

Abstract
The 1921 discovery of insulin was a Big Bang from which a vast and expanding universe of research into insulin action and resistance has issued. In the intervening century, some discoveries have matured, coalescing into solid and fertile ground for clinical application; others remain incompletely investigated and scientifically controversial. Here, we attempt to synthesize this work to guide further mechanistic investigation and to inform the development of novel therapies for type 2 diabetes (T2D). The rational development of such therapies necessitates detailed knowledge of one of the key pathophysiological processes involved in T2D: insulin resistance. Understanding insulin resistance, in turn, requires knowledge of normal insulin action. In this review, both the physiology of insulin action and the pathophysiology of insulin resistance are described, focusing on three key insulin target tissues: skeletal muscle, liver, and white adipose tissue. We aim to develop an integrated physiological perspective, placing the intricate signaling effectors that carry out the cell-autonomous response to insulin in the context of the tissue-specific functions that generate the coordinated organismal response. First, in section II, the effectors and effects of direct, cell-autonomous insulin action in muscle, liver, and white adipose tissue are reviewed, beginning at the insulin receptor and working downstream. Section III considers the critical and underappreciated role of tissue crosstalk in whole body insulin action, especially the essential interaction between adipose lipolysis and hepatic gluconeogenesis. The pathophysiology of insulin resistance is then described in section IV. Special attention is given to which signaling pathways and functions become insulin resistant in the setting of chronic overnutrition, and an alternative explanation for the phenomenon of ‟selective hepatic insulin resistanceˮ is presented. Sections V, VI, and VII critically examine the evidence for and against several putative mediators of insulin resistance. Section V reviews work linking the bioactive lipids diacylglycerol, ceramide, and acylcarnitine to insulin resistance; section VI considers the impact of nutrient stresses in the endoplasmic reticulum and mitochondria on insulin resistance; and section VII discusses non-cell autonomous factors proposed to induce insulin resistance, including inflammatory mediators, branched-chain amino acids, adipokines, and hepatokines. Finally, in section VIII, we propose an integrated model of insulin resistance that links these mediators to final common pathways of metabolite-driven gluconeogenesis and ectopic lipid accumulation.
I. INTRODUCTION
Type 2 diabetes mellitus (T2D) is one of the defining medical challenges of the 21st century (960). Overconsumption of relatively inexpensive, calorically dense, inadequately satiating, highly palatable food in industrialized nations has led to unprecedented increases in obesity. In the United States, the combined prevalence of diabetes and prediabetes is over 50% (538). Although only a subset of obese people develops T2D, obesity is a major risk factor for T2D, and rates of T2D prevalence have paralleled those of obesity (381). The fasting hyperglycemia that defines T2D is largely secondary to inadequate action of the major glucose-lowering hormone: insulin. Understanding the mechanisms of insulin action is therefore essential for the continued development of effective therapeutic strategies to combat T2D.
Insulin is an endocrine peptide hormone that binds plasma membrane-bound receptors in target cells to orchestrate an integrated anabolic response to nutrient availability. In all animals, insulin or insulin-like peptides (ILPs) have been identified (120). In invertebrates, ILPs provide mitogenic signaling input, but their effects on metabolic processes and fuel selection are less significant (917). Leveraging gene duplication events through evolutionary time, mammals developed specialized functions for the related peptide hormones insulin, insulin-like growth factor (IGF)-1 and IGF-2 (120). IGF-1 and IGF-2 promote cell growth and differentiation in mammals; in contrast, insulin primarily controls metabolic fluxes (204). However, the blurriness of these functional distinctions is highlighted by the high homology between the insulin and IGF-1 receptors, which form hybrid heterodimers in many cell types and share many downstream effectors (41, 770). The overlap in signaling functions between insulin and IGF-1 likely also contributes to the well-established relationship between hyperinsulinemia and several cancers (631). In this review, we focus on physiological effects of mammalian insulin binding to the insulin receptor and molecular mechanisms by which insulin’s effects are attenuated in the insulin-resistant state that heralds and accompanies T2D.
Although many somatic cell types express insulin receptors, the role of insulin in glucose homeostasis is typified by insulin’s direct effects on skeletal muscle, liver, and white adipocytes. These tissues perform distinct roles in metabolic homeostasis, necessitating tissue-specific insulin signal transduction pathways. For example, in skeletal muscle, insulin promotes glucose utilization and storage by increasing glucose transport and net glycogen synthesis. In liver, insulin activates glycogen synthesis, increases lipogenic gene expression, and decreases gluconeogenic gene expression. In white adipocyte tissue (WAT), insulin suppresses lipolysis and increases glucose transport and lipogenesis. Despite these diverse effects, the proximal components involved in insulin signal transduction are remarkably similar in all insulin-responsive cells. The diversity of physiological insulin responses in different cell types largely owes to distinct distal effectors. The cell-autonomous effects of insulin in skeletal muscle, liver, and WAT, with an emphasis on signal transduction events linked to physiological regulation of metabolic fluxes, will be explored in section II.
In addition to these direct effects, insulin also exerts important indirect effects on target tissues. Because of the integrated, context-specific nature of these indirect effects, they are difficult to model in cultured cells and are consequently less well understood than direct, cell-autonomous effects of insulin. An example of indirect insulin action is the effect of insulin suppression of WAT lipolysis to decrease hepatic acetyl-CoA content, in turn allosterically decreasing pyruvate carboxylase activity. This mechanism, together with suppression of glycerol turnover, enables insulin suppression of WAT lipolysis to suppress hepatic gluconeogenesis (684, 903). Insulin suppression of glucagon secretion through paracrine signaling in the pancreatic islet and insulin action in the central nervous system (CNS) represent other important pathways of indirect insulin action. These physiological processes will be examined in section III.
When higher circulating insulin levels are necessary to achieve the integrated glucose-lowering response described above, a subject is considered insulin resistant. A variety of clinical entities–prediabetes, lipodystrophy (642), polycystic ovarian syndrome (202), nonalcoholic fatty liver disease (520)–are accompanied by increased fasting plasma insulin concentrations. This increased work load for the endocrine pancreas, and consequent β-cell decompensation, is a major mechanism for the development of overt T2D (380, 389, 750). However, the importance of insulin resistance in the pathogenesis of T2D is highlighted by prospective human studies that have revealed insulin resistance as the best predictor of future T2D diagnosis (481, 884). Because insulin action serves different functions in different cell types, insulin resistance has diverse functional ramifications in the various insulin target tissues. The cellular and molecular physiology of insulin resistance will be explored in section IV, with special attention to specific molecular sites of blockade, contributions of indirect insulin action, and the proposed entity of pathway-selective hepatic insulin resistance, wherein some signaling pathways downstream of the insulin receptor appear to retain insulin responsiveness while others manifest insulin resistance (99, 921).
Having described the phenomenon of insulin resistance in section IV, we proceed to examine its mechanistic basis. Mechanisms of insulin resistance are most helpfully categorized using the molecular mediators, pathways, and networks involved. The remainder of this review examines the experimental support for several proposed mechanisms of cellular insulin resistance using this paradigm.
Several lipid moieties, including diacylglycerol (DAG), ceramides, and acylcarnitines, have been implicated in the pathogenesis of liver and skeletal muscle insulin resistance (127, 561, 724). The mechanistic pathways elucidated, with varying levels of experimental support, largely run parallel to one another such that the involvement of one mediator does not preclude the involvement of another. The putative mediators, pathways, and networks involved in lipid-induced liver and muscle insulin resistance are discussed in section V.
A substantial literature describes cellular mechanisms for insulin resistance that are thought to be independent of lipotoxicity. These include endoplasmic reticulum stress and the unfolded protein response (481), reactive oxygen intermediates acting in various subcellular compartments (37), and substrate competition between glucose and fatty acids (397, 677). Section VI examines the experimental evidence for involvement of each of these pathways in typical obesity-associated insulin resistance, with consideration of their role in an integrated physiological framework.
Finally, increasing recognition of the integrated nature of metabolic physiology has sparked investigation of mechanisms of insulin resistance that involve crosstalk between insulin-responsive tissues. Inflammatory signaling has emerged as a key paracrine/endocrine driver of insulin resistance; for example, activated adipose tissue macrophages have been strongly linked to metabolic dysfunction (331, 446, 594). The mechanisms by which inflammation promotes insulin resistance are under intense investigation. Additionally, the last two decades have yielded the identification of dozens of endogenous circulating bioactive peptide hormones with putative effects on insulin sensitivity and have also revealed that circulating branched-chain amino acids may be a predictive biomarker of insulin resistance (577). Rather than providing a catalog entry for each of these circulating factors, section VII focuses on those with established mechanistic links to cellular mechanisms of insulin action and resistance: retinol binding protein-4 (RBP4), adiponectin, fetuin-A, and fibroblast growth factor 21 (FGF21).
In offering this review, we hope that our comprehensive treatment of both insulin action and inaction presents a unified framework for understanding the physiology of this critically important signaling axis in health and disease and that it provides context for future discoveries that will facilitate the prevention and treatment of T2D. We attempt to develop such a unified summary in section VIII.
II. DIRECT INSULIN ACTION
A. Proximal Insulin Signaling: The Insulin Receptor and Its Direct Substrates
Insulin exerts all of its known physiological effects by binding to the insulin receptor (INSR) on the plasma membrane of target cells (297). INSR is a heterotetrameric receptor tyrosine kinase formed from two extracellular α subunits, which bind insulin, and two membrane-spanning β subunits, each of which contains a tyrosine kinase domain (343). There are two INSR isoforms, A and B, but the B isoform is much more specific for insulin; is the primary isoform expressed in differentiated liver, muscle, and WAT; and is thus thought to mediate most metabolic effects of insulin (44). The A isoform, differentiated by the splicing out of exon 11, is expressed highly in fetal development, when its high affinity for IGF-2 is particularly useful (44). INSR has two insulin binding sites but exhibits negative cooperativity, meaning that insulin binding at one site decreases insulin binding affinity in the other site (186). Thus available evidence indicates that at physiological concentrations, one insulin molecule binds and activates one INSR (186, 343). The induced conformational change in the β subunit relieves cis-autoinhibition in the kinase activation loop and permits trans-autophosphorylation of the activation loop tyrosines Tyr1162, Tyr1158, and Tyr1163, in that order (344, 889). The β subunit, thus activated by tris-phosphorylation, undergoes further tyrosine phosphorylation on residues including Tyr972 in the juxtamembrane region; these additional events are important for recruitment of INSR substrates (941). Signaling events downstream of INSR activation can be grossly functionally divided into mitogenic and metabolic signals. The mitogenic signals primarily involve activation of the mitogen-activated protein kinase (MAPK) pathway common to many receptor tyrosine kinases; this signaling axis has been reviewed extensively (41, 400, 535, 616). The insulin concentrations necessary to stimulate metabolic responses are lower than those needed for mitogenic responses; this relationship is reversed for the IGF-1 receptor (41). This review focuses on the INSR-activated pathways that regulate metabolism.
In all cell types, activated INSR initiates downstream metabolic signaling by first recruiting phosphotyrosine-binding scaffold proteins, which in turn activate downstream effectors (FIGURE 1) (826). This is in contrast to many other receptor tyrosine kinases, which phosphorylate cytoplasmic substrates directly. The recruitment of diverse phosphotyrosine-binding proteins to INSR permits early ramification of insulin signaling to activate multiple functional modules. INSR can engage several phosphotyrosine-binding proteins. SHC interacts through its phosphotyrosine-binding (PTB) domain with INSR pTyr972 (343). SH2B1, SH2B2/APS, GRB10, and GRB14 interact through their Src homology 2 (SH2) domains with the activated, tris-phosphorylated INSR activation loop (190, 343). These substrates can serve critical regulatory functions (190, 343). For example, GRB10 phosphorylation and stabilization by mTORC1, which is itself activated by insulin signaling, provides feedback inhibition of INSR activity (339). Other INSR substrates, such as GRB2 and SHC, are involved in the mitogenic arm of insulin signaling (41), while SH2B2/APS helps to initiate the metabolic insulin response, at least in some cell types (471). Attenuation of this proximal phosphotyrosine-based insulin signaling is carried out in part by receptor internalization and dephosphorylation. One key regulator of INSR internalization is CEACAM1, which is itself an INSR substrate (568, 660). INSR dephosphorylation is performed by protein tyrosine phosphatases (PTPases), especially PTP1B. However, this attenuation likely occurs with a time delay, after INSR internalization (808). Immediately after activation, INSR inhibits PTP1B activity by activating NAD(P)H oxidase 4 (NOX4). NOX4-derived H2O2 in turn inhibits PTP1B activity, providing feedforward amplification in the early phase of insulin signaling (515, 921).
FIGURE 1.

Proximal insulin signaling. Upon insulin binding, the insulin receptor (INSR) autophosphorylates and recruits diverse substrates. The two major arms of insulin signaling are mitogenic (initiated by GRB2 and SHC) and metabolic [initiated by insulin receptor substrate (IRS) proteins and SH2B2/APS]. Insulin signaling is also characterized by feedback mechanisms, both positive [GIV potentiation of phosphoinositide-3-kinase (PI3K)-AKT signaling, and phosphatase inhibition by NAD(P)H oxidase 4 (NOX4)-derived H2O2] and negative (stabilization and recruitment of GRB10 to the INSR, and activation of S6 kinase 1 (S6K1) to phosphorylate and inhibit IRS proteins). Green circles and arrows represent activating events; red circles and arrows represent inhibitory events.
Although the aforementioned INSR substrates play important and incompletely understood roles, the best-described class of INSR scaffolds is the insulin receptor substrate (IRS) family. Although there are six IRS isoforms (IRS1–6), IRS1 and IRS2 are thought to mediate most of the metabolic effects of INSR activation; either muscle-specific or liver-specific deletion of both Irs1 and Irs2 in mice phenocopies Insr deletion in those tissues (83, 196, 498). IRS proteins have NH2-terminal pleckstrin homology (PH) and PTB domains that target them to activated INSR, and their long COOH-terminal tails are replete with tyrosine and serine/threonine phosphorylation sites (900). After binding of the IRS PTB domain to INSR pTyr972, INSR phosphorylates multiple IRS tyrosine residues, which in turn recruit downstream signaling effectors to propagate and amplify the insulin response (343). The many (>70) COOH-terminal serine/threonine phosphorylation sites of IRS proteins affect IRS activity and protein stability, allowing them to mediate feedback inhibition of insulin signaling, most prominently by S6 kinase (S6K) (169, 554, 899). As we will consider in later sections, IRS phosphorylation is also a major mechanism by which several stimuli are thought to cause insulin resistance.
Tyrosine-phosphorylated IRS proteins then recruit phosphoinositide-3-kinase (PI3K) heterodimers containing a regulatory p85 subunit and a catalytic p110 subunit. Specifically, tyrosine-phosphorylated IRS YXXM motifs recruit the SH2 domain of the PI3K regulatory subunit (826). The five distinct PI3K regulatory subunit isoforms are encoded by three genes (Pik3r1, Pik3r2, and Pik3r3); there are three PI3K catalytic subunit isoforms encoded by three genes (Pik3ca, Pik3cb, Pik3cd). Not surprisingly, the combinations of PI3K heterodimer composition have complicated investigations of isoform-specific functions. However, the critical importance of PI3K activity in insulin action is well-established: pharmacological PI3K inhibition abolishes insulin stimulation of glucose transport and DNA synthesis, and various PI3K subunit knockout models generally support the classification of PI3K as an essential node in insulin signaling (88, 128, 150, 240, 790, 826). PI3K catalyzes the production of phosphatidylinositol-3,4,5-trisphosphate (PIP3) from phosphatidylinositol-4,5-bisphosphate (PIP2). The reverse reaction is catalyzed by phosphatase and tensin homolog deleted on chromosome 10 (PTEN), and PTEN activity is inhibited by insulin through incompletely understood mechanisms that may involve PTEN interaction with the PIP3-Rac exchanger 2 (P-REX2) (319). This coordinated activation of PI3K and inhibition of PTEN enables net accumulation of PIP3 to propagate and amplify insulin signaling. PIP3 then recruits proteins with PH domains to the plasma membrane, helping to colocalize downstream signaling effectors. Two such effectors are the phosphoinositide-dependent kinase 1 (PDK1) and AKT. After binding to PIP3, AKT is activated by phosphorylation in its activation loop (Thr308 in AKT1) by PDK1 (16, 800), and in its hydrophobic motif (Ser473 in AKT1) by mechanistic target of rapamycin complex 2 (mTORC2) (729). Importantly, although AKT Ser473 phosphorylation is perhaps the most commonly used readout of cellular insulin action, the precise signaling cascade linking INSR activation to AKT Ser473 phosphorylation is unknown. mTORC2 phosphorylation of AKT Ser473 is partially IRS-independent; insulin still stimulates AKT Ser473 phosphorylation in mice lacking both IRS1 and IRS2, although to a lesser extent than normal (195). Activated AKT phosphorylates many downstream substrates in diverse functional pathways, making it a key node in the ramification of insulin signaling. The importance of AKT for normal insulin action is highlighted by the identification of a partial loss-of-function mutation in AKT2 in ~1% of the Finnish population that impairs insulin-stimulated glucose uptake in muscle and adipose tissue and increases endogenous glucose production (454). PI3K-AKT signaling is potentiated by INSR-mediated tyrosine phosphorylation of the guanine exchange factor GIV/Girdin, providing feedforward amplification to proximal insulin action (500, 514).
These proximal insulin signaling events–insulin receptor activation and recruitment/phosphorylation of signaling proteins, most prominently IRS, PI3K, and AKT isoforms–are largely conserved in insulin target tissues and initiate the insulin response at the plasma membrane. We now consider key insulin-responsive cell types individually to better describe how specific downstream effectors produce tissue-specific physiological responses.
B. Skeletal Muscle Insulin Signaling: Effectors and Effects
Skeletal muscle is an energy-consuming tissue; any energy the myocyte stores is mostly for its own later use with the exception of 3-carbon units (lactate, alanine) generated by glycolysis that are released by skeletal muscle and mostly cycled to the liver. Insulin signals to skeletal muscle that glucose is abundant; accordingly, the myocyte insulin signaling cascade is specialized to promote glucose uptake and net glycogen synthesis. The absolute requirement of the myocellular insulin receptor for these processes was demonstrated by hyperinsulinemic-euglycemic clamp studies of muscle-specific INSR knockout (MIRKO) mice, which displayed impairments in insulin-stimulated muscle glucose uptake and muscle glycogen synthesis (407). Muscle-specific knockout of Grb10 in mice, which results in loss of its feedback inhibition on INSR as discussed previously, enhances myocellular insulin sensitivity and increases muscle size (329). Although both IRS1 and IRS2 are expressed in skeletal muscle, the primary INSR substrate in muscle appears to be IRS1. IRS1 knockdown, but not IRS2 knockdown, causes defective insulin-stimulated glucose transport in L6 rat myotubes and human primary myotubes (87, 340, 837). Additionally, isolated soleus muscles from Irs2−/− mice have normal dose-dependent insulin stimulation of glucose uptake (316). Irs2 may be important for insulin control of lipid metabolism in the myocyte (87). Both of the major isoforms of the PI3K catalytic subunit, p110α and p110β, are expressed in skeletal muscle. Of the five PI3K regulatory subunit splice isoforms, p85α, p85β, and p55α are thought to be most relevant in skeletal muscle (826), as mice with muscle-specific deletion of these isoforms have impaired (although not abolished) insulin-stimulated glucose uptake and glycogen synthesis (505). Increases in membrane PIP3 content cause the membrane recruitment of the PH domain-containing kinases PDK1 and AKT (471). Both AKT1 and AKT2 are present in skeletal muscle, but AKT2 appears to be more important for insulin-stimulated glucose metabolism. RNA interference of Akt2 in primary human myotubes abrogated insulin stimulation of glucose uptake and glycogen synthesis, while Akt1 knockdown had no effect on these parameters (87). In support of this paradigm, Akt2−/− mice are severely glucose intolerant (141), while Akt1−/− mice display normal glucose tolerance, although a severe growth defect complicates metabolic phenotyping in Akt1−/− mice (142).
Perhaps the best studied functional effect of the myocellular insulin signaling cascade is increased glucose transport activity. This is accomplished through highly coordinated translocation and fusion of the glucose transporter GLUT4, packaged in GLUT4 storage vesicles (GSVs), to the plasma membrane (471). Current understanding of this process in muscle stands somewhat in contrast to that in adipocytes, for which PI3K-dependent and PI3K-independent pathways have been described. Future research may identify essential PI3K-independent mechanisms for insulin-stimulated muscle glucose uptake, but current evidence primarily implicates PI3K-dependent control (505, 818). Unexpectedly, Pik3r1−/− mice displayed paradoxical increases in insulin-stimulated glucose transport, but this effect likely owed to compensation from other PI3K regulatory subunits (833). Mice lacking both Pik3r1 and Pik3r2 in skeletal muscle (Pik3r1 mKO Pik3r2−/− mice) exhibited impaired insulin-stimulated glucose transport (505). The magnitude of this impairment in Pik3r1 mKO Pik3r2−/− mice was smaller than what would be expected if insulin-stimulated glucose transport was entirely PI3K-dependent, and PI3K activation per se may not be sufficient to cause GLUT4 translocation in L6 myotubes, suggesting that PI3K-independent mechanisms may be operative (352, 471, 505). However, the (incompletely specific) PI3K inhibitor wortmannin can completely abolish insulin-stimulated muscle glucose uptake (274, 818). PI3K control of GLUT4 translocation is mediated through parallel signaling through AKT and the Rho GTPase RAC1 and involves the coordinated action of many proteins involved in GSV trafficking and fusion (140, 471, 818).
AKT phosphorylates several proteins involved in myocellular glucose uptake. The best characterized of these AKT substrates are the GTPase-activating protein (GAP) AKT substrate of 160 kDa (AS160), also known as TBC1D4, and the related GAP TBC1D1 (384, 727, 831). Phosphorylation by AKT blocks TBC1D4/TBC1D1 inactivation of small Rab GTPase protein switches that control vesicle trafficking; the net effect is to promote GSV translocation (413). RAB8, RAB10, and RAB14 have variously been implicated as targets of TBC1D4/TBC1D1 (471). TBC1D4 Thr649 is a physiologically important AKT substrate; mice homozygous for a Thr649Ala knock-in mutation have impaired insulin-stimulated myocellular GLUT4 translocation and are glucose intolerant (131). The physiological relevance of AS160 was further confirmed by the identification of a family carrying a truncating mutation in TBC1D4 that resulted in profound insulin resistance (178). Although TBC1D1 is better characterized as an AMP-activated kinase (AMPK) target than as an AKT target (831), mice with muscle-specific TBC1D1 deletion also have impaired insulin-stimulated muscle glucose uptake (194). The relative physiological importance of TBC1D4 versus TBC1D1 for insulin-stimulated GSV translocation in human muscle remains unclear and may vary by muscle fiber type (118, 540). Germline deletion of both Tbc1d1 and Tbc1d4 in mice totally abrogates insulin-stimulated muscle glucose uptake, resulting in glucose intolerance more severe than in either Tbc1d1−/− or Tbc1d4−/− single knockout mice (118). AKT also phosphorylates target proteins involved in GSV membrane targeting and fusion, but these processes are better understood in adipocytes than myocytes and thus will be discussed later. In general, AKT phosphorylation of TBC1D1/TBC1D4 can be thought of as insulin “releasing the brakes” on GLUT4 translocation (413).
The Rho GTPase RAC1 coordinates a second PI3K-dependent signaling mechanism for insulin-stimulated glucose uptake in skeletal muscle. RAC1 signaling promotes GLUT4 translocation by inducing cortical actin reorganization (47, 140, 818). Direct RAC1 targets include the p21-associated kinase (PAK); insulin promotes the GTP-bound form of RAC1, which stimulates PAK phosphorylation by relieving PAK autoinhibition (140, 818). Muscle-specific knockout of RAC1 severely impairs insulin-stimulated glucose uptake despite preserved AKT activation (817), and forced overexpression of constitutively active RAC1 in muscle causes GLUT4 translocation even in the absence of insulin stimulation (858). The specific mechanisms by which RAC1-mediated cortical actin reorganization promotes GLUT4 translocation are an area of continued investigation but may involve tethering of GSVs beneath the plasma membrane and changes in membrane tension (413).
The glucose that enters the myocyte upon insulin stimulation has two major possible fates: glycolysis or glycogen synthesis. The principal pathway of insulin-stimulated glucose disposal in both healthy and type 2 diabetic human muscle is glycogen synthesis (~75%), consistent with the general teleological role of insulin as an energy storage hormone (182, 768). However, glucose oxidation also increases as increased substrate availability drives glycolytic flux; in fasting rat soleus muscle, insulin per se increases relative glucose oxidation (VPDH/VTCA) from ~5 to ~60%, the remainder reflecting fatty acid oxidation (D. Song, T. Alves, R. Perry, and G. Shulman, unpublished data). Although acute insulin-stimulated increases in skeletal muscle glycolytic flux and glycogen synthesis are primarily a consequence of increased glucose transport activity and subsequent allosteric regulation by glucose metabolites, insulin independently regulates both glycolysis and glycogen synthesis (152, 689, 769). Insulin positively regulates the transcription of hexokinase II, the primary skeletal muscle isoform of the first glycolytic enzyme, thus providing relatively slow, coarse control of glycolytic capacity (589).
In contrast, glycogen synthesis is subject to acute regulation by insulin of both anabolic [glycogen synthase (GS)] and catabolic [glycogen phosphorylase (GP)] fluxes (158). This acute regulation occurs through both covalent modification (insulin promotes the dephosphorylation of both GS and GP) and allostery (by glucose-6-phosphate). We first consider glycogen synthase, in 1960 the first enzyme shown to be regulated by insulin (872). Phosphorylation-based GS regulation by insulin occurs in part through AKT phosphorylation and inactivation of glycogen synthase kinase 3 (GSK3) at Ser21 and Ser9 on the α and β isoforms, respectively, of GSK3 (157, 171, 172, 214, 813). Thus inactivated, GSK3 kinase activity toward GS is diminished; dephosphorylated GS is in turn more active. Simultaneously, insulin activation of protein phosphatase 1 (PP1) promotes dephosphorylation of GS (578, 613). Because cells harness the phosphatase activity of PP1 for many targets in diverse pathways, specificity for GS is conferred by four glycogen-targeting regulatory subunits of PP1 (578). These regulatory subunits contain binding domains for PP1, GS, and glycogen and thus serve as metabolic scaffolds (374). In skeletal muscle, GM is the most highly expressed regulatory subunit; mice lacking GM display decreased muscle glycogen stores (90, 185, 815). Insulin promotes PP1 targeting to glycogen particles and increases PP1 activity towards GS, but the specific molecular mechanisms responsible for this activity are incompletely understood (374). Canonically, the combination of inactive GSK3 and active PP1 promotes the formation of active, dephosphorylated muscle GS, thus facilitating glycogen synthesis (159). However, studies of knock-in mice with GSK3α/β Ser21/Ser9 mutated to alanine, thus rendering GSK3 insensitive to insulin, have cast serious doubts on the importance of GSK3 in glycogen synthesis (84, 85). These mice have normal insulin-stimulated glycogen synthesis and normal muscle glycogen content (84). Interestingly, mice with muscle glycogen synthase engineered to be insensitive to allosteric activation by glucose-6-phosphate displayed severely impaired insulin-stimulated glycogen synthesis and lower muscle glycogen content (85). These data suggest that the acute regulation of GS by insulin occurs primarily through allosteric glucose-6-phosphate control, thus functionally coupling insulin-stimulated glucose uptake to insulin-stimulated glycogen synthesis. The phosphorylation status of GS, then, serves to modulate the enzyme’s affinity for glucose-6-phosphate. Dephosphorylated GS is more sensitive to glucose-6-phosphate allostery, facilitating activation of insulin-stimulated glycogen synthesis (85, 769).
However, increasing GS activity alone is insufficient for insulin to promote net glycogenesis. Glycogen phosphorylase activity must simultaneously be reduced to prevent glycogen cycling (640). On the catabolic side of glycogen metabolism, glycogen phosphorylase activity is regulated by insulin by largely similar mechanisms as GS: phosphorylation and allostery (97). In a classic mechanism, active phosphorylase kinase activates glycogen phosphorylase through phosphorylation of Ser15; insulin promotes the dephosphorylation and inactivation of phosphorylase kinase, and consequently the dephosphorylation and inactivation of glycogen phosphorylase (433, 949). In addition, insulin targeting of PP1 to the glycogen particle increases its activity towards glycogen phosphorylase, dephosphorylating Ser15 and thereby decreasing phosphorylase activity (949, 951). Both mechanisms thus enable insulin to decrease glycogenolysis and promote net glycogen synthesis. And just as with glycogen synthase, allosteric control of phosphorylase through inhibition by glucose-6-phosphate is a critical mechanism for insulin control of glycogenolysis (97). Future studies perturbing insulin regulation of glycogen phosphorylase, analogous to those that have been performed for glycogen synthase, are needed to provide a fuller understanding of muscle glycogen metabolism in vivo.
Muscle insulin action is thus a tightly coordinated relay that serves to promote glucose utilization and storage (FIGURE 2). While these physiological outcomes–glucose uptake and glycogen synthesis–have long been appreciated, their molecular basis is still being elucidated. A bewildering array of protein mediators have been implicated in insulin-stimulated glucose uptake in particular, and so only a primer is offered above. The study of muscle glycogen metabolism has a storied history reaching back to the origins of biochemistry; those investigators using modern tools to yield new and surprising insights about its regulation are indeed standing on the shoulders of giants.
FIGURE 2.

The insulin signaling cascade in skeletal muscle. Insulin receptor (INSR) activation has two major metabolic functions in the skeletal myocyte: glucose uptake and glycogen storage. Insulin stimulation of glucose uptake occurs through translocation of GLUT4-containing storage vesicles (GSVs) to the plasma membrane. The resultant increase in intracellular glucose-6-phosphate production, together with a coordinated dephosphorylation of glycogen metabolic proteins, enables net glycogen synthesis. Green circles and arrows represent activating events; red circles and arrows represent inhibitory events. GSK3, glycogen synthase kinase 3; PI3K, phosphoinositide-3-kinase; PP1, protein phosphatase 1.
C. Hepatic Insulin Signaling: Effectors and Effects
Insulin from the endocrine pancreas is secreted into the portal vein, so the liver is exposed to insulin concentrations two- to threefold higher than those in the general circulation (136). Portal venous insulin measurements, especially in rodents, are difficult and infrequently performed, but investigators studying hepatic insulin action by infusing insulin peripherally must keep in mind that the increment in plasma insulin concentration measured from a peripheral site is not equal to the increment in portal vein insulin concentration “seen” by the liver.
The diverse anabolic ramifications of insulin action are exemplified by the hepatic insulin signaling cascade. Insulin promotes the synthesis of all major classes of metabolic macromolecules: glycogen, lipids, and proteins. Additionally, insulin rapidly and potently reduces hepatic glucose production (HGP) (136). Because increased fasting HGP and insensitivity of this parameter to insulin are hallmarks of T2D, measurement of insulin suppression of HGP is a commonly reported physiological readout of hepatic insulin sensitivity. However, insulin’s immediate suppression of HGP has both direct and indirect components, as we will explore here and in section III (105, 136, 504, 661, 704). Because extrahepatic control of HGP is quantitatively significant, the purest experimental readouts of direct hepatocellular insulin action are insulin-stimulated glycogen synthesis, insulin-regulated transcripts, and phosphorylation events within the insulin signaling cascade. It is with this latter aspect of hepatic insulin action that we begin our discussion, for it is these phosphorylation events that enable insulin regulation of physiological processes such as gene transcription and glycogen metabolism.
Hepatic insulin signaling begins, as in all cell types, with INSR trans-autophosphorylation, activation, and recruitment of scaffold signaling proteins. The major IRS isoforms expressed in hepatocytes are IRS1 and IRS2 (196). Various genetic perturbations of hepatic Irs1 and Irs2 expression have not clearly defined distinct roles for either isoform; rather, available evidence suggests that Irs1 and Irs2 serve functionally similar roles in liver (195, 196, 662, 827, 907). Irs1 may play a larger role than Irs2 in normal glucose homeostasis: liver-specific Irs1−/− mice have more pronounced glucose intolerance than liver-specific Irs2−/− mice (195) The mildly defective insulin signaling of Irs1−/− mice was worsened considerably by concomitant liver-specific Irs2 deletion, and liver-specific Irs2 deletion alone produced only mild glucose intolerance with preserved hepatocellular insulin signaling; absence of both isoforms was necessary to produce a severe metabolic phenotype with blunted insulin stimulation of PI3K and AKT activity and marked fasting hyperglycemia (196). Similarly, acute short hairpin RNA-mediated 70–80% knockdown of either Irs1 or Irs2 in mouse liver produced mild phenotypes, with no impairment in downstream PI3K or AKT activity (827). Only after co-deletion of Irs1 and Irs2 did mice manifest impaired glucose tolerance and blunted insulin stimulation of PI3K and AKT activity (827). Attempts to assign preferential pathway control to hepatic IRS1 or IRS2 have yielded inconsistent results (195, 827). However, it remains quite possible that IRS1 and IRS2 perform at least partially distinct functions in hepatic insulin signaling; this hypothesis is supported by 1) potent insulin regulation of Irs2, but not Irs1, transcription in liver; and 2) the unique KRLB motif of IRS2, which binds to the INSR tyrosine kinase domain and may limit its activity (915, 950). The major PI3K catalytic subunit in hepatocellular insulin signaling is p110α; liver-specific deletion of this isoform severely impairs insulin-stimulated PIP3 generation, AKT activation, and suppression of glucose production in liver (790).
The pathway diversification of hepatic insulin signaling appears to occur largely distal to AKT activation. AKT substrates include GSK3 (regulating glycogen synthesis), the transcription factor forkhead box O1 (FOXO1, regulating gluconeogenic gene transcription), and multiple regulators of mTORC1 activity, which in turn control a large anabolic program upregulating lipogenic gene expression and protein synthesis (141, 504, 604). Although direct hepatocellular insulin signaling for metabolic control may not be entirely AKT-dependent, alternative pathways are yet to be described (504). The considerable functional redundancy between insulin signaling and nutrient sensing pathways, especially mTOR signaling, has challenged attempts to prove the existence of alternative insulin signaling pathways in hepatocytes (339, 504). With this in mind, we now consider the aforementioned physiological branches of hepatocellular insulin signaling in turn.
The stimulation of net glycogen synthesis is a major, direct physiological function of postprandial insulin on the hepatocyte. In humans, half-maximal stimulation of net hepatic glycogen synthetic rate under hyperglycemic, hypoglucagonemic conditions occurs at portal vein insulin concentrations of 20–25 μU/ml (697). As in skeletal muscle, liver glycogen synthesis is regulated through both phosphorylation and allostery, with allostery of critical importance (702). Glucose transport in the hepatocyte is not insulin-regulated, and therefore, insulin exerts less complete control over glycogen synthetic rates than in skeletal muscle (440, 640). For example, hyperglycemia is sufficient to inactivate liver glycogen phosphorylase by glucose allostery and thereby promote net hepatic glycogen synthesis (109, 440, 640). Hyperglycemia also causes the translocation of glucokinase from the nucleus to the cytoplasm, enabling glucose-G6P flux (359). However, hepatic insulin signaling through the INSR is required for normal glycogen synthesis; rats with acute antisense oligonucleotide knockdown of hepatic Insr have markedly decreased hepatic glycogen synthesis under hyperglycemic conditions (R. J. Perry and G. I. Shulman, unpublished observations). Insulin stimulation of net hepatic glycogen synthesis may occur through several mechanisms. Although glucose transport is not under insulin control in liver, insulin still regulates glycogen synthase (GYS2) allostery by G6P. GYS2 Arg582 is necessary for its allosteric activation by G6P, and mice heterozygous for a GYS2 R582A mutation displayed reduced hepatic glycogen deposition in fasting-refeeding experiments (872a). Glucokinase translocation is facilitated by insulin, and the Gck gene is also under rapid and potent positive transcriptional control by insulin (9, 295, 359, 360, 451, 504). Increased glucokinase expression is critical for hepatic insulin action not only because it increases G6P allostery at GYS2, but because it controls hepatic glucose utilization and storage. Metabolic control analysis has demonstrated that glucokinase expression is a major site of rate control for glycogen synthetic flux (9), and glucokinase activity also drives de novo lipogenesis through substrate push (295). Interestingly, humans with T2D have been reported to display decreased glucokinase expression; the extent of this transcriptional repression was correlated with fasting glycemia (294). AKT phosphorylation and inactivation of GSK3, which favors dephosphorylation of GYS2, may also contribute to insulin stimulation of GYS2 activity. However, in mice lacking Akt1 and Akt2 in liver (AKT DLKO mice), fasting-refeeding failed to stimulate net glycogen synthesis despite paradoxically preserved stimulation of GSK3 phosphorylation, indicating that AKT is necessary and GSK3 phosphorylation is insufficient to drive net hepatic glycogen synthesis (504). Interestingly, glucokinase expression was minimal in AKT DLKO mice and unresponsive to insulin; AKT DLKO mice also displayed decreased glucose-G6P cycling (504). Insulin activation of GYS2 also involves activation of PP1 activity; the critical regulatory phosphorylation site on GYS2 is Ser7 (90, 702). Mice overexpressing GYS2 with S7A and S644A mutations had increased liver glycogen in both fed and fasted conditions (703). Taken together, these data point to the primacy of allosteric and substrate control of net hepatic glycogen synthesis by glucose metabolites, through both insulin-dependent and insulin-independent mechanisms.
Insulin control of hepatic glycogen metabolism also involves suppression of glycogenolytic flux. The mechanisms involved are similar to those described above for muscle. Liver phosphorylase is 79% sequence identical to muscle phosphorylase in humans, and some modes of its regulation are therefore similar. For example, phosphorylation of the NH2-terminal Ser15 strongly activates phosphorylase activity, and insulin inhibition of phosphorylase kinase and activation of protein phosphatase-1 are therefore key mechanisms for insulin suppression of glycogenolysis (579, 682). Insulin inactivation of phosphorylase can be mimicked by expression of constitutively active AKT, indicating that canonical insulin signaling does contribute to suppression of glycogenolysis (13). Control of glycogenolysis is also tightly linked to control of glycogen synthesis: the liver-type glycogen targeting subunit of PP1 (GL) is bound and inhibited by active phosphorylase, a safeguard against simultaneous activation of phosphorylase and GYS2 (9, 15). However, phosphorylase is also under potent allosteric control and, as discussed above, glucose allostery is sufficient to inhibit glycogenolysis. Liver phosphorylase, unlike the muscle isoform, is relatively insensitive to allostery by AMP or glucose-6-phosphate (579). Rather, allosteric inhibition by glucose itself is of particular regulatory importance for liver phosphorylase (579, 682). Given the rapid, non-insulin-regulated equilibration of glucose across the hepatocellular plasma membrane, and the role of hepatic glycogenolysis in maintaining euglycemia, glucose makes excellent teleological sense as the main allosteric controller of liver phosphorylase activity.
By these mechanisms and most likely by others that remain to be elucidated, insulin and glucose work in concert to regulate liver glycogen metabolism. Although physiological data support a model in which hyperinsulinemia is necessary and sufficient to increase liver glycogen synthetic flux, hyperglycemia is necessary and sufficient to suppress liver glycogenolysis, and both hyperinsulinemia and hyperglycemia are necessary to promote net liver glycogen synthesis (640), mechanistic investigations have revealed roles for insulin and glucose in both glycogenolysis and glycogen synthesis (78). In addition to the permissive role of insulin in glycogen synthesis, mediated by phosphorylation-based changes in enzyme activity, insulin also controls GS allostery and substrate availability through transcriptional regulation of glucokinase. Because allostery and substrate availability are central to the regulation of hepatic glycogen metabolism (641), the ability of insulin to modulate glycogen metabolic flux through protein phosphorylation, allostery, and substrate availability renders it a powerful regulator of net glycogen synthesis and thus of hepatic glucose production.
Another key mechanism by which insulin responds to the fed state is the transcriptional repression of gluconeogenic genes, mediated most prominently by FOXO transcription factors. FOXO1 is a particularly well-characterized AKT target with important physiological functions in the hepatocyte (103, 195, 484, 720, 857). AKT phosphorylates three residues on FOXO1: Thr24, Ser256, and Ser319, although other kinases can also target these sites (103, 857). Phosphorylated FOXO1 is excluded from the nucleus, disabling its transcription factor activity (103). Active, nuclear FOXO1 binds the transcriptional coactivator peroxisome proliferative activated receptor-γ coactivator 1-α (PGC1α) to coordinate a gluconeogenic transcriptional program involving increased expression of glucose-6-phosphatase (G6pc) and cytosolic phosphoenolpyruvate carboxykinase (Pck1) (569, 664). Active FOXO1 also binds the co-repressor SIN3A to decrease expression of glucokinase, further favoring glucose export (451). The potency of the FOXO1 gluconeogenic transcriptional program has been highlighted by studies of mice with genetic defects in hepatic FOXO1 regulation. Mice lacking hepatic FOXO1 display fasting hypoglycemia and decreased HGP (526). Even a 40% reduction in hepatic Foxo1 mRNA expression in high-fat-fed mice was sufficient to decrease basal HGP (720). Triple knockout of Foxo1, Foxo3, and Foxo4 causes particularly severe fasting hypoglycemia (295, 296). Interestingly, in several models of impaired proximal insulin signaling with increased basal HGP and impaired insulin suppression of HGP, including liver-specific Irs1−/− Irs2−/− mice, liver-specific Insr−/− mice, and liver-specific Akt1−/− Akt2−/− mice, ablation of Foxo1 was sufficient to normalize fasting HGP and resensitize HGP to insulin (195, 504, 603, 840). This remarkable phenotypic rescue likely reflects the disastrous gluconeogenic consequences of unrestrained FOXO1 activity in these models of total hepatic insulin resistance, and more importantly points to the dispensability of direct hepatic insulin action for insulin’s acute suppression of gluconeogenesis in fasted rodents subjected to hyperinsulinemia-euglycemia.
In addition to the FOXO transcription factors described above, a transcriptional complex including the cAMP response element binding protein (CREB), CREB binding protein (CBP), and CREB-regulated transcription coactivator 2 (CRTC2) controls gluconeogenic gene expression in an insulin-dependent manner (421). The CREB/CRTC2 and FOXO1/PGC1α modules appear to be nonredundant and differentially regulated: the CREB/CRTC2 module has been shown to be critical for gluconeogenic gene expression in the first several hours of fasting, while the FOXO1/PGC1α module is more critical during longer fasts (493). Upregulation of PGC1α by CREB/CRTC2 may contribute to this fascinating phenomenon (421). Just as FOXO1 is regulated by phosphorylation-induced nuclear exclusion, CRTC2 is phosphorylated at Ser171 by salt-inducible kinase 2 (SIK2) in response to insulin (188). CRTC2 phosphorylation promotes its export from the nucleus, leading to polyubiquitination and degradation and thus disabling the CRTC2 gluconeogenic program (188). Mice with severe disruptions in this axis display altered glucose homeostasis: CRTC2 knockout mice are hypoglycemic during fasting, and mice overexpressing a constitutively active CRTC2 mutant are hyperglycemic (321, 882).
The FOXO1/PGC1α and CREB/CRTC2 transcriptional modules are well described and elegant mechanisms. Pharmacological PGC1α inhibition has even been shown to reduce gluconeogenic gene expression, fasting glycemia, and hepatic insulin sensitivity in obese high-fat-fed mice (755). But the effect of these transcriptional modules on hepatic gluconeogenesis in modern human daily life, where fasting rarely exceeds 16 h in duration, has been proposed to be relatively minor (419). For example, even 2 h of insulin stimulation is insufficient to cause detectable decreases in G6pc protein levels (620). Rather, computational models indicate that nontranscriptional mechanisms exert high control over glucose metabolic fluxes (419). These include changes in substrate availability, allostery, redox state, and posttranslational modifications. As discussed above, insulin control of hepatic glycogen metabolism by such mechanisms is well described. Nontranscriptional insulin regulation of hepatic gluconeogenesis also occurs but has received less attention in recent years. As will be described later, indirect control of hepatic gluconeogenesis through white adipocyte lipolysis is critical to insulin’s acute suppression of gluconeogenesis. However, insulin can also directly regulate hepatic gluconeogenesis by counteracting cAMP-induced phosphorylation of phosphofructokinase-2/fructose-2,6-bisphosphatase-2 (PFK-2/FBPase-2) Ser36; this dephosphorylation promotes FBPase-2 activity, decreasing fructose-2,6-bisphosphate levels and thereby disinhibiting the gluconeogenic enzyme FBPase-1 (691, 914). Interestingly, PFK-2/FBPase-2 dephosphorylation may also inhibit glucokinase by promoting its nuclear translocation (173). This mechanism is likely most operative in states of high glucagon/catecholamine tone, and its role in normal postprandial suppression of gluconeogenesis requires further study.
As mentioned above, the acute suppression of HGP is one of the most commonly employed physiological readouts of hepatic insulin action (33, 54, 105, 112, 115, 484, 504, 722). The question of whether this effect is direct (i.e., hepatocyte-autonomous) or indirect has attracted considerable attention (135, 136, 208, 472, 504, 620). A key methodogical consideration in this regard is the relative contributions of glycogenolysis and gluconeogenesis to HGP. Hepatic glycogen content decays exponentially with fasting duration (implying that its derivative, the rate of net hepatic glycogenolysis, also decays exponentially with fasting), with near-total depletion after 12 h in rats and 48 h in humans (628, 704). In contrast, absolute rates of hepatic gluconeogenesis remain relatively constant during the first 48 h of the fast, until substrate (i.e., lactate, alanine) limitation results in decreased gluconeogenic flux (FIGURE 3) (628, 643, 704). Since plasma glucose concentrations in a fasting subject reflect HGP, an interesting implication of these observations is that the plasma glucose concentration (and, by extension, the plasma insulin concentration) during a fast is a key systemic signal reflecting hepatic glycogen content (i.e., available carbohydrate reserves available for the CNS and other obligate glucose-utilizing tissues). Because, as this and subsequent sections explore, direct and indirect hepatic insulin action have different effects on net hepatic glycogenolysis and gluconeogenesis, understanding the relative contributions of these fluxes is critical to the design and interpretation of experiments on this subject.
FIGURE 3.
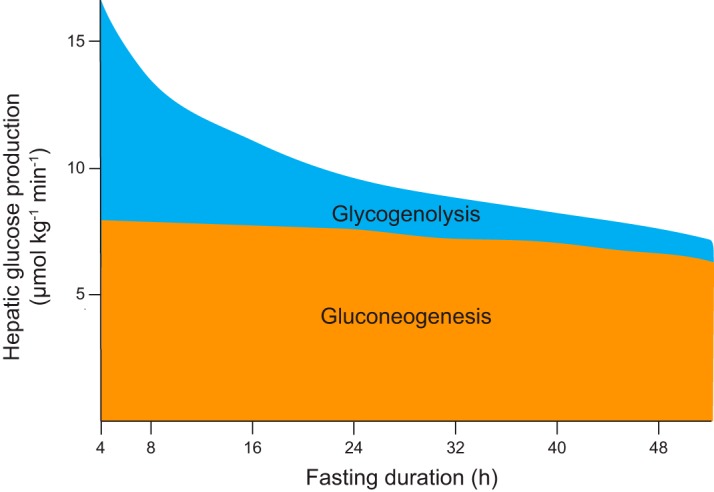
Sources of hepatic glucose production during fasting in humans. During the early postprandial period (not shown), the liver performs net glucose uptake as ingested glucose is stored as liver glycogen. Gluconeogenic flux continues but is diverted into glycogen storage. After this period, gluconeogenic flux contributes to hepatic glucose production and continues at a relatively constant rate for ~48 h, eventually decreasing due to declining substrate availability. Net hepatic glycogenolysis, in contrast, initially contributes about half of hepatic glucose production, but its rate decreases exponentially in concordance with hepatic glycogen content. Hepatic glycogenolysis still contributes appreciably to hepatic glucose production after 24 h of fasting, but is nearly depleted by 48 h. Because plasma glucose concentrations reflect rates of hepatic glucose production during a fast, the plasma glucose concentration is a systemic signal of hepatic glycogen content during fasting. [Data from Rothman et al. (704).]
With this in mind, direct insulin suppression of net hepatic glycogenolysis is certainly a physiologically important mediator of insulin suppression of HGP (208). Indeed, during the first 22 h of a fast in humans, hepatic glycogenolysis contributes an estimated ~40% of HGP (704). But in the fasted, glycogen-depleted rodents often used in hyperinsulinemic-euglycemic clamp experiments, the primary source of HGP is hepatic gluconeogenesis (620). Because insulin suppresses hepatic gluconeogenesis within minutes, long before any changes in gluconeogenic protein levels occur, the transcriptional mechanisms described above cannot account for the acute suppression of hepatic gluconeogenesis by insulin (218, 484, 620). Additionally, because multiple genetic models of total hepatic insulin resistance suppress HGP normally in response to insulin, the possible existence of an alternative AKT-independent direct hepatocellular insulin signaling pathway involved in acute gluconeogenic suppression would be an insufficient physiological explanation (105, 195, 504). Instead, insulin’s acute inhibition of hepatic gluconeogenic flux appears to be a largely indirect effect, mediated primarily through insulin suppression of WAT lipolysis (54, 136, 620, 684). This mechanism will be discussed in detail in section III; here, we merely wish to emphasize the inadequacy of measurements of insulin suppression of hepatic glucose production to specifically assess direct hepatic insulin action in the fasted state.
Insulin also has direct hepatocellular effects on lipid metabolism. Most prominent among these effects is transcriptional upregulation of several genes of de novo lipogenesis (DNL), though increased triglyceride-rich lipoprotein clearance and decreased very-low-density lipoprotein (VLDL) export have also been reported (457). The overall effect is to promote lipid storage in the hepatocyte and decrease the availability of fatty acids for oxidation by other tissues. Indeed, plasma triglyceride concentrations decrease precipitously within 15 min of insulin infusion, although in the setting of a mixed meal, absorbed triglycerides will negate this effect.
DNL, like gluconeogenesis, is under slow but potent transcriptional control by a PI3K/AKT-dependent mechanism. However, unlike gluconeogenesis, DNL is also acutely regulated by insulin-stimulated phosphorylation of lipogenic enzymes. Although DNL flux has been estimated to only account for ~25% of hepatic lipogenic flux (compared with ~60% from esterification of circulating fatty acids and 15% from dietary lipids), insulin stimulation of DNL is consistent with its overall anabolic effect (197).
The master transcriptional regulator of hepatic DNL is sterol regulatory element binding protein 1c (SREBP-1c), which promotes DNL by enhancing the transcription of several lipogenic enzymes, notably acetyl-CoA carboxylase 1 (Acaca), fatty acid synthase (Fasn), and glycerol-3-phosphate acyltransferase 1 (Gpam) (215, 442). Liver-specific overexpression of SREBP-1c is sufficient to cause hepatic steatosis (368). Insulin acts on SREBP-1c primarily by upregulating its transcription, but insulin also promotes SREBP-1c cleavage and nuclear translocation: the canonical mechanisms of SREBP activation (207, 330). These effects can be blocked by PI3K, AKT, or mTORC1 inhibition, suggesting that those kinases lie upstream of SREBP-1c (487). In particular, the observation that liver-specific Akt2−/− mice do not develop hepatic steatosis even on a leptin-deficient ob/ob background suggests that insulin regulation of lipid metabolism largely occurs downstream of AKT (458). The mTORC1 substrate S6K is required for SREBP-1c processing but not its transcriptional upregulation (487, 604). It is important to note, however, that transcriptional activation of the DNL program by insulin is particularly slow: in one study of primary rat hepatocytes, SREBP-1 was not detectable in nuclear extracts until 8 h after insulin treatment (283). A faster transcriptional mechanism by which insulin increases DNL flux is induction of glucokinase (163, 295), which increases lipogenic substrate availability.
In addition to transcriptional upregulation of DNL, insulin also acutely activates DNL flux by regulating the phosphorylation of lipogenic enzymes, although the specific signal transduction pathways involved are incompletely understood. For example, ACC is rapidly activated in response to insulin (909), likely through both dephosphorylation and phosphorylation events (100). Insulin promotes the dephosphorylation of Ser79 (on ACC1) and Ser212 (on ACC2), perhaps through inhibition of AMPK which normally phosphorylates these sites (845, 908). Knock-in mice with both ACC1 Ser79 and ACC2 Ser212 mutated to alanine have constitutively active hepatic ACC and consequent increased hepatic lipogenesis, demonstrating the physiological importance of these sites (250). Insulin may also increase the phosphorylation of other ACC residues, but it has not been shown that these modifications alter ACC activity (305). Insulin also regulates the phosphorylation of ATP citrate lyase (ACLY). ACLY converts the tricarboxylic acid cycle intermediate citrate to the lipogenic precursor acetyl CoA, thereby linking glucose metabolism to DNL. Three ACLY phosphorylation sites are insulin-responsive; Ser455 is an AKT substrate, while Thr446 and Ser450 are GSK3 substrates (57, 345). ACLY phosphorylation activates the enzyme by preventing its allosteric inhibition by citrate (658). However, it is not clear that insulin serves to increase ACLY activity (189). For example, ACLY Ser455 is also phosphorylated by protein kinase A (PKA); PKA activity opposes insulin action in most instances (658). Additionally, GSK3 activity is inhibited by insulin action, which is inconsistent with a model in which insulin promotes ACLY phosphorylation to increase its activity. Thus the physiological role of insulin-stimulated ACLY phosphorylation is uncertain. Large phosphoproteomic data sets may reveal other insulin-regulated phosphorylation events within the lipogenic pathway; the challenge will be to determine which of these are capable of altering lipogenic flux.
Thus far, we have considered how insulin regulates hepatocellular synthesis of two major classes of biological macromolecules: glycogen and lipids. We end our discussion with a third macromolecule: proteins. Insulin regulation of protein synthesis is largely mediated by signaling into the mammalian target of rapamycin (mTOR) network. mTOR is a large protein kinase which depending on its binding partners can form two mutually exclusive functional complexes, mTORC1 and mTORC2 (752). Both mTORC1 and mTORC2 interact with the insulin signaling cascade, but mTORC1 effects are better studied. Insulin-stimulated protein synthesis is mediated through mTOR in many insulin-responsive cell types, including hepatocytes, adipocytes, and myocytes, but we include it in our discussion of hepatic insulin signaling because of the hepatocyte’s particularly high rates of protein synthesis and because of mTORC1’s role in modulating insulin-stimulated de novo lipogenesis (discussed above).
Insulin activation of mTOR is highly integrated with the PI3K-AKT pathway in a bidirectional manner. AKT activation of mTORC1 is incompletely understood but may involve AKT phosphorylation and inactivation of tuberous sclerosis complex 2 (TSC2) and/or proline-rich AKT substrate of 40 kDa (PRAS40), inhibitors of mTORC1 activation (349, 866b). Activated mTORC1 phosphorylates components of the translational machinery, including S6K and the eukaryotic translation initiation factor binding proteins 1 and 2 (4EBP1/2); the overall effect is to induce a broad translational program characterized by transcripts with 5′ terminal oligopyrimidine (TOP) motifs (508a, 752, 839). In addition to these downstream effects, mTORC1 signaling also exerts negative feedback on proximal insulin signaling by promoting S6K phosphorylation and destabilization of IRS1 as well as phosphorylation and stabilization of the adapter protein GRB10, which in turn binds and inhibits INSR (339, 944). mTORC1 also regulates the synthesis of non-protein macromolecules, including the phosphatidylcholine needed for VLDL-triglyceride secretion (665). Finally, mTOR signaling positively regulates AKT. mTORC2 phosphorylation of AKT Ser473 is probably the most commonly employed readout of cellular insulin signaling, although it remains unclear precisely how mTORC2 is activated by insulin (453, 729). Ser473 phosphorylation is an activating event that increases AKT kinase activity and may alter its substrate specificity (16, 288, 362). Through these well-described mechanisms and likely through as-yet-unidentified ones, mTOR signaling affects all functional branches of insulin signaling either through direct signal propagation (as for protein synthesis) or indirect tuning (as for its feedback on INSR, IRS1, and AKT). Importantly, mTOR permits integration of other anabolic signals (e.g., amino acid availability) with insulin signaling (453).
As the above discussion demonstrates, hepatocellular insulin signaling is a richly ramified cascade with links to all branches of macronutrient anabolism (FIGURE 4). The principal direct actions of insulin on liver are to stimulate glycogen synthesis and to transcriptionally regulate gluconeogenesis, de novo lipogenesis, and protein anabolism. It is unfortunate, then, that the most common readouts used to experimentally interrogate insulin action in the liver are 1) a partially indirect action of insulin on the hepatocyte (suppression of hepatic glucose production) or 2) a phosphorylation event with multiple physiological inputs and indirect insulin control but for which excellent commercial antibodies are available (AKT Ser473). In section IV, we attempt to synthesize what is known about hepatic insulin resistance and what is known about hepatic insulin action to suggest physiologically meaningful experimental strategies.
FIGURE 4.

Hepatic insulin signaling. AKT signaling is central to hepatocellular insulin action. Fast effects include activation of the glycogen and protein synthetic machinery. Slower transcriptionally mediated effects include upregulation of glucokinase, dimunition of gluconeogenic capacity, and stimulation of de novo lipogenic capacity. Green circles and arrows represent activating events; red circles and arrows represent inhibitory events. IRS, insulin receptor substrate; GSK3, glycogen synthase kinase 3; PI3K, phosphoinositide-3-kinase; PP1, protein phosphatase 1; GPAT, glycerol-3-phosphate acyltransferase; G6PC, glucose-6-phosphatase; PCK1, phosphoenolpyruvate carboxykinase; SREBP1c, sterol regulatory element binding protein 1c; FAS, fatty acid synthase.
D. White Adipocyte Insulin Signaling: Effectors and Effects
The white adipocyte is exquisitely sensitive to insulin in vivo. The potency of insulin to control plasma nonesterified fatty acid (NEFA) levels is critical to the maintenance of euglycemia; suppression of lipolysis is an important physiological function of insulin in WAT (620, 684). Suppression of WAT lipolysis shows a steep dependence on plasma insulin levels; the ED50 in humans is ~20 μU/mL (684). Because plasma insulin levels in healthy nondiabetic humans range only from ~5 to 60 μU/mL (655, 684), physiological insulin regulation of WAT lipolysis is able to access a much larger portion of its dynamic range compared with insulin regulation of whole-body glucose uptake, which has an ED50 of ~60 μU/mL and only reaches maximal levels at supraphysiological insulin concentrations of >200 μU/mL (694). Stimulation of glucose transport is another main function of insulin in the adipocyte, although WAT only accounts for a small fraction of whole-body glucose disposal (430). We now consider the effectors involved in insulin regulation of lipolysis and glucose uptake in white adipocytes (FIGURE 5).
FIGURE 5.

Insulin signaling in the white adipocyte. The most critical physiological functions of insulin action in white adipose tissue are suppression of lipolysis and stimulation of glucose uptake. Suppression of lipolysis requires phosphodiesterase 3B (PDE3B) and occurs largely through attenuation of cAMP-stimulated events such as perilipin (PLIN) and hormone-sensitive lipase (HSL) phosphorylation. Insulin stimulation of glucose uptake occurs through phosphoinositide-3-kinase (PI3K)-dependent [left insulin receptor (INSR)] and PI3K-independent (right INSR) pathways using numerous effectors to promote translocation, docking, and fusion of GLUT-containing storage vesicles (GSVs) with the plasma membrane. Green circles and arrows represent activating events; red circles and arrows represent inhibitory events. IRS, insulin receptor substrate; PP, protein phosphatase; SREBP-1c, sterol regulatory element binding protein 1c; LPL, lipoprotein lipase.
Insulin suppression of plasma NEFA levels occurs through rapid inhibition of triglyceride lipolysis in adipocytes. Insulin is the most potent antilipolytic hormone and acts rapidly; rat plasma NEFA levels are suppressed by ~90% within 5 min of raising insulin to postprandial levels (285, 477). This rapid action is facilitated by the short half-life of plasma NEFA: 2–4 min (206). The best understood mechanisms for insulin suppression of lipolysis involve the attenuation or reversal of adrenergic signaling through cAMP and protein kinase A (PKA) (203, 367). To understand insulin regulation of WAT lipolysis, we therefore begin by summarizing these cAMP/PKA-dependent mechanisms. PKA phosphorylates two key proteins involved in WAT lipolysis: hormone-sensitive lipase (HSL) and perilipin (PLIN) (367, 869). HSL is phosphorylated on three COOH-terminal serine residues (Ser563, Ser659, Ser660), causing its translocation from the cytosol to the lipid droplet surface (328, 820). The importance of HSL in the hormonal control of WAT lipolysis was highlighted by the identification of a human HSL frameshift mutation (14). Patients homozygous for the mutation expressed no HSL and had severely impaired control of lipolysis; both isoproterenol stimulation and insulin suppression of lipolysis were markedly defective (14, 947). Similarly, Hsl deletion in mice results in severely impaired adrenergic stimulation of lipolysis (293, 557, 602). However, HSL serves primarily as a DAG lipase, with adipose triglyceride lipase (ATGL) catalyzing the initial TAG hydrolysis (210, 946, 959). Complete hormonal control of lipolytic rate also requires the lipid droplet-coating protein perilipin. Perilipins are abundant, and five PLIN isoforms perform tissue-specific functions (93). PLIN1 is highly expressed in white adipocytes and is phosphorylated by PKA at several serine residues (93, 531, 946). The precise functions of PLIN phosphorylation in lipolytic control are not fully understood, but are thought to involve at least three major mechanisms. First, PLIN phosphorylation decreases its affinity for the ATGL cofactor CGI-58, enabling CGI-58 to bind ATGL and increase ATGL activity ~20-fold (279, 946). Second, PLIN phosphorylation is important for the full activation of HSL at the lipid droplet surface (546, 820). Third, PLIN phosphorylation has been shown to increase the lipid droplet surface area-to-volume ratio by stimulating budding of lipid microvesicles; this may increase lipase access to substrate but requires prolonged exposure to adrenergic stimulation and thus is likely not involved in the acute lipolytic response (521). Plin1−/− mice have elevated basal lipolysis that is unresponsive to adrenergic stimulation, highlighting that PLIN is not merely a passive barrier to lipase access but rather an active controller of stimulated lipolysis (522, 713, 828). Further work is needed to fully understand the mechanisms by which PLIN orchestrates lipolysis; it is possible that PLIN scaffolds a large interactome of lipases and cofactors to coordinate and amplify the lipolytic response to adrenergic stimulation.
Insulin acts largely through phosphodiesterase 3B (PDE3B) to suppress lipolysis. PDE3B degrades cAMP to attenuate pro-lipolytic PKA signaling toward HSL and PLIN (147, 367, 670, 781). Stimulated lipolysis in adipocytes lacking Pde3b is not suppressed by insulin, and Pde3b−/− mice have impaired suppression of plasma NEFA levels during glucose tolerance tests (147). Interestingly, the mechanisms for PDE3B activation by insulin are incompletely defined. PDE3B Ser273 is activated through phosphorylation by AKT in a 14-3-3 protein-dependent manner after insulin stimulation (411, 483, 598, 701). However, AKT does not seem to be necessary for insulin suppression of lipolysis; Akt2−/− mice suppress lipolysis normally in response to feeding and near-normally during insulin tolerance tests and hyperinsulinemic-euglycemic clamps (423), and pharmacological AKT inhibitors do not abolish insulin suppression of lipolysis in cultured adipocytes (192). Additionally, the AKT phosphorylation site on PDE3B, Ser273, is dispensable for insulin suppression of lipolysis in cultured adipocytes (192). Several other PDE3B serine residues, including Ser296, are also phosphorylated; the functional importance of these events is uncertain though Ser296, a PKA substrate, has also been shown to be unnecessary for insulin suppression of lipolysis in vitro (192, 483, 671). Rather than regulating PDE3B activity by modulating its phosphorylation, an emerging paradigm posits that insulin primarily activates PDE3B by promoting the formation of signaling complexes or ‟signalosomesˮ (11, 192).
Despite strong evidence that insulin activation of PDE3B mediates the attenuation of cAMP/PKA-mediated lipolysis (743), it is not clear whether this mechanism–extinguishing adrenergic input–is necessary and sufficient to explain insulin suppression of WAT lipolysis under all physiological conditions. In particular, this mechanism may be less important in situations of low adrenergic tone. For example, insulin causes dephosphorylation of HSL even in the absence of detectable PKA activity, implying that insulin stimulates HSL phosphatase activity (804). Protein phosphatase 2A (PP2A) appears to be the chief mediator of this effect, although other phosphatases also act on HSL; the full mechanism remains obscure (911). Insulin also appears to have PI3K-dependent but AKT-independent effects, such as the dephosphorylation of perilipin (146, 209). In contrast to HSL, protein phosphatase 1 (PP1) has been identified as the main perilipin phosphatase in adipocytes; PP1 regulatory subunit phosphorylation and activity both increase in response to insulin (43, 151).
In summary, although the mechanisms by which insulin suppresses lipolysis are not understood in full detail, a functional model in which insulin both attenuates adrenergic kinase activity through PDE3B activation and actively dephosphorylates lipolytic regulatory proteins through protein phosphatase activation may be sufficient to account for experimental observations. A picture of the cellular physiology of WAT lipolysis is emerging in which hormone-stimulated assembly of lipolytic or antilipolytic complexes at the lipid droplet defines the net direction of lipolysis (11, 192). Ongoing investigation will undoubtedly define the relevant mediators and interactions in fuller detail (54, 620, 684).
Just as net hepatic glycogen storage depends on the balance between glycogenolysis and glycogen synthesis, net adipose lipolysis is the sum of fluxes from lipolysis and re-esterification of liberated fatty acids (474). Re-esterification can act on fatty acids originating from within the adipocyte or from the circulation (447). During fasting, almost no adipocyte re-esterification occurs, but glucose infusion induces substantial re-esterification (165). Additionally, under-replacement of insulin in type 1 diabetics impairs the postprandial storage of dietary fatty acids; these findings suggest a role for insulin in promoting adipose fatty acid esterification (475). Insulin-stimulated glucose uptake provides a source of glycerol-3-phosphate to which fatty acids can be esterified, and insulin activates lipoprotein lipase activity in adipose tissue endothelium (225, 267). Furthermore, insulin promotes the translocation of fatty acid transport proteins FATP1 and FATP4 in 3T3-L1 adipocytes (794). However, in healthy adults, insulin did not stimulate systemically derived fatty acid esterification to a greater extent than could be achieved by niacin suppression of lipolysis (17). Paired with the observation that rates of re-esterification of fatty acids liberated within the adipocyte are not dependent on insulin (110), a reasonable interpretation is that rates of adipocyte fatty acid esterification are more dependent on substrate availability and concentration gradients than on acute insulin stimulation of a pro-esterification enzymatic activity. Insulin has other pro-lipogenic functions in the adipocyte; it activates SREBP-1c and its lipogenic transcriptional program just as it does in hepatocytes (398). However, de novo lipogenesis accounts for a very small fraction of adipocyte lipogenesis; esterification of preformed fatty acids is the predominant lipogenic pathway (474). Insulin also stimulates adipogenesis through the transcription factor peroxisome proliferator-activated receptor-γ (PPARγ) (693).
Insulin regulation of cellular glucose uptake has been well studied in cultured 3T3-L1 adipocytes. While these cells derive from a fibroblast line and may not fully recapitulate all features of bona fide white adipocytes, their ease in manipulation has revealed much about the molecular mediators of insulin signaling (73, 282). Additionally, the study of insulin-stimulated glucose uptake has been facilitated by the large and productive scientific community investigating vesicle trafficking. The seed planted by the 1980 discovery that insulin stimulates the translocation of a glucose transporting activity to the plasma membrane has blossomed into a fruitful tree with branches sprouting to describe each step of the process, from GSV budding to transport to tethering to docking to fusion, in molecular detail (73, 174, 814). In adipocytes, GLUT4 translocation involves many but not all of the same effectors discussed above for myocellular glucose uptake. We now briefly consider the molecular mediators of insulin-stimulated glucose uptake in the adipocyte, though interested readers are referred to several excellent reviews for more detail on this subject (47, 73, 276, 365, 413, 471).
Insulin-stimulated glucose uptake in the adipocyte, as in muscle, is critically dependent on the IRS1-PI3K-AKT axis. Antisense knockdown of IRS1 severely impairs insulin-stimulated glucose uptake in primary rat adipocytes (666). IRS2 also participates in adipocyte insulin signaling; IRS2 Ser388 phosphorylation by insulin-activated cyclin-dependent kinase 4 (CDK4) may be a positive feedback mechanism maintaining adipocyte insulin signaling (448). The importance of PI3K activation in adipocyte insulin action is highlighted by studies of mice with inducible deletion of the PIP3 phosphatase PTEN in mature adipocytes; these mice have profoundly enhanced insulin sensitivity even on a regular chow diet (555). Synthetic or optogenetic activation of PI3K or AKT2 is sufficient to increase glucose uptake in 3T3-L1 adipocytes, and loss of Tbc1d4 (AS160) in mice completely abrogates insulin-stimulated adipocyte glucose uptake (118, 581, 930). The AS160-related Rab GAP TBC1D1, although important in skeletal muscle, is expressed at low levels in adipose tissue, and Tbc1d1−/− mice have normal insulin-stimulated glucose uptake (118). The major AS160 substrate in the adipocyte (i.e., the Rab GTPase that AS160 maintains in the inactive GDP-bound state) is thought to be RAB10, which regulates GSV exocytosis (94, 726). The related TBC1D13-RAB35 pair also supports GLUT4 trafficking to the plasma membrane, as may several other Rabs (179, 365). Another GAP-GTPase axis involving the GAP complex RGC1/2 and the GTPase RalA also participates in insulin-stimulated targeting of GSVs to the adipocyte plasma membrane; RGC2 is an AKT substrate (132, 133).
Beyond regulating vesicle trafficking through GAP-Rab interactions, AKT also promotes GLUT4 translocation in the adipocyte by phosphorylating targets involved in vesicle tethering, docking, and fusion. One such substrate is SYNIP, which when phosphorylated dissociates from the t-SNARE syntaxin-4 to enable GSV docking and fusion at the plasma membrane (544, 931). CDP138 is another AKT substrate involved in GSV fusion, although its precise function remains incompletely defined (926). Finally, the motor protein myosin 5A is an AKT substrate that aids in GLUT4 navigation of the cortical actin network to reach the plasma membrane (939). The continued identification of novel AKT substrates involved in GSV trafficking suggests that current understanding of PI3K-dependent GLUT4 translocation is incomplete but solidifies the role of AKT as its master controller, with input to many effectors in all phases of the process.
Although the Rho-family GTPase RAC1 does not appear as critical for insulin-stimulated GLUT4 trafficking in the adipocyte as it is in the skeletal myocyte, another Rho GTPase, TC10α, has been identified as an AKT-independent mediator in the adipocyte (732). TC10α is activated upon insulin stimulation, and its knockdown by siRNA results in impaired GLUT4 translocation and decreased insulin-stimulated glucose uptake (119). By binding EXO70 in the exocyst complex, TC10 promotes tethering of GSVs at the plasma membrane (350). Additionally, TC10 acts through the protein PIST to promote the proteolytic cleavage of TUG, a protein tether that sequesters GSVs intracellularly at the Golgi network (47, 73–75). TUG cleavage thus enables the mobilization of GSVs to the plasma membrane upon insulin stimulation (943). An additional AKT-independent pathway for insulin-stimulated glucose uptake in adipocytes involves direct INSR tyrosine phosphorylation of MUNC18C, which modulates GSV fusion at the plasma membrane through interactions with SNAREs (369).
The pathways described in this section controlling adipocyte glucose uptake and the pathways described above for muscle glucose uptake overlap to a significant extent. Mechanisms with strong evidence in 3T3-L1 adipocytes but weaker evidence in myocytes have been the focus of this section. It is important to note that a large subset of the aforementioned pathways (e.g., AKT/TBC1D4 signaling, TUG cleavage) is known to be operative in both adipocytes and myocytes. Furthermore, other tissues which exhibit insulin-stimulated glucose uptake, such as cardiac muscle and brown adipose tissue, likely employ a subset of these molecular mechanisms (342, 776, 777).
And as we will examine next, adipocyte insulin action has ramifications beyond fat, potently controlling hepatic gluconeogenesis.
III. INDIRECT INSULIN ACTION
A. Physiological Relevance of Indirect Insulin Action
Insulin action evolved not in an isolated, homogeneous cellular population but rather in the context of complex multicellular organisms with specialized cell types. It is not surprising, then, that several critical elements of whole-body insulin action are not cell-autonomous: they require tissue crosstalk. Because these phenomena are difficult to model in cultured cells and by definition cannot be modeled in a homogeneous, isolated cell population, progress in understanding their molecular basis has lagged behind progress in the study of direct insulin action. However, the indirect effects of insulin make quantitatively significant contributions to overall tissue insulin action; in some cases, indirect effects predominate over direct effects (6, 620, 686). Indirect insulin action thus cannot be ignored in studies of intact organisms (841). A particularly well-studied example involves insulin’s effect to suppress hepatic gluconeogenesis by inhibiting lipolysis in the adipose tissue (6, 54, 89, 135, 136, 620, 684, 686, 774). Several other indirect actions of insulin have been demonstrated. For example, insulin action in pancreatic α-cells strengthens the integrated insulin response by suppressing glucagon secretion, and a sizable literature has emerged implicating CNS insulin action in peripheral metabolic control. Because the scope of this review encompasses primarily the peripheral insulin-responsive tissues rather than the endocrine pancreas or the CNS, and because excellent recent reviews on these latter topics are available (2, 201, 307, 392, 409, 444, 459, 652, 749, 863), we focus this section on the insulin-adipocyte-hepatocyte axis and its control of hepatic glucose production and only briefly review these other important indirect actions of insulin.
B. The Adipocyte-Hepatocyte Axis: Lipolytic Control of Hepatic Gluconeogenesis
It has long been appreciated that fatty acid substrate availability can regulate hepatic glucose production. Using perfused, glycogen-depleted rat liver, several groups showed in the 1960s that oleic acid oxidation increased gluconeogenesis from alanine or lactate (315, 835, 903, 905, 906), although this was controversial (217). Because of the requirement for gluconeogenic substrate for fatty acid activation of gluconeogenesis, and because increases in mitochondrial acetyl CoA content were observed, these workers proposed that allosteric activation of pyruvate carboxylase (PC) by acetyl CoA mediated the stimulatory effect of fatty acid oxidation on hepatic gluconeogenesis (7, 434, 864, 902, 903). However, methodological limitations stemming from acetyl-CoA’s low hepatocellular concentrations and rapid degradation ex vivo thwarted accurate measurement in liver samples. Furthermore, studies in isolated hepatocytes were unable to demonstrate any direct effect of insulin on PC activity (138). But in vitro liver preparations do not enable studies of the effect of insulin to control fatty acid availability through adipose tissue lipolysis. In vivo evidence for an indirect effect of insulin on HGP can be traced to the 1966 report that in pancreatectomized dogs, inhibiting lipolysis with nicotinic acid decreased both plasma NEFA levels and HGP (615). Twenty years later, it was reported that a low-dose insulin infusion which increased peripheral but not portal insulin levels and did not alter glucagon was nevertheless effective in suppressing HGP in humans, pointing to key extrahepatic effects of insulin on HGP suppression (661). However, demonstrating the relevance of the lipolysis-acetyl CoA-PC-HGP axis conclusively in vivo was complicated by the confounding effects of NEFA to increase insulin secretion and decrease glucagon secretion, as well as methodological limitations including the measurement of hepatic acetyl-CoA content and rates of hepatic pyruvate carboxylase flux (229). Additionally, lipolysis releases glycerol, which is converted to glucose at a redox-modulated but largely substrate-dependent rate (34, 511, 620). Skilled use of the somatostatin pancreatic clamp, better measurements of portal versus peripheral hormone concentrations, and the development of tracer methods to measure fatty acid and glycerol turnover eventually yielded insights into the physiological relevance of these mechanisms. Several groups subsequently reported that the effect of insulin to suppress HGP was partially mediated by its suppression of plasma NEFA levels (6, 476, 686, 774). Bergman and co-workers (686) hypothesized that this effect of NEFA to modulate HGP could be attributed to NEFA modulation of transendothelial insulin transport. Particularly convincing evidence for NEFA modulation of HGP came from work in fasted dogs, in which NEFA infusion during hyperinsulinemic-euglycemic pancreatic clamps totally prevented insulin suppression of both plasma NEFA levels and HGP. Variation in the magnitude of this effect in other studies can partially be explained by some combination of species differences, incomplete control of portal and peripheral insulin and glucagon, and the direct effect of insulin to suppress net hepatic glycogenolysis; as discussed in section II, glycogenolysis contributes a highly variable fraction of HGP depending on species and fasting duration (136, 428, 476, 477, 773). Recently, using novel liquid chromatography-tandem mass spectrometry methods to assess hepatic acetyl-CoA content and rates of hepatic pyruvate carboxylase flux in vivo, Perry and co-workers (620, 630) tested the hypothesis that insulin acutely suppresses hepatic gluconeogenesis by suppressing WAT lipolysis and hepatic acetyl CoA content and hepatic pyruvate carboxylase activity in free-ranging rats. During hyperinsulinemic-euglycemic clamps, insulin was shown to rapidly decrease palmitate and glycerol turnover (i.e., lipolysis), hepatic acetyl CoA content, hepatic pyruvate carboxylase flux, and hepatic HGP in tight temporal alignment (620). Preventing this decrease in hepatic acetyl CoA by acetate infusion, while also replacing glycerol to match basal turnover rates, completely abrogated insulin suppression of hepatic pyruvate carboxylase flux and HGP (620). Importantly, these studies were performed in fasted rats with glycogen-depleted livers reliant on gluconeogenesis for virtually 100% of HGP.
Studies of genetically modified rodents also support a key role for the adipocyte-hepatocyte axis in insulin suppression of HGP. Studies of mice with liver-specific ablation of Akt1, Akt2, and Foxo1 (TLKO mice) offer particularly strong evidence for this mechanism (504, 620, 842). TLKO mice, despite lacking three critical effectors of the hepatocellular insulin response, suppressed hepatic acetyl CoA and HGP normally in hyperinsulinemic-euglycemic clamp studies (504). Neither vagotomy nor glucagon blockade altered this phenotype (842). However, when the effects of insulin suppression of WAT lipolysis to decrease hepatic acetyl CoA content and glycerol flux to liver were prevented by infusions of acetate and glycerol, both TLKO and wild-type mice were unable to decrease pyruvate carboxylase flux and hepatic gluconeogenesis in response to insulin (620). In a separate study, infusion of Intralipid and heparin during a clamp to maintain plasma NEFA concentrations at their basal levels prevented insulin suppression of HGP in TLKO mice, although not in wild-type mice (842). Similarly, rats with antisense oligonucleotide-mediated Insr ablation in liver and WAT were only able to suppress HGP in hyperinsulinemic-euglycemic clamp studies when adipose insulin action was functionally restored using the ATGL inhibitor atglistatin (620). These studies of rodents lacking canonical hepatocellular insulin signaling provide strong evidence that hepatic gluconeogenesis is controlled independently of direct hepatocellular insulin action (620). Rather, these data support a model wherein insulin regulation of hepatic gluconeogenesis is mostly dependent on WAT lipolysis through acetyl CoA allostery and glycerol substrate availability (620). This paradigm also explains the surprising reports that 1) whole-body Insr knockout mice failed to regain intact insulin suppression of HGP when Insr expression was transgenically rescued in liver, and 2) acute liver-specific Insr ablation failed to impair insulin suppression of HGP (105, 592). Modifying lipolysis at the level of the adipocyte can also affect insulin suppression of HGP. Mice lacking Atgl in adipose tissue displayed decreased fatty acid turnover, which was associated with decreased hepatic acetyl CoA and improved HGP suppression compared with wild-type controls (12, 620). Finally, the insulin response in Pde3b−/− mice was characterized by failure to suppress plasma NEFA levels and a corresponding impairment in HGP suppression, although these mice also developed hepatocellular insulin signaling defects (147). Taken together, available evidence supports the hypothesis that insulin’s acute suppression of hepatic gluconeogenesis involves suppression of WAT lipolysis, which decreases both conversion of glycerol to glucose and acetyl CoA activation of pyruvate carboxylase.
Unraveling the relative importance of direct versus indirect effects of insulin on HGP still requires further investigation, particularly because these relative contributions are likely to vary significantly in different physiological and pathophysiological conditions. However, a useful simplification is to conceive of glycogenolytic contributions to HGP as controlled by direct hepatic insulin action and gluconeogenic contributions to HGP as controlled by indirect insulin action, largely through effects on WAT lipolysis (FIGURE 6). Accordingly, the direct effects of insulin on HGP will predominate in a glycogen-replete (fed) liver, whereas the indirect effects of insulin on HGP will predominate in a glycogen-depleted (fasted) liver. This hypothesis might explain the discordant literature on this topic, emphasizing the importance of species differences. Humans and dogs break down their hepatic glycogen stores more slowly than rodents; therefore, direct effects of insulin on hepatic glucose metabolism might be expected to predominate following an overnight fast. In contrast, rats and mice are almost totally devoid of hepatic glycogen following an overnight fast; therefore, indirect effects of insulin on hepatic glucose metabolism would be expected to dominate under these conditions. The quantitative significance of this latter effect in humans remains to be further explored, but it is interesting to note that the increased HGP of poorly controlled type 2 diabetic humans is accounted for entirely by increases in hepatic gluconeogenesis rather than net hepatic glycogenolysis (513, 704). The link between increased NEFA levels and hyperglycemia in T2D has long been appreciated; it is hoped that increasing mechanistic evidence for their causal linkage will spur therapeutic innovation targeting WAT lipolysis.
FIGURE 6.

The adipocyte-hepatocyte axis and insulin suppression of gluconeogenesis. A: in the fasted state, the adipocyte releases nonesterified fatty acids (NEFA) into the circulation. Within the hepatocyte, NEFA are oxidized to mitochondrial acetyl CoA, an allosteric activator of pyruvate carboxylase (PC). PC drives gluconeogenic flux. This, together with net glycogenolysis, facilitates hepatic glucose production (HGP) during fasting. B: during insulin stimulation (e.g., postprandially), both direct and indirect effects of insulin suppress HGP. Adipocyte insulin signaling suppresses lipolysis, decreasing plasma NEFA concentrations, hepatic mitochondrial acetyl CoA concentrations, PC activity, and gluconeogenic flux. Simultaneously, direct insulin action on the hepatocyte promotes net glycogen synthesis. Both processes enable insulin to rapidly and potently suppress net HGP.
C. Insulin Suppression of Glucagon Secretion
Glucagon action is important for normal glucose homeostasis (871), and given its potent anti-insulin effects on the liver in particular, it is not surprising that an elegant and tightly controlled reciprocal paracrine regulatory system has evolved to favor either insulin or glucagon secretion, but not both, in any given metabolic state (863). It was shown in 1970 that insulin-deficient diabetes was a state of relative hyperglucagonemia and α-cell hyperresponsiveness to the glucagon secretagogue arginine (861), and subsequent studies confirmed that insulin acts in a paracrine manner to suppress glucagon release from the islet (29, 280, 523, 890). Insulin per se is sufficient to suppress glucagon levels during euglycemia and hypoglycemia in humans with type 1 diabetes (T1D), and fasting glucagon levels are correlated with insulin resistance in nondiabetic humans (164, 223, 756). Mice lacking the insulin receptor in α-cells provided direct genetic evidence for this mechanism; these mice exhibited hyperglucagonemia in the fed state and during insulin tolerance tests (393). The mechanism for insulin suppression of α-cell glucagon secretion is incompletely elucidated but has been shown to involve PI3K activity and phosphodiesterase-mediated degradation of cAMP (213, 683). Overall, the importance of glucagon in insulin action and diabetes is underappreciated (862); the inability of mice lacking the glucagon receptor to develop diabetes upon β-cell destruction is only one of many striking illustrations of this (468).
D. Peripheral Effects of CNS Insulin Action
A role for the CNS in the regulation of glucose metabolism has been appreciated since the 1850s (55), but the subject has experienced a renaissance in the past 20 yr (444). Insulin is transcytosed across the blood-brain barrier (40) and both neurons and glial cells express insulin receptors (652). A major function of insulin in the brain is the suppression of appetite (48, 912). Neuron-specific Insr deletion predisposes mice to diet-induced obesity and concomitant hepatic insulin resistance, likely through appetite modulation (104). Neuron-specific rescue of Insr expression in whole-body Insr−/− mice extends lifespan from a few days to a few weeks, although the rescued mice still develop profound diabetes, indicating that the full beneficial effects of brain insulin action require intact peripheral insulin receptors (591). Indeed, CNS signaling has been demonstrated to regulate hepatic insulin action through mechanisms independent of energy balance. The 2002 finding that intracerebroventricular insulin administration was sufficient to suppress hepatic glucose production in rodents (after a >1 h time delay) (588) spurred further investigation in the field. Subsequently, insulin action in certain hypothalamic nuclei was reported to potently suppress hepatic glucose production (588, 653), promote muscle glucose uptake (415), suppress adipose tissue lipolysis (415, 737), and suppress glucagon secretion (612) in rodents. Nasal insulin administration, which disproportionately increases cerebrospinal fluid insulin concentrations, has been shown to enhance insulin suppression of hepatic glucose production in hyperinsulinemic-euglycemic clamp studies in lean humans (308). However, a physiological increase in brain insulin levels per se is insufficient to alter hepatic glucose production in dogs (208, 675). The mechanisms linking brain and peripheral insulin action are elusive, but likely involve sympathetic and parasympathetic outflow and possibly the hypothalamus-pituitary-adrenal (HPA) axis. For example, hepatic vagotomy was shown in one study to block the effects of CNS insulin on HGP (653), although the normal hepatic glucose metabolism of mice lacking hepatic muscarinic acetylcholine receptors (480) and the normal hepatic insulin action of denervated rodent liver (551) and transplanted human liver (which remains denervated for at least 2 yr postoperatively) (744) challenges this hypothesis (673). Other important concerns have been raised challenging the physiological generalizability of much of the experimental work in this field. These include the unphysiological dose and route of administration of intracerebroventricular insulin, failure to preserve the physiological ~3:1 portal-peripheral insulin gradient and consequent overemphasis of extrahepatic insulin action, failure to control glucagon levels between experimental groups, and species differences in the sources and control of hepatic glucose production between rodents and larger mammals (673). An elegant attempt to address these concerns utilized the somatostatin pancreatic clamp with basal glucagon replacement and portal vein insulin infusion in conscious dogs; the PI3K inhibitor LY249002 was infused intracerebroventricularly to assess the contribution of brain insulin action to the peripheral effects of physiological hyperinsulinemia (674). The experiments revealed no effect of acute brain insulin signaling inhibition on insulin suppression of hepatic glucose production or insulin-stimulated whole-body glucose uptake, but did reveal a modest blunting in the induction of hepatic glucokinase expression, of unclear physiological significance (674). Whether chronic brain insulin action or brain insulin resistance significantly affects peripheral insulin action is an open question (444).
Aside from serving as a direct site of insulin action, the brain is also a site of signal integration for various peripherally secreted hormones. Of these, leptin has been of particular interest as a centrally acting adipokine with pleiotropic effects on energy balance and metabolism (537, 626). The mechanisms of leptin action are many and are reviewed elsewhere (166, 236, 537, 565, 626), but one mechanism that links directly to the adipocyte-hepatocyte axis discussed previously pertains to leptin’s role in starvation and diabetic ketoacidosis (623, 628). In rats with these conditions, plasma leptin concentrations are low and the HPA axis is activated with resultant corticosteroid-driven WAT lipolysis, ketogenesis, and hepatic gluconeogenesis; physiological replacement of leptin suppresses the HPA axis and prevents these effects. In this way, leptin participates in the control of substrate switching from glucose to fatty acids during starvation, an effect previously thought to be chiefly mediated by insulinopenia. Interestingly, leptin displays a hormetic effect; at low concentrations, it suppresses HPA axis-driven lipolysis, but at high concentrations, it promotes catecholamine-driven WAT lipolysis (628). A second key mechanism that contributes heavily to the antidiabetic effect of chronic leptin therapy in T1D is leptin suppression of glucagon secretion, which can be achieved through either peripheral or CNS leptin administration (249, 877). The fascinating observation that either chronic leptin treatment or insulin are sufficient to reverse the hyperglucagonemia, hyperglycemia, and ketoacidosis of florid T1D has advanced the hypothesis that the symptoms of diabetes are best conceptualized as consequences of glucagon excess rather than of insulin deficiency (862). Importantly, however, leptin reverses hyperglycemic diabetic ketoacidosis before correction of hyperglucagonemia; this acute effect occurs by suppression of HPA axis-driven WAT lipolysis (630).
Several experiments have also suggested that a gut-brain-liver axis regulates hepatic glucose metabolism in rodents. The gut is an endocrine organ in its own right, with enteroendocrine cells that produce hormones including ghrelin, incretins, FGF19, and cholecystokinin (CCK) (409). FGF19 (also known as FGF15 in rodents) is secreted by the distal small bowel in response to bile acid sensing and, in addition to its originally defined role in the enterohepatic control of bile acid synthesis, acts centrally to enhance glucose tolerance by suppression of the HPA axis in rodents (410, 556, 622). The gut microbiome may also participate in CNS control of peripheral insulin action; in rats, microbially produced acetate was shown to enhance glucose-stimulated insulin secretion through direct activation of the parasympathetic nervous system (624). It is unknown whether this mechanism is operative in humans. Direct nutrient sensing is another proposed mechanism by which the gut might prompt CNS regulation of peripheral metabolism. Duodenal fatty acid esterification has been shown in rats to act through parasympathetic activation to suppress hepatic glucose production upon upper intestinal lipid delivery (879), but similar experiments in humans (low-dose duodenal lipid infusion under pancreatic clamp conditions) revealed no effect of duodenal lipid on plasma glucose or hepatic glucose production (925). In contrast, a role for duodenal lipid delivery in suppressing food intake through CCK secretion is uncontroversial (529). The complexity of these integrated physiological mechanisms uncovered in rodents requires careful confirmation in humans, but further work on gut-brain signaling pathways is likely to yield continued insights into therapeutically relevant areas such as mechanisms of bariatric surgery efficacy and incretin physiology (201).
IV. PATHOPHYSIOLOGY OF INSULIN RESISTANCE
A. What Is Insulin Resistance?
The notion of insulin resistance can be traced to the observations of Himsworth (317), who noted that concurrent injection of glucose and insulin in diabetic patients produced one of two outcomes. Some diabetics responded to the challenge with stable or decreased blood glucose; these were termed insulin-sensitive. In others, the challenge increased blood glucose markedly; these were considered insulin-insensitive. We now appreciate that these latter patients typify the insulin resistance of the metabolic syndrome: at a normal plasma insulin level, target tissues are unable to mount a normal coordinated glucose-lowering response involving suppression of endogenous glucose production, suppression of lipolysis, cellular uptake of available plasma glucose, and net glycogen synthesis (377, 378, 381, 595, 684). This insulin resistance necessitates increased insulin secretion to compensate, so fasting plasma insulin levels increase (176, 380). The real-time feedback circuit linking insulin sensitivity and insulin secretion complicates the ‟chicken-eggˮ problem of identifying the primary defect; what is clear is that defects in both insulin target tissues and β-cells are required for the development of fasting hyperglycemia and T2D (380). A diverse cadre of bioactive factors is capable of impairing insulin sensitivity, as is chronic hyperinsulinemia per se (595, 724). Although these defects in tissue insulin action are readily reversible (even in patients with T2D) by weight loss and hypocaloric regimens, continuous overnutrition in the setting of insulin resistance creates a vicious cycle of hyperinsulinemia and insulin resistance that culminates in eventual β-cell failure, likely due to glucose and lipid toxicity and other factors leading to overt T2D (380, 637).
The integrated physiology of insulin resistance owes to defective insulin action at target cells. Attempts to localize the defect in cellular insulin action have been driven by the identification of new effectors. In the 1970s and early 1980s, when INSR was the only known molecular effector of insulin action, several groups used insulin dose-response curves and 125I-insulin binding studies to relate surface INSR content to physiological insulin action and resistance (26, 272, 378, 417, 420, 595, 787). The central question at the time was whether insulin resistance owed to “receptor defects” (i.e., decreased INSR expression at the cell surface) or “postreceptor defects” (i.e., impaired signal transduction) (FIGURE 7) (417, 595). Receptor defects were identified in obese and diabetic rodents and humans, in adipocytes as well as in other cell types (26, 272, 379, 418, 786, 787). Compensatory downregulation of surface INSR content in the face of sustained hyperinsulinemia is likely to partially explain the phenomenon, although mechanisms for active regulation of surface INSR expression and dysregulation in insulin resistance are beginning to emerge. For example, the ubiquitin ligase MARCH1 ubiquitinates INSR to decrease the number of receptors at the cell surface (566). MARCH1 is transcriptionally regulated through FOXO1, and MARCH1 expression is increased in WAT from obese mice and humans, consistent with insulin resistance to FOXO1 inhibition (566). The effect of decreasing surface INSR expression on insulin action is a right-shift of the insulin dose-response curve; because cells have spare receptors, no decrease in maximal response is observed unless the cell surface INSR content drops below 5–10% of normal (378).
FIGURE 7.

Insulin resistance in dose-response curves. A: in a hypothetical cell with decreased surface insulin receptor (INSR) content, the dose-response curve is right-shifted but the maximal biological response is not decreased unless >90% of surface receptors are lost. B: in a cell with an insulin signal transduction (“post-receptor”) defect, or a combined receptor/post-receptor defect, both a right shift and decreased maximal response are observed. The right graph typifies human obesity-associated insulin resistance in muscle, liver, and adipose tissues.
When the insulin resistance of human T2D was shown to involve both a right-shifted dose-response curve and a decreased maximal insulin response with respect to whole-body glucose uptake despite <90% loss of surface receptor content, it was inferred that both receptor and ‟postreceptorˮ defects contribute to insulin resistance (417, 595). As a result, the hypothesis that decreased insulin receptor binding accounts for typical obesity-associated insulin resistance has long since given way to a model in which insulin signal transduction defects are central (35). Additionally, the signal transduction (postreceptor) defects of typical obesity-associated insulin resistance have long been appreciated to involve the insulin receptor itself in addition to postreceptor effectors, confusing the semantics of ‟receptorˮ versus ‟postreceptorˮ defects (77, 247, 595, 821). Nearly all mechanistic work in this field in the last 30 yr, after the discovery that the INSR is a tyrosine kinase (390, 391), has focused on identifying defects in insulin signal transduction. Nevertheless, it is clear that both decreased surface INSR content and impaired insulin signal transduction contribute to typical obesity-associated insulin resistance.
Although distinguishing insulin “resistance,” an increase in insulin EC50, from decreased insulin “responsiveness,” a decreased maximal effect, can be useful (378), it is no longer common practice in the field. Properly distinguishing insulin resistance from responsiveness requires the construction of insulin dose-response curves, which can be cumbersome for in vivo studies. Insulin resistance in the commonly used sense, and therefore throughout the remainder of this review, is thus defined as an insulin dose-response curve with increased EC50, with or without decreased maximal response. Importantly, insulin resistance is not a binary switching-off of insulin signaling. For this reason, hyperinsulinemia is an effective compensatory mechanism that preserves insulin action in mild and moderate insulin resistance. Because insulin resistance displays tissue-specific functional consequences, we now consider the particular nature of insulin resistance in skeletal muscle, liver, and WAT, with attention to which signaling effectors and which physiological functions are impaired in typical obesity-associated insulin resistance.
B. Pathophysiology of Skeletal Muscle Insulin Resistance
The principal function of insulin in the skeletal muscle is to promote cellular glucose uptake, a process controlled by GLUT4 translocation. Insulin-stimulated muscle glucose uptake is highly susceptible to insulin resistance and is indeed a principal component of typical obesity-associated insulin resistance and T2D (182, 767). Because skeletal muscle is a major site of insulin-stimulated glucose disposal (70–80% during a hyperinsulinemic euglycemic clamp, although only 25–30% in the postprandial state where glucose appearance site, glucose concentrations, and tissue glucose demand all differ from the clamped state), muscle insulin resistance has a large effect on whole body glucose turnover (182, 428). Insulin stimulation of glycogen synthesis and glycolysis both require intact insulin-stimulated glucose uptake to furnish substrate, so these effects also become resistant to insulin action (152, 768).
The mechanisms for insulin resistance to muscle glucose disposal have been the subject of extensive investigation (FIGURE 8). Early work suggested that nonoxidative glucose metabolism (i.e., glycogen synthesis) was the major fate of myocellular glucose, and this was later directly confirmed in humans using 13C magnetic resonance spectroscopy (MRS) (58, 180, 768). The demonstration that insulin-stimulated muscle glycogen synthesis was markedly (~50%) impaired both in patients with T2D and in lean, healthy insulin-resistant offspring of patients with T2D provided a functional description of muscle insulin resistance, but did not localize the site of blockade (633, 768). Although many other studies demonstrated decreased glucose transport in insulin-resistant and T2D muscle, these results similarly could have resulted from primary blockade at the level of glycogen synthetic activity, hexokinase activity, or glucose transport, ‟backing upˮ the system with the final effect of decreasing glucose transport (193, 255, 767). Eventually, 13C and 31P MRS measurements of intracellular glucose and G6P concentrations revealed that the major rate-controlling step responsible for reduced insulin-stimulated muscle glycogen synthesis in diabetic patients was indeed glucose transport (152, 285, 633, 698). These physiological studies complemented cell biological observations that insulin potently controlled myocellular GLUT4 translocation to the plasma membrane and t-tubules and that this translocation was defective in human insulin resistance (258, 651, 881). Together, these studies focused the problem of muscle insulin resistance: the defect(s) impinge upon the signal transduction cascade linking insulin-INSR binding and GLUT4 translocation.
FIGURE 8.

Functional consequences of skeletal muscle insulin resistance. A: an insulin-sensitive skeletal myocyte activates the metabolic insulin receptor substrate 1 (IRS1)-phosphoinositide-3-kinase (PI3K)-AKT arm of the insulin signaling cascade to increase glucose uptake and glycogen synthesis. B: an insulin-resistant myocyte exhibits impairments in proximal insulin signaling events, blunting insulin’s ability to stimulate GLUT4 translocation and glycogen synthesis.
Skeletal muscle insulin resistance is traceable to defects at the most proximal levels of insulin signaling: INSR, IRS1, PI3K, and AKT activity. Early work revealed defective INSR tyrosine kinase activity in purified INSR from the skeletal muscle of obese mice; this provided the first demonstration of impaired intracellular insulin signaling and validated the prediction that surface insulin receptor downregulation was not the only defect in obesity-associated insulin resistance (417, 470, 595). This observation was later extended to obese and diabetic humans, in which two INSR defects were present: decreased surface INSR content and decreased INSR kinase (IRK) activity from purified receptors (117). Decreased IRK activity was also observed in skeletal muscle from diabetic rats, women with gestational diabetes, and young lean relatives of patients with T2D (66, 107, 301). However, IRK activity is now rarely measured from tissue samples. Instead, INSR activation is most often assessed by phosphotyrosine immunoblotting. This experiment is technically easier but does not directly assess INSR signaling; the multiplicity of tyrosine phosphorylation sites on INSR also complicates the relationship between these parameters (83, 117, 248, 470). Defective IRK activity is also suggested by the decrease in IRS1 tyrosine phosphorylation consistently observed in insulin-resistant skeletal muscle (175, 248, 285, 436, 942). Blunted insulin stimulation of IRS1-associated PI3K activity is also reproducibly observed in muscle insulin resistance, whether induced acutely or chronically (175, 198, 285, 441, 942). Interestingly, skeletal muscle from obese or type 2 diabetic humans does not develop insulin resistance to mitogenic signaling through MAPK (175). Although impaired insulin activation of other distal effectors, including AKT, is often seen in muscle insulin resistance, the simultaneous presence of proximal insulin signaling defects makes it difficult to determine whether these distal defects have an independent origin or are merely secondary to proximal defects. While it is likely that insulin resistance involves dysregulation of multiple signaling effectors, it is also possible that proximal signaling defects are sufficient to account for the entire impairment in insulin stimulation of glucose uptake seen in obesity-associated insulin resistance. Advances in computational modeling of signal transduction pathways will enable deconvolution of the relative contributions of specific signaling defects to the final functional defect of impaired insulin-stimulated glucose uptake (81, 402, 745).
C. Pathophysiology of Hepatic Insulin Resistance
As discussed in section II, hepatic insulin action affects the metabolism of all macronutrients. It has both acute and chronic, and both direct and indirect, components. Here we consider insulin regulation of glucose and lipid metabolism in the liver, with attention to which processes become resistant in obesity and T2D.
Insulin, in combination with adequate substrate supply, orchestrates a switch from net glucose production to net glucose uptake in the liver. This involves the coordinated suppression of gluconeogenesis and glycogenolysis, and the activation of glycogen synthesis. Suppression of gluconeogenesis can be divided into acute and chronic processes, with very different mechanisms. The acute suppression of hepatic gluconeogenesis (within 10 min of insulin stimulation in the rat) appears to be primarily mediated by extrahepatic insulin action (6, 208, 620, 686, 774). Suppression of lipolysis in adipose tissue reduces fatty acid delivery to liver, which in turn reduces hepatic β-oxidative flux. As a result, hepatic acetyl CoA levels are reduced, allosterically decreasing PC activity. Together with the decreased glycerol turnover caused by suppression of lipolysis, gluconeogenic flux is diminished (620). This mechanism has implications for the use of impaired insulin suppression of HGP as a surrogate for hepatic insulin resistance. In glycogen-depleted livers where gluconeogenesis accounts for most of HGP, impaired insulin suppression of HGP may signal adipose insulin resistance rather than hepatic insulin resistance. This paradigm reconciles the many surprising genetically modified rodent models in which severe perturbations in hepatic insulin signaling do not predict insulin’s ability to suppress HGP during hyperinsulinemic-euglycemic clamp studies (105, 135, 504, 592, 603, 620, 840). The most striking studies of this phenomenon have ablated the hepatic insulin receptor by either conventional gene targeting or antisense oligonucleotide treatment. Liver-specific Insr−/− (LIRKO) mice totally lack hepatocellular insulin signaling and are severely glucose intolerant; they do not suppress HGP in hyperinsulinemic-euglycemic clamp studies (234, 539). However, this defect can be totally rescued: not by replacing the hepatic INSR, but by concomitantly ablating hepatic Foxo1 to correct the unrestrained gluconeogenic enzyme expression (and glucokinase repression) produced by constitutive FOXO1 activation in LIRKO mice (592, 603, 840). Similarly, antisense oligonucleotide ablation of INSR in liver and WAT prevents insulin suppression of HGP in rats, but this defect is rescued by simulating WAT insulin action with the lipolysis inhibitor atglistatin (620). Both models of hepatic INSR ablation dissociate the hepatocellular insulin signaling pathway from normal insulin suppression of gluconeogenesis, suggesting that measurements of HGP suppression should be used with caution as readouts of hepatic insulin resistance.
Rates of hepatic gluconeogenesis are increased in T2D and are the proximate cause of the fasting hyperglycemia that defines the disease (259, 346, 513). But because gluconeogenesis is regulated by diverse mechanisms (e.g., allostery, redox, substrate-driven, transcriptional, posttranslational) (647), it does not necessarily follow that insulin’s direct hepatocellular effects on gluconeogenesis are dysregulated in T2D. The chronic suppression of gluconeogenesis by insulin, better observed in fasting-refeeding studies than acute insulin stimulation studies, is a direct hepatocellular effect mediated by inhibition of the FOXO transcription factors, especially FOXO1. Does increased FOXO1 activity have any role in the increased hepatic gluconeogenesis of T2D? FOXO1 mRNA expression and nuclear localization (a surrogate for activity) have been reported to be increased in humans with nonalcoholic steatohepatitis, correlated with the homeostatic model assessment of insulin resistance (HOMA-IR) score (866). Additionally, as discussed above, ablation of the hepatic insulin receptor is associated with unrestrained FOXO1 activity, which is necessary and sufficient to drive pathological gluconeogenesis (603, 840). This phenotype is also seen in mice with severe hepatic insulin resistance owing to ablation of hepatic Irs1 and Irs2, in which concomitant Foxo1 deletion normalizes glucose tolerance (195). Furthermore, antisense oligonucleotide knockdown of FOXO1 in liver and WAT improves hepatic insulin action in chronically fat-fed mice (720). Mouse models of profound hepatic insulin resistance, such as ob/ob and lipodystrophic mice, display increased G6pc expression suggestive of increased FOXO1 activity (764). However, the hepatic insulin resistance of both short-duration (5 day) and medium-duration (4 wk) fat-fed rats is not accompanied by alterations in gluconeogenic enzyme content, arguing against a role for FOXO1-driven gluconeogenic gene transcription in genetically normal rodents (620, 629). Furthermore, reducing hepatic Pck1 expression by >90% in mice caused only a modest ~40% reduction in gluconeogenic flux, suggesting that oscillations in Pck1 expression exert limited control over gluconeogenesis (108). Finally, a study of humans with T2D found no increase in the hepatic expression of FOXO1 targets G6pc and Pck1, despite fasting hyperglycemia (719). In summary, it is clear that if totally and chronically derepressed, FOXO1 can drive pathological gluconeogenesis in rodents, but there is no strong evidence that this is an operative mechanism for fasting hyperglycemia in typical human hepatic insulin resistance. The multifaceted control of hepatic gluconeogenesis means that modest transcriptionally mediated increases in ‟gluconeogenic capacityˮ may not be sufficient to drive gluconeogenesis in genetically normal subjects. Further studies are needed to adequately describe the relative contribution of FOXO1 dysregulation to the increased hepatic gluconeogenesis of humans with T2D.
Insulin suppression of hepatic glycogenolysis and stimulation of glycogenesis is a direct effect that requires intact hepatocellular insulin action as discussed in section II. The question of whether insulin control of hepatic glycogen metabolism becomes resistant in diabetes is confounded by the potent allosteric control of GS and phosphorylase by G6P and glucose, and by the insulin-independent transport of glucose across the hepatocellular plasma membrane by GLUT2. Likely owing to this, some studies disagree on the contribution of defective hepatic glycogen metabolic regulation to hepatic insulin resistance and T2D. For example, in hyperinsulinemic-euglycemic clamp studies of streptozotocin-induced diabetic rats, insulin-stimulated hepatic GS activity was not impaired, while similar studies in 3-day high-fat-fed rats found impairments (439, 721). However, the bulk of available data suggest that hepatic insulin resistance is accompanied by defective hepatic glycogen metabolism. In humans, MRS measurements of hepatic glycogen content revealed that T2D was associated with lower 4-h postprandial glycogen content, suggesting a defect in glycogen synthesis (513). Subjects with T2D were further found to display lower fasting hepatic glycogen content as well as diminished glycogen synthesis under both postprandial and hyperinsulinemic-hyperglycemic clamp conditions (437). Furthermore, glycogenolysis was found to be decreased and incompletely suppressed by insulin in people with T2D (38, 69, 513). The picture of daily hepatic glycogen metabolism in T2D is thus one of decreased amplitude, in which dampened insulin control of both glycogen synthesis and mobilization lead to relatively static hepatic glycogen content instead of the large oscillations characteristic of normal feeding and fasting. This likely owes to impaired insulin action on both posttranslational modifications of the glycogen synthetic machinery, transcriptional regulation of glucokinase activity, and translocation from the nucleus to the cytoplasm.
Just as understanding acute hepatic insulin action on glucose metabolism requires separating the direct hepatocellular effects on glycogen metabolism from the largely indirect metabolite-mediated effects on gluconeogenesis, understanding hepatic insulin resistance requires distinguishing these components from one another (647). True hepatic insulin resistance should impair insulin-stimulated glycogen synthesis, shifting the insulin dose-response curve for this function rightward and possibly decreasing its maximum. But isolated hepatic insulin resistance would not be expected to block the ability of insulin to suppress gluconeogenesis, because insulin suppression of gluconeogenesis has a large indirect component. This prediction has been borne out dramatically in multiple rodent models of ablated hepatocellular insulin signaling as discussed above.
D. Selective Hepatic Insulin Resistance
In addition to glucose handling, liver insulin action also powerfully controls lipid metabolism, largely through SREBP-1c. If insulin normally promotes net hepatic lipogenesis, insulin-resistant subjects might be expected to display decreased lipogenesis. This is indeed the case in models of genetic total hepatic insulin resistance (i.e., ablation of the hepatic insulin receptor), which display decreased plasma triglycerides and decreased hepatic DNL (60, 868). But hepatic insulin resistance in genetically normal rodents and humans is highly associated with hepatic steatosis; net lipogenesis is consistently elevated in insulin-resistant livers. This phenomenon has been termed “selective hepatic insulin resistance” (99) or “pathway-selective insulin resistance and responsiveness” (920), with proponents of these models arguing that “insulin-resistant” liver is in fact only resistant to glucose handling (i.e., the FOXO transcriptional program), not to lipid handling (i.e., the SREBP-1c transcriptional program) (764). Efforts to mechanistically rationalize selective hepatic insulin resistance have focused on potential insulin signaling bifurcations that would enable the lipid-handling arm of insulin signaling to remain intact while the glucose-handling arm becomes resistant. Early reports suggesting that hepatic IRS1 might be more responsible for glucose handling while IRS2 regulated lipid handling have not been corroborated by subsequent investigations (195, 196, 443, 662, 827). Yet another alternative hypothesis has focused on potential differences in substrate specificity for singly phosphorylated (pThr308) versus doubly phosphorylated (pThr308, pSer473) AKT (487, 920, 921). Ser473 phosphorylation, in the hydrophobic motif of AKT, may enable activity toward some AKT substrates (e.g., the NH2-terminal FOXO site) while AKT activity toward other substrates (e.g., GSK3β, TSC2) may only require Thr308 phosphorylation by PDK1 (288, 362). Because some models of hepatic insulin resistance, like the db/db mouse, appear to display preserved insulin stimulation of Thr308 but not Ser473, this is an intriguing hypothesis (921). However, direct experimental support is thus far lacking and the mulitiplicity of nutrient-sensitive inputs into AKT Ser473 phosphorylation confounds easy interpretation. A final possible mechanism for continued insulin activation of SREBP-1c despite FOXO1 unresponsiveness in hepatic insulin resistance is that these pathways have different intrinsic sensitivities to insulin. In support of this model, fourfold higher concentrations of an INSR inhibitor are necessary to cause resistance to SREBP-1c activation than resistance to FOXO1 inactivation (162).
A unifying hypothesis for the supposed paradox of pathway-selective hepatic insulin resistance has been elusive in part because of the many branch points involved in insulin signaling and the involvement of some effectors, such as AKT and FOXO1, in both glucose and lipid handling (99, 457). However, a possible resolution to the paradox is that hepatic insulin resistance is not selective, but chronic overnutrition activates several insulin-independent drivers of lipogenesis. First, substrate-push re-esterification of circulating fatty acids, facilitated by adipose insulin resistance, is likely a major source of liver triglyceride in nonalcoholic fatty liver disease (NAFLD). Evidence for a role of fatty acid re-esterification in NAFLD includes the recent demonstration that an increase in plasma fatty acids, independent of hepatic insulin signaling or transcriptional effects, is fully sufficient to drive hepatic triglyceride accumulation by re-esterification through a substrate push mechanism (868). Of note, re-esterification, rather than DNL, is the primary lipogenic flux in insulin-resistant humans (197). Second, upregulation of the DNL transcriptional program does not require insulin. Nutrient induction of the DNL program involves both the carbohydrate response element binding protein (ChREBP; stimulated by glucose) (657) and SREBP-1c (stimulated by amino acid activation of mTORC1) (487). Fructose is also a particularly potent stimulator of lipogenesis, acting through multiple mechanisms including substrate push acutely and activation of both ChREBP and SREBP-1c chronically (311, 718). Because insulin resistance is often accompanied by nutrient oversupply, activation of these nutrient-sensitive pathways is an attractive explanation for the coexistence of hepatic steatosis and hepatic insulin resistance. In this conception, although hepatic insulin resistance blunts the effect of insulin per se on SREBP-1c, alternative nutrient-sensitive pathways are sufficient to drive lipogenesis. This mechanism explains the increased DNL that has been observed in humans with NAFLD (347, 449, 868). Interestingly, de novo lipogenic flux has been found to be decreased in high-fat-fed rats, consistent with this pathway becoming insulin resistant rather than remaining insulin sensitive (184, 200, 868). Importantly, fat feeding would not be expected to strongly activate the alternative nutrient-sensitive drivers of the DNL program (ChREBP, mTORC1). It is also useful to bear in mind that insulin resistance is merely a shifted insulin dose-response curve; the portal hyperinsulinemia that accompanies peripheral insulin resistance is capable of activating SREBP-1c, perhaps even to normal levels given the exquisite sensitivity of this pathway to insulin (162). Given these considerations–insulin-independent esterification of circulating fatty acids, nutrient activation of DNL transcriptional programs, and hyperinsulinemia–it may be unnecessary to invoke the paradox of selective hepatic insulin resistance to understand how insulin resistant humans develop NAFLD (FIGURE 9).
FIGURE 9.

Mechanisms for the development of nonalcoholic fatty liver disease (NAFLD) despite hepatic insulin resistance. A: insulin normally activates de novo lipogenesis through sterol regulatory element binding protein 1c (SREBP-1c). B: the seemingly paradoxical coexistence of NAFLD and hepatic insulin resistance has spawned the hypothesis of selective hepatic insulin resistance, wherein insulin activation of lipogenesis is preserved despite impaired insulin regulation of glucose metabolism. However, hepatic de novo lipogenesis has multiple inputs, including ChREBP and mTORC1/SREBP-1c, both of which are activated in states of chronic overnutrition. Additionally, the primary pathway for hepatic triglyceride synthesis is re-esterification of preformed fatty acids, which are readily available in states of chronic overnutrition owing both to dietary supply and to adipose insulin resistance. Even if insulin receptor (INSR) activation of SREBP-1c is impaired by hepatic insulin resistance, these other inputs are likely capable of supporting the lipogenic fluxes that lead to NAFLD. NEFA, nonesterified fatty acid; WAT, white adipose tissue.
Pathway-selective hepatic insulin resistance is also difficult to reconcile with the finding of impaired proximal insulin signaling in several models of liver insulin resistance. In the late 1980s, defects in IRK activity were appreciated in liver samples from high-fat-fed rats (886) and humans with T2D (116). Decreased IRK activity and IRS2 tyrosine phosphorylation were also observed in insulin-resistant livers of rats fed a 3-day high-fat diet (722). These lines of evidence argue that the primary defect in hepatic insulin resistance is not in a downstream ‟branch pointˮ effector but rather in the insulin receptor: the most proximal and powerful locus of control in insulin action.
A final important and INSR-related component of hepatic insulin resistance is decreased hepatic insulin clearance (80, 222, 650). Although the molecular determinants of this phenomenon are incompletely understood, the reduced cell surface INSR content observed in insulin-resistant hepatocytes (116, 787) may involve regulators of the surface INSR pool including CEACAM1 (660), MARCH1 (566), CHIP (830), and p31comet (145). Regardless of mechanism, the net effect of decreased hepatic insulin clearance is to promote hyperinsulinemia. The fasting plasma insulin concentration is thus often used as a general readout of hepatic insulin sensitivity in algorithms such as the HOMA-IR and the quantitative insulin sensitivity check index (QUICK-I), with the obvious caveats that β-cell function and peripheral insulin sensitivity also provide input to the plasma glucose-insulin circuit.
In summary, insulin resistance in liver is traceable to defects at the level of the insulin receptor and therefore probably affects all arms of hepatocellular insulin signaling. The variable functional impact of hepatic insulin resistance in different signaling arms is not surprising given the varied and complex inputs to each arm. The most popular physiological readouts of hepatic insulin resistance, suppression of HGP and fasting plasma insulin, have both direct and indirect components and are therefore imperfect assessments of hepatocellular insulin action. Yet these readouts will, and should, continue to be used widely, especially in human studies, because 1) they can be determined noninvasively and 2) they give a useful integrated picture of the direct and indirect components of hepatic insulin action. Accurate measurement of hepatic insulin resistance is best performed using multiple complementary readouts, summarized in TABLE 1. The use of readouts with no indirect component is preferable if the investigator seeks to make claims about hepatocellular insulin action per se. Although these readouts can be used to determine the degree and nature of hepatic insulin resistance, they offer only limited insight into causative factors. Experimental considerations are discussed in section IVF.
Table 1.
Readouts of hepatic insulin resistance
| Readout | Change in Insulin Resistance | Direct/Indirect | Acute/Chronic | Notes |
|---|---|---|---|---|
| Activation of net hepatic glycogen synthesis | ↓ | Direct | Acute | Requires both hyperinsulinemia and hyperglycemia. |
| Suppression of hepatic gluconeogenesis | ↓ | Indirect | Acute | Related to suppression of WAT lipolysis leading to decreased NEFA and glycerol turnover and hepatic acetyl-CoA content. There may also be a small direct effect of insulin by suppression of intrahepatic lipolysis. |
| Suppression of hepatic glucose production | ↓ | Direct and indirect | Acute | Variable contributions of gluconeogenesis and glycogenolysis depending on species and fasting duration. |
| INSR Tyr phosphorylation and IRK activity | ↓ | Direct | Acute | |
| IRS Tyr phosphorylation | ↓ | Direct | Acute | |
| AKT Ser/Thr phosphorylation | ↓ | Direct | Acute | Multiple inputs to Ser473 phosphorylation. |
| Gluconeogenic gene expression | ↑ | Direct | Chronic | Especially G6pc, Pck1. |
| De novo lipogenesis | ↓ | Direct | Chronic | Multiple inputs including mTORC1, ChREBP. |
| Fasting plasma insulin | ↑ | Direct and indirect | Chronic | Crude surrogate of hepatic insulin resistance, useful in large epidemiologic studies. |
INSR, insulin receptor; IRK, INSR kinase; IRS, insulin receptor substrate; WAT, white adipose tissue; NEFA, nonesterified fatty acid.
E. Pathophysiology of Adipose Insulin Resistance
Interest in adipocyte insulin resistance has seen a resurgence in recent years as investigators unravel the spectacular complexity of adipose tissue as both nutrient sink and endocrine organ (735). As with muscle and liver, adipose insulin receptor tyrosine kinase activity is diminished in humans with T2D (246, 247). This defect, paired with decreased plasma membrane insulin receptor content (378, 379, 418, 595, 786), may be sufficient from a signaling perspective to account for the manifestations of adipose insulin resistance. Indeed, weight loss corrects both adipose insulin resistance and defective adipocyte IRK activity (247). Unfortunately, however, the mechanistic basis for these adipocyte insulin receptor defects remains largely obscure.
The adipocyte performs insulin-stimulated glucose uptake; as in skeletal muscle, this function becomes resistant in obesity and T2D (377). However, WAT is not a quantitatively significant site of insulin-stimulated glucose disposal, accounting for <5% of an oral glucose load in humans (363, 428). Despite this, adipose-specific deletion of GLUT4 in mice results in liver and skeletal muscle insulin resistance without altering adiposity or body weight (1), pointing to indirect, physiologically significant consequences of insulin resistance to glucose uptake in adipocytes (725). The global insulin-sensitizing effects of adipocyte glucose uptake may partially be mediated through glucose activation of ChREBP. ChREBP activation promotes lipogenic gene expression, enabling adipose tissue to serve as a nutrient sink which in turn decreases substrate delivery to liver and muscle (310). Adipocyte glucose uptake also generates glycerol-3-phosphate for fatty acid esterification, facilitating eutopic lipid storage in WAT and decreasing lipid delivery to liver and muscle. Alternatively, the link between adipose GLUT4 expression and global insulin sensitivity has also been ascribed to adipokines such as retinol binding protein 4 (RBP4; see sect. VII) (935) and to altered expression of nicotinamide N-methyltransferase (432).
Physiologically, an extremely significant function of WAT insulin action is the suppression of lipolysis. As discussed in section II, adipose lipolysis is exquisitely sensitive to insulin. The suppressive effect is rapid (~10 min) in rodents and humans (620). Because the major source of plasma NEFA is adipose tissue, because lipolytic substrate release is a critical regulator of hepatic gluconeogenesis on a minute-to-minute basis, and because increased gluconeogenesis is a key driver of the fasting hyperglycemia that defines T2D, resistance to insulin’s antilipolytic effects in the adipocyte is of immense pathophysiological importance.
If adipocyte insulin resistance impairs suppression of lipolysis, one might expect that plasma NEFA concentrations would be elevated in T2D. In poorly controlled T2D, this is indeed the case; multiple studies have observed significantly increased plasma NEFA concentrations in patients with poorly controlled T2D compared with lean nondiabetic controls (134, 245, 266, 685, 816). In lean, healthy relatives of patients with T2D, postprandial suppression of plasma NEFA concentrations is impaired (31). Moreover, increased plasma NEFA concentrations predict incident T2D (121, 611). The extent of the increase in plasma NEFA is much more modest and sometimes nonexistent in patients with well-controlled T2D (245, 286, 685, 816). Yet these concentration measurements belie much larger differences in the more physiologically relevant parameter: flux. Under hyperinsulinemic-euglycemic clamp conditions, plasma NEFA turnover was in relative terms much more elevated than plasma NEFA concentration in T2D patients compared with controls (286). Fasting glycerol turnover is also increased in T2D, as is the rate of gluconeogenesis from glycerol (586, 663). Furthermore, in patients undergoing bariatric surgery, decreases in basal lipolysis correlate with improvements in HOMA-IR (265). Importantly, obesity per se is not correlated with plasma NEFA concentration in humans, indicating that a functional defect (i.e., adipocyte insulin resistance), rather than a simple mass effect, mediates the increased plasma NEFA of T2D (388). Consistent with this interpretation of adipose insulin resistance, insulin suppression of lipolytic flux as measured by glycerol turnover is impaired in obese insulin-resistant human adolescents, in diabetic humans, and in first degree relatives of type 2 diabetic humans (263, 309, 370, 696). Adipose insulin resistance, assessed as the product of fasting plasma insulin and fasting plasma NEFA concentrations, increased continuously in the progression from normal glucose tolerance to impaired glucose tolerance to T2D in a cohort of over 300 subjects, suggesting that adipose insulin resistance contributes to the pathogenesis of T2D (261). Using the product of fasting plasma insulin and plasma NEFA concentrations as a practical index of adipose insulin resistance was recently validated against the multistep pancreatic clamp (789).
Whether insulin resistance to suppression of adipose lipolysis involves primarily increased lipolysis per se, decreased re-esterification of fatty acids, or both is not settled. A provocative study using [14C]palmitate to trace and model fatty acid kinetics during a hyperinsulinemic-euglycemic clamp concluded that net lipolysis was not different between lean and obese T2D subjects, but that the obese T2D subjects exhibited higher rates of fatty acid escape from tissue uptake, and that this latter finding accounted for the defect in insulin suppression of plasma NEFA levels in the diabetics (692). Along these lines, a study of obese nondiabetic and T2D subjects found that plasma glycerol and NEFA levels were similar between groups during an oral glucose tolerance test, and that ex vivo suppression of isoproterenol-stimulated lipolysis was similar between groups, but that expression of the fatty acid transporter FABP4 was decreased in T2D; the investigators concluded that lipid storage rather than lipolysis was subject to insulin resistance (618).
Just as glycogen metabolism in the insulin-resistant liver exhibits a decreased amplitude in its net magnitude during both fasting and feeding, so there is evidence that insulin-resistant adipose tissue exhibits an inability to maximally stimulate lipolysis during fasting and maximally promote lipid storage during feeding. A multiple meal-ingestion study of lean healthy men compared with men with abdominal obesity highlighted this phenomenon: the direction of net fatty acid flux across the adipose tissue capillary bed favored release during fasting and favored storage postprandially, and in both cases was smaller in magnitude in the obese men (536). The net effect was a significant decrement in the percentage of ingested fat properly stored in WAT in the obese subjects, presumably facilitating lipid storage in nonadipose tissues.
Interestingly, in contrast to skeletal muscle and liver, the specific molecular defects mediating adipocyte insulin resistance are largely uncharacterized. Rather, most progress in understanding the pathogenesis of adipocyte insulin resistance has focused on autocrine or paracrine inflammatory cytokines which may impair insulin signaling (27, 289). These mechanisms are considered in detail in section VII. Yet resistance to insulin suppression of adipose lipolysis is evident after only 3 days of high-fat feeding in rats (722)–before significant inflammation develops–suggesting that noninflammatory mechanisms are likely responsible for the initial defects in adipocyte insulin resistance.
The specific roles of proteins involved in lipolysis–ATGL, HSL, PLIN, FSP27–in adipocyte insulin resistance are unclear. But impaired suppression of net adipose lipolysis could also result from defective NEFA esterification. Indeed, one interesting potential mechanism for insulin resistance to suppression of lipolysis involves NEFA esterification and offers a mechanistic link to insulin-resistant glucose uptake. Insulin-stimulated glucose uptake promotes glycolysis, producing the three-carbon precursors that can yield glycerol-3-phosphate. Glycerol-3-phosphate is in turn required for fatty acid esterification and proper insulin-stimulated lipid storage (474). Insulin resistance to glucose uptake might by this mechanism prevent the adipocyte from switching from net lipid export to net lipid storage upon insulin stimulation.
In this way, the exquisite sensitivity of the white adipocyte to insulin may be a double-edged sword. This sensitivity enables agile and appropriate postprandial nutrient storage. Yet the status of WAT as ‟first responderˮ to nutritional oversupply may render it vulnerable to nutrient stress-induced insulin resistance. Even modest adipocyte insulin resistance will, due to the steepness of the insulin dose-response curve for lipolytic suppression, increase fatty acid delivery to liver and skeletal muscle and thereby promote insulin resistance in those tissues (see sect. V). The absolute importance of WAT insulin action for whole-body glucose homeostasis is powerfully demonstrated by the extreme, but reversible, insulin resistance of lipodystrophy (79, 406, 642, 714, 785). Far from being an inert storage depot, WAT may in fact be the linchpin of whole-body insulin sensitivity and resistance.
F. Integrated Physiology of Insulin Resistance and Experimental Considerations
Because insulin action and resistance are best understood in this integrated physiological context, experimental methods assessing insulin action require careful design and interpretation (18). The Himsworth “glucose-insulin tolerance test” described above (317) highlights a critical consideration in experimental tests of insulin action. To directly measure insulin resistance, the investigator needs to carefully distinguish insulin action and insulin secretion. This distinction is easily illustrated by the standard glucose tolerance test (GTT) (23, 33, 534). FIGURE 10 shows hypothetical results of an intraperitoneal GTT performed with mice fed a regular chow diet, mice fed a high-fat diet (HFD) for 8 wk, and nonobese diabetic (NOD) mice, a model of autoimmune insulitis, studied after the onset of insulitis but before the development of overt type 1 diabetes (358, 361). Although plasma glucose excursions are increased in both HFD-fed mice and NOD mice, examination of the plasma insulin time course reveals that the impaired glucose tolerance of the NOD mice represents β-cell dysfunction, not impaired insulin action. Conversely, the HFD-fed mice exhibit increased insulin levels accompanying their hyperglycemia, indicating that the primary cause of their impaired glucose tolerance is insulin resistance. Importantly, improved glucose tolerance can similarly be the result of either exaggerated insulin secretion or hypersensitivity to insulin action. The former situation can be modeled by treatment with an insulin secretagogue such as a sulfonylurea (688), and the latter by treatment with an insulin-sensitizing agent such as FGF21 (112, 401). In a prescient observation, Himsworth noted in his classic study (317) that for essentially the reasons outlined above, a glucose challenge without exogenous insulin would be insufficient to distinguish the two types of diabetics. In the modern setting, this ambiguity can be experimentally sidestepped by employing the hyperinsulinemic-euglycemic clamp method developed by Andres (33, 534). This technique largely negates insulin secretion differences by matching insulin levels among study subjects. At a constant, elevated insulin concentration, the glucose infusion rate needed to maintain euglycemia reflects whole-body insulin sensitivity (181). When combined with infusions of isotopically labeled glucose and glucose analogs to trace whole-body glucose turnover, endogenous glucose production (EGP), and tissue-specific glucose uptake, the hyperinsulinemic-euglycemic clamp represents the most powerful in vivo experimental assessment of insulin action in target tissues and is therefore widely used both in humans and animal models (33, 534, 760).
FIGURE 10.

Interpretation of glucose tolerance tests requires measurement of plasma insulin concentrations. A: diet-induced obese and prediabetic nonobese diabetic (NOD) mice both display glucose intolerance compared with lean chow-fed control mice, but the causes differ. The diet-induced obese mice mount a normal or even heightened insulin secretory response but are hyperglycemic owing to insulin resistance. The prediabetic NOD mice are glucose intolerant owing to defective insulin secretion. B: improved glucose tolerance can similarly result from increased insulin sensitivity [fibroblast growth factor 21 (FGF21)-treated mice] or from increased insulin secretion (sulfonylurea-treated mice).
Although accurate and useful, EGP measurements in hyperinsulinemic-euglycemic clamp studies have limitations. For one, although often assumed to be identical to HGP, EGP reflects the rate of appearance of glucose in the plasma from all sources, which in fasted subjects also includes a small contribution from renal gluconeogenesis. Furthermore, measurements of insulin suppression of EGP cannot distinguish between direct effects of insulin on liver glycogen synthesis and indirect effects of insulin on hepatic gluconeogenesis. Glycogen synthetic flux in liver, although perhaps a better readout of direct hepatic insulin action than EGP suppression, is more difficult to assess in vivo because, as discussed in section II, hepatic glycogen metabolism is exquisitely sensitive to glucose availability. Hyperglycemia, tightly matched between subjects, is necessary to accurately compare insulin stimulation of glycogen synthesis between experimental groups (640). Simple measurements of liver glycogen content after insulin stimulation require extensive fasting to minimize basal glycogen content; these extended fasts are often undesirable in rodents because they do not represent a normal physiological state (32, 33, 534). One method that has been successfully used to circumvent the problem of highly variable basal hepatic glycogen content is measurement of uniformly labeled 13C-glucose incorporation into hepatic glycogen during hyperinsulinemic-hyperglycemic clamps. Knowledge of the m+6 mole fraction in both liver glycogen and plasma glucose, and of the total hepatic glycogen concentration, permits calculation of the absolute rate of glycogen deposition via the direct pathway during the infusion (153, 645). Measurement of the dilution in the m+6 mole fraction from plasma glucose to hepatic UDP-glucose reveals the relative contributions of direct and indirect pathways to glycogen synthesis (645). Knowledge of both the absolute rate of direct pathway glycogen synthesis and the percent contribution of the direct pathway to total glycogen synthetic flux enables calculation of the total glycogen synthetic rate during a hyperinsulinemic-hyperglycemic clamp. In humans, noninvasive MRS approaches have been used to measure both glycogen synthetic and glycogenolytic fluxes (640, 641, 704, 832). However, it must be emphasized that insulin merely plays a permissive role in regulating hepatic glycogen synthesis; the key driver is the plasma (portal vein) glucose concentration. Without careful matching of the portal vein glucose concentration in all subjects, assessing the effect of insulin per se on hepatic glycogen metabolism is difficult.
Effective methods for in vivo assessment of insulin control of lipid metabolism are also available. Insulin suppression of lipolysis can be traced during hyperinsulinemic-euglycemic clamp studies using labeled palmitate and glycerol (620, 910). Insulin control of hepatic de novo lipogenesis is a transcriptionally mediated effect and therefore cannot be assessed in acute infusion studies, but effective tracer methods, such as deuterated water supplementation, enable measurement of DNL over a period of several days and reveal decreased DNL in insulin-resistant models (868, 910). A commonly employed protocol to measure insulin upregulation of the lipogenic transcriptional program is fasting (e.g., 24 h)–refeeding (e.g., 6 h). It is important to note, however, that insulin is not the only relevant variable in such studies; nutrient activation of mTOR is a major consideration as are changes in other hormones (452, 764).
One commonality of the various methods outlined above and in FIGURE 11 is the use of isotopic tracers to calculate flux through metabolic pathways. Although tracer studies are generally more resource-intensive than metabolite concentration studies, they are also likely to provide more relevant assessments of insulin action. Although advances in metabolomics have facilitated hypothesis generation through rapid, accurate, and automated measurement of hundreds of metabolites, insulin acts by modifying metabolic fluxes. These changes in flux may favor the accumulation or diminution of specific metabolites, but concentration differences are always a consequence of flux differences. It is ultimately flux measurements that have yielded and will continue to yield the most potent mechanistic insights to the integrated physiology of metabolism.
FIGURE 11.

Isotope tracer methods to assess insulin resistance in vivo. Six example methods are illustrated. The experimental protocol and mode of tracer delivery are italicized, the tracer is bolded, and the effect of insulin resistance is at bottom right. A: several glucose isotopomers can be used to trace whole-body glucose turnover, Rd. During a hyperinsulinemic-euglycemic clamp, where Rd and the glucose infusion rate F are both known, endogenous glucose production can be calculated by subtracting F from Rd. Insulin suppression of endogenous glucose production is impaired in insulin-resistant subjects. B: under hyperinsulinemic-euglycemic clamp conditions, 70–80% of Rd is accounted for by skeletal muscle glucose uptake, so skeletal muscle insulin resistance is often accompanied by decreased Rd. C: the nonmetabolizable glucose analogue 2-deoxyglucose (2-DG) is phosphorylated and trapped inside tissues which lack glucose-6-phosphatase (e.g., skeletal muscle and adipose tissue, but not liver). Tissue 2-DG-6-phosphate levels can thus be used to estimate insulin-stimulated glucose uptake, which is decreased in insulin resistance. D: the negligible natural abundance of m+6 glucose makes [U-13C]glucose a useful tracer of hepatic glycogen synthesis, which is decreased in hepatic insulin resistance. To stimulate net hepatic glycogen synthesis, both hyperinsulinemia and hyperglycemia are necessary. E: the incorporation of deuterated or tritiated water into hepatic palmitate yields a measurement of hepatic de novo lipogenesis (DNL) but requires several days of administration to reach isotopic steady state. DNL is decreased in some models of insulin resistance, such as the high-fat-fed rodent. Because insulin regulation of DNL is a slow, transcriptionally mediated process, this method is compatible with the physiology being studied. F: several palmitate and glycerol tracers, including [U-13C]palmitate and [1,1,2,3,3-2H]glycerol, can be used to trace lipolysis. Insulin suppression of lipolysis from white adipose tissue is impaired in adipose insulin resistance. Notably, Ra glycerol under fasting conditions is likely a more accurate measure of lipolysis than Ra palmitate, because palmitate can be re-esterified within the adipocyte.
G. Insulin Resistance: The ‟Whatˮ and the ‟Whyˮ
Section IV has attempted to describe the central characteristics of insulin resistance in the skeletal muscle, liver, and WAT, focusing on the effectors of insulin signaling and the direct metabolic effects of insulin that become insulin resistant. There are certainly more pathophysiological attributes of insulin-resistant tissue than are described here; insulin signaling interfaces with diverse cellular functions, and myriad signaling pathways have been identified as altered in insulin resistance. Additionally, each of the more than 20 well-characterized animal models of obesity and T2D exhibits a distinct pathophysiology of its metabolic disease that has some overlap with human obesity-associated T2D but certainly fails to recapitulate all aspects of the human disease it attempts to model (412).
A challenge for investigators studying a process with such protean manifestations as insulin resistance is to distinguish cause from effect, primary defect from secondary consequence. In sections V–VII, we review efforts to pinpoint the initial insults responsible for the impairments in insulin action described in this section. It is not enough for investigators to identify candidate genes or metabolites whose activity or levels are altered in the insulin-resistant state. Perturbations assessing whether a candidate mediator is necessary, sufficient, or both necessary and sufficient for insulin resistance are required to infer causality. Human studies are also required to interrogate whether a candidate pathway is operative in typical obesity-associated human insulin resistance and thus might be a viable therapeutic target. In rodent studies, approaches seeking to identify the earliest defects in insulin action in response to overnutrition are the most likely to yield insights into the mechanistic basis of insulin resistance (548, 721, 854); efforts to characterize the myriad derangements of chronic metabolic disease may yield therapeutic targets but are unlikely to elucidate the root cause of cellular insulin resistance (788). It is important to acknowledge that a defect which is a secondary consequence of insulin resistance may nonetheless further exacerbate insulin resistance and therefore be a viable therapeutic target. Here, however, we focus primarily on efforts to define the primary insult(s) of insulin resistance; the underlying assumption is that if these primary defects can be identified and ameliorated, then all secondary consequences will be prevented. We begin by reviewing the oldest and most extensively investigated hypothesis of insulin resistance: defective insulin action as a direct consequence of altered lipid metabolic fluxes.
V. LIPID-INDUCED INSULIN RESISTANCE
A. Introduction to Lipid-Induced Insulin Resistance
The phenomenon of lipid-induced insulin resistance was perhaps first described in a 1941 report describing insensitivity to insulin-induced hypoglycemia after intravenous lipid infusion in rabbits (940). In the early 1960s, a series of reports linked elevated NEFA or ketone body levels to impaired insulin-stimulated glucose uptake (257, 677, 765, 904). These findings were synthesized by Randle and co-workers (677–679), who proposed a glucose-fatty acid cycle controlling oxidative substrate selection in skeletal and cardiac muscle. The glucose-fatty acid cycle hypothesis posited that oxidative substrate selection followed a reciprocal relationship controlled by substrate supply: fatty acid availability promotes fat oxidation while inhibiting glucose oxidation, and vice versa (676). The proposed mechanisms were allosteric in nature: increased fat oxidation drives accumulation of mitochondrial acetyl CoA and NADH, which inhibit PDH to limit entry of pyruvate into the mitochondrion for oxidation. Increased cytoplasmic citrate would also slow glycolytic flux through allosteric feedback inhibition of phosphofructokinase-1. The resultant increase in glucose-6-phosphate would in turn allosterically inhibit hexokinase and lead to an increase in intramyocellular glucose concentration.
The Randle glucose-fatty acid cycle has been extensively studied using the most straightforward and popular experimental model of lipid-induced insulin resistance: the acute lipid infusion. Such studies typically use a 20% triglyceride emulsion with heparin added to activate lipoprotein lipase and further raise plasma NEFA concentrations. These infusions effectively induce insulin resistance to muscle glucose uptake–though only after several hours of infusion–and afford the experimenter full control over the duration and extent of lipid exposure (68, 198, 229, 285, 464, 698, 836). Although this technique can be used to achieve supraphysiologic plasma NEFA concentrations of 2 meq/l or greater, even infusions targeting high physiological concentrations such as 0.75 meq/l are able to impair insulin-stimulated glucose uptake (68). The acute lipid infusion is an attractive experimental model for investigating mechanisms of lipid-induced insulin resistance in large part because it circumvents the confounding physiological compensations seen in models of chronic dietary lipid-induced insulin resistance–basal hyperinsulinemia, inflammation, altered body composition.
MRS studies of skeletal muscle glucose metabolism during acute lipid infusions have enabled in vivo testing of the Randle hypothesis (FIGURE 12). Specifically, MRS measurements of intramyocellular glucose-6-phosphate (G6P) and glucose concentrations, as well as glycolytic and glycogen synthetic rates, directly tested the Randle prediction that lipid oxidation would decrease glycolytic flux and increase G6P concentrations. These studies revealed that contrary to the increases predicted by the Randle hypothesis, G6P and glucose levels actually decreased in acute lipid-induced insulin resistance (198, 698). Glycolysis and glycogen synthesis also decreased, but this owed to impaired glucose transport rather than Randle allostery (198, 698). These findings dovetailed with the elucidation of the molecular mechanisms connecting insulin receptor activation to GLUT4 translocation and observations that GLUT4 translocation and glucose transport were defective in insulin resistance and T2D (152, 258, 633, 847). A key implication for future mechanistic studies of lipid-induced muscle insulin resistance was the requirement that they link lipid exposure to inhibition of insulin signaling.
FIGURE 12.

The Randle hypothesis and lipid-induced skeletal muscle insulin resistance. A: Randle and co-workers proposed that lipid-induced impairments in muscle glucose oxidation are secondary to substrate competition with fatty acids. Increased fatty acid oxidation would increase the mitochondrial acetyl CoA/CoA and NADH/NAD+ ratios. This would allosterically inhibit pyruvate dehydrogenase (PDH) and phosphofructokinase (PFK; through increases in its allosteric inhibitor citrate), decreasing glycolytic flux. The ensuing increase in glucose-6-phosphate concentration would in turn allosterically inhibit hexokinase (HK), leading to increased intracellular glucose concentrations. B: measurements of intramyocellular glucose and glucose-6-phosphate during acute lipid infusions revealed that concentrations of these metabolites are decreased, not increased, in this setting. Together with studies linking muscle insulin resistance with impaired GLUT4 translocation, these data implicated impaired glucose transport as the chief defect in lipid-induced muscle insulin resistance.
Other in vivo models have challenged the relevance of the glucose-fatty acid cycle to lipid-induced insulin resistance. For example, mice lacking Pdk2 and Pdk4 (Pdk2/4 DKO) have constitutively dephosphorylated, active PDH in skeletal muscle and therefore preferentially oxidize glucose in an inflexible manner (669). The glucose-fatty acid cycle is thus inoperative in Pdk2/4 DKO mice. However, these mice develop profound muscle insulin resistance associated with increased intramyocellular lipid, implying that the glucose-fatty acid cycle is not necessary for lipid-induced muscle insulin resistance (669).
Overall, available evidence indicates that the glucose-fatty acid cycle accounts neither for the pronounced impairment in insulin-stimulated muscle glucose uptake that develops after several hours of lipid infusion (68, 71, 698), nor for the insulin resistance of human obesity/T2D. However, the glucose-fatty acid cycle is undoubtedly nevertheless a physiologically relevant process for controlling oxidative substrate selection (244). For example, in the first 3 h of an acute lipid infusion, before profound insulin resistance develops, intramyocellular G6P concentrations are increased and glycolytic flux is decreased, matching the predictions of the Randle hypothesis (372, 373). The allosteric mechanisms proposed by Randle are likely sufficient to explain this phenomenon. The reciprocal control mechanism–inhibition of fatty acid oxidation by glucose through malonyl CoA inhibition of carnitine palmitoyltransferase-1–highlights the integrated nature of this substrate-selection circuit (533). The glucose-fatty acid cycle can thus be thought of as a cell-autonomous response to whole-body control of substrate availability, enabling efficient oxidation of glucose in the fed state and fatty acids in the fasted state.
The broad relevance of the lipid infusion model of muscle insulin resistance is highlighted by the well-established association between increased plasma NEFA turnover and insulin resistance in humans (134, 245, 266, 632, 684, 685, 816). However, the increment in plasma NEFA turnover and/or concentrations observed in human insulin resistance are modest (632). In addition to often achieving supraphysiological NEFA concentrations, the acute intravenous lipid exposure fails to accurately model obesity-associated insulin resistance in several other obvious ways: the lipid exposure is acute rather than chronic, the lipid delivery is intravenous rather than enteral or from liver/adipose tissue, and plasma fatty acid concentrations are static rather than dynamic. As a result, the acute lipid exposure is typically only employed in direct mechanistic investigations of lipid-induced insulin resistance. Most studies testing the effect of a given experimental perturbation on tissue insulin sensitivity appropriately use chronic interventions.
The most popular model of typical, obesity-associated, lipid-induced insulin resistance is the high-fat-fed rodent. While mouse and rat strains significantly differ in their susceptibility to diet-induced obesity and insulin resistance, susceptible strains such as the C57BL/6J mouse and the Sprague-Dawley rat provide tractable model systems (239, 548, 898). However, although these rodent strains may broadly mimic humans in their susceptibility to obesity, there are several key physiological differences to bear in mind when interpreting studies of fat-fed rodents (428). For example, short-term fat feeding protocols in rodents can produce marked hepatic lipid accumulation and consequent hepatic insulin resistance without appreciable skeletal muscle insulin resistance (721, 722, 854), but in humans with the metabolic syndrome skeletal muscle insulin resistance is thought to precede hepatic insulin resistance (182, 639, 725). The remarkable propensity for some mouse strains to develop obesity and glucose intolerance with high-fat feeding while others remain lean and metabolically healthy highlights the integrated nature of glucose homeostasis, with energy expenditure, pancreatic function, and tissue insulin sensitivity all serving important roles in generating the phenotype (428). Some commonly used rodent models of diet-induced obesity, including the ob/ob and db/db mice and the Zucker fa/fa rat, leverage naturally occurring mutations in the leptin satiety axis to achieve marked hyperphagia and consequent obesity. These models can be useful, but must be interpreted with the knowledge that leptin is far more than simply a satiety switch (341, 801) and that the consequences of the mutation are protean. Additionally, the hyperphagic rodent models are typically studied as morbidly obese adults, long after the development of insulin resistance. Their phenotype thus renders them unsuitable for studies attempting to define the initial insult(s) of insulin resistance. For such studies, the high-fat-fed rodent is useful because it can be maintained with a normal body composition and metabolic phenotype until the experimental challenge is begun. However, both the high-fat-fed rodent and the genetically hyperphagic rodent share one key phenotypic similarity with human obesity-associated insulin resistance: increased ectopic lipid deposition in liver and skeletal muscle. The ectopic lipid hypothesis of liver and muscle insulin resistance is described below and in FIGURE 13.
FIGURE 13.

Ectopic lipid hypothesis of liver and muscle insulin resistance. The central organizing principle is that lipid storage in white adipose tissue, especially in subcutaneous depots, is physiologically appropriate, but lipid storage in liver and skeletal muscle is inappropriate and metabolically harmful. Lipid accumulation at these ‟ectopicˮ sites is associated with insulin resistance. Human phenotypes are listed above silhouettes, and corresponding mouse models, described in the main text, are below.
One of the most reproducible associations in human insulin resistance is with intrahepatic triglyceride (IHTG) (96, 627). NAFLD, defined as increased IHTG without excessive alcohol intake, is the most common liver disease in industrialized nations (870). Approximately two-thirds of obese people, and nearly all obese people with T2D, have NAFLD (45, 772, 843, 883). Similarly, IHTG is an exceptionally strong predictor of insulin resistance (106, 422, 520, 728). Causal evidence for a role of IHTG in hepatic insulin resistance derives from both human and rodent studies. In diabetic humans, decreasing IHTG content by modest weight loss normalizes insulin suppression of hepatic glucose production and fasting glycemia without significant peripheral effects (637). Although visceral fat has been proposed to contribute to hepatic insulin resistance, it is more likely a good biomarker for IHTG (260); IHTG is a much stronger predictor of hepatic insulin resistance and surgical removal of omental fat without concomitant reduction in IHTG does not correct hepatic insulin resistance (220, 221). Furthermore, lipodystrophic individuals have profound increases in IHTG, which is associated with severe hepatic insulin resistance in the absence of any visceral fat (536). Visceral fat is also a quantitatively small contributor to whole-body fatty acid turnover (388). In people with NAFLD but without T2D, ameliorating NAFLD decreases T2D risk even after controlling for changes in weight (933). Even a single oral fat bolus in humans is sufficient to increase IHTG and impair hepatic insulin sensitivity (313).
A corollary proposition of the finding that IHTG, not adipose tissue mass, predicts insulin resistance is that redistribution of fat from liver to WAT should correct insulin resistance. Indeed, numerous models support this proposition. In severely insulin-resistant lipodystrophic A-ZIP/F-1 mice, transplantation of WAT normalizes liver insulin action associated with marked decreases in IHTG and intramyocellular lipid (262, 406). Lipodystrophic humans with severe NAFLD and hepatic insulin resistance, as well as lipodystrophic mice, achieve massive reductions in IHTG and normalization of hepatic insulin action when treated with leptin (79, 642, 763). This ‟redistributionˮ hypothesis extends beyond models of lipodystrophy. Mice with enhanced adipocyte insulin sensitivity owing to inducible Pten deletion accumulate fat in adipose tissue rather than liver when fed a high-fat diet and are totally protected from diet-induced hepatic insulin resistance (555). Adipose-specific overexpression of Pck1 (PEPCK) in mice facilitates glyceroneogenesis and re-esterification of fatty acids within the adipocyte, decreasing plasma NEFA levels; consequently, these mice are obese but insulin-sensitive in liver and skeletal muscle (242). Redistribution of fat has also been proposed to mediate the insulin-sensitizing effects of the thiazolidinedione class of antidiabetic drugs, which promote adipogenesis through activation of PPARγ (723). Perhaps most impressively, adiponectin overexpression in leptin-deficient ob/ob mice (Ad Tg ob/ob) results in massive redistribution of fat such that the Ad Tg ob/ob mice weigh up to twice as much as obese ob/ob controls yet display reduced IHTG (408). Remarkably, these enormous mice display fasting glucose and insulin levels similar to lean chow-fed mice and have normal insulin sensitivity (408). In a dramatic example of reverse redistribution, adipose-specific re-expression of the leptin receptor in db/db mice prevents obesity but results in IHTG accumulation, hyperinsulinemia, and the development of diabetes at 4 wk of age, compared with 14 wk in the db/db controls (878).
A variety of interventional models support the hypothesis that IHTG is casually related to hepatic insulin resistance. Mice ectopically expressing malonyl CoA decarboxylase in liver are protected from hepatic lipid accumulation upon high-fat feeding, associated with preserved hepatic insulin sensitivity (24). Similar phenotypes have been observed in mouse models as diverse as estrogen-treated ovariectomized female mice (111) and mice treated with FGF21 (112). Two independent studies have reported that overexpression of the triacylglycerol hydrolase CES2 in db/db or high-fat-fed mice reduced IHTG in conjunction with improved glucose tolerance (495, 705). Furthermore, mice with liver-specific overexpression of SREBP-1c display liver-specific triglyceride accumulation and liver-specific insulin resistance (368). Perhaps the most direct tests of the relationship between IHTG and hepatic insulin action in vivo have been carried out using the mitochondrial protonophore 2,4-dinitrophenol (DNP) to rapidly decrease IHTG stores. In rats with isolated hepatic insulin resistance produced by short-term fat feeding, DNP treatment normalizes both IHTG and hepatic insulin action (721). The latter result has been extended using pharmacologically distinct forms of DNP to show drastic improvements in both IHTG and hepatic insulin action in more extreme forms of hepatic insulin resistance including the streptozotocin-induced diabetic rat, the Zucker diabetic fatty rat, lipodystrophic mice (3), and rats fed a methionine/choline-deficient diet to induce nonalcoholic steatohepatitis (621, 629).
Lipoprotein lipase (LpL) acts locally at target tissues to release fatty acids from circulating triglycerides for tissue uptake. Experiments modulating LpL activity have enabled instructive testing of the hypothesis that hepatic lipid accumulation is causally linked to hepatic insulin resistance. Although LpL is not endogenously expressed in liver, hepatic overexpression of LpL is sufficient to increase IHTG and cause liver-specific insulin resistance (404). Antisense oligonucleotide inhibition of the adipose-derived LpL inhibitor angiopoietin-like 8 (Angptl8) or adipose-specific deletion of the LpL inhibitor angiopoietin-like 4 (Angptl4) in rodents facilitates WAT lipid uptake and prevents IHTG accumulation and hepatic insulin resistance (28, 867). Antisense oligonucleotide inhibition of the LpL activator ApoA5 in mice generates an interesting phenotype in which plasma triglycerides are threefold elevated compared with controls but IHTG is decreased and hepatic insulin action is preserved upon high-fat feeding (113). In lean South Asian men, a common variant in the gene encoding apolipoprotein C3 (APOC3), also an LpL inhibitor, results in higher plasma ApoC3 levels, hypertriglyceridemia, and predisposes them to increased IHTG, and hepatic insulin resistance at a relatively low body mass index (638). Similarly, mice overexpressing apolipoprotein C3 (ApoC3 Tg mice) have similar body composition to wild-type controls but display increased hepatic triglyceride and hepatic insulin resistance (462).
Despite this preponderance of data from both human and rodent studies supporting a causal link between IHTG and hepatic insulin resistance, several intriguing studies have dissociated the two (224). In humans, the common PNPLA3 I148M mutant allele is strongly associated with IHTG, but studies disagree with respect to whether the polymorphism is associated with insulin resistance (386, 607, 700, 875). Mice homozygous for a knockin I148M allele displayed normal glucose tolerance despite increased IHTG on a high-sucrose diet, although hepatic insulin action was not directly assessed (778). Interestingly, the Pnpla3I148M knockin mice did not develop worsened hepatic steatosis on a high-fat diet compared with wild-type controls and accordingly displayed similar glycemia, fasting insulin, and glucose (in)tolerance (778). Other rodent models have challenged the IHTG-hepatic insulin resistance connection as well. Various mice with liver-specific disruptions in insulin signaling display, by definition, insulin resistance, yet do not develop fatty liver; this dissociation does not, however, pose a logical challenge to the hypothesis that IHTG causes hepatic insulin resistance (811). Several other mice with defects in hepatic fatty acid oxidation accumulate IHTG yet are hypoglycemic upon fasting with high insulin sensitivity and glucose tolerance; these models highlight the well-established role of fatty acid oxidation in supporting gluconeogenesis which confounds metabolic phenotyping (216, 399, 587). The same problem of physiological confounding applies to mice with defects in gluconeogenesis, which have a similar phenotype (273, 563, 829, 882, 952). A particularly interesting mouse model that also dissociates IHTG from hepatic insulin resistance is the liver-specific Scap−/− mouse (550). The SCAP protein is required for SREBP processing; Scap ablation blocks the SREBP lipogenic program (550). Scap deletion in ob/ob mice normalized IHTG but only slightly improved fasting glucose and insulin concentrations (550). When Scap was inducibly deleted in liver followed by 16 wk of high-fat feeding, IHTG did not increase during the HFD but fasting glucose and insulin levels were similar to those in wild-type fat-fed controls (550). Although hepatic insulin action was not directly assessed, these models are provocative and indicate that in the setting of chronic overnutrition, reversal or prevention of hepatic lipid accumulation is likely not sufficient to overcome the many other forms of metabolic dysregulation–especially, perhaps, adipose insulin resistance and excess lipolysis–that accompany such states.
The link between skeletal muscle lipid accumulation and insulin resistance is more complex and controversial than in liver. In the late 1990s, pioneering 1H-MRS studies revealed that in normal-weight, nondiabetic humans, intramyocellular lipid (IMCL) was an extremely strong predictor of insulin sensitivity during hyperinsulinemic euglycemic clamps, stronger even than plasma NEFA concentration (438). In nondiabetic Pima Indians, muscle triglyceride, but not body mass index, was negatively correlated with whole-body glucose uptake (610). Additionally, the correction of muscle insulin resistance–in lipodystrophic A-ZIP/F-1 mice by WAT transplantation or in lipodystrophic humans by leptin treatment–is associated with marked decreases in muscle triglyceride (406, 642). In addition, mice lacking LpL selectively in skeletal muscle (and thus unable to use plasma triglycerides as fuel) are protected from muscle insulin resistance (876). In the converse experiment, mice overexpressing LpL selectively in skeletal muscle display increased IMCL and develop skeletal muscle insulin resistance (404). However, assigning a causal role to IMCL in muscle insulin resistance is complicated by the long-appreciated “athlete’s paradox,” wherein highly trained, insulin-sensitive endurance athletes display IMCL levels greater than or equal to levels observed in people with T2D (275, 488, 560). Although the pathways by which T2D and endurance athletes develop increased IMCL stores differ markedly, the athlete’s paradox provides evidence against a causal role for stored triglyceride in the pathogenesis of muscle insulin resistance (866e).
The proposition that lipid accumulation in nonadipose tissues is causally associated with insulin resistance in those tissues is known as the ectopic lipid hypothesis of insulin resistance. Although, as discussed below, the ectopic lipid hypothesis implicates tissue- and cell-autonomous processes in nonadipose tissues, adipose tissue also plays a central role in ectopic lipid-induced insulin resistance. If all excess energy could be stored in WAT, ectopic lipid-induced insulin resistance could not develop. The transgenic adiponectin-overexpressing ob/ob mouse discussed above, which weighs up to twice as much as the obese ob/ob mouse yet retains normal glucose tolerance, is a dramatic example of this principle (408). Indirect human genetic evidence for the ectopic lipid hypothesis includes the strong association of genomic variants limiting peripheral adipose storage capacity with insulin resistance as assessed by multiple parameters including fasting insulin, euglycemic clamp data, and glucose tolerance testing (501).
Recognizing that stored triglyceride per se was unlikely to directly impair cellular insulin action, researchers have long sought to identify lipid moieties that could mechanistically link lipid accumulation and insulin resistance (766). We now examine the three such lipid classes that have received the most attention: diacylglycerol, ceramides, and acylcarnitines.
B. The DAG-Novel Protein Kinase C Axis and Insulin Resistance
Early work linking DAG to insulin resistance preceded the development of the MRS methods that linked intracellular lipid content to insulin resistance. Rather, these initial investigations derived from the observation that phorbol esters, tumorigenic analogues of sn-1,2-DAG, caused insulin resistance in vitro and in vivo (821, 822, 866c). This observation prompted measurements of DAG in muscle, heart, and liver, which revealed increases in all tissues in obese or diabetic rats (593, 852). As the penultimate intermediate in triacylglycerol (TAG) synthesis, DAG levels track with TAG levels in muscle and liver from nearly all rodent and human models with intact lipid handling (112, 113, 445, 621, 627, 629, 669, 724, 767). As a consequence, the associations between IMCL/muscle insulin resistance and IHTG/hepatic insulin resistance discussed above are also associations between DAG and insulin resistance. To date, five human studies have observed significant positive correlations between hepatic DAG and hepatic insulin resistance, as measured by either HOMA-IR (445, 506, 705) or suppression of HGP during hyperinsulinemic-euglycemic clamp studies (512, 834).
Because DAG was known to be a bioactive signaling lipid, requisite for activation of protein kinase C (PKC), attention soon turned to the potential role of PKC activity in insulin resistance. However, early work in this field was hampered by incomplete knowledge of the several classes and isoforms of PKC present in mammalian cells, as well as by highly nonspecific pharmacological inhibitors. As a result, literature in this field from the 1980s and early 1990s contains contradictory reports describing both stimulatory and inhibitory effects of PKCs on insulin action (160, 353–355, 387, 558, 866c).
Advances in molecular biology and careful biochemical characterization gradually facilitated the separation of PKCs into three major classes: conventional PKCs (cPKC; isoforms α, β, γ) require both Ca2+ and DAG for full activation, novel PKCs (nPKC; isoforms δ, ε, θ, η) require only DAG, and atypical PKCs (aPKC; isoforms ζ, λ, ι) require neither Ca2+ nor DAG (799). Conventional PKC isoforms are activated rapidly and participate largely in phospholipase C-mediated signaling, which produces the parallel spikes in Ca2+ and DAG necessary for full cPKC activation (580, 799). In contrast, nPKCs display a slow, sustained activation by DAG, a consequence of a single W/Y amino acid replacement in the DAG-binding C1 domains of nPKCs versus cPKCs that lends nPKCs twofold greater affinity for DAG (199). This property appears a priori to position nPKCs as the most suitable PKC isoforms to mediate the insulin resistance of chronic cellular lipid accumulation.
Indeed, activation of nPKCs has been consistently observed in insulin-resistant skeletal muscle and liver. Skeletal muscle from high-fat-fed rats displayed translocation of PKCε and PKCθ, but not PKCα or PKCζ; this study was also notable for identifying a positive linear relationship between muscle TAG and DAG content and PKCθ translocation (739). Skeletal muscle PKCθ and PKCε translocation were subsequently measured in obese, insulin-resistant, and/or diabetic rat muscle by several groups and found to be increased in some (285, 348, 356, 456, 667), but not all (357), models. In human studies, PKCθ (819) and PKCε (619) translocation have both been found to be increased in T2D muscle compared with lean controls. Efforts to identify and characterize PKC isoforms involved in hepatic insulin resistance revealed a somewhat different picture than in muscle. PKCθ is not significantly expressed in hepatocytes, so investigators pursued other isoforms. Early work in obese people with T2D and Zucker diabetic fatty rats revealed increases in membrane-associated PKCε and PKCζ compared with lean controls (161). Intralipid-heparin infusions in the rat were reported to increase PKCδ translocation in liver, although translocation of other PKC isoforms was not reported (450). In 2004, Samuel et al., using the 3-day high-fat-fed rat as a model of acute hepatic insulin resistance, measured translocation of all PKC isoforms highly expressed in liver–α, β, δ, ε, and ζ–and found that only PKCε translocation was increased in this model (721). Hepatic PKCε translocation has subsequently been observed in dozens of high-fat-fed rodent models (646). The finding of isolated PKCε activation was later replicated in liver biopsies from obese humans (445). Interestingly, PKCε translocation was strongly correlated with both hepatocellular DAG content and insulin resistance as assessed by HOMA-IR, the only PKC isoform of six tested to show this relationship (445). However, increased mRNA expression of both PKCε and PKCδ has also been reported in obese human liver (59).
One approach to determine whether nPKCs cause tissue insulin resistance has relied on correlation, demonstrating that upon an intervention, changes in insulin sensitivity track with changes in nPKC translocation. Treatment of fat-fed rats with the thiazolidinedione insulin sensitizer rosiglitazone decreased muscle DAG content, and nPKC translocation decreased in parallel (742). Similarly, feeding a single low-fat meal to chronically high-fat-fed rats decreased PKCθ (and not PKCε) translocation in muscle and normalized insulin-stimulated muscle glucose uptake (46). Intermittent fasting in diabetes-prone NZO mice prevented the development of hyperglycemia, associated with reduced skeletal muscle DAG, reduced hepatic DAG, and reduced PKCε activation compared with ad lib-fed controls (39). The acute muscle insulin resistance of lipid/heparin infusion has also been a productive model system for this line of inquiry. A salient and highly reproducible feature of acute lipid-induced muscle insulin resistance is that it requires a time delay of 3–5 h to take effect (71, 942). In one study, acute lipid infusion impaired insulin signaling and glucose uptake in muscle in temporal parallel with marked translocation of PKCθ, but not PKCε (285). In another, the 4–5 h of lipid infusion needed to induce insulin resistance correlated with peaks in intramyocellular long-chain acyl CoA, DAG content, and PKCθ activation, but not with ceramide or triglyceride content (942).
Studies investigating the dynamic reciprocal relationship between nPKC translocation and insulin sensitivity have also been pursued in humans. Acute lipid administered either orally or parenterally reduced insulin-stimulated muscle glucose uptake and was accompanied by increased PKCθ translocation in muscle (585, 819). The delayed and transient rise in myocellular DAG during acute lipid infusions observed in rodents has also been observed in humans (819).
These correlative studies provided strong impetus for further testing of the DAG/nPKC hypothesis using the power of rodent genetic manipulation. Many such models have been used to this end, with somewhat varying results. These can broadly be divided into models testing the necessity and/or sufficiency of DAG for lipid-induced insulin resistance and models testing the necessity and/or sufficiency of nPKCs for lipid-induced insulin resistance. We now consider these two major categories sequentially.
In the Kennedy pathway, the major route for triacylglycerol synthesis in liver and muscle, fatty acyl-CoA moieties are sequentially added to sn-glycerol-3-phosphate to form first lysophosphatidic acid via glycerol-3-phosphate acyltransferase (GPAT) and then phosphatidic acid via acylglycerophosphate acyltransferase (AGPAT). The phosphate at the 3-position is removed by phosphatidic acid phosphatase (PAP, also known as lipin) to yield sn-1,2-diacylglycerol (DAG), which is finally acylated by diacylglycerol acyltransferase (DGAT) to produce triacylglycerol (TAG). This pathway provided a logical launching point to test the DAG hypothesis of cellular insulin resistance by genetic modification. Mice lacking mitochondrial GPAT (mtGPAT), a key hepatic GPAT, displayed increased fatty acyl-CoA levels and decreased hepatic DAG and TAG levels after high-fat feeding, as predicted (575). The decrease in hepatic DAG was accompanied by decreased PKCε translocation (575). Consistent with the DAG/nPKC hypothesis, mtGPAT−/− mice were protected from HFD-induced hepatic insulin resistance as demonstrated both by enhanced HGP suppression during hyperinsulinemic-euglycemic clamp studies and by increased hepatic insulin signaling: IRS2-associated PI3K activity and AKT activity (575). Similarly, mice with adenoviral hepatic overexpression of mtGPAT displayed increased hepatic DAG, PKCε translocation, and hepatic insulin resistance (567). Whole-body deletion of either AGPAT2 or lipin-1 causes profound lipodystrophy in mice and humans, confounding efforts to test the DAG/nPKC hypothesis with these models (170, 634). Analysis of mice with germline deletion of the major hepatic lipin, lipin-2, is also confounded by compensatory lipin-1 upregulation (205). However, acute shRNA-mediated lipin-1 or lipin-2 knockdown in HFD-fed mice improves glucose tolerance and decreases hepatic DAG and TAG content (707, 708). Similarly, acute adenoviral lipin-2 overexpression in liver is sufficient to impair glucose tolerance, associated with increased hepatic DAG and TAG (708). Mice overexpressing lipin-1 in skeletal muscle develop profound obesity and insulin resistance, although the cellular mechanisms responsible in this model were not completely defined (649). Finally, several genetic perturbations of DGAT in mice have yielded somewhat conflicting results. Mice overexpressing DGAT1 in skeletal muscle display the expected increase in myocellular TAG and decrease in myocellular DAG and are protected from HFD-induced insulin resistance (491). Although Dgat1−/− mice are unexpectedly protected from HFD-induced obesity and insulin resistance, this may owe to increased energy expenditure; isolated lipid-treated soleus muscles from Dgat1−/− mice displayed blunted insulin-stimulated 2-deoxyglucose uptake, consistent with the DAG/nPKC hypothesis (491, 782). DGAT2 perturbations also yield complex phenotypes. Muscle-specific DGAT2-overexpressing mice exhibit decreased DAG (only in relatively insulin-insensitive glycolytic muscle fibers, however) and surprisingly display modest glucose intolerance, associated with increased ceramide content (473). In contrast, mice overexpressing DGAT2 in liver exhibit increases in both TAG and, unexpectedly, DAG content (371, 547). These mice have increased hepatic PKCε translocation and develop severe hepatic insulin resistance, as evidenced by profoundly impaired HGP suppression during hyperinsulinemic-euglycemic clamp studies (371). Although one of the two studies performed on the liver-specific DGAT2-overexpressing mice did not observe differences in clamp EGP between the transgenic mice and wild-type controls, interpretation of those experiments is difficult because both the control and the transgenic mice displayed clamp EGP rates consistent with hepatic insulin resistance (547). Consistent with this, high-fat-fed rats with antisense oligonucleotide-mediated hepatic knockdown of DGAT2 displayed decreases in liver DAG and PKCε translocation associated with improved hepatic insulin sensitivity (144). Although not part of the Kennedy pathway, monoacylglycerol acyltransferases (MGAT) are another lipogenic source of DAG. Mgat1 was found to be a PPARγ-regulated driver of hepatic steatosis; Mgat1 knockdown by adenoviral shRNA or antisense oligonucleotide decreased hepatosteatosis and improved glucose tolerance in HFD-fed mice (466, 791). However, a second study using the MGAT1 ASO found that the improved glucose tolerance was not accompanied by decreased IHTG in HFD-fed or ob/ob mice but unexpectedly was accompanied by increased DAG (298). Although the PKCε membrane/cytosol ratio was not formally determined in this study, membrane PKCε content was decreased by MGAT1 ASO, suggesting that while total hepatic DAG was increased, the DAG pool available for PKCε activation was not (298). Interestingly, rates of de novo lipogenesis were decreased in the MGAT1 ASO-treated mice despite increased DAG levels, suggesting that lipogenic flux may be more important than steady-state DAG levels in mediating insulin resistance (791). In summary, mouse models perturbing the lipogenic pathway (FIGURE 14) are frequently complicated by unexpected physiological compensations but are nevertheless generally consistent with the DAG/nPKC hypothesis of lipid-induced insulin resistance.
FIGURE 14.

Rodent models of perturbed hepatic triglyceride synthesis. Various genetic loss-of-function and gain-of function rodent models have been generated to test the role of diacylglycerol (DAG) in lipid-induced hepatic insulin resistance. These models are broadly consistent with the DAG/protein kinase C (PKC)ε hypothesis of lipid-induced hepatic insulin resistance. TAG, triacylglycerol; HFD, high-fat diet, KO, knockout. See text for details and references.
Several other models of genetically altered lipid metabolism address the DAG/nPKC hypothesis. Mice lacking the fatty acid elongase Elovl6 were susceptible to hepatosteaosis when fed a HFD, but did not display increased hepatic DAG or PKCε translocation and were protected from HFD-induced hepatic insulin resistance (527). Perturbations in diacylglycerol kinase (DAGK), which converts DAG to PA (the reverse reaction of lipin), have also been studied. There are 10 mammalian DAGK isoforms, with different substrate specificities, tissue expression profiles, and subcellular localizations, and data regarding their roles in lipid metabolism and insulin resistance are only beginning to emerge (50, 517, 518, 571, 573). DAGKδ was found to be decreased in subjects with T2D, and Dgkd+/− mice displayed increased myocellular DAG and impaired muscle insulin signaling and insulin-stimulated glucose uptake (137). Additionally, DAGKζ knockdown impaired insulin-stimulated GLUT4 translocation in 3T3-L1 adipocytes (492). Curiously, DAGKζ knockout mice exhibited improvements in muscle insulin sensitivity on a HFD despite elevated muscle DAG, but these mice also suffered from growth defects, reduced adiposity, and altered fuel selection, complicating interpretation of their phenotype (50). The physiological consequences of DAGKε ablation are also uncertain, with one group reporting protection from, and another reporting increased susceptibility to, HFD-induced glucose intolerance (518, 571).
Mice with genetic perturbations in triglyceride lipolysis have yielded diverse and fascinating phenotypes. Overexpression of a TAG hydrolase, carboxylesterase 2 (CES2), reduced hepatic DAG in high-fat-fed mice in association with improved glucose tolerance (705). Adipose-specific Atgl−/− mice provide another example of a model consistent with the DAG hypothesis; these mice displayed blunted adipose fatty acid oxidation and decreased hepatocellular DAG, accompanied by improved insulin suppression of hepatic glucose production during hyperinsulinemic euglycemic clamps (12). Similarly, adenoviral overexpression of Atgl in HFD-fed mouse liver reduced hepatic DAG and improved hepatic insulin signaling (855). Liver-specific Atgl−/− mice develop profound hepatosteatosis but display normal glucose and insulin tolerance, consistent with the hypothesis that IHTG per se does not induce hepatic insulin resistance (916). Similarly, mice treated with an adenovirally delivered shRNA targeting Atgl for 12 wk developed increased hepatic TAG and DAG but maintained normal glucose tolerance and displayed lower fasting glycemia than controls (597). These phenotypes likely reflect the importance of intrahepatic lipolysis for gluconeogenic support (e.g., through glycerol entry into gluconeogenesis and acetyl CoA activation of PC).
Together, at least two dozen rodent studies in which a genetic or pharmacological intervention was used to alter hepatic insulin action have observed an inverse relationship between hepatic DAG content and hepatic insulin sensitivity (646). Yet in addition to those discussed above (298, 547, 597), several other genetically modified mouse models have also yielded phenotypes that appear to dissociate DAG accumulation from hepatic insulin resistance, engendering skepticism toward the DAG-PKC hypothesis of lipid-induced insulin resistance (20, 231). Mice overexpressing ChREBP predictably displayed upregulation of several lipogenic genes and developed hepatic steatosis with increased hepatic DAG (49). Yet the ChREBP-overexpressing mice had normal glucose and insulin tolerance on regular chow diet and were even protected from HFD-induced defects in glucose tolerance and hepatic insulin signaling (49). Although PKCε activation was not determined in this study, it challenges the sufficiency of lipogenic DAG accumulation for hepatic insulin resistance and points to potential benefits of expedient lipid storage (496). Another important mouse model inconsistent with the DAG hypothesis of hepatic insulin resistance is the liver-specific microsomal triglyceride transfer protein (Mttp) knockout mouse (543). These mice have a defect in very-low-density lipoprotein (VLDL) secretion and accordingly accumulate hepatic lipids, including DAG, ceramides, and TAG, despite normal body weight and adipose tissue mass (543). Despite hepatic lipid accumulation, the liver-specific Mttp−/− mice displayed normal hepatic insulin sensitivity during hyperinsulinemic-euglycemic clamp studies (543). PKCε activation was not assessed in these mice, and their normal adiposity suggests that indirect hepatic insulin action (mediated by suppression of adipose lipolysis) was likely intact; this may account for the normal suppression of hepatic glucose production observed during clamps (543). Another provocative model is the liver-specific Hdac3−/− mouse, which develops a distinctive hepatosteatosis characterized by very small lipid droplets and a reduced acyl CoA pool, and displays decreased fasting insulin, improved glucose tolerance, and improved insulin tolerance on regular chow diet (812). Here, although hepatic DAG was increased severalfold on regular chow, PKCε activation was not observed in the Hdac3−/− mice, another example in which discordance between hepatic lipids and hepatic insulin action is accompanied by concordance between hepatic PKCε activation and hepatic insulin action (812).
A prominent challenge to the DAG/nPKC hypothesis that led to an important advance in the field came from mice with antisense oligonucleotide knockdown of the ATGL coactivator CGI-58. These mice manifest profound hepatosteatosis and increased hepatic DAG content, yet retain normal hepatic insulin signaling and suppress hepatic glucose production normally during hyperinsulinemic euglycemic clamps (98, 115). Although one explanation for these data is that increased DAG is insufficient to induce hepatic insulin resistance, more nuanced interpretations are also possible. For example, PKCε was observed to translocate to the lipid droplet in CGI-58 ASO-treated mice, rather than to the membrane-associated fraction as typically observed in lipid-induced insulin resistance (115). It is possible that preferential lipid droplet DAG/nPKC activity constitutes a sequestration of PKCε away from the compartments involved in insulin receptor trafficking and signaling, such as the secretory apparatus and the plasma membrane. A similar phenotype in mice with adenoviral hepatic overexpression of perilipin 5, characterized by increased hepatic TAG and DAG with preserved hepatic insulin sensitivity, may involve a similar mechanism (848). As a result of increasing recognition of the importance of DAG localization, it has become standard practice in recent years to measure DAG levels in multiple subcellular compartments.
Another way to subdivide cellular DAG is by acyl chain composition: length and saturation. In vitro, different DAG species may activate PKC isoforms with modestly different potencies, although no clear molecular basis for such differences has been identified (510). In vivo, various studies have observed positive, negative, or no association between the degree of acyl group saturation and insulin resistance (21, 53, 819, 866d). DAG composition is largely a function of circulating fatty acid availability, which may limit the utility of studies done using acute lipid infusions or synthetic rodent HFDs to address this question. Careful correction for multiple comparisons is also essential for studies in which correlation coefficients are calculated for many lipid species.
A final important distinction to consider pertains to DAG stereoisomers. DAG exists as one of three stereoisomers: sn-1,2, sn-2,3, or sn-1,3 depending on the placement of the two fatty acyl chains along the three-carbon glycerol backbone. Only sn-1,2-DAG is capable of activating PKC isoforms (680). This stereospecificity has important physiological implications. The lipolytic enzyme ATGL preferentially generates sn-1,3-DAG rather than the PKC-activating sn-1,2-DAG, arguing against intracellular lipolysis as a driver of nPKC-mediated cellular insulin resistance (210). The relevance of DAG stereoisomers in lipolysis is also highlighted by studies of HSL knockout mice, which displayed increased insulin-stimulated glucose uptake despite increased muscle sn-1,3-DAG content; sn-1,2-DAG content was unaffected by HSL loss (754). It is plausible that lipogenic DAG flux, rather than lipolytic DAG flux, drives DAG/nPKC-mediated insulin resistance. Interestingly, mRNA expression of Dgat2 is decreased in the insulin-sensitive steatotic livers of mice with liver-specific deletion of either Atgl or CGI-58 as well as in CGI-58 ASO-treated mice, suggesting antilipogenic compensation (98, 290, 916). Stereoisomer-specific DAG measurements are not frequently reported, and spontaneous acyl migration tends to convert sn-1,2- to sn-1,3-DAG during sample preparation. However, a recent study reported muscle DAG concentrations subdivided by stereoisomer, compartment, and species in a human cohort that included athletes, lean controls, obese subjects, and subjects with T2D (619). These workers found increased total sn-1,2-DAG in T2D subjects compared with lean controls in the total cellular lysate and the sarcolemmal fraction, but not in the mitochondrial/ER, nuclear, or cytosolic fractions (619). In related experiments, Pesta et al. have found that insulin-sensitive athletes have reduced PKCθ translocation in muscle compared with obese insulin-resistant subjects, suggesting that differences in the compartmentation and/or stereoisomer distribution of DAGs may account for differences in muscle insulin sensitivity between these populations despite similar increases in total muscle TAG and DAG content (D. Pesta, D. Zhang, G. Shulman, M. Roden, unpublished results). Further work, both in methodological refinement and standardization, and in data collection across multiple populations, will likely emerge in this area.
It is also important to recognize that changes in the DAG/nPKC axis measured in all models must be interpreted with caution because the practical methods currently available for quantitating DAG levels and nPKC activation are relatively crude, lacking organellar spatial resolution and any temporal resolution. For example, the ‟membraneˮ or ‟particulateˮ fraction typically used to interrogate DAG compartmentation or PKC translocation contains plasma membrane, mitochondrial membranes, endoplasmic reticulum, Golgi membranes, and other endomembranes (115). The investigator typically cannot be certain that the pattern of DAG/nPKC perturbation in a given mouse model corresponds to the pattern observed in typical human insulin resistance. It is also not certain that the relevant compartment for DAG/PKC axis activation is the plasma membrane, although this is widely assumed. The DAG-PKCε interaction has been shown in 3T3 fibroblasts to occur primarily at the Golgi, where its anchoring protein RACK2 is localized (469, 648). A single tryptophan residue in the DAG-binding C1 domain of nPKCs also appears to facilitate targeting to the Golgi (199). Although Golgi membranes are a component of the ‟membraneˮ fraction typically employed to interrogate the DAG/PKC axis, no studies have specifically examined DAG/nPKC axis activation at the Golgi in cellular insulin resistance. Formation of sn-1,2-DAG is best studied in other organelles: at the plasma membrane by phospholipase C cleavage of phosphatidylinositol 4,5-bisphophate (a major mechanism for activation of classical PKC isoforms such as PKCα, β1, β2, and γ), and in the endoplasmic reticulum by phosphatidic acid phosphatase in the Kennedy pathway or by MGAT activity (211). However, sn-1,2-DAG is also present in the Golgi, where it signals in membrane protein trafficking (36, 730). Additionally, RACK2 is a component of the coatomer complex that mediates vesicle trafficking between the ER and Golgi, highlighting the extensive crosstalk between these structures (648). Overall, given what is known about how different DAG moieties may participate in insulin resistance, a reasonable refinement of the initial hypothesis that DAGs activate nPKC isoforms may be that lipogenic sn-1,2-DAGs act at the Golgi to activate nPKC isoforms (FIGURE 15).
FIGURE 15.

Subcellular localization and stereoisomers of diacylglycerol (DAG). DAG is produced by the action of various enzymes at multiple intracellular sites, only a few of which are depicted. At the plasma membrane, phospholipase C (PLC) generates sn-1,2-DAG from phosphatidylinositol-4,5-bisphosphate (PIP2); this DAG is particularly critical for the activation of the classical protein kinase C (PKC) isoforms that also require the Ca2+ liberated downstream of PLC activity. The lipogenic enzymes of the Kennedy pathway are localized to the endoplasmic reticulum (ER) and produce sn-1,2-DAG. The novel PKC isoforms display significant localization to the Golgi. For PKCε in particular, this partially owes to the Golgi localization of its adapter protein RACK2. RACK2 also participates in vesicle transport between the ER and Golgi, and Golgi DAGs regulate protein trafficking to the plasma membrane. Notably, lipolytic DAG generated by the action of adipose triglyceride lipase (ATGL) at the lipid droplet is of the sn-1,3 stereoisomer and would not be predicted to activate PKC isoforms.
Having considered several models designed to examine the role of DAG in insulin resistance, we now turn our attention to models generated to genetically test the role of nPKC activation in lipid-induced insulin resistance. The first such model reported was a mouse ectopically expressing a dominant-negative, kinase-dead PKCθ specifically in skeletal muscle (753). Unexpectedly, these mice developed age-associated obesity accompanied by glucose intolerance and impaired muscle insulin signaling (753). Similarly, whole-body PKCθ knockout mice were more susceptible to HFD-induced obesity and insulin resistance owing to decreased physical activity and energy expenditure (256). However, when subjected to the simplest test of lipid-induced insulin resistance–the acute lipid infusion–the PKCθ knockout mice were completely protected from skeletal muscle insulin resistance (405). PKCθ overexpression is also sufficient to cause insulin resistance in cultured myocytes (292). These disparate results suggest that in addition to a possible pathophysiological role in lipid-induced insulin resistance, PKCθ may have an as-yet-undetermined physiological role in normal skeletal muscle insulin action and/or energy balance. They also indicate that while PKCθ is likely involved in lipid-induced insulin resistance, it is not totally necessary for obesity-associated muscle insulin resistance.
The role of PKCε in lipid-induced insulin resistance has also been tested genetically. Antisense oligonucleotide knockdown of PKCε specifically in liver and WAT protects rats from hepatic insulin resistance when fed a 3-day HFD (722). Additionally, whole-body PKCε knockout mice are totally protected from glucose intolerance when fed a 1-wk HFD despite increased liver TAG and DAG content (668). With longer durations of high-fat feeding in PKCε knockout mice, assessment of the liver phenotype is confounded by improvements in β-cell function, but the improvements in glucose tolerance persist (741). Thus available genetic models are consistent with a role for PKCε in the pathophysiology of lipid-induced hepatic insulin resistance.
The contribution of PKCδ activation to lipid-induced insulin resistance is incompletely understood. PKCδ translocation has been observed in the lipid-infused rat liver (450), and mouse models also support a deleterious role for this nPKC in liver insulin action. PKCδ knockout mice, whether whole body or liver specific, have diminished transcriptional programs for gluconeogenesis and lipogenesis and displayed improved hepatic insulin signaling, while mice adenovirally overexpressing PKCδ in liver are glucose intolerant (59). However, neither humans with NAFLD nor 3-day high-fat-fed rats display increased hepatic PKCδ translocation, challenging the physiological relevance of these observations (445, 721). The decreased lipogenic capacity of PKCδ knockout mice may in turn decrease DAG/PKCε axis activation, directly mediating the observed improvements in hepatic insulin action. The role of PKCδ in skeletal muscle insulin resistance is complex. In vitro work has suggested that PKCδ activation may actually enhance myocellular insulin action (91, 92). However, this hypothesis is not supported by in vivo data. In skeletal muscle of young mice, PKCδ expression is low and, interestingly, decreases with fat feeding (482). Accordingly, muscle-specific PKCδ deletion (M-PKCδKO mice) does not confer protection from HFD-induced muscle insulin resistance in young mice (482). However, skeletal muscle PKCδ expression increases with age, and old M-PKCδKO mice display protection from age-related glucose intolerance (482). Understanding the potential role of PKCδ in lipid-induced insulin resistance will thus require further study.
Although these genetic models have provided important clues to the tissue-specific roles of various nPKCs in lipid-induced insulin resistance, establishing the pathophysiological significance of the DAG/nPKC axis necessitates elucidating the molecular mechanisms by which activated nPKCs impair insulin action. If the DAG/nPKC axis mediates lipid-induced insulin resistance, the most straightforward mechanism would be direct serine/threonine phosphorylation of insulin signaling mediators. This hypothesis has been long pursued, but conclusive evidence linking specific serine/threonine phosphorylation sites to lipid-induced insulin resistance has only recently begun to emerge.
Early work investigating PKC inhibition of insulin signaling employed phorbol esters, especially phorbol-12-myristate-13-acetate (PMA) [also known as 12-O-tetradecanoylphorbol-13-acetate (TPA)]. Phorbol esters were first observed to decrease INSR tyrosine phosphorylation and insulin-stimulated glycogen synthesis in 1984 (822). This inhibition was associated with serine and threonine phosphorylation of INSR and was reversed by alkaline phosphatase treatment (821, 822). These lines of evidence, together with the 1986 demonstration that purified PKC preparations (albeit with Ca2+-dependent activity) could phosphorylate purified INSR (77), suggested that PKC could directly inhibit IRK activity. The search for specific sites of serine and threonine phosphorylation on INSR yielded several candidates, mostly in the relatively unstructured COOH-terminal tail of the receptor. These sites included Thr1348 (478), Ser1327 (156), Ser1305/Ser1306 (479), Ser1006 (802), and Ser1035/Ser1037 (489, 802). However, phorbol esters are nonspecific activators of multiple PKC isoforms, and most of the above studies used either phorbol esters or cPKC isoforms such as PKCα or PKCβII. Despite careful study, these candidate sites have ultimately not been implicated in insulin resistance in vivo (82, 394, 564). One possible exception to this paradigm is Ser994, which is an in vitro substrate of PKCα, β, and ζ isoforms as well as TANK-binding kinase 1; hepatic INSR Ser994 phosphorylation has been shown to increase in the basal state in several rodent models of obesity-associated insulin resistance (155, 559, 802). However, mechanistic insight to the role of Ser994 phosphorylation in INSR kinase activity is lacking.
With the observations that PKCθ activation was involved in lipid-induced skeletal muscle insulin resistance and that PKCε was involved in lipid-induced hepatic insulin resistance, mechanistic investigations turned to these specific isoforms. In 2004, in vitro kinase assays and cell-based experiments identified IRS1 Ser1101 as a PKCθ substrate in myocytes (494). IRS1 Ser1101 is phosphorylated within 15 min of insulin stimulation and impairs IRS1 tyrosine phosphorylation, suggesting that it may act in an acute negative feedback circuit to attenuate insulin action (494). Increased IRS1 Ser1101 phosphorylation in muscle has been reported in acutely lipid-infused humans, associated with the development of muscle insulin resistance (819). However, other stimuli, including PMA, tumor necrosis factor (TNF)-α, arachidonic acid, and oleic acid, were also observed to increase IRS1 Ser1101 phosphorylation (494). In addition, the amino acid-activated kinase S6 kinase 1 (S6K1) was later shown to phosphorylate IRS1 Ser1101 in response to nutrient status, suggesting complex and integrated regulation of this functionally important phosphorylation site (846). IRS1 can be phosphorylated on more than 50 serine/threonine sites, most of which are dynamically regulated by insulin and other metabolic stimuli; the structural basis by which a given serine/threonine phosphorylation event may impair IRS1 tyrosine phosphorylation is largely unknown (169, 300). The difficulty of assigning causal roles to specific IRS phosphorylation sites in insulin sensitivity or resistance is typified by the case of IRS1 Ser307, long considered a marker for inflammation-induced insulin resistance through the c-Jun NH2-terminal kinase (JNK; see sect. VII) (465). Despite extensive evidence suggesting that IRS1 Ser307 phosphorylation mediates insulin resistance, mice homozygous for a Ser307Ala mutation surprisingly were more, not less, susceptible to diet-induced insulin resistance (167). Similarly, alanine knock-in mice for another well-studied IRS1 phosphorylation site, Ser302, displayed no defects in muscle insulin action (168). The bewildering complexity of IRS1 serine/threonine phosphorylation seems to cast doubt on the hypothesis that single IRS phosphorylation sites can exhibit on/off control over IRS signaling intensity; rather, a more viable model treats IRS phosphorylation events as signaling modulators that serve individually minor but collectively major roles in attenuating and/or inhibiting IRS signaling (169). Although the specific importance of PKCθ phosphorylation of IRS1 Ser1101 is uncertain, there have been reports of other potential mediators of PKCθ-induced muscle insulin resistance. PKCθ has been reported to phosphorylate phosphoinositide-dependent kinase 1 (PDK1) Ser504 and Ser532 in palmitate-treated myotubes, with these phosphorylation events associated with impaired PDK1-mediated Akt Thr308 phosphorylation (873). A final intriguing PKCθ target is the guanine exchange factor GIV/Girdin, which interacts with INSR, IRS1, and PI3K and is required for insulin-stimulated glucose uptake (514); PKCθ inhibits GIV through Ser1689 phosphorylation, decreasing PI3K-AKT signaling (500). GIV was shown to be necessary for palmitate-induced insulin resistance to glucose uptake in L6 myotubes, expression of a phosphomimetic Ser1689Asp GIV mutant was sufficient to abolish insulin-stimulated glucose uptake in L6 myotubes, and GIV Ser1689 phosphorylation was profoundly decreased by pioglitazone treatment in women with polycystic ovarian syndrome (514). Though promising, a full understanding of the physiological significance of this phosphorylation event requires future study, and still other PKCθ substrates within the proximal insulin signaling cascade may yet be uncovered.
Efforts to understand the mechanistic link between PKCε and hepatic insulin resistance have also yielded interesting results. Because the signaling defects of hepatic insulin resistance can be traced as far proximally as IRK activity (116, 155, 161, 722), direct inhibition of IRK activity by PKCε was hypothesized. PKCε was shown to coimmunoprecipitate with INSRβ in liver, suggesting direct interaction (722). Furthermore, recombinant PKCε dose-dependently inhibited recombinant IRK activity in vitro, and hepatic INSR immunoprecipitated from rats treated with an antisense oligonucleotide (ASO) targeting PKCε displayed complete protection from HFD-induced impairments in IRK activity (722). However, the mechanistic basis of PKCε inhibition of IRK activity was unknown until recently. Phosphopeptide mass spectrometry of in vitro PKCε/IRK kinase assays revealed a novel phosphorylation site within the IRK activation loop, Thr1160 (645). This threonine is conserved in metazoans as distantly related as Drosophila, and its phosphorylation is predicted to disrupt the normal configuration of the active IRK through steric hindrance and electrostatic repulsion (645). Indeed, phosphomimetic Thr1160Glu mutation produces a nearly kinase-dead INSR (645). Conversely, Thr1160Ala mutation abolished IRK inhibition by PKCε in vitro (645). InsrT1150A knock-in mice were protected from HFD-induced hepatic insulin resistance, indicating that Thr1160 phosphorylation is a physiologically relevant mechanism for lipid-induced hepatic insulin resistance (645). These studies support a model of lipid-induced hepatic insulin resistance wherein activation of the DAG/PKCε axis promotes direct inhibitory phosphorylation of INSR Thr1160 (FIGURE 16). Importantly, although this defect localizes to the insulin receptor, it is not a decrease in receptor number and is therefore best classified as a post-receptor defect, shifting the insulin dose-response curve rightward and downward. Yet for a significant portion of this shifted insulin dose-response curve, DAG/PKCε/INSR-mediated hepatic insulin resistance can be overcome by the portal hyperinsulinemia that often accompanies mild-moderate insulin resistance. This concept should apply to both insulin-stimulated hepatic glycogen synthesis and insulin-stimulated de novo lipogenesis as discussed in section IV. Although this model is sufficient to account for the signaling patterns typically observed in hepatic insulin resistance (e.g., activation of PKCε, inhibition of IRK, and all downstream effectors), it is nevertheless likely that additional PKCε-dependent or -independent mechanisms for lipid-induced hepatic insulin resistance are operative.
FIGURE 16.

The diacylglycerol (DAG)/protein kinase C (PKC)ε axis in lipid-induced hepatic insulin resistance. Chronic overnutrition promotes intrahepatic lipid accumulation. Although this lipid is primarily stored as relatively inert triglyceride, levels of the bioactive lipid DAG, the penultimate intermediate in triglyceride synthesis, increase as well. DAG activates PKC isoforms by promoting PKC translocation to cellular membranes, and PKCε translocation in particular is chronically and reproducibly increased in the setting of lipid-induced hepatic insulin resistance. PKCε impairs insulin action by directly phosphorylating and inhibiting the insulin receptor (INSR) at Thr1160 in the activation loop of its tyrosine kinase domain.
Together, these studies have both shed light and cast doubt upon the DAG/nPKC hypothesis, enabling it to become more focused and specific. Genetic and pharmacological manipulations have yielded strong evidence for a causal role of DAG/nPKC axis activation in lipid-induced insulin resistance, while correlative studies in “wild-type” rodents and humans suggest physiological relevance. The era of the genetically modified mouse has both bolstered and challenged the hypothesis, but with the ever-present caveat of unphysiological compensations. The application of advanced cell biology and analytical chemistry, especially in cells and tissues with intact lipid handling pathways, will be necessary to drive further progress in understanding if, when, where, and how diacylglycerols activate nPKCs to impair insulin signaling.
C. Ceramides and Insulin Resistance
The sphingolipids, which derive from the condensation of serine and, primarily, palmitoyl CoA, encompass hundreds of distinct lipid species (325). Initial suggestions that sphingolipids might interact with insulin action came from in vitro reports that sphinganine and sphingosine blocked insulin-stimulated 2-deoxyglucose uptake in 3T3-L1 fibroblasts (574). Sphingosine was also shown to impair maximal insulin-stimulated glucose uptake and lipogenesis in cultured adipocytes (695, 779). Early studies of ceramides, produced by the covalent addition of a fatty acyl group to sphingosine, used short-chain (C2, C6) cell-permeable ceramides, often at supraphysiological concentrations. These studies largely agreed that adding ceramide to the culture medium inhibited insulin action, but reported different sites of blockade. Some groups reported inhibition of INSR tyrosine kinase activity by ceramides (385, 617), while others found no impairment in proximal insulin signaling (574, 740, 809, 874, 956). The 1998 reports that C2-ceramides inhibited insulin action at the level of AKT–distal to INSR-IRS-PI3K activation–were particularly influential in guiding consensus for the mechanism of ceramide-induced insulin resistance (809, 956). The mechanism for C2-ceramide inhibition of AKT has been linked to both increased PP2A activity (715, 748, 806, 961) and defective insulin-stimulated AKT translocation (805) through activation of atypical PKCζ (67, 241, 659, 806). Subsequent studies of ceramide-induced insulin resistance have focused on AKT inhibition as the primary mechanism.
Short-chain ceramides provided a convenient early experimental tool for in vitro studies, but the most prevalent endogenous ceramides incorporate palmitate or other long-chain saturated fatty acids (SFAs). Accordingly, most recent studies of ceramide-induced insulin resistance have focused on the effects of SFAs. Palmitate potently induces insulin resistance in cultured myocytes and hepatocytes, especially when AKT Ser473 phosphorylation is used as the primary readout of insulin resistance (126, 740). However, when functional readouts of insulin resistance such as insulin-stimulated glycogen synthesis, glucose uptake, and HGP suppression are used, palmitate induces insulin resistance to a similar extent as unsaturated fatty acids (USFAs) such as oleate and linoleate (323, 740). Because USFAs do not increase ceramide levels, it follows that ceramides cannot be the only mediator of lipid-induced insulin resistance (124).
A potential role for ceramides in SFA-induced muscle insulin resistance has been investigated in several in vitro and in vivo studies in which ceramide synthesis is perturbed and insulin action is assessed. Many of these studies have used the natural fungal product myriocin, which inhibits the first step in ceramide biosynthesis: serine palmitoyltransferase-1 activity (127, 545). For example, palmitate-rich lard oil infusion caused acute muscle insulin resistance in rats, associated with increases in both myocellular DAG and ceramides; myriocin treatment partially prevented this insulin resistance, associated with abrogation of ceramide but not DAG accumulation (322, 323). Additionally, myriocin pretreatment in C2C12 myotubes totally prevented palmitate-induced, but not linoleate-induced, insulin resistance (323). However, another study found that myriocin treatment did not reverse glucose intolerance in SFA-fed mice despite normalization of muscle ceramide content (243). A key confounder in many studies of ceramide-induced insulin resistance, even those designed to specifically modify ceramide biosynthesis, is concomitant changes throughout the lipidome, including changes in other putative mediators of insulin resistance such as DAG (101, 102). For example, myriocin, described as the “workhorse” of ceramide studies (127), has been shown to alter energy balance, weight gain, and ectopic lipid accumulation in multiple models of obesity (934). The relative contribution of each of these known modulators of muscle insulin sensitivity to the overall phenotype of myriocin-treated rodents is challenging to assess.
Ceramides have also been investigated for a potential role in lipid-induced hepatic insulin resistance. Mice lacking one copy of dihydroceramide synthase 1 (Des1+/−) displayed improved insulin sensitivity in insulin tolerance tests (although whole-body ceramide levels were similar to wild-type littermates and glucose tolerance was not altered) (323). Mice with haploinsufficiency for ceramide synthase 2 (CerS2+/−), which produces very-long-chain (C22/C24) ceramides, did not display altered total hepatic ceramide content (672). However, CerS2+/− mice shifted the acyl chain composition of their hepatic ceramides to increase C16:0 (palmitoyl) ceramides, and this was associated with worsening of hepatic steatosis and insulin resistance; the steatosis at least partially owed to mitochondrial lipid oxidation defects (672). The coexistence of hepatic steatosis in many experimental models of perturbed ceramide biosynthesis (61, 323) complicates efforts to identify the mediator(s) responsible. Even in models genetically perturbing lipid metabolism, rodents and humans with elevated liver triglyceride also generally exhibit elevated DAG (115, 144, 371, 547). Additionally, a recent human lipidomic study noted positive associations between hepatic ceramides and HOMA-IR score, but observed similar associations with hepatic DAG (506). However, recent studies of mice with tissue-specific overexpression of the ceramide-degrading enzyme acid ceramidase challenge this paradigm. Liver-specific inducible overexpression of acid ceramidase (Alb-AC mice) decreased hepatic ceramide content and was associated with protection from HFD-induced hepatic insulin resistance despite increased hepatic DAG content (922). Interestingly, however, HFD-fed Alb-AC mice also displayed marked protection from hepatic steatosis (~3-fold lower liver TAG content) (922). The mechanistic basis for this unusual dissociation of hepatic DAG and TAG content, also observed in mice with adipose-specific overexpression of acid ceramidase (922), is not clear. Interestingly, reversal of hepatic insulin resistance upon acid ceramidase induction paralleled reversal of hepatic steatosis in this study (922). Although ceramide content was only one of many physiological parameters altered by tissue-specific acid ceramidase overexpression, this study is certainly consistent with a role for ceramides in hepatic insulin resistance.
Ultimately, the questions of whether and how ceramides impair cellular insulin action derive their significance from the extent to which ceramide biosynthesis and ceramide levels are altered in insulin-resistant states. Many studies weigh in on this latter question, with varying conclusions. In the earliest such report, obese Zucker fa/fa rats were found to have increased liver and skeletal muscle ceramide content (852). Ceramides were also elevated in liver and muscle from Zucker diabetic fatty (ZDF) rats (323), lard-oil infused rat liver and muscle (323), ob/ob mouse liver (8), and obese human muscle (4, 21, 803). However, because these common models of insulin resistance are all characterized by generalized elevations in tissue lipid content, they do not permit assessment of the specific role of ceramides in insulin resistance. Indeed, many models of lipid-induced insulin resistance in both muscle and liver are not associated with increased ceramide levels. For example, 8 wk of high-fat feeding was associated with unchanged muscle ceramide content and decreased hepatic ceramide content in C57BL/6J mice (548). Acute Liposyn II infusion potently induced muscle insulin resistance without altering muscle ceramides (942). Mice lacking pyruvate dehydrogenase kinase 2 and 4 (Pdk2/4−/−) constitutively oxidized glucose in muscle and consequently developed lipid-induced muscle insulin resistance, but muscle ceramide levels were not increased (669). Neither SFA-rich nor USFA-rich 3-day HFDs raised liver ceramide content in rats, despite the presence of profound hepatic insulin resistance (253). In four independent studies of obese, nondiabetic humans, hepatic ceramide content was not correlated with insulin resistance in three; one study did observe a positive association (445, 506, 512, 834). In mice with reduced tissue lipid delivery secondary to knockdown of ApoA5, improved muscle and liver insulin sensitivity were not associated with differences in tissue ceramide content (113). In fructose-fed liver-specific Xbp1−/− mice with decreased de novo lipogenesis relative to wild-type fructose-fed controls, hepatic insulin sensitivity was increased but hepatic ceramide levels were increased (375). Furthermore, the insulin-sensitizing effects of estradiol in ovariectomized female mice were associated with correction of DAG/nPKC axis activation but no changes in liver or muscle ceramides (111). These models suggest that increased liver or muscle ceramide levels are not necessary for lipid-induced insulin resistance.
Just as studies of diacylglycerol-mediated insulin resistance have increasingly subdivided their measurements by acyl chain length, stereoisomer, and subcellular localization, so several ceramide studies have focused on specific ceramide species and their subcellular localization. Muscle C18:0 ceramides in particular have been identified as inversely correlated with insulin sensitivity during hyperinsulinemic-euglycemic clamps in three human studies (52, 619, 844), although this relationship is not observed in all studies (21, 149, 819). Interestingly, one recent study employing subcellular fractionation found that this C18:0 ceramide relationship was observed in the sarcolemmal, mitochondrial/ER, and nuclear compartments alike (619). However, another fractionation study that separated muscle biopsies into subsarcolemmal and intramyofibrillar fractions observed strong correlations with HOMA-IR for only C16:0 and C18:1 ceramides (149). Because of its saturated acyl chain, C18:0 ceramide has been hypothesized to impair insulin action by decreasing membrane fluidity in addition to the PP2A/AKT mechanism discussed above, but these hypotheses have not yet been directly tested (52). Intriguingly, however, C18:0 ceramides are the first muscle ceramide species to increase during high-fat feeding in mice (854).
The role of ceramides in adipose insulin resistance is unclear, but research in this area is accelerating. Human studies have reported correlation of adipose ceramide content with HOMA-IR (64) and increased adipose ceramides in obese diabetic compared with obese nondiabetic subjects (122). Additionally, the whole-body insulin sensitization associated with adipose-specific induction of ceramide degradation suggests a role for adipose ceramides in insulin action, whether direct or via tissue cross-talk (922). Indeed, C16:0 ceramides have been reported to be increased in adipose tissue from obese subjects, in concert with increased mRNA expression of the relevant synthetic enzyme CerS6 (686). High-fat feeding has also been reported to increase adipose ceramides in mice (856). Inhibition of adipocyte ceramide biosynthesis in WAT-specific serine palmitoyltransferase (Sptlc2) knockout mice protected mice from HFD-induced weight gain and hyperglycemia, although, surprisingly, this genetic perturbation did not change the abundance of most WAT ceramide species (122). As with many rodent models investigating the role of ceramides in lipid-induced hepatic insulin resistance, the global alterations in energetics confound efforts to ascribe the metabolic improvements seen in these mice to any one mechanism.
Another important question for assessment of the relevance of ceramide-induced insulin resistance pertains to the site(s) of signal transduction blockade in insulin resistance. If ceramides are responsible for typical obesity-associated insulin resistance, and ceramides induce insulin resistance through AKT inhibition, then one would predict that proximal insulin signaling would be intact and all detectable defects would be downstream of AKT. While defects in AKT Ser473 phosphorylation are certainly detected in both muscle and liver insulin resistance (113, 115, 669, 921), proximal insulin signaling defects are also prominent (117, 470, 722). Interestingly, glucosylceramides such as GM3 ganglioside have been suggested to impair proximal insulin signaling through altered INSR membrane microdomain localization (125, 376), and specific inhibition of glucosylceramide synthase improves insulin sensitivity in obese rodents (8); more work is needed to understand the role of specific glucosylceramides in insulin resistance. However, the observed impairment in proximal insulin signaling in typical insulin resistance poses a problem for the hypothesis that ceramide inhibition of AKT is a central defect in insulin resistance; a primary defect at the level of AKT might actually be predicted to enhance proximal insulin signaling through the loss of AKT-regulated negative-feedback mechanisms such as GRB10 stabilization (339, 944). Additionally, increasing AKT phosphorylation through acute PP2A inhibition–blocking one of the proposed mechanisms for ceramide-induced insulin resistance–is insufficient to restore insulin sensitivity in fat-fed rats and unexpectedly exacerbates muscle insulin resistance (254). Unless ceramides also impair proximal insulin signaling through currently unidentified mechanisms, it seems unlikely that ceramide signaling to AKT can fully account for typical obesity-associated insulin resistance.
Taken together, the mechanistic evidence for ceramide-induced insulin resistance is strongest in skeletal muscle, with more equivocal data for liver and WAT (644, 810). The relative significance of ceramide-induced insulin resistance in typical obesity-associated tissue insulin resistance is unclear; ceramide signaling may be sufficient to drive insulin resistance in some models but does not appear to be necessary for lipid-induced insulin resistance.
D. Acylcarnitines, Metabolic Inflexibility, and Insulin Resistance
The DAG/nPKC and ceramide/AKT models of skeletal muscle insulin resistance both implicate inappropriate anabolic shunting of excess lipid to bioactive moieties. However, an alternative hypothesis posits that inappropriate catabolism of excess lipid, mismatched to tricarboxylic acid (TCA) cycle flux, can also impair insulin action in skeletal muscle. A key observation fueling this hypothesis was that rates of fatty acid oxidation (FAO) were increased by high-fat feeding in ex vivo skeletal muscle homogenates, suggesting that the fate of excess lipid in the myocyte is not simply ectopic storage (427). However, this increase in FAO was not accompanied by increases in CO2 production (i.e., complete oxidation) but instead by increases in myocellular acylcarnitines:fatty acids bound to carnitine to enable mitochondrial entry, a marker of incomplete FAO (427). Muscle acylcarnitine profiling in profoundly insulin-resistant Zucker diabetic fatty rats revealed striking increases in long-chain acylcarnitines, especially those with chain length >10 carbons (427). These increases, coupled with measurements of decreased TCA cycle metabolites, were interpreted as reflecting FAO mismatched to TCA flux and consequent mitochondrial stress (427). This result complemented the earlier observation that PGC-1α, a transcriptional coactivator activated by exercise, promoted complete oxidation of lipids and was diminished by high-fat feeding (426). However, muscle-specific PGC-1α overexpression was later shown to unexpectedly cause muscle insulin resistance despite increased rates of FAO; these mice also displayed increased muscle DAG content and PKCθ activation (143). Additionally, the addition of carnitine to the culture medium was reported to facilitate the ability of palmitate to induce insulin resistance in L6 myotubes, suggesting a role for FAO in lipid-induced insulin resistance (427). These results, though intriguing, are somewhat difficult to reconcile with other reports associating carnitine supplementation with increased muscle insulin sensitivity in high-fat-fed rodents (583, 866a).
Acylcarnitines are measured in plasma to noninvasively probe inborn errors of FAO (746). The mechanisms of acylcarnitine appearance in plasma are incompletely understood, but plasma acylcarnitine concentrations are thought to reflect intracellular levels (654, 746). Could plasma acylcarnitine levels thus be used as a biomarker for incomplete FAO in muscle? The major problem with this proposition is that plasma acylcarnitine levels are a function of FAO rates in not just skeletal muscle, but in all tissues. The liver in particular preferentially oxidizes lipids and is likely a major contributor to plasma acylcarnitine levels (19, 746). Interestingly, in Pdk2/4−/− mice with constitutive glucose oxidation in skeletal muscle, myocellular medium- and long-chain acylcarnitines were markedly decreased but plasma levels were unchanged compared with wild-type controls (669). This dissociation of muscle and plasma acylcarnitine profiles, also reported in humans (784), indicates that plasma acylcarnitines probably cannot be used to interrogate incomplete FAO in skeletal muscle, although several groups have attempted to do so (5, 541).
How might incomplete FAO in general, or acylcarnitines in particular, mediate insulin resistance? One possibility is that acylcarnitines directly impair insulin signaling. Treatment of C2C12 myotubes with physiologically relevant concentrations (5–25 μM) of C4:0, C14:0, or C16:0 acylcarnitine impaired insulin-stimulated AKT Ser473 phosphorylation, although the effect was modest (20–30% decrease) and not dose dependent (10). Proximal insulin signaling effectors were not assessed. Incomplete FAO has also been proposed to induce muscle insulin resistance by increasing reactive oxygen species (ROS) production. Palmitate treatment increases ROS production in association with impaired insulin-stimulated glucose uptake, but pharmacological inhibition of FAO with mildronate prevents both effects (10). However, acylcarnitines per se have not directly been shown to increase ROS production. Rather, acylcarnitine levels may reflect the degree to which FAO exceeds cellular ATP demand, a condition in which ROS production would be greater (235). Putative mechanisms of ROS-induced insulin resistance are discussed in section VI.
The physiological rationale for acylcarnitine-induced insulin resistance is unclear. Muscle acylcarnitine concentrations are regulated by nutritional status in insulin-sensitive rats, and HFD-induced muscle insulin resistance is accompanied by loss of this nutritional regulation but does not cause absolute increases in acylcarnitine concentrations compared with levels in fasted, chow-fed rodents (427). This indicates that if acylcarnitines impair insulin action in fat-fed rodent muscle, they might also be expected to do so during the fasted state in insulin-sensitive rodents; this is difficult to reconcile with the known insulin-sensitizing effects of fasting in rodent skeletal muscle (32).
An alternative explanation for the increase in muscle acylcarnitines observed in some models of insulin resistance is that they simply reflect relative oxidative substrate selection. The pattern of muscle acylcarnitine levels observed in fasted and fed rats fed chow or HFD, described above, is completely consistent with isotopic measurements of oxidative substrate selection (VPDH/VTCA) in soleus muscle of chow- and high-fat-fed rats, in which relative rates of FAO are similar in fasted chow-fed rats to those in high-fat-fed rats both basally and after insulin stimulation (T. Alves, R. Perry, Y. Rahimi, and G. Shulman, unpublished data). Similar results have been reported in lean and obese insulin-resistant human skeletal muscle (395). A simple interpretation of these data is that they reflect metabolic inflexibility: the inability of insulin-resistant skeletal muscle to increase relative glucose utilization upon transition to the fed, or insulin-stimulated, state. The concept of metabolic inflexibility in muscle insulin resistance has its roots in the Randle glucose-fatty acid cycle, but incorporates modern models of lipid-induced insulin resistance (397, 561). The mechanism for metabolic inflexibility in lipid-induced insulin resistance is incompletely elucidated, but an explanation consistent with available mechanistic data is that bioactive lipid moieties such as DAG or ceramide impair insulin signaling to decrease insulin-stimulated GLUT4 translocation, thereby impairing insulin-mediated increases in glucose availability for oxidative metabolism. This occurs in parallel with increased lipid availability, resulting in increased relative rates of FAO in fat-fed, insulin-resistant muscle. Metabolic inflexibility, in this conception, would be a consequence rather than a cause of lipid-induced insulin resistance. If acylcarnitine levels primarily reflect relative FAO flux, then their association with muscle insulin resistance may also be secondary to the primary defect of lipid-induced impairments in insulin signaling.
Unravelling the mechanisms of lipid-induced insulin resistance has proven a difficult task. Indeed, even the core premise that ectopic lipid accumulation impairs insulin action in liver and muscle remains controversial. While only one fully defined mechanistic framework linking a specific bioactive lipid to impaired hepatocellular insulin signaling (the DAG/PKCε/INSR axis) has been described, lipid-induced skeletal muscle insulin resistance has received more mechanistic attention. However, although many partial mechanisms have been elucidated, each has weaknesses and more work is needed to define their respective roles in human disease. As currently understood, however, the proposed mechanisms linking the lipid mediators discussed in this section–DAG, ceramide, and acylcarnitines–to impaired skeletal muscle insulin signaling are summarized in FIGURE 17.
FIGURE 17.

Proposed molecular mechanisms of lipid-induced skeletal muscle insulin resistance. A: diacylglycerol (DAG) has been proposed to cause muscle insulin resistance by activating protein kinase C-θ (PKCθ). The targets of PKCθ within the insulin signaling cascade are incompletely defined but may include insulin receptor substrate 1 (IRS1) and GIV. PI3K, phosphoinositide-3-kinase. B: ceramides have been proposed to mediate skeletal muscle insulin resistance by decreasing AKT activity through at least two mechanisms. PP2A, protein phosphatase 2A. C: incomplete mitochondrial fatty acid oxidation has been proposed to mediate skeletal muscle insulin resistance, either through direct effects of the resultant acylcarnitine species or through production of reactive oxygen species, which modulate various cellular processes.
VI. CELLULAR NUTRIENT STRESS AND INSULIN RESISTANCE
A. Endoplasmic Reticulum Stress and Insulin Resistance
The underlying cause of obesity-associated insulin resistance is nutrient oversupply. The mechanisms for lipid-induced insulin resistance described in section V outlined cellular and molecular responses to chronic elevations in one major class of biological macromolecule: fats. However, other cell-autonomous responses to nutrient overload have also been implicated in the pathogenesis of insulin resistance. One of these is the unfolded protein response (UPR), activated by endoplasmic reticulum (ER) stress.
The UPR is an intricate, elegant, and well-understood mechanism that allows the cell to match protein synthetic demand to protein synthetic capacity (191). The three branches of the UPR are controlled by three integral ER membrane proteins: PKR-like eukaryotic initiation factor 2α kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6) (331). In the normal, unstressed ER, these three proteins are kept inactive by binding of the chaperone BiP/GRP78 to PERK, IRE1, and ATF6; the accumulation of misfolded proteins leads to BiP dissociation and UPR activation (331). Key downstream effectors in the ER stress response with proposed roles in metabolic regulation include NF-κB and c-Jun NH2-terminal kinase (JNK).
In 2004, the first report directly linking ER stress to obesity and insulin resistance in vivo was published (605). This landmark study first noted that in both high-fat-fed and ob/ob mice, markers of ER stress such as phosphorylated eIF2α, PERK, and JNK were strongly increased in liver (605). Acute chemical induction of ER stress impaired insulin signaling at the level of IRS1, associated with JNK-dependent serine phosphorylation of IRS1 (465, 605). Taken together with the same group’s earlier report that JNK activity was increased in obese insulin resistant mice (318), these studies advanced the hypothesis that ER stress induces insulin resistance by promoting inhibitory phosphorylation of IRS1 by JNK. Curiously, whole-body Jnk deletion was associated with improved INSR signaling in obese mouse liver in the first study (318), but JNK activation was not associated with impaired INSR signaling in the later study (605). Given the known role of direct INSR defects in hepatic insulin resistance (116, 722), the hypothesis that JNK inhibits insulin signaling at the level of IRS1 implies that other defects must be present to account for INSR inhibition. Additionally, the role of JNK activation in hepatocellular insulin resistance has been called into question by the finding that liver-specific Jnk−/− mice are more, not less, prone to hepatosteatosis and hepatic insulin resistance (709).
The initial observation of an activated UPR in obesity has been repeatedly confirmed in mouse liver (572, 792) and in human liver (284, 445) and adipose tissue (70, 284, 757). Mechanistic work describing the relationship between ER stress and insulin resistance has primarily been carried out in liver, a logical candidate tissue to develop ER stress during nutrient oversupply.
A major model system for investigating the role for ER stress in hepatic insulin resistance has been mice with defects in X-box binding protein 1 (XBP1). The mRNA splice variant of XBP1, XBP1s, is a transcription factor and a key effector of the UPR (237, 938). Whole-body Xbp1+/− mice display ER stress (e.g., PERK and JNK activation), associated with glucose intolerance and impaired insulin action that is apparent as far proximally as the INSR (605). However, the Xbp1+/− mice also gain more weight than wild-type controls, raising the possibility that other obesity-associated mechanisms such as ectopic lipid-induced insulin resistance may contribute to the phenotype (605). Indeed, an alternative hypothesis for the relationship between ER stress and insulin resistance emerged from the 2008 report that XBP1 activates the de novo lipogenic transcriptional program (460). Mice with liver-specific Xbp1 deletion displayed markedly defective DNL, which was particularly apparent on a lipogenic high-fructose diet (460). However, liver-specific deletion of Xbp1 unexpectedly led to increased levels of its upstream regulator IRE1, which in turn increased JNK activation (375). Other signs of ER stress, including BiP/GRP78 levels and eIF2α phosphorylation, were also observed in this model (375). Together, these observations comprised a novel phenotype: a mouse with increased hepatic ER stress but decreased hepatic lipids. When subjected to hyperinsulinemic-euglycemic clamp studies, the fructose-fed liver-specific Xbp1−/− mice displayed increased hepatic insulin sensitivity (375). These data indicate that UPR activation and JNK activity are insufficient to cause hepatic insulin resistance and suggest that in many models of ER stress-associated insulin resistance, XBP1-mediated lipogenesis may be a contributing factor, unifying ER stress with the DAG/nPKC model of lipid-induced insulin resistance (FIGURE 18A). Interestingly, adenoviral overexpression of Xbp1 in high-fat-fed mice also prevented hepatosteatosis, PKCε activation, and hepatic insulin resistance, indicating that the relationship between XBP1 and lipogenesis may be more complex than previously thought (314). Similarly, activation of IKKβ, which phosphorylates and activates XBP1, reversed diet-induced hepatosteatosis and hepatic insulin resistance in multiple mouse models (490). The finding that both XBP1 inhibition and activation improve hepatic insulin action is not easily reconciled.
FIGURE 18.

Nutrient stress and insulin resistance. A: endoplasmic reticulum (ER) stress, manifested as the unfolded protein response (UPR), is activated in multiple models of liver insulin resistance. Accumulation of misfolded proteins in the ER lumen initiates the UPR, activating PKR-like eukaryotic initiation factor 2α kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6). Key effectors of the UPR include c-Jun NH2-terminal kinase (JNK) and the mRNA splice variant of X-box binding protein 1 (XBP1s). JNK may directly impair proximal insulin signaling, although this is controversial. XBP1s transcriptionally activates the de novo lipogenic program and promotes hepatic steatosis, which may in turn drive hepatic insulin resistance through diacylglycerol (DAG)/protein kinase C (PKC)ε signaling. B: muscle insulin resistance is associated with mitochondrial dysfunction, although cause and effect relationships are not clearly defined. The red pathway describes observations made in humans: older adults and the young lean insulin-resistant offspring of parents with type 2 diabetes (T2D) display reduced mitochondrial ATP synthesis in skeletal muscle, probably reflective of reduced ATP demand as resting mitochondria operate at submaximal ATP synthetic rates. This decrease in substrate oxidation promotes lipid storage, increasing intramyocellular lipid (IMCL). IMCL accumulation may then impair proximal insulin signaling through activation of the DAG/PKCθ axis, accounting for the reduced insulin-stimulated glucose uptake observed in these individuals. Alternatively, chronic overnutrition and/or lipid oversupply increases the mitochondrial capacity for fatty acid oxidation. However, because ATP demand is relatively inflexible, this increase is not enough to match supply, leading to IMCL accumulation and increased rates of incomplete fatty acid oxidation. Incomplete fatty acid oxidation produces acylcarnitine species and reactive oxygen species (ROS). Acylcarnitines have been hypothesized to impair insulin signaling through undefined mechanisms, and ROS have broad cellular effects, including impairment of mitochondrial function. ROS-induced mitochondrial damage would then exacerbate these effects, further promoting IMCL accumulation and ROS production.
Overall, it has been difficult to elucidate whether ER stress per se is a significant primary defect in human hepatic insulin resistance. Some markers of ER stress (eIF2α phosphorylation, CHOP induction) are associated with insulin resistance in human liver, but the most mechanistically critical readouts of ER stress (XBP1 splicing, JNK phosphorylation) are not (445). Furthermore, the interaction between ER stress and lipid metabolism means that many models of ER stress are also models of ectopic lipid deposition. Does ectopic lipid induce insulin resistance through activation of the UPR, or is ER stress merely a secondary consequence of a primary, ectopic lipid-mediated defect? There is evidence that excess lipid accumulation, especially oversupply of saturated fatty acids, induces ER stress (251), but the triad of increased hepatic insulin sensitivity, decreased hepatic lipid accumulation, and increased ER stress in liver-specific Xbp1−/− mice ultimately indicates that ER stress may require ectopic lipid accumulation to induce hepatic insulin resistance.
However, while ER stress may not be the primary defect in hepatic insulin resistance, it may well exacerbate primary lipotoxic mechanisms. In support of this hypothesis, ameliorating ER stress in ob/ob mice by BiP/GRP78 overexpression decreases hepatic steatosis and increases hepatic insulin sensitivity (382). In addition, ER stress has been shown to increase the expression of lipin-2 (which catalyzes the synthesis of DAG from PA); lipin-2 overexpression impairs hepatic insulin signaling by activating the DAG/PKCε axis, while lipin-2 knockdown in high-fat-fed mice improves hepatic insulin signaling (708).
WAT ER stress is less well studied than hepatic ER stress, but a small but intriguing literature links ER stress in WAT to increased lipolysis. The ER-associated triglyceride synthetic enzyme DGAT1 serves to re-esterify liberated fatty acids to triglyceride, and adipose-specific Dgat1−/− mice developed WAT ER stress during cold exposure and fasting (139). This activation of the UPR was associated with adipose tissue macrophage infiltration, pointing to links between ER stress and inflammation in WAT (139, 331). The prolipolytic effects of ER stress are potentially mediated by PKA activation and consequent perilipin phosphorylation (76, 187, 958). Given the primacy of lipolysis in adipocyte insulin action, understanding the contribution of ER stress to overall lipolytic tone has major implications. Even if proximal insulin signaling is intact, insulin’s ability to suppress cAMP-mediated lipolysis could be thwarted by ER stress-mediated activation of cAMP-mediated lipolysis, a functional insulin resistance.
The role of ER stress in muscle insulin resistance remains uncertain. The landmark 2004 study of ER stress and insulin resistance in liver reported no induction of ER stress markers in skeletal muscle of high-fat-fed mice (605). However, others have observed mild increases in some (BiP, CHOP expression), but not all (IRE1α or JNK activation) markers of ER stress in human muscle insulin resistance (416). Still others report no induction of any ER stress markers (BiP, CHOP, IRE1α, PERK expression) in skeletal muscle after 6 wk of high-fat feeding in humans despite inducing glucose intolerance associated with increased intramyocellular lipid content (183). Furthermore, neither expression of a constitutively active JNK mutant specifically in skeletal muscle nor skeletal muscle-specific ablation of JNK altered muscle insulin action or glucose homeostasis (606a).
B. Mitochondrial Energetics, Oxidative Stress, and Insulin Resistance
The ER is not the only organelle placed under nutrient stress during chronic caloric excess. We now consider the pathogenesis and significance of mitochondrial oxidative stress in the context of nutrient, especially lipid, oversupply. In broad terms, the two major fates of fatty acids once transported into the myocyte are oxidation and storage. The evidence linking excess lipid storage to myocellular insulin resistance (see sect. V) has prompted intense investigation of the other side of the coin: lipid oxidation (561). An early hypothesis in this field posited that IMCL accumulation and consequent insulin resistance might be driven by inappropriately decreased lipid oxidation. Indeed, there is clear evidence for decreased mitochondrial activity (i.e., ATP synthesis as measured by 31P-MRS) in insulin-resistant human populations such as the elderly, the young lean offspring of parents with T2D, and prediabetic subjects, providing a potential etiological clue to the myocellular energetics underlying T2D (219, 503, 635, 636). There is also strong evidence for structural damage and decreased total content of skeletal muscle mitochondria in insulin-resistant humans (148, 396, 552, 614). This does not necessarily imply mitochondrial insufficiency: resting mitochondria are typically not operating at their maximal oxidative capacity (699). Furthermore, mitochondrial activity is primarily dictated by cellular ATP demand rather than by changes in substrate availability (326). Regardless of mechanism, however, the decreased mitochondrial activity of insulin-resistant skeletal muscle could significantly alter whole-body caloric demand, facilitating intracellular nutrient oversupply and lipid-induced insulin resistance.
Recently, human genetic studies have identified metabolic risk alleles with mechanisms that link T2D to mitochondrial energetics in a way consistent with the lipid storage versus utilization paradigm. One such gene, SLC16A11, is a monocarboxylate (i.e., pyruvate, lactate) transporter localized to the endoplasmic reticulum and plasma membrane; it is expressed particularly highly in liver (706, 872a). The SLC16A11 risk haplotype is particularly prevalent (~30% allele frequency) in individuals of Mexican descent and is estimated to account for ~20% of the increased T2D risk in that population (872a). The several variants in the SLC16A11 T2D risk haplotype decrease SLC16A11 expression and impair its plasma membrane localization; hepatocytes with siRNA-mediated SLC16A11 knockdown displayed accumulation of acylcarnitines, DAG, and TAG, indicative of decreased FAO. These studies suggest that the SLC16A11 mutations in the T2D risk haplotype increase diabetes risk by promoting intracellular lipid storage, which results from impaired mitochondrial fatty acid oxidation. A second interesting risk allele lies in the N-acetyltransferase 2 (NAT2) gene, which emerged in a genome-wide association study searching for genes associated with insulin resistance (414). Mice with whole-body deletion of the mouse ortholog Nat1 displayed a decreased basal metabolic rate, decreased energy expenditure, and mitochondrial dysfunction in diverse tissues including WAT, brown adipose tissue, liver, heart, and skeletal muscle (114, 130). As a result, these mice were predisposed to lipid-induced liver and muscle insulin resistance associated with DAG-induced PKCε and PKCθ activation in liver and muscle, respectively, when placed on a HFD (114).
However, the relationship between mitochondrial activity and insulin action is more complex than the simple model outlined above, in which insulin-resistant myocytes store rather than oxidize lipid. Interestingly, after several weeks of high-fat feeding, rodents develop skeletal muscle insulin resistance in parallel with increased capacity for fatty acid oxidation ex vivo (427, 548, 853). This may reflect an adaptive response that insufficiently compensates for increased lipid availability, leading to IMCL accumulation and insulin resistance (549). Indeed, even potent activation of FAO, by pharmacological AMPK activation or acetyl-CoA carboxylase 2 deletion, is insufficient to protect mice from HFD-induced muscle insulin resistance, highlighting the primacy of ATP demand rather than substrate supply in mitochondrial energetics (320, 596). Alternatively, there is evidence that increased relative β-oxidative flux may itself drive insulin resistance in skeletal muscle, contrary to the initial hypothesis that impaired lipid oxidation drives muscle insulin resistance. This hypothesis is supported by several models in which blocking β-oxidative flux improves muscle insulin sensitivity, even despite IMCL accumulation (230, 287, 427). However, increased FAO is certainly not necessary for lipid-induced muscle insulin resistance, as demonstrated by the muscle insulin resistance of constitutively glucose-oxidizing Pdk2/4−/− mice (669).
How might β-oxidative flux impair muscle insulin action? One proposed mediator is the acylcarnitine species produced by incomplete FAO, discussed in section V. Another potential link is ROS, which serve as an ‟electron release valveˮ when substrate oxidation exceeds ATP demand (i.e., in states of nutritional oversupply) (37, 235). Although all ROS derive from superoxide (O2·−), the key bioactive ROS is thought to be hydrogen peroxide (H2O2) which serves as a second messenger to communicate mitochondrial redox status (235). High-fat feeding increases mitochondrial H2O2 production within 3 days, the effect persists in chronically fat-fed mice, and this oxidized redox state is detectable throughout the cell as a decreased ratio of reduced to oxidized glutathione (GSH/GSSG ratio) (22). Furthermore, skeletal muscle from obese humans displays increased mitochondrial H2O2 production and decreased GSH/GSSG ratio (22). Because the activity of many protein kinases and phosphatases is regulated by the redox status of cysteine thiols, it has been proposed that a more oxidized cellular environment may favor the serine/threonine phosphorylation events that characterize normal negative feedback of insulin action (235). For example, ROS activation of stress kinases such as JNK has been observed (570). Importantly, cellular redox state is highly dynamic; models linking increased H2O2 production to impaired insulin action propose that in states of chronic overnutrition, the tendency to oxidize the cellular environment postprandially outweighs the tendency to reduce the cellular environment during times of relative fasting, preventing this normally self-correcting system from restoring homeostasis (235). Redox regulation of cell signaling is also likely to depend critically on spatiotemporal factors; for example, local H2O2 production by NADPH oxidase 4 (NOX4) is increased upon insulin receptor activation to inactivate local phosphatases (e.g., PTP1B, PTEN) and amplify proximal insulin signaling (37, 515, 920). This may account for the phenotype of mice lacking the ROS scavenger glutathione peroxidase 1, which were protected from HFD-induced insulin resistance (497).
There is experimental evidence for the hypothesis that blocking mitochondrial ROS production can prevent muscle insulin resistance. Scavenging mitochondrial H2O2, either through pharmacological means or by transgenically expressing catalase in mitochondria (MCAT mice), protects against HFD-induced muscle insulin resistance (22, 338, 747). Notably, the MCAT mice were also protected from age-induced muscle insulin resistance, associated with preserved mitochondrial activity and decreased DAG/PKCθ axis activation compared with aged wild-type mice (463). However, the therapeutic potential of targeting oxidative stress remains controversial; studies of antioxidant supplementation have yielded conflicting results (177, 549), and a systematic review of 78 randomized clinical trials of human antioxidant supplementation revealed no effect on mortality (63).
Together, these studies suggest a unified model in which, faced with chronic lipid oversupply, myocytes attempt to compensate by becoming metabolically inflexible and increasing relative lipid utilization. However, because cellular ATP demand does not increase to match substrate supply, the rate of lipid utilization is limited and is insufficient to prevent IMCL deposition. In parallel, increased relative FAO drives ROS production, which damages the mitochondria, inducing the reduced mitochondrial activity of insulin resistant muscle. These reductions in mitochondrial activity exacerbate the tendency towards positive energy balance, further favoring IMCL deposition and muscle insulin resistance (FIGURE 18B). Interestingly, several transgenic mouse models have challenged this model by inducing severe mitochondrial dysfunction and yet observing improvements in muscle glucose uptake (327). These models include mice with deletion of the mitochondrial transcription factor Tfam, mice with deletion of the mitochondrial apoptosis-inducing factor Aif, and mice with deletion of the transcriptional co-activators PGC-1α and PGC-1β (656, 913, 945). Such models force a re-evaluation of the hypothesis that decreased mitochondrial function leads to intramyocellular lipid accumulation and consequent muscle insulin resistance. One possible reconciliation is that these mechanisms predominate in the setting of relatively mild (<40%) reductions in mitochondrial oxidative phosphorylation, whereas severe mitochondrial dysfunction as seen in the aforementioned transgenic models may lead to increased anaerobic glycolysis, decreased fat oxidation, and an increase in the ADP:ATP ratio that activates AMPK-dependent glucose transport, apparently increasing insulin sensitivity. Differences in the parameter being assessed as mitochondrial function, whether in vivo ATP synthetic function, total oxidative capacity, mitochondrial density, or another readout, can also affect the conclusions drawn (42).
VII. INTEGRATED PHYSIOLOGICAL MECHANISMS OF INSULIN RESISTANCE
A. The Macrophage-Adipocyte Interaction and Inflammatory Signaling in Insulin Resistance
Human obesity is characterized by expansion of the adipose tissue; both hyperplasia and hypertrophy of adipocytes contribute to this effect. However, this expansion is a homeostatic stress and is associated with increased adipocyte cell death (562). Chemotactic signals from stressed adipocytes recruit cellular pioneers: bone marrow-derived macrophages (892, 928). These adipose tissue macrophages (ATMs) deposit in “crownlike structures” around dead adipocytes and secrete cytokines with autocrine, paracrine, and endocrine effects (562, 594). Obesity-stimulated ATMs display characteristic activation patterns that differ from those observed in the classically activated (‟M1-polarizedˮ) macrophages seen in response to, for example, bacterial infection (86, 431). Importantly, studies of mice with impaired acute inflammatory capability have revealed that acute adipose tissue inflammation is critical for proper tissue remodeling and nutrient storage in WAT; loss of this capability increases ectopic lipid storage and worsens diet-induced insulin resistance (896). Metabolically activated ATMs exocytose lysosomes to help clear dead adipocytes and employ a PPARγ-driven transcriptional program to buffer excess fatty acids (154, 431). It is chronic inflammation, then, that is the maladaptive condition associated with insulin resistance. Although it is now well-established that human obesity is a chronic inflammatory state that alters metabolic homeostasis (424, 467), the mechanisms by which inflammation may cause insulin resistance in various tissues and the importance of these processes to the development of insulin resistance and T2D remain areas of active investigation (446, 717). Here, we focus on proposed mechanistic links between inflammation and insulin resistance, primarily in WAT.
The basic model for inflammation-induced insulin resistance is a “two-hit” model in which macrophage activation is followed by macrophage elaboration of paracrine and/or endocrine factors which induce insulin resistance in target cells such as adipocytes or hepatocytes (594). Both processes have been well studied and are complex.
Both WAT resident macrophages and monocyte-derived macrophages are activated in inflamed WAT. However, multiple other immune cell types contribute to the inflammatory milieu in obese adipose tissue. Indeed, the first inflammatory cells to infiltrate WAT during high-fat feeding are not macrophages but neutrophils (212, 823). Neutrophils may serve a key role in recruiting and activating ATMs during high-fat feeding (783). Adipose B2 lymphocytes also accumulate in obese adipose tissue; their depletion mitigates HFD-induced insulin resistance in part by impairing ATM activation (937). The recruitment of ATMs may also involve the chemokine MCP-1 and its receptor CCR2 (731); genetic or pharmacological inhibition of CCR2 or MCP-1 in mice has been associated with decreased obesity-associated ATM infiltration and improved insulin sensitivity (383, 891), although another study of Ccr2−/− mice unexpectedly observed that ATM infiltration was unaltered and glucose tolerance was worsened (351). Metabolically activated ATMs are not easily classified using the traditional M1-pro-inflammatory/M2-anti-inflammatory paradigm, but rather appear to adopt a third polarization state that can be recapitulated in vitro by palmitate exposure (431). ATM activation may also be driven in part by natural killer (NK) cells residing in visceral adipose depots, which detect adipocyte stress and promote macrophage activation, possibly through interferon-γ (IFN-γ) or TNF-α (461, 895). Blocking the adipocyte-NK cell interaction, depleting NK cells, or ablating IFN-γ signaling protected mice against HFD-induced ATM activation and glucose intolerance (461, 601, 895). As discussed above, the characterization of metabolically activated ATMs as purely deleterious is an oversimplification (927); metabolically beneficial ATM functions such as lipid storage and dead adipocyte clearance are also upregulated in obesity (154, 929).
Ultimately, the mechanism by which ATM activation (the “first hit”) promotes insulin resistance is presumed to require the “second hit”–elaboration of inflammatory cytokines (594). The cytokines most commonly implicated in insulin resistance include TNF-α and interleukin (IL)-1β, although others including leukotriene B4 and galectin-3 have been the subject of recent investigation (332, 336, 364, 485, 486, 894). TNF-α was the first inflammatory cytokine revealed to be increased in obese adipose tissue in rodents and humans (332, 336). Neutralization of TNF-α improved whole-body insulin-stimulated glucose uptake in obese fa/fa rats, suggesting a mechanistic role for TNF-α in obesity-associated insulin resistance (336). Furthermore, Tnfa−/− mice were protected from HFD-induced insulin resistance, and TNF-α receptor ablation partially protected ob/ob mice from insulin resistance (865). However, more recent results demonstrating that local inhibition of TNF-α action impairs glucose tolerance indicate that the relationship between TNF-α and insulin resistance is more complex than initially suspected (896).
The leading hypothesis linking cytokines such as TNF-α to insulin resistance has long been that cytokine receptor activation in insulin target cells activates signaling pathways that directly or indirectly impair insulin action. In vitro, several cytokines including TNF-α induce insulin resistance through direct inhibition of IRK activity (333–335, 485), but the doses required to achieve inhibitory effects are often orders of magnitude higher than those measured in plasma from insulin-resistant subjects (332, 334, 335). Although paracrine effects (macrophage to adipocyte, or Kupffer cell to hepatocyte) are likely operative, quantifying paracrine signaling in vivo is difficult.
Efforts to close the mechanistic circuit linking cytokine receptor activation and impaired insulin signaling have largely converged on a pleiotropic effector of both cytokine signaling and ER stress: JNK. JNK induces a complex proinflammatory transcriptional program but also directly phosphorylates IRS1. JNK activity is increased in obese insulin-resistant liver, WAT, and skeletal muscle (318); in liver, JNK activation is present as early as 3 days after beginning a HFD (721). Jnk1−/− mice were protected from HFD-induced obesity and insulin resistance, but their increased energy expenditure, implied by their increased core body temperature, prevents attribution of their insulin-sensitive phenotype to direct effects of JNK on the insulin signaling pathway (318). A link between Jnk inhibition and increased energy expenditure is also supported by experiments using a dominant-negative JNK to block JNK activity in adipocytes and macrophages (849) and experiments using JNK-specific antisense oligonucleotides (775).
However, tissue-specific Jnk knockout mice have provided more nuanced insights into JNK biology and suggest that in some cases, insulin-sensitizing effects of Jnk deletion can be dissociated from effects on body composition (711). Adipocyte-specific Jnk deletion did not prevent body weight gain in HFD-fed mice, but did preserve hepatic insulin sensitivity, associated with protection from hepatic lipid accumulation (710). The observation that Jnk1/Jnk2 ablation in macrophages (ΦKO mice) prevented ATM infiltration and whole-body insulin resistance in high-fat-fed mice pointed to a key role for JNK in establishing the proinflammatory state in macrophages and provided further evidence for the importance of macrophage activation in HFD-induced insulin resistance (302). However, ΦKO mice displayed similar improvements in whole-body insulin sensitivity on regular chow diet (302). Because increased tissue JNK activity and ATM infiltration are characteristics of high-fat feeding (302, 318), it is not clear why the metabolic phenotype of ΦKO mice was similarly pronounced on both regular chow and high-fat diets. Nevertheless, one interpretation of these data is that JNK activation in adipocytes and macrophages contributes to HFD-induced insulin resistance in distant tissues such as liver.
In skeletal muscle, JNK activation is not sufficient for obesity-associated insulin resistance (606a), and reports conflict as to whether skeletal muscle-specific Jnk deletion protects from obesity-associated insulin resistance (606a, 712). In the one study of muscle-specific Jnk deletion that did observe protection from HFD-induced muscle insulin resistance, muscle LpL expression was also decreased, raising the possibility that the insulin-sensitizing phenotype might be attributable to protection from lipotoxicity rather than direct effects of JNK to inhibit proximal insulin signaling (712). Additionally, liver-specific Jnk deletion does not protect mice from hepatic insulin resistance but instead promotes hepatosteatosis and glucose intolerance (709). Furthermore, the canonical mechanism by which JNK activation is proposed to impair cellular insulin signaling–IRS1 Ser307 phosphorylation–is unlikely to mediate insulin resistance in vivo. Knock-in IRS1 Ser307Ala mice display worsened, not preserved, insulin action (167); the phosphomimetic IRS1 Ser307Asp mutation also fails to impair insulin signaling (888). Alternative mechanisms for JNK-induced insulin resistance, including transcriptional mechanisms, require further study. Together, the emerging picture linking JNK to insulin resistance is not one of direct insulin signaling blockade in target tissues like muscle and liver, but rather, perhaps, one of indirect effects involving the macrophage-adipocyte-hepatocyte axis.
Indeed, an emerging paradigm posits that cytokine-induced lipolysis mediates the link between inflammation and insulin resistance. HFD-fed ΦKO mice displayed reduced palmitate and glycerol turnover (i.e., reduced lipolysis) during hyperinsulinemic-euglycemic clamps compared with HFD-fed wild-type controls, associated with improved suppression of hepatic glucose production (620). TNF-α may promote lipolysis by decreasing expression of perilipin and/or fat-specific protein 27 (FSP27), lipid droplet proteins thought to control the access of lipases to the adipocyte lipid droplet (455, 681). Furthermore, the increased plasma NEFA concentrations of ob/ob mice were rescued to the levels of lean mice by TNF-α loss (865). Importantly, however, the increased fatty acid turnover of WAT in chronic obesity likely owes not only to cytokine-mediated lipolysis but also to fatty acid spillage from dead adipocytes. The importance of this latter mechanism was highlighted by studies of whole-body and myeloid-specific Nox2−/− mice, which lack functional metabolically activated ATMs (154). These mice accumulated dead adipocytes and developed profound hepatosteatosis and insulin resistance (154). Overall, adipose lipolysis, whether nutrient stress induced or inflammation induced, may cause insulin resistance by increasing lipid delivery to skeletal muscle and liver, activating pathways of lipid-induced insulin resistance (e.g., the DAG/nPKC axis) and also promoting hepatic gluconeogenesis through acetyl CoA activation of pyruvate carboxylase (620). This hypothesis unifies the paradigms of lipid-induced insulin resistance and inflammation-induced insulin resistance and points to an integrated physiological mechanism regulating the cell-autonomous defects of lipid-induced insulin resistance.
Is inflammation a primary insult in T2D, or rather an exacerbating factor? Several lines of evidence suggest that adipose tissue inflammation is not necessary for adipose insulin resistance. For example, adipose tissue insulin resistance is detectable in rodents even after 1 wk of high-fat feeding (115, 722), but adipocyte death and ATM infiltration are minimal even after 4 wk of HFD and do not become prominent until 12 wk of HFD (582, 807). Genomic evidence also argues against the classification of T2D as an inflammatory disease. Large-scale analysis of disease-related single nucleotide polymorphisms (SNPs) cross-referenced to cell type-specific epigenetic regulatory activity revealed robust clustering of all known autoimmune and inflammatory diseases examined (226). T2D SNPs decidedly did not share this enrichment in lymphoid or myeloid cell types, indicating that the strong heritability of T2D risk (599) is likely not mediated by genetic effects in inflammatory cells such as macrophages (226). Adipose tissue inflammation is also not required for insulin resistance; multiple models of partial and complete lipodystrophy manifest severe insulin resistance in the absence of significant ATM activation (406, 642, 957). A particularly interesting case of this phenomenon involves mice lacking FSP27, also known as cell death-inducing DFFA-like effector c (CIDEC) (825, 957). When subjected to energy storage stresses (high-fat feeding, leptin deficiency, or lack of brown adipose tissue), Fsp27−/− mice failed to expand their adipose depots or develop significant ATM infiltration but nevertheless developed severe hepatic insulin resistance with profound hepatic steatosis (825, 957). Furthermore, adipose insulin resistance induced by adipose-specific deletion of the obligate mTORC2 component Rictor led to increased MCP-1 expression and macrophage infiltration, suggesting that adipose insulin resistance is sufficient to induce adipose tissue inflammation (762). Together, these lines of evidence suggest that although adipose tissue inflammation can accompany and exacerbate obesity-associated insulin resistance, it is likely not the primary defect.
Several anti-inflammatory agents in clinical use have been evaluated for efficacy in T2D (271). Marked species differences between rodent and human inflammation and integrated insulin action may underlie the wildly disparate results obtained with some such agents in mice and men. For example, the TNF-α antagonists etanercept and infliximab have not shown a consistent insulin-sensitizing effect in human trials despite good efficacy in mice (25, 56, 467, 795, 885). Although the salicylate prodrug salsalate has achieved modest and consistent glucose lowering in randomized human trials, the ability of salicylate to directly activate AMPK and induce mitochondrial uncoupling renders an anti-inflammatory mechanism unnecessary for its antihyperglycemic effects (268–270, 303, 780). The anti-inflammatory agent amlexanox, which inhibits the obesity-upregulated kinases IKKε and TBK1, reverses adipose tissue inflammation and obesity in mice by increasing energy expenditure, consistent with the role of TBK1 as a negative regulator of AMPK and energy balance (687, 955). In a small randomized double-blind clinical trial in patients with T2D, 12 wk of amlexanox achieved small (<0.5%) but statistically significant reductions in glycated hemoglobin (600). Interestingly, response to amlexanox was strongly related to reductions in IHTG (600). The relative contributions of reductions in adipose tissue inflammation and increases in energy expenditure to the glycemic improvements observed with amlexanox is uncertain, although C-reactive protein levels tended to be higher at baseline in subjects who had a good response to amlexanox treatment (600). However, serum IL-6 levels were also increased in responders, a finding at odds with the frequently (though not universally) metabolically deleterious effects of this cytokine (403). This example highlights the complex interplay between the metabolically beneficial and metabolically harmful effects of inflammation, which may complicate ongoing efforts to test other anti-inflammatory agents for antidiabetic effects (271, 467, 716).
B. Circulating Branched-Chain Amino Acids and Insulin Resistance
In 1969, measurements of all amino acids in human plasma revealed that concentrations of all three branched-chain amino acids (BCAAs; valine, leucine, and isoleucine) were elevated in obese subjects compared with lean controls and positively correlated with fasting insulin (228). The interaction between plasma amino acids and glucose homeostasis is complex, with effects on insulin secretion, hepatic glucose production, and peripheral glucose disposal all well-established (72, 238, 435). Interest in the link between BCAAs and insulin resistance has been reinvigorated in the 21st century by metabolomics methods that have confirmed a strong association between the HOMA-IR score and circulating BCAA concentration (129, 516, 577, 918). Furthermore, plasma BCAA concentrations predict future T2D risk (608, 880, 919), and genomic variants that increased BCAA levels were associated with T2D in a Mendelian randomization study (502). Adipose tissue BCAA catabolic enzyme expression is consistently decreased in rodent and human models of obesity, providing a putative etiology for the obesity-associated increase in circulating BCAAs (312, 508, 751, 759).
With these associations established, a key question became whether BCAAs actively modulate or passively reflect insulin sensitivity. This is an area of active investigation and controversy (508, 576). BCAA supplementation alone is insufficient to induce insulin resistance in regular chow-fed rats, but contributes to insulin resistance in high-fat-fed rats (577). Two main mechanisms have been proposed by which BCAAs may impair insulin action. In one, chronic BCAA activation of mTOR, which senses leucine (733), provides negative feedback to insulin signaling at the level of IRS1 (577). The mTORC1-activated ribosomal S6K1 is chronically activated in obese and diabetic rodents and humans (846) and is a known negative regulator of IRS signaling (339, 846, 859, 860, 948). Furthermore, the insulin resistance of high-fat-fed/BCAA-supplemented rats was reversed by treatment with the mTORC1 inhibitor rapamycin (577). However, activation of the BCAA/mTORC1 axis has also been associated with improved insulin sensitivity (e.g., exercise or BCAA supplementation) (508, 509, 954). Additionally, mice with deletion of mitochondrial BCAA transaminase (BCATm) display profoundly increased plasma BCAA concentrations but are protected from HFD-induced obesity and insulin resistance (758), arguing that BCAA-mediated effects such as mTOR activation alone are insufficient to produce insulin resistance.
In the other proposed mechanism for BCAA-induced insulin resistance, BCAAs themselves are not the culprit. Rather, the bioactive moieties are proposed to be BCAA catabolic products (e.g., propionyl CoA, succinyl CoA, branched-chain ketoacids). In one hypothesis, increased BCAA turnover drives the production of toxic mitochondrial BCAA catabolites, which are in turn proposed to impair mitochondrial oxidative metabolism (508, 576) and thereby induce mitochondrial stress, activating stress kinases such as JNK that have been linked to insulin resistance (576, 711). Evidence for this hypothesis, especially knowledge of precisely how BCAA catabolism is altered in human insulin resistance, is currently limited. Recently, the BCAA catabolic product 3-hydroxyisobutyrate (3-HIB; a valine catabolite) has been identified as a paracrine positive regulator of myocellular fatty acid uptake (366). 3-HIB was shown to be secreted from myocytes and to promote transendothelial fatty acid transport and myocellular lipid uptake; mice given 3-HIB-supplemented drinking water developed DAG/PKCθ axis activation in skeletal muscle and impaired glucose tolerance (366). 3-HIB levels were shown to be elevated in muscle from db/db mice and T2D humans, suggesting physiological relevance (366). This intriguing mechanism thus links BCAA-induced insulin resistance with lipid-induced insulin resistance.
An alternative hypothesis proposes that elevated plasma BCAAs are a consequence of insulin resistance. This hypothesis is supported by a human genetic study in which risk alleles for insulin resistance were associated with higher plasma BCAA levels, but risk alleles for higher BCAA levels were not associated with higher HOMA-IR scores (516). Mechanistically, a potentially trivial explanation is that resistance to insulin’s anabolic effects on protein synthesis results in increased circulating BCAA levels; however, insulin suppression of leucine turnover has been reported to be similar in T2D patients and controls (299, 507). It has also been proposed that, rather than BCAA catabolism promoting oxidative stress, lipid availability and usage may induce oxidative stress that inhibits BCAA catabolic enzymes (508). It is also possible that the decreased adipose expression of BCAA catabolic enzymes which drives the obesity-associated increase in plasma BCAAs is secondary to insulin resistance (508). Additionally, because BCAA catabolism provides acetyl CoA substrate for lipogenesis (an estimated 30% of lipogenic acetyl CoA in differentiated 3T3-L1 adipocytes is derived from BCAA catabolism) (281), the decreased adipose BCAA catabolic enzyme expression of obesity may impair proper nutrient storage in adipose tissue, promoting ectopic lipid deposition in tissues such as skeletal muscle with preserved BCAA catabolic capacity in obesity. Although this latter mechanism is attractive for its unification of the BCAA metabolic signature with established lipotoxic mechanisms of insulin resistance, it lacks direct experimental support. With many important questions currently unanswered, this field is ripe for future investigation.
C. Adipokines and Hepatokines in Insulin Resistance
The explosion of newly identified secreted peptide hormones in the past 20 yr calls to mind the early 20th century, when investigators in the nascent discipline of endocrinology were rewarded with the discoveries of insulin, sex steroids, thyroxine, and other fundamental hormones (65). The identification of new hormones, now as then, is often received with great excitement and therapeutic hope. For many new hormones, however, pathophysiological significance and therapeutic potential are still uncertain. Here, we briefly highlight several of the best-studied new hormones, focusing on the evidence for their role in human insulin resistance.
In this review, we have highlighted the role of WAT and liver as master controllers of substrate storage and delivery. However, they are also prolific endocrine organs, and several adipokines and hepatokines have been implicated in human insulin resistance. Here we limit our discussion to retinol binding protein 4 (RBP4), adiponectin, fetuin-A (FetA), and FGF21, four circulating mediators with particularly strong evidence of relevance to human insulin resistance.
RBP4 was identified in 2005 as a gene with reciprocal transcriptional regulation in adipose GLUT4-overexpressing and adipose GLUT4-null mice (935). RBP4 is a secreted protein produced by both liver and WAT, but the liver is the source of nearly all circulating RBP4 in mice (838). In mice, transgenic overexpression of Rbp4 or chronic RBP4 administration induces whole-body insulin resistance, and Rbp4 deletion improves insulin action (935). In three distinct human cohorts, serum RBP4 levels correlated with body mass index (BMI), fasting plasma insulin, and impaired insulin-stimulated peripheral glucose uptake during hyperinsulinemic-euglycemic clamp studies (278). Furthermore, the insulin-sensitizing effect of exercise was highly correlated with the extent of the decrease in serum RBP4 concentration (278, 425). The increased adipose RBP4 expression of human obesity is associated with an approximate doubling of serum RBP4 concentration (277, 278). The mechanism by which RBP4 impairs peripheral insulin action is not clear. Proposed mechanisms include direct activation of hepatocellular lipogenic programs (924) and activation of adipose tissue macrophages (ATMs) (553, 584). The latter mechanism is supported by studies employing transfer of RBP4-treated dendritic cells into wild-type mice; this intervention increased ATM infiltration and worsened glucose tolerance (553). This paradigm unifies RBP4 with lipolysis-driven peripheral and hepatic insulin resistance.
Adiponectin was discovered in 1995 as an adipocyte-specific secreted protein that circulates in high-molecular-weight oligomers at a concentration of 5–10 μg/ml (51, 736). Although this concentration is quite high by endocrine hormone standards, adiponectin’s oligomeric quarternary structure means that its molar concentration is only 10–30 nM (51). Adiponectin concentrations are consistently lower in obese humans, but generally remain in the 5–10 μg/ml range (337, 850). Acute elevation of plasma adiponectin concentrations >100 μg/ml reduces hepatic glucose production, but the physiological significance of this effect is unclear (51). The modestly decreased circulating adiponectin levels in humans with metabolic disease are a reproducible finding and are widely used as a biomarker for adipose tissue dysfunction, although it is unclear to what extent these small changes contribute to systemic metabolic disease. For example, the lean offspring of parents with T2D are insulin resistant but display unaltered plasma adiponectin levels (636). Mechanistically, attempts to attribute the glucose-lowering effects of adiponectin to AMPK activation (932) are challenged by observations that AMPK activation is insufficient to suppress HGP (511) and that adiponectin still suppresses HGP in mice lacking the AMPK activator LKB1 in liver (324, 542).
An intriguing link between adiponectin and other paradigms of insulin resistance was revealed in the 2011 report that activation of the adiponectin receptors AdipoR1 and AdipoR2 induces a ceramidase (i.e., ceramide-degrading) activity (324). Although likely not sufficient to explain the acute suppression of HGP by adiponectin, liver-specific ceramidase activation in the chronic setting is sufficient to prevent hepatosteatosis and HFD-induced hepatic insulin resistance (922). These findings provide one possible mechanism for the many models linking adiponectin to insulin sensitivity, such as the ob/ob adiponectin transgenic mice, which display approximately threefold increased circulating adiponectin and normal insulin sensitivity despite weighing up to twice as much as obese ob/ob controls (408). Remarkably, acute inducible deletion of adiponectin is sufficient to rapidly (within 2 wk) induce hepatic insulin resistance, in concert with increased hepatic lipid uptake and increased hepatic ceramides (923). Together, these results suggest that AdipoR1/2 agonism might be an effective strategy to treat insulin resistance, and indeed, such an agonist, AdipoRon, has been reported to have beneficial effects in high-fat-fed and db/db mice (590). Although AdipoRon did not affect weight gain in these models, it improved glucose tolerance and insulin sensitivity associated with reduced liver and muscle triglyceride content; ceramide levels were not specifically reported (590). The pleiotropic effects of adiponectin are truly staggering (851, 936), and potential intersections with insulin resistance include modulation of ectopic lipid content, ceramide levels, and adipose tissue inflammation. Although the molecular mechanisms of adiponectin action remain incompletely understood, and the contribution of reduced adiponectin levels to human disease remains uncertain, adiponectin agonism may nevertheless be a promising space for therapeutic innovation.
We previously considered SFAs in section V as ceramide precursors. In that context, SFAs have been proposed to promote insulin resistance by providing substrate for ceramide biosynthesis (323). However, another proposed mechanism for SFA-induced insulin resistance is activation of Toll-like receptor 4 (TLR4) signaling (761). Mechanistically, the link between SFA-TLR4 signaling and insulin resistance has been proposed to involve transcriptional upregulation of ceramide biosynthetic enzymes (62). Although SFAs are not ligands for TLR4 (734), the fatty acid-binding glycoprotein FetA was later shown to be such a ligand and to thus link SFAs to TLR4 activation (606). FetA is a hepatokine, produced by the liver and proposed to activate SFA-TLR4 signaling in adipocytes; this in turn is thought to promote inflammatory cytokine production (606). In support of this hypothesis, mice lacking FetA are protected from diet-induced insulin resistance (525). In humans, serum FetA and NEFA concentrations show a statistical interaction; when either FetA or NEFA levels are high; higher levels of the other predict impaired glucose tolerance (798). By this mechanism, adipocyte-hepatocyte crosstalk is proposed to be bidirectional: the hepatokine FetA induces adipokines like IL-6 and TNF-α, which may in turn modulate hepatocellular signaling pathways (797). Although an intriguing paradigm, the overall pathophysiological significance of the SFA-TLR4 axis is still uncertain. In one study, female Tlr4−/− mice were protected from HFD-induced insulin resistance (though males were not) (761). In a separate report, neither Tlr4−/− mice nor mice with knockdown of TLR4 or adapter protein MyD88 were protected from SFA-induced ceramide accumulation, DAG/PKCε axis activation, or hepatic insulin resistance (253). Additionally, FetA has been shown to directly impair INSR tyrosine kinase activity (indeed, it was discovered in this context) (30, 524, 793), so it may not be necessary to invoke TLR4 signaling to explain the effect of FetA on cellular insulin action.
Finally, the hepatokine FGF21 has received tremendous interest for its ability to increase FAO and energy expenditure and to decrease plasma lipids (738, 797). Pharmacological doses of FGF21 improve insulin sensitivity even in lean, chow-fed mice and reverse insulin resistance in fat-fed mice, effects attributable in part to reduced ectopic lipid accumulation (112). But with species-specific, tissue-specific, and dose-dependent roles in seemingly contradictory physiological processes, a complete understanding of FGF21 physiology is still elusive (233). For example, although rodent studies have largely focused on FGF21 as a mediator of the physiological response to fasting, plasma FGF21 concentrations in humans are not increased until 10 days of fasting (227, 233). Additionally, FGF21 levels are paradoxically increased in obese and insulin-resistant humans, suggesting that obesity may be an FGF21-resistant state (123, 953). Indeed, FGF21 resistance, as measured by signaling responses, is apparent in obese mice (232). Because the fatty acid-regulated transcription factor PPARα positively regulates FGF21 expression, it has been proposed that the hepatosteatosis of obesity drives increases in circulating FGF21, which in turn promotes the FGF21 resistance of obesity (233). However, just as pharmacological insulin dosing achieves glycemic control despite insulin resistance in people with T2D, FGF21 and FGF21 analogs may have potential for the treatment of hepatic steatosis and nonalcoholic steatohepatitis (NASH) in humans despite FGF21 resistance (252, 264, 824). Unfortunately, osteopenia is a major on-target adverse effect that may limit the clinical utility of FGF21 or its analogues (824, 893).
VIII. SUMMARY
The sheer complexity of biological systems means that any effort to understand insulin resistance with a unified, succinct, and straightforward model may be a fool’s errand. Certainly normal insulin action, despite sharing important effectors among different cell types, performs myriad functions that are not particularly amenable to encapsulation. In particular, understanding the intricate relationship between insulin control of both lipid and carbohydrate metabolism has proved a worthy challenge for generations of investigators (532). But in considering the several putative mediators of insulin resistance discussed in the preceding sections, it is tempting both to note potential areas of unification and to veer into teleological speculation.
The fundamental element linking all putative mediators of insulin resistance is a relationship to nutrient oversupply. Each mechanism discussed in this review is proposed to cause insulin resistance by either increasing nutrient-derived toxic metabolites (DAG, ceramide, acylcarnitine, BCAA), overdriving nutrient utilization processes (ER stress, oxidative stress), or responding to nutrient stress-mediated cellular toxicity (inflammation). Moreover, the pathophysiology of insulin resistance driven by cellular stress pathways and by inflammation shares common threads with the insulin resistance induced by bioactive lipids. ER stress promotes de novo lipogenesis. The mitochondrial dysfunction of aged and insulin-resistant humans facilitates positive energy balance and ectopic lipid storage. Adipose tissue inflammation drives lipolysis, increasing substrate delivery to nonadipose tissues. We therefore propose an integrated model of insulin resistance in which several simultaneous responses to nutrient oversupply converge and collide to facilitate ectopic lipid accumulation and consequent insulin resistance in skeletal muscle and liver (FIGURE 19).
FIGURE 19.

An integrated physiological perspective on tissue insulin resistance. Chronic overnutrition is the ultimate cause of systemic insulin resistance and promotes insulin resistance by both tissue-autonomous and crosstalk-dependent mechanisms. Chronic overnutrition promotes lipid accumulation in skeletal muscle and liver, which causes insulin resistance in those tissues. Additionally, chronic overnutrition poses a nutrient stress to adipocytes, resulting in adipocyte insulin resistance and adipocyte death. Increases in the adipokine RBP4 and other proinflammatory signals lead to the recruitment of macrophages to white adipose tissue. Inflammatory signaling in macrophages, including activation of c-Jun NH2-terminal kinase (JNK), leads to the elaboration of paracrine mediators such as tumor necrosis factor-α (TNFα), interleukin-1β (IL-1β), and others. These inflammatory cytokines may increase adipocyte lipolysis either directly or indirectly by impairing insulin signaling. The increased adipocyte lipolysis of inflammation increases nonesterified fatty acid (NEFA) and glycerol turnover. This has direct (glycerol conversion to glucose) and indirect [NEFA-derived acetyl CoA activation of pyruvate carboxylase (PC)] stimulatory effects on gluconeogenesis, and also promotes accumulation of intrahepatic triglyceride (IHTG) and consequent lipid-induced hepatic insulin resistance, which impairs insulin stimulation of net hepatic glycogen synthesis. Together, these effects increase hepatic glucose production. Chronically increased lipolysis may also facilitate the accumulation of intramyocellular lipid (IMCL) and consequent lipid-induced muscle insulin resistance. The decreased glucose disposal of muscle insulin resistance increases glucose availability for the liver, which in turn promotes IHTG accumulation and worsens hepatic insulin resistance.
If overnutrition is the central driver of all these metabolic defects, then the most obvious therapeutic option is calorie restriction. Although the cellular effects of caloric restriction are complex and incompletely understood, the physiological effects of applying a hypocaloric diet to an obese insulin-resistant subject represent a useful test of the hypotheses presented in this review. Recently, Perry et al. (625) catalogued the metabolic consequences of a 3-day very-low-calorie diet (VLCD; 25% of normal caloric intake) in a rat model of insulin-resistant T2D (4 wk of high-fat feeding or Western diet followed by low-dose streptozotocin/nicotinamide to achieve fasting hyperglycemia). Without significantly reducing body weight, VLCD achieved near-normalization of plasma glucose and insulin levels. This was associated with reductions in IHTG, hepatic acetyl CoA, hepatic membrane-associated DAG, and hepatic PKCε activation; parameters that did not change included hepatic ceramides, plasma glucagon, a panel of inflammatory cytokines, plasma FGF21, plasma BCAAs, and hepatic ER stress markers (625). In hyperinsulinemic-euglycemic clamp studies, VLCD resulted in increased AKT activation and insulin suppression of HGP (625). Interestingly, both direct and indirect components of hepatic insulin action were improved by VLCD; the improvements in HGP seen with VLCD could be abrogated by acetate infusion (to prevent VLCD-induced decreases in hepatic acetyl CoA) or recapitulated by a glycogen phosphorylase inhibitor (to simulate VLCD-induced improvements in insulin-stimulated hepatic glycogen synthesis) (625). The utility of this rapid intervention is that it helps to distinguish the parameters that drive hyperglycemia from those that are secondary consequences or exacerbating factors. The results incriminate hepatic DAG-PKCε axis activation and metabolite-driven gluconeogenesis.
Yet all studies have limitations, and a major limitation of the above study is one shared by much of the work cited in this review: the use of a rodent model to draw inferences about human pathophysiology. One of the rodent-human differences most germane to the study of insulin resistance is the order in which tissues develop insulin resistance upon overnutrition. In rodents, just a few days of high-fat feeding is sufficient to cause hepatic steatosis and hepatic insulin resistance; skeletal muscle insulin resistance requires several weeks to develop (429). In those weeks, meanwhile, WAT expands and eventually becomes inflamed, stimulating lipolysis and in turn hepatic gluconeogenesis (620). In humans, available evidence points to skeletal muscle insulin resistance as the first defect; the young, healthy, lean offspring of type 2 diabetics display skeletal muscle insulin resistance but normal IHTG and normal hepatic insulin action (639). Muscle insulin resistance promotes hepatic lipogenesis, however, and eventually NAFLD and hepatic insulin resistance develop. How adipose insulin resistance fits into this paradigm in humans remains relatively uncertain.
Indeed, adipose tissue insulin resistance is a particularly exciting topic of active exploration (176). WAT is adapted to store excess energy and can do so prolifically without inducing metabolic derangements [evidenced most dramatically by the adiponectin transgenic ob/ob mouse, which remains normally insulin sensitive despite morbid obesity (408)]. As a result, some paradigms of nutrient stress well characterized in skeletal muscle and liver, such as lipid-induced insulin resistance, do not obviously translate to the white adipocyte. Gross measurement of tissue lipids such as DAG in a cell type that is at least 50% lipid by mass (528) is unlikely to reveal meaningful information. Yet chronic overnutrition clearly constitutes a stress for adipocytes; the crownlike structures that blemish obese WAT are adipocyte tombstones. The development of new techniques to study the spatiotemporal compartmentalization of metabolic fluxes may enable investigation of the seemingly paradoxical hypothesis that a cell specialized for lipid storage could also be vulnerable to lipotoxicity. Inflammation is a prominent putative mediator of adipose insulin resistance, but HFDs seem to cause adipose insulin resistance before the development of detectable adipose inflammation. ER stress is present in obese WAT and appears related to lipolysis, but this potential link requires further study. The essential role of adipose tissue in the integrated physiology of insulin action has been a major theme of metabolic research in the 21st century (735), yet a deeper mechanistic understanding of the underpinnings of adipose tissue insulin resistance is urgently needed.
Physiological systems seek homeostasis (424), so the teleology of insulin resistance should be viewed through a homeostatic lens. However, because evolution did not occur in an environment of permanent caloric surplus, it is also possible that the physiological consequences of insulin resistance at best are meant to be temporary and at worst are adventitious. Mechanistically, the phosphorylation events and other signaling phenomena that produce insulin resistance may represent appropriately activated negative-feedback mechanisms. More insidiously, the phosphorylation events mediating insulin resistance could result from a coopting of normal negative-feedback mechanisms by pathologically activated kinases not normally essential to the insulin response (e.g., nPKCs) but evolutionarily similar enough to the kinases mediating normal negative feedback (e.g., S6K1) to phosphorylate similar substrates in the right context. This could only have occurred if a permanently overnourished cellular milieu was rare enough in evolutionary history to not exert significant selection pressure against this adventitious pathological use of physiological negative-feedback mechanisms. Although this scenario is possible, there are nevertheless several viable hypotheses treating insulin resistance as an appropriate, nonaccidental homeostatic mechanism.
Plausibly, insulin resistance could have evolved as a cell-autonomous protective response to nutrient oversupply. The organellar nutrient stress responses–mitochondrial oxidative stress, ER stress–highlight the threat to cell survival that nutritional oversupply poses. Just as ER stress induces signaling cascades that restore homeostasis by promoting membrane synthesis and reducing new protein synthesis, so insulin resistance might be an appropriate response to nutrient oversupply that limits glucose utilization, macromolecule synthesis, and other anabolic processes. In a nutrient-replete cell, continued anabolism may have greater costs (e.g., oxidative and ER stresses) than benefits.
Another possibility is that insulin resistance represents a multi-organ physiological response that benefits the organism by promoting calorie storage in WAT in times of nutritional plenty. Shutting down insulin-stimulated anabolic fluxes in muscle and liver should shunt substrate to alternative destinations such as WAT. The chief flaw in this conception is that WAT also becomes insulin resistant upon overnutrition, although perhaps through different mechanisms than liver and skeletal muscle.
A final interesting teleology that applies to lipid-induced insulin resistance in particular treats insulin resistance as a beneficial adaptation to fasting. Fasting promotes lipolysis and increases relative lipid utilization; as a result, fasting induces triglyceride accumulation in liver and skeletal muscle (306, 796). Fasting has been shown to induce profound skeletal muscle insulin resistance in humans (519, 887); this does not occur in mice (306), but mouse studies are complicated by the significant body composition changes (~10% body weight loss) induced by even overnight fasting. Lipid-induced insulin resistance in liver and skeletal muscle could serve a useful purpose during fasting by favoring gluconeogenesis, minimizing glycogen storage, and conserving glucose for the CNS. Consistent with this hypothesis, DAG accumulation, PKC activation, and inhibition of IRK activity have been observed in starved rat liver (387, 628). Recently, a remarkable study of river-dwelling and cave-dwelling populations of the Mexican tetra fish provided evolutionary support for the starvation-adaptation hypothesis of insulin resistance (690). Fish adapted to the nutrient-scarce cave environment were markedly insulin resistant, in many cases due to a partial loss-of-function mutation in the insulin receptor (690). The cavefish develop larger fat depots and are better able to maintain their body weight during starvation (690). Although the cavefish have other possibly independent metabolic adaptations such as decreased metabolic rate and altered circadian periodicity, these observations suggest that insulin resistance is evolutionarily selected for in nutrient-limited environments. However, other evidence for this hypothesis is lacking, partially due to the marked dissimilarity in fasting responses between mice and humans. Additionally, fasting is a hypoinsulinemic state in which the physiological relevance of insulin resistance is not obvious beyond serving to prime the response to refeeding.
Regardless of its physiological provenance, insulin resistance is maladaptive in the setting of chronic overnutrition. Understanding insulin action and resistance more completely will facilitate the intelligent use of existing antidiabetic therapies, enable the development of new therapeutics, and, perhaps most importantly, inform prevention strategies to stem the tide of type 2 diabetes.
GRANTS
M. C. Petersen is supported by National Institutes of Health (NIH) Medical Scientist Training Program Grant T32 GM-007205 and National Research Service Award F30 DK-104596. G. I. Shulman is supported by NIH Grants R01 DK116774, R01 DK113984, R01 DK114793, and P30 DK-45735.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
ACKNOWLEDGMENTS
We are grateful to Brandon Gassaway, Daniel Vatner, Michael Jurczak, and Varman Samuel for helpful discussions. We apologize to colleagues whose work was not discussed owing to the large scope of the review.
Address for reprint requests and other correspondence: G. I. Shulman, Depts. of Internal Medicine and Cellular & Molecular Physiology, Howard Hughes Medical Institute, Yale University School of Medicine, New Haven, CT 06520 (e-mail: gerald.shulman@yale.edu).
REFERENCES
- 1.Abel ED, Peroni O, Kim JK, Kim YB, Boss O, Hadro E, Minnemann T, Shulman GI, Kahn BB. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 409: 729–733, 2001. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- 2.Abraham MA, Filippi BM, Kang GM, Kim M-S, Lam TKT. Insulin action in the hypothalamus and dorsal vagal complex. Exp Physiol 99: 1104–1109, 2014. doi: 10.1113/expphysiol.2014.079962. [DOI] [PubMed] [Google Scholar]
- 3.Abulizi A, Perry RJ, Camporez JPG, Jurczak MJ, Petersen KF, Aspichueta P, Shulman GI. A controlled-release mitochondrial protonophore reverses hypertriglyceridemia, nonalcoholic steatohepatitis, and diabetes in lipodystrophic mice. FASEB J 31: 2916–2924, 2017. doi: 10.1096/fj.201700001R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adams JM II, Pratipanawatr T, Berria R, Wang E, DeFronzo RA, Sullards MC, Mandarino LJ. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 53: 25–31, 2004. doi: 10.2337/diabetes.53.1.25. [DOI] [PubMed] [Google Scholar]
- 5.Adams SH, Hoppel CL, Lok KH, Zhao L, Wong SW, Minkler PE, Hwang DH, Newman JW, Garvey WT. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid β-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J Nutr 139: 1073–1081, 2009. doi: 10.3945/jn.108.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ader M, Bergman RN. Peripheral effects of insulin dominate suppression of fasting hepatic glucose production. Am J Physiol Endocrinol Metab 258: E1020–E1032, 1990. [DOI] [PubMed] [Google Scholar]
- 7.Adina-Zada A, Zeczycki TN, St Maurice M, Jitrapakdee S, Cleland WW, Attwood PV. Allosteric regulation of the biotin-dependent enzyme pyruvate carboxylase by acetyl-CoA. Biochem Soc Trans 40: 567–572, 2012. doi: 10.1042/BST20120041. [DOI] [PubMed] [Google Scholar]
- 8.Aerts JM, Ottenhoff R, Powlson AS, Grefhorst A, van Eijk M, Dubbelhuis PF, Aten J, Kuipers F, Serlie MJ, Wennekes T, Sethi JK, O’Rahilly S, Overkleeft HS. Pharmacological inhibition of glucosylceramide synthase enhances insulin sensitivity. Diabetes 56: 1341–1349, 2007. doi: 10.2337/db06-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agius L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem J 414: 1–18, 2008. doi: 10.1042/BJ20080595. [DOI] [PubMed] [Google Scholar]
- 10.Aguer C, McCoin CS, Knotts TA, Thrush AB, Ono-Moore K, McPherson R, Dent R, Hwang DH, Adams SH, Harper M-E. Acylcarnitines: potential implications for skeletal muscle insulin resistance. FASEB J 29: 336–345, 2015. doi: 10.1096/fj.14-255901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahmad F, Degerman E, Manganiello VC. Cyclic nucleotide phosphodiesterase 3 signaling complexes. Horm Metab Res 44: 776–785, 2012. doi: 10.1055/s-0032-1312646. [DOI] [PubMed] [Google Scholar]
- 12.Ahmadian M, Abbott MJ, Tang T, Hudak CSS, Kim Y, Bruss M, Hellerstein MK, Lee H-Y, Samuel VT, Shulman GI, Wang Y, Duncan RE, Kang C, Sul HS. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab 13: 739–748, 2011. doi: 10.1016/j.cmet.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aiston S, Hampson LJ, Arden C, Iynedjian PB, Agius L. The role of protein kinase B/Akt in insulin-induced inactivation of phosphorylase in rat hepatocytes. Diabetologia 49: 174–182, 2006. doi: 10.1007/s00125-005-0068-4. [DOI] [PubMed] [Google Scholar]
- 14.Albert JS, Yerges-Armstrong LM, Horenstein RB, Pollin TI, Sreenivasan UT, Chai S, Blaner WS, Snitker S, O’Connell JR, Gong D-W, Breyer RJ III, Ryan AS, McLenithan JC, Shuldiner AR, Sztalryd C, Damcott CM. Null mutation in hormone-sensitive lipase gene and risk of type 2 diabetes. N Engl J Med 370: 2307–2315, 2014. doi: 10.1056/NEJMoa1315496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alemany S, Cohen P. Phosphorylase a is an allosteric inhibitor of the glycogen and microsomal forms of rat hepatic protein phosphatase-1. FEBS Lett 198: 194–202, 1986. doi: 10.1016/0014-5793(86)80404-5. [DOI] [PubMed] [Google Scholar]
- 16.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15: 6541–6551, 1996. [PMC free article] [PubMed] [Google Scholar]
- 17.Ali AH, Mundi M, Koutsari C, Bernlohr DA, Jensen MD. Adipose Tissue Free Fatty Acid Storage In Vivo: Effects of Insulin Versus Niacin as a Control for Suppression of Lipolysis. Diabetes 64: 2828–2835, 2015. doi: 10.2337/db14-1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alquier T, Poitout V. Considerations and guidelines for mouse metabolic phenotyping in diabetes research. Diabetologia 61: 526–538, 2018. doi: 10.1007/s00125-017-4495-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alves TC, Befroy DE, Kibbey RG, Kahn M, Codella R, Carvalho RA, Falk Petersen K, Shulman GI. Regulation of hepatic fat and glucose oxidation in rats with lipid-induced hepatic insulin resistance. Hepatology 53: 1175–1181, 2011. doi: 10.1002/hep.24170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amati F. Revisiting the diacylglycerol-induced insulin resistance hypothesis. Obes Rev 13, Suppl 2: 40–50, 2012. doi: 10.1111/j.1467-789X.2012.01036.x. [DOI] [PubMed] [Google Scholar]
- 21.Amati F, Dubé JJ, Alvarez-Carnero E, Edreira MM, Chomentowski P, Coen PM, Switzer GE, Bickel PE, Stefanovic-Racic M, Toledo FGS, Goodpaster BH. Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: another paradox in endurance-trained athletes? Diabetes 60: 2588–2597, 2011. doi: 10.2337/db10-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin C-T, Price JW III, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119: 573–581, 2009. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andrikopoulos S, Blair AR, Deluca N, Fam BC, Proietto J. Evaluating the glucose tolerance test in mice. Am J Physiol Endocrinol Metab 295: E1323–E1332, 2008. doi: 10.1152/ajpendo.90617.2008. [DOI] [PubMed] [Google Scholar]
- 24.An J, Muoio DM, Shiota M, Fujimoto Y, Cline GW, Shulman GI, Koves TR, Stevens R, Millington D, Newgard CB. Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver and whole-animal insulin resistance. Nat Med 10: 268–274, 2004. doi: 10.1038/nm995. [DOI] [PubMed] [Google Scholar]
- 25.Araújo EP, De Souza CT, Ueno M, Cintra DE, Bertolo MB, Carvalheira JB, Saad MJ, Velloso LA. Infliximab restores glucose homeostasis in an animal model of diet-induced obesity and diabetes. Endocrinology 148: 5991–5997, 2007. doi: 10.1210/en.2007-0132. [DOI] [PubMed] [Google Scholar]
- 26.Archer JA, Gorden P, Roth J. Defect in insulin binding to receptors in obese man. Amelioration with calorie restriction. J Clin Invest 55: 166–174, 1975. doi: 10.1172/JCI107907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arner P. The adipocyte in insulin resistance: key molecules and the impact of the thiazolidinediones. Trends Endocrinol Metab 14: 137–145, 2003. doi: 10.1016/S1043-2760(03)00024-9. [DOI] [PubMed] [Google Scholar]
- 28.Aryal B, Singh AK, Zhang X, Varela L, Rotllan N, Goedeke L, Chaube B, Camporez JP, Vatner DF, Horvath TL, Shulman GI, Suárez Y, Fernández-Hernando C. Absence of ANGPTL4 in adipose tissue improves glucose tolerance and attenuates atherogenesis. JCI Insight 3: e97918, 2018. doi: 10.1172/jci.insight.97918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Asplin CM, Paquette TL, Palmer JP. In vivo inhibition of glucagon secretion by paracrine beta cell activity in man. J Clin Invest 68: 314–318, 1981. doi: 10.1172/JCI110251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Auberger P, Falquerho L, Contreres JO, Pages G, Le Cam G, Rossi B, Le Cam A. Characterization of a natural inhibitor of the insulin receptor tyrosine kinase: cDNA cloning, purification, and anti-mitogenic activity. Cell 58: 631–640, 1989. doi: 10.1016/0092-8674(89)90098-6. [DOI] [PubMed] [Google Scholar]
- 31.Axelsen M, Smith U, Eriksson JW, Taskinen MR, Jansson PA. Postprandial hypertriglyceridemia and insulin resistance in normoglycemic first-degree relatives of patients with type 2 diabetes. Ann Intern Med 131: 27–31, 1999. doi: 10.7326/0003-4819-131-1-199907060-00006. [DOI] [PubMed] [Google Scholar]
- 32.Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes 55: 390–397, 2006. doi: 10.2337/diabetes.55.02.06.db05-0686. [DOI] [PubMed] [Google Scholar]
- 33.Ayala JE, Samuel VT, Morton GJ, Obici S, Croniger CM, Shulman GI, Wasserman DH, McGuinness OP; NIH Mouse Metabolic Phenotyping Center Consortium . Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis Model Mech 3: 525–534, 2010. doi: 10.1242/dmm.006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baba H, Zhang XJ, Wolfe RR. Glycerol gluconeogenesis in fasting humans. Nutrition 11: 149–153, 1995. [PubMed] [Google Scholar]
- 35.Bak JF, Pedersen O. Insulin Receptors and Diabetes. In: Diabetes: Clinical Science in Practice, edited by Leslie RDG, Robbins DC. Cambridge, UK: Cambridge Univ. Press, 1995, p. 55. [Google Scholar]
- 36.Baron CL, Malhotra V. Role of diacylglycerol in PKD recruitment to the TGN and protein transport to the plasma membrane. Science 295: 325–328, 2002. doi: 10.1126/science.1066759. [DOI] [PubMed] [Google Scholar]
- 37.Bashan N, Kovsan J, Kachko I, Ovadia H, Rudich A. Positive and negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiol Rev 89: 27–71, 2009. doi: 10.1152/physrev.00014.2008. [DOI] [PubMed] [Google Scholar]
- 38.Basu R, Chandramouli V, Dicke B, Landau B, Rizza R. Obesity and type 2 diabetes impair insulin-induced suppression of glycogenolysis as well as gluconeogenesis. Diabetes 54: 1942–1948, 2005. doi: 10.2337/diabetes.54.7.1942. [DOI] [PubMed] [Google Scholar]
- 39.Baumeier C, Kaiser D, Heeren J, Scheja L, John C, Weise C, Eravci M, Lagerpusch M, Schulze G, Joost H-G, Schwenk RW, Schürmann A. Caloric restriction and intermittent fasting alter hepatic lipid droplet proteome and diacylglycerol species and prevent diabetes in NZO mice. Biochim Biophys Acta 1851: 566–576, 2015. doi: 10.1016/j.bbalip.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 40.Baura GD, Foster DM, Porte D Jr, Kahn SE, Bergman RN, Cobelli C, Schwartz MW. Saturable transport of insulin from plasma into the central nervous system of dogs in vivo. A mechanism for regulated insulin delivery to the brain. J Clin Invest 92: 1824–1830, 1993. doi: 10.1172/JCI116773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bedinger DH, Adams SH. Metabolic, anabolic, and mitogenic insulin responses: a tissue-specific perspective for insulin receptor activators. Mol Cell Endocrinol 415: 143–156, 2015. doi: 10.1016/j.mce.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 42.Befroy DE, Rothman DL, Petersen KF, Shulman GI. 31P-magnetization transfer magnetic resonance spectroscopy measurements of in vivo metabolism. Diabetes 61: 2669–2678, 2012. doi: 10.2337/db12-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Begum N. Stimulation of protein phosphatase-1 activity by insulin in rat adipocytes. Evaluation of the role of mitogen-activated protein kinase pathway. J Biol Chem 270: 709–714, 1995. doi: 10.1074/jbc.270.2.709. [DOI] [PubMed] [Google Scholar]
- 44.Belfiore A, Malaguarnera R, Vella V, Lawrence MC, Sciacca L, Frasca F, Morrione A, Vigneri R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr Rev 38: 379–431, 2017. doi: 10.1210/er.2017-00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis 28: 155–161, 2010. doi: 10.1159/000282080. [DOI] [PubMed] [Google Scholar]
- 46.Bell KS, Schmitz-Peiffer C, Lim-Fraser M, Biden TJ, Cooney GJ, Kraegen EW. Acute reversal of lipid-induced muscle insulin resistance is associated with rapid alteration in PKC-theta localization. Am J Physiol Endocrinol Metab 279: E1196–E1201, 2000. doi: 10.1152/ajpendo.2000.279.5.E1196. [DOI] [PubMed] [Google Scholar]
- 47.Belman JP, Habtemichael EN, Bogan JS. A proteolytic pathway that controls glucose uptake in fat and muscle. Rev Endocr Metab Disord 15: 55–66, 2014. doi: 10.1007/s11154-013-9276-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Benedict C, Kern W, Schultes B, Born J, Hallschmid M. Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin. J Clin Endocrinol Metab 93: 1339–1344, 2008. doi: 10.1210/jc.2007-2606. [DOI] [PubMed] [Google Scholar]
- 49.Benhamed F, Denechaud P-D, Lemoine M, Robichon C, Moldes M, Bertrand-Michel J, Ratziu V, Serfaty L, Housset C, Capeau J, Girard J, Guillou H, Postic C. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest 122: 2176–2194, 2012. doi: 10.1172/JCI41636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benziane B, Borg ML, Tom RZ, Riedl I, Massart J, Björnholm M, Gilbert M, Chibalin AV, Zierath JR. DGKζ deficiency protects against peripheral insulin resistance and improves energy metabolism. J Lipid Res 58: 2324–2333, 2017. doi: 10.1194/jlr.M079723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med 7: 947–953, 2001. doi: 10.1038/90992. [DOI] [PubMed] [Google Scholar]
- 52.Bergman BC, Brozinick JT, Strauss A, Bacon S, Kerege A, Bui HH, Sanders P, Siddall P, Wei T, Thomas MK, Kuo MS, Perreault L. Muscle sphingolipids during rest and exercise: a C18:0 signature for insulin resistance in humans. Diabetologia 59: 785–798, 2016. doi: 10.1007/s00125-015-3850-y. [DOI] [PubMed] [Google Scholar]
- 53.Bergman BC, Hunerdosse DM, Kerege A, Playdon MC, Perreault L. Localisation and composition of skeletal muscle diacylglycerol predicts insulin resistance in humans. Diabetologia 55: 1140–1150, 2012. doi: 10.1007/s00125-011-2419-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bergman RN. Non-esterified fatty acids and the liver: why is insulin secreted into the portal vein? Diabetologia 43: 946–952, 2000. doi: 10.1007/s001250051474. [DOI] [PubMed] [Google Scholar]
- 55.Bernard C. Leçons de physiologie expérimentale appliquée a la médecine. Paris: J.-B. Baillière, 1855. [Google Scholar]
- 56.Bernstein LE, Berry J, Kim S, Canavan B, Grinspoon SK. Effects of etanercept in patients with the metabolic syndrome. Arch Intern Med 166: 902–908, 2006. doi: 10.1001/archinte.166.8.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berwick DC, Hers I, Heesom KJ, Moule SK, Tavaré JM. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J Biol Chem 277: 33895–33900, 2002. doi: 10.1074/jbc.M204681200. [DOI] [PubMed] [Google Scholar]
- 58.Best CH, Dale HH, Hoet JP, Marks HP. Oxidation and Storage of Glucose under the Action of Insulin. Proc R Soc Lond B Biol Sci 100: 55–71, 1926. doi: 10.1098/rspb.1926.0033. [DOI] [Google Scholar]
- 59.Bezy O, Tran TT, Pihlajamäki J, Suzuki R, Emanuelli B, Winnay J, Mori MA, Haas J, Biddinger SB, Leitges M, Goldfine AB, Patti ME, King GL, Kahn CR. PKCδ regulates hepatic insulin sensitivity and hepatosteatosis in mice and humans. J Clin Invest 121: 2504–2517, 2011. doi: 10.1172/JCI46045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Biddinger SB, Hernandez-Ono A, Rask-Madsen C, Haas JT, Alemán JO, Suzuki R, Scapa EF, Agarwal C, Carey MC, Stephanopoulos G, Cohen DE, King GL, Ginsberg HN, Kahn CR. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab 7: 125–134, 2008. doi: 10.1016/j.cmet.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bikman BT, Summers SA. Sphingolipids and hepatic steatosis. Adv Exp Med Biol 721: 87–97, 2011. doi: 10.1007/978-1-4614-0650-1_6. [DOI] [PubMed] [Google Scholar]
- 62.Bikman BT, Summers SA. Ceramides as modulators of cellular and whole-body metabolism. J Clin Invest 121: 4222–4230, 2011. doi: 10.1172/JCI57144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. In: Cochrane Database of Systematic Reviews , edited by The Cochrane Collaboration New York: Wiley, 2016. http://doi.wiley.com/10.1002/14651858.CD007176.pub2. [DOI] [PubMed] [Google Scholar]
- 64.Błachnio-Zabielska AU, Baranowski M, Hirnle T, Zabielski P, Lewczuk A, Dmitruk I, Górski J. Increased bioactive lipids content in human subcutaneous and epicardial fat tissue correlates with insulin resistance. Lipids 47: 1131–1141, 2012. doi: 10.1007/s11745-012-3722-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bliss M. The Discovery of Insulin. Chicago: Univ. of Chicago Press, 1984. [Google Scholar]
- 66.Block NE, Komori K, Dutton SL, Robinson KA, Buse MG. Skeletal muscle insulin-receptor kinase. Effects of substrate inhibition and diabetes. Diabetes 40: 1691–1700, 1991. doi: 10.2337/diab.40.12.1691. [DOI] [PubMed] [Google Scholar]
- 67.Blouin CM, Prado C, Takane KK, Lasnier F, Garcia-Ocana A, Ferré P, Dugail I, Hajduch E. Plasma membrane subdomain compartmentalization contributes to distinct mechanisms of ceramide action on insulin signaling. Diabetes 59: 600–610, 2010. doi: 10.2337/db09-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boden G, Chen X, Ruiz J, White JV, Rossetti L. Mechanisms of fatty acid-induced inhibition of glucose uptake. J Clin Invest 93: 2438–2446, 1994. doi: 10.1172/JCI117252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boden G, Chen X, Stein TP. Gluconeogenesis in moderately and severely hyperglycemic patients with type 2 diabetes mellitus. Am J Physiol Endocrinol Metab 280: E23–E30, 2001. doi: 10.1152/ajpendo.2001.280.1.E23. [DOI] [PubMed] [Google Scholar]
- 70.Boden G, Duan X, Homko C, Molina EJ, Song W, Perez O, Cheung P, Merali S. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes 57: 2438–2444, 2008. doi: 10.2337/db08-0604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boden G, Jadali F, White J, Liang Y, Mozzoli M, Chen X, Coleman E, Smith C. Effects of fat on insulin-stimulated carbohydrate metabolism in normal men. J Clin Invest 88: 960–966, 1991. doi: 10.1172/JCI115399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boden G, Tappy L. Effects of amino acids on glucose disposal. Diabetes 39: 1079–1084, 1990. doi: 10.2337/diab.39.9.1079. [DOI] [PubMed] [Google Scholar]
- 73.Bogan JS. Regulation of glucose transporter translocation in health and diabetes. Annu Rev Biochem 81: 507–532, 2012. doi: 10.1146/annurev-biochem-060109-094246. [DOI] [PubMed] [Google Scholar]
- 74.Bogan JS, Hendon N, McKee AE, Tsao T-S, Lodish HF. Functional cloning of TUG as a regulator of GLUT4 glucose transporter trafficking. Nature 425: 727–733, 2003. doi: 10.1038/nature01989. [DOI] [PubMed] [Google Scholar]
- 75.Bogan JS, Rubin BR, Yu C, Löffler MG, Orme CM, Belman JP, McNally LJ, Hao M, Cresswell JA. Endoproteolytic cleavage of TUG protein regulates GLUT4 glucose transporter translocation. J Biol Chem 287: 23932–23947, 2012. doi: 10.1074/jbc.M112.339457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bogdanovic E, Kraus N, Patsouris D, Diao L, Wang V, Abdullahi A, Jeschke MG. Endoplasmic reticulum stress in adipose tissue augments lipolysis. J Cell Mol Med 19: 82–91, 2015. doi: 10.1111/jcmm.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bollag GE, Roth RA, Beaudoin J, Mochly-Rosen D, Koshland DE Jr. Protein kinase C directly phosphorylates the insulin receptor in vitro and reduces its protein-tyrosine kinase activity. Proc Natl Acad Sci USA 83: 5822–5824, 1986. doi: 10.1073/pnas.83.16.5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bollen M, Keppens S, Stalmans W. Specific features of glycogen metabolism in the liver. Biochem J 336: 19–31, 1998. doi: 10.1042/bj3360019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bolze F, Bast A, Mocek S, Morath V, Yuan D, Rink N, Schlapschy M, Zimmermann A, Heikenwalder M, Skerra A, Klingenspor M. Treatment of diet-induced lipodystrophic C57BL/6J mice with long-acting PASylated leptin normalises insulin sensitivity and hepatic steatosis by promoting lipid utilisation. Diabetologia 59: 2005–2012, 2016. doi: 10.1007/s00125-016-4004-6. [DOI] [PubMed] [Google Scholar]
- 80.Bonora E, Zavaroni I, Coscelli C, Butturini U. Decreased hepatic insulin extraction in subjects with mild glucose intolerance. Metabolism 32: 438–446, 1983. doi: 10.1016/0026-0495(83)90004-5. [DOI] [PubMed] [Google Scholar]
- 81.Borisov N, Aksamitiene E, Kiyatkin A, Legewie S, Berkhout J, Maiwald T, Kaimachnikov NP, Timmer J, Hoek JB, Kholodenko BN. Systems-level interactions between insulin-EGF networks amplify mitogenic signaling. Mol Syst Biol 5: 256, 2009. doi: 10.1038/msb.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bossenmaier B, Strack V, Stoyanov B, Krützfeldt J, Beck A, Lehmann R, Kellerer M, Klein H, Ullrich A, Lammers R, Häring HU. Serine residues 1177/78/82 of the insulin receptor are required for substrate phosphorylation but not autophosphorylation. Diabetes 49: 889–895, 2000. doi: 10.2337/diabetes.49.6.889. [DOI] [PubMed] [Google Scholar]
- 83.Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol 6: a009191, 2014. doi: 10.1101/cshperspect.a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bouskila M, Hirshman MF, Jensen J, Goodyear LJ, Sakamoto K. Insulin promotes glycogen synthesis in the absence of GSK3 phosphorylation in skeletal muscle. Am J Physiol Endocrinol Metab 294: E28–E35, 2008. doi: 10.1152/ajpendo.00481.2007. [DOI] [PubMed] [Google Scholar]
- 85.Bouskila M, Hunter RW, Ibrahim AFM, Delattre L, Peggie M, van Diepen JA, Voshol PJ, Jensen J, Sakamoto K. Allosteric regulation of glycogen synthase controls glycogen synthesis in muscle. Cell Metab 12: 456–466, 2010. doi: 10.1016/j.cmet.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 86.Boutens L, Hooiveld GJ, Dhingra S, Cramer RA, Netea MG, Stienstra R. Unique metabolic activation of adipose tissue macrophages in obesity promotes inflammatory responses. Diabetologia 61: 942–953, 2018. doi: 10.1007/s00125-017-4526-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bouzakri K, Zachrisson A, Al-Khalili L, Zhang BB, Koistinen HA, Krook A, Zierath JR. siRNA-based gene silencing reveals specialized roles of IRS-1/Akt2 and IRS-2/Akt1 in glucose and lipid metabolism in human skeletal muscle. Cell Metab 4: 89–96, 2006. doi: 10.1016/j.cmet.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 88.Brachmann SM, Ueki K, Engelman JA, Kahn RC, Cantley LC. Phosphoinositide 3-kinase catalytic subunit deletion and regulatory subunit deletion have opposite effects on insulin sensitivity in mice. Mol Cell Biol 25: 1596–1607, 2005. doi: 10.1128/MCB.25.5.1596-1607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bradley DC, Poulin RA, Bergman RN. Dynamics of hepatic and peripheral insulin effects suggest common rate-limiting step in vivo. Diabetes 42: 296–306, 1993. doi: 10.2337/diab.42.2.296. [DOI] [PubMed] [Google Scholar]
- 90.Brady MJ, Saltiel AR. The role of protein phosphatase-1 in insulin action. Recent Prog Horm Res 56: 157–173, 2001. doi: 10.1210/rp.56.1.157. [DOI] [PubMed] [Google Scholar]
- 91.Braiman L, Alt A, Kuroki T, Ohba M, Bak A, Tennenbaum T, Sampson SR. Protein kinase Cdelta mediates insulin-induced glucose transport in primary cultures of rat skeletal muscle. Mol Endocrinol 13: 2002–2012, 1999. [DOI] [PubMed] [Google Scholar]
- 92.Braiman L, Alt A, Kuroki T, Ohba M, Bak A, Tennenbaum T, Sampson SR. Insulin induces specific interaction between insulin receptor and protein kinase C delta in primary cultured skeletal muscle. Mol Endocrinol 15: 565–574, 2001. [DOI] [PubMed] [Google Scholar]
- 93.Brasaemle DL. Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J Lipid Res 48: 2547–2559, 2007. doi: 10.1194/jlr.R700014-JLR200. [DOI] [PubMed] [Google Scholar]
- 94.Brewer PD, Habtemichael EN, Romenskaia I, Mastick CC, Coster ACF. Glut4 Is Sorted from a Rab10 GTPase-independent Constitutive Recycling Pathway into a Highly Insulin-responsive Rab10 GTPase-dependent Sequestration Pathway after Adipocyte Differentiation. J Biol Chem 291: 773–789, 2016. doi: 10.1074/jbc.M115.694919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brouwers B, Schrauwen-Hinderling VB, Jelenik T, Gemmink A, Havekes B, Bruls Y, Dahlmans D, Roden M, Hesselink MKC, Schrauwen P. Metabolic disturbances of non-alcoholic fatty liver resemble the alterations typical for type 2 diabetes. Clin Sci (Lond) 131: 1905–1917, 2017. doi: 10.1042/CS20170261. [DOI] [PubMed] [Google Scholar]
- 97.Browner MF, Fletterick RJ. Phosphorylase: a biological transducer. Trends Biochem Sci 17: 66–71, 1992. doi: 10.1016/0968-0004(92)90504-3. [DOI] [PubMed] [Google Scholar]
- 98.Brown JM, Betters JL, Lord C, Ma Y, Han X, Yang K, Alger HM, Melchior J, Sawyer J, Shah R, Wilson MD, Liu X, Graham MJ, Lee R, Crooke R, Shulman GI, Xue B, Shi H, Yu L. CGI-58 knockdown in mice causes hepatic steatosis but prevents diet-induced obesity and glucose intolerance. J Lipid Res 51: 3306–3315, 2010. doi: 10.1194/jlr.M010256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab 7: 95–96, 2008. doi: 10.1016/j.cmet.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 100.Brownsey RW, Boone AN, Elliott JE, Kulpa JE, Lee WM. Regulation of acetyl-CoA carboxylase. Biochem Soc Trans 34: 223–227, 2006. doi: 10.1042/BST0340223. [DOI] [PubMed] [Google Scholar]
- 101.Bruce CR, Risis S, Babb JR, Yang C, Kowalski GM, Selathurai A, Lee-Young RS, Weir JM, Yoshioka K, Takuwa Y, Meikle PJ, Pitson SM, Febbraio MA. Overexpression of sphingosine kinase 1 prevents ceramide accumulation and ameliorates muscle insulin resistance in high-fat diet-fed mice. Diabetes 61: 3148–3155, 2012. doi: 10.2337/db12-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bruce CR, Risis S, Babb JR, Yang C, Lee-Young RS, Henstridge DC, Febbraio MA. The sphingosine-1-phosphate analog FTY720 reduces muscle ceramide content and improves glucose tolerance in high fat-fed male mice. Endocrinology 154: 65–76, 2013. doi: 10.1210/en.2012-1847. [DOI] [PubMed] [Google Scholar]
- 103.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857–868, 1999. doi: 10.1016/S0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 104.Brüning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Müller-Wieland D, Kahn CR. Role of brain insulin receptor in control of body weight and reproduction. Science 289: 2122–2125, 2000. doi: 10.1126/science.289.5487.2122. [DOI] [PubMed] [Google Scholar]
- 105.Buettner C, Patel R, Muse ED, Bhanot S, Monia BP, McKay R, Obici S, Rossetti L. Severe impairment in liver insulin signaling fails to alter hepatic insulin action in conscious mice. J Clin Invest 115: 1306–1313, 2005. doi: 10.1172/JCI23109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bugianesi E, Gastaldelli A, Vanni E, Gambino R, Cassader M, Baldi S, Ponti V, Pagano G, Ferrannini E, Rizzetto M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: sites and mechanisms. Diabetologia 48: 634–642, 2005. doi: 10.1007/s00125-005-1682-x. [DOI] [PubMed] [Google Scholar]
- 107.Burant CF, Treutelaar MK, Buse MG. Diabetes-induced functional and structural changes in insulin receptors from rat skeletal muscle. J Clin Invest 77: 260–270, 1986. doi: 10.1172/JCI112285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Burgess SC, He T, Yan Z, Lindner J, Sherry AD, Malloy CR, Browning JD, Magnuson MA. Cytosolic phosphoenolpyruvate carboxykinase does not solely control the rate of hepatic gluconeogenesis in the intact mouse liver. Cell Metab 5: 313–320, 2007. doi: 10.1016/j.cmet.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Buschiazzo H, Exton JH, Park CR. Effects of glucose on glycogen synthetase, phosphorylase, and glycogen deposition in the perfused rat liver. Proc Natl Acad Sci USA 65: 383–387, 1970. doi: 10.1073/pnas.65.2.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Campbell PJ, Carlson MG, Hill JO, Nurjhan N. Regulation of free fatty acid metabolism by insulin in humans: role of lipolysis and reesterification. Am J Physiol Endocrinol Metab 263: E1063–E1069, 1992. [DOI] [PubMed] [Google Scholar]
- 111.Camporez JPG, Jornayvaz FR, Lee H-Y, Kanda S, Guigni BA, Kahn M, Samuel VT, Carvalho CRO, Petersen KF, Jurczak MJ, Shulman GI. Cellular mechanism by which estradiol protects female ovariectomized mice from high-fat diet-induced hepatic and muscle insulin resistance. Endocrinology 154: 1021–1028, 2013. doi: 10.1210/en.2012-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Camporez JPG, Jornayvaz FR, Petersen MC, Pesta D, Guigni BA, Serr J, Zhang D, Kahn M, Samuel VT, Jurczak MJ, Shulman GI. Cellular mechanisms by which FGF21 improves insulin sensitivity in male mice. Endocrinology 154: 3099–3109, 2013. doi: 10.1210/en.2013-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Camporez JPG, Kanda S, Petersen MC, Jornayvaz FR, Samuel VT, Bhanot S, Petersen KF, Jurczak MJ, Shulman GI. ApoA5 knockdown improves whole-body insulin sensitivity in high-fat-fed mice by reducing ectopic lipid content. J Lipid Res 56: 526–536, 2015. doi: 10.1194/jlr.M054080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Camporez JP, Wang Y, Faarkrog K, Chukijrungroat N, Petersen KF, Shulman GI. Mechanism by which arylamine N-acetyltransferase 1 ablation causes insulin resistance in mice. Proc Natl Acad Sci USA 114: E11285–E11292, 2017. doi: 10.1073/pnas.1716990115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cantley JL, Yoshimura T, Camporez JPG, Zhang D, Jornayvaz FR, Kumashiro N, Guebre-Egziabher F, Jurczak MJ, Kahn M, Guigni BA, Serr J, Hankin J, Murphy RC, Cline GW, Bhanot S, Manchem VP, Brown JM, Samuel VT, Shulman GI. CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proc Natl Acad Sci USA 110: 1869–1874, 2013. doi: 10.1073/pnas.1219456110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Caro JF, Ittoop O, Pories WJ, Meelheim D, Flickinger EG, Thomas F, Jenquin M, Silverman JF, Khazanie PG, Sinha MK. Studies on the mechanism of insulin resistance in the liver from humans with noninsulin-dependent diabetes. Insulin action and binding in isolated hepatocytes, insulin receptor structure, and kinase activity. J Clin Invest 78: 249–258, 1986. doi: 10.1172/JCI112558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Caro JF, Sinha MK, Raju SM, Ittoop O, Pories WJ, Flickinger EG, Meelheim D, Dohm GL. Insulin receptor kinase in human skeletal muscle from obese subjects with and without noninsulin dependent diabetes. J Clin Invest 79: 1330–1337, 1987. doi: 10.1172/JCI112958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chadt A, Immisch A, de Wendt C, Springer C, Zhou Z, Stermann T, Holman GD, Loffing-Cueni D, Loffing J, Joost H-G, Al-Hasani H. Deletion of both Rab-GTPase-activating proteins TBC14KO and TBC1D4 in mice eliminates insulin- and AICAR-stimulated glucose transport. Diabetes 2015;64:746-759. Diabetes 64: 1492, 2015. doi: 10.2337/db15-er04. [DOI] [PubMed] [Google Scholar]
- 119.Chang L, Chiang S-H, Saltiel AR. TC10alpha is required for insulin-stimulated glucose uptake in adipocytes. Endocrinology 148: 27–33, 2007. doi: 10.1210/en.2006-1167. [DOI] [PubMed] [Google Scholar]
- 120.Chan SJ, Steiner DF. Insulin Through the Ages: Phylogeny of a Growth Promoting and Metabolic Regulatory Hormone. Am Zool 40: 213–222, 2000. [Google Scholar]
- 121.Charles MA, Eschwège E, Thibult N, Claude JR, Warnet JM, Rosselin GE, Girard J, Balkau B. The role of non-esterified fatty acids in the deterioration of glucose tolerance in Caucasian subjects: results of the Paris Prospective Study. Diabetologia 40: 1101–1106, 1997. doi: 10.1007/s001250050793. [DOI] [PubMed] [Google Scholar]
- 122.Chaurasia B, Kaddai VA, Lancaster GI, Henstridge DC, Sriram S, Galam DLA, Gopalan V, Prakash KNB, Velan SS, Bulchand S, Tsong TJ, Wang M, Siddique MM, Yuguang G, Sigmundsson K, Mellet NA, Weir JM, Meikle PJ, Bin M Yassin MS, Shabbir A, Shayman JA, Hirabayashi Y, Shiow ST, Sugii S, Summers SA. Adipocyte Ceramides Regulate Subcutaneous Adipose Browning, Inflammation, and Metabolism. Cell Metab 24: 820–834, 2016. doi: 10.1016/j.cmet.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 123.Chavez AO, Molina-Carrion M, Abdul-Ghani MA, Folli F, Defronzo RA, Tripathy D. Circulating fibroblast growth factor-21 is elevated in impaired glucose tolerance and type 2 diabetes and correlates with muscle and hepatic insulin resistance. Diabetes Care 32: 1542–1546, 2009. doi: 10.2337/dc09-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chavez JA, Knotts TA, Wang L-P, Li G, Dobrowsky RT, Florant GL, Summers SA. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J Biol Chem 278: 10297–10303, 2003. doi: 10.1074/jbc.M212307200. [DOI] [PubMed] [Google Scholar]
- 125.Chavez JA, Siddique MM, Wang ST, Ching J, Shayman JA, Summers SA. Ceramides and glucosylceramides are independent antagonists of insulin signaling. J Biol Chem 289: 723–734, 2014. doi: 10.1074/jbc.M113.522847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chavez JA, Summers SA. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch Biochem Biophys 419: 101–109, 2003. doi: 10.1016/j.abb.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 127.Chavez JA, Summers SA. A ceramide-centric view of insulin resistance. Cell Metab 15: 585–594, 2012. doi: 10.1016/j.cmet.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 128.Cheatham B, Vlahos CJ, Cheatham L, Wang L, Blenis J, Kahn CR. Phosphatidylinositol 3-kinase activation is required for insulin stimulation of pp70 S6 kinase, DNA synthesis, and glucose transporter translocation. Mol Cell Biol 14: 4902–4911, 1994. doi: 10.1128/MCB.14.7.4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cheng S, Rhee EP, Larson MG, Lewis GD, McCabe EL, Shen D, Palma MJ, Roberts LD, Dejam A, Souza AL, Deik AA, Magnusson M, Fox CS, O’Donnell CJ, Vasan RS, Melander O, Clish CB, Gerszten RE, Wang TJ. Metabolite profiling identifies pathways associated with metabolic risk in humans. Circulation 125: 2222–2231, 2012. doi: 10.1161/CIRCULATIONAHA.111.067827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chennamsetty I, Coronado M, Contrepois K, Keller MP, Carcamo-Orive I, Sandin J, Fajardo G, Whittle AJ, Fathzadeh M, Snyder M, Reaven G, Attie AD, Bernstein D, Quertermous T, Knowles JW. Nat1 Deficiency Is Associated with Mitochondrial Dysfunction and Exercise Intolerance in Mice. Cell Reports 17: 527–540, 2016. doi: 10.1016/j.celrep.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chen S, Wasserman DH, MacKintosh C, Sakamoto K. Mice with AS160/TBC1D4-Thr649Ala knockin mutation are glucose intolerant with reduced insulin sensitivity and altered GLUT4 trafficking. Cell Metab 13: 68–79, 2011. doi: 10.1016/j.cmet.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen X-W, Leto D, Chiang S-H, Wang Q, Saltiel AR. Activation of RalA is required for insulin-stimulated Glut4 trafficking to the plasma membrane via the exocyst and the motor protein Myo1c. Dev Cell 13: 391–404, 2007. doi: 10.1016/j.devcel.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 133.Chen X-W, Leto D, Xiong T, Yu G, Cheng A, Decker S, Saltiel AR. A Ral GAP complex links PI 3-kinase/Akt signaling to RalA activation in insulin action. Mol Biol Cell 22: 141–152, 2011. doi: 10.1091/mbc.e10-08-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chen YD, Golay A, Swislocki AL, Reaven GM. Resistance to insulin suppression of plasma free fatty acid concentrations and insulin stimulation of glucose uptake in noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab 64: 17–21, 1987. doi: 10.1210/jcem-64-1-17. [DOI] [PubMed] [Google Scholar]
- 135.Cherrington AD. The role of hepatic insulin receptors in the regulation of glucose production. J Clin Invest 115: 1136–1139, 2005. doi: 10.1172/JCI200525152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cherrington AD, Edgerton D, Sindelar DK. The direct and indirect effects of insulin on hepatic glucose production in vivo. Diabetologia 41: 987–996, 1998. doi: 10.1007/s001250051021. [DOI] [PubMed] [Google Scholar]
- 137.Chibalin AV, Leng Y, Vieira E, Krook A, Björnholm M, Long YC, Kotova O, Zhong Z, Sakane F, Steiler T, Nylén C, Wang J, Laakso M, Topham MK, Gilbert M, Wallberg-Henriksson H, Zierath JR. Downregulation of diacylglycerol kinase delta contributes to hyperglycemia-induced insulin resistance. Cell 132: 375–386, 2008. doi: 10.1016/j.cell.2007.12.035. [DOI] [PubMed] [Google Scholar]
- 138.Chisholm AB, Allan EH, Titheradge MA. Regulation of mitochondrial pyruvate carboxylation in isolated hepatocytes by acute insulin treatment. Biochem J 214: 451–458, 1983. doi: 10.1042/bj2140451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chitraju C, Mejhert N, Haas JT, Diaz-Ramirez LG, Grueter CA, Imbriglio JE, Pinto S, Koliwad SK, Walther TC, Farese RV Jr. Triglyceride Synthesis by DGAT1 Protects Adipocytes from Lipid-Induced ER Stress during Lipolysis. Cell Metab 26: 407–418.e3, 2017. doi: 10.1016/j.cmet.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chiu TT, Jensen TE, Sylow L, Richter EA, Klip A. Rac1 signalling towards GLUT4/glucose uptake in skeletal muscle. Cell Signal 23: 1546–1554, 2011. doi: 10.1016/j.cellsig.2011.05.022. [DOI] [PubMed] [Google Scholar]
- 141.Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB III, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB β). Science 292: 1728–1731, 2001. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 142.Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem 276: 38349–38352, 2001. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 143.Choi CS, Befroy DE, Codella R, Kim S, Reznick RM, Hwang Y-J, Liu Z-X, Lee H-Y, Distefano A, Samuel VT, Zhang D, Cline GW, Handschin C, Lin J, Petersen KF, Spiegelman BM, Shulman GI. Paradoxical effects of increased expression of PGC-1alpha on muscle mitochondrial function and insulin-stimulated muscle glucose metabolism. Proc Natl Acad Sci USA 105: 19926–19931, 2008. doi: 10.1073/pnas.0810339105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Choi CS, Savage DB, Kulkarni A, Yu XX, Liu Z-X, Morino K, Kim S, Distefano A, Samuel VT, Neschen S, Zhang D, Wang A, Zhang X-M, Kahn M, Cline GW, Pandey SK, Geisler JG, Bhanot S, Monia BP, Shulman GI. Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J Biol Chem 282: 22678–22688, 2007. doi: 10.1074/jbc.M704213200. [DOI] [PubMed] [Google Scholar]
- 145.Choi E, Zhang X, Xing C, Yu H. Mitotic Checkpoint Regulators Control Insulin Signaling and Metabolic Homeostasis. Cell 166: 567–581, 2016. doi: 10.1016/j.cell.2016.05.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Choi SM, Tucker DF, Gross DN, Easton RM, DiPilato LM, Dean AS, Monks BR, Birnbaum MJ. Insulin regulates adipocyte lipolysis via an Akt-independent signaling pathway. Mol Cell Biol 30: 5009–5020, 2010. doi: 10.1128/MCB.00797-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Choi YH, Park S, Hockman S, Zmuda-Trzebiatowska E, Svennelid F, Haluzik M, Gavrilova O, Ahmad F, Pepin L, Napolitano M, Taira M, Sundler F, Stenson Holst L, Degerman E, Manganiello VC. Alterations in regulation of energy homeostasis in cyclic nucleotide phosphodiesterase 3B-null mice. J Clin Invest 116: 3240–3251, 2006. doi: 10.1172/JCI24867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chomentowski P, Coen PM, Radiková Z, Goodpaster BH, Toledo FGS. Skeletal muscle mitochondria in insulin resistance: differences in intermyofibrillar versus subsarcolemmal subpopulations and relationship to metabolic flexibility. J Clin Endocrinol Metab 96: 494–503, 2011. doi: 10.1210/jc.2010-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chung JO, Koutsari C, Blachnio-Zabielska AU, Hames KC, Jensen MD. Intramyocellular Ceramides: Subcellular Concentrations and Fractional De Novo Synthesis in Postabsorptive Humans. Diabetes 66: 2082–2091, 2017. doi: 10.2337/db17-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Clarke JF, Young PW, Yonezawa K, Kasuga M, Holman GD. Inhibition of the translocation of GLUT1 and GLUT4 in 3T3-L1 cells by the phosphatidylinositol 3-kinase inhibitor, wortmannin. Biochem J 300: 631–635, 1994. doi: 10.1042/bj3000631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Clifford GM, McCormick DKT, Londos C, Vernon RG, Yeaman SJ. Dephosphorylation of perilipin by protein phosphatases present in rat adipocytes. FEBS Lett 435: 125–129, 1998. doi: 10.1016/S0014-5793(98)01052-7. [DOI] [PubMed] [Google Scholar]
- 152.Cline GW, Petersen KF, Krssak M, Shen J, Hundal RS, Trajanoski Z, Inzucchi S, Dresner A, Rothman DL, Shulman GI. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N Engl J Med 341: 240–246, 1999. doi: 10.1056/NEJM199907223410404. [DOI] [PubMed] [Google Scholar]
- 153.Cline GW, Shulman GI. Mass and positional isotopomer analysis of glucose metabolism in periportal and pericentral hepatocytes. J Biol Chem 270: 28062–28067, 1995. doi: 10.1074/jbc.270.47.28062. [DOI] [PubMed] [Google Scholar]
- 154.Coats BR, Schoenfelt KQ, Barbosa-Lorenzi VC, Peris E, Cui C, Hoffman A, Zhou G, Fernandez S, Zhai L, Hall BA, Haka AS, Shah AM, Reardon CA, Brady MJ, Rhodes CJ, Maxfield FR, Becker L. Metabolically Activated Adipose Tissue Macrophages Perform Detrimental and Beneficial Functions during Diet-Induced Obesity. Cell Reports 20: 3149–3161, 2017. doi: 10.1016/j.celrep.2017.08.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Coba MP, Muñoz MC, Dominici FP, Toblli JE, Peña C, Bartke A, Turyn D. Increased in vivo phosphorylation of insulin receptor at serine 994 in the liver of obese insulin-resistant Zucker rats. J Endocrinol 182: 433–444, 2004. doi: 10.1677/joe.0.1820433. [DOI] [PubMed] [Google Scholar]
- 156.Coghlan MP, Pillay TS, Tavaré JM, Siddle K. Site-specific anti-phosphopeptide antibodies: use in assessing insulin receptor serine/threonine phosphorylation state and identification of serine-1327 as a novel site of phorbol ester-induced phosphorylation. Biochem J 303: 893–899, 1994. doi: 10.1042/bj3030893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cohen P. The Croonian Lecture 1998. Identification of a protein kinase cascade of major importance in insulin signal transduction. Philos Trans R Soc Lond B Biol Sci 354: 485–495, 1999. doi: 10.1098/rstb.1999.0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Cohen P. The twentieth century struggle to decipher insulin signalling. Nat Rev Mol Cell Biol 7: 867–873, 2006. doi: 10.1038/nrm2043. [DOI] [PubMed] [Google Scholar]
- 159.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol 2: 769–776, 2001. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 160.Considine RV, Caro JF. Protein kinase C: mediator or inhibitor of insulin action? J Cell Biochem 52: 8–13, 1993. doi: 10.1002/jcb.240520103. [DOI] [PubMed] [Google Scholar]
- 161.Considine RV, Nyce MR, Allen LE, Morales LM, Triester S, Serrano J, Colberg J, Lanza-Jacoby S, Caro JF. Protein kinase C is increased in the liver of humans and rats with non-insulin-dependent diabetes mellitus: an alteration not due to hyperglycemia. J Clin Invest 95: 2938–2944, 1995. doi: 10.1172/JCI118001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cook JR, Langlet F, Kido Y, Accili D. Pathogenesis of selective insulin resistance in isolated hepatocytes. J Biol Chem 290: 13972–13980, 2015. doi: 10.1074/jbc.M115.638197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cook JR, Matsumoto M, Banks AS, Kitamura T, Tsuchiya K, Accili D. A mutant allele encoding DNA binding-deficient FoxO1 differentially regulates hepatic glucose and lipid metabolism. Diabetes 64: 1951–1965, 2015. doi: 10.2337/db14-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Cooperberg BA, Cryer PE. Insulin reciprocally regulates glucagon secretion in humans. Diabetes 59: 2936–2940, 2010. doi: 10.2337/db10-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Coppack SW, Persson M, Judd RL, Miles JM. Glycerol and nonesterified fatty acid metabolism in human muscle and adipose tissue in vivo. Am J Physiol Endocrinol Metab 276: E233–E240, 1999. [DOI] [PubMed] [Google Scholar]
- 166.Coppari R, Bjørbæk C. Leptin revisited: its mechanism of action and potential for treating diabetes. Nat Rev Drug Discov 11: 692–708, 2012. doi: 10.1038/nrd3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Copps KD, Hancer NJ, Opare-Ado L, Qiu W, Walsh C, White MF. Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metab 11: 84–92, 2010. doi: 10.1016/j.cmet.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Copps KD, Hançer NJ, Qiu W, White MF. Serine 302 Phosphorylation of Mouse Insulin Receptor Substrate 1 (IRS1) Is Dispensable for Normal Insulin Signaling and Feedback Regulation by Hepatic S6 Kinase. J Biol Chem 291: 8602–8617, 2016. doi: 10.1074/jbc.M116.714915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 55: 2565–2582, 2012. doi: 10.1007/s00125-012-2644-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cortés VA, Curtis DE, Sukumaran S, Shao X, Parameswara V, Rashid S, Smith AR, Ren J, Esser V, Hammer RE, Agarwal AK, Horton JD, Garg A. Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2-deficient mouse model of congenital generalized lipodystrophy. Cell Metab 9: 165–176, 2009. doi: 10.1016/j.cmet.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378: 785–789, 1995. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 172.Cross DA, Alessi DR, Vandenheede JR, McDowell HE, Hundal HS, Cohen P. The inhibition of glycogen synthase kinase-3 by insulin or insulin-like growth factor 1 in the rat skeletal muscle cell line L6 is blocked by wortmannin, but not by rapamycin: evidence that wortmannin blocks activation of the mitogen-activated protein kinase pathway in L6 cells between Ras and Raf. Biochem J 303: 21–26, 1994. doi: 10.1042/bj3030021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Cullen KS, Al-Oanzi ZH, O’Harte FPM, Agius L, Arden C. Glucagon induces translocation of glucokinase from the cytoplasm to the nucleus of hepatocytes by transfer between 6-phosphofructo 2-kinase/fructose 2,6-bisphosphatase-2 and the glucokinase regulatory protein. Biochim Biophys Acta 1843: 1123–1134, 2014. doi: 10.1016/j.bbamcr.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Cushman SW, Wardzala LJ. Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. Apparent translocation of intracellular transport systems to the plasma membrane. J Biol Chem 255: 4758–4762, 1980. [PubMed] [Google Scholar]
- 175.Cusi K, Maezono K, Osman A, Pendergrass M, Patti ME, Pratipanawatr T, DeFronzo RA, Kahn CR, Mandarino LJ. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J Clin Invest 105: 311–320, 2000. doi: 10.1172/JCI7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med 23: 804–814, 2017. doi: 10.1038/nm.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Czernichow S, Vergnaud A-C, Galan P, Arnaud J, Favier A, Faure H, Huxley R, Hercberg S, Ahluwalia N. Effects of long-term antioxidant supplementation and association of serum antioxidant concentrations with risk of metabolic syndrome in adults. Am J Clin Nutr 90: 329–335, 2009. doi: 10.3945/ajcn.2009.27635. [DOI] [PubMed] [Google Scholar]
- 178.Dash S, Sano H, Rochford JJ, Semple RK, Yeo G, Hyden CSS, Soos MA, Clark J, Rodin A, Langenberg C, Druet C, Fawcett KA, Tung YCL, Wareham NJ, Barroso I, Lienhard GE, O’Rahilly S, Savage DB. A truncation mutation in TBC1D4 in a family with acanthosis nigricans and postprandial hyperinsulinemia. Proc Natl Acad Sci USA 106: 9350–9355, 2009. doi: 10.1073/pnas.0900909106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Davey JR, Humphrey SJ, Junutula JR, Mishra AK, Lambright DG, James DE, Stöckli J. TBC1D13 is a RAB35 specific GAP that plays an important role in GLUT4 trafficking in adipocytes. Traffic 13: 1429–1441, 2012. doi: 10.1111/j.1600-0854.2012.01397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.DeFronzo RA, Jacot E, Jequier E, Maeder E, Wahren J, Felber JP. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 30: 1000–1007, 1981. doi: 10.2337/diab.30.12.1000. [DOI] [PubMed] [Google Scholar]
- 181.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol Endocrinol Metab 237: E214–E223, 1979. [DOI] [PubMed] [Google Scholar]
- 182.DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32, Suppl 2: S157–S163, 2009. doi: 10.2337/dc09-S302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Deldicque L, Van Proeyen K, Francaux M, Hespel P. The unfolded protein response in human skeletal muscle is not involved in the onset of glucose tolerance impairment induced by a fat-rich diet. Eur J Appl Physiol 111: 1553–1558, 2011. doi: 10.1007/s00421-010-1783-1. [DOI] [PubMed] [Google Scholar]
- 184.Delgado TC, Pinheiro D, Caldeira M, Castro MMCA, Geraldes CFGC, López-Larrubia P, Cerdán S, Jones JG. Sources of hepatic triglyceride accumulation during high-fat feeding in the healthy rat. NMR Biomed 22: 310–317, 2009. doi: 10.1002/nbm.1327. [DOI] [PubMed] [Google Scholar]
- 185.Delibegovic M, Armstrong CG, Dobbie L, Watt PW, Smith AJH, Cohen PTW. Disruption of the striated muscle glycogen targeting subunit PPP1R3A of protein phosphatase 1 leads to increased weight gain, fat deposition, and development of insulin resistance. Diabetes 52: 596–604, 2003. doi: 10.2337/diabetes.52.3.596. [DOI] [PubMed] [Google Scholar]
- 186.De Meyts P. The insulin receptor: a prototype for dimeric, allosteric membrane receptors? Trends Biochem Sci 33: 376–384, 2008. doi: 10.1016/j.tibs.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 187.Deng J, Liu S, Zou L, Xu C, Geng B, Xu G. Lipolysis response to endoplasmic reticulum stress in adipose cells. J Biol Chem 287: 6240–6249, 2012. doi: 10.1074/jbc.M111.299115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Dentin R, Liu Y, Koo S-H, Hedrick S, Vargas T, Heredia J, Yates J III, Montminy M. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature 449: 366–369, 2007. doi: 10.1038/nature06128. [DOI] [PubMed] [Google Scholar]
- 189.Denton RM, Brownsey RW, Yeaman SJ. The role of phosphorylation in the regulation of fatty acid synthesis by insulin and other hormones. Philos Trans R Soc Lond B Biol Sci 302: 33–45, 1983. doi: 10.1098/rstb.1983.0036. [DOI] [PubMed] [Google Scholar]
- 190.Desbuquois B, Carré N, Burnol A-F. Regulation of insulin and type 1 insulin-like growth factor signaling and action by the Grb10/14 and SH2B1/B2 adaptor proteins. FEBS J 280: 794–816, 2013. [DOI] [PubMed] [Google Scholar]
- 191.Dillin A. Profile of Kazutoshi Mori and Peter Walter, 2014 Lasker Basic Medical Research awardees: the unfolded protein response. Proc Natl Acad Sci USA 111: 17696–17697, 2014. [Correction in Proc Natl Acad Sci USA 112: E2112, 2105.] 10.1073/pnas.1419343111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.DiPilato LM, Ahmad F, Harms M, Seale P, Manganiello V, Birnbaum MJ. The Role of PDE3B Phosphorylation in the Inhibition of Lipolysis by Insulin. Mol Cell Biol 35: 2752–2760, 2015. doi: 10.1128/MCB.00422-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Dohm GL, Tapscott EB, Pories WJ, Dabbs DJ, Flickinger EG, Meelheim D, Fushiki T, Atkinson SM, Elton CW, Caro JF. An in vitro human muscle preparation suitable for metabolic studies. Decreased insulin stimulation of glucose transport in muscle from morbidly obese and diabetic subjects. J Clin Invest 82: 486–494, 1988. doi: 10.1172/JCI113622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Dokas J, Chadt A, Nolden T, Himmelbauer H, Zierath JR, Joost H-G, Al-Hasani H. Conventional knockout of Tbc1d1 in mice impairs insulin- and AICAR-stimulated glucose uptake in skeletal muscle. Endocrinology 154: 3502–3514, 2013. doi: 10.1210/en.2012-2147. [DOI] [PubMed] [Google Scholar]
- 195.Dong XC, Copps KD, Guo S, Li Y, Kollipara R, DePinho RA, White MF. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab 8: 65–76, 2008. doi: 10.1016/j.cmet.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Dong X, Park S, Lin X, Copps K, Yi X, White MF. Irs1 and Irs2 signaling is essential for hepatic glucose homeostasis and systemic growth. J Clin Invest 116: 101–114, 2006. doi: 10.1172/JCI25735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 115: 1343–1351, 2005. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, Slezak LA, Andersen DK, Hundal RS, Rothman DL, Petersen KF, Shulman GI. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest 103: 253–259, 1999. doi: 10.1172/JCI5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Dries DR, Gallegos LL, Newton AC. A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. J Biol Chem 282: 826–830, 2007. doi: 10.1074/jbc.C600268200. [DOI] [PubMed] [Google Scholar]
- 200.Duarte JAG, Carvalho F, Pearson M, Horton JD, Browning JD, Jones JG, Burgess SC. A high-fat diet suppresses de novo lipogenesis and desaturation but not elongation and triglyceride synthesis in mice. J Lipid Res 55: 2541–2553, 2014. doi: 10.1194/jlr.M052308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Duca FA, Bauer PV, Hamr SC, Lam TKT. Glucoregulatory Relevance of Small Intestinal Nutrient Sensing in Physiology, Bariatric Surgery, and Pharmacology. Cell Metab 22: 367–380, 2015. doi: 10.1016/j.cmet.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 202.Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev 18: 774–800, 1997. [DOI] [PubMed] [Google Scholar]
- 203.Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS. Regulation of lipolysis in adipocytes. Annu Rev Nutr 27: 79–101, 2007. doi: 10.1146/annurev.nutr.27.061406.093734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Dupont J, Khan J, Qu BH, Metzler P, Helman L, LeRoith D. Insulin and IGF-1 induce different patterns of gene expression in mouse fibroblast NIH-3T3 cells: identification by cDNA microarray analysis. Endocrinology 142: 4969–4975, 2001. doi: 10.1210/endo.142.11.8476. [DOI] [PubMed] [Google Scholar]
- 205.Dwyer JR, Donkor J, Zhang P, Csaki LS, Vergnes L, Lee JM, Dewald J, Brindley DN, Atti E, Tetradis S, Yoshinaga Y, De Jong PJ, Fong LG, Young SG, Reue K. Mouse lipin-1 and lipin-2 cooperate to maintain glycerolipid homeostasis in liver and aging cerebellum. Proc Natl Acad Sci USA 109: E2486–E2495, 2012. doi: 10.1073/pnas.1205221109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Eaton RP, Berman M, Steinberg D. Kinetic studies of plasma free fatty acid and triglyceride metabolism in man. J Clin Invest 48: 1560–1579, 1969. doi: 10.1172/JCI106122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Eberlé D, Hegarty B, Bossard P, Ferré P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86: 839–848, 2004. doi: 10.1016/j.biochi.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 208.Edgerton DS, Lautz M, Scott M, Everett CA, Stettler KM, Neal DW, Chu CA, Cherrington AD. Insulin’s direct effects on the liver dominate the control of hepatic glucose production. J Clin Invest 116: 521–527, 2006. doi: 10.1172/JCI27073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Egan JJ, Greenberg AS, Chang MK, Londos C. Control of endogenous phosphorylation of the major cAMP-dependent protein kinase substrate in adipocytes by insulin and beta-adrenergic stimulation. J Biol Chem 265: 18769–18775, 1990. [PubMed] [Google Scholar]
- 210.Eichmann TO, Kumari M, Haas JT, Farese RV Jr, Zimmermann R, Lass A, Zechner R. Studies on the substrate and stereo/regioselectivity of adipose triglyceride lipase, hormone-sensitive lipase, and diacylglycerol-O-acyltransferases. J Biol Chem 287: 41446–41457, 2012. doi: 10.1074/jbc.M112.400416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Eichmann TO, Lass A. DAG tales: the multiple faces of diacylglycerol–stereochemistry, metabolism, and signaling. Cell Mol Life Sci 72: 3931–3952, 2015. doi: 10.1007/s00018-015-1982-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Elgazar-Carmon V, Rudich A, Hadad N, Levy R. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J Lipid Res 49: 1894–1903, 2008. doi: 10.1194/jlr.M800132-JLR200. [DOI] [PubMed] [Google Scholar]
- 213.Elliott AD, Ustione A, Piston DW. Somatostatin and insulin mediate glucose-inhibited glucagon secretion in the pancreatic α-cell by lowering cAMP. Am J Physiol Endocrinol Metab 308: E130–E143, 2015. doi: 10.1152/ajpendo.00344.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem 107: 519–527, 1980. doi: 10.1111/j.1432-1033.1980.tb06059.x. [DOI] [PubMed] [Google Scholar]
- 215.Ericsson J, Jackson SM, Kim JB, Spiegelman BM, Edwards PA. Identification of glycerol-3-phosphate acyltransferase as an adipocyte determination and differentiation factor 1- and sterol regulatory element-binding protein-responsive gene. J Biol Chem 272: 7298–7305, 1997. doi: 10.1074/jbc.272.11.7298. [DOI] [PubMed] [Google Scholar]
- 216.Estall JL, Kahn M, Cooper MP, Fisher FM, Wu MK, Laznik D, Qu L, Cohen DE, Shulman GI, Spiegelman BM. Sensitivity of lipid metabolism and insulin signaling to genetic alterations in hepatic peroxisome proliferator-activated receptor-gamma coactivator-1alpha expression. Diabetes 58: 1499–1508, 2009. doi: 10.2337/db08-1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Exton JH, Corbin JG, Park CR. Control of gluconeogenesis in liver. IV. Differential effects of fatty acids and glucagon on ketogenesis and gluconeogenesis in the perfused rat liver. J Biol Chem 244: 4095–4102, 1969. [PubMed] [Google Scholar]
- 218.Exton JH, Park CR. Control of gluconeogenesis in liver. I. General features of gluconeogenesis in the perfused livers of rats. J Biol Chem 242: 2622–2636, 1967. [PubMed] [Google Scholar]
- 219.Fabbri E, Chia CW, Spencer RG, Fishbein KW, Reiter DA, Cameron D, Zane AC, Moore ZA, Gonzalez-Freire M, Zoli M, Studenski SA, Kalyani RR, Egan JM, Ferrucci L. Insulin Resistance Is Associated With Reduced Mitochondrial Oxidative Capacity Measured by 31P-Magnetic Resonance Spectroscopy in Participants Without Diabetes From the Baltimore Longitudinal Study of Aging. Diabetes 66: 170–176, 2017. doi: 10.2337/db16-0754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Fabbrini E, Magkos F, Mohammed BS, Pietka T, Abumrad NA, Patterson BW, Okunade A, Klein S. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc Natl Acad Sci USA 106: 15430–15435, 2009. doi: 10.1073/pnas.0904944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Fabbrini E, Tamboli RA, Magkos F, Marks-Shulman PA, Eckhauser AW, Richards WO, Klein S, Abumrad NN. Surgical removal of omental fat does not improve insulin sensitivity and cardiovascular risk factors in obese adults. Gastroenterology 139: 448–455, 2010. doi: 10.1053/j.gastro.2010.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Faber OK, Christensen K, Kehlet H, Madsbad S, Binder C. Decreased insulin removal contributes to hyperinsulinemia in obesity. J Clin Endocrinol Metab 53: 618–621, 1981. doi: 10.1210/jcem-53-3-618. [DOI] [PubMed] [Google Scholar]
- 223.Færch K, Vistisen D, Pacini G, Torekov SS, Johansen NB, Witte DR, Jonsson A, Pedersen O, Hansen T, Lauritzen T, Jørgensen ME, Ahrén B, Holst JJ. Insulin Resistance Is Accompanied by Increased Fasting Glucagon and Delayed Glucagon Suppression in Individuals With Normal and Impaired Glucose Regulation. Diabetes 65: 3473–3481, 2016. doi: 10.2337/db16-0240. [DOI] [PubMed] [Google Scholar]
- 224.Farese RV Jr, Zechner R, Newgard CB, Walther TC. The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab 15: 570–573, 2012. doi: 10.1016/j.cmet.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Farese RV Jr, Yost TJ, Eckel RH. Tissue-specific regulation of lipoprotein lipase activity by insulin/glucose in normal-weight humans. Metabolism 40: 214–216, 1991. doi: 10.1016/0026-0495(91)90178-Y. [DOI] [PubMed] [Google Scholar]
- 226.Farh KK-H, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJH, Shishkin AA, Hatan M, Carrasco-Alfonso MJ, Mayer D, Luckey CJ, Patsopoulos NA, De Jager PL, Kuchroo VK, Epstein CB, Daly MJ, Hafler DA, Bernstein BE. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518: 337–343, 2015. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Fazeli PK, Lun M, Kim SM, Bredella MA, Wright S, Zhang Y, Lee H, Catana C, Klibanski A, Patwari P, Steinhauser ML. FGF21 and the late adaptive response to starvation in humans. J Clin Invest 125: 4601–4611, 2015. doi: 10.1172/JCI83349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Felig P, Marliss E, Cahill GF Jr. Plasma amino acid levels and insulin secretion in obesity. N Engl J Med 281: 811–816, 1969. doi: 10.1056/NEJM196910092811503. [DOI] [PubMed] [Google Scholar]
- 229.Ferrannini E, Barrett EJ, Bevilacqua S, DeFronzo RA. Effect of fatty acids on glucose production and utilization in man. J Clin Invest 72: 1737–1747, 1983. doi: 10.1172/JCI111133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Finck BN, Bernal-Mizrachi C, Han DH, Coleman T, Sambandam N, LaRiviere LL, Holloszy JO, Semenkovich CF, Kelly DP. A potential link between muscle peroxisome proliferator-activated receptor-alpha signaling and obesity-related diabetes. Cell Metab 1: 133–144, 2005. doi: 10.1016/j.cmet.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 231.Finck BN, Hall AM. Does Diacylglycerol Accumulation in Fatty Liver Disease Cause Hepatic Insulin Resistance? BioMed Res Int 2015: 104132, 2015. doi: 10.1155/2015/104132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Fisher FM, Chui PC, Antonellis PJ, Bina HA, Kharitonenkov A, Flier JS, Maratos-Flier E. Obesity is a fibroblast growth factor 21 (FGF21)-resistant state. Diabetes 59: 2781–2789, 2010. doi: 10.2337/db10-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Fisher FM, Maratos-Flier E. Understanding the Physiology of FGF21. Annu Rev Physiol 78: 223–241, 2016. doi: 10.1146/annurev-physiol-021115-105339. [DOI] [PubMed] [Google Scholar]
- 234.Fisher SJ, Kahn CR. Insulin signaling is required for insulin’s direct and indirect action on hepatic glucose production. J Clin Invest 111: 463–468, 2003. doi: 10.1172/JCI16426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Fisher-Wellman KH, Neufer PD. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol Metab 23: 142–153, 2012. doi: 10.1016/j.tem.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Flak JN, Myers MG Jr. Minireview: CNS Mechanisms of Leptin Action. Mol Endocrinol 30: 3–12, 2016. doi: 10.1210/me.2015-1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Flamment M, Hajduch E, Ferré P, Foufelle F. New insights into ER stress-induced insulin resistance. Trends Endocrinol Metab 23: 381–390, 2012. doi: 10.1016/j.tem.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 238.Floyd JC Jr, Fajans SS, Conn JW, Knopf RF, Rull J. Stimulation of insulin secretion by amino acids. J Clin Invest 45: 1487–1502, 1966. doi: 10.1172/JCI105456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Fontaine DA, Davis DB. Attention to Background Strain Is Essential for Metabolic Research: C57BL/6 and the International Knockout Mouse Consortium. Diabetes 65: 25–33, 2016. doi: 10.2337/db15-0982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Foukas LC, Claret M, Pearce W, Okkenhaug K, Meek S, Peskett E, Sancho S, Smith AJH, Withers DJ, Vanhaesebroeck B. Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature 441: 366–370, 2006. doi: 10.1038/nature04694. [DOI] [PubMed] [Google Scholar]
- 241.Fox TE, Houck KL, O’Neill SM, Nagarajan M, Stover TC, Pomianowski PT, Unal O, Yun JK, Naides SJ, Kester M. Ceramide recruits and activates protein kinase C zeta (PKC zeta) within structured membrane microdomains. J Biol Chem 282: 12450–12457, 2007. doi: 10.1074/jbc.M700082200. [DOI] [PubMed] [Google Scholar]
- 242.Franckhauser S, Muñoz S, Pujol A, Casellas A, Riu E, Otaegui P, Su B, Bosch F. Increased fatty acid re-esterification by PEPCK overexpression in adipose tissue leads to obesity without insulin resistance. Diabetes 51: 624–630, 2002. doi: 10.2337/diabetes.51.3.624. [DOI] [PubMed] [Google Scholar]
- 243.Frangioudakis G, Garrard J, Raddatz K, Nadler JL, Mitchell TW, Schmitz-Peiffer C. Saturated- and n-6 polyunsaturated-fat diets each induce ceramide accumulation in mouse skeletal muscle: reversal and improvement of glucose tolerance by lipid metabolism inhibitors. Endocrinology 151: 4187–4196, 2010. doi: 10.1210/en.2010-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Frayn KN. The glucose-fatty acid cycle: a physiological perspective. Biochem Soc Trans 31: 1115–1119, 2003. doi: 10.1042/bst0311115. [DOI] [PubMed] [Google Scholar]
- 245.Fraze E, Donner CC, Swislocki AL, Chiou YA, Chen YD, Reaven GM. Ambient plasma free fatty acid concentrations in noninsulin-dependent diabetes mellitus: evidence for insulin resistance. J Clin Endocrinol Metab 61: 807–811, 1985. doi: 10.1210/jcem-61-5-807. [DOI] [PubMed] [Google Scholar]
- 246.Freidenberg GR, Henry RR, Klein HH, Reichart DR, Olefsky JM. Decreased kinase activity of insulin receptors from adipocytes of non-insulin-dependent diabetic subjects. J Clin Invest 79: 240–250, 1987. doi: 10.1172/JCI112789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Freidenberg GR, Reichart D, Olefsky JM, Henry RR. Reversibility of defective adipocyte insulin receptor kinase activity in non-insulin-dependent diabetes mellitus. Effect of weight loss. J Clin Invest 82: 1398–1406, 1988. doi: 10.1172/JCI113744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Fröjdö S, Vidal H, Pirola L. Alterations of insulin signaling in type 2 diabetes: a review of the current evidence from humans. Biochim Biophys Acta 1792: 83–92, 2009. doi: 10.1016/j.bbadis.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 249.Fujikawa T, Chuang J-C, Sakata I, Ramadori G, Coppari R. Leptin therapy improves insulin-deficient type 1 diabetes by CNS-dependent mechanisms in mice. Proc Natl Acad Sci USA 107: 17391–17396, 2010. doi: 10.1073/pnas.1008025107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen Z-P, O’Neill HM, Ford RJ, Palanivel R, O’Brien M, Hardie DG, Macaulay SL, Schertzer JD, Dyck JRB, van Denderen BJ, Kemp BE, Steinberg GR. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med 19: 1649–1654, 2013. doi: 10.1038/nm.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Fu S, Watkins SM, Hotamisligil GS. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab 15: 623–634, 2012. doi: 10.1016/j.cmet.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 252.Gaich G, Chien JY, Fu H, Glass LC, Deeg MA, Holland WL, Kharitonenkov A, Bumol T, Schilske HK, Moller DE. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab 18: 333–340, 2013. doi: 10.1016/j.cmet.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 253.Galbo T, Perry RJ, Jurczak MJ, Camporez J-PG, Alves TC, Kahn M, Guigni BA, Serr J, Zhang D, Bhanot S, Samuel VT, Shulman GI. Saturated and unsaturated fat induce hepatic insulin resistance independently of TLR-4 signaling and ceramide synthesis in vivo. Proc Natl Acad Sci USA 110: 12780–12785, 2013. doi: 10.1073/pnas.1311176110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Galbo T, Perry RJ, Nishimura E, Samuel VT, Quistorff B, Shulman GI. PP2A inhibition results in hepatic insulin resistance despite Akt2 activation. Aging (Albany NY) 5: 770–781, 2013. doi: 10.18632/aging.100611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Galuska D, Ryder J, Kawano Y, Charron MJ, Zierath JR. Insulin signaling and glucose transport in insulin resistant skeletal muscle. Special reference to GLUT4 transgenic and GLUT4 knockout mice. Adv Exp Med Biol 441: 73–85, 1998. doi: 10.1007/978-1-4899-1928-1_7. [DOI] [PubMed] [Google Scholar]
- 256.Gao Z, Wang Z, Zhang X, Butler AA, Zuberi A, Gawronska-Kozak B, Lefevre M, York D, Ravussin E, Berthoud H-R, McGuinness O, Cefalu WT, Ye J. Inactivation of PKCtheta leads to increased susceptibility to obesity and dietary insulin resistance in mice. Am J Physiol Endocrinol Metab 292: E84–E91, 2007. doi: 10.1152/ajpendo.00178.2006. [DOI] [PubMed] [Google Scholar]
- 257.Garland PB, Newsholme EA, Randle PJ. Effect of fatty acids, ketone bodies, diabetes and starvation on pyruvate metabolism in rat heart and diaphragm muscle. Nature 195: 381–383, 1962. doi: 10.1038/195381a0. [DOI] [PubMed] [Google Scholar]
- 258.Garvey WT, Maianu L, Zhu JH, Brechtel-Hook G, Wallace P, Baron AD. Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J Clin Invest 101: 2377–2386, 1998. doi: 10.1172/JCI1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Gastaldelli A, Baldi S, Pettiti M, Toschi E, Camastra S, Natali A, Landau BR, Ferrannini E. Influence of obesity and type 2 diabetes on gluconeogenesis and glucose output in humans: a quantitative study. Diabetes 49: 1367–1373, 2000. doi: 10.2337/diabetes.49.8.1367. [DOI] [PubMed] [Google Scholar]
- 260.Gastaldelli A, Cusi K, Pettiti M, Hardies J, Miyazaki Y, Berria R, Buzzigoli E, Sironi AM, Cersosimo E, Ferrannini E, Defronzo RA. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology 133: 496–506, 2007. doi: 10.1053/j.gastro.2007.04.068. [DOI] [PubMed] [Google Scholar]
- 261.Gastaldelli A, Gaggini M, DeFronzo RA. Role of Adipose Tissue Insulin Resistance in the Natural History of Type 2 Diabetes: Results From the San Antonio Metabolism Study. Diabetes 66: 815–822, 2017. doi: 10.2337/db16-1167. [DOI] [PubMed] [Google Scholar]
- 262.Gavrilova O, Marcus-Samuels B, Graham D, Kim JK, Shulman GI, Castle AL, Vinson C, Eckhaus M, Reitman ML. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J Clin Invest 105: 271–278, 2000. doi: 10.1172/JCI7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Gelding SV, Coldham N, Niththyananthan R, Anyaoku V, Johnston DG. Insulin resistance with respect to lipolysis in non-diabetic relatives of European patients with type 2 diabetes. Diabet Med 12: 66–73, 1995. doi: 10.1111/j.1464-5491.1995.tb02065.x. [DOI] [PubMed] [Google Scholar]
- 264.Gimeno RE, Moller DE. FGF21-based pharmacotherapy–potential utility for metabolic disorders. Trends Endocrinol Metab 25: 303–311, 2014. doi: 10.1016/j.tem.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 265.Girousse A, Tavernier G, Valle C, Moro C, Mejhert N, Dinel A-L, Houssier M, Roussel B, Besse-Patin A, Combes M, Mir L, Monbrun L, Bézaire V, Prunet-Marcassus B, Waget A, Vila I, Caspar-Bauguil S, Louche K, Marques M-A, Mairal A, Renoud M-L, Galitzky J, Holm C, Mouisel E, Thalamas C, Viguerie N, Sulpice T, Burcelin R, Arner P, Langin D. Partial inhibition of adipose tissue lipolysis improves glucose metabolism and insulin sensitivity without alteration of fat mass. PLoS Biol 11: e1001485, 2013. doi: 10.1371/journal.pbio.1001485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Golay A, Swislocki AL, Chen YD, Reaven GM. Relationships between plasma-free fatty acid concentration, endogenous glucose production, and fasting hyperglycemia in normal and non-insulin-dependent diabetic individuals. Metabolism 36: 692–696, 1987. doi: 10.1016/0026-0495(87)90156-9. [DOI] [PubMed] [Google Scholar]
- 267.Goldberg IJ, Eckel RH, Abumrad NA. Regulation of fatty acid uptake into tissues: lipoprotein lipase- and CD36-mediated pathways. J Lipid Res 50, Suppl: S86–S90, 2009. doi: 10.1194/jlr.R800085-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Goldfine AB, Conlin PR, Halperin F, Koska J, Permana P, Schwenke D, Shoelson SE, Reaven PD. A randomised trial of salsalate for insulin resistance and cardiovascular risk factors in persons with abnormal glucose tolerance. Diabetologia 56: 714–723, 2013. doi: 10.1007/s00125-012-2819-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Goldfine AB, Fonseca V, Jablonski KA, Chen Y-DI, Tipton L, Staten MA, Shoelson SE; Targeting Inflammation Using Salsalate in Type 2 Diabetes Study Team . Salicylate (salsalate) in patients with type 2 diabetes: a randomized trial. Ann Intern Med 159: 1–12, 2013. doi: 10.7326/0003-4819-159-1-201307020-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE; TINSAL-T2D (Targeting Inflammation Using Salsalate in Type 2 Diabetes) Study Team . The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Intern Med 152: 346–357, 2010. doi: 10.7326/0003-4819-152-6-201003160-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Goldfine AB, Shoelson SE. Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk. J Clin Invest 127: 83–93, 2017. doi: 10.1172/JCI88884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Goldfine ID, Kahn CR, Neville DM Jr, Roth J, Garrison MM, Bates RW. Decreased binding of insulin to its receptors in rats with hormone induced insulin resistance. Biochem Biophys Res Commun 53: 852–857, 1973. doi: 10.1016/0006-291X(73)90171-X. [DOI] [PubMed] [Google Scholar]
- 273.Gómez-Valadés AG, Méndez-Lucas A, Vidal-Alabró A, Blasco FX, Chillon M, Bartrons R, Bermúdez J, Perales JC. Pck1 gene silencing in the liver improves glycemia control, insulin sensitivity, and dyslipidemia in db/db mice. Diabetes 57: 2199–2210, 2008. doi: 10.2337/db07-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Gonzalez E, McGraw TE. Insulin signaling diverges into Akt-dependent and -independent signals to regulate the recruitment/docking and the fusion of GLUT4 vesicles to the plasma membrane. Mol Biol Cell 17: 4484–4493, 2006. doi: 10.1091/mbc.e06-07-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Goodpaster BH, He J, Watkins S, Kelley DE. Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab 86: 5755–5761, 2001. doi: 10.1210/jcem.86.12.8075. [DOI] [PubMed] [Google Scholar]
- 276.Govers R. Molecular mechanisms of GLUT4 regulation in adipocytes. Diabetes Metab 40: 400–410, 2014. doi: 10.1016/j.diabet.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 277.Graham TE, Wason CJ, Blüher M, Kahn BB. Shortcomings in methodology complicate measurements of serum retinol binding protein (RBP4) in insulin-resistant human subjects. Diabetologia 50: 814–823, 2007. doi: 10.1007/s00125-006-0557-0. [DOI] [PubMed] [Google Scholar]
- 278.Graham TE, Yang Q, Blüher M, Hammarstedt A, Ciaraldi TP, Henry RR, Wason CJ, Oberbach A, Jansson P-A, Smith U, Kahn BB. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N Engl J Med 354: 2552–2563, 2006. doi: 10.1056/NEJMoa054862. [DOI] [PubMed] [Google Scholar]
- 279.Granneman JG, Moore H-PH, Krishnamoorthy R, Rathod M. Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl). J Biol Chem 284: 34538–34544, 2009. doi: 10.1074/jbc.M109.068478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Greenbaum CJ, Havel PJ, Taborsky GJ Jr, Klaff LJ. Intra-islet insulin permits glucose to directly suppress pancreatic A cell function. J Clin Invest 88: 767–773, 1991. doi: 10.1172/JCI115375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Green CR, Wallace M, Divakaruni AS, Phillips SA, Murphy AN, Ciaraldi TP, Metallo CM. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat Chem Biol 12: 15–21, 2016. doi: 10.1038/nchembio.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Gregoire FM, Smas CM, Sul HS. Understanding adipocyte differentiation. Physiol Rev 78: 783–809, 1998. doi: 10.1152/physrev.1998.78.3.783. [DOI] [PubMed] [Google Scholar]
- 283.Gregori C, Guillet-Deniau I, Girard J, Decaux J-F, Pichard A-L. Insulin regulation of glucokinase gene expression: evidence against a role for sterol regulatory element binding protein 1 in primary hepatocytes. FEBS Lett 580: 410–414, 2006. doi: 10.1016/j.febslet.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 284.Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS, Klein S. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes 58: 693–700, 2009. doi: 10.2337/db08-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D, Goodyear LJ, Kraegen EW, White MF, Shulman GI. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 48: 1270–1274, 1999. doi: 10.2337/diabetes.48.6.1270. [DOI] [PubMed] [Google Scholar]
- 286.Groop LC, Bonadonna RC, DelPrato S, Ratheiser K, Zyck K, Ferrannini E, DeFronzo RA. Glucose and free fatty acid metabolism in non-insulin-dependent diabetes mellitus. Evidence for multiple sites of insulin resistance. J Clin Invest 84: 205–213, 1989. doi: 10.1172/JCI114142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Guerre-Millo M, Rouault C, Poulain P, André J, Poitout V, Peters JM, Gonzalez FJ, Fruchart JC, Reach G, Staels B. PPAR-alpha-null mice are protected from high-fat diet-induced insulin resistance. Diabetes 50: 2809–2814, 2001. doi: 10.2337/diabetes.50.12.2809. [DOI] [PubMed] [Google Scholar]
- 288.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 11: 859–871, 2006. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 289.Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol 9: 367–377, 2008. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Guo F, Ma Y, Kadegowda AKG, Betters JL, Xie P, Liu G, Liu X, Miao H, Ou J, Su X, Zheng Z, Xue B, Shi H, Yu L. Deficiency of liver Comparative Gene Identification-58 causes steatohepatitis and fibrosis in mice. J Lipid Res 54: 2109–2120, 2013. doi: 10.1194/jlr.M035519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Haasch D, Berg C, Clampit JE, Pederson T, Frost L, Kroeger P, Rondinone CM. PKCtheta is a key player in the development of insulin resistance. Biochem Biophys Res Commun 343: 361–368, 2006. doi: 10.1016/j.bbrc.2006.02.177. [DOI] [PubMed] [Google Scholar]
- 293.Haemmerle G, Zimmermann R, Hayn M, Theussl C, Waeg G, Wagner E, Sattler W, Magin TM, Wagner EF, Zechner R. Hormone-sensitive lipase deficiency in mice causes diglyceride accumulation in adipose tissue, muscle, and testis. J Biol Chem 277: 4806–4815, 2002. doi: 10.1074/jbc.M110355200. [DOI] [PubMed] [Google Scholar]
- 294.Haeusler RA, Camastra S, Astiarraga B, Nannipieri M, Anselmino M, Ferrannini E. Decreased expression of hepatic glucokinase in type 2 diabetes. Mol Metab 4: 222–226, 2014. doi: 10.1016/j.molmet.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Haeusler RA, Hartil K, Vaitheesvaran B, Arrieta-Cruz I, Knight CM, Cook JR, Kammoun HL, Febbraio MA, Gutierrez-Juarez R, Kurland IJ, Accili D. Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Nat Commun 5: 5190, 2014. doi: 10.1038/ncomms6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Haeusler RA, Kaestner KH, Accili D. FoxOs function synergistically to promote glucose production. J Biol Chem 285: 35245–35248, 2010. doi: 10.1074/jbc.C110.175851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Haeusler RA, McGraw TE, Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol 19: 31–44, 2018. doi: 10.1038/nrm.2017.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Hall AM, Soufi N, Chambers KT, Chen Z, Schweitzer GG, McCommis KS, Erion DM, Graham MJ, Su X, Finck BN. Abrogating monoacylglycerol acyltransferase activity in liver improves glucose tolerance and hepatic insulin signaling in obese mice. Diabetes 63: 2284–2296, 2014. doi: 10.2337/db13-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Halvatsiotis P, Short KR, Bigelow M, Nair KS. Synthesis rate of muscle proteins, muscle functions, and amino acid kinetics in type 2 diabetes. Diabetes 51: 2395–2404, 2002. doi: 10.2337/diabetes.51.8.2395. [DOI] [PubMed] [Google Scholar]
- 300.Hançer NJ, Qiu W, Cherella C, Li Y, Copps KD, White MF. Insulin and metabolic stress stimulate multisite serine/threonine phosphorylation of insulin receptor substrate 1 and inhibit tyrosine phosphorylation. J Biol Chem 289: 12467–12484, 2014. doi: 10.1074/jbc.M114.554162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Handberg A, Vaag A, Vinten J, Beck-Nielsen H. Decreased tyrosine kinase activity in partially purified insulin receptors from muscle of young, non-obese first degree relatives of patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 36: 668–674, 1993. doi: 10.1007/BF00404079. [DOI] [PubMed] [Google Scholar]
- 302.Han MS, Jung DY, Morel C, Lakhani SA, Kim JK, Flavell RA, Davis RJ. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 339: 218–222, 2013. doi: 10.1126/science.1227568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green KA, Mustard KJ, Kemp BE, Sakamoto K, Steinberg GR, Hardie DG. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 336: 918–922, 2012. doi: 10.1126/science.1215327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Heesom KJ, Moule SK, Denton RM. Purification and characterisation of an insulin-stimulated protein-serine kinase which phosphorylates acetyl-CoA carboxylase. FEBS Lett 422: 43–46, 1998. doi: 10.1016/S0014-5793(97)01597-4. [DOI] [PubMed] [Google Scholar]
- 306.Heijboer AC, Donga E, Voshol PJ, Dang Z-C, Havekes LM, Romijn JA, Corssmit EPM. Sixteen hours of fasting differentially affects hepatic and muscle insulin sensitivity in mice. J Lipid Res 46: 582–588, 2005. doi: 10.1194/jlr.M400440-JLR200. [DOI] [PubMed] [Google Scholar]
- 307.Heni M, Kullmann S, Preissl H, Fritsche A, Häring H-U. Impaired insulin action in the human brain: causes and metabolic consequences. Nat Rev Endocrinol 11: 701–711, 2015. doi: 10.1038/nrendo.2015.173. [DOI] [PubMed] [Google Scholar]
- 308.Heni M, Wagner R, Kullmann S, Gancheva S, Roden M, Peter A, Stefan N, Preissl H, Häring H-U, Fritsche A. Hypothalamic and Striatal Insulin Action Suppresses Endogenous Glucose Production and May Stimulate Glucose Uptake During Hyperinsulinemia in Lean but Not in Overweight Men. Diabetes 66: 1797–1806, 2017. doi: 10.2337/db16-1380. [DOI] [PubMed] [Google Scholar]
- 309.Heptulla RA, Stewart A, Enocksson S, Rife F, Ma TY-Z, Sherwin RS, Tamborlane WV, Caprio S. In situ evidence that peripheral insulin resistance in adolescents with poorly controlled type 1 diabetes is associated with impaired suppression of lipolysis: a microdialysis study. Pediatr Res 53: 830–835, 2003. doi: 10.1203/01.PDR.0000059552.08913.B7. [DOI] [PubMed] [Google Scholar]
- 310.Herman MA, Peroni OD, Villoria J, Schön MR, Abumrad NA, Blüher M, Klein S, Kahn BB. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 484: 333–338, 2012. doi: 10.1038/nature10986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Herman MA, Samuel VT. The Sweet Path to Metabolic Demise: Fructose and Lipid Synthesis. Trends Endocrinol Metab 27: 719–730, 2016. doi: 10.1016/j.tem.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Herman MA, She P, Peroni OD, Lynch CJ, Kahn BB. Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J Biol Chem 285: 11348–11356, 2010. doi: 10.1074/jbc.M109.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Hernández EÁ, Kahl S, Seelig A, Begovatz P, Irmler M, Kupriyanova Y, Nowotny B, Nowotny P, Herder C, Barosa C, Carvalho F, Rozman J, Neschen S, Jones JG, Beckers J, de Angelis MH, Roden M. Acute dietary fat intake initiates alterations in energy metabolism and insulin resistance. J Clin Invest 127: 695–708, 2017. doi: 10.1172/JCI89444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Herrema H, Zhou Y, Zhang D, Lee J, Salazar Hernandez MA, Shulman GI, Ozcan U. XBP1s Is an Anti-lipogenic Protein. J Biol Chem 291: 17394–17404, 2016. doi: 10.1074/jbc.M116.728949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Herrera MG, Kamm D, Ruderman N, Cahill GF Jr. Non-hormonal factors in the control of gluconeogenesis. Adv Enzyme Regul 4: 225–235, 1966. doi: 10.1016/0065-2571(66)90017-3. [DOI] [PubMed] [Google Scholar]
- 316.Higaki Y, Wojtaszewski JF, Hirshman MF, Withers DJ, Towery H, White MF, Goodyear LJ. Insulin receptor substrate-2 is not necessary for insulin- and exercise-stimulated glucose transport in skeletal muscle. J Biol Chem 274: 20791–20795, 1999. doi: 10.1074/jbc.274.30.20791. [DOI] [PubMed] [Google Scholar]
- 317.Himsworth HP. Diabetes mellitus: its differentiation into insulin-sensitive and insulin-insensitive types. Lancet 227: 127–130, 1936. doi: 10.1016/S0140-6736(01)36134-2. [DOI] [PubMed] [Google Scholar]
- 318.Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature 420: 333–336, 2002. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 319.Hodakoski C, Hopkins BD, Barrows D, Mense SM, Keniry M, Anderson KE, Kern PA, Hawkins PT, Stephens LR, Parsons R. Regulation of PTEN inhibition by the pleckstrin homology domain of P-REX2 during insulin signaling and glucose homeostasis. Proc Natl Acad Sci USA 111: 155–160, 2014. doi: 10.1073/pnas.1213773111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Hoehn KL, Turner N, Swarbrick MM, Wilks D, Preston E, Phua Y, Joshi H, Furler SM, Larance M, Hegarty BD, Leslie SJ, Pickford R, Hoy AJ, Kraegen EW, James DE, Cooney GJ. Acute or chronic upregulation of mitochondrial fatty acid oxidation has no net effect on whole-body energy expenditure or adiposity. Cell Metab 11: 70–76, 2010. doi: 10.1016/j.cmet.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Hogan MF, Ravnskjaer K, Matsumura S, Huising MO, Hull RL, Kahn SE, Montminy M. Hepatic Insulin Resistance Following Chronic Activation of the CREB Coactivator CRTC2. J Biol Chem 290: 25997–26006, 2015. doi: 10.1074/jbc.M115.679266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Holland WL, Bikman BT, Wang L-P, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MR, Pagliassotti MJ, Scherer PE, Summers SA. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest 121: 1858–1870, 2011. doi: 10.1172/JCI43378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Holland WL, Brozinick JT, Wang L-P, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ, Summers SA. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 5: 167–179, 2007. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 324.Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM, Wade MR, Tenorio VM, Kuo M-S, Brozinick JT, Zhang BB, Birnbaum MJ, Summers SA, Scherer PE. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med 17: 55–63, 2011. doi: 10.1038/nm.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Holland WL, Summers SA. Sphingolipids, insulin resistance, and metabolic disease: new insights from in vivo manipulation of sphingolipid metabolism. Endocr Rev 29: 381–402, 2008. doi: 10.1210/er.2007-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Holloszy JO. Skeletal muscle “mitochondrial deficiency” does not mediate insulin resistance. Am J Clin Nutr 89: 463S–466S, 2009. doi: 10.3945/ajcn.2008.26717C. [DOI] [PubMed] [Google Scholar]
- 327.Holloszy JO. “Deficiency” of mitochondria in muscle does not cause insulin resistance. Diabetes 62: 1036–1040, 2013. doi: 10.2337/db12-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Holm C. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochem Soc Trans 31: 1120–1124, 2003. doi: 10.1042/bst0311120. [DOI] [PubMed] [Google Scholar]
- 329.Holt LJ, Brandon AE, Small L, Suryana E, Preston E, Wilks D, Mokbel N, Coles CA, White JD, Turner N, Daly RJ, Cooney GJ. Ablation of Grb10 Specifically in Muscle Impacts Muscle Size and Glucose Metabolism in Mice. Endocrinology 159: 1339–1351, 2018. doi: 10.1210/en.2017-00851. [DOI] [PubMed] [Google Scholar]
- 330.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109: 1125–1131, 2002. doi: 10.1172/JCI0215593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140: 900–917, 2010. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 95: 2409–2415, 1995. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Hotamisligil GS, Budavari A, Murray D, Spiegelman BM. Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. Central role of tumor necrosis factor-alpha. J Clin Invest 94: 1543–1549, 1994. doi: 10.1172/JCI117495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci USA 91: 4854–4858, 1994. doi: 10.1073/pnas.91.11.4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 271: 665–670, 1996. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 336.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259: 87–91, 1993. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 337.Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, Iwahashi H, Kuriyama H, Ouchi N, Maeda K, Nishida M, Kihara S, Sakai N, Nakajima T, Hasegawa K, Muraguchi M, Ohmoto Y, Nakamura T, Yamashita S, Hanafusa T, Matsuzawa Y. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol 20: 1595–1599, 2000. doi: 10.1161/01.ATV.20.6.1595. [DOI] [PubMed] [Google Scholar]
- 338.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440: 944–948, 2006. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 339.Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, Marto JA, Sabatini DM. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332: 1317–1322, 2011. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Huang C, Thirone ACP, Huang X, Klip A. Differential contribution of insulin receptor substrates 1 versus 2 to insulin signaling and glucose uptake in l6 myotubes. J Biol Chem 280: 19426–19435, 2005. doi: 10.1074/jbc.M412317200. [DOI] [PubMed] [Google Scholar]
- 341.Huang L, Li C. Leptin: a multifunctional hormone. Cell Res 10: 81–92, 2000. doi: 10.1038/sj.cr.7290038. [DOI] [PubMed] [Google Scholar]
- 342.Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab 5: 237–252, 2007. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 343.Hubbard SR. The insulin receptor: both a prototypical and atypical receptor tyrosine kinase. Cold Spring Harb Perspect Biol 5: a008946, 2013. doi: 10.1101/cshperspect.a008946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Hubbard SR, Wei L, Hendrickson WA. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 372: 746–754, 1994. doi: 10.1038/372746a0. [DOI] [PubMed] [Google Scholar]
- 345.Hughes K, Ramakrishna S, Benjamin WB, Woodgett JR. Identification of multifunctional ATP-citrate lyase kinase as the alpha-isoform of glycogen synthase kinase-3. Biochem J 288: 309–314, 1992. doi: 10.1042/bj2880309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Hundal RS, Krssak M, Dufour S, Laurent D, Lebon V, Chandramouli V, Inzucchi SE, Schumann WC, Petersen KF, Landau BR, Shulman GI. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes 49: 2063–2069, 2000. doi: 10.2337/diabetes.49.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci USA 101: 7281–7286, 2004. doi: 10.1073/pnas.0401516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Ikeda Y, Olsen GS, Ziv E, Hansen LL, Busch AK, Hansen BF, Shafrir E, Mosthaf-Seedorf L. Cellular mechanism of nutritionally induced insulin resistance in Psammomys obesus: overexpression of protein kinase Cepsilon in skeletal muscle precedes the onset of hyperinsulinemia and hyperglycemia. Diabetes 50: 584–592, 2001. doi: 10.2337/diabetes.50.3.584. [DOI] [PubMed] [Google Scholar]
- 349.Inoki K, Li Y, Zhu T, Wu J, Guan K-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4: 648–657, 2002. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 350.Inoue M, Chang L, Hwang J, Chiang S-H, Saltiel AR. The exocyst complex is required for targeting of Glut4 to the plasma membrane by insulin. Nature 422: 629–633, 2003. doi: 10.1038/nature01533. [DOI] [PubMed] [Google Scholar]
- 351.Inouye KE, Shi H, Howard JK, Daly CH, Lord GM, Rollins BJ, Flier JS. Absence of CC chemokine ligand 2 does not limit obesity-associated infiltration of macrophages into adipose tissue. Diabetes 56: 2242–2250, 2007. doi: 10.2337/db07-0425. [DOI] [PubMed] [Google Scholar]
- 352.Isakoff SJ, Taha C, Rose E, Marcusohn J, Klip A, Skolnik EY. The inability of phosphatidylinositol 3-kinase activation to stimulate GLUT4 translocation indicates additional signaling pathways are required for insulin-stimulated glucose uptake. Proc Natl Acad Sci USA 92: 10247–10251, 1995. doi: 10.1073/pnas.92.22.10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Ishizuka T, Cooper DR, Arnold T, Hernandez H, Farese RV. Downregulation of protein kinase C and insulin-stimulated 2-deoxyglucose uptake in rat adipocytes by phorbol esters, glucose, and insulin. Diabetes 40: 1274–1281, 1991. doi: 10.2337/diab.40.10.1274. [DOI] [PubMed] [Google Scholar]
- 354.Ishizuka T, Cooper DR, Farese RV. Insulin stimulates the translocation of protein kinase C in rat adipocytes. FEBS Lett 257: 337–340, 1989. doi: 10.1016/0014-5793(89)81565-0. [DOI] [PubMed] [Google Scholar]
- 355.Ishizuka T, Cooper DR, Hernandez H, Buckley D, Standaert M, Farese RV. Effects of insulin on diacylglycerol-protein kinase C signaling in rat diaphragm and soleus muscles and relationship to glucose transport. Diabetes 39: 181–190, 1990. doi: 10.2337/diab.39.2.181. [DOI] [PubMed] [Google Scholar]
- 356.Itani SI, Pories WJ, Macdonald KG, Dohm GL. Increased protein kinase C theta in skeletal muscle of diabetic patients. Metabolism 50: 553–557, 2001. doi: 10.1053/meta.2001.22512. [DOI] [PubMed] [Google Scholar]
- 357.Itani SI, Zhou Q, Pories WJ, MacDonald KG, Dohm GL. Involvement of protein kinase C in human skeletal muscle insulin resistance and obesity. Diabetes 49: 1353–1358, 2000. doi: 10.2337/diabetes.49.8.1353. [DOI] [PubMed] [Google Scholar]
- 358.Ito M, Kondo Y, Nakatani A, Hayashi K, Naruse A. Characterization of low dose streptozotocin-induced progressive diabetes in mice. Environ Toxicol Pharmacol 9: 71–78, 2001. doi: 10.1016/S1382-6689(00)00064-8. [DOI] [PubMed] [Google Scholar]
- 359.Iynedjian PB. Molecular physiology of mammalian glucokinase. Cell Mol Life Sci 66: 27–42, 2009. doi: 10.1007/s00018-008-8322-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Iynedjian PB, Jotterand D, Nouspikel T, Asfari M, Pilot PR. Transcriptional induction of glucokinase gene by insulin in cultured liver cells and its repression by the glucagon-cAMP system. J Biol Chem 264: 21824–21829, 1989. [PubMed] [Google Scholar]
- 361.Ize-Ludlow D, Lightfoot YL, Parker M, Xue S, Wasserfall C, Haller MJ, Schatz D, Becker DJ, Atkinson MA, Mathews CE. Progressive erosion of β-cell function precedes the onset of hyperglycemia in the NOD mouse model of type 1 diabetes. Diabetes 60: 2086–2091, 2011. doi: 10.2337/db11-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127: 125–137, 2006. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 363.Jackson RA, Roshania RD, Hawa MI, Sim BM, DiSilvio L. Impact of glucose ingestion on hepatic and peripheral glucose metabolism in man: an analysis based on simultaneous use of the forearm and double isotope techniques. J Clin Endocrinol Metab 63: 541–549, 1986. doi: 10.1210/jcem-63-3-541. [DOI] [PubMed] [Google Scholar]
- 364.Jager J, Grémeaux T, Cormont M, Le Marchand-Brustel Y, Tanti J-F. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology 148: 241–251, 2007. doi: 10.1210/en.2006-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Jaldin-Fincati JR, Pavarotti M, Frendo-Cumbo S, Bilan PJ, Klip A. Update on GLUT4 Vesicle Traffic: A Cornerstone of Insulin Action. Trends Endocrinol Metab 28: 597–611, 2017. doi: 10.1016/j.tem.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 366.Jang C, Oh SF, Wada S, Rowe GC, Liu L, Chan MC, Rhee J, Hoshino A, Kim B, Ibrahim A, Baca LG, Kim E, Ghosh CC, Parikh SM, Jiang A, Chu Q, Forman DE, Lecker SH, Krishnaiah S, Rabinowitz JD, Weljie AM, Baur JA, Kasper DL, Arany Z. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat Med 22: 421–426, 2016. doi: 10.1038/nm.4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Jaworski K, Sarkadi-Nagy E, Duncan RE, Ahmadian M, Sul HS. Regulation of triglyceride metabolism. IV. Hormonal regulation of lipolysis in adipose tissue. Am J Physiol Gastrointest Liver Physiol 293: G1–G4, 2007. doi: 10.1152/ajpgi.00554.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Jelenik T, Kaul K, Séquaris G, Flögel U, Phielix E, Kotzka J, Knebel B, Fahlbusch P, Hörbelt T, Lehr S, Reinbeck AL, Müller-Wieland D, Esposito I, Shulman GI, Szendroedi J, Roden M. Mechanisms of Insulin Resistance in Primary and Secondary Nonalcoholic Fatty Liver. Diabetes 66: 2241–2253, 2017. doi: 10.2337/db16-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Jewell JL, Oh E, Ramalingam L, Kalwat MA, Tagliabracci VS, Tackett L, Elmendorf JS, Thurmond DC. Munc18c phosphorylation by the insulin receptor links cell signaling directly to SNARE exocytosis. J Cell Biol 193: 185–199, 2011. doi: 10.1083/jcb.201007176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Jocken JWE, Goossens GH, Boon H, Mason RR, Essers Y, Havekes B, Watt MJ, van Loon LJ, Blaak EE. Insulin-mediated suppression of lipolysis in adipose tissue and skeletal muscle of obese type 2 diabetic men and men with normal glucose tolerance. Diabetologia 56: 2255–2265, 2013. doi: 10.1007/s00125-013-2995-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Jornayvaz FR, Birkenfeld AL, Jurczak MJ, Kanda S, Guigni BA, Jiang DC, Zhang D, Lee H-Y, Samuel VT, Shulman GI. Hepatic insulin resistance in mice with hepatic overexpression of diacylglycerol acyltransferase 2. Proc Natl Acad Sci USA 108: 5748–5752, 2011. doi: 10.1073/pnas.1103451108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Jucker BM, Barucci N, Shulman GI. Metabolic control analysis of insulin-stimulated glucose disposal in rat skeletal muscle. Am J Physiol Endocrinol Metab 277: E505–E512, 1999. [DOI] [PubMed] [Google Scholar]
- 373.Jucker BM, Rennings AJ, Cline GW, Shulman GI. 13C and 31P NMR studies on the effects of increased plasma free fatty acids on intramuscular glucose metabolism in the awake rat. J Biol Chem 272: 10464–10473, 1997. doi: 10.1074/jbc.272.16.10464. [DOI] [PubMed] [Google Scholar]
- 374.Jurczak MJ, Danos AM, Rehrmann VR, Brady MJ. The role of protein translocation in the regulation of glycogen metabolism. J Cell Biochem 104: 435–443, 2008. doi: 10.1002/jcb.21634. [DOI] [PubMed] [Google Scholar]
- 375.Jurczak MJ, Lee A-H, Jornayvaz FR, Lee H-Y, Birkenfeld AL, Guigni BA, Kahn M, Samuel VT, Glimcher LH, Shulman GI. Dissociation of inositol-requiring enzyme (IRE1α)-mediated c-Jun N-terminal kinase activation from hepatic insulin resistance in conditional X-box-binding protein-1 (XBP1) knock-out mice. J Biol Chem 287: 2558–2567, 2012. doi: 10.1074/jbc.M111.316760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Kabayama K, Sato T, Saito K, Loberto N, Prinetti A, Sonnino S, Kinjo M, Igarashi Y, Inokuchi J. Dissociation of the insulin receptor and caveolin-1 complex by ganglioside GM3 in the state of insulin resistance. Proc Natl Acad Sci USA 104: 13678–13683, 2007. doi: 10.1073/pnas.0703650104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest 106: 473–481, 2000. doi: 10.1172/JCI10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Kahn CR. Insulin resistance, insulin insensitivity, and insulin unresponsiveness: a necessary distinction. Metabolism 27, Suppl 2: 1893–1902, 1978. doi: 10.1016/S0026-0495(78)80007-9. [DOI] [PubMed] [Google Scholar]
- 379.Kahn CR, Neville DM Jr, Roth J. Insulin-receptor interaction in the obese-hyperglycemic mouse. A model of insulin resistance. J Biol Chem 248: 244–250, 1973. [PubMed] [Google Scholar]
- 380.Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 46: 3–19, 2003. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- 381.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444: 840–846, 2006. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 382.Kammoun HL, Chabanon H, Hainault I, Luquet S, Magnan C, Koike T, Ferré P, Foufelle F. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest 119: 1201–1215, 2009. doi: 10.1172/JCI37007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, Kasuga M. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 116: 1494–1505, 2006. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Kane S, Sano H, Liu SCH, Asara JM, Lane WS, Garner CC, Lienhard GE. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem 277: 22115–22118, 2002. doi: 10.1074/jbc.C200198200. [DOI] [PubMed] [Google Scholar]
- 385.Kanety H, Hemi R, Papa MZ, Karasik A. Sphingomyelinase and ceramide suppress insulin-induced tyrosine phosphorylation of the insulin receptor substrate-1. J Biol Chem 271: 9895–9897, 1996. doi: 10.1074/jbc.271.17.9895. [DOI] [PubMed] [Google Scholar]
- 386.Kantartzis K, Peter A, Machicao F, Machann J, Wagner S, Königsrainer I, Königsrainer A, Schick F, Fritsche A, Häring H-U, Stefan N. Dissociation between fatty liver and insulin resistance in humans carrying a variant of the patatin-like phospholipase 3 gene. Diabetes 58: 2616–2623, 2009. doi: 10.2337/db09-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Karasik A, Rothenberg PL, Yamada K, White MF, Kahn CR. Increased protein kinase C activity is linked to reduced insulin receptor autophosphorylation in liver of starved rats. J Biol Chem 265: 10226–10231, 1990. [PubMed] [Google Scholar]
- 388.Karpe F, Dickmann JR, Frayn KN. Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes 60: 2441–2449, 2011. doi: 10.2337/db11-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Kasuga M. Insulin resistance and pancreatic beta cell failure. J Clin Invest 116: 1756–1760, 2006. doi: 10.1172/JCI29189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Kasuga M, Karlsson FA, Kahn CR. Insulin stimulates the phosphorylation of the 95,000-dalton subunit of its own receptor. Science 215: 185–187, 1982. doi: 10.1126/science.7031900. [DOI] [PubMed] [Google Scholar]
- 391.Kasuga M, Zick Y, Blithe DL, Crettaz M, Kahn CR. Insulin stimulates tyrosine phosphorylation of the insulin receptor in a cell-free system. Nature 298: 667–669, 1982. doi: 10.1038/298667a0. [DOI] [PubMed] [Google Scholar]
- 392.Kawamori D, Kulkarni RN. Insulin modulation of glucagon secretion: the role of insulin and other factors in the regulation of glucagon secretion. Islets 1: 276–279, 2009. doi: 10.4161/isl.1.3.9967. [DOI] [PubMed] [Google Scholar]
- 393.Kawamori D, Kurpad AJ, Hu J, Liew CW, Shih JL, Ford EL, Herrera PL, Polonsky KS, McGuinness OP, Kulkarni RN. Insulin signaling in alpha cells modulates glucagon secretion in vivo. Cell Metab 9: 350–361, 2009. doi: 10.1016/j.cmet.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Kellerer M, Coghlan M, Capp E, Mühlhöfer A, Kroder G, Mosthaf L, Galante P, Siddle K, Häring HU. Mechanism of insulin receptor kinase inhibition in non-insulin-dependent diabetes mellitus patients. Phosphorylation of serine 1327 or threonine 1348 is unaltered. J Clin Invest 96: 6–11, 1995. doi: 10.1172/JCI118073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Kelley DE, Goodpaster B, Wing RR, Simoneau JA. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am J Physiol Endocrinol Metab 277: E1130–E1141, 1999. [DOI] [PubMed] [Google Scholar]
- 396.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51: 2944–2950, 2002. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 397.Kelley DE, Mandarino LJ. Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes 49: 677–683, 2000. doi: 10.2337/diabetes.49.5.677. [DOI] [PubMed] [Google Scholar]
- 398.Kersten S. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep 2: 282–286, 2001. doi: 10.1093/embo-reports/kve071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest 103: 1489–1498, 1999. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Keshet Y, Seger R. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol Biol 661: 3–38, 2010. doi: 10.1007/978-1-60761-795-2_1. [DOI] [PubMed] [Google Scholar]
- 401.Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, Hammond LJ, Moyers JS, Owens RA, Gromada J, Brozinick JT, Hawkins ED, Wroblewski VJ, Li D-S, Mehrbod F, Jaskunas SR, Shanafelt AB. FGF-21 as a novel metabolic regulator. J Clin Invest 115: 1627–1635, 2005. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Kholodenko BN. Cell-signalling dynamics in time and space. Nat Rev Mol Cell Biol 7: 165–176, 2006. doi: 10.1038/nrm1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Kim J-H, Bachmann RA, Chen J. Interleukin-6 and insulin resistance. Vitam Horm 80: 613–633, 2009. doi: 10.1016/S0083-6729(08)00621-3. [DOI] [PubMed] [Google Scholar]
- 404.Kim JK, Fillmore JJ, Chen Y, Yu C, Moore IK, Pypaert M, Lutz EP, Kako Y, Velez-Carrasco W, Goldberg IJ, Breslow JL, Shulman GI. Tissue-specific overexpression of lipoprotein lipase causes tissue-specific insulin resistance. Proc Natl Acad Sci USA 98: 7522–7527, 2001. doi: 10.1073/pnas.121164498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Kim JK, Fillmore JJ, Sunshine MJ, Albrecht B, Higashimori T, Kim D-W, Liu Z-X, Soos TJ, Cline GW, O’Brien WR, Littman DR, Shulman GI. PKC-theta knockout mice are protected from fat-induced insulin resistance. J Clin Invest 114: 823–827, 2004. doi: 10.1172/JCI200422230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem 275: 8456–8460, 2000. doi: 10.1074/jbc.275.12.8456. [DOI] [PubMed] [Google Scholar]
- 407.Kim JK, Michael MD, Previs SF, Peroni OD, Mauvais-Jarvis F, Neschen S, Kahn BB, Kahn CR, Shulman GI. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J Clin Invest 105: 1791–1797, 2000. doi: 10.1172/JCI8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Kim J-Y, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, Jelicks LA, Mehler MF, Hui DY, Deshaies Y, Shulman GI, Schwartz GJ, Scherer PE. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest 117: 2621–2637, 2007. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Kim K-S, Seeley RJ, Sandoval DA. Signalling from the periphery to the brain that regulates energy homeostasis. Nat Rev Neurosci 19: 185–196, 2018. doi: 10.1038/nrn.2018.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Kir S, Beddow SA, Samuel VT, Miller P, Previs SF, Suino-Powell K, Xu HE, Shulman GI, Kliewer SA, Mangelsdorf DJ. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science 331: 1621–1624, 2011. doi: 10.1126/science.1198363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Kitamura T, Kitamura Y, Kuroda S, Hino Y, Ando M, Kotani K, Konishi H, Matsuzaki H, Kikkawa U, Ogawa W, Kasuga M. Insulin-induced phosphorylation and activation of cyclic nucleotide phosphodiesterase 3B by the serine-threonine kinase Akt. Mol Cell Biol 19: 6286–6296, 1999. doi: 10.1128/MCB.19.9.6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Kleinert M, Clemmensen C, Hofmann SM, Moore MC, Renner S, Woods SC, Huypens P, Beckers J, de Angelis MH, Schürmann A, Bakhti M, Klingenspor M, Heiman M, Cherrington AD, Ristow M, Lickert H, Wolf E, Havel PJ, Müller TD, Tschöp MH. Animal models of obesity and diabetes mellitus. Nat Rev Endocrinol 14: 140–162, 2018. doi: 10.1038/nrendo.2017.161. [DOI] [PubMed] [Google Scholar]
- 413.Klip A, Sun Y, Chiu TT, Foley KP. Signal transduction meets vesicle traffic: the software and hardware of GLUT4 translocation. Am J Physiol Cell Physiol 306: C879–C886, 2014. doi: 10.1152/ajpcell.00069.2014. [DOI] [PubMed] [Google Scholar]
- 414.Knowles JW, Xie W, Zhang Z, Chennamsetty I, Assimes TL, Paananen J, Hansson O, Pankow J, Goodarzi MO, Carcamo-Orive I, Morris AP, Chen YD, Mäkinen VP, Ganna A, Mahajan A, Guo X, Abbasi F, Greenawalt DM, Lum P, Molony C, Lind L, Lindgren C, Raffel LJ, Tsao PS, Schadt EE, Rotter JI, Sinaiko A, Reaven G, Yang X, Hsiung CA, Groop L, Cordell HJ, Laakso M, Hao K, Ingelsson E, Frayling TM, Weedon MN, Walker M, Quertermous T; RISC (Relationship between Insulin Sensitivity and Cardiovascular Disease) ConsortiumEUGENE2 (European Network on Functional Genomics of Type 2 Diabetes) StudyGUARDIAN (Genetics UndeRlying DIAbetes in HispaNics) Consortium; SAPPHIRe (Stanford Asian and Pacific Program for Hypertension and Insulin Resistance) Study . Identification and validation of N-acetyltransferase 2 as an insulin sensitivity gene. J Clin Invest 125: 1739–1751, 2015. doi: 10.1172/JCI74692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Koch L, Wunderlich FT, Seibler J, Könner AC, Hampel B, Irlenbusch S, Brabant G, Kahn CR, Schwenk F, Brüning JC. Central insulin action regulates peripheral glucose and fat metabolism in mice. J Clin Invest 118: 2132–2147, 2008. doi: 10.1172/JCI31073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Koh H-J, Toyoda T, Didesch MM, Lee M-Y, Sleeman MW, Kulkarni RN, Musi N, Hirshman MF, Goodyear LJ. Tribbles 3 mediates endoplasmic reticulum stress-induced insulin resistance in skeletal muscle. Nat Commun 4: 1871, 2013. doi: 10.1038/ncomms2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Kolterman OG, Gray RS, Griffin J, Burstein P, Insel J, Scarlett JA, Olefsky JM. Receptor and postreceptor defects contribute to the insulin resistance in noninsulin-dependent diabetes mellitus. J Clin Invest 68: 957–969, 1981. doi: 10.1172/JCI110350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Kolterman OG, Reaven GM, Olefsky JM. Relationship between in vivo insulin resistance and decreased insulin receptors in obese man. J Clin Endocrinol Metab 48: 487–494, 1979. doi: 10.1210/jcem-48-3-487. [DOI] [PubMed] [Google Scholar]
- 419.König M, Bulik S, Holzhütter H-G. Quantifying the contribution of the liver to glucose homeostasis: a detailed kinetic model of human hepatic glucose metabolism. PLOS Comput Biol 8: e1002577, 2012. doi: 10.1371/journal.pcbi.1002577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Kono T, Barham FW. The relationship between the insulin-binding capacity of fat cells and the cellular response to insulin. Studies with intact and trypsin-treated fat cells. J Biol Chem 246: 6210–6216, 1971. [PubMed] [Google Scholar]
- 421.Koo S-H, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, Takemori H, Montminy M. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437: 1109–1111, 2005. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- 422.Korenblat KM, Fabbrini E, Mohammed BS, Klein S. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology 134: 1369–1375, 2008. doi: 10.1053/j.gastro.2008.01.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Koren S, DiPilato LM, Emmett MJ, Shearin AL, Chu Q, Monks B, Birnbaum MJ. The role of mouse Akt2 in insulin-dependent suppression of adipocyte lipolysis in vivo. Diabetologia 58: 1063–1070, 2015. doi: 10.1007/s00125-015-3532-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell 160: 816–827, 2015. doi: 10.1016/j.cell.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Kotnik P, Fischer-Posovszky P, Wabitsch M. RBP4: a controversial adipokine. Eur J Endocrinol 165: 703–711, 2011. doi: 10.1530/EJE-11-0431. [DOI] [PubMed] [Google Scholar]
- 426.Koves TR, Li P, An J, Akimoto T, Slentz D, Ilkayeva O, Dohm GL, Yan Z, Newgard CB, Muoio DM. Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J Biol Chem 280: 33588–33598, 2005. doi: 10.1074/jbc.M507621200. [DOI] [PubMed] [Google Scholar]
- 427.Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JRB, Newgard CB, Lopaschuk GD, Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab 7: 45–56, 2008. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 428.Kowalski GM, Bruce CR. The regulation of glucose metabolism: implications and considerations for the assessment of glucose homeostasis in rodents. Am J Physiol Endocrinol Metab 307: E859–E871, 2014. doi: 10.1152/ajpendo.00165.2014. [DOI] [PubMed] [Google Scholar]
- 429.Kraegen EW, Clark PW, Jenkins AB, Daley EA, Chisholm DJ, Storlien LH. Development of muscle insulin resistance after liver insulin resistance in high-fat-fed rats. Diabetes 40: 1397–1403, 1991. doi: 10.2337/diab.40.11.1397. [DOI] [PubMed] [Google Scholar]
- 430.Kraegen EW, James DE, Jenkins AB, Chisholm DJ. Dose-response curves for in vivo insulin sensitivity in individual tissues in rats. Am J Physiol Endocrinol Metab 248: E353–E362, 1985. [DOI] [PubMed] [Google Scholar]
- 431.Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, Landerholm RW, Crouthamel M, Gozal D, Hwang S, Singh PK, Becker L. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab 20: 614–625, 2014. doi: 10.1016/j.cmet.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Kraus D, Yang Q, Kong D, Banks AS, Zhang L, Rodgers JT, Pirinen E, Pulinilkunnil TC, Gong F, Wang YC, Cen Y, Sauve AA, Asara JM, Peroni OD, Monia BP, Bhanot S, Alhonen L, Puigserver P, Kahn BB. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 508: 258–262, 2014. doi: 10.1038/nature13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Krebs EG, Love DS, Bratvold GE, Trayser KA, Meyer WL, Fischer EH. Purification and properties of rabbit skeletal muscle phosphorylase B kinase. Biochemistry 3: 1022–1033, 1964. doi: 10.1021/bi00896a003. [DOI] [PubMed] [Google Scholar]
- 434.Krebs HA, Speake RN, Hems R. Acceleration of renal gluconeogenesis by ketone bodies and fatty acids. Biochem J 94: 712–720, 1965. doi: 10.1042/bj0940712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Krebs M, Krssak M, Bernroider E, Anderwald C, Brehm A, Meyerspeer M, Nowotny P, Roth E, Waldhäusl W, Roden M. Mechanism of amino acid-induced skeletal muscle insulin resistance in humans. Diabetes 51: 599–605, 2002. doi: 10.2337/diabetes.51.3.599. [DOI] [PubMed] [Google Scholar]
- 436.Krook A, Björnholm M, Galuska D, Jiang XJ, Fahlman R, Myers MG Jr, Wallberg-Henriksson H, Zierath JR. Characterization of signal transduction and glucose transport in skeletal muscle from type 2 diabetic patients. Diabetes 49: 284–292, 2000. doi: 10.2337/diabetes.49.2.284. [DOI] [PubMed] [Google Scholar]
- 437.Krssak M, Brehm A, Bernroider E, Anderwald C, Nowotny P, Dalla Man C, Cobelli C, Cline GW, Shulman GI, Waldhäusl W, Roden M. Alterations in postprandial hepatic glycogen metabolism in type 2 diabetes. Diabetes 53: 3048–3056, 2004. doi: 10.2337/diabetes.53.12.3048. [DOI] [PubMed] [Google Scholar]
- 438.Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, Roden M, Shulman GI. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia 42: 113–116, 1999. doi: 10.1007/s001250051123. [DOI] [PubMed] [Google Scholar]
- 439.Kruszynska YT, Home PD. Liver and muscle insulin sensitivity, glycogen concentration and glycogen synthase activity in a rat model of non-insulin-dependent diabetes. Diabetologia 31: 304–309, 1988. [DOI] [PubMed] [Google Scholar]
- 440.Kruszynska YT, Home PD, Alberti KGMM. In vivo regulation of liver and skeletal muscle glycogen synthase activity by glucose and insulin. Diabetes 35: 662–667, 1986. doi: 10.2337/diab.35.6.662. [DOI] [PubMed] [Google Scholar]
- 441.Kruszynska YT, Worrall DS, Ofrecio J, Frias JP, Macaraeg G, Olefsky JM. Fatty acid-induced insulin resistance: decreased muscle PI3K activation but unchanged Akt phosphorylation. J Clin Endocrinol Metab 87: 226–234, 2002. doi: 10.1210/jcem.87.1.8187. [DOI] [PubMed] [Google Scholar]
- 442.Krycer JR, Sharpe LJ, Luu W, Brown AJ. The Akt-SREBP nexus: cell signaling meets lipid metabolism. Trends Endocrinol Metab 21: 268–276, 2010. doi: 10.1016/j.tem.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 443.Kubota N, Kubota T, Kajiwara E, Iwamura T, Kumagai H, Watanabe T, Inoue M, Takamoto I, Sasako T, Kumagai K, Kohjima M, Nakamuta M, Moroi M, Sugi K, Noda T, Terauchi Y, Ueki K, Kadowaki T. Differential hepatic distribution of insulin receptor substrates causes selective insulin resistance in diabetes and obesity. Nat Commun 7: 12977, 2016. doi: 10.1038/ncomms12977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Kullmann S, Heni M, Hallschmid M, Fritsche A, Preissl H, Häring H-U. Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol Rev 96: 1169–1209, 2016. doi: 10.1152/physrev.00032.2015. [DOI] [PubMed] [Google Scholar]
- 445.Kumashiro N, Erion DM, Zhang D, Kahn M, Beddow SA, Chu X, Still CD, Gerhard GS, Han X, Dziura J, Petersen KF, Samuel VT, Shulman GI. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci USA 108: 16381–16385, 2011. doi: 10.1073/pnas.1113359108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Lackey DE, Olefsky JM. Regulation of metabolism by the innate immune system. Nat Rev Endocrinol 12: 15–28, 2016. doi: 10.1038/nrendo.2015.189. [DOI] [PubMed] [Google Scholar]
- 447.Lafontan M, Langin D. Lipolysis and lipid mobilization in human adipose tissue. Prog Lipid Res 48: 275–297, 2009. doi: 10.1016/j.plipres.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 448.Lagarrigue S, Lopez-Mejia IC, Denechaud P-D, Escoté X, Castillo-Armengol J, Jimenez V, Chavey C, Giralt A, Lai Q, Zhang L, Martinez-Carreres L, Delacuisine B, Annicotte J-S, Blanchet E, Huré S, Abella A, Tinahones FJ, Vendrell J, Dubus P, Bosch F, Kahn CR, Fajas L. CDK4 is an essential insulin effector in adipocytes. J Clin Invest 126: 335–348, 2016. doi: 10.1172/JCI81480. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 449.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 146: 726–735, 2014. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Lam TKT, Yoshii H, Haber CA, Bogdanovic E, Lam L, Fantus IG, Giacca A. Free fatty acid-induced hepatic insulin resistance: a potential role for protein kinase C-delta. Am J Physiol Endocrinol Metab 283: E682–E691, 2002. doi: 10.1152/ajpendo.00038.2002. [DOI] [PubMed] [Google Scholar]
- 451.Langlet F, Haeusler RA, Lindén D, Ericson E, Norris T, Johansson A, Cook JR, Aizawa K, Wang L, Buettner C, Accili D. Selective Inhibition of FOXO1 Activator/Repressor Balance Modulates Hepatic Glucose Handling. Cell 171: 824–835.e18, 2017. doi: 10.1016/j.cell.2017.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Laplante M, Sabatini DM. mTORC1 activates SREBP-1c and uncouples lipogenesis from gluconeogenesis. Proc Natl Acad Sci USA 107: 3281–3282, 2010. [Correction in ProcNatl Acad Sci USA 107: 7616, 2010.] 10.1073/pnas.1000323107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 149: 274–293, 2012. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Latva-Rasku A, Honka M-J, Stančáková A, Koistinen HA, Kuusisto J, Guan L, Manning AK, Stringham H, Gloyn AL, Lindgren CM, Collins FS, Mohlke KL, Scott LJ, Karjalainen T, Nummenmaa L, Boehnke M, Nuutila P, Laakso M; T2D-GENES Consortium . A Partial Loss-of-Function Variant in AKT2 Is Associated With Reduced Insulin-Mediated Glucose Uptake in Multiple Insulin-Sensitive Tissues: A Genotype-Based Callback Positron Emission Tomography Study. Diabetes 67: 334–342, 2018. doi: 10.2337/db17-1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Laurencikiene J, van Harmelen V, Arvidsson Nordström E, Dicker A, Blomqvist L, Näslund E, Langin D, Arner P, Rydén M. NF-kappaB is important for TNF-alpha-induced lipolysis in human adipocytes. J Lipid Res 48: 1069–1077, 2007. doi: 10.1194/jlr.M600471-JLR200. [DOI] [PubMed] [Google Scholar]
- 456.Laybutt DR, Schmitz-Peiffer C, Saha AK, Ruderman NB, Biden TJ, Kraegen EW. Muscle lipid accumulation and protein kinase C activation in the insulin-resistant chronically glucose-infused rat. Am J Physiol Endocrinol Metab 277: E1070–E1076, 1999. [DOI] [PubMed] [Google Scholar]
- 457.Leavens KF, Birnbaum MJ. Insulin signaling to hepatic lipid metabolism in health and disease. Crit Rev Biochem Mol Biol 46: 200–215, 2011. doi: 10.3109/10409238.2011.562481. [DOI] [PubMed] [Google Scholar]
- 458.Leavens KF, Easton RM, Shulman GI, Previs SF, Birnbaum MJ. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab 10: 405–418, 2009. doi: 10.1016/j.cmet.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Lechin F, Dijs B, Pardey-Maldonado B. Insulin versus glucagon crosstalk: central plus peripheral mechanisms. Am J Ther 20: 349–362, 2013. doi: 10.1097/MJT.0b013e318235f295. [DOI] [PubMed] [Google Scholar]
- 460.Lee A-H, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320: 1492–1496, 2008. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Lee B-C, Kim M-S, Pae M, Yamamoto Y, Eberlé D, Shimada T, Kamei N, Park H-S, Sasorith S, Woo JR, You J, Mosher W, Brady HJM, Shoelson SE, Lee J. Adipose Natural Killer Cells Regulate Adipose Tissue Macrophages to Promote Insulin Resistance in Obesity. Cell Metab 23: 685–698, 2016. doi: 10.1016/j.cmet.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Lee H-Y, Birkenfeld AL, Jornayvaz FR, Jurczak MJ, Kanda S, Popov V, Frederick DW, Zhang D, Guigni B, Bharadwaj KG, Choi CS, Goldberg IJ, Park J-H, Petersen KF, Samuel VT, Shulman GI. Apolipoprotein CIII overexpressing mice are predisposed to diet-induced hepatic steatosis and hepatic insulin resistance. Hepatology 54: 1650–1660, 2011. doi: 10.1002/hep.24571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Lee H-Y, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ, Zhang D, Woo DK, Shadel GS, Ladiges W, Rabinovitch PS, Santos JH, Petersen KF, Samuel VT, Shulman GI. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab 12: 668–674, 2010. doi: 10.1016/j.cmet.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Lee KU, Lee HK, Koh CS, Min HK. Artificial induction of intravascular lipolysis by lipid-heparin infusion leads to insulin resistance in man. Diabetologia 31: 285–290, 1988. [DOI] [PubMed] [Google Scholar]
- 465.Lee YH, Giraud J, Davis RJ, White MF. c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J Biol Chem 278: 2896–2902, 2003. doi: 10.1074/jbc.M208359200. [DOI] [PubMed] [Google Scholar]
- 466.Lee YJ, Ko EH, Kim JE, Kim E, Lee H, Choi H, Yu JH, Kim HJ, Seong J-K, Kim K-S, Kim JW. Nuclear receptor PPARγ-regulated monoacylglycerol O-acyltransferase 1 (MGAT1) expression is responsible for the lipid accumulation in diet-induced hepatic steatosis. Proc Natl Acad Sci USA 109: 13656–13661, 2012. doi: 10.1073/pnas.1203218109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Lee YS, Wollam J, Olefsky JM. An Integrated View of Immunometabolism. Cell 172: 22–40, 2018. doi: 10.1016/j.cell.2017.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Lee Y, Wang M-Y, Du XQ, Charron MJ, Unger RH. Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes 60: 391–397, 2011. doi: 10.2337/db10-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Lehel C, Olah Z, Jakab G, Anderson WB. Protein kinase C epsilon is localized to the Golgi via its zinc-finger domain and modulates Golgi function. Proc Natl Acad Sci USA 92: 1406–1410, 1995. doi: 10.1073/pnas.92.5.1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Le Marchand-Brustel Y, Grémeaux T, Ballotti R, Van Obberghen E. Insulin receptor tyrosine kinase is defective in skeletal muscle of insulin-resistant obese mice. Nature 315: 676–679, 1985. doi: 10.1038/315676a0. [DOI] [PubMed] [Google Scholar]
- 471.Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol 13: 383–396, 2012. doi: 10.1038/nrm3351. [DOI] [PubMed] [Google Scholar]
- 472.Levine R, Fritz IB. The relation of insulin to liver metabolism. Diabetes 5: 209–222, 1956. doi: 10.2337/diab.5.3.209. [DOI] [PubMed] [Google Scholar]
- 473.Levin MC, Monetti M, Watt MJ, Sajan MP, Stevens RD, Bain JR, Newgard CB, Farese RV Sr, Farese RV Jr. Increased lipid accumulation and insulin resistance in transgenic mice expressing DGAT2 in glycolytic (type II) muscle. Am J Physiol Endocrinol Metab 293: E1772–E1781, 2007. doi: 10.1152/ajpendo.00158.2007. [DOI] [PubMed] [Google Scholar]
- 474.Lewis GF, Carpentier A, Adeli K, Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev 23: 201–229, 2002. doi: 10.1210/edrv.23.2.0461. [DOI] [PubMed] [Google Scholar]
- 475.Lewis GF, O’Meara NM, Cabana VG, Blackman JD, Pugh WL, Druetzler AF, Lukens JR, Getz GS, Polonsky KS. Postprandial triglyceride response in type 1 (insulin-dependent) diabetes mellitus is not altered by short-term deterioration in glycaemic control or level of postprandial insulin replacement. Diabetologia 34: 253–259, 1991. doi: 10.1007/BF00405084. [DOI] [PubMed] [Google Scholar]
- 476.Lewis GF, Vranic M, Harley P, Giacca A. Fatty acids mediate the acute extrahepatic effects of insulin on hepatic glucose production in humans. Diabetes 46: 1111–1119, 1997. doi: 10.2337/diab.46.7.1111. [DOI] [PubMed] [Google Scholar]
- 477.Lewis GF, Zinman B, Groenewoud Y, Vranic M, Giacca A. Hepatic glucose production is regulated both by direct hepatic and extrahepatic effects of insulin in humans. Diabetes 45: 454–462, 1996. doi: 10.2337/diab.45.4.454. [DOI] [PubMed] [Google Scholar]
- 478.Lewis RE, Cao L, Perregaux D, Czech MP. Threonine 1336 of the human insulin receptor is a major target for phosphorylation by protein kinase C. Biochemistry 29: 1807–1813, 1990. doi: 10.1021/bi00459a020. [DOI] [PubMed] [Google Scholar]
- 479.Lewis RE, Wu GP, MacDonald RG, Czech MP. Insulin-sensitive phosphorylation of serine 1293/1294 on the human insulin receptor by a tightly associated serine kinase. J Biol Chem 265: 947–954, 1990. [PubMed] [Google Scholar]
- 480.Li JH, Gautam D, Han S-J, Guettier J-M, Cui Y, Lu H, Deng C, O’Hare J, Jou W, Gavrilova O, Buettner C, Wess J. Hepatic muscarinic acetylcholine receptors are not critically involved in maintaining glucose homeostasis in mice. Diabetes 58: 2776–2787, 2009. doi: 10.2337/db09-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Lillioja S, Mott DM, Spraul M, Ferraro R, Foley JE, Ravussin E, Knowler WC, Bennett PH, Bogardus C. Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus. Prospective studies of Pima Indians. N Engl J Med 329: 1988–1992, 1993. doi: 10.1056/NEJM199312303292703. [DOI] [PubMed] [Google Scholar]
- 482.Li M, Vienberg SG, Bezy O, O’Neill BT, Kahn CR. Role of PKCδ in Insulin Sensitivity and Skeletal Muscle Metabolism. Diabetes 64: 4023–4032, 2015. doi: 10.2337/db14-1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Lindh R, Ahmad F, Resjö S, James P, Yang JS, Fales HM, Manganiello V, Degerman E. Multisite phosphorylation of adipocyte and hepatocyte phosphodiesterase 3B. Biochim Biophys Acta 1773: 584–592, 2007. doi: 10.1016/j.bbamcr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 484.Lin HV, Accili D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab 14: 9–19, 2011. doi: 10.1016/j.cmet.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Li P, Liu S, Lu M, Bandyopadhyay G, Oh D, Imamura T, Johnson AMF, Sears D, Shen Z, Cui B, Kong L, Hou S, Liang X, Iovino S, Watkins SM, Ying W, Osborn O, Wollam J, Brenner M, Olefsky JM. Hematopoietic-Derived Galectin-3 Causes Cellular and Systemic Insulin Resistance. Cell 167: 973–984.e12, 2016. doi: 10.1016/j.cell.2016.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Li P, Oh DY, Bandyopadhyay G, Lagakos WS, Talukdar S, Osborn O, Johnson A, Chung H, Maris M, Ofrecio JM, Taguchi S, Lu M, Olefsky JM. LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat Med 21: 239–247, 2015. doi: 10.1038/nm.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci USA 107: 3441–3446, 2010. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Lithell H, Orlander J, Schéle R, Sjödin B, Karlsson J. Changes in lipoprotein-lipase activity and lipid stores in human skeletal muscle with prolonged heavy exercise. Acta Physiol Scand 107: 257–261, 1979. doi: 10.1111/j.1748-1716.1979.tb06471.x. [DOI] [PubMed] [Google Scholar]
- 489.Liu F, Roth RA. Identification of serines-1035/1037 in the kinase domain of the insulin receptor as protein kinase C alpha mediated phosphorylation sites. FEBS Lett 352: 389–392, 1994. doi: 10.1016/0014-5793(94)00996-1. [DOI] [PubMed] [Google Scholar]
- 490.Liu J, Ibi D, Taniguchi K, Lee J, Herrema H, Akosman B, Mucka P, Salazar Hernandez MA, Uyar MF, Park SW, Karin M, Ozcan U. Inflammation Improves Glucose Homeostasis through IKKβ-XBP1s Interaction. Cell 167: 1052–1066.e18, 2016. doi: 10.1016/j.cell.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Liu L, Zhang Y, Chen N, Shi X, Tsang B, Yu Y-H. Upregulation of myocellular DGAT1 augments triglyceride synthesis in skeletal muscle and protects against fat-induced insulin resistance. J Clin Invest 117: 1679–1689, 2007. doi: 10.1172/JCI30565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Liu T, Yu B, Kakino M, Fujimoto H, Ando Y, Hakuno F, Takahashi S-I. A novel IRS-1-associated protein, DGKζ regulates GLUT4 translocation in 3T3-L1 adipocytes. Sci Rep 6: 35438, 2016. doi: 10.1038/srep35438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Liu Y, Dentin R, Chen D, Hedrick S, Ravnskjaer K, Schenk S, Milne J, Meyers DJ, Cole P, Yates J III, Olefsky J, Guarente L, Montminy M. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature 456: 269–273, 2008. doi: 10.1038/nature07349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Li Y, Soos TJ, Li X, Wu J, Degennaro M, Sun X, Littman DR, Birnbaum MJ, Polakiewicz RD. Protein kinase C θ inhibits insulin signaling by phosphorylating IRS1 at Ser(1101). J Biol Chem 279: 45304–45307, 2004. doi: 10.1074/jbc.C400186200. [DOI] [PubMed] [Google Scholar]
- 495.Li Y, Zalzala M, Jadhav K, Xu Y, Kasumov T, Yin L, Zhang Y. Carboxylesterase 2 prevents liver steatosis by modulating lipolysis, endoplasmic reticulum stress, and lipogenesis and is regulated by hepatocyte nuclear factor 4 alpha in mice. Hepatology 63: 1860–1874, 2016. doi: 10.1002/hep.28472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Lodhi IJ, Wei X, Semenkovich CF. Lipoexpediency: de novo lipogenesis as a metabolic signal transmitter. Trends Endocrinol Metab 22: 1–8, 2011. doi: 10.1016/j.tem.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Loh K, Deng H, Fukushima A, Cai X, Boivin B, Galic S, Bruce C, Shields BJ, Skiba B, Ooms LM, Stepto N, Wu B, Mitchell CA, Tonks NK, Watt MJ, Febbraio MA, Crack PJ, Andrikopoulos S, Tiganis T. Reactive oxygen species enhance insulin sensitivity. Cell Metab 10: 260–272, 2009. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Long YC, Cheng Z, Copps KD, White MF. Insulin receptor substrates Irs1 and Irs2 coordinate skeletal muscle growth and metabolism via the Akt and AMPK pathways. Mol Cell Biol 31: 430–441, 2011. [Correction in Mol Cell Biol 37: e00232-17, 2017.] 10.1128/MCB.00983-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.López-Sánchez I, Garcia-Marcos M, Mittal Y, Aznar N, Farquhar MG, Ghosh P. Protein kinase C-theta (PKCθ) phosphorylates and inhibits the guanine exchange factor, GIV/Girdin. Proc Natl Acad Sci USA 110: 5510–5515, 2013. doi: 10.1073/pnas.1303392110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Lotta LA, Gulati P, Day FR, Payne F, Ongen H, van de Bunt M, Gaulton KJ, Eicher JD, Sharp SJ, Luan J, De Lucia Rolfe E, Stewart ID, Wheeler E, Willems SM, Adams C, Yaghootkar H, Forouhi NG, Khaw K-T, Johnson AD, Semple RK, Frayling T, Perry JRB, Dermitzakis E, McCarthy MI, Barroso I, Wareham NJ, Savage DB, Langenberg C, O’Rahilly S, Scott RA; EPIC-InterAct Consortium; Cambridge FPLD1 Consortium . Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet 49: 17–26, 2017. [Corrigendum in NatGenet 49: 317, 2017.] 10.1038/ng.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Lotta LA, Scott RA, Sharp SJ, Burgess S, Luan J, Tillin T, Schmidt AF, Imamura F, Stewart ID, Perry JRB, Marney L, Koulman A, Karoly ED, Forouhi NG, Sjögren RJO, Näslund E, Zierath JR, Krook A, Savage DB, Griffin JL, Chaturvedi N, Hingorani AD, Khaw K-T, Barroso I, McCarthy MI, O’Rahilly S, Wareham NJ, Langenberg C. Genetic Predisposition to an Impaired Metabolism of the Branched-Chain Amino Acids and Risk of Type 2 Diabetes: A Mendelian Randomisation Analysis. PLoS Med 13: e1002179, 2016. doi: 10.1371/journal.pmed.1002179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science 307: 384–387, 2005. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 504.Lu M, Wan M, Leavens KF, Chu Q, Monks BR, Fernandez S, Ahima RS, Ueki K, Kahn CR, Birnbaum MJ. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med 18: 388–395, 2012. doi: 10.1038/nm.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Luo J, Sobkiw CL, Hirshman MF, Logsdon MN, Li TQ, Goodyear LJ, Cantley LC. Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell Metab 3: 355–366, 2006. doi: 10.1016/j.cmet.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 506.Luukkonen PK, Zhou Y, Sädevirta S, Leivonen M, Arola J, Orešič M, Hyötyläinen T, Yki-Järvinen H. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J Hepatol 64: 1167–1175, 2016. doi: 10.1016/j.jhep.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 507.Luzi L, Petrides AS, De Fronzo RA. Different sensitivity of glucose and amino acid metabolism to insulin in NIDDM. Diabetes 42: 1868–1877, 1993. doi: 10.2337/diab.42.12.1868. [DOI] [PubMed] [Google Scholar]
- 508.Lynch CJ, Adams SH. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat Rev Endocrinol 10: 723–736, 2014. doi: 10.1038/nrendo.2014.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508a.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 10: 307–318, 2009. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 509.Macotela Y, Emanuelli B, Bång AM, Espinoza DO, Boucher J, Beebe K, Gall W, Kahn CR. Dietary leucine–an environmental modifier of insulin resistance acting on multiple levels of metabolism. PLoS One 6: e21187, 2011. doi: 10.1371/journal.pone.0021187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Madani S, Hichami A, Legrand A, Belleville J, Khan NA. Implication of acyl chain of diacylglycerols in activation of different isoforms of protein kinase C. FASEB Jl 15: 2595–2601, 2001. doi: 10.1096/fj.01-0753int. [DOI] [PubMed] [Google Scholar]
- 511.Madiraju AK, Erion DM, Rahimi Y, Zhang X-M, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, Jurczak MJ, Camporez J-P, Lee H-Y, Cline GW, Samuel VT, Kibbey RG, Shulman GI. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 510: 542–546, 2014. doi: 10.1038/nature13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Magkos F, Su X, Bradley D, Fabbrini E, Conte C, Eagon JC, Varela JE, Brunt EM, Patterson BW, Klein S. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 142: 1444–1446.e2, 2012. doi: 10.1053/j.gastro.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Magnusson I, Rothman DL, Katz LD, Shulman RG, Shulman GI. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J Clin Invest 90: 1323–1327, 1992. doi: 10.1172/JCI115997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Ma GS, Lopez-Sanchez I, Aznar N, Kalogriopoulos N, Pedram S, Midde K, Ciaraldi TP, Henry RR, Ghosh P. Activation of G proteins by GIV-GEF is a pivot point for insulin resistance and sensitivity. Mol Biol Cell 26: 4209–4223, 2015. doi: 10.1091/mbc.e15-08-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Mahadev K, Motoshima H, Wu X, Ruddy JM, Arnold RS, Cheng G, Lambeth JD, Goldstein BJ. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol 24: 1844–1854, 2004. doi: 10.1128/MCB.24.5.1844-1854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Mahendran Y, Jonsson A, Have CT, Allin KH, Witte DR, Jørgensen ME, Grarup N, Pedersen O, Kilpeläinen TO, Hansen T. Genetic evidence of a causal effect of insulin resistance on branched-chain amino acid levels. Diabetologia 60: 873–878, 2017. doi: 10.1007/s00125-017-4222-6. [DOI] [PubMed] [Google Scholar]
- 517.Mannerås-Holm L, Kirchner H, Björnholm M, Chibalin AV, Zierath JR. mRNA expression of diacylglycerol kinase isoforms in insulin-sensitive tissues: effects of obesity and insulin resistance. Physiol Rep 3: e12372, 2015. doi: 10.14814/phy2.12372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Mannerås-Holm L, Schönke M, Brozinick JT, Vetterli L, Bui H-H, Sanders P, Nascimento EBM, Björnholm M, Chibalin AV, Zierath JR. Diacylglycerol kinase ε deficiency preserves glucose tolerance and modulates lipid metabolism in obese mice. J Lipid Res 58: 907–915, 2017. doi: 10.1194/jlr.M074443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Mansell PI, Macdonald IA. The effect of starvation on insulin-induced glucose disposal and thermogenesis in humans. Metabolism 39: 502–510, 1990. doi: 10.1016/0026-0495(90)90009-2. [DOI] [PubMed] [Google Scholar]
- 520.Marchesini G, Brizi M, Morselli-Labate AM, Bianchi G, Bugianesi E, McCullough AJ, Forlani G, Melchionda N. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med 107: 450–455, 1999. doi: 10.1016/S0002-9343(99)00271-5. [DOI] [PubMed] [Google Scholar]
- 521.Marcinkiewicz A, Gauthier D, Garcia A, Brasaemle DL. The phosphorylation of serine 492 of perilipin a directs lipid droplet fragmentation and dispersion. J Biol Chem 281: 11901–11909, 2006. doi: 10.1074/jbc.M600171200. [DOI] [PubMed] [Google Scholar]
- 522.Martinez-Botas J, Anderson JB, Tessier D, Lapillonne A, Chang BH-J, Quast MJ, Gorenstein D, Chen K-H, Chan L. Absence of perilipin results in leanness and reverses obesity in Lepr(db/db) mice. Nat Genet 26: 474–479, 2000. doi: 10.1038/82630. [DOI] [PubMed] [Google Scholar]
- 523.Maruyama H, Hisatomi A, Orci L, Grodsky GM, Unger RH. Insulin within islets is a physiologic glucagon release inhibitor. J Clin Invest 74: 2296–2299, 1984. doi: 10.1172/JCI111658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Mathews ST, Chellam N, Srinivas PR, Cintron VJ, Leon MA, Goustin AS, Grunberger G. α2-HSG, a specific inhibitor of insulin receptor autophosphorylation, interacts with the insulin receptor. Mol Cell Endocrinol 164: 87–98, 2000. doi: 10.1016/S0303-7207(00)00237-9. [DOI] [PubMed] [Google Scholar]
- 525.Mathews ST, Singh GP, Ranalletta M, Cintron VJ, Qiang X, Goustin AS, Jen K-LC, Charron MJ, Jahnen-Dechent W, Grunberger G. Improved insulin sensitivity and resistance to weight gain in mice null for the Ahsg gene. Diabetes 51: 2450–2458, 2002. doi: 10.2337/diabetes.51.8.2450. [DOI] [PubMed] [Google Scholar]
- 526.Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab 6: 208–216, 2007. doi: 10.1016/j.cmet.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 527.Matsuzaka T, Shimano H, Yahagi N, Kato T, Atsumi A, Yamamoto T, Inoue N, Ishikawa M, Okada S, Ishigaki N, Iwasaki H, Iwasaki Y, Karasawa T, Kumadaki S, Matsui T, Sekiya M, Ohashi K, Hasty AH, Nakagawa Y, Takahashi A, Suzuki H, Yatoh S, Sone H, Toyoshima H, Osuga J, Yamada N. Crucial role of a long-chain fatty acid elongase, Elovl6, in obesity-induced insulin resistance. Nat Med 13: 1193–1202, 2007. doi: 10.1038/nm1662. [DOI] [PubMed] [Google Scholar]
- 528.Mattacks CA, Sadler D, Pond CM. The cellular structure and lipid/protein composition of adipose tissue surrounding chronically stimulated lymph nodes in rats. J Anat 202: 551–561, 2003. doi: 10.1046/j.1469-7580.2003.00188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Matzinger D, Gutzwiller JP, Drewe J, Orban A, Engel R, D’Amato M, Rovati L, Beglinger C. Inhibition of food intake in response to intestinal lipid is mediated by cholecystokinin in humans. Am J Physiol Regul Integr Comp Physiol 277: R1718–R1724, 1999. [DOI] [PubMed] [Google Scholar]
- 531.McDonough PM, Maciejewski-Lenoir D, Hartig SM, Hanna RA, Whittaker R, Heisel A, Nicoll JB, Buehrer BM, Christensen K, Mancini MG, Mancini MA, Edwards DP, Price JH. Differential phosphorylation of perilipin 1A at the initiation of lipolysis revealed by novel monoclonal antibodies and high content analysis. PLoS One 8: e55511, 2013. doi: 10.1371/journal.pone.0055511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.McGarry JD. What if Minkowski had been ageusic? An alternative angle on diabetes. Science 258: 766–770, 1992. doi: 10.1126/science.1439783. [DOI] [PubMed] [Google Scholar]
- 533.McGarry JD, Mannaerts GP, Foster DW. A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J Clin Invest 60: 265–270, 1977. doi: 10.1172/JCI108764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.McGuinness OP, Ayala JE, Laughlin MR, Wasserman DH. NIH experiment in centralized mouse phenotyping: the Vanderbilt experience and recommendations for evaluating glucose homeostasis in the mouse. Am J Physiol Endocrinol Metab 297: E849–E855, 2009. doi: 10.1152/ajpendo.90996.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene 26: 3113–3121, 2007. doi: 10.1038/sj.onc.1210394. [DOI] [PubMed] [Google Scholar]
- 536.McQuaid SE, Hodson L, Neville MJ, Dennis AL, Cheeseman J, Humphreys SM, Ruge T, Gilbert M, Fielding BA, Frayn KN, Karpe F. Downregulation of adipose tissue fatty acid trafficking in obesity: a driver for ectopic fat deposition? Diabetes 60: 47–55, 2011. doi: 10.2337/db10-0867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537.Meek TH, Morton GJ. The role of leptin in diabetes: metabolic effects. Diabetologia 59: 928–932, 2016. doi: 10.1007/s00125-016-3898-3. [DOI] [PubMed] [Google Scholar]
- 538.Menke A, Casagrande S, Geiss L, Cowie CC. Revalence of and trends in diabetes among adults in the united states, 1988–2012. JAMA 314: 1021–1029, 2015. doi: 10.1001/jama.2015.10029. [DOI] [PubMed] [Google Scholar]
- 539.Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA, Kahn CR. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell 6: 87–97, 2000. doi: 10.1016/S1097-2765(05)00015-8. [DOI] [PubMed] [Google Scholar]
- 540.Middelbeek RJW, Chambers MA, Tantiwong P, Treebak JT, An D, Hirshman MF, Musi N, Goodyear LJ. Insulin stimulation regulates AS160 and TBC1D1 phosphorylation sites in human skeletal muscle. Nutr Diabetes 3: e74, 2013. doi: 10.1038/nutd.2013.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Mihalik SJ, Goodpaster BH, Kelley DE, Chace DH, Vockley J, Toledo FGS, DeLany JP. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity (Silver Spring) 18: 1695–1700, 2010. doi: 10.1038/oby.2009.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Miller RA, Chu Q, Le Lay J, Scherer PE, Ahima RS, Kaestner KH, Foretz M, Viollet B, Birnbaum MJ. Adiponectin suppresses gluconeogenic gene expression in mouse hepatocytes independent of LKB1-AMPK signaling. J Clin Invest 121: 2518–2528, 2011. doi: 10.1172/JCI45942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Minehira K, Young SG, Villanueva CJ, Yetukuri L, Oresic M, Hellerstein MK, Farese RV Jr, Horton JD, Preitner F, Thorens B, Tappy L. Blocking VLDL secretion causes hepatic steatosis but does not affect peripheral lipid stores or insulin sensitivity in mice. J Lipid Res 49: 2038–2044, 2008. doi: 10.1194/jlr.M800248-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Min J, Okada S, Kanzaki M, Elmendorf JS, Coker KJ, Ceresa BP, Syu LJ, Noda Y, Saltiel AR, Pessin JE. Synip: a novel insulin-regulated syntaxin 4-binding protein mediating GLUT4 translocation in adipocytes. Mol Cell 3: 751–760, 1999. doi: 10.1016/S1097-2765(01)80007-1. [DOI] [PubMed] [Google Scholar]
- 545.Miyake Y, Kozutsumi Y, Nakamura S, Fujita T, Kawasaki T. Serine palmitoyltransferase is the primary target of a sphingosine-like immunosuppressant, ISP-1/myriocin. Biochem Biophys Res Commun 211: 396–403, 1995. doi: 10.1006/bbrc.1995.1827. [DOI] [PubMed] [Google Scholar]
- 546.Miyoshi H, Souza SC, Zhang H-H, Strissel KJ, Christoffolete MA, Kovsan J, Rudich A, Kraemer FB, Bianco AC, Obin MS, Greenberg AS. Perilipin promotes hormone-sensitive lipase-mediated adipocyte lipolysis via phosphorylation-dependent and -independent mechanisms. J Biol Chem 281: 15837–15844, 2006. doi: 10.1074/jbc.M601097200. [DOI] [PubMed] [Google Scholar]
- 547.Monetti M, Levin MC, Watt MJ, Sajan MP, Marmor S, Hubbard BK, Stevens RD, Bain JR, Newgard CB, Farese RV Sr, Hevener AL, Farese RV Jr. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab 6: 69–78, 2007. doi: 10.1016/j.cmet.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 548.Montgomery MK, Hallahan NL, Brown SH, Liu M, Mitchell TW, Cooney GJ, Turner N. Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding. Diabetologia 56: 1129–1139, 2013. doi: 10.1007/s00125-013-2846-8. [DOI] [PubMed] [Google Scholar]
- 549.Montgomery MK, Turner N. Mitochondrial dysfunction and insulin resistance: an update. Endocr Connect 4: R1–R15, 2015. doi: 10.1530/EC-14-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Moon Y-A, Liang G, Xie X, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, Brown MS, Goldstein JL, Horton JD. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab 15: 240–246, 2012. doi: 10.1016/j.cmet.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Moore MC, Shulman GI, Giaccari A, Pagliassotti MJ, Cline G, Neal D, Rossetti L, Cherrington AD. Effect of hepatic nerves on disposition of an intraduodenal glucose load. Am J Physiol Endocrinol Metab 265: E487–E496, 1993. [DOI] [PubMed] [Google Scholar]
- 552.Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273, 2003. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 553.Moraes-Vieira PM, Yore MM, Dwyer PM, Syed I, Aryal P, Kahn BB. RBP4 activates antigen-presenting cells, leading to adipose tissue inflammation and systemic insulin resistance. Cell Metab 19: 512–526, 2014. doi: 10.1016/j.cmet.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554.Morino K, Neschen S, Bilz S, Sono S, Tsirigotis D, Reznick RM, Moore I, Nagai Y, Samuel V, Sebastian D, White M, Philbrick W, Shulman GI. Muscle-specific IRS-1 Ser->Ala transgenic mice are protected from fat-induced insulin resistance in skeletal muscle. Diabetes 57: 2644–2651, 2008. doi: 10.2337/db06-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Morley TS, Xia JY, Scherer PE. Selective enhancement of insulin sensitivity in the mature adipocyte is sufficient for systemic metabolic improvements. Nat Commun 6: 7906, 2015. doi: 10.1038/ncomms8906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Morton GJ, Matsen ME, Bracy DP, Meek TH, Nguyen HT, Stefanovski D, Bergman RN, Wasserman DH, Schwartz MW. FGF19 action in the brain induces insulin-independent glucose lowering. J Clin Invest 123: 4799–4808, 2013. doi: 10.1172/JCI70710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Mulder H, Sörhede-Winzell M, Contreras JA, Fex M, Ström K, Ploug T, Galbo H, Arner P, Lundberg C, Sundler F, Ahrén B, Holm C. Hormone-sensitive lipase null mice exhibit signs of impaired insulin sensitivity whereas insulin secretion is intact. J Biol Chem 278: 36380–36388, 2003. doi: 10.1074/jbc.M213032200. [DOI] [PubMed] [Google Scholar]
- 558.Müller HK, Kellerer M, Ermel B, Mühlhöfer A, Obermaier-Kusser B, Vogt B, Häring HU. Prevention by protein kinase C inhibitors of glucose-induced insulin-receptor tyrosine kinase resistance in rat fat cells. Diabetes 40: 1440–1448, 1991. doi: 10.2337/diab.40.11.1440. [DOI] [PubMed] [Google Scholar]
- 559.Muñoz MC, Giani JF, Mayer MA, Toblli JE, Turyn D, Dominici FP. TANK-binding kinase 1 mediates phosphorylation of insulin receptor at serine residue 994: a potential link between inflammation and insulin resistance. J Endocrinol 201: 185–197, 2009. doi: 10.1677/JOE-08-0276. [DOI] [PubMed] [Google Scholar]
- 560.Muoio DM. Revisiting the connection between intramyocellular lipids and insulin resistance: a long and winding road. Diabetologia 55: 2551–2554, 2012. doi: 10.1007/s00125-012-2597-y. [DOI] [PubMed] [Google Scholar]
- 561.Muoio DM, Neufer PD. Lipid-induced mitochondrial stress and insulin action in muscle. Cell Metab 15: 595–605, 2012. doi: 10.1016/j.cmet.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, Cinti S. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J Lipid Res 49: 1562–1568, 2008. doi: 10.1194/jlr.M800019-JLR200. [DOI] [PubMed] [Google Scholar]
- 563.Mutel E, Abdul-Wahed A, Ramamonjisoa N, Stefanutti A, Houberdon I, Cavassila S, Pilleul F, Beuf O, Gautier-Stein A, Penhoat A, Mithieux G, Rajas F. Targeted deletion of liver glucose-6 phosphatase mimics glycogen storage disease type 1a including development of multiple adenomas. J Hepatol 54: 529–537, 2011. doi: 10.1016/j.jhep.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 564.Myers MG, Backer JM, Siddle K, White MF. The insulin receptor functions normally in Chinese hamster ovary cells after truncation of the C terminus. J Biol Chem 266: 10616–10623, 1991. [PubMed] [Google Scholar]
- 565.Myers MG, Cowley MA, Münzberg H. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol 70: 537–556, 2008. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- 566.Nagarajan A, Petersen MC, Nasiri AR, Butrico G, Fung A, Ruan H-B, Kursawe R, Caprio S, Thibodeau J, Bourgeois-Daigneault M-C, Sun L, Gao G, Bhanot S, Jurczak MJ, Green MR, Shulman GI, Wajapeyee N. MARCH1 regulates insulin sensitivity by controlling cell surface insulin receptor levels. Nat Commun 7: 12639, 2016. doi: 10.1038/ncomms12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Nagle CA, An J, Shiota M, Torres TP, Cline GW, Liu Z-X, Wang S, Catlin RL, Shulman GI, Newgard CB, Coleman RA. Hepatic overexpression of glycerol-sn-3-phosphate acyltransferase 1 in rats causes insulin resistance. J Biol Chem 282: 14807–14815, 2007. doi: 10.1074/jbc.M611550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Najjar SM. Regulation of insulin action by CEACAM1. Trends Endocrinol Metab 13: 240–245, 2002. doi: 10.1016/S1043-2760(02)00608-2. [DOI] [PubMed] [Google Scholar]
- 569.Nakae J, Kitamura T, Silver DL, Accili D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest 108: 1359–1367, 2001. doi: 10.1172/JCI200112876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Nakano H, Nakajima A, Sakon-Komazawa S, Piao J-H, Xue X, Okumura K. Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell Death Differ 13: 730–737, 2006. doi: 10.1038/sj.cdd.4401830. [DOI] [PubMed] [Google Scholar]
- 571.Nakano T, Seino K, Wakabayashi I, Stafforini DM, Topham MK, Goto K. Deletion of diacylglycerol kinase ε confers susceptibility to obesity via reduced lipolytic activity in murine adipocytes. FASEB J fj201701050R, 2018. doi: 10.1096/fj.201701050R. [DOI] [PubMed] [Google Scholar]
- 572.Nakatani Y, Kaneto H, Kawamori D, Yoshiuchi K, Hatazaki M, Matsuoka TA, Ozawa K, Ogawa S, Hori M, Yamasaki Y, Matsuhisa M. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem 280: 847–851, 2005. doi: 10.1074/jbc.M411860200. [DOI] [PubMed] [Google Scholar]
- 573.Nascimento EBM, Mannerås-Holm L, Chibalin AV, Björnholm M, Zierath JR. Diacylglycerol kinase α deficiency alters inflammation markers in adipose tissue in response to a high-fat diet. J Lipid Res 59: 273–282, 2018. doi: 10.1194/jlr.M079517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Nelson DH, Murray DK. Sphingolipids inhibit insulin and phorbol ester stimulated uptake of 2-deoxyglucose. Biochem Biophys Res Commun 138: 463–467, 1986. doi: 10.1016/0006-291X(86)90303-7. [DOI] [PubMed] [Google Scholar]
- 575.Neschen S, Morino K, Hammond LE, Zhang D, Liu Z-X, Romanelli AJ, Cline GW, Pongratz RL, Zhang X-M, Choi CS, Coleman RA, Shulman GI. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metab 2: 55–65, 2005. doi: 10.1016/j.cmet.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 576.Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab 15: 606–614, 2012. doi: 10.1016/j.cmet.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, Rochon J, Gallup D, Ilkayeva O, Wenner BR, Yancy WS Jr, Eisenson H, Musante G, Surwit RS, Millington DS, Butler MD, Svetkey LP. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 9: 311–326, 2009. doi: 10.1016/j.cmet.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Newgard CB, Brady MJ, O’Doherty RM, Saltiel AR. Organizing glucose disposal: emerging roles of the glycogen targeting subunits of protein phosphatase-1. Diabetes 49: 1967–1977, 2000. doi: 10.2337/diabetes.49.12.1967. [DOI] [PubMed] [Google Scholar]
- 579.Newgard CB, Hwang PK, Fletterick RJ. The family of glycogen phosphorylases: structure and function. Crit Rev Biochem Mol Biol 24: 69–99, 1989. doi: 10.3109/10409238909082552. [DOI] [PubMed] [Google Scholar]
- 580.Newton AC. Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab 298: E395–E402, 2010. doi: 10.1152/ajpendo.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Ng Y, Ramm G, Lopez JA, James DE. Rapid activation of Akt2 is sufficient to stimulate GLUT4 translocation in 3T3-L1 adipocytes. Cell Metab 7: 348–356, 2008. doi: 10.1016/j.cmet.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 582.Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, Yoshimura K, Kadowaki T, Nagai R. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 15: 914–920, 2009. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 583.Noland RC, Koves TR, Seiler SE, Lum H, Lust RM, Ilkayeva O, Stevens RD, Hegardt FG, Muoio DM. Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. J Biol Chem 284: 22840–22852, 2009. doi: 10.1074/jbc.M109.032888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584.Norseen J, Hosooka T, Hammarstedt A, Yore MM, Kant S, Aryal P, Kiernan UA, Phillips DA, Maruyama H, Kraus BJ, Usheva A, Davis RJ, Smith U, Kahn BB. Retinol-binding protein 4 inhibits insulin signaling in adipocytes by inducing proinflammatory cytokines in macrophages through a c-Jun N-terminal kinase- and toll-like receptor 4-dependent and retinol-independent mechanism. Mol Cell Biol 32: 2010–2019, 2012. doi: 10.1128/MCB.06193-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Nowotny B, Zahiragic L, Krog D, Nowotny PJ, Herder C, Carstensen M, Yoshimura T, Szendroedi J, Phielix E, Schadewaldt P, Schloot NC, Shulman GI, Roden M. Mechanisms underlying the onset of oral lipid-induced skeletal muscle insulin resistance in humans. Diabetes 62: 2240–2248, 2013. doi: 10.2337/db12-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586.Nurjhan N, Consoli A, Gerich J. Increased lipolysis and its consequences on gluconeogenesis in non-insulin-dependent diabetes mellitus. J Clin Invest 89: 169–175, 1992. doi: 10.1172/JCI115558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587.Nyman LR, Tian L, Hamm DA, Schoeb TR, Gower BA, Nagy TR, Wood PA. Long term effects of high fat or high carbohydrate diets on glucose tolerance in mice with heterozygous carnitine palmitoyltransferase-1a (CPT-1a) deficiency: diet influences on CPT1a deficient mice. Nutr Diabetes 1: e14, 2011. doi: 10.1038/nutd.2011.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med 8: 1376–1382, 2002. doi: 10.1038/nm1202-798. [DOI] [PubMed] [Google Scholar]
- 589.O’Brien RM, Granner DK. Regulation of gene expression by insulin. Physiol Rev 76: 1109–1161, 1996. doi: 10.1152/physrev.1996.76.4.1109. [DOI] [PubMed] [Google Scholar]
- 590.Okada-Iwabu M, Yamauchi T, Iwabu M, Honma T, Hamagami K, Matsuda K, Yamaguchi M, Tanabe H, Kimura-Someya T, Shirouzu M, Ogata H, Tokuyama K, Ueki K, Nagano T, Tanaka A, Yokoyama S, Kadowaki T. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature 503: 493–499, 2013. doi: 10.1038/nature12656. [DOI] [PubMed] [Google Scholar]
- 591.Okamoto H, Nakae J, Kitamura T, Park B-C, Dragatsis I, Accili D. Transgenic rescue of insulin receptor-deficient mice. J Clin Invest 114: 214–223, 2004. doi: 10.1172/JCI200421645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Okamoto H, Obici S, Accili D, Rossetti L. Restoration of liver insulin signaling in Insr knockout mice fails to normalize hepatic insulin action. J Clin Invest 115: 1314–1322, 2005. doi: 10.1172/JCI200523096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Okumura K, Akiyama N, Hashimoto H, Ogawa K, Satake T. Alteration of 1,2-diacylglycerol content in myocardium from diabetic rats. Diabetes 37: 1168–1172, 1988. doi: 10.2337/diab.37.9.1168. [DOI] [PubMed] [Google Scholar]
- 594.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 72: 219–246, 2010. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 595.Olefsky JM, Kolterman OG, Scarlett JA. Insulin action and resistance in obesity and noninsulin-dependent type II diabetes mellitus. Am J Physiol Endocrinol Metab 243: E15–E30, 1982. [DOI] [PubMed] [Google Scholar]
- 596.Olson DP, Pulinilkunnil T, Cline GW, Shulman GI, Lowell BB. Gene knockout of Acc2 has little effect on body weight, fat mass, or food intake. Proc Natl Acad Sci USA 107: 7598–7603, 2010. doi: 10.1073/pnas.0913492107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Ong KT, Mashek MT, Bu SY, Mashek DG. Hepatic ATGL knockdown uncouples glucose intolerance from liver TAG accumulation. FASEB J 27: 313–321, 2013. doi: 10.1096/fj.12-213454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598.Onuma H, Osawa H, Yamada K, Ogura T, Tanabe F, Granner DK, Makino H. Identification of the insulin-regulated interaction of phosphodiesterase 3B with 14-3-3 β protein. Diabetes 51: 3362–3367, 2002. doi: 10.2337/diabetes.51.12.3362. [DOI] [PubMed] [Google Scholar]
- 599.O’Rahilly S, Barroso I, Wareham NJ. Genetic factors in type 2 diabetes: the end of the beginning? Science 307: 370–373, 2005. doi: 10.1126/science.1104346. [DOI] [PubMed] [Google Scholar]
- 600.Oral EA, Reilly SM, Gomez AV, Meral R, Butz L, Ajluni N, Chenevert TL, Korytnaya E, Neidert AH, Hench R, Rus D, Horowitz JF, Poirier B, Zhao P, Lehmann K, Jain M, Yu R, Liddle C, Ahmadian M, Downes M, Evans RM, Saltiel AR. Inhibition of IKKɛ and TBK1 Improves Glucose Control in a Subset of Patients with Type 2 Diabetes. Cell Metab 26: 157–170.e7, 2017. doi: 10.1016/j.cmet.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.O’Rourke RW, Metcalf MD, White AE, Madala A, Winters BR, Maizlin II, Jobe BA, Roberts CT Jr, Slifka MK, Marks DL. Depot-specific differences in inflammatory mediators and a role for NK cells and IFN-gamma in inflammation in human adipose tissue. Int J Obes 33: 978–990, 2009. doi: 10.1038/ijo.2009.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Osuga J, Ishibashi S, Oka T, Yagyu H, Tozawa R, Fujimoto A, Shionoiri F, Yahagi N, Kraemer FB, Tsutsumi O, Yamada N. Targeted disruption of hormone-sensitive lipase results in male sterility and adipocyte hypertrophy, but not in obesity. Proc Natl Acad Sci USA 97: 787–792, 2000. doi: 10.1073/pnas.97.2.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603.O'Sullivan I, Zhang W, Wasserman DH, Liew CW, Liu J, Paik J, DePinho RA, Stolz DB, Kahn CR, Schwartz MW, Unterman TG. FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat Commun 6: 7079, 2015. doi: 10.1038/ncomms8079. [Erratum in Nat Commun 6: 7861, 2015.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604.Owen JL, Zhang Y, Bae S-H, Farooqi MS, Liang G, Hammer RE, Goldstein JL, Brown MS. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc Natl Acad Sci USA 109: 16184–16189, 2012. doi: 10.1073/pnas.1213343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Özcan U, Cao Q, Yilmaz E, Lee A-H, Iwakoshi NN, Özdelen E, Tuncman G, Görgün C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306: 457–461, 2004. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 606.Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, Ray S, Majumdar SS, Bhattacharya S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med 18: 1279–1285, 2012. doi: 10.1038/nm.2851. [DOI] [PubMed] [Google Scholar]
- 606a.Pal M, Wunderlich CM, Spohn G, Brönneke HS, Schmidt-Supprian M, Wunderlich FT. Alteration of JNK-1 signaling in skeletal muscle fails to affect glucose homeostasis and obesity-associated insulin resistance in mice. PLoS One 8: e54247, 2013. doi: 10.1371/journal.pone.0054247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Palmer CNA, Maglio C, Pirazzi C, Burza MA, Adiels M, Burch L, Donnelly LA, Colhoun H, Doney AS, Dillon JF, Pearson ER, McCarthy M, Hattersley AT, Frayling T, Morris AD, Peltonen M, Svensson P-A, Jacobson P, Borén J, Sjöström L, Carlsson LMS, Romeo S. Paradoxical lower serum triglyceride levels and higher type 2 diabetes mellitus susceptibility in obese individuals with the PNPLA3 148M variant. PLoS One 7: e39362, 2012. doi: 10.1371/journal.pone.0039362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 608.Palmer ND, Stevens RD, Antinozzi PA, Anderson A, Bergman RN, Wagenknecht LE, Newgard CB, Bowden DW. Metabolomic profile associated with insulin resistance and conversion to diabetes in the Insulin Resistance Atherosclerosis Study. J Clin Endocrinol Metab 100: E463–E468, 2015. doi: 10.1210/jc.2014-2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610.Pan DA, Lillioja S, Kriketos AD, Milner MR, Baur LA, Bogardus C, Jenkins AB, Storlien LH. Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes 46: 983–988, 1997. doi: 10.2337/diab.46.6.983. [DOI] [PubMed] [Google Scholar]
- 611.Paolisso G, Tataranni PA, Foley JE, Bogardus C, Howard BV, Ravussin E. A high concentration of fasting plasma non-esterified fatty acids is a risk factor for the development of NIDDM. Diabetologia 38: 1213–1217, 1995. doi: 10.1007/BF00422371. [DOI] [PubMed] [Google Scholar]
- 612.Paranjape SA, Chan O, Zhu W, Horblitt AM, McNay EC, Cresswell JA, Bogan JS, McCrimmon RJ, Sherwin RS. Influence of insulin in the ventromedial hypothalamus on pancreatic glucagon secretion in vivo. Diabetes 59: 1521–1527, 2010. doi: 10.2337/db10-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613.Parker PJ, Caudwell FB, Cohen P. Glycogen synthase from rabbit skeletal muscle; effect of insulin on the state of phosphorylation of the seven phosphoserine residues in vivo. Eur J Biochem 130: 227–234, 1983. doi: 10.1111/j.1432-1033.1983.tb07140.x. [DOI] [PubMed] [Google Scholar]
- 614.Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci USA 100: 8466–8471, 2003. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615.Paul P, Issekutz B Jr, Miller HI. Interrelationship of free fatty acids and glucose metabolism in the dog. Am J Physiol 211: 1313–1320, 1966. [DOI] [PubMed] [Google Scholar]
- 616.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22: 153–183, 2001. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 617.Peraldi P, Hotamisligil GS, Buurman WA, White MF, Spiegelman BM. Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. J Biol Chem 271: 13018–13022, 1996. doi: 10.1074/jbc.271.22.13018. [DOI] [PubMed] [Google Scholar]
- 618.Pereira MJ, Skrtic S, Katsogiannos P, Abrahamsson N, Sidibeh CO, Dahgam S, Månsson M, Risérus U, Kullberg J, Eriksson JW. Impaired adipose tissue lipid storage, but not altered lipolysis, contributes to elevated levels of NEFA in type 2 diabetes. Degree of hyperglycemia and adiposity are important factors. Metabolism 65: 1768–1780, 2016. doi: 10.1016/j.metabol.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 619.Perreault L, Newsom SA, Strauss A, Kerege A, Kahn DE, Harrison KA, Snell-Bergeon JK, Nemkov T, D’Alessandro A, Jackman MR, MacLean PS, Bergman BC. Intracellular localization of diacylglycerols and sphingolipids influences insulin sensitivity and mitochondrial function in human skeletal muscle. JCI Insight 3: e96805, 2018. doi: 10.1172/jci.insight.96805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620.Perry RJ, Camporez JG, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang X-M, Ruan H-B, Yang X, Caprio S, Kaech SM, Sul HS, Birnbaum MJ, Davis RJ, Cline GW, Petersen KF, Shulman GI. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 160: 745–758, 2015. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 621.Perry RJ, Kim T, Zhang X-M, Lee H-Y, Pesta D, Popov VB, Zhang D, Rahimi Y, Jurczak MJ, Cline GW, Spiegel DA, Shulman GI. Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab 18: 740–748, 2013. doi: 10.1016/j.cmet.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622.Perry RJ, Lee S, Ma L, Zhang D, Schlessinger J, Shulman GI. FGF1 and FGF19 reverse diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nat Commun 6: 6980, 2015. doi: 10.1038/ncomms7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623.Perry RJ, Peng L, Abulizi A, Kennedy L, Cline GW, Shulman GI. Mechanism for leptin’s acute insulin-independent effect to reverse diabetic ketoacidosis. J Clin Invest 127: 657–669, 2017. doi: 10.1172/JCI88477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624.Perry RJ, Peng L, Barry NA, Cline GW, Zhang D, Cardone RL, Petersen KF, Kibbey RG, Goodman AL, Shulman GI. Acetate mediates a microbiome-brain-β-cell axis to promote metabolic syndrome. Nature 534: 213–217, 2016. doi: 10.1038/nature18309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625.Perry RJ, Peng L, Cline GW, Wang Y, Rabin-Court A, Song JD, Zhang D, Zhang X-M, Nozaki Y, Dufour S, Petersen KF, Shulman GI. Mechanisms by which a Very-Low-Calorie Diet Reverses Hyperglycemia in a Rat Model of Type 2 Diabetes. Cell Metab 27: 210–217.e3, 2018. doi: 10.1016/j.cmet.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 626.Perry RJ, Petersen KF, Shulman GI. Pleotropic effects of leptin to reverse insulin resistance and diabetic ketoacidosis. Diabetologia 59: 933–937, 2016. doi: 10.1007/s00125-016-3909-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627.Perry RJ, Samuel VT, Petersen KF, Shulman GI. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 510: 84–91, 2014. doi: 10.1038/nature13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628.Perry RJ, Wang Y, Cline GW, Rabin-Court A, Song JD, Dufour S, Zhang XM, Petersen KF, Shulman GI. Leptin Mediates a Glucose-Fatty Acid Cycle to Maintain Glucose Homeostasis in Starvation. Cell 172: 234–248.e17, 2018. doi: 10.1016/j.cell.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629.Perry RJ, Zhang D, Zhang X-M, Boyer JL, Shulman GI. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347: 1253–1256, 2015. doi: 10.1126/science.aaa0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630.Perry RJ, Zhang X-M, Zhang D, Kumashiro N, Camporez J-PG, Cline GW, Rothman DL, Shulman GI. Leptin reverses diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nat Med 20: 759–763, 2014. doi: 10.1038/nm.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Perseghin G, Calori G, Lattuada G, Ragogna F, Dugnani E, Garancini MP, Crosignani P, Villa M, Bosi E, Ruotolo G, Piemonti L. Insulin resistance/hyperinsulinemia and cancer mortality: the Cremona study at the 15th year of follow-up. Acta Diabetol 49: 421–428, 2012. doi: 10.1007/s00592-011-0361-2. [DOI] [PubMed] [Google Scholar]
- 632.Perseghin G, Ghosh S, Gerow K, Shulman GI. Metabolic defects in lean nondiabetic offspring of NIDDM parents: a cross-sectional study. Diabetes 46: 1001–1009, 1997. doi: 10.2337/diab.46.6.1001. [DOI] [PubMed] [Google Scholar]
- 633.Perseghin G, Price TB, Petersen KF, Roden M, Cline GW, Gerow K, Rothman DL, Shulman GI. Increased glucose transport-phosphorylation and muscle glycogen synthesis after exercise training in insulin-resistant subjects. N Engl J Med 335: 1357–1362, 1996. doi: 10.1056/NEJM199610313351804. [DOI] [PubMed] [Google Scholar]
- 634.Péterfy M, Phan J, Xu P, Reue K. Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat Genet 27: 121–124, 2001. doi: 10.1038/83685. [DOI] [PubMed] [Google Scholar]
- 635.Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300: 1140–1142, 2003. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 636.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 350: 664–671, 2004. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637.Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 54: 603–608, 2005. doi: 10.2337/diabetes.54.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 638.Petersen KF, Dufour S, Hariri A, Nelson-Williams C, Foo JN, Zhang X-M, Dziura J, Lifton RP, Shulman GI. Apolipoprotein C3 gene variants in nonalcoholic fatty liver disease. N Engl J Med 362: 1082–1089, 2010. doi: 10.1056/NEJMoa0907295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 639.Petersen KF, Dufour S, Savage DB, Bilz S, Solomon G, Yonemitsu S, Cline GW, Befroy D, Zemany L, Kahn BB, Papademetris X, Rothman DL, Shulman GI. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci USA 104: 12587–12594, 2007. doi: 10.1073/pnas.0705408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640.Petersen KF, Laurent D, Rothman DL, Cline GW, Shulman GI. Mechanism by which glucose and insulin inhibit net hepatic glycogenolysis in humans. J Clin Invest 101: 1203–1209, 1998. doi: 10.1172/JCI579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 641.Petersen KF, Laurent D, Yu C, Cline GW, Shulman GI. Stimulating effects of low-dose fructose on insulin-stimulated hepatic glycogen synthesis in humans. Diabetes 50: 1263–1268, 2001. doi: 10.2337/diabetes.50.6.1263. [DOI] [PubMed] [Google Scholar]
- 642.Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, Cline GW, DePaoli AM, Taylor SI, Gorden P, Shulman GI. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest 109: 1345–1350, 2002. doi: 10.1172/JCI0215001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.Petersen KF, Price T, Cline GW, Rothman DL, Shulman GI. Contribution of net hepatic glycogenolysis to glucose production during the early postprandial period. Am J Physiol Endocrinol Metab 270: E186–E191, 1996. [DOI] [PubMed] [Google Scholar]
- 644.Petersen MC, Jurczak MJ. CrossTalk opposing view: Intramyocellular ceramide accumulation does not modulate insulin resistance. J Physiol 594: 3171–3174, 2016. doi: 10.1113/JP271677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 645.Petersen MC, Madiraju AK, Gassaway BM, Marcel M, Nasiri AR, Butrico G, Marcucci MJ, Zhang D, Abulizi A, Zhang X-M, Philbrick W, Hubbard SR, Jurczak MJ, Samuel VT, Rinehart J, Shulman GI. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J Clin Invest 126: 4361–4371, 2016. doi: 10.1172/JCI86013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 646.Petersen MC, Shulman GI. Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol Sci 38: 649–665, 2017. doi: 10.1016/j.tips.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647.Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol 13: 572–587, 2017. doi: 10.1038/nrendo.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 648.Peterson TA, Stamnes M. ARF1-regulated coatomer directs the steady-state localization of protein kinase C epsilon at the Golgi apparatus. Biochim Biophys Acta 1833: 487–493, 2013. doi: 10.1016/j.bbamcr.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 649.Phan J, Reue K. Lipin, a lipodystrophy and obesity gene. Cell Metab 1: 73–83, 2005. doi: 10.1016/j.cmet.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 650.Pivovarova O, Bernigau W, Bobbert T, Isken F, Möhlig M, Spranger J, Weickert MO, Osterhoff M, Pfeiffer AFH, Rudovich N. Hepatic insulin clearance is closely related to metabolic syndrome components. Diabetes Care 36: 3779–3785, 2013. doi: 10.2337/dc12-1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651.Ploug T, van Deurs B, Ai H, Cushman SW, Ralston E. Analysis of GLUT4 distribution in whole skeletal muscle fibers: identification of distinct storage compartments that are recruited by insulin and muscle contractions. J Cell Biol 142: 1429–1446, 1998. doi: 10.1083/jcb.142.6.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 652.Plum L, Schubert M, Brüning JC. The role of insulin receptor signaling in the brain. Trends Endocrinol Metab 16: 59–65, 2005. doi: 10.1016/j.tem.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 653.Pocai A, Lam TKT, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J, Aguilar-Bryan L, Rossetti L. Hypothalamic K(ATP) channels control hepatic glucose production. Nature 434: 1026–1031, 2005. doi: 10.1038/nature03439. [DOI] [PubMed] [Google Scholar]
- 654.Pochini L, Oppedisano F, Indiveri C. Reconstitution into liposomes and functional characterization of the carnitine transporter from renal cell plasma membrane. Biochim Biophys Acta 1661: 78–86, 2004. doi: 10.1016/j.bbamem.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 655.Polonsky KS, Given BD, Van Cauter E. Twenty-four-hour profiles and pulsatile patterns of insulin secretion in normal and obese subjects. J Clin Invest 81: 442–448, 1988. doi: 10.1172/JCI113339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656.Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, Esterbauer H, Kozlov A, Kahn CR, Kroemer G, Rustin P, Burcelin R, Penninger JM. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 131: 476–491, 2007. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- 657.Postic C, Dentin R, Denechaud P-D, Girard J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu Rev Nutr 27: 179–192, 2007. doi: 10.1146/annurev.nutr.27.061406.093618. [DOI] [PubMed] [Google Scholar]
- 658.Potapova IA, El-Maghrabi MR, Doronin SV, Benjamin WB. Phosphorylation of recombinant human ATP:citrate lyase by cAMP-dependent protein kinase abolishes homotropic allosteric regulation of the enzyme by citrate and increases the enzyme activity. Allosteric activation of ATP:citrate lyase by phosphorylated sugars. Biochemistry 39: 1169–1179, 2000. doi: 10.1021/bi992159y. [DOI] [PubMed] [Google Scholar]
- 659.Powell DJ, Hajduch E, Kular G, Hundal HS. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Mol Cell Biol 23: 7794–7808, 2003. doi: 10.1128/MCB.23.21.7794-7808.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 660.Poy MN, Yang Y, Rezaei K, Fernström MA, Lee AD, Kido Y, Erickson SK, Najjar SM. CEACAM1 regulates insulin clearance in liver. Nat Genet 30: 270–276, 2002. doi: 10.1038/ng840. [DOI] [PubMed] [Google Scholar]
- 661.Prager R, Wallace P, Olefsky JM. Direct and indirect effects of insulin to inhibit hepatic glucose output in obese subjects. Diabetes 36: 607–611, 1987. doi: 10.2337/diab.36.5.607. [DOI] [PubMed] [Google Scholar]
- 662.Previs SF, Withers DJ, Ren J-M, White MF, Shulman GI. Contrasting effects of IRS-1 versus IRS-2 gene disruption on carbohydrate and lipid metabolism in vivo. J Biol Chem 275: 38990–38994, 2000. doi: 10.1074/jbc.M006490200. [DOI] [PubMed] [Google Scholar]
- 663.Puhakainen I, Koivisto VA, Yki-Järvinen H. Lipolysis and gluconeogenesis from glycerol are increased in patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab 75: 789–794, 1992. [DOI] [PubMed] [Google Scholar]
- 664.Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D, Spiegelman BM. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature 423: 550–555, 2003. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 665.Quinn WJ III, Wan M, Shewale SV, Gelfer R, Rader DJ, Birnbaum MJ, Titchenell PM. mTORC1 stimulates phosphatidylcholine synthesis to promote triglyceride secretion. J Clin Invest 127: 4207–4215, 2017. doi: 10.1172/JCI96036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 666.Quon MJ, Butte AJ, Zarnowski MJ, Sesti G, Cushman SW, Taylor SI. Insulin receptor substrate 1 mediates the stimulatory effect of insulin on GLUT4 translocation in transfected rat adipose cells. J Biol Chem 269: 27920–27924, 1994. [PubMed] [Google Scholar]
- 667.Qu X, Seale JP, Donnelly R. Tissue and isoform-selective activation of protein kinase C in insulin-resistant obese Zucker rats - effects of feeding. J Endocrinol 162: 207–214, 1999. doi: 10.1677/joe.0.1620207. [DOI] [PubMed] [Google Scholar]
- 668.Raddatz K, Turner N, Frangioudakis G, Liao BM, Pedersen DJ, Cantley J, Wilks D, Preston E, Hegarty BD, Leitges M, Raftery MJ, Biden TJ, Schmitz-Peiffer C. Time-dependent effects of Prkce deletion on glucose homeostasis and hepatic lipid metabolism on dietary lipid oversupply in mice. Diabetologia 54: 1447–1456, 2011. doi: 10.1007/s00125-011-2073-0. [DOI] [PubMed] [Google Scholar]
- 669.Rahimi Y, Camporez J-PG, Petersen MC, Pesta D, Perry RJ, Jurczak MJ, Cline GW, Shulman GI. Genetic activation of pyruvate dehydrogenase alters oxidative substrate selection to induce skeletal muscle insulin resistance. Proc Natl Acad Sci USA 111: 16508–16513, 2014. doi: 10.1073/pnas.1419104111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 670.Rahn T, Ridderstråle M, Tornqvist H, Manganiello V, Fredrikson G, Belfrage P, Degerman E. Essential role of phosphatidylinositol 3-kinase in insulin-induced activation and phosphorylation of the cGMP-inhibited cAMP phosphodiesterase in rat adipocytes. Studies using the selective inhibitor wortmannin. FEBS Lett 350: 314–318, 1994. doi: 10.1016/0014-5793(94)00797-7. [DOI] [PubMed] [Google Scholar]
- 671.Rahn T, Rönnstrand L, Leroy MJ, Wernstedt C, Tornqvist H, Manganiello VC, Belfrage P, Degerman E. Identification of the site in the cGMP-inhibited phosphodiesterase phosphorylated in adipocytes in response to insulin and isoproterenol. J Biol Chem 271: 11575–11580, 1996. doi: 10.1074/jbc.271.19.11575. [DOI] [PubMed] [Google Scholar]
- 672.Raichur S, Wang ST, Chan PW, Li Y, Ching J, Chaurasia B, Dogra S, Öhman MK, Takeda K, Sugii S, Pewzner-Jung Y, Futerman AH, Summers SA. CerS2 haploinsufficiency inhibits β-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab 20: 687–695, 2014. [Erratum in Cell Metab 20: 919, 2014.] 10.1016/j.cmet.2014.09.015. [DOI] [PubMed] [Google Scholar]
- 673.Ramnanan CJ, Edgerton DS, Cherrington AD. Evidence against a physiologic role for acute changes in CNS insulin action in the rapid regulation of hepatic glucose production. Cell Metab 15: 656–664, 2012. doi: 10.1016/j.cmet.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 674.Ramnanan CJ, Kraft G, Smith MS, Farmer B, Neal D, Williams PE, Lautz M, Farmer T, Donahue EP, Cherrington AD, Edgerton DS. Interaction between the central and peripheral effects of insulin in controlling hepatic glucose metabolism in the conscious dog. Diabetes 62: 74–84, 2013. doi: 10.2337/db12-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 675.Ramnanan CJ, Saraswathi V, Smith MS, Donahue EP, Farmer B, Farmer TD, Neal D, Williams PE, Lautz M, Mari A, Cherrington AD, Edgerton DS. Brain insulin action augments hepatic glycogen synthesis without suppressing glucose production or gluconeogenesis in dogs. J Clin Invest 121: 3713–3723, 2011. doi: 10.1172/JCI45472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676.Randle PJ. Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev 14: 263–283, 1998. doi: 10.1002/(SICI)1099-0895(199812)14:4<263::AID-DMR233>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 677.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1: 785–789, 1963. doi: 10.1016/S0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 678.Randle PJ, Garland PB, Newsholme EA, Hales CN. The glucose fatty acid cycle in obesity and maturity onset diabetes mellitus. Ann N Y Acad Sci 131: 324–333, 1965. doi: 10.1111/j.1749-6632.1965.tb34800.x. [DOI] [PubMed] [Google Scholar]
- 679.Randle PJ, Newsholme EA, Garland PB. Regulation of glucose uptake by muscle. 8. Effects of fatty acids, ketone bodies and pyruvate, and of alloxan-diabetes and starvation, on the uptake and metabolic fate of glucose in rat heart and diaphragm muscles. Biochem J 93: 652–665, 1964. doi: 10.1042/bj0930652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 680.Rando RR, Young N. The stereospecific activation of protein kinase C. Biochem Biophys Res Commun 122: 818–823, 1984. doi: 10.1016/S0006-291X(84)80107-2. [DOI] [PubMed] [Google Scholar]
- 681.Ranjit S, Boutet E, Gandhi P, Prot M, Tamori Y, Chawla A, Greenberg AS, Puri V, Czech MP. Regulation of fat specific protein 27 by isoproterenol and TNF-α to control lipolysis in murine adipocytes. J Lipid Res 52: 221–236, 2011. doi: 10.1194/jlr.M008771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 682.Rath VL, Ammirati M, LeMotte PK, Fennell KF, Mansour MN, Danley DE, Hynes TR, Schulte GK, Wasilko DJ, Pandit J. Activation of human liver glycogen phosphorylase by alteration of the secondary structure and packing of the catalytic core. Mol Cell 6: 139–148, 2000. doi: 10.1016/S1097-2765(05)00006-7. [DOI] [PubMed] [Google Scholar]
- 683.Ravier MA, Rutter GA. Glucose or insulin, but not zinc ions, inhibit glucagon secretion from mouse pancreatic alpha-cells. Diabetes 54: 1789–1797, 2005. doi: 10.2337/diabetes.54.6.1789. [DOI] [PubMed] [Google Scholar]
- 684.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 37: 1595–1607, 1988. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 685.Reaven GM, Hollenbeck C, Jeng CY, Wu MS, Chen YD. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes 37: 1020–1024, 1988. doi: 10.2337/diab.37.8.1020. [DOI] [PubMed] [Google Scholar]
- 686.Rebrin K, Steil GM, Mittelman SD, Bergman RN. Causal linkage between insulin suppression of lipolysis and suppression of liver glucose output in dogs. J Clin Invest 98: 741–749, 1996. doi: 10.1172/JCI118846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 687.Reilly SM, Chiang S-H, Decker SJ, Chang L, Uhm M, Larsen MJ, Rubin JR, Mowers J, White NM, Hochberg I, Downes M, Yu RT, Liddle C, Evans RM, Oh D, Li P, Olefsky JM, Saltiel AR. An inhibitor of the protein kinases TBK1 and IKK-ɛ improves obesity-related metabolic dysfunctions in mice. Nat Med 19: 313–321, 2013. doi: 10.1038/nm.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 688.Remedi MS, Nichols CG. Chronic antidiabetic sulfonylureas in vivo: reversible effects on mouse pancreatic β-cells. PLoS Med 5: e206, 2008. doi: 10.1371/journal.pmed.0050206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 689.Ren JM, Marshall BA, Gulve EA, Gao J, Johnson DW, Holloszy JO, Mueckler M. Evidence from transgenic mice that glucose transport is rate-limiting for glycogen deposition and glycolysis in skeletal muscle. J Biol Chem 268: 16113–16115, 1993. [PubMed] [Google Scholar]
- 690.Riddle MR, Aspiras AC, Gaudenz K, Peuß R, Sung JY, Martineau B, Peavey M, Box AC, Tabin JA, McGaugh S, Borowsky R, Tabin CJ, Rohner N. Insulin resistance in cavefish as an adaptation to a nutrient-limited environment. Nature 555: 647–651, 2018. doi: 10.1038/nature26136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 691.Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J 381: 561–579, 2004. doi: 10.1042/BJ20040752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 692.Riemens SC, Sluiter WJ, Dullaart RPF. Enhanced escape of non-esterified fatty acids from tissue uptake: its role in impaired insulin-induced lowering of total rate of appearance in obesity and Type II diabetes mellitus. Diabetologia 43: 416–426, 2000. doi: 10.1007/s001250051324. [DOI] [PubMed] [Google Scholar]
- 693.Rieusset J, Andreelli F, Auboeuf D, Roques M, Vallier P, Riou JP, Auwerx J, Laville M, Vidal H. Insulin acutely regulates the expression of the peroxisome proliferator-activated receptor-gamma in human adipocytes. Diabetes 48: 699–705, 1999. doi: 10.2337/diabetes.48.4.699. [DOI] [PubMed] [Google Scholar]
- 694.Rizza RA, Mandarino LJ, Gerich JE. Dose-response characteristics for effects of insulin on production and utilization of glucose in man. Am J Physiol Endocrinol Metab 240: E630–E639, 1981. [DOI] [PubMed] [Google Scholar]
- 695.Robertson DG, DiGirolamo M, Merrill AH Jr, Lambeth JD. Insulin-stimulated hexose transport and glucose oxidation in rat adipocytes is inhibited by sphingosine at a step after insulin binding. J Biol Chem 264: 6773–6779, 1989. [PubMed] [Google Scholar]
- 696.Robinson C, Tamborlane WV, Maggs DG, Enoksson S, Sherwin RS, Silver D, Shulman GI, Caprio S. Effect of insulin on glycerol production in obese adolescents. Am J Physiol Endocrinol Metab 274: E737–E743, 1998. [DOI] [PubMed] [Google Scholar]
- 697.Roden M, Perseghin G, Petersen KF, Hwang JH, Cline GW, Gerow K, Rothman DL, Shulman GI. The roles of insulin and glucagon in the regulation of hepatic glycogen synthesis and turnover in humans. J Clin Invest 97: 642–648, 1996. doi: 10.1172/JCI118460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 698.Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW, Shulman GI. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest 97: 2859–2865, 1996. doi: 10.1172/JCI118742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 699.Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev 77: 731–758, 1997. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- 700.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 40: 1461–1465, 2008. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 701.Rondinone CM, Carvalho E, Rahn T, Manganiello VC, Degerman E, Smith UP. Phosphorylation of PDE3B by phosphatidylinositol 3-kinase associated with the insulin receptor. J Biol Chem 275: 10093–10098, 2000. doi: 10.1074/jbc.275.14.10093. [DOI] [PubMed] [Google Scholar]
- 702.Ros S, García-Rocha M, Domínguez J, Ferrer JC, Guinovart JJ. Control of liver glycogen synthase activity and intracellular distribution by phosphorylation. J Biol Chem 284: 6370–6378, 2009. doi: 10.1074/jbc.M808576200. [DOI] [PubMed] [Google Scholar]
- 703.Ros S, Zafra D, Valles-Ortega J, García-Rocha M, Forrow S, Domínguez J, Calbó J, Guinovart JJ. Hepatic overexpression of a constitutively active form of liver glycogen synthase improves glucose homeostasis. J Biol Chem 285: 37170–37177, 2010. doi: 10.1074/jbc.M110.157396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 704.Rothman DL, Magnusson I, Katz LD, Shulman RG, Shulman GI. Quantitation of hepatic glycogenolysis and gluconeogenesis in fasting humans with 13C NMR. Science 254: 573–576, 1991. doi: 10.1126/science.1948033. [DOI] [PubMed] [Google Scholar]
- 705.Ruby MA, Massart J, Hunerdosse DM, Schönke M, Correia JC, Louie SM, Ruas JL, Näslund E, Nomura DK, Zierath JR. Human Carboxylesterase 2 Reverses Obesity-Induced Diacylglycerol Accumulation and Glucose Intolerance. Cell Reports 18: 636–646, 2017. doi: 10.1016/j.celrep.2016.12.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 706.Rusu V, Hoch E, Mercader JM, Tenen DE, Gymrek M, Hartigan CR, DeRan M, von Grotthuss M, Fontanillas P, Spooner A, Guzman G, Deik AA, Pierce KA, Dennis C, Clish CB, Carr SA, Wagner BK, Schenone M, Ng MCY, Chen BH, Centeno-Cruz F, Zerrweck C, Orozco L, Altshuler DM, Schreiber SL, Florez JC, Jacobs SBR, Lander ES, Ng MCY, Shriner D, Chen BH, Li J, Chen W-M, Guo X, Liu J, Bielinski SJ, Yanek LR, Nalls MA, Comeau ME, Rasmussen-Torvik LJ, Jensen RA, Evans DS, Sun YV, An P, Patel SR, Lu Y, Long J, Armstrong LL, Wagenknecht L, Yang L, Snively BM, Palmer ND, Mudgal P, Langefeld CD, Keene KL, Freedman BI, Mychaleckyj JC, Nayak U, Raffel LJ, Goodarzi MO, Chen Y-DI, Taylor HA Jr, Correa A, Sims M, Couper D, Pankow JS, Boerwinkle E, Adeyemo A, Doumatey A, Chen G, Mathias RA, Vaidya D, Singleton AB, Zonderman AB, Igo RP Jr, Sedor JR, Kabagambe EK, Siscovick DS, McKnight B, Rice K, Liu Y, Hsueh W-C, Zhao W, Bielak LF, Kraja A, Province MA, Bottinger EP, Gottesman O, Cai Q, Zheng W, Blot WJ, Lowe WL, Pacheco JA, Crawford DC, Grundberg E, Rich SS, Hayes MG, Shu X-O, Loos RJF, Borecki IB, Peyser PA, Cummings SR, Psaty BM, Fornage M, Iyengar SK, Evans MK, Becker DM, Kao WHL, Wilson JG, Rotter JI, Sale MM, Liu S, Rotimi CN, Bowden DW, Mercader JM, Huerta-Chagoya A, García-Ortiz H, Moreno-Macías H, Manning A, Caulkins L, Burtt NP, Flannick J, Patterson N, Aguilar-Salinas CA, Tusié-Luna T, Altshuler D, Florez JC, Martínez-Hernández A, Centeno-Cruz F, Barajas-Olmos FM, Zerrweck C, Contreras-Cubas C, Mendoza-Caamal E, Revilla-Monsalve C, Islas-Andrade S, Córdova E, Soberón X, Orozco L, González-Villalpando C, González-Villalpando ME, Haiman CA, Wilkens L, Le Marchand L, Monroe K, Kolonel L, Arellano-Campos O, Ordóñez-Sánchez ML, Rodríguez-Torres M, Segura-Kato Y, Rodríguez-Guillén R, Cruz-Bautista I, Muñoz-Hernandez LL, Sáenz T, Gómez D, Alvirde U, Almeda-Valdés P, Cortes ML; MEDIA Consortium; SIGMA T2D Consortium . Type 2 Diabetes Variants Disrupt Function of SLC16A11 through Two Distinct Mechanisms. Cell 170: 199–212.e20, 2017. doi: 10.1016/j.cell.2017.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 707.Ryu D, Oh K-J, Jo H-Y, Hedrick S, Kim Y-N, Hwang Y-J, Park T-S, Han J-S, Choi CS, Montminy M, Koo S-H. TORC2 regulates hepatic insulin signaling via a mammalian phosphatidic acid phosphatase, LIPIN1. Cell Metab 9: 240–251, 2009. doi: 10.1016/j.cmet.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 708.Ryu D, Seo W-Y, Yoon Y-S, Kim Y-N, Kim SS, Kim H-J, Park T-S, Choi CS, Koo S-H. Endoplasmic reticulum stress promotes LIPIN2-dependent hepatic insulin resistance. Diabetes 60: 1072–1081, 2011. doi: 10.2337/db10-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 709.Sabio G, Cavanagh-Kyros J, Ko HJ, Jung DY, Gray S, Jun JY, Barrett T, Mora A, Kim JK, Davis RJ. Prevention of steatosis by hepatic JNK1. Cell Metab 10: 491–498, 2009. doi: 10.1016/j.cmet.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 710.Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, Barrett T, Kim JK, Davis RJ. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 322: 1539–1543, 2008. doi: 10.1126/science.1160794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 711.Sabio G, Davis RJ. cJun NH2-terminal kinase 1 (JNK1): roles in metabolic regulation of insulin resistance. Trends Biochem Sci 35: 490–496, 2010. doi: 10.1016/j.tibs.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 712.Sabio G, Kennedy NJ, Cavanagh-Kyros J, Jung DY, Ko HJ, Ong H, Barrett T, Kim JK, Davis RJ. Role of muscle c-Jun NH2-terminal kinase 1 in obesity-induced insulin resistance. Mol Cell Biol 30: 106–115, 2010. doi: 10.1128/MCB.01162-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 713.Saha PK, Kojima H, Martinez-Botas J, Sunehag AL, Chan L. Metabolic adaptations in the absence of perilipin: increased β-oxidation and decreased hepatic glucose production associated with peripheral insulin resistance but normal glucose tolerance in perilipin-null mice. J Biol Chem 279: 35150–35158, 2004. doi: 10.1074/jbc.M405499200. [DOI] [PubMed] [Google Scholar]
- 714.Sakaguchi M, Fujisaka S, Cai W, Winnay JN, Konishi M, O’Neill BT, Li M, García-Martín R, Takahashi H, Hu J, Kulkarni RN, Kahn CR. Adipocyte Dynamics and Reversible Metabolic Syndrome in Mice with an Inducible Adipocyte-Specific Deletion of the Insulin Receptor. Cell Metab 25: 448–462, 2017. doi: 10.1016/j.cmet.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 715.Salinas M, López-Valdaliso R, Martín D, Alvarez A, Cuadrado A. Inhibition of PKB/Akt1 by C2-ceramide involves activation of ceramide-activated protein phosphatase in PC12 cells. Mol Cell Neurosci 15: 156–169, 2000. doi: 10.1006/mcne.1999.0813. [DOI] [PubMed] [Google Scholar]
- 716.Saltiel AR. New therapeutic approaches for the treatment of obesity. Sci Transl Med 8: 323rv2, 2016. doi: 10.1126/scitranslmed.aad1811. [DOI] [PubMed] [Google Scholar]
- 717.Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest 127: 1–4, 2017. doi: 10.1172/JCI92035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 718.Samuel VT. Fructose induced lipogenesis: from sugar to fat to insulin resistance. Trends Endocrinol Metab 22: 60–65, 2011. doi: 10.1016/j.tem.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 719.Samuel VT, Beddow SA, Iwasaki T, Zhang X-M, Chu X, Still CD, Gerhard GS, Shulman GI. Fasting hyperglycemia is not associated with increased expression of PEPCK or G6Pc in patients with Type 2 Diabetes. Proc Natl Acad Sci USA 106: 12121–12126, 2009. doi: 10.1073/pnas.0812547106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 720.Samuel VT, Choi CS, Phillips TG, Romanelli AJ, Geisler JG, Bhanot S, McKay R, Monia B, Shutter JR, Lindberg RA, Shulman GI, Veniant MM. Targeting foxo1 in mice using antisense oligonucleotide improves hepatic and peripheral insulin action. Diabetes 55: 2042–2050, 2006. doi: 10.2337/db05-0705. [DOI] [PubMed] [Google Scholar]
- 721.Samuel VT, Liu Z-X, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem 279: 32345–32353, 2004. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 722.Samuel VT, Liu Z-X, Wang A, Beddow SA, Geisler JG, Kahn M, Zhang XM, Monia BP, Bhanot S, Shulman GI. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest 117: 739–745, 2007. doi: 10.1172/JCI30400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 723.Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unravelling the mechanism. Lancet 375: 2267–2277, 2010. doi: 10.1016/S0140-6736(10)60408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 724.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell 148: 852–871, 2012. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 725.Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest 126: 12–22, 2016. doi: 10.1172/JCI77812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 726.Sano H, Eguez L, Teruel MN, Fukuda M, Chuang TD, Chavez JA, Lienhard GE, McGraw TE. Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab 5: 293–303, 2007. doi: 10.1016/j.cmet.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 727.Sano H, Kane S, Sano E, Mîinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem 278: 14599–14602, 2003. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- 728.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 120: 1183–1192, 2001. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 729.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307: 1098–1101, 2005. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 730.Sarri E, Sicart A, Lázaro-Diéguez F, Egea G. Phospholipid synthesis participates in the regulation of diacylglycerol required for membrane trafficking at the Golgi complex. J Biol Chem 286: 28632–28643, 2011. doi: 10.1074/jbc.M111.267534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 731.Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci USA 100: 7265–7270, 2003. doi: 10.1073/pnas.1133870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 732.Satoh T. Rho GTPases in insulin-stimulated glucose uptake. Small GTPases 5: e28102, 2014. doi: 10.4161/sgtp.28102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 733.Saxton RA, Knockenhauer KE, Wolfson RL, Chantranupong L, Pacold ME, Wang T, Schwartz TU, Sabatini DM. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science 351: 53–58, 2016. doi: 10.1126/science.aad2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 734.Schaeffler A, Gross P, Buettner R, Bollheimer C, Buechler C, Neumeier M, Kopp A, Schoelmerich J, Falk W. Fatty acid-induced induction of Toll-like receptor-4/nuclear factor-kappaB pathway in adipocytes links nutritional signalling with innate immunity. Immunology 126: 233–245, 2009. doi: 10.1111/j.1365-2567.2008.02892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 735.Scherer PE. The Multifaceted Roles of Adipose Tissue-Therapeutic Targets for Diabetes and Beyond: The 2015 Banting Lecture. Diabetes 65: 1452–1461, 2016. doi: 10.2337/db16-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 736.Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem 270: 26746–26749, 1995. doi: 10.1074/jbc.270.45.26746. [DOI] [PubMed] [Google Scholar]
- 737.Scherer T, O’Hare J, Diggs-Andrews K, Schweiger M, Cheng B, Lindtner C, Zielinski E, Vempati P, Su K, Dighe S, Milsom T, Puchowicz M, Scheja L, Zechner R, Fisher SJ, Previs SF, Buettner C. Brain insulin controls adipose tissue lipolysis and lipogenesis. Cell Metab 13: 183–194, 2011. doi: 10.1016/j.cmet.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 738.Schlein C, Talukdar S, Heine M, Fischer AW, Krott LM, Nilsson SK, Brenner MB, Heeren J, Scheja L. FGF21 Lowers Plasma Triglycerides by Accelerating Lipoprotein Catabolism in White and Brown Adipose Tissues. Cell Metab 23: 441–453, 2016. doi: 10.1016/j.cmet.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 739.Schmitz-Peiffer C, Browne CL, Oakes ND, Watkinson A, Chisholm DJ, Kraegen EW, Biden TJ. Alterations in the expression and cellular localization of protein kinase C isozymes epsilon and theta are associated with insulin resistance in skeletal muscle of the high-fat-fed rat. Diabetes 46: 169–178, 1997. doi: 10.2337/diab.46.2.169. [DOI] [PubMed] [Google Scholar]
- 740.Schmitz-Peiffer C, Craig DL, Biden TJ. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J Biol Chem 274: 24202–24210, 1999. doi: 10.1074/jbc.274.34.24202. [DOI] [PubMed] [Google Scholar]
- 741.Schmitz-Peiffer C, Laybutt DR, Burchfield JG, Gurisik E, Narasimhan S, Mitchell CJ, Pedersen DJ, Braun U, Cooney GJ, Leitges M, Biden TJ. Inhibition of PKCepsilon improves glucose-stimulated insulin secretion and reduces insulin clearance. Cell Metab 6: 320–328, 2007. doi: 10.1016/j.cmet.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 742.Schmitz-Peiffer C, Oakes ND, Browne CL, Kraegen EW, Biden TJ. Reversal of chronic alterations of skeletal muscle protein kinase C from fat-fed rats by BRL-49653. Am J Physiol Endocrinol Metab 273: E915–E921, 1997. [DOI] [PubMed] [Google Scholar]
- 743.Schmitz-Peiffer C, Reeves ML, Denton RM. Characterization of the cyclic nucleotide phosphodiesterase isoenzymes present in rat epididymal fat cells. Cell Signal 4: 37–49, 1992. doi: 10.1016/0898-6568(92)90006-T. [DOI] [PubMed] [Google Scholar]
- 744.Schneiter P, Gillet M, Chioléro R, Wauters JP, Berger M, Tappy L. Postprandial hepatic glycogen synthesis in liver transplant recipients. Transplantation 69: 978–981, 2000. doi: 10.1097/00007890-200003150-00052. [DOI] [PubMed] [Google Scholar]
- 745.Schoeberl B, Eichler-Jonsson C, Gilles ED, Müller G. Computational modeling of the dynamics of the MAP kinase cascade activated by surface and internalized EGF receptors. Nat Biotechnol 20: 370–375, 2002. doi: 10.1038/nbt0402-370. [DOI] [PubMed] [Google Scholar]
- 746.Schooneman MG, Vaz FM, Houten SM, Soeters MR. Acylcarnitines: reflecting or inflicting insulin resistance? Diabetes 62: 1–8, 2013. doi: 10.2337/db12-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 747.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308: 1909–1911, 2005. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 748.Schubert KM, Scheid MP, Duronio V. Ceramide inhibits protein kinase B/Akt by promoting dephosphorylation of serine 473. J Biol Chem 275: 13330–13335, 2000. doi: 10.1074/jbc.275.18.13330. [DOI] [PubMed] [Google Scholar]
- 749.Schwartz MW, Seeley RJ, Tschöp MH, Woods SC, Morton GJ, Myers MG, D’Alessio D. Cooperation between brain and islet in glucose homeostasis and diabetes. Nature 503: 59–66, 2013. doi: 10.1038/nature12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 750.Schwartz SS, Epstein S, Corkey BE, Grant SFA, Gavin Iii JR, Aguilar RB, Herman ME. A Unified Pathophysiological Construct of Diabetes and its Complications. Trends Endocrinol Metab 28: 645–655, 2017. doi: 10.1016/j.tem.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 751.Sears DD, Hsiao G, Hsiao A, Yu JG, Courtney CH, Ofrecio JM, Chapman J, Subramaniam S. Mechanisms of human insulin resistance and thiazolidinedione-mediated insulin sensitization. Proc Natl Acad Sci USA 106: 18745–18750, 2009. doi: 10.1073/pnas.0903032106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 752.Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell 40: 310–322, 2010. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 753.Serra C, Federici M, Buongiorno A, Senni MI, Morelli S, Segratella E, Pascuccio M, Tiveron C, Mattei E, Tatangelo L, Lauro R, Molinaro M, Giaccari A, Bouché M. Transgenic mice with dominant negative PKC-theta in skeletal muscle: a new model of insulin resistance and obesity. J Cell Physiol 196: 89–97, 2003. doi: 10.1002/jcp.10278. [DOI] [PubMed] [Google Scholar]
- 754.Serup AK, Alsted TJ, Jordy AB, Schjerling P, Holm C, Kiens B. Partial Disruption of Lipolysis Increases Postexercise Insulin Sensitivity in Skeletal Muscle Despite Accumulation of DAG. Diabetes 65: 2932–2942, 2016. doi: 10.2337/db16-0655. [DOI] [PubMed] [Google Scholar]
- 755.Sharabi K, Lin H, Tavares CDJ, Dominy JE, Camporez JP, Perry RJ, Schilling R, Rines AK, Lee J, Hickey M, Bennion M, Palmer M, Nag PP, Bittker JA, Perez J, Jedrychowski MP, Ozcan U, Gygi SP, Kamenecka TM, Shulman GI, Schreiber SL, Griffin PR, Puigserver P. Selective Chemical Inhibition of PGC-1α Gluconeogenic Activity Ameliorates Type 2 Diabetes. Cell 169: 148–160.e15, 2017. doi: 10.1016/j.cell.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 756.Sharma A, Varghese RT, Shah M, Man CD, Cobelli C, Rizza RA, Bailey KR, Vella A. Impaired Insulin Action Is Associated With Increased Glucagon Concentrations in Nondiabetic Humans. J Clin Endocrinol Metab 103: 314–319, 2018. doi: 10.1210/jc.2017-01197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 757.Sharma NK, Das SK, Mondal AK, Hackney OG, Chu WS, Kern PA, Rasouli N, Spencer HJ, Yao-Borengasser A, Elbein SC. Endoplasmic reticulum stress markers are associated with obesity in nondiabetic subjects. J Clin Endocrinol Metab 93: 4532–4541, 2008. doi: 10.1210/jc.2008-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 758.She P, Reid TM, Bronson SK, Vary TC, Hajnal A, Lynch CJ, Hutson SM. Disruption of BCATm in mice leads to increased energy expenditure associated with the activation of a futile protein turnover cycle. Cell Metab 6: 181–194, 2007. doi: 10.1016/j.cmet.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 759.She P, Van Horn C, Reid T, Hutson SM, Cooney RN, Lynch CJ. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am J Physiol Endocrinol Metab 293: E1552–E1563, 2007. doi: 10.1152/ajpendo.00134.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 760.Sherwin RS, Kramer KJ, Tobin JD, Insel PA, Liljenquist JE, Berman M, Andres R. A model of the kinetics of insulin in man. J Clin Invest 53: 1481–1492, 1974. doi: 10.1172/JCI107697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 761.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116: 3015–3025, 2006. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 762.Shimobayashi M, Albert V, Woelnerhanssen B, Frei IC, Weissenberger D, Meyer-Gerspach AC, Clement N, Moes S, Colombi M, Meier JA, Swierczynska MM, Jenö P, Beglinger C, Peterli R, Hall MN. Insulin resistance causes inflammation in adipose tissue. J Clin Invest 128: 1538–1550, 2018. doi: 10.1172/JCI96139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 763.Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature 401: 73–76, 1999. doi: 10.1038/43448. [DOI] [PubMed] [Google Scholar]
- 764.Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell 6: 77–86, 2000. doi: 10.1016/S1097-2765(05)00010-9. [DOI] [PubMed] [Google Scholar]
- 765.Shipp JC, Opie LH, Challoner D. Fatty Acid and Glucose Metabolism in the Perfused Heart. Nature 189: 1018–1019, 1961. doi: 10.1038/1891018a0. [DOI] [Google Scholar]
- 766.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest 106: 171–176, 2000. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 767.Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med 371: 1131–1141, 2014. doi: 10.1056/NEJMra1011035. [DOI] [PubMed] [Google Scholar]
- 768.Shulman GI, Rothman DL, Jue T, Stein P, DeFronzo RA, Shulman RG. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N Engl J Med 322: 223–228, 1990. doi: 10.1056/NEJM199001253220403. [DOI] [PubMed] [Google Scholar]
- 769.Shulman RG, Rothman DL. Enzymatic phosphorylation of muscle glycogen synthase: a mechanism for maintenance of metabolic homeostasis. Proc Natl Acad Sci USA 93: 7491–7495, 1996. doi: 10.1073/pnas.93.15.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 770.Siddle K. Signalling by insulin and IGF receptors: supporting acts and new players. J Mol Endocrinol 47: R1–R10, 2011. doi: 10.1530/JME-11-0022. [DOI] [PubMed] [Google Scholar]
- 772.Silverman JF, O’Brien KF, Long S, Leggett N, Khazanie PG, Pories WJ, Norris HT, Caro JF. Liver pathology in morbidly obese patients with and without diabetes. Am J Gastroenterol 85: 1349–1355, 1990. [PubMed] [Google Scholar]
- 773.Sindelar DK, Balcom JH, Chu CA, Neal DW, Cherrington AD. A comparison of the effects of selective increases in peripheral or portal insulin on hepatic glucose production in the conscious dog. Diabetes 45: 1594–1604, 1996. doi: 10.2337/diab.45.11.1594. [DOI] [PubMed] [Google Scholar]
- 774.Sindelar DK, Chu CA, Rohlie M, Neal DW, Swift LL, Cherrington AD. The role of fatty acids in mediating the effects of peripheral insulin on hepatic glucose production in the conscious dog. Diabetes 46: 187–196, 1997. doi: 10.2337/diab.46.2.187. [DOI] [PubMed] [Google Scholar]
- 775.Singh R, Wang Y, Xiang Y, Tanaka KE, Gaarde WA, Czaja MJ. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology 49: 87–96, 2009. doi: 10.1002/hep.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 776.Slot JW, Geuze HJ, Gigengack S, James DE, Lienhard GE. Translocation of the glucose transporter GLUT4 in cardiac myocytes of the rat. Proc Natl Acad Sci USA 88: 7815–7819, 1991. doi: 10.1073/pnas.88.17.7815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 777.Slot JW, Geuze HJ, Gigengack S, Lienhard GE, James DE. Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J Cell Biol 113: 123–135, 1991. doi: 10.1083/jcb.113.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 778.Smagris E, BasuRay S, Li J, Huang Y, Lai KM, Gromada J, Cohen JC, Hobbs HH. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 61: 108–118, 2015. doi: 10.1002/hep.27242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 779.Smal J, De Meyts P. Sphingosine, an inhibitor of protein kinase C, suppresses the insulin-like effects of growth hormone in rat adipocytes. Proc Natl Acad Sci USA 86: 4705–4709, 1989. doi: 10.1073/pnas.86.12.4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 780.Smith BK, Ford RJ, Desjardins EM, Green AE, Hughes MC, Houde VP, Day EA, Marcinko K, Crane JD, Mottillo EP, Perry CGR, Kemp BE, Tarnopolsky MA, Steinberg GR. Salsalate (Salicylate) Uncouples Mitochondria, Improves Glucose Homeostasis, and Reduces Liver Lipids Independent of AMPK-β1. Diabetes 65: 3352–3361, 2016. doi: 10.2337/db16-0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 781.Smith CJ, Vasta V, Degerman E, Belfrage P, Manganiello VC. Hormone-sensitive cyclic GMP-inhibited cyclic AMP phosphodiesterase in rat adipocytes. Regulation of insulin- and cAMP-dependent activation by phosphorylation. J Biol Chem 266: 13385–13390, 1991. [PubMed] [Google Scholar]
- 782.Smith SJ, Cases S, Jensen DR, Chen HC, Sande E, Tow B, Sanan DA, Raber J, Eckel RH, Farese RV Jr. Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking Dgat. Nat Genet 25: 87–90, 2000. doi: 10.1038/75651. [DOI] [PubMed] [Google Scholar]
- 783.Soehnlein O, Zernecke A, Eriksson EE, Rothfuchs AG, Pham CT, Herwald H, Bidzhekov K, Rottenberg ME, Weber C, Lindbom L. Neutrophil secretion products pave the way for inflammatory monocytes. Blood 112: 1461–1471, 2008. doi: 10.1182/blood-2008-02-139634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 784.Soeters MR, Sauerwein HP, Duran M, Wanders RJ, Ackermans MT, Fliers E, Houten SM, Serlie MJ. Muscle acylcarnitines during short-term fasting in lean healthy men. Clin Sci (Lond) 116: 585–592, 2009. doi: 10.1042/CS20080433. [DOI] [PubMed] [Google Scholar]
- 785.Softic S, Boucher J, Solheim MH, Fujisaka S, Haering M-F, Homan EP, Winnay J, Perez-Atayde AR, Kahn CR. Lipodystrophy Due to Adipose Tissue-Specific Insulin Receptor Knockout Results in Progressive NAFLD. Diabetes 65: 2187–2200, 2016. doi: 10.2337/db16-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 786.Soli AH, Kahn CR, Neville DM Jr, Roth J. Insulin receptor deficiency in genetic and acquired obesity. J Clin Invest 56: 769–780, 1975. doi: 10.1172/JCI108155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 787.Soll AH, Kahn CR, Neville DM Jr. Insulin binding to liver plasm membranes in the obese hyperglycemic (ob/ob) mouse. Demonstration of a decreased number of functionally normal receptors. J Biol Chem 250: 4702–4707, 1975. [PubMed] [Google Scholar]
- 788.Soltis AR, Kennedy NJ, Xin X, Zhou F, Ficarro SB, Yap YS, Matthews BJ, Lauffenburger DA, White FM, Marto JA, Davis RJ, Fraenkel E. Hepatic Dysfunction Caused by Consumption of a High-Fat Diet. Cell Reports 21: 3317–3328, 2017. doi: 10.1016/j.celrep.2017.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 789.Søndergaard E, Espinosa De Ycaza AE, Morgan-Bathke M, Jensen MD. How to Measure Adipose Tissue Insulin Sensitivity. J Clin Endocrinol Metab 102: 1193–1199, 2017. doi: 10.1210/jc.2017-00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 790.Sopasakis VR, Liu P, Suzuki R, Kondo T, Winnay J, Tran TT, Asano T, Smyth G, Sajan MP, Farese RV, Kahn CR, Zhao JJ. Specific roles of the p110alpha isoform of phosphatidylinsositol 3-kinase in hepatic insulin signaling and metabolic regulation. Cell Metab 11: 220–230, 2010. doi: 10.1016/j.cmet.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 791.Soufi N, Hall AM, Chen Z, Yoshino J, Collier SL, Mathews JC, Brunt EM, Albert CJ, Graham MJ, Ford DA, Finck BN. Inhibiting monoacylglycerol acyltransferase 1 ameliorates hepatic metabolic abnormalities but not inflammation and injury in mice. J Biol Chem 289: 30177–30188, 2014. doi: 10.1074/jbc.M114.595850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 792.Sreejayan N, Dong F, Kandadi MR, Yang X, Ren J. Chromium alleviates glucose intolerance, insulin resistance, and hepatic ER stress in obese mice. Obesity (Silver Spring) 16: 1331–1337, 2008. doi: 10.1038/oby.2008.217. [DOI] [PubMed] [Google Scholar]
- 793.Srinivas PR, Wagner AS, Reddy LV, Deutsch DD, Leon MA, Goustin AS, Grunberger G. Serum alpha 2-HS-glycoprotein is an inhibitor of the human insulin receptor at the tyrosine kinase level. Mol Endocrinol 7: 1445–1455, 1993. [DOI] [PubMed] [Google Scholar]
- 794.Stahl A, Evans JG, Pattel S, Hirsch D, Lodish HF. Insulin causes fatty acid transport protein translocation and enhanced fatty acid uptake in adipocytes. Dev Cell 2: 477–488, 2002. doi: 10.1016/S1534-5807(02)00143-0. [DOI] [PubMed] [Google Scholar]
- 795.Stanley TL, Zanni MV, Johnsen S, Rasheed S, Makimura H, Lee H, Khor VK, Ahima RS, Grinspoon SK. TNF-alpha antagonism with etanercept decreases glucose and increases the proportion of high molecular weight adiponectin in obese subjects with features of the metabolic syndrome. J Clin Endocrinol Metab 96: E146–E150, 2011. doi: 10.1210/jc.2010-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 796.Stannard SR, Thompson MW, Fairbairn K, Huard B, Sachinwalla T, Thompson CH. Fasting for 72 h increases intramyocellular lipid content in nondiabetic, physically fit men. Am J Physiol Endocrinol Metab 283: E1185–E1191, 2002. doi: 10.1152/ajpendo.00108.2002. [DOI] [PubMed] [Google Scholar]
- 797.Stefan N, Häring H-U. The role of hepatokines in metabolism. Nat Rev Endocrinol 9: 144–152, 2013. doi: 10.1038/nrendo.2012.258. [DOI] [PubMed] [Google Scholar]
- 798.Stefan N, Häring H-U. Circulating fetuin-A and free fatty acids interact to predict insulin resistance in humans. Nat Med 19: 394–395, 2013. doi: 10.1038/nm.3116. [DOI] [PubMed] [Google Scholar]
- 799.Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev 88: 1341–1378, 2008. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 800.Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P, Coadwell J, Hawkins PT. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science 279: 710–714, 1998. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- 801.Stern JH, Rutkowski JM, Scherer PE. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab 23: 770–784, 2016. doi: 10.1016/j.cmet.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 802.Strack V, Hennige AM, Krützfeldt J, Bossenmaier B, Klein HH, Kellerer M, Lammers R, Häring HU. Serine residues 994 and 1023/25 are important for insulin receptor kinase inhibition by protein kinase C isoforms beta2 and theta. Diabetologia 43: 443–449, 2000. doi: 10.1007/s001250051327. [DOI] [PubMed] [Google Scholar]
- 803.Straczkowski M, Kowalska I, Nikolajuk A, Dzienis-Straczkowska S, Kinalska I, Baranowski M, Zendzian-Piotrowska M, Brzezinska Z, Gorski J. Relationship between insulin sensitivity and sphingomyelin signaling pathway in human skeletal muscle. Diabetes 53: 1215–1221, 2004. doi: 10.2337/diabetes.53.5.1215. [DOI] [PubMed] [Google Scholar]
- 804.Strålfors P, Honnor RC. Insulin-induced dephosphorylation of hormone-sensitive lipase. Correlation with lipolysis and cAMP-dependent protein kinase activity. Eur J Biochem 182: 379–385, 1989. doi: 10.1111/j.1432-1033.1989.tb14842.x. [DOI] [PubMed] [Google Scholar]
- 805.Stratford S, DeWald DB, Summers SA. Ceramide dissociates 3′-phosphoinositide production from pleckstrin homology domain translocation. Biochem J 354: 359–368, 2001. doi: 10.1042/bj3540359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 806.Stratford S, Hoehn KL, Liu F, Summers SA. Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J Biol Chem 279: 36608–36615, 2004. doi: 10.1074/jbc.M406499200. [DOI] [PubMed] [Google Scholar]
- 807.Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW II, DeFuria J, Jick Z, Greenberg AS, Obin MS. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 56: 2910–2918, 2007. doi: 10.2337/db07-0767. [DOI] [PubMed] [Google Scholar]
- 808.Stuible M, Tremblay ML. In control at the ER: PTP1B and the down-regulation of RTKs by dephosphorylation and endocytosis. Trends Cell Biol 20: 672–679, 2010. doi: 10.1016/j.tcb.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 809.Summers SA, Garza LA, Zhou H, Birnbaum MJ. Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol Cell Biol 18: 5457–5464, 1998. doi: 10.1128/MCB.18.9.5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 810.Summers SA, Goodpaster BH. CrossTalk proposal: Intramyocellular ceramide accumulation does modulate insulin resistance. J Physiol 594: 3167–3170, 2016. doi: 10.1113/JP271676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 811.Sun Z, Lazar MA. Dissociating fatty liver and diabetes. Trends Endocrinol Metab 24: 4–12, 2013. doi: 10.1016/j.tem.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 812.Sun Z, Miller RA, Patel RT, Chen J, Dhir R, Wang H, Zhang D, Graham MJ, Unterman TG, Shulman GI, Sztalryd C, Bennett MJ, Ahima RS, Birnbaum MJ, Lazar MA. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat Med 18: 934–942, 2012. doi: 10.1038/nm.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 813.Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J 296: 15–19, 1993. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 814.Suzuki K, Kono T. Evidence that insulin causes translocation of glucose transport activity to the plasma membrane from an intracellular storage site. Proc Natl Acad Sci USA 77: 2542–2545, 1980. doi: 10.1073/pnas.77.5.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 815.Suzuki Y, Lanner C, Kim J-H, Vilardo PG, Zhang H, Yang J, Cooper LD, Steele M, Kennedy A, Bock CB, Scrimgeour A, Lawrence JC Jr, DePaoli-Roach AA. Insulin control of glycogen metabolism in knockout mice lacking the muscle-specific protein phosphatase PP1G/RGL. Mol Cell Biol 21: 2683–2694, 2001. doi: 10.1128/MCB.21.8.2683-2694.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 816.Swislocki AL, Chen YD, Golay A, Chang MO, Reaven GM. Insulin suppression of plasma-free fatty acid concentration in normal individuals and patients with type 2 (non-insulin-dependent) diabetes. Diabetologia 30: 622–626, 1987. [DOI] [PubMed] [Google Scholar]
- 817.Sylow L, Jensen TE, Kleinert M, Højlund K, Kiens B, Wojtaszewski J, Prats C, Schjerling P, Richter EA. Rac1 signaling is required for insulin-stimulated glucose uptake and is dysregulated in insulin-resistant murine and human skeletal muscle. Diabetes 62: 1865–1875, 2013. doi: 10.2337/db12-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 818.Sylow L, Kleinert M, Pehmøller C, Prats C, Chiu TT, Klip A, Richter EA, Jensen TE. Akt and Rac1 signaling are jointly required for insulin-stimulated glucose uptake in skeletal muscle and downregulated in insulin resistance. Cell Signal 26: 323–331, 2014. doi: 10.1016/j.cellsig.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 819.Szendroedi J, Yoshimura T, Phielix E, Koliaki C, Marcucci M, Zhang D, Jelenik T, Müller J, Herder C, Nowotny P, Shulman GI, Roden M. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc Natl Acad Sci USA 111: 9597–9602, 2014. doi: 10.1073/pnas.1409229111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 820.Sztalryd C, Xu G, Dorward H, Tansey JT, Contreras JA, Kimmel AR, Londos C. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. J Cell Biol 161: 1093–1103, 2003. [Correction in J Cell Biol 162: 353, 2003.] 10.1083/jcb.200210169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 821.Takayama S, White MF, Kahn CR. Phorbol ester-induced serine phosphorylation of the insulin receptor decreases its tyrosine kinase activity. J Biol Chem 263: 3440–3447, 1988. [PubMed] [Google Scholar]
- 822.Takayama S, White MF, Lauris V, Kahn CR. Phorbol esters modulate insulin receptor phosphorylation and insulin action in cultured hepatoma cells. Proc Natl Acad Sci USA 81: 7797–7801, 1984. doi: 10.1073/pnas.81.24.7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 823.Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, Ofrecio J, Lin M, Brenner MB, Olefsky JM. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med 18: 1407–1412, 2012. doi: 10.1038/nm.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 824.Talukdar S, Zhou Y, Li D, Rossulek M, Dong J, Somayaji V, Weng Y, Clark R, Lanba A, Owen BM, Brenner MB, Trimmer JK, Gropp KE, Chabot JR, Erion DM, Rolph TP, Goodwin B, Calle RA. A Long-Acting FGF21 Molecule, PF-05231023, Decreases Body Weight and Improves Lipid Profile in Non-human Primates and Type 2 Diabetic Subjects. Cell Metab 23: 427–440, 2016. doi: 10.1016/j.cmet.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 825.Tanaka N, Takahashi S, Matsubara T, Jiang C, Sakamoto W, Chanturiya T, Teng R, Gavrilova O, Gonzalez FJ. Adipocyte-specific disruption of fat-specific protein 27 causes hepatosteatosis and insulin resistance in high-fat diet-fed mice. J Biol Chem 290: 3092–3105, 2015. doi: 10.1074/jbc.M114.605980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 826.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7: 85–96, 2006. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 827.Taniguchi CM, Ueki K, Kahn R. Complementary roles of IRS-1 and IRS-2 in the hepatic regulation of metabolism. J Clin Invest 115: 718–727, 2005. doi: 10.1172/JCI23187. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 828.Tansey JT, Sztalryd C, Gruia-Gray J, Roush DL, Zee JV, Gavrilova O, Reitman ML, Deng CX, Li C, Kimmel AR, Londos C. Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc Natl Acad Sci USA 98: 6494–6499, 2001. doi: 10.1073/pnas.101042998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 829.Tao R, Wei D, Gao H, Liu Y, DePinho RA, Dong XC. Hepatic FoxOs regulate lipid metabolism via modulation of expression of the nicotinamide phosphoribosyltransferase gene. J Biol Chem 286: 14681–14690, 2011. doi: 10.1074/jbc.M110.201061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 830.Tawo R, Pokrzywa W, Kevei É, Akyuz ME, Balaji V, Adrian S, Höhfeld J, Hoppe T. The Ubiquitin Ligase CHIP Integrates Proteostasis and Aging by Regulation of Insulin Receptor Turnover. Cell 169: 470–482.e13, 2017. doi: 10.1016/j.cell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 831.Taylor EB, An D, Kramer HF, Yu H, Fujii NL, Roeckl KSC, Bowles N, Hirshman MF, Xie J, Feener EP, Goodyear LJ. Discovery of TBC1D1 as an insulin-, AICAR-, and contraction-stimulated signaling nexus in mouse skeletal muscle. J Biol Chem 283: 9787–9796, 2008. doi: 10.1074/jbc.M708839200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 832.Taylor R, Magnusson I, Rothman DL, Cline GW, Caumo A, Cobelli C, Shulman GI. Direct assessment of liver glycogen storage by 13C nuclear magnetic resonance spectroscopy and regulation of glucose homeostasis after a mixed meal in normal subjects. J Clin Invest 97: 126–132, 1996. doi: 10.1172/JCI118379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 833.Terauchi Y, Tsuji Y, Satoh S, Minoura H, Murakami K, Okuno A, Inukai K, Asano T, Kaburagi Y, Ueki K, Nakajima H, Hanafusa T, Matsuzawa Y, Sekihara H, Yin Y, Barrett JC, Oda H, Ishikawa T, Akanuma Y, Komuro I, Suzuki M, Yamamura K, Kodama T, Suzuki H, Yamamura K, Kodama T, Suzuki H, Koyasu S, Aizawa S, Tobe K, Fukui Y, Yazaki Y, Kadowaki T. Increased insulin sensitivity and hypoglycaemia in mice lacking the p85 α subunit of phosphoinositide 3-kinase. Nat Genet 21: 230–235, 1999. doi: 10.1038/6023. [DOI] [PubMed] [Google Scholar]
- 834.Ter Horst KW, Gilijamse PW, Versteeg RI, Ackermans MT, Nederveen AJ, la Fleur SE, Romijn JA, Nieuwdorp M, Zhang D, Samuel VT, Vatner DF, Petersen KF, Shulman GI, Serlie MJ. Hepatic Diacylglycerol-Associated Protein Kinase Cε Translocation Links Hepatic Steatosis to Hepatic Insulin Resistance in Humans. Cell Reports 19: 1997–2004, 2017. doi: 10.1016/j.celrep.2017.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 835.Teufel H, Menahan LA, Shipp JC, Böning S, Wieland O. Effect of oleic acid on the oxidation and gluconeogenesis from [1-14C]pyruvate in the perfused rat liver. Eur J Biochem 2: 182–186, 1967. doi: 10.1111/j.1432-1033.1967.tb00124.x. [DOI] [PubMed] [Google Scholar]
- 836.Thiébaud D, DeFronzo RA, Jacot E, Golay A, Acheson K, Maeder E, Jéquier E, Felber JP. Effect of long chain triglyceride infusion on glucose metabolism in man. Metabolism 31: 1128–1136, 1982. doi: 10.1016/0026-0495(82)90163-9. [DOI] [PubMed] [Google Scholar]
- 837.Thirone ACP, Huang C, Klip A. Tissue-specific roles of IRS proteins in insulin signaling and glucose transport. Trends Endocrinol Metab 17: 72–78, 2006. doi: 10.1016/j.tem.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 838.Thompson SJ, Sargsyan A, Lee S-A, Yuen JJ, Cai J, Smalling R, Ghyselinck N, Mark M, Blaner WS, Graham TE. Hepatocytes Are the Principal Source of Circulating RBP4 in Mice. Diabetes 66: 58–63, 2017. doi: 10.2337/db16-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 839.Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485: 109–113, 2012. doi: 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 840.Titchenell PM, Chu Q, Monks BR, Birnbaum MJ. Hepatic insulin signalling is dispensable for suppression of glucose output by insulin in vivo. Nat Commun 6: 7078, 2015. doi: 10.1038/ncomms8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 841.Titchenell PM, Lazar MA, Birnbaum MJ. Unraveling the Regulation of Hepatic Metabolism by Insulin. Trends Endocrinol Metab 28: 497–505, 2017. doi: 10.1016/j.tem.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 842.Titchenell PM, Quinn WJ, Lu M, Chu Q, Lu W, Li C, Chen H, Monks BR, Chen J, Rabinowitz JD, Birnbaum MJ. Direct Hepatocyte Insulin Signaling Is Required for Lipogenesis but Is Dispensable for the Suppression of Glucose Production. Cell Metab 23: 1154–1166, 2016. doi: 10.1016/j.cmet.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 843.Tolman KG, Fonseca V, Dalpiaz A, Tan MH. Spectrum of liver disease in type 2 diabetes and management of patients with diabetes and liver disease. Diabetes Care 30: 734–743, 2007. doi: 10.2337/dc06-1539. [DOI] [PubMed] [Google Scholar]
- 844.Tonks KT, Coster AC, Christopher MJ, Chaudhuri R, Xu A, Gagnon-Bartsch J, Chisholm DJ, James DE, Meikle PJ, Greenfield JR, Samocha-Bonet D. Skeletal muscle and plasma lipidomic signatures of insulin resistance and overweight/obesity in humans. Obesity (Silver Spring) 24: 908–916, 2016. doi: 10.1002/oby.21448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 845.Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res 100: 328–341, 2007. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- 846.Tremblay F, Brûlé S, Hee Um S, Li Y, Masuda K, Roden M, Sun XJ, Krebs M, Polakiewicz RD, Thomas G, Marette A. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc Natl Acad Sci USA 104: 14056–14061, 2007. doi: 10.1073/pnas.0706517104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 847.Tremblay F, Lavigne C, Jacques H, Marette A. Defective insulin-induced GLUT4 translocation in skeletal muscle of high fat-fed rats is associated with alterations in both Akt/protein kinase B and atypical protein kinase C (ζ/λ) activities. Diabetes 50: 1901–1910, 2001. doi: 10.2337/diabetes.50.8.1901. [DOI] [PubMed] [Google Scholar]
- 848.Trevino MB, Mazur-Hart D, Machida Y, King T, Nadler J, Galkina EV, Poddar A, Dutta S, Imai Y. Liver Perilipin 5 Expression Worsens Hepatosteatosis But Not Insulin Resistance in High Fat-Fed Mice. Mol Endocrinol 29: 1414–1425, 2015. doi: 10.1210/me.2015-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 849.Tuncman G, Hirosumi J, Solinas G, Chang L, Karin M, Hotamisligil GS. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc Natl Acad Sci USA 103: 10741–10746, 2006. doi: 10.1073/pnas.0603509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 850.Turer AT, Khera A, Ayers CR, Turer CB, Grundy SM, Vega GL, Scherer PE. Adipose tissue mass and location affect circulating adiponectin levels. Diabetologia 54: 2515–2524, 2011. doi: 10.1007/s00125-011-2252-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 851.Turer AT, Scherer PE. Adiponectin: mechanistic insights and clinical implications. Diabetologia 55: 2319–2326, 2012. doi: 10.1007/s00125-012-2598-x. [DOI] [PubMed] [Google Scholar]
- 852.Turinsky J, O’Sullivan DM, Bayly BP. 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J Biol Chem 265: 16880–16885, 1990. [PubMed] [Google Scholar]
- 853.Turner N, Bruce CR, Beale SM, Hoehn KL, So T, Rolph MS, Cooney GJ. Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle: evidence against a role for reduced fatty acid oxidation in lipid-induced insulin resistance in rodents. Diabetes 56: 2085–2092, 2007. doi: 10.2337/db07-0093. [DOI] [PubMed] [Google Scholar]
- 854.Turner N, Kowalski GM, Leslie SJ, Risis S, Yang C, Lee-Young RS, Babb JR, Meikle PJ, Lancaster GI, Henstridge DC, White PJ, Kraegen EW, Marette A, Cooney GJ, Febbraio MA, Bruce CR. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 56: 1638–1648, 2013. doi: 10.1007/s00125-013-2913-1. [DOI] [PubMed] [Google Scholar]
- 855.Turpin SM, Hoy AJ, Brown RD, Rudaz CG, Honeyman J, Matzaris M, Watt MJ. Adipose triacylglycerol lipase is a major regulator of hepatic lipid metabolism but not insulin sensitivity in mice. Diabetologia 54: 146–156, 2011. doi: 10.1007/s00125-010-1895-5. [DOI] [PubMed] [Google Scholar]
- 856.Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM, Mauer J, Xu E, Hammerschmidt P, Brönneke HS, Trifunovic A, LoSasso G, Wunderlich FT, Kornfeld J-W, Blüher M, Krönke M, Brüning JC. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab 20: 678–686, 2014. doi: 10.1016/j.cmet.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 857.Tzivion G, Dobson M, Ramakrishnan G. FoxO transcription factors: regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta 1813: 1938–1945, 2011. doi: 10.1016/j.bbamcr.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 858.Ueda S, Kitazawa S, Ishida K, Nishikawa Y, Matsui M, Matsumoto H, Aoki T, Nozaki S, Takeda T, Tamori Y, Aiba A, Kahn CR, Kataoka T, Satoh T. Crucial role of the small GTPase Rac1 in insulin-stimulated translocation of glucose transporter 4 to the mouse skeletal muscle sarcolemma. FASEB J 24: 2254–2261, 2010. doi: 10.1096/fj.09-137380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 859.Um SH, D’Alessio D, Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab 3: 393–402, 2006. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 860.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431: 200–205, 2004. [Corrigendum in Nature 431: 485, 2004.] 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 861.Unger RH, Aguilar-Parada E, Müller WA, Eisentraut AM. Studies of pancreatic alpha cell function in normal and diabetic subjects. J Clin Invest 49: 837–848, 1970. doi: 10.1172/JCI106297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 862.Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest 122: 4–12, 2012. doi: 10.1172/JCI60016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 863.Unger RH, Orci L. Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci USA 107: 16009–16012, 2010. doi: 10.1073/pnas.1006639107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 864.Utter MF, Keech DB. Pyruvate carboxylase. I. Nature of the reaction. J Biol Chem 238: 2603–2608, 1963. [PubMed] [Google Scholar]
- 865.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 389: 610–614, 1997. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- 866.Valenti L, Rametta R, Dongiovanni P, Maggioni M, Fracanzani AL, Zappa M, Lattuada E, Roviaro G, Fargion S. Increased expression and activity of the transcription factor FOXO1 in nonalcoholic steatohepatitis. Diabetes 57: 1355–1362, 2008. doi: 10.2337/db07-0714. [DOI] [PubMed] [Google Scholar]
- 866a.Van den Broek NMA, Ciapaite J, De Feyter HMML, Houten SM, Wanders RJA, Jeneson JAL, Nicolay K, Prompers JJ. Increased mitochondrial content rescues in vivo muscle oxidative capacity in long-term high-fat-diet-fed rats. FASEB J 24: 1354–1364, 2010. doi: 10.1096/fj.09-143842. [DOI] [PubMed] [Google Scholar]
- 866b.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim D-H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 9: 316–323, 2007. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 866c.Van de Werve G, Zaninetti D, Lang U, Vallotton MB, Jeanrenaud B. Identification of a major defect in insulin-resistant tissues of genetically obese (fa/fa) rats. Impaired protein kinase C. Diabetes 36: 310–314, 1987. doi: 10.2337/diab.36.3.310. [DOI] [PubMed] [Google Scholar]
- 866d.Van Hees AMJ, Jans A, Hul GB, Roche HM, Saris WHM, Blaak EE. Skeletal muscle fatty acid handling in insulin resistant men. Obesity (Silver Spring) 19: 1350–1359, 2011. doi: 10.1038/oby.2011.10. [DOI] [PubMed] [Google Scholar]
- 866e.Van Loon LJC, Goodpaster BH. Increased intramuscular lipid storage in the insulin-resistant and endurance-trained state. Pflugers Arch 451: 606–616, 2006. doi: 10.1007/s00424-005-1509-0. [DOI] [PubMed] [Google Scholar]
- 867.Vatner DF, Goedeke L, Camporez JG, Lyu K, Nasiri AR, Zhang D, Bhanot S, Murray SF, Still CD, Gerhard GS, Shulman GI, Samuel VT. Angptl8 antisense oligonucleotide improves adipose lipid metabolism and prevents diet-induced NAFLD and hepatic insulin resistance in rodents. Diabetologia 61: 1435–1446, 2018. doi: 10.1007/s00125-018-4579-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 868.Vatner DF, Majumdar SK, Kumashiro N, Petersen MC, Rahimi Y, Gattu AK, Bears M, Camporez J-PG, Cline GW, Jurczak MJ, Samuel VT, Shulman GI. Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proc Natl Acad Sci USA 112: 1143–1148, 2015. doi: 10.1073/pnas.1423952112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 869.Vaughan M, Berger JE, Steinberg D. Hormone-sensitive lipase and monoglyceride lipase activities in adipose tissue. J Biol Chem 239: 401–409, 1964. [PubMed] [Google Scholar]
- 870.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther 34: 274–285, 2011. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 871.Vidnes J, Oyasaeter S. Glucagon deficiency causing severe neonatal hypoglycemia in a patient with normal insulin secretion. Pediatr Res 11: 943–949, 1977. doi: 10.1203/00006450-197709000-00001. [DOI] [PubMed] [Google Scholar]
- 872.Villar-Palasi C, Larner J. Insulin-mediated effect on the activity of UDPG-glycogen transglucosylase of muscle. Biochim Biophys Acta 39: 171–173, 1960. doi: 10.1016/0006-3002(60)90142-6. [DOI] [PubMed] [Google Scholar]
- 872a.Von Wilamowitz-Moellendorff A, Hunter RW, García-Rocha M, Kang L, López-Soldado I, Lantier L, Patel K, Peggie MW, Martínez-Pons C, Voss M, Calbó J, Cohen PTW, Wasserman DH, Guinovart JJ, Sakamoto K. Glucose-6-phosphate-mediated activation of liver glycogen synthase plays a key role in hepatic glycogen synthesis. Diabetes 62: 4070–4082, 2013. doi: 10.2337/db13-0880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 873.Wang C, Liu M, Riojas RA, Xin X, Gao Z, Zeng R, Wu J, Dong LQ, Liu F. Protein kinase C theta (PKCtheta)-dependent phosphorylation of PDK1 at Ser504 and Ser532 contributes to palmitate-induced insulin resistance. J Biol Chem 284: 2038–2044, 2009. doi: 10.1074/jbc.M806336200. [DOI] [PubMed] [Google Scholar]
- 874.Wang CN, O’Brien L, Brindley DN. Effects of cell-permeable ceramides and tumor necrosis factor-alpha on insulin signaling and glucose uptake in 3T3-L1 adipocytes. Diabetes 47: 24–31, 1998. doi: 10.2337/diab.47.1.24. [DOI] [PubMed] [Google Scholar]
- 875.Wang C-W, Lin H-Y, Shin S-J, Yu M-L, Lin Z-Y, Dai C-Y, Huang J-F, Chen S-C, Li SS-L, Chuang W-L. The PNPLA3 I148M polymorphism is associated with insulin resistance and nonalcoholic fatty liver disease in a normoglycaemic population. Liver Int 31: 1326–1331, 2011. doi: 10.1111/j.1478-3231.2011.02526.x. [DOI] [PubMed] [Google Scholar]
- 876.Wang H, Knaub LA, Jensen DR, Young Jung D, Hong E-G, Ko H-J, Coates AM, Goldberg IJ, de la Houssaye BA, Janssen RC, McCurdy CE, Rahman SM, Soo Choi C, Shulman GI, Kim JK, Friedman JE, Eckel RH. Skeletal muscle-specific deletion of lipoprotein lipase enhances insulin signaling in skeletal muscle but causes insulin resistance in liver and other tissues. Diabetes 58: 116–124, 2009. [Erratum in Diabetes 59: 756, 2010.] 10.2337/db07-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 877.Wang MY, Chen L, Clark GO, Lee Y, Stevens RD, Ilkayeva OR, Wenner BR, Bain JR, Charron MJ, Newgard CB, Unger RH. Leptin therapy in insulin-deficient type I diabetes. Proc Natl Acad Sci USA 107: 4813–4819, 2010. doi: 10.1073/pnas.0909422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 878.Wang M-Y, Grayburn P, Chen S, Ravazzola M, Orci L, Unger RH. Adipogenic capacity and the susceptibility to type 2 diabetes and metabolic syndrome. Proc Natl Acad Sci USA 105: 6139–6144, 2008. doi: 10.1073/pnas.0801981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 879.Wang PYT, Caspi L, Lam CKL, Chari M, Li X, Light PE, Gutierrez-Juarez R, Ang M, Schwartz GJ, Lam TKT. Upper intestinal lipids trigger a gut-brain-liver axis to regulate glucose production. Nature 452: 1012–1016, 2008. doi: 10.1038/nature06852. [DOI] [PubMed] [Google Scholar]
- 880.Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, Lewis GD, Fox CS, Jacques PF, Fernandez C, O’Donnell CJ, Carr SA, Mootha VK, Florez JC, Souza A, Melander O, Clish CB, Gerszten RE. Metabolite profiles and the risk of developing diabetes. Nat Med 17: 448–453, 2011. doi: 10.1038/nm.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 881.Wang W, Hansen PA, Marshall BA, Holloszy JO, Mueckler M. Insulin unmasks a COOH-terminal Glut4 epitope and increases glucose transport across T-tubules in skeletal muscle. J Cell Biol 135: 415–430, 1996. doi: 10.1083/jcb.135.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 882.Wang Y, Inoue H, Ravnskjaer K, Viste K, Miller N, Liu Y, Hedrick S, Vera L, Montminy M. Targeted disruption of the CREB coactivator Crtc2 increases insulin sensitivity. Proc Natl Acad Sci USA 107: 3087–3092, 2010. doi: 10.1073/pnas.0914897107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 883.Wanless IR, Lentz JS. Fatty liver hepatitis (steatohepatitis) and obesity: an autopsy study with analysis of risk factors. Hepatology 12: 1106–1110, 1990. doi: 10.1002/hep.1840120505. [DOI] [PubMed] [Google Scholar]
- 884.Warram JH, Martin BC, Krolewski AS, Soeldner JS, Kahn CR. Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offspring of diabetic parents. Ann Intern Med 113: 909–915, 1990. doi: 10.7326/0003-4819-113-12-909. [DOI] [PubMed] [Google Scholar]
- 885.Wascher TC, Lindeman JHN, Sourij H, Kooistra T, Pacini G, Roden M. Chronic TNF-α neutralization does not improve insulin resistance or endothelial function in “healthy” men with metabolic syndrome. Mol Med 17: 189–193, 2011. doi: 10.2119/molmed.2010.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 886.Watarai T, Kobayashi M, Takata Y, Sasaoka T, Iwasaki M, Shigeta Y. Alteration of insulin-receptor kinase activity by high-fat feeding. Diabetes 37: 1397–1404, 1988. doi: 10.2337/diab.37.10.1397. [DOI] [PubMed] [Google Scholar]
- 887.Webber J, Taylor J, Greathead H, Dawson J, Buttery PJ, Macdonald IA. Effects of fasting on fatty acid kinetics and on the cardiovascular, thermogenic and metabolic responses to the glucose clamp. Clin Sci (Lond) 87: 697–706, 1994. doi: 10.1042/cs0870697. [DOI] [PubMed] [Google Scholar]
- 888.Weigert C, Kron M, Kalbacher H, Pohl AK, Runge H, Häring H-U, Schleicher E, Lehmann R. Interplay and effects of temporal changes in the phosphorylation state of serine-302, -307, and -318 of insulin receptor substrate-1 on insulin action in skeletal muscle cells. Mol Endocrinol 22: 2729–2740, 2008. doi: 10.1210/me.2008-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 889.Wei L, Hubbard SR, Hendrickson WA, Ellis L. Expression, characterization, and crystallization of the catalytic core of the human insulin receptor protein-tyrosine kinase domain. J Biol Chem 270: 8122–8130, 1995. doi: 10.1074/jbc.270.14.8122. [DOI] [PubMed] [Google Scholar]
- 890.Weir GC, Knowlton SD, Atkins RF, McKennan KX, Martin DB. Glucagon secretion from the perfused pancreas of streptozotocin-treated rats. Diabetes 25: 275–282, 1976. doi: 10.2337/diab.25.4.275. [DOI] [PubMed] [Google Scholar]
- 891.Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, Ferrante AW Jr. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest 116: 115–124, 2006. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 892.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796–1808, 2003. doi: 10.1172/JCI200319246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 893.Wei W, Dutchak PA, Wang X, Ding X, Wang X, Bookout AL, Goetz R, Mohammadi M, Gerard RD, Dechow PC, Mangelsdorf DJ, Kliewer SA, Wan Y. Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator-activated receptor γ. Proc Natl Acad Sci USA 109: 3143–3148, 2012. doi: 10.1073/pnas.1200797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 894.Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT-H, Brickey WJ, Ting JP-Y. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 12: 408–415, 2011. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 895.Wensveen FM, Jelenčić V, Valentić S, Šestan M, Wensveen TT, Theurich S, Glasner A, Mendrila D, Štimac D, Wunderlich FT, Brüning JC, Mandelboim O, Polić B. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat Immunol 16: 376–385, 2015. doi: 10.1038/ni.3120. [DOI] [PubMed] [Google Scholar]
- 896.Wernstedt Asterholm I, Tao C, Morley TS, Wang QA, Delgado-Lopez F, Wang ZV, Scherer PE. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab 20: 103–118, 2014. doi: 10.1016/j.cmet.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 898.West DB, Boozer CN, Moody DL, Atkinson RL. Dietary obesity in nine inbred mouse strains. Am J Physiol Regul Integr Comp Physiol 262: R1025–R1032, 1992. [DOI] [PubMed] [Google Scholar]
- 899.White MF. The IRS-signalling system: a network of docking proteins that mediate insulin action. Mol Cell Biochem 182: 3–11, 1998. doi: 10.1023/A:1006806722619. [DOI] [PubMed] [Google Scholar]
- 900.White MF. Mechanisms of Insulin Action. In: Atlas of Diabetes, edited by Skyler J. New York: Springer, p. 19–38. [7 Nov. 2013], http://link.springer.com/chapter/10.1007/978-1-4614-1028-7_2. [Google Scholar]
- 901.Williams AL, Jacobs SBR, Moreno-Macías H, Huerta-Chagoya A, Churchhouse C, Márquez-Luna C, García-Ortíz H, Gómez-Vázquez MJ, Burtt NP, Aguilar-Salinas CA, González-Villalpando C, Florez JC, Orozco L, Haiman CA, Tusié-Luna T, Altshuler D; SIGMA Type 2 Diabetes Consortium . Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature 506: 97–101, 2014. doi: 10.1038/nature12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 902.Williamson JR. Mechanism for the stimulation in vivo of hepatic gluconeogenesis by glucagon. Biochem J 101: 11C–14C, 1966. doi: 10.1042/bj1010011C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 903.Williamson JR, Browning ET, Olson M. Interrelations between fatty acid oxidation and the control of gluconeogenesis in perfused rat liver. Adv Enzyme Regul 6: 67–100, 1968. doi: 10.1016/0065-2571(68)90008-3. [DOI] [PubMed] [Google Scholar]
- 904.Williamson JR, Krebs HA. Acetoacetate as fuel of respiration in the perfused rat heart. Biochem J 80: 540–547, 1961. doi: 10.1042/bj0800540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 905.Williamson JR, Kreisberg RA, Felts PW. Mechanism for the stimulation of gluconeogenesis by fatty acids in perfused rat liver. Proc Natl Acad Sci USA 56: 247–254, 1966. doi: 10.1073/pnas.56.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 906.Williamson JR, Wright PH, Malaisse WJ, Ashmore J. Control of gluconeogenesis by acetyl CoA in rats treated with glucagon and anti-insulin serum. Biochem Biophys Res Commun 24: 765–770, 1966. doi: 10.1016/0006-291X(66)90391-3. [DOI] [PubMed] [Google Scholar]
- 907.Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren J-M, Previs S, Zhang Y, Bernal D, Pons S, Shulman GI, Bonner-Weir S, White MF. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391: 900–904, 1998. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- 908.Witters LA, Kemp BE. Insulin activation of acetyl-CoA carboxylase accompanied by inhibition of the 5′-AMP-activated protein kinase. J Biol Chem 267: 2864–2867, 1992. [PubMed] [Google Scholar]
- 909.Witters LA, Watts TD, Daniels DL, Evans JL. Insulin stimulates the dephosphorylation and activation of acetyl-CoA carboxylase. Proc Natl Acad Sci USA 85: 5473–5477, 1988. doi: 10.1073/pnas.85.15.5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 910.Wolfe RR, Chinkes DL. Isotope Tracers in Metabolic Research: Principles and Practice of Kinetic Analysis. New York: Wiley, 2005. [Google Scholar]
- 911.Wood SL, Emmison N, Borthwick AC, Yeaman SJ. The protein phosphatases responsible for dephosphorylation of hormone-sensitive lipase in isolated rat adipocytes. Biochem J 295: 531–535, 1993. doi: 10.1042/bj2950531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 912.Woods SC, Lotter EC, McKay LD, Porte D Jr. Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 282: 503–505, 1979. doi: 10.1038/282503a0. [DOI] [PubMed] [Google Scholar]
- 913.Wredenberg A, Freyer C, Sandström ME, Katz A, Wibom R, Westerblad H, Larsson N-G. Respiratory chain dysfunction in skeletal muscle does not cause insulin resistance. Biochem Biophys Res Commun 350: 202–207, 2006. doi: 10.1016/j.bbrc.2006.09.029. [DOI] [PubMed] [Google Scholar]
- 914.Wu C, Khan SA, Peng L-J, Li H, Carmella SG, Lange AJ. Perturbation of glucose flux in the liver by decreasing F26P2 levels causes hepatic insulin resistance and hyperglycemia. Am J Physiol Endocrinol Metab 291: E536–E543, 2006. doi: 10.1152/ajpendo.00126.2006. [DOI] [PubMed] [Google Scholar]
- 915.Wu J, Tseng YD, Xu C-F, Neubert TA, White MF, Hubbard SR. Structural and biochemical characterization of the KRLB region in insulin receptor substrate-2. Nat Struct Mol Biol 15: 251–258, 2008. doi: 10.1038/nsmb.1388. [DOI] [PubMed] [Google Scholar]
- 916.Wu JW, Wang SP, Alvarez F, Casavant S, Gauthier N, Abed L, Soni KG, Yang G, Mitchell GA. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology 54: 122–132, 2011. doi: 10.1002/hep.24338. [DOI] [PubMed] [Google Scholar]
- 917.Wu Q, Brown MR. Signaling and function of insulin-like peptides in insects. Annu Rev Entomol 51: 1–24, 2006. doi: 10.1146/annurev.ento.51.110104.151011. [DOI] [PubMed] [Google Scholar]
- 918.Würtz P, Mäkinen V-P, Soininen P, Kangas AJ, Tukiainen T, Kettunen J, Savolainen MJ, Tammelin T, Viikari JS, Rönnemaa T, Kähönen M, Lehtimäki T, Ripatti S, Raitakari OT, Järvelin M-R, Ala-Korpela M. Metabolic signatures of insulin resistance in 7,098 young adults. Diabetes 61: 1372–1380, 2012. doi: 10.2337/db11-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 919.Würtz P, Soininen P, Kangas AJ, Rönnemaa T, Lehtimäki T, Kähönen M, Viikari JS, Raitakari OT, Ala-Korpela M. Branched-chain and aromatic amino acids are predictors of insulin resistance in young adults. Diabetes Care 36: 648–655, 2013. doi: 10.2337/dc12-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 920.Wu X, Chen K, Williams KJ. The role of pathway-selective insulin resistance and responsiveness in diabetic dyslipoproteinemia. Curr Opin Lipidol 23: 334–344, 2012. doi: 10.1097/MOL.0b013e3283544424. [DOI] [PubMed] [Google Scholar]
- 921.Wu X, Williams KJ. NOX4 pathway as a source of selective insulin resistance and responsiveness. Arterioscler Thromb Vasc Biol 32: 1236–1245, 2012. doi: 10.1161/ATVBAHA.111.244525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 922.Xia JY, Holland WL, Kusminski CM, Sun K, Sharma AX, Pearson MJ, Sifuentes AJ, McDonald JG, Gordillo R, Scherer PE. Targeted Induction of Ceramide Degradation Leads to Improved Systemic Metabolism and Reduced Hepatic Steatosis. Cell Metab 22: 266–278, 2015. doi: 10.1016/j.cmet.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 923.Xia JY, Sun K, Hepler C, Ghaben AL, Gupta RK, An YA, Holland WL, Morley TS, Adams AC, Gordillo R, Kusminski CM, Scherer PE. Acute loss of adipose tissue-derived adiponectin triggers immediate metabolic deterioration in mice. Diabetologia 61: 932–941, 2018. doi: 10.1007/s00125-017-4516-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 924.Xia M, Liu Y, Guo H, Wang D, Wang Y, Ling W. Retinol binding protein 4 stimulates hepatic sterol regulatory element-binding protein 1 and increases lipogenesis through the peroxisome proliferator-activated receptor-γ coactivator 1β-dependent pathway. Hepatology 58: 564–575, 2013. doi: 10.1002/hep.26227. [DOI] [PubMed] [Google Scholar]
- 925.Xiao C, Dash S, Morgantini C, Koulajian K, Lewis GF. Evaluation of the Effect of Enteral Lipid Sensing on Endogenous Glucose Production in Humans. Diabetes 64: 2939–2943, 2015. doi: 10.2337/db15-0148. [DOI] [PubMed] [Google Scholar]
- 926.Xie X, Gong Z, Mansuy-Aubert V, Zhou QL, Tatulian SA, Sehrt D, Gnad F, Brill LM, Motamedchaboki K, Chen Y, Czech MP, Mann M, Krüger M, Jiang ZY. C2 domain-containing phosphoprotein CDP138 regulates GLUT4 insertion into the plasma membrane. Cell Metab 14: 378–389, 2011. doi: 10.1016/j.cmet.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 927.Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, Ganesan H, Nino-Castro A, Mallmann MR, Labzin L, Theis H, Kraut M, Beyer M, Latz E, Freeman TC, Ulas T, Schultze JL. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40: 274–288, 2014. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 928.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112: 1821–1830, 2003. doi: 10.1172/JCI200319451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 929.Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, Ferrante AW Jr. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab 18: 816–830, 2013. doi: 10.1016/j.cmet.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 930.Xu Y, Nan D, Fan J, Bogan JS, Toomre D. Optogenetic activation reveals distinct roles of PIP3 and Akt in adipocyte insulin action. J Cell Sci 129: 2085–2095, 2016. doi: 10.1242/jcs.174805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 931.Yamada E, Okada S, Saito T, Ohshima K, Sato M, Tsuchiya T, Uehara Y, Shimizu H, Mori M. Akt2 phosphorylates Synip to regulate docking and fusion of GLUT4-containing vesicles. J Cell Biol 168: 921–928, 2005. doi: 10.1083/jcb.200408182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 932.Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 8: 1288–1295, 2002. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 933.Yamazaki H, Tsuboya T, Tsuji K, Dohke M, Maguchi H. Independent Association Between Improvement of Nonalcoholic Fatty Liver Disease and Reduced Incidence of Type 2 Diabetes. Diabetes Care 38: 1673–1679, 2015. doi: 10.2337/dc15-0140. [DOI] [PubMed] [Google Scholar]
- 934.Yang G, Badeanlou L, Bielawski J, Roberts AJ, Hannun YA, Samad F. Central role of ceramide biosynthesis in body weight regulation, energy metabolism, and the metabolic syndrome. Am J Physiol Endocrinol Metab 297: E211–E224, 2009. doi: 10.1152/ajpendo.91014.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 935.Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, Kotani K, Quadro L, Kahn BB. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 436: 356–362, 2005. doi: 10.1038/nature03711. [DOI] [PubMed] [Google Scholar]
- 936.Ye R, Scherer PE. Adiponectin, driver or passenger on the road to insulin sensitivity? Mol Metab 2: 133–141, 2013. doi: 10.1016/j.molmet.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 937.Ying W, Wollam J, Ofrecio JM, Bandyopadhyay G, El Ouarrat D, Lee YS, Oh DY, Li P, Osborn O, Olefsky JM. Adipose tissue B2 cells promote insulin resistance through leukotriene LTB4/LTB4R1 signaling. J Clin Invest 127: 1019–1030, 2017. doi: 10.1172/JCI90350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 938.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107: 881–891, 2001. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 939.Yoshizaki T, Imamura T, Babendure JL, Lu J-C, Sonoda N, Olefsky JM. Myosin 5a is an insulin-stimulated Akt2 (protein kinase Bbeta) substrate modulating GLUT4 vesicle translocation. Mol Cell Biol 27: 5172–5183, 2007. doi: 10.1128/MCB.02298-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 940.Young FG. The Croonian Lecture: On Insulin and Its Action. Proc R Soc Lond B Biol Sci 157: 1–26, 1962. doi: 10.1098/rspb.1962.0059. [DOI] [Google Scholar]
- 941.Youngren JF. Regulation of insulin receptor function. Cell Mol Life Sci 64: 873–891, 2007. doi: 10.1007/s00018-007-6359-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 942.Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen EW, Shulman GI. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem 277: 50230–50236, 2002. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- 943.Yu C, Cresswell J, Löffler MG, Bogan JS. The glucose transporter 4-regulating protein TUG is essential for highly insulin-responsive glucose uptake in 3T3-L1 adipocytes. J Biol Chem 282: 7710–7722, 2007. doi: 10.1074/jbc.M610824200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 944.Yu Y, Yoon S-O, Poulogiannis G, Yang Q, Ma XM, Villén J, Kubica N, Hoffman GR, Cantley LC, Gygi SP, Blenis J. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 332: 1322–1326, 2011. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 945.Zechner C, Lai L, Zechner JF, Geng T, Yan Z, Rumsey JW, Collia D, Chen Z, Wozniak DF, Leone TC, Kelly DP. Total skeletal muscle PGC-1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metab 12: 633–642, 2010. doi: 10.1016/j.cmet.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 946.Zechner R. FAT FLUX: enzymes, regulators, and pathophysiology of intracellular lipolysis. EMBO Mol Med 7: 359–362, 2015. doi: 10.15252/emmm.201404846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 947.Zechner R, Langin D. Hormone-sensitive lipase deficiency in humans. Cell Metab 20: 199–201, 2014. doi: 10.1016/j.cmet.2014.07.018. [DOI] [PubMed] [Google Scholar]
- 948.Zhang J, Gao Z, Yin J, Quon MJ, Ye J. S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(alpha) signaling through IKK2. J Biol Chem 283: 35375–35382, 2008. doi: 10.1074/jbc.M806480200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 949.Zhang JN, Hiken J, Davis AE, Lawrence JC Jr. Insulin stimulates dephosphorylation of phosphorylase in rat epitrochlearis muscles. J Biol Chem 264: 17513–17523, 1989. [PubMed] [Google Scholar]
- 950.Zhang J, Ou J, Bashmakov Y, Horton JD, Brown MS, Goldstein JL. Insulin inhibits transcription of IRS-2 gene in rat liver through an insulin response element (IRE) that resembles IREs of other insulin-repressed genes. Proc Natl Acad Sci USA 98: 3756–3761, 2001. doi: 10.1073/pnas.071054598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 951.Zhang T, Wang S, Lin Y, Xu W, Ye D, Xiong Y, Zhao S, Guan K-L. Acetylation negatively regulates glycogen phosphorylase by recruiting protein phosphatase 1. Cell Metab 15: 75–87, 2012. doi: 10.1016/j.cmet.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 952.Zhang W, Patil S, Chauhan B, Guo S, Powell DR, Le J, Klotsas A, Matika R, Xiao X, Franks R, Heidenreich KA, Sajan MP, Farese RV, Stolz DB, Tso P, Koo S-H, Montminy M, Unterman TG. FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem 281: 10105–10117, 2006. doi: 10.1074/jbc.M600272200. [DOI] [PubMed] [Google Scholar]
- 953.Zhang X, Yeung DCY, Karpisek M, Stejskal D, Zhou Z-G, Liu F, Wong RLC, Chow W-S, Tso AWK, Lam KSL, Xu A. Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes 57: 1246–1253, 2008. doi: 10.2337/db07-1476. [DOI] [PubMed] [Google Scholar]
- 954.Zhang Y, Guo K, LeBlanc RE, Loh D, Schwartz GJ, Yu Y-H. Increasing dietary leucine intake reduces diet-induced obesity and improves glucose and cholesterol metabolism in mice via multimechanisms. Diabetes 56: 1647–1654, 2007. doi: 10.2337/db07-0123. [DOI] [PubMed] [Google Scholar]
- 955.Zhao P, Wong KI, Sun X, Reilly SM, Uhm M, Liao Z, Skorobogatko Y, Saltiel AR. TBK1 at the Crossroads of Inflammation and Energy Homeostasis in Adipose Tissue. Cell 172: 731–743.e12, 2018. doi: 10.1016/j.cell.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 956.Zhou H, Summers SA, Birnbaum MJ, Pittman RN. Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis. J Biol Chem 273: 16568–16575, 1998. doi: 10.1074/jbc.273.26.16568. [DOI] [PubMed] [Google Scholar]
- 957.Zhou L, Park S-Y, Xu L, Xia X, Ye J, Su L, Jeong K-H, Hur JH, Oh H, Tamori Y, Zingaretti CM, Cinti S, Argente J, Yu M, Wu L, Ju S, Guan F, Yang H, Choi CS, Savage DB, Li P. Insulin resistance and white adipose tissue inflammation are uncoupled in energetically challenged Fsp27-deficient mice. Nat Commun 6: 5949, 2015. doi: 10.1038/ncomms6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 958.Zhou QG, Zhou M, Hou FF, Peng X. Asymmetrical dimethylarginine triggers lipolysis and inflammatory response via induction of endoplasmic reticulum stress in cultured adipocytes. Am J Physiol Endocrinol Metab 296: E869–E878, 2009. doi: 10.1152/ajpendo.91011.2008. [DOI] [PubMed] [Google Scholar]
- 959.Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306: 1383–1386, 2004. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- 960.Zimmet P, Alberti KGMM, Shaw J. Global and societal implications of the diabetes epidemic. Nature 414: 782–787, 2001. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 961.Zinda MJ, Vlahos CJ, Lai MT. Ceramide induces the dephosphorylation and inhibition of constitutively activated Akt in PTEN negative U87mg cells. Biochem Biophys Res Commun 280: 1107–1115, 2001. doi: 10.1006/bbrc.2000.4248. [DOI] [PubMed] [Google Scholar]