
Figure 1.
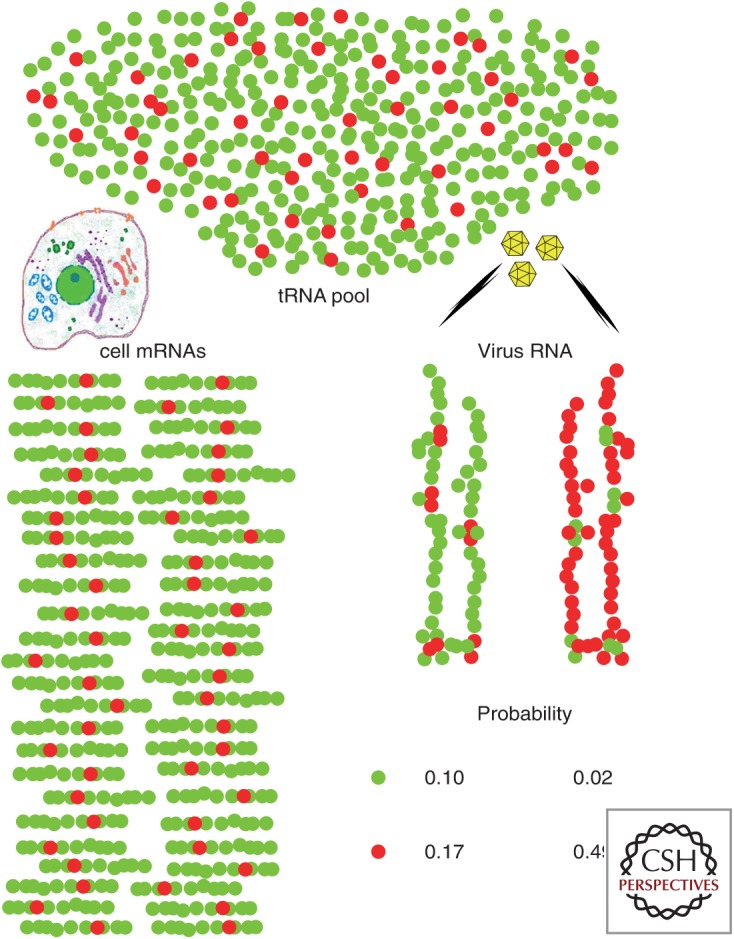
Probability that hepatitis A virus (HAV) RNA will pair successfully with the abundant or nonabundant transfer RNAs (tRNAs) required for its translation under two different codon usage simulations, considered within the framework of a very simplistic model for cellular codon usage involving only a single amino acid encoded by either a major abundant codon (green balls) or a minor rare codon (red balls). Three assumptions are made: (1) HAV is a very slowly replicating virus (inefficient internal ribosome entry site [IRES]) with few, but very lengthy translating RNAs compared to cellular messenger RNAs (mRNAs) (the model assumes one translating HAV RNA for every 50 cellular mRNAs per cell, and that it is 6× longer than the average cellular mRNA), (2) HAV is unable to shut down cellular protein synthesis, and (3) tRNA pools are limiting and adapted to the cellular mRNAs. In a hypothetical situation in which HAV adopts the same codon usage as the cell (optimized: viral RNA on the left), the probabilities of tRNA pairing with the most abundant codon and tRNA pairing with the less abundant codon are 0.1 and 0.17, respectively. In contrast, with codon usage opposite with respect to the cell (deoptimized: viral RNA on the right), these probabilities are 0.02 and 0.49. The latter scenario provides an overall advantageous outcome for the virus, with a higher probability of getting tRNAs pairing with its most abundant codons, while accomplishing the goal of using incidentally very low abundance tRNAs to slow down translation at certain positions.