small round-cell tumor and its variants. J Clin Oncol. 1998;16(9):3028–36. doi: 10.1200/JCO.1998.16.9.3028. [DOI] [PubMed] [Google Scholar]
- 6.Gerald WL, Haber DA. The EWS-WT1 gene fusion in desmoplastic small round cell tumor. Seminars in cancer biology. 2005;15(3):197–205. doi: 10.1016/j.semcancer.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Lettieri CK, Garcia-Filion P, Hingorani P. Incidence and outcomes of desmoplastic small round cell tumor: results from the surveillance, epidemiology, and end results database. Journal of cancer epidemiology. 2014;2014:680126. doi: 10.1155/2014/680126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Honore C, Amroun K, Vilcot L, Mir O, Domont J, Terrier P, et al. Abdominal Desmoplastic Small Round Cell Tumor: Multimodal Treatment Combining Chemotherapy, Surgery, and Radiotherapy is the Best Option. Annals of surgical oncology. 2014 doi: 10.1245/s10434-014-4123-6. [DOI] [PubMed] [Google Scholar]
- 9.Kushner BH, LaQuaglia MP, Wollner N, Meyers PA, Lindsley KL, Ghavimi F, et al. Desmoplastic small round-cell tumor: prolonged progression-free survival with aggressive multimodality therapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1996;14(5):1526–31. doi: 10.1200/JCO.1996.14.5.1526. [DOI] [PubMed] [Google Scholar]
- 10.Aurias A, Rimbaut C, Buffe D, Dubousset J, Mazabraud A. [Translocation of chromosome 22 in Ewing's sarcoma] Comptes rendus des seances de l'Academie des sciences Serie III, Sciences de la vie. 1983;296(23):1105–7. [PubMed] [Google Scholar]
- 11.Aurias A, Rimbaut C, Buffe D, Zucker JM, Mazabraud A. Translocation involving chromosome 22 in Ewing's sarcoma. A cytogenetic study of four fresh tumors. Cancer Genet Cytogenet. 1984;12(1):21–5. doi: 10.1016/0165-4608(84)90003-7. [DOI] [PubMed] [Google Scholar]
- 12.Mora J, Modak S, Cheung NK, Meyers P, de Alava E, Kushner B, et al. Desmoplastic small round cell tumor 20 years after its discovery. Future Oncol. 2015;11(7):1071–81. doi: 10.2217/fon.15.32. [DOI] [PubMed] [Google Scholar]
- 13.Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348(8):694–701. doi: 10.1056/NEJMoa020890348/8/694. [pii] [DOI] [PubMed] [Google Scholar]
- 14.Schwarz RE, Gerald WL, Kushner BH, Coit DG, Brennan MF, La Quaglia MP. Desmoplastic small round cell tumors: prognostic indicators and results of surgical management. Annals of surgical oncology. 1998;5(5):416–22. doi: 10.1007/BF02303860. [DOI] [PubMed] [Google Scholar]
- 15.Pinnix CC, Fontanilla HP, Hayes-Jordan A, Subbiah V, Bilton SD, Chang EL, et al. Whole Abdominopelvic Intensity-Modulated Radiation Therapy for Desmoplastic Small Round Cell Tumor after Surgery. International journal of radiation oncology, biology, physics. 2011 doi: 10.1016/j.ijrobp.2011.06.1985. doi S0360-3016(11)02915-4 [pii] 10.1016/j.ijrobp.2011.06.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Groisberg R, Hong DS, Behrang A, Hess K, Janku F, Piha-Paul S, et al. Characteristics and outcomes of patients with advanced sarcoma enrolled in early phase immunotherapy trials. J Immunother Cancer. 2017;5(1):100. doi: 10.1186/s40425-017-0301-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rossig C. Cellular immunotherapy strategies for Ewing sarcoma. Immunotherapy. 2014;6(5):611–21. doi: 10.2217/imt.14.36. [DOI] [PubMed] [Google Scholar]
- 18.Tsukahara T, Emori M, Murata K, Mizushima E, Shibayama Y, Kubo T, et al. The future of immunotherapy for sarcoma. Expert Opin Biol Ther. 2016;16(8):1049–57. doi: 10.1080/14712598.2016.1188075. [DOI] [PubMed] [Google Scholar]
- 19.Philippe-Chomette P, Kabbara N, Andre N, Pierron G, Coulomb A, Laurence V, et al. Desmoplastic small round cell tumors with EWS-WT1 fusion transcript in children and young adults. Pediatric blood & cancer. 2012;58(6):891–7. doi: 10.1002/pbc.23403. [DOI] [PubMed] [Google Scholar]
- 20.Wong HH, Hatcher HM, Benson C, Al-Muderis O, Horan G, Fisher C, et al. Desmoplastic small round cell tumour: characteristics and prognostic factors of 41 patients and review of the literature. Clinical sarcoma research. 2013;3(1):14. doi: 10.1186/2045-3329-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Xu H, Ren F, Yang Y, Chen B, Zhang F. Analysis of clinicopathological features and prognostic factors of desmoplastic small round cell tumor. Pathology oncology research : POR. 2014;20(1):161–8. doi: 10.1007/s12253-013-9679-0. [DOI] [PubMed] [Google Scholar]
- 22.Wagner MJ, Gopalakrishnan V, Ravi V, Livingston JA, Conley AP, Araujo D, et al. Vincristine, Ifosfamide, and Doxorubicin for Initial Treatment of Ewing Sarcoma in Adults. Oncologist. 2017 doi: 10.1634/theoncologist.2016-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez-Gonzalez A, Cantos B, Tejerina E, Provencio M. Activity of trabectidin in desmoplastic small round cell tumor. Medical oncology. 2011;28(Suppl 1):S644–6. doi: 10.1007/s12032-010-9687-9. [DOI] [PubMed] [Google Scholar]
- 24.Frezza AM, Whelan JS, Dileo P. Trabectedin for desmoplastic small round cell tumours: a possible treatment option? Clinical sarcoma research. 2014;4:3. doi: 10.1186/2045-3329-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brunetti AE, Delcuratolo S, Lorusso V, Palermo L, Di Giorgio A, Pisconti S, et al. Third-line trabectedin for a metastatic desmoplastic small round cell tumour treated with multimodal therapy. Anticancer research. 2014;34(7):3683–8. [PubMed] [Google Scholar]
- 26.Italiano A, Kind M, Cioffi A, Maki RG, Bui B. Clinical activity of sunitinib in patients with advanced desmoplastic round cell tumor: a case series. Targeted oncology. 2013;8(3):211–3. doi: 10.1007/s11523-012-0251-8. [DOI] [PubMed] [Google Scholar]
- 27.Glade Bender JL, Lee A, Reid JM, Baruchel S, Roberts T, Voss SD, et al. Phase I pharmacokinetic and pharmacodynamic study of pazopanib in children with soft tissue sarcoma and other refractory solid tumors: a children's oncology group phase I consortium report. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31(24):3034–43. doi: 10.1200/JCO.2012.47.0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Menegaz BA, Cuglievan B, Benson J, Camacho P, Lamhamedi-Cherradi SE, Leung CH, et al. Clinical Activity of Pazopanib in Patients with Advanced Desmoplastic Small Round Cell Tumor. Oncologist. 2018;23(3):360–6. doi: 10.1634/theoncologist.2017-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Modak S, Gerald W, Cheung NK. Disialoganglioside GD2 and a novel tumor antigen: potential targets for immunotherapy of desmoplastic small round cell tumor. Med Pediatr Oncol. 2002;39(6):547–51. doi: 10.1002/mpo.10151. [DOI] [PubMed] [Google Scholar]
- 30.Modak S, Kramer K, Gultekin SH, Guo HF, Cheung NK. Monoclonal antibody 8H9 targets a novel cell surface antigen expressed by a wide spectrum of human solid tumors. Cancer research. 2001;61(10):4048–54. [PubMed] [Google Scholar]
- 31.Chao J, Budd GT, Chu P, Frankel P, Garcia D, Junqueira M, et al. Phase II clinical trial of imatinib mesylate in therapy of KIT and/or PDGFRalpha-expressing Ewing sarcoma family of tumors and desmoplastic small round cell tumors. Anticancer research. 2010;30(2):547–52. [PubMed] [Google Scholar]
- 32.Subbiah V, Brown RE, Jiang Y, Buryanek J, Hayes-Jordan A, Kurzrock R, et al. Morphoproteomic profiling of the mammalian target of rapamycin (mTOR) signaling pathway in desmoplastic small round cell tumor (EWS/WT1), Ewing's sarcoma (EWS/FLI1) and Wilms' tumoRWT1) PloS one. 2013;8(7):e68985. doi: 10.1371/journal.pone.0068985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thijs AM, van der Graaf WT, van Herpen CM. Temsirolimus for metastatic desmoplastic small round cell tumor. Pediatric blood & cancer. 2010;55(7):1431–2. doi: 10.1002/pbc.22755. [DOI] [PubMed] [Google Scholar]
- 34.Naing A, LoRusso P, Fu S, Hong DS, Anderson P, Benjamin RS, et al. Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing's sarcoma family tumors. Clin Cancer Res. 2012;18(9):2625–31. doi: 10.1158/1078-0432.CCR-12-0061. doi 1078-0432.CCR-12-0061 [pii] 10.1158/1078-0432.CCR-12-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fine RL, Shah SS, Moulton TA, Yu IR, Fogelman DR, Richardson M, et al. Androgen and c-Kit receptors in desmoplastic small round cell tumors resistant to chemotherapy: novel targets for therapy. Cancer chemotherapy and pharmacology. 2007;59(4):429–37. doi: 10.1007/s00280-006-0280-z. [DOI] [PubMed] [Google Scholar]
- 36.Lim SJ, Cormier JN, Feig BW, Mansfield PF, Benjamin RS, Griffin JR, et al. Toxicity and outcomes associated with surgical cytoreduction and hyperthermic intraperitoneal chemotherapy (HIPEC) for patients with sarcomatosis. Annals of surgical oncology. 2007;14(8):2309–18. doi: 10.1245/s10434-007-9463-z. [DOI] [PubMed] [Google Scholar]
- 37.Goodman KA, Wolden SL, La Quaglia MP, Kushner BH. Whole abdominopelvic radiotherapy for desmoplastic small round-cell tumor. International journal of radiation oncology, biology, physics. 2002;54(1):170–6. doi: 10.1016/s0360-3016(02)02871-7. [DOI] [PubMed] [Google Scholar]
- 38.Pinnix CC, Fontanilla HP, Hayes-Jordan A, Subbiah V, Bilton SD, Chang EL, et al. Whole abdominopelvic intensity-modulated radiation therapy for desmoplastic small round cell tumor after surgery. International journal of radiation oncology, biology, physics. 2012;83(1):317–26. doi: 10.1016/j.ijrobp.2011.06.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Osborne EM, Briere TM, Hayes-Jordan A, Levy LB, Huh WW, Mahajan A, et al. Survival and toxicity following sequential multimodality treatment including whole abdominopelvic radiotherapy for patients with desmoplastic small round cell tumor. Radiother Oncol. 2016;119(1):40–4. doi: 10.1016/j.radonc.2015.10.016. [DOI] [PubMed] [Google Scholar]
- 40.Lal DR, Su WT, Wolden SL, Loh KC, Modak S, La Quaglia MP. Results of multimodal treatment for desmoplastic small round cell tumors. Journal of pediatric surgery. 2005;40(1):251–5. doi: 10.1016/j.jpedsurg.2004.09.046. [DOI] [PubMed] [Google Scholar]
- 41.Ordonez NG. Desmoplastic small round cell tumor: II: an ultrastructural and immunohistochemical study with emphasis on new immunohistochemical markers. The American journal of surgical pathology. 1998;22(11):1314–27. doi: 10.1097/00000478-199811000-00002. [DOI] [PubMed] [Google Scholar]
- 42.Ordonez NG. Desmoplastic small round cell tumor: I: a histopathologic study of 39 cases with emphasis on unusual histological patterns. The American journal of surgical pathology. 1998;22(11):1303–13. doi: 10.1097/00000478-199811000-00001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.