

Abstract
Background
Flowering is a critical reproductive process in higher plants. Timing of optimal flowering depends upon the coordination among seasonal environmental cues. For cool season grasses, such as Dactylis glomerata, vernalization induced by low temperature provides competence to initiate flowering after prolonged cold. We combined analyses of the transcriptome and microRNAs (miRNAs) to generate a comprehensive resource for regulatory miRNAs and their target circuits during vernalization and heading stages.
Results
A total of 3,846 differentially expressed genes (DEGs) and 69 differentially expressed miRNAs were identified across five flowering stages. The expression of miR395, miR530, miR167, miR396, miR528, novel_42, novel_72, novel_107, and novel_123 demonstrated significant variations during vernalization. These miRNA targeted genes were involved in phytohormones, transmembrane transport, and plant morphogenesis in response to vernalization. The expression patterns of DEGs related to plant hormones, stress responses, energy metabolism, and signal transduction changed significantly in the transition from vegetative to reproductive phases.
Conclusions
Five hub genes, c136110_g1 (BRI1), c131375_g1 (BZR1), c133350_g1 (VRN1), c139830_g1 (VIN3), and c125792_g2 (FT), might play central roles in vernalization response. Our comprehensive analyses have provided a useful platform for investigating consecutive transcriptional and post-transcriptional regulation of critical phases in D. glomerata and provided insights into the genetic engineering of flowering-control in cereal crops.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-5104-0) contains supplementary material, which is available to authorized users.
Keywords: Dactylis glomerata, Flowering, miRNA, Orchardgrass, Transcriptome, Vernalization-response
Background
Flowering is a crucial physiological event for plant reproduction. The appropriate flowering time is essential for the avoidance of adverse natural conditions allowing for a successful completion of reproduction. Typically, cold-induced vernalization and long day periods following vernalization are the two major factors driving the transition from vegetative to reproductive development. Vernalization provides competence to flower after prolonged cold exposure [1]. In Arabidopsis thaliana, several genes conduct a regulatory loop for the initiation of flowering. Prior to vernalization, FRIGIDA (FRI) up-regulates the expression of FLOWERING LOCUS C (FLC), which represses the flowering promoter FLOWERING LOCUS T (FT) and lead to flowering delay [2]. When plants are exposed to cold conditions, the expression of FLC is inhibited by the high expression of VERNALIZATION 1 (VRN1), VERNALIZATION 2(VRN2), and VERNALIZATION INSENSITIVE 3(VIN3) [3], thus flowering is stimulated. For winter cereals, vernalization induced by low temperatures is the major driving factor for developmental phase transition and flowering initiation. Lacking a clear genetic background, information from A. thaliana has been used to infer possible genes and gene interactions involved in flowering pathways in cereals. Subsequent studies identified VRN1, VRN2, VIN3, and VRN3 (an FT ortholog) in wheat and barley [4]. The functions of flowering time orthologues have likely been modified due to gene duplication in the complex genome of cereal crops, and the mode of vernalization response may be discrepant [5, 6]. In cereals, vernalization response is confined by the expression level of VRN2 [7]. Before vernalization, highly expressed VRN2 represses the expression of VRN3 and causes delayed flowering. Vernalization accelerates flowering via suppressing expression of VRN2 by the accumulation of VRN1 product, and thus FT expression is stimulated. However, while these studies have provided rich information on the control of flowering time, they have primarily focused on annual crop species. In perennial grasses, the genetic mechanism of flowering is still largely unknown.
It has been demonstrated that miRNAs and their targets play important regulatory roles in plant development such as flowering stage and adaptation [8, 9]. For example, miRNA156 and miRNA172 antagonistically regulate phase transitions by negatively regulating their target SQUAMOSApromoter binding proteinlike (SPLs) and APETALA2-LIKE (AP2), respectively [10]. AP2-like transcription factor levels are reduced when miRNA172 expression is induced by SPLs from the vegetative to the reproductive phase. In another study, miRNA157 was predicted to cleave SPLs to facilitate vegetative-reproductive transition [11]. Past research has suggested that several miRNAs modulate plant development by targeting AUXIN RESPONSE FACTORS (ARFs) such as miRNA160 and miRNA167 [12, 13]. Similarly, blocking miRNA396 dramatically affects inflorescence development by inducing the GRF6 and subsequently activating the downstream biological clades including AUXIN (IAA) biosynthesis and ARFs [14, 15]. In addition, the expression of miRNA159, 164, 319, and 444 were also found to be altered in a manner correlated with plant development and flowering [16–20]. However, previous studies of miRNAs regulating flowering mostly emphasized potential interactions of miRNA-targets. The existing results were insufficient to elaborate the functional shift and dynamic functional variations of miRNAs throughout the reproductive cycle.
Temperate grasses are important source for human food and animal feed [21]. Orchardgrass (Dactylis glomerata) is one of the most economically important forage cereals due to its high biomass, good shade tolerance, and high nutritional value [22]. Vernalization is required for orchardgrass to flower [23]. However, few attempts have been made to review sequential miRNA expression profiles in orchardgrass among critical developmental stages in vernalization. In addition, the whole transcriptome reflecting the vernalization, booting, and heading process have not been previously described. The ever-accelerating improvement of sequencing technology has made it possible systematically conduct analyses of both mRNAs and miRNAs in non-model plants [24]. Thus, deep sequencing of miRNAs and mRNAs was performed to investigate the continuous molecular dynamics and identify potential regulatory miRNA targeted circuit responses to vernalization and flowering. Furthermore, a coexpression network was constructed to detect the key regulators during vernalization. This research will facilitate the understanding of vernalization and flowering mechanisms of D. glomerata and provide valuable information regarding flowering control in perennial grasses. In addition, this research may supply more foundation information for the grass family (Poaceae) including economically important cereals.
Results
Transcriptome and small RNA sequencing in five developmental stages
A total of 905.9 million raw reads were generated from 15 libraries. After trimming, 865.8 million clean reads were retrieved, accounting for 129.87 Gb of sequencing data (Additional file 1: Table S1 and Table S2). More than 193.7 million raw reads were obtained from 15 small RNA libraries, and the raw reads ranged between 10.6 million and 17.8 million per library (Additional file 1: Table S3). After the removal of low-quality sequences, 188.3 million clean reads remained (Additional file 1: Table S4). In each sample, read lengths ranging from 18 to 30 bp were mapped to D. glomerata transcriptome data, and the average percentage of total clean reads mapped was 65.33% (Additional file 1: Table S5). The lengths of D. glomerata miRNAs ranged from 18 to 30 nt, and 24 nt miRNAs were the most frequent size class among the 15 libraries (Additional file 1: Table S6). The unique sequences were mapped to species precursors in miRBase20.0 (http://microrna.sanger.ac.uk/) by BLAST to identify known miRNAs. A total of 109 known miRNAs were identified in the 15 libraries, and 21 nt miRNAs accounted for 75.23% of the known unique miRNAs (Additional file 1: Table S7). In addition, 163 novel miRNAs were detected with lengths ranging from 18 to 25 nt. The most commonly found novel miRNA length was 21 nt (Additional file 1: Table S7). The frequency of the first base of mature miRNAs showed that the known miRNAs of 24 and 25 nt preferentially started with ‘A’ (96% and 54.41%, respectively). Other miRNAs preferentially started with ‘U’ (Additional file 2: Figure S1A). Among novel miRNAs, 24 and 25 bp novel miRNAs preferentially started with ‘A’ (70.98% and 60.56%, respectively), the 29 bp novel miRNAs preferentially started with ‘C’ (55.56%), and other novel miRNAs preferentially started with ‘U’ (Additional file 2: Figure S1B).
Differentially expressed miRNAs and correlation analysis of expression profiles of miRNA-targets
To investigate the expression dynamics of the major miRNAs associated with the transition from vegetative period to reproductive period, differentially expressed miRNAs were detected in the following five comparisons: VE-BV (Vernalization-Before Vernalization), AV-VE (After Vernalization-Vernalization), BH-AV (Before Heading-After Vernalization), and HT-BH (Heading-Before Heading). In total, 53 miRNAs showed differential expression patterns among the five comparisons including 30 known miRNAs and 23 novel miRNAs. Of the 53 miRNAs, 11 were identified in the VE-BV comparison, 27 in AV-VE, nine in BH-AV, and six in HT-BH. Among these 53 differentially expressed miRNAs, 24 miRNAs were up-regulated while 29 miRNAs were down-regulated (Fig. 1a, Additional file 1: Table S8). The hierarchical clustering analysis showed that most miRNAs were preferentially expressed in a certain stage such as miR528, miR408, miR156, miR166, and miR1432 at the BV-stage; miR395, novel_72, novel_42, and novel_107 at VE stage; and novel_105, miR5072, and miR172 at the HT stage. In addition, the expression of several miRNAs significantly varied among different stages; for instance, miR166, novel_173, miR444, miR168, novel_16, miR398, miR397, and miR397 at the BV and VE stages; and miR1601, miR399, and miR394 at the BV and HT stages.
Fig. 1.
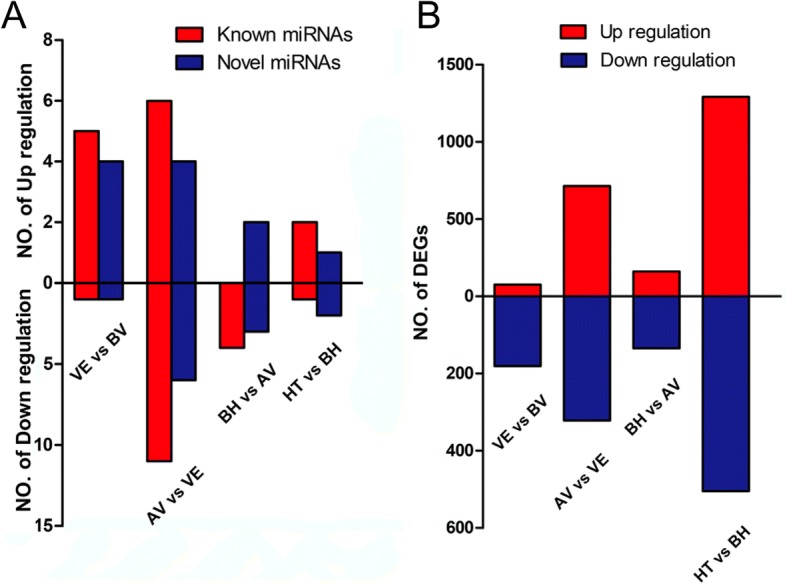
Summary of differentially expressed miRNAs and genes. a Differential expressed miRNAs in five comparisons. b Differential expressed genes in five comparisons. The abscissa represents the different stages, ordinate represents the gene number
An expression analysis of miRNAs in combination with mRNA was performed to investigate the potential miRNA-target pairs with a significant influence on vernalization and floral development. A total of 71 DEGs were targeted by 39 differentially expressed miRNAs with a negative correlation in expression level [25] (Fig. 2a and b, Additional file 1: Table S9), of which several interesting miRNA-target pairs (miR156/172-SPLs/AP2s, miR396-GRFs, miR160/167-ARFs, and miR398-HSFs/HSPs) indicated that a transcriptional repression may be mediated by their corresponding miRNAs (Fig. 2c, d, e, and f).In addition, the qRT-PCR validation indicated the differentially expressed miRNAs and DEGs showed similar expression pattern to the sequencing data (Fig. 3).
Fig. 2.

A combined view of expressions levels between differentially expressed miRNAs and their target genes in Dactylis glomerata L. at five developmental stages. a The differential expression of miRNAs and (b) their targets. c The expression profile of miR156, miR172, SPL family and AP2 family genes. d The expression profile of miR396 and GRF family genes. e The expression profile of miR160, miR167 and ARF family genes. f The expression profile of miR398, HSFs and HSPs family genes. The horizontal axis showed the different developmental stages and the vertical axis showed the miRNAs and genes. The separate heatmap labeled by “Tem” indicated the change of temperature
Fig. 3.

Verification by qRT-PCR of DEGs and differential expressed miRNAs. GAPDH and U6 were used as reference genes. The expression levels of each mRNA and miRNA were normalized by comparison with their expression with BV stage. The abscissa represents the different stages, ordinate represents the relative expression. The bar with oblique stripes represents the relative expression base on qRT-PCR results and the bar with faillette base on sequencing results. Figure a-h indicated the expression of eight DEGs and fig. i-p indicated 8 differential expressed miRNAs, the bottom title represents the gene and miRNA name, respectively
Function annotation and enrichment analysis of targets for miRNAs
The targets of specific miRNAs at the BV, VE, and AV stages were predicted to capture the functional properties related to vernalization, and 345 transcripts were anticipated to be the targets of 27 miRNAs (Additional file 3). The functional annotations indicated that several transcription factors were involved in growth regulation, such as TCP2, PCF6, GAMYB, SPL16/17, MADS47/57, AIL1, and LHY. Targets related to auxin efflux carrier components and calcium-transporters were also identified, which influence plant growth and flowering time by modulating the directional distribution of the phytohormone auxin and Ca2+ storage capacity within chloroplasts [26, 27]. The gene ontology (GO) analysis indicated that targets were significantly enriched (P < 0.05) in 122 GO terms including 68 biological process categories, 24 cellular component categories, and 30 molecular function categories (Additional file 3). In biological processes, the most frequent category was “transmembrane transport.” The results indicated that there were a series of transporters, sugar phosphate/phosphate translocator, metal-nicotianamine transporter, high-affinity nitrate transporter, and high-affinity iron ion transmembrane transport, which play essential roles in allowing plant response to intricate environmental signals [27, 28]. In addition, GO terms related to plant morphogenesis and functional differentiation, photosynthesis, immune system processes, and carbohydrate metabolism were also involved. This suggested that the miRNA targets related to transmembrane transport, carbohydrate metabolism, photosynthesis, immune system processes, and plant morphogenesis respond to vernalization and floral development.
DEGs of transcriptome sequencing
A total of 3,384 DEGs were detected including 256 DEGs in VE-BV, 1,036 DEGs in AV-VE, 295 DEGs in BH-AV, and 1,796 in HT-BH (Fig. 1b and Additional file 1: Table S10). Interestingly, 29 DEGs (including two ice recrystallization inhibition protein 4, an FT-like protein, and an ethylene-responsive element binding protein 2) were found up-regulated during vernalization and repressed afterwards (DEGs detected in VE-BV-UP and AV-VE-DOWN group). Seventy-nine DEGs (including a negative regulator of gibberellin signal DELLA protein GAI, three bHLH transcription factors, three heat stress transcription factors, and two heat shock proteins) were detected in both VE-BV-DOWN and AV-VE-UP (Fig. 4, Additional file 3).
Fig. 4.
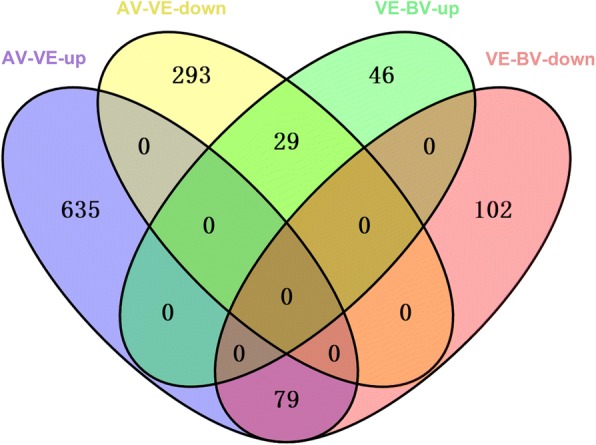
Relationships of changes in gene expression based on transcriptome sequencing. AV-VE-up, up-regulated genes at after vernalization stage compared with vernalization stage. AV-VE-down, down-regulated genes at after vernalization stage compared with vernalization stage. VE-BV-up, up-regulated genes at vernalization stage compared with before vernalization stage. VE-BV-down, down-regulated genes at vernalization stage compared with before vernalization stage
Low temperature is the primary external factor of vernalization induction in temperate grasses. The expression of genes in response to external stimulation was up-regulated in VE-BV (such as ice recrystallization inhibition protein 4, light-inducible protein CPRF2-like, those related to plant hormones, such as cytokinin oxidase/dehydrogenase 2, ethylene-responsive transcription factor 73 (ERF073), ERF3, ethylene-responsive element binding protein 2 and AUXIN-related genes, and two flowering-related genes FT-like and flowering-promoting factor 1-like). The expression of 181 DEGs significantly declined (P < 0.01) from the BV stage to the VE stage including 25 DEGs for heat response such as HSP20 family proteins and heat shock transcription factors (Additional file 3). In the VE stage, the expression of two HSP20 family proteins, c125612_g1 and c112012_g1, decreased significantly (P < 0.01) with log2FC values relative to the BV stage of − 5.9908 and − 5.5984, respectively (Additional file 3). In VE-BV, the expression of seven plant hormone-related DEGs changed significantly (P < 0.01). The functions of two gibberellin-related genes with altered expression included gibberellin 2-beta-dioxygenase 8 and DELLA protein GAI. Ethylene-related genes with altered expression included ERF024, ERF025, ERF027, and ERF1 (Additional file 3). A cytokinin-related gene, cytokinin riboside 5′-monophosphate phosphoribohydrolase, also decreased. In addition, several TFs were detected including WRKY18, bHLH92, bHLH35, GATA transcription factor 5, and NAC domain-containing protein 74 (NAC74). Remarkably, the expression of a flowering-delayed gene, CONSTANS-LIKE 9 (c144305_g1), decreased significantly (P < 0.01) (Additional file 3).
The temperature increased from stage VE to stage AV after a long period of chilling, causing a rapid acceleration of the growth of orchardgrass (Fig. 5). Our results showed 10 cellulose synthesis-related DEGs were up-regulated in the AV stage compared to the VE stage, most of which were annotated as cellulose synthase A. Meanwhile, the expression of seven DEGs with annotation of germin-like protein significantly (P < 0.01) increased in AV-VE (Additional file 3). It was also noted that the expression of NAC members elevated from the VE to AV stage (Additional file 3). Furthermore, nine AUXIN-related genes were detected among the up-regulated DEGs including IAA-amino acid hydrolase ILR1-like 2, auxin-responsive protein IAA31, indole-3-acetic acid-induced protein ARG7, indole-3-pyruvate monooxygenase YUCCA11, and indole-3-glycerol phosphate lyase. In contrast with AUXIN-related genes, the gibberellin-related genes gibberellin 3-beta-dioxygenase 2 and gibberellin 3-beta-dioxygenase 8 showed significant suppression in the AV stage (Additional file 3). Analogously, the expression of flowering-related genes CONSTANS-LIKE 10, HEADING DATE 3A/3B, and flowering-promoting factor 1-like protein 5 were also suppressed in the AV stage. The above results indicated that genes involved in stimuli-response; the plant hormones gibberellin, ethylene, and cytokinin; and flowering-related genes FT-like and FPF1 were up-regulated in VE-BV, and genes-related to plant growth and AUXIN significantly varied between the AV-VE stages.
Fig. 5.

The floral primordium and young inflorescence of Dactylis glomerata after vernalization stimulation. a and (b) The floral primordium. c The young inflorescence. Arrows in (a) and (b) point to the floral primordium; arrows in (c) point to the young inflorescence
Gene coexpression network analysis
After excluding DEGs with low expression, 3,301 DEGs were used to construct a coexpression network to determine the genes with common expression trends across different samples in this study. The resulting network was composed of 12 distinct modules (labeled by different colors) (Additional file 4: Figure S2A). The size of the constructed genes for the 12 modules ranged from 1,506 (turquoise module) to 9 genes (grey module). The module-trait relationships analysis indicated that the pink module, black module, and brown module were highly correlated with the VE stage (Additional file 4: Figure S2B). To investigate the gene connection during vernalization, a subnetwork was identified with five hub genes in the brown module (Fig. 6, Additional file 3), in which two genes were related to brassinosteroid (BR) signal (BZR1and BRI1) and three genes were flowering regulators (VRN1, VIN3-like, and FT-like) relevant to the integration of veranlization and flowering pathways. The hub gene with the highest edge number (104 edges) was VRN1, a crucial regulator in vernalization response. In this subnetwork, 114 genes were involved in categories of transport process, transcription factors, stress response, protein modification, plant hormones, oxidation-reduction process, metabolic process, and carbohydrate metabolic process. Of the 114 genes, 14 genes were in the transport process category. Several potassium channel-related genes KOR1 and AKT1 were detected, which may function in perceiving and transferring environmental signals. In the stress response category, several genes related to environmental stimuli clustered together such as ice recrystallization inhibition protein 4, dehydrin, and heat stress transcription factor. In addition, genes in the plant hormone category, including 16 genes related to ABA, IAA, GA, ethylene, cytokinin, auxin, and brassinolide, may indicate the critical roles of plant hormones during vernalization. Furthermore, two floral homeotic proteins, APETALA 2, and a MADS-box protein SOC1-like, were also identified in this subnetwork.
Fig. 6.

The coexpression subnetwork of BRI1, BZR1, VRN1, VIN3 and FT. The rectangular frame indicated the different functional categories. The dotted arrow showed the potential miRNA-target regulation
Discussion
Differentially expressed miRNAs in developmental stages of D. glomerata
Vernalization is a process of phase transitions from vegetative growth to reproductive growth under ambient stimulation, specifically by low temperatures [29] and is programed and controlled by a large number of dynamically expressed genes including miRNA. Three up-regulated miRNAs (miR395, miR530, and miR167) and two down-regulated miRNAs (miR396 and miR528) in the VE-BV stage were most likely the main players in controlling vernalization. The expression of miR395 was negatively correlated with temperature and altered significantly in response to chilling stress. A previous study has demonstrated that miR395 was involved in BR signal response [30]. One target of miR395, ABA-receptor (ABAR), is a crucial element in the ABA signaling pathway [31]. Our results showed that the expression of miR395 and two BR signaling components, BRI1 and BZR1, were up-regulated while ABAR was down-regulated from the BV to VE stage. This supports that miR395 is probably involved in vernalization through regulating BR and ABA signaling. Compared with miR395, the expression of miR530 was up-regulated in both the VE-BV and HT-BH comparisons. Nine miR530 targets were identified, including an indole biosynthetic process related gene (Enolase 1, ENO1) (Additional file 3). Loss of function of ENO1 exhibited retarded vegetative growth, disturbed flower development, and seed abortion [32]. Thus, flowering time may be influenced by the inhibition of ENO1 through highly expressed miR530 in the VE stage.
MiR160 and miR167 are two critical miRNAs via crosstalk with the hormone auxin in plants [33]. MiR167 is thought be involved in floral development and the transition of vegetative and generative development [34], and miR160 plays a role in maintaining the process of normal developmental programs [35, 36]. MiR160 and miR167 converge on ARFs, which are involved in auxin and defense responses and are involved in many developmental stages. Specifically, miR167 mediates floral development by targeting ARF6 and ARF8 [37, 38], while miR160 negatively regulates ARF10, ARF16, ARF17, and ARF18 in Arabidopsis [12, 39–41]. During the transition from the BV stage to the VE stage, miR160 and miR167 were oppositely expressed, and both of these miRNAs has varied expression in the VE stage. Even more noteworthy, the expression level of miR160 was positively correlated with temperature change, while miR167 was inversely correlated with temperature change. Out of 24 ARFs identified in our transcriptome data, the pattern of expression level changes of ARF11, ARF25, ARF1, ARF12, ARF5, ARF7, and ARF17 were similar to the expression level changes of ARF6, indicating their possible functions in controlling flowering time. Therefore, we assumed that temperature regulated ARFs through miR160 and miR167, thereby affecting flowering time.
MiR396 promotes cell proliferation by targeting the GROWTH-REGULATING FACTOR (GRFs) in various plant organs [42, 43]. Meanwhile, miR396 was a positive regulator in response to drought stress in tobacco [44]. Recent research revealed that when GRF4 relieved suppression by osamiR396, the expression of GRF4 accumulates to a proper extent and increases grain size by activating specific BR responses [45]. The suppression of GRF by miR396 was no longer present after vernalization, which probably accelerates cell division and differentiation with the increase in temperature during the transition from the VE stage to the AV stage. A study conducted by Yuan et al., [46] suggested that the potential crosstalk of miR528, miR172, miR156, and miR396 could contribute to orchestrating response to stress, plant phase transition, and flowering. It is possible that both the up-regulation of miR395, miR530, and miR167 and the down-regulation of miR396 and miR528 are mediated by temperature and associated with vernalization, determining the transition under adverse conditions in D. glomerata.
MiR156 and miR172 are the two key regulators inducing juvenile-to-adult transition that have antagonistic regulation [47]. MiR156 is required for the expression of juvenile leaf characteristics and repressing the expression of SPLs to prevent the change from the vegetative phase [10]. The MiR156-SPL3 module regulates FT to control ambient temperature-responsive flowering [48]. Recently, Hyun et al., [49] has demonstrated that SPL15 cooperated with SOC1 to coordinate floral initiation by activating the transcription of target genes. In turn, miR172 is activated by SPLs to promote the adult phase via the repression of APETALA2-like genes such as AP2, TOE1, TOE2, SNZ, and SMZ [50, 51]. In D. glomerata, the expression of miR156 was higher than that of miR172 at the BV and VE stages, while it was suppressed by miR172 at the AV, BH, and HT stages. The expression of miR156 gradually decreased during the transition from the BV stage to the HT stage, which is inversely correlated with the expression of miR172. In our data, five AP2-like unigenes were identified as floral homeotic protein APETALA2 with a high expression level at the VE stage. The expression levels of nine SPL transcription factors (SPL2, SPL3, SPL7, SPL8, SPL9, SPL13, SPL15, SPL16, and SPL17) showed inverse correlation with the expression level of miR156, except for SPL7 and SPL9. MiR166 and miR168 are also related to growth and developmental phase shift as identified in this study and reported previously [52, 53]. These observations indicated that four miRNAs, miR156, miR172, miR160, and miR168, may have a critical function in the transition from vegetative to reproductive phase after vernalization.
Three differentially expressed miRNAs, miR162, miR397, and miR398, related to stress response were observed in the comparison of the AV-VE stages. MiR162 is known to be involved in drought and salinity response in Arabidopsis [54]. Meanwhile, miR397 and miR398 were involved in the induction of freezing tolerance. Over-expression of miR397 improved plant tolerance to chilling stress by affecting the expression of CBF genes and COR genes [55]. In rice, the overexpression of osamiR397 increases grain size by down-regulating LAC, which negatively regulates the sensitivity to BR signaling [56]. The expression of miR398 was regulated by the inducer of CBF expression (ICE) family genes, and its target genes copper/zinc superoxide dismutase 1 (CSD1) and CSD2 inducible expression under acclimation at 16 °C [57]. Subsequent studies have shown that miR398 was heat-inhibitive and negatively regulated the expression of HSFs and HSPs [58]. In some plants, both chilling and heating was necessary for flowering [59]. Recent research has demonstrated that HSP90 plays an extensive and essential role during the transition from the vegetative phase to the reproductive phase and flower development [60]. In our data, 54 HSFs and HSPs were detected in HT-BH. The expression profile of these DEGs was consistent with temperature change and the highest expression levels were found during the heading stage. This result indicated that the HSF and HSP gene families may participate in flowering in D. golmerata.
Gene coexpression during vernalization
Five hub genes, BRI1, BZR1, VRN1, VIN3, and FT, might play significant roles in regulation of vernalization response. Genes related to transporting, stress response, and plant hormones were detected in a subnetwork, which may have direct or indirect connections. Previous research has indicated that flowering time can be affected by BR signaling [61]. BRI1 and BZR1 are two vital nodes in the BR signaling pathway. BRI1 mediated BR signaling leads to dephosphorylation and accumulation of BZR1 in the nucleus [62]. In Arabidopsis, BRI1 decreased the FLC expression by suppressing FRI to elevate the levels of the FT transcripts, thus accelerating floral transition [63]. Similarly, our investigation indicated that the expression of BRI1 and FT were up-regulated while FRI was inhibited in vernalization response (Fig. 7). BRI1 and BZR1 were potentially relevant to vernalization in D. golmerata. BR-regulated processes largely interact with other plant hormone signaling [64]. For instance, ABSCISIC ACID-INSENSITIVE 5 (ABI5) is dual modulated by BIN2 and BZR1 in BR signaling [65]. BIN2 phosphorylates and stabilizes ABI5 while BZR1 suppresses the expression of ABI5, which makes plants less sensitive to ABA [66]. Wang et al. reported that the direct binding of ABI5 to the promoter of FLC is a critical step for promoting FLC transcription, which inhibits FT expression and causes the postponement of flowering time [67]. ABI5, as the mutual downstream target, suggested a possible cross-regulatory relation between BR and ABA signaling in regulating flowering [68]. Gibberellins (GAs) play an important role in regulating diverse developmental processes in plants such as vegetative growth, floral induction, and flower development [69]. In GA signaling, GIBBERELLIN INSENSITIVE DWARF1 (GID1) and DELLAs constitute negative feedback due to the influence of DELLAs on the expression of several genes [70, 71]. The BR and GA signaling processes converge at the BZR1 and DELLA interaction by the formation of a BZR1-DELLA complex, which reduces the transcriptional ability of BZR1 [71]. In our data, DELLA protein SLN1 and GAI coexpressed with BZR1 in the VE stage, providing evidence for BR-GA cross-talk. Hormone signaling has been shown to be interdependent and synergistic in many plant processes [64], and our research further illustrated the extensive cross-talk between different hormones in vernalization response.
Fig. 7.
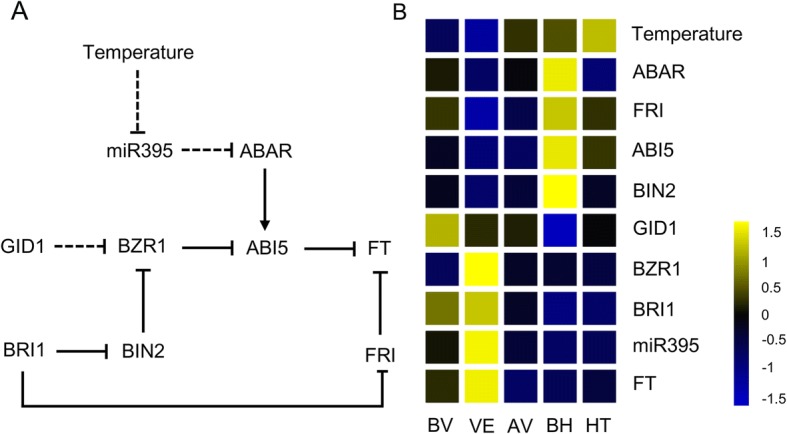
Analysis of gene expression in plant hormones pathways from growth stages BV to HT. a The putative network between GA, ABA and BR pathway. Line with arrows indicate positive regulation and line with blunt end indicated negative regulation. Dashed indicated the potential regulation. b The heatmap showed the gene expression involved in the network
Timely flowering initiation was control by the complex endogenous triggers and external environment. In temperate cereals, VRN1 was homologous to the Arabidopsis meristem identity genes APETALA1 and VIN3-like, which were also identified in diploid wheat (Triticum monococcum L.) [4, 72]. In this study, three critical flowering-related genes VRN1, VIN3, and FT were identified in D. glomerata with up-regulated pattern when exposed to low temperature, suggests a certain degree of evolutionary conservation of flowering network between annuals and perennials. However, past research has revealed a differentiation in vernalization response between A. thaliana and cereal crops [3]. The vernalization response in temperate grasses is conferred by high expression levels of VRN2 [7], which represses the FT ortholog VRN3 [73, 74]. A more critical difference in the vernalization pathways is the absence of clear homologues of VRN2 in A. thaliana [75]. In addition, homologue of FLC in temperate cereals also not been identified. Based on genome synteny studies, several putative FLC-like genes have been detected in monocots [76], but less functional verification of FLC orthologs reported in cereals thus far [77]. This study may supply useful information for identifying genes related to vernalization response in temperate grasses.
Conclusions
Our study displayed a dynamic variation of transcriptional and post-transcriptional regulation in vernalization and the heading stage of perennial grass. A total of 3,846 significant DEGs and 69 differentially expressed miRNAs were identified among five stages. The target genes for vernalization-responsive miRNAs functioned in transmembrane transport, signal transduction, and plant morphogenesis. One hundred and eighteen coexpressed genes formed a subnetwork, of which five hub targets, BRI1, BZR1, VRN1, VIN3, and FT, might provide new insights in the control of flowering time. These results promote a comprehensive understanding and elucidation of miRNA-mediated molecular mechanisms during vernalization, which may facilitate the molecular breeding in cereal crops and forages.
Methods
Plant materials
The seeds of orchardgrass variety BAOXING (Registered No. 197) were obtained from the Department of Grassland Science, Sichuan Agricultural University. The plants were grown in pots under natural environmental conditions in Sichuan Agricultural University, Chengdu, Sichuan Province, on 21 September 2016. Mixed samples of young leaves were collected and immediately frozen in liquid nitrogen. Five sampling points were chosen: before vernalization (BV, 4 January 2016), vernalization (VE, 2 February 2016), after vernalization (AV, 2 March 2016), before heading (BH, 24 March 2016), and heading (HT, 9 April 2016) (Additional file 5: Figure S3). At each sampling point, three biological replicates were collected. Total RNA was extracted from the mixed samples using the RNAprep Pure Plant Kit (Tiangen Biochemical Technology Company, Beijing, China). RNA purity and concentration was measured via the NanoPhotometer® spectrophotometer (IMPLEN, CA, USA) and Qubit® RNA Assay Kit in Qubit® 2.0 Flurometer (Life Technologies, CA, USA). RNA integrity was assessed using the RNA Nano 6000 Assay Kit and Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Floral primordium was collected to investigate the inflorescence formation after the induction of vernalization. The floral primordium was excised and immediately fixed in Carnoy’s Fluid. After fixation, tissues were routinely dehydrated and embedded with paraffin. Thin sections (5 μm) of each tissue were sliced from each block and mounted on glass. Then, the sections were stained with hematoxylin. Images were recorded using a Leica DM4 B-microscope system (Leica, Germany).
Transcriptome sequencing and de novo assembly analysis
Approximately 3 μg RNA per sample was used for transcriptome library preparation, and a total of 15 libraries were generated using NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer’s protocols. First strand cDNA was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase (RNase H-), then second strand cDNA was synthesized using DNA polymerase I and RNase H. The AMPure XP system (Beckman Coulter, Beverly, USA) was used to select cDNA fragments preferentially 150–200 bp in length. Products were purified using the AMPure XP system and library quality was assessed on Agilent Bioanalyzer 2100 system. Transcriptome sequencing was performed on an Illumina HiSeqTM 2000 platform. Trinity (http://trinityrnaseq.github.io/) was used to assemble high-quality reads into nonredundant unigenes. All the assembled unigenes were searched and annotated against the publicly available protein databases including Nr, Nt, Pfam, KOG/COG(EuKaryotic Orthologous Groups/ Clusters of Orthologous Groups, Swiss-prot, KEGG(Kyoto Encyclopedia of Genes and Genomes), and GO (Gene Ontology), using BLASTx analysis with an E-value cut-off of 1.0E-05. Genes were thus tentatively identified according to the best hits against known sequences. The expected number of fragments per kilobase of transcript sequence per million base pairs sequenced (FPKM) was used to estimate the gene expression levels. Differentially expressed genes (DEGs) of five pairwise sample comparisons including VE vs BV, AV vs VE, BH vs AV and HT vs BH were identified applying the cutoff |log2FC| > 1, P < 0.01 using DESeq Rpackage (1.18.0) software [78, 79].
Small RNA sequencing and identification of microRNAs
To generate small RNA libraries, approximately 3 μg of RNA (RIN number > 7.0) per sample from the five sampling points was used. Sequencing libraries were generated using NEBNext® Multiplex Small RNA Library Prep Set for Illumina® (NEB, USA) following the manufacturer’s protocols. After PCR amplification, the products were purified on an 8% polyacrylamide gel (100 V, 80 min). Fragments corresponding to 140–160 bp were recovered and dissolved in 8 μL elution buffer. Library quality was assessed on the Agilent Bioanalyzer 2100 system using DNA High Sensitivity Chips. Sequencing was performed on Illumina Hiseq 2500/2000 platform for raw reads in 15 sRNA libraries. Clean reads were obtained by removing the contaminated sequences (5′ adapters), reads without 3′ adapters and insert, low-quality sequences (the reads with sQ ≤ 5 base more than 50%), reads containing poly-N(> 10%), and poly-A/T/G/C. Reads smaller than 18 nt also removed through Illumina’s Genome Analyzer Pipeline V1.5. After trim the 3′ adapters, The remaining clean reads were mapped to D. glomerata reference sequence to analyze their expression and distribution by Bowtie [80]. MiRBase20.0 (http://www.mirbase.org/) was used to look for known miRNA. MiREvo and mirdeep2 were integrated to predict novel miRNA [81, 82]. The identified, known miRNAs were searched against miFam.dat (http://www.mirbase.org/ftp.shtml) to identify their families. Novel miRNA precursors were submitted to Rfam (http://xfam.org/) to look for Rfam families. Target genes of miRNA were predicted using psRobot_tar in psRobot [83]. MiRNA expression levels were estimated by TPM (transcript per million) using the following formula: Normalized expression = mapped read count/ (total reads*1,000,000) [84]. Differential expression analysis was performed using DESeq in R (1.8.3) [78]. The P-values were adjusted according to the Benjamini and Hochberg method. The significantly differentially expressed genes were identified based on the following thresholds: P-value ≤0.01, false discovery rate ≤ 0.01, and |log2 ratio| ≥ 1. Goseq and KOBAS software were implemented for enrichment analyses (http://www.genome.jp/kegg/) [85, 86]. Weighted gene coexpression network analysis was performed by WGCNA package in R (v3.3.0) as previously described [87]. Transcripts with FPKM values above 0 in more than two duplications were chosen for the WGCNA analysis [88, 89]. Gene expression adjacency matrix was constructed to analyze the network topology with the soft threshold power set to 10, TOMType as unsigned, minModuleSize as 30, and mergeCutHeight as 0.25 in the analysis. The blockwiseModules were used to obtain the modules using the default setting. The networks were visualized using Cytoscape v.3.0.0. The heat map was obtained using OmicShare tools (www.omicshare.com/tools). All the sequencing data was deposited into the NCBI SRA database under the accession numbers SRP131840 and SRP131899.
Verification by qRT-PCR
The validity of transcriptome and miRNA sequences was verified by quantitative real-time PCR (qRT-PCR). Eight flowering related genes (LHY (c146679_g3), FRI (c132929_g2), LHP1 (c143664_g4), FD (c128431_g1), VRN1 (c147469_g1), SPL9 (c150483_g1), GI (c151406_g1), and SPL3 (c134262_g2)) and eight differentially expressed miRNAs (miR528, miR408, miR399, miR398, miR397, miR166, miR167, and miR172) were selected for validation. Primers were designed from the candidate gene sequences using the online Primer BLAST program (https://www.ncbi.nlm.nih.gov/tools/primer-blast) (Additional file 3). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and U6 were selected as reference genes [90, 91]. The detailed methods have been described in our previous study [92]. First-strand cDNAs of miRNAs were synthesized using Mir-X™ miRNA FirstStrand Synthesis Kit (Code No. 638315) (Takara, Japan), and the qRT-PCR reaction was performed using Bio-Rad CFX96 following the instructions for the SYBR® Premix Ex Taq™ II (Tli RNaseH Plus) (Code No.RR820Q) (Takara, Japan). Three replicates were performed on each sample. Relative quantitative level were calculated based on 2-ΔΔCT method [93].
Additional files
Table S1. Summary statistics of RNA sequencing in different samples. Table S2. Summary of transcriptome sequencing for D. glomerata. Table S3. Summary statistics of the quality of sRNA sequencing. Table S4. Summary statistics of sRNA sequencing in samples from different stages. Table S5. Summary statistics of sRNA mapping results. Table S6. Length distribution of total sRNA. Table S7. Summary statistics and Length distribution of known miRNAs and novel miRNAs. Table S8. Statistics of differentially expressed miRNAs. Table S9. The targets of differentially expressed miRNAs. Table S10. Statistics for significant DEGs. (XLSX 313 kb)
Figure S1. Characterization of identified miRNAs from D. glomerata. (A) The first nucleotide of known miRNAs. (B) The first nucleotide of novel miRNAs. (TIF 1545 kb)
Table S11. The primers used for qRT-PCR. Table S12. Targets of miRNAs in the BV stage, VE stage, and AV stage. Table S13. Statistics for GO enrichment results. Table S14. DEGs in the VE-BV-UP vs AV-VE-DOWN and VE-BV-DOWN vs AV-VE-UP comparisons. Table S15. Concerned DEGs in the VE-BV, AV-VE, and HT-BH comparison. Table S16. The genes in the brown-module. Table S17. The targets of miR530. (XLSX 36 kb)
Figure S2. WGCNA of DEGs in different stages. (A) Hierarchical cluster tree showing coexpression modules identified by WGCNA. Each leaf in the tree is one gene. The major tree branches constitute 12 modules labeled by different colors. (B) Module-stage association. Each row corresponds to a module. Each column corresponds to a specific stage. The color of each cell at the row-column intersection indicates the correlation coefficient between the module and the stage. A high degree of correlation between a specific module and the stage is indicated by dark red. (TIF 10484 kb)
Figure S3. The definition of sampling point and the photographs of D.glomerata at five developmental stages. (TIF 11006 kb)
Acknowledgements
We are grateful to Xia Wang from the Sichuan Agricultural University for the help in samples collection.
Funding
This research was funded by the National Basic Research Program (973 program) in China (No.2014CB138705), the earmarked fund for Modern Agro-industry Technology Research System (No. CARS-34) and the National Natural Science Foundation of China (NSFC 31872997). The funding bodies had no role in study design, data collection, analysis and interpretation, decision to public, or manuscript preparation.
Availability of data and materials
All the data pertaining to the present study has been included in table and/or figure form in the manuscript and authors are pleased to share analyzed/raw data and plant materials upon reasonable request; The raw RNA-seq and small RNA reads have been deposited in the NCBI database (accession SRP131840 and SRP131899, https://www.ncbi.nlm.nih.gov/sra). The plant materials were provided by Department of Grassland Science, College of Animal Science and Technology, Sichuan Agricultural University, Chengdu, China.
Abbreviations
- ABA
Abscisic acid
- ABI5
ABSCISIC ACID-INSENSITIVE 5
- ARF
Auxin response factor
- AV
After vernalization
- BH
Before heading
- bHLH
Basic helix-loop-helix
- BR
Brassinosteroid
- BRI1
BRASSINOSTEROID INSENSITIVE1
- BV
Before vernalization
- BZR1
Brassinazole BRASSINAZOLE RESISTANT 1
- CIR1
Circadian 1
- CO
CONSTANS
- COG
Cluster of orthologous group
- DEGs
Differentially expressed genes
- ERF
Ethylene-responsive transcription factor
- FLC
Flowering locus C
- FPKM
Fragments Per Kilobase of transcript sequence per Million base pairs sequenced
- FRI
FRIGIDA
- FT
Flowering locus T
- GA
Gibberellin
- GO
Gene Ontology
- HSP
Heat shock protein
- HT
Heading stage
- IAA
Indole-3-acetic acid
- ICE1
Inducer of CBF expression 1
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KOBAS
Kegg Orthology Based Annotation System
- KOG
Eukaryotic orthologous groups
- LAF1
Long after far-red light 1
- MADS
MCM1, AGAMOUS, DEFICIENS and SRF
- MYB
Myeloblastosis, a transcription factor family
- NAC
NAM, ATAF1,2, CUC2
- Nr
Non-redundant protein sequence
- Pfam
Protein family
- qRT-PCR
Quantitative real-time PCR
- SOC1
Suppressor of overexpression of CO1
- SPL3
SBP-box gene family squamosa promoter binding protein-like3
- Swiss-prot
Annotated protein sequence database
- TFs
Transcription factors
- TPM
Transcript per million
- VE
vernalization
- VIN3
Vernalization insensitive 3
- VRN
Vernalization
- WGCNA
Weighted correlation network analysis
Authors’ contributions
XL, HG, ZY participated the laboratory experiment. XL participated in sampling and extracting and quantifying RNA; HG participated in synthesis of cDNA and performed qRT-PCR; ZY participated in observation of floral primordium section. GF, HY, and WX analyzed, interpreted the data, and wrote manuscript. GF is the major contributor in drafting the manuscript; WX guide data analyses and interpreted data; HY performed most in bioinformatics analyses. JW and BS guided interpreted data and revising the manuscript. XZ, LH and NG conceived and designed the experiments. XZ conceived and coordinated the study and provided materials and resources; LH guided the study and designed the experiments; NG revising and perfecting the experiments, interpreted data and wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Guangyan Feng, Email: fg62586336@163.com.
Lei Xu, Email: 1466356142@qq.com.
Jianping Wang, Email: wangjp@ufl.edu.
Gang Nie, Email: nieganggrass@hotmail.com.
Bradley Shaun Bushman, Email: shaun.bushman@ars.usda.gov.
Wengang Xie, Email: xiewg@lzu.edu.cn.
Haidong Yan, Email: yanhd@vt.edu.
Zhongfu Yang, Email: yangzf211@163.com.
Hao Guan, Email: guan0427@stu.sicau.edu.cn.
Linkai Huang, Email: huanglinkai@sicau.edu.cn.
Xinquan Zhang, Email: zhangxq@sicau.edu.cn.
References
- 1.Woods Daniel P., McKeown Meghan A., Dong Yinxin, Preston Jill C., Amasino Richard M. Evolution ofVRN2/Ghd7-Like Genes in Vernalization-Mediated Repression of Grass Flowering. Plant Physiology. 2016;170(4):2124–2135. doi: 10.1104/pp.15.01279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang J, Tian L, Lee HS, Chen ZJ. Nonadditive regulation of FRI and FLC loci mediates flowering-time variation in Arabidopsis allopolyploids. Genetics. 2006;173(2):965–974. doi: 10.1534/genetics.106.056580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blumel M, Dally N, Jung C. Flowering time regulation in crops-what did we learn from Arabidopsis? Curr Opin Biotechnol. 2015;32:121–129. doi: 10.1016/j.copbio.2014.11.023. [DOI] [PubMed] [Google Scholar]
- 4.Fu D, Dunbar M, Dubcovsky J. Wheat VIN3-like PHD finger genes are up-regulated by vernalization. Mol Genet Genomics. 2007;277(3):301–313. doi: 10.1007/s00438-006-0189-6. [DOI] [PubMed] [Google Scholar]
- 5.Ober D. Seeing double: gene duplication and diversification in plant secondary metabolism. Trends Plant Sci. 2005;10(9):444–449. doi: 10.1016/j.tplants.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Kong H, Landherr LL, Frohlich MW, Leebens-Mack J, Ma H, Depamphilis CW. Patterns of gene duplication in the plant SKP1 gene family in angiosperms: evidence for multiple mechanisms of rapid gene birth. Plant J. 2010;50(5):873–885. doi: 10.1111/j.1365-313X.2007.03097.x. [DOI] [PubMed] [Google Scholar]
- 7.Ream TS, Woods DP, Amasino RM. The molecular basis of vernalization in different plant groups. Cold Spring Harb Symp Quant Biol. 2012;77:105–115. doi: 10.1101/sqb.2013.77.014449. [DOI] [PubMed] [Google Scholar]
- 8.Axtell MJ, Bowman JL. Evolution of plant microRNAs and their targets. Trends Plant Sci. 2008;13(7):343–349. doi: 10.1016/j.tplants.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 9.Chen X. Small RNAs and their roles in plant development. Annu Rev Cell Dev Biol. 2009;25:21–44. doi: 10.1146/annurev.cellbio.042308.113417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu G, Park MY, Conway SR, Wang JW, Weigel D, Poethig RS. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell. 2009;138(4):750–759. doi: 10.1016/j.cell.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen W, Kong J, Lai T, Manning K, Wu C, Wang Y, Qin C, Li B, Yu Z, Zhang X, et al. Tuning LeSPL-CNR expression by SlymiR157 affects tomato fruit ripening. Sci Rep. 2015;5:7852. doi: 10.1038/srep07852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang J, Li Z, Zhao D. Deregulation of the OsmiR160 target gene OsARF18 causes growth and developmental defects with an alteration of auxin signaling in Rice. Sci Rep. 2016;6:29938. doi: 10.1038/srep29938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Li K, Chen L, Zou Y, Liu H, Tian Y, Li D, Wang R, Zhao F, Ferguson BJ. microRNA167-directed regulation of the auxin response factors, GmARF8a and GmARF8b, is required for soybean (Glycine max L.) nodulation and lateral root development. Plant Physiol. 2015;168(3):984. doi: 10.1104/pp.15.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao F, Wang K, Liu Y, Chen Y, Chen P, Shi Z, Luo J, Jiang D, Fan F, Zhu Y, et al. Blocking miR396 increases rice yield by shaping inflorescence architecture. Nature Plants. 2015;2:15196. doi: 10.1038/nplants.2015.196. [DOI] [PubMed] [Google Scholar]
- 15.Liu H, Guo S, Xu Y, Li C, Zhang Z, Zhang D, Xu S, Zhang C, Chong K. OsmiR396d-regulated OsGRFs function in floral organogenesis in rice through binding to their targets OsJMJ706 and OsCR4. Plant Physiol. 2014;165(1):160–174. doi: 10.1104/pp.114.235564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Sun F, Cao H, Peng H, Ni Z, Sun Q, Yao Y. TamiR159 directed wheat TaGAMYB cleavage and its involvement in anther development and heat response. PLoS One. 2012;7(11):e48445. doi: 10.1371/journal.pone.0048445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang Y, Xie K, Xiong L. Conserved miR164-targeted NAC genes negatively regulate drought resistance in rice. J Exp Bot. 2014;65(8):2119–2135. doi: 10.1093/jxb/eru072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ori N, Cohen AR, Etzioni A, Brand A, Yanai O, Shleizer S, Menda N, Amsellem Z, Efroni I, Pekker I, et al. Regulation of LANCEOLATE by miR319 is required for compound-leaf development in tomato. Nat Genet. 2007;39(6):787–791. doi: 10.1038/ng2036. [DOI] [PubMed] [Google Scholar]
- 19.Yang C, Li D, Mao D, Liu X, Ji C, Li X, Zhao X, Cheng Z, Chen C, Zhu L. Overexpression of microRNA319 impacts leaf morphogenesis and leads to enhanced cold tolerance in rice (Oryza sativa L.) Plant Cell Environ. 2013;36(12):2207–2218. doi: 10.1111/pce.12130. [DOI] [PubMed] [Google Scholar]
- 20.Yan Y, Wang H, Hamera S, Chen X, Fang R. miR444a has multiple functions in the rice nitrate-signaling pathway. Plant J. 2014;78(1):44–55. doi: 10.1111/tpj.12446. [DOI] [PubMed] [Google Scholar]
- 21.Campoli C, von Korff M. Genetic control of reproductive development in temperate cereals.Aav Bot Res. 2014;72:131–58.
- 22.Huang LK, Yan HD, Zhao XX, Zhang XQ, Wang J, Frazier T, Yin G, Huang X, Yan DF, Zang WJ. Identifying differentially expressed genes under heat stress and developing molecular markers in orchardgrass (Dactylis glomerata L.) through transcriptome analysis. Mol Ecol Resour. 2015;15(6):1497–1509. doi: 10.1111/1755-0998.12418. [DOI] [PubMed] [Google Scholar]
- 23.Amasino R. Seasonal and developmental timing of flowering. Plant J. 2010;61(6):1001–1013. doi: 10.1111/j.1365-313X.2010.04148.x. [DOI] [PubMed] [Google Scholar]
- 24.Mutz KO, Heilkenbrinker A, Lonne M, Walter JG, Stahl F. Transcriptome analysis using next-generation sequencing. Curr Opin Biotechnol. 2013;24(1):22–30. doi: 10.1016/j.copbio.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP. Prediction of plant microRNA targets. Cell. 2002;110(4):513–520. doi: 10.1016/S0092-8674(02)00863-2. [DOI] [PubMed] [Google Scholar]
- 26.Weller B, Zourelidou M, Frank L, Barbosa IC, Fastner A, Richter S, Jurgens G, Hammes UZ, Schwechheimer C. Dynamic PIN-FORMED auxin efflux carrier phosphorylation at the plasma membrane controls auxin efflux-dependent growth. Proc Natl Acad Sci U S A. 2017;114(5):E887–E896. doi: 10.1073/pnas.1614380114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang D, Xu Y, Li Q, Hao X, Cui K, Sun F, Zhu Y. Transgenic expression of a putative calcium transporter affects the time of Arabidopsis flowering. Plant J. 2003;33(2):285–292. doi: 10.1046/j.1365-313X.2003.01627.x. [DOI] [PubMed] [Google Scholar]
- 28.Dyson BC, Allwood JW, Feil R, Xu Y, Miller M, Bowsher CG, Goodacre R, Lunn JE, Johnson GN. Acclimation of metabolism to light in Arabidopsis thaliana: the glucose 6-phosphate/phosphate translocator GPT2 directs metabolic acclimation. Plant Cell Environ. 2015;38(7):1404–1417. doi: 10.1111/pce.12495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huijser P, Schmid M. The control of developmental phase transitions in plants. Development. 2011;138(19):4117–4129. doi: 10.1242/dev.063511. [DOI] [PubMed] [Google Scholar]
- 30.Lin LL, Wu CC, Huang HC, Chen HJ, Hsieh HL, Juan HF. Identification of microRNA 395a in 24-epibrassinolide-regulated root growth of Arabidopsis thaliana using microRNA arrays. Int J Mol Sci. 2013;14(7):14270–14286. doi: 10.3390/ijms140714270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang W, Deng Z, Osesprieto JA, Suzuki N, Zhu S, Zhang X, Burlingame AL, Wang ZY. Proteomics studies of brassinosteroid signal transduction using prefractionation and two-dimensional DIGE. Mol Cell Proteomics. 2008;7(4):728. doi: 10.1074/mcp.M700358-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prabhakar V, Lottgert T, Geimer S, Dormann P, Kruger S, Vijayakumar V, Schreiber L, Gobel C, Feussner K, Feussner I, et al. Phosphoenolpyruvate provision to plastids is essential for gametophyte and sporophyte development in Arabidopsis thaliana. Plant Cell. 2010;22(8):2594–2617. doi: 10.1105/tpc.109.073171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang J, Chu C. MicroRNAs in crop improvement: fine-tuners for complex traits. Nat Plants. 2017;3:17077. doi: 10.1038/nplants.2017.77. [DOI] [PubMed] [Google Scholar]
- 34.Glazinska P, Wojciechowski W, Wilmowicz E, Zienkiewicz A, Frankowski K, Kopcewicz J. The involvement of InMIR167 in the regulation of expression of its target gene InARF8, and their participation in the vegetative and generative development of Ipomoea nil plants. J Plant Physiol. 2014;171(3–4):225–234. doi: 10.1016/j.jplph.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 35.Damodharan S, Zhao D, Arazi T. A common miRNA160-based mechanism regulates ovary patterning, floral organ abscission and lamina outgrowth in tomato. Plant J. 2016;86(6):458–471. doi: 10.1111/tpj.13127. [DOI] [PubMed] [Google Scholar]
- 36.Nizampatnam NR, Schreier SJ, Damodaran S, Adhikari S, Subramanian S. microRNA160 dictates stage-specific auxin and cytokinin sensitivities and directs soybean nodule development. Plant J. 2015;84(1):140–153. doi: 10.1111/tpj.12965. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Li K, Chen L, Zou Y, Liu H, Tian Y, Li D, Wang R, Zhao F, Ferguson BJ, et al. MicroRNA167-directed regulation of the auxin response factors GmARF8a and GmARF8b is required for soybean nodulation and lateral root development. Plant Physiol. 2015;168(3):984–999. doi: 10.1104/pp.15.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu N, Wu S, Van Houten J, Wang Y, Ding B, Fei Z, Clarke TH, Reed JW, van der Knaap E. Down-regulation of AUXIN RESPONSE FACTORS 6 and 8 by microRNA 167 leads to floral development defects and female sterility in tomato. J Exp Bot. 2014;65(9):2507–2520. doi: 10.1093/jxb/eru141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu PP, Montgomery TA, Fahlgren N, Kasschau KD, Nonogaki H, Carrington JC. Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J. 2007;52(1):133–146. doi: 10.1111/j.1365-313X.2007.03218.x. [DOI] [PubMed] [Google Scholar]
- 40.Wang JW, Wang LJ, Mao YB, Cai WJ, Xue HW, Chen XY. Control of root cap formation by MicroRNA-targeted auxin response factors in Arabidopsis. Plant Cell. 2005;17(8):2204–2216. doi: 10.1105/tpc.105.033076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mallory AC, Bartel DP, Bartel B. MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell. 2005;17(5):1360–1375. doi: 10.1105/tpc.105.031716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baucher M, Moussawi J, Vandeputte OM, Monteyne D, Mol A, Perez-Morga D, El Jaziri M. A role for the miR396/GRF network in specification of organ type during flower development, as supported by ectopic expression of Populus trichocarpa miR396c in transgenic tobacco. Plant Biol (Stuttg) 2013;15(5):892–898. doi: 10.1111/j.1438-8677.2012.00696.x. [DOI] [PubMed] [Google Scholar]
- 43.Ercoli MF, Rojas AM, Debernardi JM, Palatnik JF, Rodriguez RE. Control of cell proliferation and elongation by miR396. Plant Signal Behav. 2016;11(6):e1184809. doi: 10.1080/15592324.2016.1184809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang FX, Yu DQ. Overexpression of Arabidopsis MiR396 enhances drought tolerance in transgenic tobacco plants. Acta Bot Yunnanica. 2009;31(5):421–426. doi: 10.3724/SP.J.1143.2009.09044. [DOI] [Google Scholar]
- 45.Che R, Tong H, Shi B, Liu Y, Fang S, Liu D, Xiao Y, Hu B, Liu L, Wang H, et al. Control of grain size and rice yield by GL2-mediated brassinosteroid responses. Nature Plants. 2015;2:15195. doi: 10.1038/nplants.2015.195. [DOI] [PubMed] [Google Scholar]
- 46.Yuan S, Li Z, Li D, Yuan N, Hu Q, Luo H. Constitutive expression of Rice MicroRNA528 alters plant development and enhances tolerance to salinity stress and nitrogen starvation in creeping Bentgrass. Plant Physiol. 2015;169(1):576–593. doi: 10.1104/pp.15.00899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tripathi RK, Bregitzer P, Singh J. Genome-wide analysis of the SPL/miR156 module and its interaction with the AP2/miR172 unit in barley. Sci Rep. 2018;8:7085. [DOI] [PMC free article] [PubMed]
- 48.Kim JJ, Lee JH, Kim W, Jung HS, Huijser P, Ahn JH. The microRNA156-SQUAMOSA PROMOTER BINDING PROTEIN-LIKE3 module regulates ambient temperature-responsive flowering via FLOWERING LOCUS T in Arabidopsis. Plant Physiol. 2012;159(1):461–478. doi: 10.1104/pp.111.192369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hyun Y, Richter R, Vincent C, Martinez-Gallegos R, Porri A, Coupland G. Multi-layered regulation of SPL15 and cooperation with SOC1 integrate endogenous flowering pathways at the Arabidopsis shoot meristem. Dev Cell. 2016;37(3):254–266. doi: 10.1016/j.devcel.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 50.Zhu QH, Helliwell CA. Regulation of flowering time and floral patterning by miR172. J Exp Bot. 2011;62(2):487–495. doi: 10.1093/jxb/erq295. [DOI] [PubMed] [Google Scholar]
- 51.Mathieu J, Yant LJ, Mürdter F, Küttner F, Schmid M. Repression of flowering by the miR172 target SMZ. PLoS Biol. 2009;7(7):e1000148. doi: 10.1371/journal.pbio.1000148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu H, Hu F, Wang R, Zhou X, Sze SH, Liou LW, Barefoot A, Dickman M, Zhang X. Arabidopsis Argonaute10 specifically sequesters miR166/165 to regulate shoot apical meristem development. Cell. 2011;145(2):242–256. doi: 10.1016/j.cell.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xian Z, Huang W, Yang Y, Tang N, Zhang C, Ren M, Li Z. miR168 influences phase transition, leaf epinasty, and fruit development via SlAGO1s in tomato. J Exp Bot. 2014;65(22):6655–6666. doi: 10.1093/jxb/eru387. [DOI] [PubMed] [Google Scholar]
- 54.Barciszewska-Pacak M, Knop K, Jarmolowski A, Szweykowska-Kulinska Z. Arabidopsis thaliana microRNA162 level is posttranscriptionally regulated via splicing and polyadenylation site selection. Acta Biochim Pol. 2016;63(4):811–816. doi: 10.18388/abp.2016_1349. [DOI] [PubMed] [Google Scholar]
- 55.Dong C-H, Pei H. Over-expression of miR397 improves plant tolerance to cold stress in Arabidopsis thaliana. J Plant Biol. 2014;57(4):209–217. doi: 10.1007/s12374-013-0490-y. [DOI] [Google Scholar]
- 56.Zhang YC, Yu Y, Wang CY, Li ZY, Liu Q, Xu J, Liao JY, Wang XJ, Qu LH, Chen F, et al. Overexpression of microRNA OsmiR397 improves rice yield by increasing grain size and promoting panicle branching. Nat Biotechnol. 2013;31(9):848–852. doi: 10.1038/nbt.2646. [DOI] [PubMed] [Google Scholar]
- 57.Yu C, Jiang J, Song A, Chen S, Hong S, Luo H, Gu C, Jing S, Lu Z, Fang W. Ambient temperature enhanced freezing tolerance of Chrysanthemum dichrum CdICE1 Arabidopsis via miR398. BMC Biol. 2013;11(1):121. doi: 10.1186/1741-7007-11-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu X, Guan Q, Zhu J. Downregulation of CSD2 by a heat-inducible miR398 is required for thermotolerance in Arabidopsis. Plant Signal Behav. 2013;8(8):e24952. [DOI] [PMC free article] [PubMed]
- 59.Greer DH, Weston C. Heat stress affects flowering, berry growth, sugar accumulation and photosynthesis of Vitis vinifera cv. Semillon grapevines grown in a controlled environment. Funct Plant Biol. 2010;37(3):206–214. doi: 10.1071/FP09209. [DOI] [Google Scholar]
- 60.Margaritopoulou T, Kryovrysanaki N, Megkoula P, Prassinos C, Samakovli D, Milioni D, Hatzopoulos P. HSP90 canonical content organizes a molecular scaffold mechanism to progress flowering. Plant J. 2016;87(2):174–187. doi: 10.1111/tpj.13191. [DOI] [PubMed] [Google Scholar]
- 61.Clouse SD. Brassinosteroid Signaling. Development. 2007;140(8):1615–1620. doi: 10.1242/dev.060590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.He JX, Gendron JM, Yang Y, Li J, Wang ZY. The GSK3-like kinase BIN2 phosphorylates and destabilizes BZR1, a positive regulator of the brassinosteroid signaling pathway in Arabidopsis. Proc Natl Acad Sci U S A. 2002;99(15):10185–10190. doi: 10.1073/pnas.152342599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Domagalska MA, Schomburg FM, Amasino RM, Vierstra RD, Nagy F, Davis SJ. Attenuation of brassinosteroid signaling enhances FLC expression and delays flowering. Development. 2007;134(15):2841–2850. doi: 10.1242/dev.02866. [DOI] [PubMed] [Google Scholar]
- 64.Depuydt S, Hardtke CS. Hormone signalling crosstalk in plant growth regulation. Curr Biol. 2011;21(9):R365–R373. doi: 10.1016/j.cub.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 65.Hu Y, Yu D. BRASSINOSTEROID INSENSITIVE2 interacts with ABSCISIC ACID INSENSITIVE5 to mediate the antagonism of brassinosteroids to abscisic acid during seed germination in Arabidopsis. Plant Cell. 2014;26(11):4394–4408. doi: 10.1105/tpc.114.130849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang X, Bai Y, Shang J, Xin R, Tang W. The antagonistic regulation of abscisic acid-inhibited root growth by brassinosteroids is partially mediated via direct suppression of ABSCISIC ACID INSENSITIVE 5 expression by BRASSINAZOLE RESISTANT 1. Plant Cell Environ. 2016;39(9):1994–2003. doi: 10.1111/pce.12763. [DOI] [PubMed] [Google Scholar]
- 67.Wang Y, Li L, Ye T, Lu Y, Chen X, Wu Y. The inhibitory effect of ABA on floral transition is mediated by ABI5 in Arabidopsis. J Exp Bot. 2013;64(2):675–684. doi: 10.1093/jxb/ers361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Skubacz A, Daszkowska-Golec A, Szarejko I. The role and regulation of ABI5 (ABA-insensitive 5) in plant development, abiotic stress responses and Phytohormone crosstalk. Front Plant Sci. 2016;7:1884. doi: 10.3389/fpls.2016.01884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Park J, Oh DH, Dassanayake M, Nguyen KT, Ogas J, Choi G, Sun TP. Gibberellin signaling requires chromatin remodeler PICKLE to promote vegetative growth and phase transitions. Plant Physiol. 2017;173(2):1463–1474. doi: 10.1104/pp.16.01471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yamaguchi S. Gibberellin metabolism and its regulation. Annu Rev Plant Biol. 2008;59:225–251. doi: 10.1146/annurev.arplant.59.032607.092804. [DOI] [PubMed] [Google Scholar]
- 71.Li QF, Wang C, Jiang L, Li S, Sun SS, He JX. An interaction between BZR1 and DELLAs mediates direct signaling crosstalk between brassinosteroids and gibberellins in Arabidopsis. Sci Signal. 2012;5(244):ra72. doi: 10.1126/scisignal.2002908. [DOI] [PubMed] [Google Scholar]
- 72.Yan L, Loukoianov A, Tranquilli G, Helguera M, Fahima T, Dubcovsky J. Positional cloning of the wheat vernalization gene VRN1. Proc Natl Acad Sci U S A. 2003;100(10):6263–6268. doi: 10.1073/pnas.0937399100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Faure S, Higgins J, Turner A, Laurie DA. The FLOWERING LOCUS T-like gene family in barley (Hordeum vulgare) Genetics. 2007;176(1):599–609. doi: 10.1534/genetics.106.069500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yan L, Fu D, Li C, Blechl A, Tranquilli G, Bonafede M, Sanchez A, Valarik M, Yasuda S, Dubcovsky J. The wheat and barley vernalization gene VRN3 is an orthologue of FT. Proc Natl Acad Sci U S A. 2006;103(51):19581–19586. doi: 10.1073/pnas.0607142103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yan L, Loukoianov A, Blechl A, Tranquilli G, Ramakrishna W, Sanmiguel P, Bennetzen JL, Echenique V, Dubcovsky J. The wheat VRN2 gene is a flowering repressor down-regulated by vernalization. Science. 2004;303(5664):1640–1644. doi: 10.1126/science.1094305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ruelens P, de Maagd RA, Proost S, Theissen G, Geuten K, Kaufmann K. FLOWERING LOCUS C in monocots and the tandem origin of angiosperm-specific MADS-box genes. Nat Commun. 2013;4:2280. doi: 10.1038/ncomms3280. [DOI] [PubMed] [Google Scholar]
- 77.Milec Z, Valarik M, Bartos J, Safar J. Can a late bloomer become an early bird? Tools for flowering time adjustment. Biotechnol Adv. 2014;32(1):200–214. doi: 10.1016/j.biotechadv.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 78.Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26(1):136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 79.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):1–12. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wen M, Shen Y, Shi S, Tang T. miREvo: an integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinformatics. 2012;13:140. doi: 10.1186/1471-2105-13-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40(1):37–52. doi: 10.1093/nar/gkr688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu HJ, Ma YK, Chen T, Wang M, Wang XJ. PsRobot: a web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012;40(Web Server issue):W22–W28. doi: 10.1093/nar/gks554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou L, Chen J, Li Z, Li X, Hu X, Huang Y, Zhao X, Liang C, Wang Y, Sun L, et al. Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS One. 2010;5(12):e15224. doi: 10.1371/journal.pone.0015224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Young MD, Wakeeld MJ, Smyth GK, Oshlack A. goseq: gene ontology testing for RNA-seq datasets. 2012. [Google Scholar]
- 86.Mao X, Cai T, Olyarchuk JG, Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21(19):3787–3793. doi: 10.1093/bioinformatics/bti430. [DOI] [PubMed] [Google Scholar]
- 87.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9(1):559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hollender CA, Kang C, Darwish O, Geretz A, Matthews BF, Slovin J, Alkharouf N, Liu Z. Floral transcriptomes in woodland strawberry uncover developing receptacle and anther gene networks. Plant Physiol. 2014;165(3):1062–1075. doi: 10.1104/pp.114.237529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Han X, Yin H, Song X, Zhang Y, Liu M, Sang J, Jiang J, Li J, Zhuo R. Integration of small RNAs, degradome and transcriptome sequencing in hyperaccumulator Sedum alfredii uncovers a complex regulatory network and provides insights into cadmium phytoremediation. Plant Biotechnol J. 2016;14(6):1470–1483. doi: 10.1111/pbi.12512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peng F, James S, Niclas O, Reid KE, Lund ST. An optimized grapevine RNA isolation procedure and statistical determination of reference genes for real-time RT-PCR during berry development. BMC Plant Biol. 2006;6(1):27. doi: 10.1186/1471-2229-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Turner Marie, Adhikari Sajag, Subramanian Senthil. Optimizing stem-loop qPCR assays through multiplexed cDNA synthesis of U6 and miRNAs. Plant Signaling & Behavior. 2013;8(8):e24918. doi: 10.4161/psb.24918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Feng G, Huang L, Li J, Wang J, Xu L, Pan L, Zhao X, Wang X, Huang T, Zhang X. Comprehensive transcriptome analysis reveals distinct regulatory programs during vernalization and floral bud development of orchardgrass (Dactylis glomerataL.) BMC Plant Biol. 2017;17(1):216. doi: 10.1186/s12870-017-1170-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schefe JH, Lehmann KE, Buschmann IR, Unger T, Funke-Kaiser H. Quantitative real-time RT-PCR data analysis: current concepts and the novel “gene expression’s CT difference” formula. J Mol Med. 2006;84(11):901–910. doi: 10.1007/s00109-006-0097-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary statistics of RNA sequencing in different samples. Table S2. Summary of transcriptome sequencing for D. glomerata. Table S3. Summary statistics of the quality of sRNA sequencing. Table S4. Summary statistics of sRNA sequencing in samples from different stages. Table S5. Summary statistics of sRNA mapping results. Table S6. Length distribution of total sRNA. Table S7. Summary statistics and Length distribution of known miRNAs and novel miRNAs. Table S8. Statistics of differentially expressed miRNAs. Table S9. The targets of differentially expressed miRNAs. Table S10. Statistics for significant DEGs. (XLSX 313 kb)
Figure S1. Characterization of identified miRNAs from D. glomerata. (A) The first nucleotide of known miRNAs. (B) The first nucleotide of novel miRNAs. (TIF 1545 kb)
Table S11. The primers used for qRT-PCR. Table S12. Targets of miRNAs in the BV stage, VE stage, and AV stage. Table S13. Statistics for GO enrichment results. Table S14. DEGs in the VE-BV-UP vs AV-VE-DOWN and VE-BV-DOWN vs AV-VE-UP comparisons. Table S15. Concerned DEGs in the VE-BV, AV-VE, and HT-BH comparison. Table S16. The genes in the brown-module. Table S17. The targets of miR530. (XLSX 36 kb)
Figure S2. WGCNA of DEGs in different stages. (A) Hierarchical cluster tree showing coexpression modules identified by WGCNA. Each leaf in the tree is one gene. The major tree branches constitute 12 modules labeled by different colors. (B) Module-stage association. Each row corresponds to a module. Each column corresponds to a specific stage. The color of each cell at the row-column intersection indicates the correlation coefficient between the module and the stage. A high degree of correlation between a specific module and the stage is indicated by dark red. (TIF 10484 kb)
Figure S3. The definition of sampling point and the photographs of D.glomerata at five developmental stages. (TIF 11006 kb)
Data Availability Statement
All the data pertaining to the present study has been included in table and/or figure form in the manuscript and authors are pleased to share analyzed/raw data and plant materials upon reasonable request; The raw RNA-seq and small RNA reads have been deposited in the NCBI database (accession SRP131840 and SRP131899, https://www.ncbi.nlm.nih.gov/sra). The plant materials were provided by Department of Grassland Science, College of Animal Science and Technology, Sichuan Agricultural University, Chengdu, China.