

Abstract
Plasmodium parasites exerted a strong selective pressure on primate genomes and mutations in genes encoding erythrocyte cytoskeleton proteins (ECP) determine protective effects against Plasmodium infection/pathogenesis. We thus hypothesized that ECP-encoding genes have evolved in response to Plasmodium-driven selection. We analyzed the evolutionary history of 15 ECP-encoding genes in primates, as well as of their Plasmodium-encoded ligands (KAHRP, MESA and EMP3). Results indicated that EPB42, SLC4A1, and SPTA1 evolved under pervasive positive selection and that episodes of positive selection tended to occur more frequently in primate species that host a larger number of Plasmodium parasites. Conversely, several genes, including ANK1 and SPTB, displayed extensive signatures of purifying selection in primate phylogenies, Homininae lineages, and human populations, suggesting strong functional constraints. Analysis of Plasmodium genes indicated adaptive evolution in MESA and KAHRP; in the latter, different positively selected sites were located in the spectrin-binding domains. Because most of the positively selected sites in alpha-spectrin localized to the domains involved in the interaction with KAHRP, we suggest that the two proteins are engaged in an arms-race scenario. This observation is relevant because KAHRP is essential for the formation of “knobs”, which represent a major virulence determinant for P. falciparum.
Introduction
Malaria is annually responsible for hundreds of thousands of deaths and millions of illnesses per year (WHO, http://www.who.int/malaria/publications/world-malaria-report-2016/). It is caused by protozoan parasites of the genus Plasmodium, and, although there are about 30 Plasmodium species that infect primates1, only five (P. falciparum, P. vivax, P. malariae, P. knowlesi, and P. ovale) cause malaria in humans. In particular, P. falciparum and P. vivax are the most prevalent, with P. falciparum causing the deadliest form of malaria (see review2).
The extreme virulence of P. falciparum and its propensity to cause severe disease symptoms are due to its ability to invade and refurbish mature human red blood cells (RBCs). In fact, during parasite asexual development, infected RBCs (iRBCs) undergo extensive phenotypic changes in their structure and function. Modification in erythrocyte surface topology, membrane permeability, stiffness, adhesiveness, and deformability lead to sequestration of iRBCs in the microvasculature preventing parasite splenic clearance and causing obstruction (reviewed in3).
P. falciparum achieves these changes in the iRBC structure by exporting parasite proteins into the erythrocytes. Some of these proteins interact with host components of the cytoskeleton and plasma membrane and lead to the formation of cytoadhesive and antigenic supramolecular protrusions (“knobs”) at the iRBC surface3,4. Knobs act as platforms for the presentation of P. falciparum erythrocyte membrane protein 1 (PfEMP1): this membrane-embedded protein is responsible for adhesion to the vascular endothelium5 and represents a major P. falciparum parasite virulence factor. PfEMP1 also interacts with P. falciparum KAHRP (Knob-associated Histidine-rich Protein), which in turn binds host ankyrin as well as spectrin α- and β-chains6–8. In addition to KAHRP, other exported P. falciparum proteins, namely erythrocyte membrane protein 3 (PfEMP3), mature parasite-infected erythrocyte surface antigen (MESA, also known as pfEMP2), Plasmodium helical interspersed subtelomeric (PHIST) proteins, and ring-infected erythrocyte surface antigen (RESA) contribute to the formation and maintenance of the knob structure. All these molecules form direct interactions with human erythrocyte cytoskeleton proteins (ECP)3,4.
ECPs are therefore central for the life cycle and proliferation of P. falciparum. In fact, alterations of the RBC cytoskeleton due to human genetic disorders such as hereditary spherocytosis (HS) and hereditary elliptocytosis (HE) (see Table 1 for associated gene defects) decrease the growth of P. falciparum in RBCs in vitro9–11. Similarly, Southeast Asian ovalocytosis (SAO), which is due to a heterozygous 27 bp deletion in the SLC4A1 gene (encoding the band 3 protein), is associated with protection from cerebral P. falciparum malaria12,13. However, SAO also represents a protective factor for P. vivax infection and for P. vivax malaria severity14. Likewise, mice carrying mutations in genes encoding ECPs are protected from rodent malaria (P. chabaudi, P. berghei, or P. yoelii infection)15–20. Thus, genetic variants in genes encoding ECPs can affect the infection success or pathogenesis of malaria caused by Plasmodium parasites other than P. falciparum.
Table 1.
Likelihood ratio test (LRT) statistics for F3x4 and F61 models in primate phylogenies.
| Gene (Protein) |
Human Disordera | N° species | Model | M1a vs M2ab | M7 vs M8b | % negatively selected sitesd | Positively selected sitese | ||
|---|---|---|---|---|---|---|---|---|---|
| −2ΔlnLc | p value | −2ΔlnLc | p value | ||||||
|
ACTB
(Actin beta) |
23 | ||||||||
| F3x4 | 0.004 | 0.998 | 0.004 | 0.998 | 35.47 | na | |||
| F61 | 0.004 | 0.998 | 0.0003 | 0.999 | |||||
|
ADD1
(Adducin 1) |
23 | ||||||||
| F3x4 | 0 | 1 | 0.22 | 0.898 | 34.19 | na | |||
| F61 | 0 | 1 | 0.02 | 0.989 | |||||
|
ADD2
(Adducin 2) |
23 | ||||||||
| F3x4 | 0.08 | 0.959 | 1.68 | 0.433 | 33.20 | na | |||
| F61 | 0 | 1 | 0.003 | 0.999 | |||||
|
ANK1
(Ankyrin-1) |
HS | 22 | |||||||
| F3x4 | 0 | 1 | 19.03 | 7.39 × 10−5 | 40.67 | na | |||
| F61 | 5.47 | 0.07 | 23.33 | 8.57 × 10−6 | |||||
|
DMTN
(Dematin) | |||||||||
| F3x4 | 0 | 1 | 2.59 | 0.27 | 26,67 | na | |||
| F61 | 0 | 1 | 2.97 | 0.23 | |||||
|
EPB41
(Protein 4.1) |
HE | 23 | |||||||
| Reg1 (241 aa) | F3x4 | 0.009 | 0.99 | 0.83 | 0.660 | 8.68 | na | ||
| F61 | 0.006 | 0.99 | 0 | 1 | |||||
| Reg2 (55 aa) | F3x4 | 3.57 | 0.17 | 5.14 | 0.077 | 9.09 | na | ||
| F61 | 3.17 | 0.21 | 4.17 | 0.124 | |||||
| Reg3 (568 aa) | F3x4 | 0.001 | 0.99 | 0 | 1 | 17.96 | na | ||
| F61 | 0 | 1 | 0 | 1 | |||||
|
EPB42
(Protein 4.2) |
HS | 23 | |||||||
| F3x4 | 40.25 | 1.82 × 10−9 | 53.78 | 2.10 × 10−12 | 12.62 | R9, P24, I102, R117, L159, Q163, R224, R243, F251, R289, L390, E487, R495, T501, H562, I572, N581, E675 | |||
| F61 | 31.63 | 1.35 × 10−7 | 37.80 | 6.19 × 10−9 | |||||
|
MPP1
(p55) |
24 | ||||||||
| F3x4 | 0.003 | 0.999 | 0.009 | 0.995 | 32.83 | na | |||
| F61 | 0 | 1 | 0.011 | 0.994 | |||||
|
RHAG
(Ammonium transporter Rh type A) |
OHS | 24 | |||||||
| F3x4 | 4.12 | 0.127 | 9.37 | 0.009 | 13.17 | na | |||
| F61 | 2.52 | 0.283 | 6.08 | 0.048 | |||||
|
SLC4A1
(Band 3) |
HS, SAO | 31 | |||||||
| F3x4 | 6.43 | 0.04 | 29.02 | 5.01 × 10−7 | 32.69 | E28, R112, E152, D235, H309, E658 | |||
| F61 | 15.76 | 3.79 × 10−4 | 32.44 | 9.02 × 10−8 | |||||
|
SPTA1
(α spectrin) |
HE, HS | 22 | |||||||
| F3x4 | 30.43 | 2.47 × 10−7 | 55.68 | 8.12 × 10−13 | 26.50 | E117, L148, V164, D430, Q434, T459, D466, I745, V1233, Q1332, Q1584 | |||
| F61 | 18.80 | 8.27 × 10−5 | 38.73 | 3.90 × 10−9 | |||||
|
SPTB
(β spectrin) |
HE, HS | 24 | |||||||
| F3x4 | 0 | 1 | 17.94 | 1.27 × 10−4 | 42.69 | na | |||
| F61 | 0 | 1 | 2.90 | 0.234 | |||||
|
TMOD1
(Tropomodulin-1) |
24 | ||||||||
| F3x4 | 0 | 1 | 0.02 | 0.991 | 20.89 | na | |||
| F61 | 0 | 1 | 0 | 1 | |||||
|
TPM1
(Tropomyosin alpha-1 chain) |
23 | ||||||||
| F3x4 | 0 | 1 | 0.05 | 0.974 | 10.92 | na | |||
| F61 | 0 | 1 | 0.16 | 0.922 | |||||
|
TPM3
(Tropomyosin alpha-1 chain) |
24 | ||||||||
| Reg1 (80 aa) | F3x4 | 0 | 1 | 0 | 1 | 15.00 | na | ||
| F61 | 0 | 1 | 0 | 1 | |||||
| Reg2 (205 aa) | F3x4 | 4.22 | 0.121 | 4.26 | 0.119 | 10.24 | na | ||
| F61 | 0.68 | 0.711 | 0.80 | 0.671 | |||||
Notes: aHuman red cell membrane disorders associated with ECP (https://www.ncbi.nlm.nih.gov/medgen/): HS, Hereditary spherocytosis; HE, hereditary elliptocytosis; OHS, overhydratate hereditary stomatocytosis; SAO, Southeast Asian ovalocytosis; bM1a is a nearly neutral model that assumes one ω class between 0 and 1 and one class with ω = 1; M2a (positive selection model) is the same as M1a plus an extra class of ω > 1; M7 is a null model that assumes that 0 < ω < 1 is beta distributed among sites; M8 (positive selection model) is the same as M7 but also includes an extra category of sites with ω > 1; c2ΔlnL: twice the difference of the natural logs of the maximum likelihood of the models being compared; dPercentage of sites evolving under negative selection by FUBAR; ePositions refer to the human sequence.
HE reaches frequencies up to 2% in African populations who live in malaria-endemic areas21 and SAO is very common in P. falciparum-endemic regions of Island Southeast Asia and Melanesia22. These observations clearly underscore the extremely strong selective pressure that Plasmodium parasites exerted on human populations23. Such selection most likely operated on a large number of human loci, including polymorphic variants in genes encoding ECPs24.
Several non-human primates (NHPs) are infected by Plasmodium parasites, and close relatives of P. falciparum and P. vivax were described in recent years in African great apes25. Indeed, P. falciparum, which belongs to the Laverania subgenus, originated from a single gorilla to human cross-species transmission event26. These observations and the notion whereby primate Plasmodium species diverged millions of years ago27 suggest that these parasites have exerted a selective pressure which was not limited to the recent history of human populations, but extended during the timing of primate (and possibly mammalian) speciation27. Herein, we thus investigate the evolutionary history of genes encoding ECPs in primates. We use different strategies to infer which parasites exerted the strongest selective pressure and to identify regions and sites that evolved in response to such pressure.
Results
Positive selection at erythrocyte cytoskeleton proteins
We first aimed to comprehensively analyze the selective pressure acting on primate genes that encode erythrocyte cytoskeleton proteins (ECP). In particular, we focused our attention on genes encoding ECPs that are involved in the remodeling of RBC during Plasmodium infection3,4. Some of these genes (ANK1, EPB41, EPB42, SLC4A1, SPTA1, and SPTB), when mutated, cause red cell membrane disorders (HE, HS, or SAO)21 (Table 1) and encode proteins that are directly bound by malaria parasite proteins.
Coding sequences were obtained for at least 22 primate species (Supplementary Table S1). These sequence data allow sufficient power to detect positive selection at primate genes28. Because recombination can be mistaken as positive selection29,30, DNA alignments were screened for the presence of recombination signals. EPB41 and TPM3 showed 3 and 1 recombination breakpoints, respectively; alignments were thus split in three and two regions (Table 1).
Pervasive positive selection was searched for using the “site models” implemented in the codeml program31. Using likelihood ratio tests (LRTs), codeml compares models of gene evolution that allow (models M2a and M8) or disallow (models M1a and M7) a class of codons to evolve with dN/dS > 1. Thus, M2a and M8 represent the positive selection models that are tested against the neutral M1a and M7 models. The latter were rejected in favor of the positive selection models for SLC4A1, SPTA1, and EPB42 (Table 1).
We next applied the Bayes Empirical Bayes (BEB) analysis, as well as the FUBAR and FEL methods (see Materials and Methods), to identify specific sites targeted by positive selection in these 3 genes. We applied a conservative strategy and called a site as positively selected only if it was detected by at least two methods.
Several positively selected sites were detected in SPTA1, SLC4A1, and EPB42 (Table 1). In SPTA1, the 11 positively selected sites are distributed along the protein sequence, although four of them localize to the α4 spectrin repeat (Fig. 1), which is involved in direct interaction with KAHRP and SBP1 (skeleton binding protein 1) of P. falciparum7,32 (Fig. 1).
Figure 1.

Domain representation of SPTA1, SPTB, and ANK1 proteins. Positively selected sites identified in the primate phylogeny (black), in Homininae lineages (human in red; chimpanzee in blue), as well as sites showing evolutionary rate shifts associated to the Laverania host state (magenta box) are reported. Mutations that cause hereditary elliptocytosis (HE) and decrease P. falciparum growth in vitro (red dots) and regions corresponding to the binding sites of Laverania-encoded proteins are also indicated. Asterisks indicate sites that were identified in different analyses (red asterisk: human lineage, blue asterisk: chimpanzee lineage, black asterisk: primate phylogeny). Plots represent the distribution of negatively selected codons across SPTA1, SPTB1, and ANK1 in Homininae lineages. In particular, the gammaMap-derived posterior probability of γ < 0 is plotted.
For SLC4A1, 5 out 6 positively selected sites localized to the cytosolic N-terminal domain, which is important as an anchoring point for erythroid cytoskeletal proteins (e.g. ankirin-1, spectrins, protein 4.1 and 4.2), denatured hemoglobin, and glycolitic enzymes33. The other positively selected site (E658) is located in the fourth extracellular loop and is required for band 3 association to glycophorin A (GPA)34,35. Both band 3 and GPA interact with P. falciparum merozoite surface protein 1 (MSP1) and band 3 is also bound by multiple P. vivax merozoite proteins15,36,37. Although the regions of band 3 involved in the interaction with merozoite proteins are located on extracellular loops different from loop 436,37, E658 may modulate RBC invasion by (de)stabilizing the GPA-band 3 complex. In this respect it is worth noting that mouse models genetically deficient for band 3, and consequently lacking the GPA-band 3-protein 4.2 complex, are fully resistant to P. yoelii15.
Finally, in EPB42 the 18 positively selected sites are distributed along the protein sequence and one of them (E487) falls in the region involved in the interaction with α spectrin38. Unfortunately, the details of the interaction between EPB42 and Plasmodium-encoded proteins are unknown.
To explore possible variations in selective pressure across the primate phylogeny for the three positively selected genes (EPB42, SLC4A1, and SPTA1), we applied the free ratio (FR) model implemented in the PAML software. In particular, we tested whether models that allow dN/dS to vary along branches had significant better fit to the data than models that assume one same dN/dS across the entire phylogeny39. Results indicated that the FR model fitted the data better than the null model for all three positively selected genes (data not shown), suggesting that the selection acted differently across the phylogeny, with some branches showing dN/dS values higher than 1. We then overlaid the selection signals over the phylogenetic tree to obtain a glimpse of whether positive selection acted on specific lineages (Fig. 2). Most of the Homininae and Cercopithecinae branches showed at least one gene with dN/dS > 1, and this is particularly abundant at the internal and external branches of the Hominini and Papionini tribes (Fig. 2). We next retrieved information on Plasmodium infection for the taxa represented in the phylogeny40–42 (Supplementary Table S1). Notably, episodes of positive selection tend to be more common for species that host a larger number of different Plasmodium parasites (Fig. 2).
Figure 2.

Free ratio model analysis. Results are shown for a restricted primate phylogeny, which includes only species with available sequence information for EPB41, SLC4A1, and SPTA1 genes. Branches showing dN/dS > 1 are marked with colored rectangles. The number of different Plasmodium species identified in each primate species are represented with blue circles.
Laverania-driven selection at SPTA1
To investigate whether some of the selective events we identified in genes encoding ECPs were due to the pressure imposed by Laverania, we used a recently developed approach (TraitRateProp) that allows testing whether a proportion of sites in a gene or region exhibit evolutionary rate shifts that are associated with the state of a binary phenotypic trait43. We thus set phenotype states for species that are or that are not natural hosts for Laverania (see Materials and Methods). Strong evidence that the rate of sequence evolution is associated with Laverania infection was obtained for SPTA1 alone (chi-squared likelihood ratio test, p value = 1.84 × 10−3; relative rate = 10). In particular, TraitRateProp identified 10 SPTA1 residues that show evolutionary rate shifts (Bayes-factor >= 8) associated to the Laverania host state (Fig. 1). Two of these sites (residues I745 and Q1584) were also identified in the codeml analysis described above.
Positive selection in Homininae lineages
To further explore the selective pattern in species that are hosts for Laverania, we took advantage of the availability of genetic diversity data for humans and great apes44,45 to search for positively selected sites in the human, chimpanzee, and gorilla lineages. Specifically, we used a population genetics-phylogenetics approach (gammaMap46) which leverages information of intra-species polymorphism and between-species divergence. This approach has higher power than those described above for selective events that occurred during the most recent evolutionary history of specific lineages.
The gammaMap method categorizes codon-wise population-scaled selection coefficients (γ) into different classes, ranging from strongly beneficial (γ = 100) to inviable (γ = −500), with γ equal to 0 indicating neutrality. We called positively selected sites as those having a cumulative probability higher than 0.8 of γ ≥ 1.
Several positively selected sites were identified in the human and chimpanzee lineages for SPTA1. In humans, positive selection also drove the evolution of a few codons in EPB41, EPB42, and TMOD1 (Table 2). In RHAG, a few positively selected codons were detected in all three great ape lineages.
Table 2.
Positively selected sites in great ape lineages.
| Positively selected sites (posterior probability ≥ 0.80)a | ||
|---|---|---|
| Humanb | Chimpanzeeb | Gorillab |
| EPB41 | ||
| Q190, T195 | — | — |
| EPB42 | ||
| Y74, L159, N172, K228 | — | V539, G577, A583 |
| RHAG | ||
| V86, K407 | K40, I46, D234,C237 | T42, I82, T139, E199, R238, N395 |
| SLC4A1 | ||
| — | A255, I262 | — |
| SPTA1 | ||
| N1501, D1508, A1531, H1546, D1549, E1553, H1556, Q1584, N1597, K1601, R1654, E1671, V1685, K1686, E1700 | Q427, D430, D455, N456, T459, D466, D546, E551, K554, D1549, E1553, H1556, G1557 | — |
| TMOD1 | ||
| M285 | I210 | — |
Note. aPosterior probability of γ > 0 as detected by gammaMap. bPositively selected site in both primate phylogeny and specific lineage are in bold. Positions refer to the human sequence.
In SPTA1, we observed that positively selected sites are clustered into two specific regions: the α4 and α13–15 spectrin repeats. Both regions were reported to directly interact with P. falciparum proteins7,8 (Fig. 1). Notably, all sites that were positively selected in the human lineage map to the distal KAHRP interaction domain, whereas the sites identified in the chimpanzee lineage localize to both KAHRP-binding regions. Five of the sites we identified with GammaMap were also identified by TraitRateProp (Fig. 1).
These data suggest that the selection signals identified in human and chimpanzee SPTA1 derive from an arms-race with KAHRP or, possibly, with SBP1.
Strong selective constraints limit ANK1 and SPTB evolution
β spectrin and ankirin-1 play very important role in stabilizing the erythrocyte membrane and they are bound by several Laverania proteins. For instance, KAHRP binds β spectrin repeats 10–14 with three-fold higher affinity compared to α spectrin 12–16 repeats8. Ankirin is also targeted by KAHRP6 and other P. falciparum proteins47,48. Indeed, a previous study that analyzed the evolution of genes that interact with Apicomplexa parasites indicated that SPTB and ANK1 display features suggestive of adaptive evolution in mammals49. However, we detected no positive selection at SPTB and ANK1, either in the entire primate phylogeny or in Homininae lineages. We thus reasoned that this finding may result from strong functional constraints that prevent amino acid replacements to accrue in response to Plasmodium-exerted selective pressures. To test this possibility, we used FUBAR to calculate the percentage of sites that are target by negative selection in ECP-encoding genes. SPTB and ANK1 showed the highest portion of negatively selected sites (Table 1). We next explored the distribution of negatively selected codons across SPTB1 and ANK1 in Homininae by plotting the gammaMap-derived posterior probability of γ < 0. Results indicated that both genes display extensive regions of selective constraint in humans and great apes (chimpanzee and gorilla) and these regions cover the binding sites of Laverania-encoded proteins (Fig. 1).
To assess whether SPTB and ANK1 were also severely constrained during to the more recent evolution of human populations, we used SnIPRE, which contrasts polymorphism and divergence data at nonsynonymous and synonymous sites, to calculate the constraint parameter f. f represents the proportion of non-synonymous mutations that are tolerated and, therefore, low values of f indicate strong constraints50. f values were calculated for 14881 human genes and a distribution was obtained (Fig. 3). SPTB and ANK1 displayed some of the lowest f values among ECP-encoding genes and their f values were well below the median for all human genes (Fig. 3). Conversely, SPTA1 showed the weakest selective constraint among ECP-encoding genes.
Figure 3.
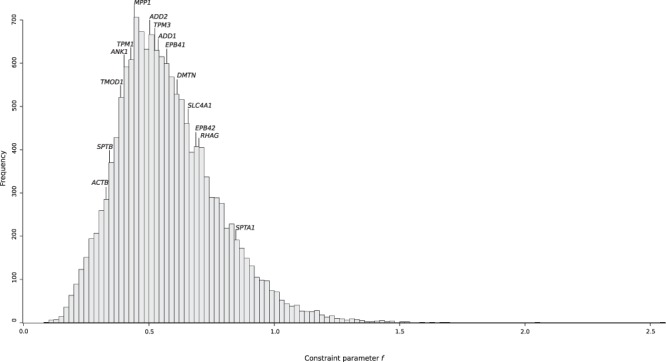
Selective constraints at ECP-encoding genes in human populations. The distribution of the constraint parameter f calculated for reference genes and ECP-encoding genes is reported.
Overall, these results suggest that functional constraint prevent SPTB and ANK1 to evolve in response to the selective pressure exerted by Plasmodium proteins that remodel the RBC cytoskeleton.
Positive selection at Laverania genes that encode proteins involved in RBC remodeling
Several Laverania proteins interact with components of the erythrocyte cytoskeleton. Among these the best known are PfEMP1, MESA, PfEMP3, PHIST proteins, RESA, and KAHRP3. PfEMP1 is encoded by several var genes, and a number of PHIST proteins were detected in Laverania3,51. These proteins cannot thus be analyzed using molecular evolution methods based on ortholog identification and alignment. Similar considerations apply to RESA proteins, which are encoded by at least three paralogs51. We thus focused on KAHRP (PF3D7_0202000), MESA (PF3D7_0500800), and EMP3 (PF3D7_0201900). MESA and EMP3 are composed of unique N-terminal regions and extensive repetitive sequences in the central and C-terminal portions. Repetitive regions could not be reliably aligned and the analysis was thus restricted to the N-terminal portions, which harbor motifs responsible for binding to ECPs52–54.
Alignments of KAHRP and the N-terminal regions of EMP3 and MESA included available Plasmodium falciparum strains, as well as the sequences of P. reichenowi and P. gaboni (this latter was not available for MESA) (Supplementary Table S2). Evidence of positive selection was searched for using the codeml LRT tests, as described above.
The neutral model was not rejected in favor of the positive selection models for EMP3. Conversely, models that allow a class of codons to evolve with dN/dS > 1 fitted the data better than the neutral model for KAHRP and MESA (Table 3). Notably, four of the six positively selected sites in KAHRP are located in a domain which binds α and β spectrins8. Some of these sites are variable across Laverania species and among Plasmodium falciparum isolates (Fig. 4). In contrast, the only selected site in MESA was not located in the protein region that binds protein 4.152,53.
Table 3.
Likelihood ratio test (LRT) statistics for F3x4 and F61 models in Plasmodium phylogenies.
| Gene | N° strain/isolates | Model | M1a vs M2aa | M7 vs M8a | Positively selected sitesc | ||
|---|---|---|---|---|---|---|---|
| −2ΔlnLb | p value | −2ΔlnLb | p value | ||||
| KAHRP | 16 | ||||||
| F3x4 | 34.81 | 2.78 × 10−08 | 35.78 | 1.70 × 10−08 | P123, K443, S467, V492, G516, S603 | ||
| F61 | 36.05 | 1.92 × 10−9 | 37 | 1.18 × 10−9 | |||
| MESA | 11 | ||||||
| F3x4 | 13.54 | 1.15 × 10−03 | 13.71 | 1.05 × 10−03 | N315 | ||
| F61 | 6.61 | 0.010 | 7.24 | 0.0071 | |||
| EMP3 | 15 | ||||||
| F3x4 | 1.70 | 0.43 | 1.76 | 0.41 | — | ||
| F61 | 1.50 | 0.22 | 6.39 | 0.011 | |||
Notes: aM1a is a nearly neutral model that assumes one ω class between 0 and 1 and one class with ω = 1; M2a (positive selection model) is the same as M1a plus an extra class of ω > 1; M7 is a null model that assumes that 0 < ω < 1 is beta distributed among sites; M8 (positive selection model) is the same as M7 but also includes an extra category of sites with ω > 1; b2ΔlnL: twice the difference of the natural logs of the maximum likelihood of the models being compared; c. Positions refer to the Pf_3D7 strain sequence.
Figure 4.

Positive selection at Laverania KAHRP genes. (A) Schematic representation of KAHRP domains. Sites targeted by pervasive positive selection in the Plasmodium phylogeny (orange) and by episodic positive selection on the branch leading to P. falciparum (red) are shown. Regions corresponding to the binding sites of ankiryn-1 and spectrin proteins are indicated. (B) Protein alignments of representative Plasmodium strains for the regions surrounding positively selected sites. (C) Phylogenetic tree of Laverania KAHRP. The branch leading to P. falciparum strains and showing evidence of episodic positive selection is in red. The results of codeml and aBS-REL are also reported.
Finally, we assessed whether episodic positive selection acted on the internal branches of the KAHRP and MESA phylogenies. Evidence of selection was detected for the branch leading to P. falciparum strains for KAHRP (Fig. 4). Only one positively selected sites was detected and is located in a 79-amino acid region that is sufficient to bind ankirin6.
Discussion
Plasmodium parasites represented one of the major selective pressures during the recent evolutionary history of human populations23,24 and, in endemic areas, malaria remains an important cause of death, especially for children and pregnant women (WHO, http://www.who.int/malaria/publications/world-malaria-report-2016/). Data from NHP populations in Africa and Asia indicated that Plasmodium infections are highly prevalent and that these animals can be simultaneously infected by multiple Plasmodium species1,55–61. Although Plasmodium infection is generally thought to result in no or very weak pathology in NHPs, recent reports have indicated that, as in humans, parasitemia is higher in young and pregnant chimpanzees62–64. In a young individual, transitory malaria-like symptoms were also described as a consequence of P. reichenowi infection65. Thus, Plasmodium parasites are potentially pathogenic for NHPs and these animals are likely to have evolved resistance mechanisms. In line with the idea that Plasmodium parasites have exerted a long-standing selective pressure on NHPs, host molecules that serve as the receptor for P. falciparum (basigin) and P. vivax/P. knowlesi (Duffy antigen) were previously shown to have evolved under positive selection in primate phylogenies66,67. It is thus conceivable that other genes that encode molecules involved in Plasmodium infection or pathogenesis have evolved to avoid or limit the fitness costs imposed on primates by these parasites. Indeed, a previous analysis with a limited number of primate species detected evidence of adaptive evolution at the SLC4A1 gene and interpreted this in terms of Laverania-driven selection68. Herein, we detected evidence of pervasive positive selection at genes encoding α spectrin, protein 4.2 and, in agreement with Steiper et al.68, band 3.
Notably, we do not imply that Plasmodium parasites represented the only selective forces that acted on primate ECP-encoding genes. In fact, evidence of adaptive evolution at SPTA1 was recently reported in a study that focused on mammals and detected positively selected sites on several branches of the phylogeny, including species that are not known to be infected by Plasmodium parasites49. However, we analyzed several RBC genes and we detected the strongest evidence of positive selection for those that encode proteins directly interacting with Plasmodium-encoded components. Also, we found that episodes of positive selection tended to occur more frequently in primate species that host a larger number of Plasmodium parasites. Finally, in the case of SPTA1, some of the sites we identified are located close to mutations that were reported to cause HE and to decrease P. falciparum growth in vitro9 (Fig. 1). For this gene, TraitRateProp supported the view that a fraction of sites shifted their evolutionary rates in response to Laverania-exerted selective pressure. Some of these sites are located in the regions bound by PfKAHRP and/or by PfSBP1 and, indeed, they partially overlap with the selection signals we detected in the human and chimpanzee lineages. These latter signals were strongly clustered in the α 4–5 and α 14–15 spectrin domains, suggesting that the recent evolution of SPTA1 was dominated in humans and chimpanzees by a selective pressure that targeted these domains. Overall, these observations suggest that malaria parasites contributed to the shaping of SPTA1 genetic diversity in primates. Experimental validation will nonetheless be required to determine whether selected sites modulate the binding of Plasmodium proteins and affect parasite replication.
We note that some controversy exists about the binding site of PfKAHRP to human α spectrin. An initial analysis indicated an interaction with spectrin repeat α 47, whereas a more recent study detected no binding with this region and indicated repeats α 16–17 (corresponding to α 15–16 spectrin domains in Fig. 1) as the interaction site with PfKAHRP8. Whereas additional experiments will be required to clarify the reasons for these discrepancies and whether they may derive from the use of different P. falciparum strains (IT and 3D7), we note that the α 4 spectrin repeat, which may not bind PfKAHRP and is nonetheless targeted by strong selection in chimpanzee, represents the interaction site with PfSBP132. SBP1 orthologs are found in P. reichenowi and P. gaboni, suggesting that the selective pressure responsible for the observed selection signals at the chimpanzee α 4 is related to SBP1 binding. Indeed, remarkable differences are observed in the selection pattern of the three ape SPTA1 genes. Whereas all selected sites in humans are located in α 13–15, selection in chimpanzees mainly targeted α 4–5, and no positively selected site was detected in gorillas. Unfortunately, the genomes of gorilla-infecting Laverania are unavailable and it is thus unknown whether they also encode KAHRP and SBP1. This is highly plausible, though, as P. preafalciparum and P. blacklocki are more closely related to P. falciparum and P. reichenowi than P. gaboni25, which encodes both proteins. The reasons why no selection signals were detected at the gorilla SPTA1 gene remain to be investigated. We nonetheless found positively selected sites in the gorilla protein 4.1 region that is bound by SBP132. Overall, these results suggest that the interactions between Laverania-encoded proteins that are exported to the erythrocyte membrane and ECPs are dynamic and possibly change over time or during parasite speciation events.
Data herein also clearly indicate that the ability of primate hosts to adapt in response to Plasmodium-exerted pressure is limited by functional constraints. Despite the high-affinity binding of PfKAHRP to β spectrin8, no signal of selection was detected at primate SPTB genes. Across the different time frames considered herein, namely primate radiation, great ape speciation and human evolution, SPTB and, to a lesser extent ANK1, showed extensive signatures of purifying selection, suggesting that most amino acid replacements cause substantial fitness loss. From the parasite’s perspective these molecules most likely represent ideal interactors, as they are not allowed to engage in molecular arms-races.
A limitation of this study is that the evolutionary analyses of Plasmodium genes were conducted using a very limited number of orthologs, as few genome sequences of Laverania species are available. Under these circumstances, the power to detect selection is limited, although false positives are not expected69. This might explain why signals of selection were not detected for EMP3 and only one selected site was identified in MESA. We also mention that, as many proteins encoded by Plasmodium parasites, those we analyzed herein contain repetitive sequences70. Whereas highly repetitive regions must be filtered out to allow reliable alignments, they most likely contribute to protein evolution and parasite adaptation by regulating protein localization, binding affinities, and structural properties70. Thus, analyses herein necessarily fail to account for an important source of variability that may contribute to modulate the binding of Plasmodium proteins to the erythrocyte cytoskeleton. Despite these limitations, we were able to identify some selected sites in KAHRP, most of which are located in the spectrin-binding domain8.
KAHRP-like proteins were described in several Plasmodium species although it is unclear whether they represent orthologs of PfKAHRP as these proteins display different domain structures71. In fact, we used PSI-BLAST to search for proteins similar to PfKAHRP and we detected significant homology over a considerable protein length for Laverania species only. The only exception was a protein encoded by Plasmodium fragile (GenBank: CAB96390.3) which showed almost 98% identity to Plasmodium falciparum KAHRP and was more closely related to PfKAHRP than PrKAHRP or PgKAHRP. This high level of identity was previously noted51. However, because P. fragile belongs to a different Plasmodium subgenus than Laverania, such high identity is surprising and the P. fragile protein was never characterized in a published manuscript. Thus, this sequence was not included in our analyses (that focuses on Laverania) and we suggest that caution should be used when inferring its real occurrence in P. fragile.
In summary, data herein indicate that SPTA1 and KAHRP have been engaged in a genetic conflict and that band 3 may also have evolved in response to the selective pressure exerted by Plasmodium parasites. Conversely, other ECPs have been strongly constrained throughout primate evolutionary history, limiting their ability to accommodate changes that may confer resistance to Plasmodium infection.
Materials and Methods
Evolutionary analysis in Primates and Laverania phylogenies
Coding sequence information for primate species were retrieved from the NCBI database (http://www.ncbi.nlm.nih.gov/) and from UCSC server (http://genome.ucsc.edu/). A complete list of species analyzed for each gene is reported in Supplementary Table S1. Sequence alignments were performed using the RevTrans 2.0 utility72.
For Laverania genes (KAHRP, MESA, and EMP3), coding sequences were retrieved from the NCBI database (http://www.ncbi.nlm.nih.gov/) or via interrogation of the European Nucleotide Archive (https://www.ebi.ac.uk/ena) using the protein ID accession. Lists of accession numbers are reported in Supplementary Table S2.
PSI-BLAST was run via the EMBL-EBI dedicated website (https://www.ebi.ac.uk/Tools/sss/psiblast/).
We used MAFFT73 to generate multiple sequence alignments and GUIDANCE274 for filtering unreliably aligned codons with a score <0.9075.
Each alignment was screened for the presence of recombination breakpoints using GARD (Genetic Algorithm Recombination Detection)76, a program that uses phylogenetic incongruence among segments of a sequence alignment to detect the best-fit number and location of recombination breakpoints. Evidence of recombination was detected for EPB41 and TPM3, whereas no breakpoints were detected for all the remaining primate genes and for Laverania genes.
To detect positive selection, we used the site models implemented in PAML31 for whole gene alignments or independently for sub-regions defined in accordance with the recombination breakpoints. Specifically, we fitted site models that allow (M2a, M8) or disallow (M1a, M7) a class of sites to evolve with ω > 1 to the data using the F3x4 and the F61 codon frequency models. Statistical significance was assessed by comparing twice the ΔlnL of the two models with a χ2 distribution with 2 degrees of freedom. We considered a gene to be positively selected if both comparisons, M1a vs M2a and M7 vs M8, were statistically significant for both codon frequency models (F3x4 and F61). Input phylogenetic trees were reconstructed using the phyML program with a maximum-likelihood approach, a General Time Reversible (GTR) model plus gamma-distributed rates and 4 substitution rate categories77.
Positively selected sites were identified using the codeml Bayes Empirical Bayes analysis (BEB, from model M8 with a cutoff of 0.90)78, the Fixed effects likelihood (FEL, with a default cutoff of 0.1)79, and the Fast Unconstrained Bayesian AppRoximation (FUBAR, with a default cutoff of 0.90)80. To limit false positives, we considered a site as positively selected if it was detected by at least two different methods.
To analyze the pattern of selection at EPB42, SLC4A1, and SPTA1 genes across the primate phylogeny we applied the free ratio (FR) model implemented in the PAML software39. In particular, the FR model was used to estimate the value of dN/dS (non-synonymous substitution/synonymous substitution rate ratio) for each branch of the phylogenies and was compared with a null model that estimates one dN/dS for the entire phylogeny. Statistical significance is assessed by comparing twice the ΔlnL of the two models with a χ2 distribution with degrees of freedom equal to the difference in model parameters.
Data on Plasmodium distribution in NHP were obtained from a previous work that used published records of Plasmodium parasites in NHPs to provide a global overview of primate malarias1. These data were updated through literature searches of studies published after 200540–42.
In order to identify specific branches with a proportion of sites evolving with dN/dS > 1 in the Plasmodium KAHRP and MESA phylogenies, we used the adaptive Branch-Site Random Effects Likelihood method (aBS-REL)81. This method applies sequential likelihood ratio tests to identify branches under positive selection without a priori knowledge about which lineages are of interest81; branches identified using this approach were cross-validated using the branch-site likelihood ratio tests from PAML (models MA and MA1). To identified sites evolving under positive selection on specific branches we used the BEB analysis from MA (with a cutoff of 0.90) and the Mixed Effects Model of Evolution (MEME) (with the default cutoff of 0.1)82. MEME allows the distribution of ω to vary from site to site and from branch to branch at a site. Again, to limit false positives, only sites confirmed by both methods were considered as positively selected.
GARD, FEL, FUBAR, MEME, and aBS-REL analyses were performed either through the DataMonkey server83 (http://www.datamonkey.org) or run locally (through HyPhy84).
The name and the localization of the domains in the protein sequences are taken from Uniprot85.
Population genetics-phylogenetics analysis in Homininae
In order to study the evolution in Homininae and to gain insight into the more recent selective events in specific lineages (human, chimpanzee, and gorilla lineages), we applied a population genetics-phylogenetics approach (gammaMap46) for ANK1, EPB41, EPB42, SLC4A1, SPTA1, and SPTB genes. Ancestral sequences were reconstructed by parsimony from the human, chimpanzee, gorilla, orangutan, gibbon, and macaque sequences.
For human analyses, genotype data from the Phase 1 of the 1000 Genomes Project were retrieved from the dedicated website (http://www.1000genomes.org/)45; in particular, SNP information were retrieved for individuals of three human populations: African (Yoruba), European, and East Asian (Chinese). For the chimpanzee and gorilla analyses, we used SNP information from 25 and 27 individuals, respectively44.
gammaMap uses intra-specific variation and inter-specific diversity to estimate the distribution of population-scaled selection coefficients (γ) along coding regions. The program classifies γ values into 12 categories, ranging from strongly beneficial (γ = 100) to inviable (γ = −500), with γ equal to 0 indicating neutrality. In the analysis, we assumed θ (neutral mutation rate per site), k (transitions/transversions ratio), and T (branch length) to vary among the gene following log-normal distributions. For p (the probability that adjacent codons share the same population-scaled selection coefficient) we assumed a uniform distribution. We set the neutral frequencies of non-STOP codons to 1/61. For population-scaled selection coefficients we considered a uniform Dirichlet distribution with the same prior weight for each selection class. For each gene, two Markov Chain Monte Carlo runs of 100,000 iterations each were run with a thinning interval of 10 iterations. Runs were compared to assess convergence and merged to obtain posterior probabilities. To be conservative, we declared a codon to be targeted by positive selection when the cumulative posterior probability of γ ≥ 1 was ≥0.80.
Purifying selection in humans
The strength of purifying selection was estimated using SnIPRE50, a tool that relies on the comparison of polymorphism and divergence data from synonymous and non-synonymous sites within genes. SnIPRE uses a generalized linear mixed model to represent the genome-wide variability among categories of mutations and to estimate its functional consequence. We estimated the degree of selective constraints at each gene using the f parameter, which is the proportion of non-synonymous mutation that are not deleterious.
The f parameter was estimated for each gene and for 14881 autosomal coding human genes used as reference.
SNP information were retrieved for individuals of all 1000 Genomes Project Phase 1 populations45. To evaluate divergence within genes, we used the liftOver tool to convert human GRCh37/hg19 genome coordinates to Pan troglodytes (CGSC 2.1.3/PanTro3) coordinates; we selected only genes that could be mapped onto chimpanzee genome (n = 14805).
Electronic supplementary material
Acknowledgements
This work was supported by the Italian Ministry of Health, grant n. RC 2016–2018 to Manuela Sironi.
Author Contributions
M.S. and R.C. conceived the study; R.C., M.S. and D.F. performed the analyses; R.C., M.S., D.F. and M.C. analyzed the data; R.C., M.S. and D.F. produced the figures; M.S. and R.C. wrote the manuscript, with critical input from M.C.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-33049-y.
References
- 1.Faust C, Dobson AP. Primate malarias: Diversity, distribution and insights for zoonotic Plasmodium. One Health. 2015;1:66–75. doi: 10.1016/j.onehlt.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.White NJ, et al. Malaria. Lancet. 2014;383:723–735. doi: 10.1016/S0140-6736(13)60024-0. [DOI] [PubMed] [Google Scholar]
- 3.Maier AG, Cooke BM, Cowman AF, Tilley L. Malaria parasite proteins that remodel the host erythrocyte. Nat. Rev. Microbiol. 2009;7:341–354. doi: 10.1038/nrmicro2110. [DOI] [PubMed] [Google Scholar]
- 4.Warncke JD, Vakonakis I, Beck HP. Plasmodium Helical Interspersed Subtelomeric (PHIST) Proteins, at the Center of Host Cell Remodeling. Microbiol. Mol. Biol. Rev. 2016;80:905–927. doi: 10.1128/MMBR.00014-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith JD, Rowe JA, Higgins MK, Lavstsen T. Malaria’s deadly grip: cytoadhesion of Plasmodium falciparum-infected erythrocytes. Cell. Microbiol. 2013;15:1976–1983. doi: 10.1111/cmi.12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weng H, et al. Interaction of Plasmodium falciparum knob-associated histidine-rich protein (KAHRP) with erythrocyte ankyrin R is required for its attachment to the erythrocyte membrane. Biochim. Biophys. Acta. 2014;1838:185–192. doi: 10.1016/j.bbamem.2013.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pei X, et al. Structural and functional studies of interaction between Plasmodium falciparum knob-associated histidine-rich protein (KAHRP) and erythrocyte spectrin. J. Biol. Chem. 2005;280:31166–31171. doi: 10.1074/jbc.M505298200. [DOI] [PubMed] [Google Scholar]
- 8.Cutts EE, et al. Structural analysis of P. falciparum KAHRP and PfEMP1 complexes with host erythrocyte spectrin suggests a model for cytoadherent knob protrusions. PLoS Pathog. 2017;13:e1006552. doi: 10.1371/journal.ppat.1006552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dhermy D, Schrevel J, Lecomte MC. Spectrin-based skeleton in red blood cells and malaria. Curr. Opin. Hematol. 2007;14:198–202. doi: 10.1097/MOH.0b013e3280d21afd. [DOI] [PubMed] [Google Scholar]
- 10.Facer CA. Erythrocytes carrying mutations in spectrin and protein 4.1 show differing sensitivities to invasion by Plasmodium falciparum. Parasitol. Res. 1995;81:52–57. doi: 10.1007/BF00932417. [DOI] [PubMed] [Google Scholar]
- 11.Schulman S, et al. Growth of Plasmodium falciparum in human erythrocytes containing abnormal membrane proteins. Proc. Natl. Acad. Sci. USA. 1990;87:7339–7343. doi: 10.1073/pnas.87.18.7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allen SJ, et al. Prevention of cerebral malaria in children in Papua New Guinea by southeast Asian ovalocytosis band 3. Am. J. Trop. Med. Hyg. 1999;60:1056–1060. doi: 10.4269/ajtmh.1999.60.1056. [DOI] [PubMed] [Google Scholar]
- 13.Taylor SM, Fairhurst RM. Malaria parasites and red cell variants: when a house is not a home. Curr. Opin. Hematol. 2014;21:193–200. doi: 10.1097/MOH.0000000000000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosanas-Urgell A, et al. Reduced risk of Plasmodium vivax malaria in Papua New Guinean children with Southeast Asian ovalocytosis in two cohorts and a case-control study. PLoS Med. 2012;9:e1001305. doi: 10.1371/journal.pmed.1001305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baldwin MR, Li X, Hanada T, Liu SC, Chishti AH. Merozoite surface protein 1 recognition of host glycophorin A mediates malaria parasite invasion of red blood cells. Blood. 2015;125:2704–2711. doi: 10.1182/blood-2014-11-611707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greth A, et al. A novel ENU-mutation in ankyrin-1 disrupts malaria parasite maturation in red blood cells of mice. PLoS One. 2012;7:e38999. doi: 10.1371/journal.pone.0038999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang HM, et al. Ankyrin-1 Gene Exhibits Allelic Heterogeneity in Conferring Protection Against Malaria. G3 (Bethesda) 2017;7:3133–3144. doi: 10.1534/g3.117.300079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang HM, et al. A novel ENU-induced ankyrin-1 mutation impairs parasite invasion and increases erythrocyte clearance during malaria infection in mice. Sci. Rep. 2016;6:37197. doi: 10.1038/srep37197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lelliott PM, et al. Erythrocyte beta spectrin can be genetically targeted to protect mice from malaria. Blood Adv. 2017;1:2624–2636. doi: 10.1182/bloodadvances.2017009274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shear HL, Roth EF, Jr, Ng C, Nagel RL. Resistance to malaria in ankyrin and spectrin deficient mice. Br. J. Haematol. 1991;78:555–560. doi: 10.1111/j.1365-2141.1991.tb04488.x. [DOI] [PubMed] [Google Scholar]
- 21.Narla J, Mohandas N. Red cell membrane disorders. Int. J. Lab. Hematol. 2017;39(Suppl 1):47–52. doi: 10.1111/ijlh.12657. [DOI] [PubMed] [Google Scholar]
- 22.Mgone CS, et al. Occurrence of the erythrocyte band 3 (AE1) gene deletion in relation to malaria endemicity in Papua New Guinea. Trans. R. Soc. Trop. Med. Hyg. 1996;90:228–231. doi: 10.1016/S0035-9203(96)90223-0. [DOI] [PubMed] [Google Scholar]
- 23.Kwiatkowski DP. How malaria has affected the human genome and what human genetics can teach us about malaria. Am. J. Hum. Genet. 2005;77:171–192. doi: 10.1086/432519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pozzoli U, et al. The role of protozoa-driven selection in shaping human genetic variability. Trends Genet. 2010;26:95–9. doi: 10.1016/j.tig.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 25.Loy DE, et al. Out of Africa: origins and evolution of the human malaria parasites Plasmodium falciparum and Plasmodium vivax. Int. J. Parasitol. 2017;47:87–97. doi: 10.1016/j.ijpara.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu W, et al. Origin of the human malaria parasite Plasmodium falciparum in gorillas. Nature. 2010;467:420–425. doi: 10.1038/nature09442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silva JC, Egan A, Arze C, Spouge JL, Harris DG. A new method for estimating species age supports the coexistence of malaria parasites and their Mammalian hosts. Mol. Biol. Evol. 2015;32:1354–1364. doi: 10.1093/molbev/msv005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McBee RM, Rozmiarek SA, Meyerson NR, Rowley PA, Sawyer SL. The effect of species representation on the detection of positive selection in primate gene data sets. Mol. Biol. Evol. 2015;32:1091–1096. doi: 10.1093/molbev/msu399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Worobey M. A novel approach to detecting and measuring recombination: new insights into evolution in viruses, bacteria, and mitochondria. Mol. Biol. Evol. 2001;18:1425–1434. doi: 10.1093/oxfordjournals.molbev.a003928. [DOI] [PubMed] [Google Scholar]
- 30.Schierup MH, Hein J. Consequences of recombination on traditional phylogenetic analysis. Genetics. 2000;156:879–891. doi: 10.1093/genetics/156.2.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 32.Kats LM, et al. Interactions between Plasmodium falciparum skeleton-binding protein 1 and the membrane skeleton of malaria-infected red blood cells. Biochim. Biophys. Acta. 2015;1848:1619–1628. doi: 10.1016/j.bbamem.2015.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang D, Kiyatkin A, Bolin JT, Low PS. Crystallographic structure and functional interpretation of the cytoplasmic domain of erythrocyte membrane band 3. Blood. 2000;96:2925–2933. [PubMed] [Google Scholar]
- 34.Young MT, Tanner MJ. Distinct regions of human glycophorin A enhance human red cell anion exchanger (band 3; AE1) transport function and surface trafficking. J. Biol. Chem. 2003;278:32954–32961. doi: 10.1074/jbc.M302527200. [DOI] [PubMed] [Google Scholar]
- 35.Williamson RC, Toye AM. Glycophorin A: Band 3 aid. Blood Cells Mol. Dis. 2008;41:35–43. doi: 10.1016/j.bcmd.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 36.Goel VK, et al. Band 3 is a host receptor binding merozoite surface protein 1 during the Plasmodium falciparum invasion of erythrocytes. Proc. Natl. Acad. Sci. USA. 2003;100:5164–5169. doi: 10.1073/pnas.0834959100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alam MS, Zeeshan M, Rathore S, Sharma YD. Multiple Plasmodium vivax proteins of Pv-fam-a family interact with human erythrocyte receptor Band 3 and have a role in red cell invasion. Biochem. Biophys. Res. Commun. 2016;478:1211–1216. doi: 10.1016/j.bbrc.2016.08.096. [DOI] [PubMed] [Google Scholar]
- 38.Mandal D, Moitra PK, Basu J. Mapping of a spectrin-binding domain of human erythrocyte membrane protein 4.2. Biochem. J. 2002;364:841–847. doi: 10.1042/bj20020195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Z, Nielsen R. Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J. Mol. Evol. 1998;46:409–418. doi: 10.1007/PL00006320. [DOI] [PubMed] [Google Scholar]
- 40.Figueiredo MAP, Di Santi SM, Manrique WG, Andre MR, Machado RZ. Identification of Plasmodium spp. in Neotropical primates of Maranhense Amazon in Northeast Brazil. PLoS One. 2017;12:e0182905. doi: 10.1371/journal.pone.0182905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Erkenswick GA, Watsa M, Pacheco MA, Escalante AA, Parker PG. Chronic Plasmodium brasilianum infections in wild Peruvian tamarins. PLoS One. 2017;12:e0184504. doi: 10.1371/journal.pone.0184504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu W, et al. Wild bonobos host geographically restricted malaria parasites including a putative new Laverania species. Nat. Commun. 2017;8:1635-017–01798-5. doi: 10.1038/s41467-017-01798-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levy Karin E, Wicke S, Pupko T, Mayrose I. An Integrated Model of Phenotypic Trait Changes and Site-Specific Sequence Evolution. Syst. Biol. 2017;66:917–933. doi: 10.1093/sysbio/syx032. [DOI] [PubMed] [Google Scholar]
- 44.Prado-Martinez J, et al. Great ape genetic diversity and population history. Nature. 2013;499:471–475. doi: 10.1038/nature12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.1000 Genomes Project Consortium et al. A map of human genome variation from population-scale sequencing. Nature467, 1061–1073 (2010). [DOI] [PMC free article] [PubMed]
- 46.Wilson DJ, Hernandez RD, Andolfatto P, Przeworski M. A population genetics-phylogenetics approach to inferring natural selection in coding sequences. PLoS Genet. 2011;7:e1002395. doi: 10.1371/journal.pgen.1002395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tellez M, Matesanz F, Alcina A. The C-terminal domain of the Plasmodium falciparum acyl-CoA synthetases PfACS1 and PfACS3 functions as ligand for ankyrin. Mol. Biochem. Parasitol. 2003;129:191–198. doi: 10.1016/S0166-6851(03)00123-3. [DOI] [PubMed] [Google Scholar]
- 48.Shakya B, Penn WD, Nakayasu ES, LaCount DJ. The Plasmodium falciparum exported protein PF3D7_0402000 binds to erythrocyte ankyrin and band 4.1. Mol. Biochem. Parasitol. 2017;216:5–13. doi: 10.1016/j.molbiopara.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ebel ER, Telis N, Venkataram S, Petrov DA, Enard D. High rate of adaptation of mammalian proteins that interact with Plasmodium and related parasites. PLoS Genet. 2017;13:e1007023. doi: 10.1371/journal.pgen.1007023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eilertson KE, Booth JG, Bustamante CD. SnIPRE: selection inference using a Poisson random effects model. PLoS Comput. Biol. 2012;8:e1002806. doi: 10.1371/journal.pcbi.1002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sargeant, T. J. et al. Lineage-specific expansion of proteins exported to erythrocytes in malaria parasites. Genome Biol. 7, R12-2006-7-2-r12. Epub 2006 Feb 20 (2006). [DOI] [PMC free article] [PubMed]
- 52.Bennett BJ, Mohandas N, Coppel RL. Defining the minimal domain of the Plasmodium falciparum protein MESA involved in the interaction with the red cell membrane skeletal protein 4.1. J. Biol. Chem. 1997;272:15299–15306. doi: 10.1074/jbc.272.24.15299. [DOI] [PubMed] [Google Scholar]
- 53.Black CG, et al. In vivo studies support the role of trafficking and cytoskeletal-binding motifs in the interaction of MESA with the membrane skeleton of Plasmodium falciparum-infected red blood cells. Mol. Biochem. Parasitol. 2008;160:143–147. doi: 10.1016/j.molbiopara.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waller KL, et al. Interactions of Plasmodium falciparum erythrocyte membrane protein 3 with the red blood cell membrane skeleton. Biochim. Biophys. Acta. 2007;1768:2145–2156. doi: 10.1016/j.bbamem.2007.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu W, et al. African origin of the malaria parasite Plasmodium vivax. Nat. Commun. 2014;5:3346. doi: 10.1038/ncomms4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krief S, et al. On the diversity of malaria parasites in African apes and the origin of Plasmodium falciparum from Bonobos. PLoS Pathog. 2010;6:e1000765. doi: 10.1371/journal.ppat.1000765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pacheco M, Cranfield Michael, Cameron Kenneth, Escalante Ananias A. Malarial parasite diversity in chimpanzees: the value of comparative approaches to ascertain the evolution of Plasmodium falciparum antigens. Malaria Journal. 2013;12(1):328. doi: 10.1186/1475-2875-12-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boundenga Larson, Ollomo Benjamin, Rougeron Virginie, Mouele Lauriane, Mve-Ondo Bertrand, Delicat-Loembet Lucrèce M, Moukodoum Nancy, Okouga Alain, Arnathau Céline, Elguero Eric, Durand Patrick, Liégeois Florian, Boué Vanina, Motsch Peggy, Le Flohic Guillaume, Ndoungouet Alphonse, Paupy Christophe, Ba Cheikh, Renaud Francois, Prugnolle Franck. Diversity of malaria parasites in great apes in Gabon. Malaria Journal. 2015;14(1):111. doi: 10.1186/s12936-015-0622-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee KS, et al. Plasmodium knowlesi: reservoir hosts and tracking the emergence in humans and macaques. PLoS Pathog. 2011;7:e1002015. doi: 10.1371/journal.ppat.1002015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Akter, R. et al. Simian malaria in wild macaques: first report from Hulu Selangor district, Selangor, Malaysia. Malar J. 14, 386-015-0856-3 (2015). [DOI] [PMC free article] [PubMed]
- 61.Zhang, X. et al. Distribution and prevalence of malaria parasites among long-tailed macaques (Macaca fascicularis) in regional populations across Southeast Asia. Malar J. 15, 450-016-1494-0 (2016). [DOI] [PMC free article] [PubMed]
- 62.Wu, D. F. et al. Seasonal and inter-annual variation of malaria parasite detection in wild chimpanzees. Malar J. 17, 38-018-2187-7 (2018). [DOI] [PMC free article] [PubMed]
- 63.De Nys Hélène M, Calvignac-Spencer Sébastien, Boesch Christophe, Dorny Pierre, Wittig Roman M, Mundry Roger, Leendertz Fabian H. Malaria parasite detection increases during pregnancy in wild chimpanzees. Malaria Journal. 2014;13(1):413. doi: 10.1186/1475-2875-13-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Nys HM, et al. Age-related effects on malaria parasite infection in wild chimpanzees. Biol. Lett. 2013;9:20121160. doi: 10.1098/rsbl.2012.1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Herbert, A. et al. Malaria-like symptoms associated with a natural Plasmodium reichenowi infection in a chimpanzee. Malar J. 14, 220-015-0743-y (2015). [DOI] [PMC free article] [PubMed]
- 66.Forni D, et al. Positive selection underlies the species-specific binding of Plasmodium falciparum RH5 to human basigin. Mol. Ecol. 2015;24:4711–4722. doi: 10.1111/mec.13354. [DOI] [PubMed] [Google Scholar]
- 67.Demogines A, Truong KA, Sawyer SL. Species-specific features of DARC, the primate receptor for Plasmodium vivax and Plasmodium knowlesi. Mol. Biol. Evol. 2012;29:445–449. doi: 10.1093/molbev/msr204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Steiper ME, Walsh F, Zichello JM. The SLC4A1 gene is under differential selective pressure in primates infected by Plasmodium falciparum and related parasites. Infect. Genet. Evol. 2012;12:1037–1045. doi: 10.1016/j.meegid.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wong WS, Yang Z, Goldman N, Nielsen R. Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics. 2004;168:1041–1051. doi: 10.1534/genetics.104.031153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Davies HM, Nofal SD, McLaughlin EJ, Osborne AR. Repetitive sequences in malaria parasite proteins. FEMS Microbiol. Rev. 2017;41:923–940. doi: 10.1093/femsre/fux046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Davies HM, Thalassinos K, Osborne AR. Expansion of Lysine-rich Repeats in Plasmodium Proteins Generates Novel Localization Sequences That Target the Periphery of the Host Erythrocyte. J. Biol. Chem. 2016;291:26188–26207. doi: 10.1074/jbc.M116.761213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wernersson R, Pedersen AG. RevTrans: Multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 2003;31:3537–3539. doi: 10.1093/nar/gkg609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sela I, Ashkenazy H, Katoh K, Pupko T. GUIDANCE2: accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 2015;43:W7–14. doi: 10.1093/nar/gkv318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Privman E, Penn O, Pupko T. Improving the performance of positive selection inference by filtering unreliable alignment regions. Mol. Biol. Evol. 2012;29:1–5. doi: 10.1093/molbev/msr177. [DOI] [PubMed] [Google Scholar]
- 76.Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SD. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 2006;23:1891–1901. doi: 10.1093/molbev/msl051. [DOI] [PubMed] [Google Scholar]
- 77.Guindon S, Delsuc F, Dufayard JF, Gascuel O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol. Biol. 2009;537:113–137. doi: 10.1007/978-1-59745-251-9_6. [DOI] [PubMed] [Google Scholar]
- 78.Anisimova M, Bielawski JP, Yang Z. Accuracy and power of bayes prediction of amino acid sites under positive selection. Mol. Biol. Evol. 2002;19:950–958. doi: 10.1093/oxfordjournals.molbev.a004152. [DOI] [PubMed] [Google Scholar]
- 79.Kosakovsky Pond SL, Frost SD. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005;22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- 80.Murrell B, et al. FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013;30:1196–1205. doi: 10.1093/molbev/mst030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Smith MD, et al. Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 2015;32:1342–1353. doi: 10.1093/molbev/msv022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Murrell B, et al. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012;8:e1002764. doi: 10.1371/journal.pgen.1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Delport W, Poon AF, Frost SD, Kosakovsky Pond SL. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics. 2010;26:2455–2457. doi: 10.1093/bioinformatics/btq429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pond SL, Frost SD, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- 85. The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 (2017). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.