

Abstract
Acetylation plays an important role in regulating the chaperone activity of heat shock protein 90 (Hsp90) during malignant transformation through the stabilization and conformational maturation of oncogenic proteins. However, the functional acetylation sites, potential anticancer drug targets, are still emerging. We found that acetylation at K292 in Hsp90α is critical for the development and treatment of breast cancer. Acetylation at K292 not only augments the affinity of Hsp90 to ATP, cochaperones, and client proteins but it also promotes cancer cell colony formation, migration, and invasion in vitro as well as tumor growth in vivo. Importantly, K292‐acetylated Hsp90 has been validated as an exciting anticancer drug target by interfering with the complex formation between K292‐acetylated Hsp90 and cochaperone Cdc37, leading to diminishment of kinase client maturation and proteasome‐dependent degradation of kinase substrates. Furthermore, we showed that simvastatin prevented, whereas LBH589 promoted, the progression of Hsp90 chaperone cycling and client maturation, resulting in an increment of cell apoptosis by the combination of simvastatin and LBH589 in a mouse xenograft model. These data suggest that simvastatin is a novel Hsp90 inhibitor to disrupt the formation of the K292‐acetylated Hsp90/Cdc37 complex in triple‐negative breast cancer cells. The combination of simvastatin with LBH589 could be used as a novel therapeutic strategy for triple‐negative breast cancer.
Keywords: acetylation, Cdc37, Hsp90, LBH589, simvastatin
1. INTRODUCTION
Heat shock protein 90 (Hsp90), an ATP‐dependent molecular chaperone, facilitates the activation and stabilization of proteins (clients) that regulate various cellular processes.1 It forms the chaperone machinery with cochaperones2, 3 for the maturation of Hsp90 clients, including receptor tyrosine kinases, signaling molecules, and cell cycle regulators. In addition to cochaperone association, ATP binding and hydrolysis, posttranslational modifications including phosphorylation, S‐nitrosylation, ethylation, and acetylation also regulate Hsp90 function.4 Generally, hyperacetylation decreases the binding of Hsp90 to ATP, cochaperone p23, and client proteins, resulting in decreased Hsp90 function.5, 6, 7
Because Hsp90 clients are frequently mutated, overexpressed, or persistently activated in tumors, cancer cells use the Hsp90 chaperone machinery to protect clients from misfolding and degradation. Although many Hsp90 inhibitors are tested as anticancer agents in clinical trials,2 none of them have been approved due to side‐effects or lacking anticancer activity by upregulating other HSPs (eg, Hsp27 and Hsp72).
Triple‐negative breast cancer (TNBC) without estrogen receptor, progesterone receptor, or human epidermal growth factor receptor 2, represents the most aggressive subtype of breast cancer.8 In TNBC, Hsp90 levels correlate with poor prognosis and drug resistance.9 Heat shock protein 90 interacts with clients that are important for breast neoplasia and metastasis.10, 11, 12 Among client proteins, protein kinases are activated and stabilized by Hsp90 in cooperation with the kinase‐specific cochaperone Cdc37.
Histone deacetylase inhibitors (HDACi) are approved by the US FDA.13, 14 Panobinostat (LBH589), a hydroxamic acid‐based deacetylase inhibitor, is used to treat multiple myeloma. Combinations of HDACi with other agents are potential therapeutic strategies for solid tumors.13, 15, 16, 17 Previously, we identified seven hyperacetylated lysine residues in Hsp90α after LBH589 treatment.6 Interestingly, only acetylation at K292 increases the binding of Hsp90α to ATP. However, the significance of this in tumorigenesis is unclear. We report here that simvastatin (Sim),18 an inhibitor of 3‐hydroxy 3‐methylglutaryl coenzyme A reductase (HMGCR), targets K292‐acetylated Hsp90 to abrogate the interaction of Hsp90 with its cochaperones, clients, and the machinery formation of K292‐acetylated Hsp90/Cdc37. We also report the combinatory effects of Sim and LBH589 on TNBC in vitro and in vivo.
2. MATERIALS AND METHODS
2.1. Plasmids, reagents, and antibodies
Plasmids expressing Flag (F)‐Hsp90, K292Q, and K292R mutant were described previously.6
The following reagents and antibodies were used: Cdc37, cyclin‐dependent kinase 4 (CDK4), Cyclin D1, Hsp27, p‐(S326)‐Heat shock factor 1, and Serine/threonine‐protein kinase 33 (Abcam, Cambridge, MA, USA); p21Cip1 (BD Biosciences, Sparks, MD, USA); AKT, p‐AKT, caspase‐3, cleaved caspase‐3, caspase‐8, active caspase‐8, caspase‐9, cleaved caspase‐9, p‐Cdc37, CDK2, CDK6, Cyclin D1, Eukaryotic elongation factor 2 kinase, p‐eEF2K (S366), Hsp70‐Hsp90 organizing protein 1, HSF1, MEK1/2, p‐MEK1/2, Erk1/2, p‐Erk1/2, and poly(ADP‐ribose) polymerase (PARP)1 (Cell Signaling Technology, Danvers, MA, USA); Hsp40/Hdj1, Hsp70, Hsp90α, and p23 (Enzo Life Sciences, New York, NY, USA); Platelet‐derived growth factor receptor β (Merck Millipore, Temecula, CA, USA); Raf‐1 (C20) and Activator of Hsp90 ATPase protein 1 (Santa Cruz Biotechnology, Dallas, TX, USA); LBH589, Sim, mevastatin (Mev), pravastatin, anti‐Myc beads (Selleck Chemicals, Houston, TX, USA); mevalonate, anti‐Flag, β‐actin, anti‐M2, and anti‐HA beads (Sigma‐Aldrich, St. Louis, MO, USA); and Transforming growth factor‐β receptor II (Thermo Fisher Scientific, Grand Island, NY, USA). Secondary antibodies were from Jackson ImmunoResearch (West Grove, PA, USA).
2.2. Cell culture, transfection, and stable cell lines
MDA‐MB‐231, MDA‐MB‐468, MDA‐MB‐453, HS578T, MCF‐10A, and HEK293 cells were cultured as described.16 Lipofectamine 2000 (Thermo Fisher Scientific) was used for transfection.
2.3. Generation of acetylated K292 Hsp90α antibody
Affinity‐purified polyclonal antibody against Ac‐K292‐hsp90α was generated against peptide QEELNK(Ac)TKPIW containing acetylated K292 (Shanghai Kanwin Biotechnology, Shanghai, China).
2.4. Colony formation evaluation
MDA‐MB‐231 cells expressing Hsp90 grew in 6‐well plates at an initial density of 200 cells per well for 2 weeks. After colony formation, cells were fixed in 4% formaldehyde at room temperature for 30 minutes followed by crystal violet staining for 1 hour. Cells were washed with PBS and dried at room temperature, and photographed by light microscope.
2.5. Wound healing evaluation
MDA‐MB‐231 cells expressing Hsp90 were seeded into 6‐well plates at a density of 2 × 105 cells/well. After 24 hours, cells were scratched with a 20‐μL pipette tip, and photographed by light microscope. Wound healing was continued for 36 hours and photographed.
2.6. Transwell assay
Transwell chambers were coated with DMEM‐diluted Matrigel and MDA‐MB‐231 cells stably expressing Hsp90 were suspended in serum‐free medium, and added to the upper chamber. Medium containing 10% FBS was added to the lower chamber. Twenty‐four hours later, the invasive cells that passed through the membrane to the lower surface were fixed and stained with crystal violet, and photographed by light microscope.
2.7. Evaluation of cytotoxicity
Cells were plated in 96‐well microplates at a density of 2 × 104 cells/well for 24 hours and were treated with fresh medium containing various concentrations of Sim or LBH589, or various concentrations of Sim with 25 nmol/L LBH589. After 72 hours of treatment, cell numbers were determined using CCK‐8 (Dojindo Molecular Tech, Rockville, MD, USA).
2.8. Determination of synergism and antagonism
Cells were plated in 96‐well microplates at a density of 2 × 104 cells/well followed by treatment with Sim, LBH589, or a combination for 72 hours. Synergism or antagonism after drug treatments was evaluated using the combination index (CI) method as described.16 Each CI ratio is the mean value of at least three independent experiments.
2.9. Immunoprecipitations and immunoblots
Cells were lysed using lysis buffer (50 mmol/L Tris‐HCl, 150 mmol/L NaCl, 2 mmol/L EDTA, 1% NP‐40, and 0.1% SDS [pH 7.4]) for immunoprecipitations and immunoblotting as described.16
2.10. Complementation‐based bioluminescence assay
Complementation‐based bioluminescence assay (SRL‐PFAC) was used to explore the interaction between K292Q Hsp90 and S13D Cdc37. The fragment (1‐229) of Renilla luciferase (RL) from pGL5 vector (Promega, Madison, WI, USA) was fused to the N terminus of Hsp90 harboring the K292Q mutation and the fragment (230‐343) of RL was fused to the C terminus of Cdc37 harboring S13D mutation. MDA‐MB‐231 cells expressing those plasmids were lysed followed by treatment with different compounds for 1 hour, and the substrate EnduRen (Promega) of RL was added to the reaction mixture. Luciferase activity was determined by M200Pro microplate reader (Tecan, Seestrasse, Männedorf, Switzerland).
2.11. ATP‐sepharose binding assay
Cell lysates were affinity precipitated using KinaseBind γ‐phosphate‐linked ATP resin (Innova Biosciences, Cambridge, UK) at 4°C for 3 hours. After washing three times with the lysis buffer, the resin was pelleted followed by SDS‐PAGE analysis.
2.12. Confocal microscopy
For immunofluorescence analysis, MDA‐MB‐468 cells were fixed in 4% formaldehyde at room temperature for 30 minutes. Cells were permeabilized using 1× PBS containing 0.1% Triton X‐100 at room temperature for 20 minutes, were blocked with 1× PBS containing 1% BSA for 1 hour, and were incubated with monoclonal anti‐Hsp90α antibody (1:300) and polyclonal anti‐Cdc37 antibody (1:300) at room temperature for 2 hours. After washing with 1× PBS, cells were incubated with secondary antibody (1:500 dilution) in the dark for 1 hour and mounted using Vectashield medium (Vector Laboratories, Burlingame, CA, USA) with DAPI. Images were captured on a Carl Zeiss LSM710 confocal microscope (Jena, Germany).
2.13. Tumor growth analysis in vivo
Exponentially growing MDA‐MB‐231 cells were harvested and suspended in 1× PBS to a concentration of 5 × 107 cells/mL. In vivo studies were carried out as described.16 To evaluate their survival, mice were kept behind a sanitary barrier in the controlled environment of a pathogen‐free animal facility with a 12:12‐hour light : dark cycle at 25 ± 1°C and a relative humidity of 40%‐70%. The resting animals were then killed. The survival curve of mice was plotted using the Kaplan‐Meier method.
2.14. Statistical methods
Data were obtained from three independent experiments, and are presented as mean ± SEM. Statistical analysis was carried out by using Prism 5.0 (La Jolla, CA, USA; Student's t test or one‐way ANOVA). Differences in survival time between animals were assessed using the Cox regression model. P < .05 was considered statistically significant, and all P‐values were two‐sided.
3. RESULTS
3.1. Acetylation status of Hsp90α at K292 correlates with cancer progression
Although K292 is in the hinge region at the beginning of the middle domain of Hsp90a, the functional significance of acetylation at K292 is yet unclear. To gain insight into the role of acetylation at K292 in the formation and stability of Hsp90 complexes with cochaperone or protein kinases, we developed an acetylation‐specific antiserum (acK292) for probing the acetylation status of Hsp90 at K292. The specificity of the antibody was confirmed by determining its ability to detect increasing amounts of the peptide harboring acetylated or non‐acetylated lysine (Figure 1A) and selectively recognizing Wt Hsp90α but not the Hsp90α containing K292A mutation after treatment with LBH589 (Figure 1B). Importantly, in paired human breast carcinoma and para‐carcinoma tissues, malignancies also showed up‐regulated Hsp90α expression and acetylation at K292 (Figure 1C). To further explore the relationship between tumor size and acetylation status of Hsp90α, we detected the acetylation of Hsp90α in MDA‐MB‐231 carcinoma mouse xenograft tumors. The volumes were positively related to the degrees of acetylation at K292 in Hsp90α, but not to the level of Hsp90α expression (Figure 1D). Therefore, the acetylation status of Hsp90α on K292 is positively associated with cancer progression.
Figure 1.
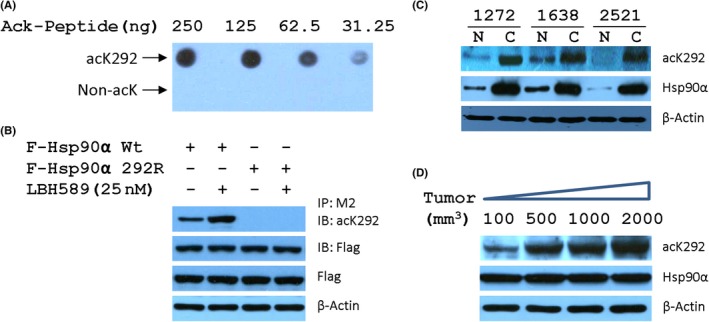
Acetylation status of heat shock protein 90α (Hsp90α) at K292 correlates with cancer progression. A, Specificity of acK292 against K292‐acetylated Hsp90 was tested by recognizing Hsp90α peptide containing either acetylated or non‐acetylated lysine. B, Antibody acK292 selectively recognized K292‐acetylated Hsp90α but not Hsp90α containing K292R mutation. IB, immunoblot; IP, immunoprecipitant. C, Acetylation status of Hsp90α at K292 were monitored in paired human breast carcinoma (C) and paracarcinoma (N) tissues. D, Levels of K292‐acetylated Hsp90α were determined in MDA‐MB‐231 xenografted tumors with distinct volumes
3.2. Acetylation at K292 in Hsp90α promotes breast cancer development
To evaluate the effect of acetylation at K292 residue on tumorigenesis, we established MDA‐MB‐231 cells stably expressing Flag‐tagged Hsp90α‐Wt, mutant mimicking the acetylated (K292Q) or nonacetylated (K292R) Hsp90, to undertake the colony formation assay, migration assay, and Matrigel‐based invasion assay. As expected, overexpression of Hsp90α‐Wt markedly increased the colony formation, migration, and invasion of MDA‐MB‐231 cells, and K292Q mutant statistically and significantly intensified the increase, whereas K292R mutant had marginal effect (Figure 2A–C). Next, we sought to investigate the role of acetylation at K292 in an in vivo setting. MDA‐MB‐231 cells stably expressing Flag‐tagged Hsp90α, K292Q, or K292R mutant were inoculated individually into the mammary fat pads of immunodeficient mice to compare their overall growth rates. As shown in Figure 2D–F MDA‐MB‐231 cells stably expressing Flag‐tagged Hsp90α or K292Q mutant showed a significant increase (>2‐fold) in tumor growth over parental MDA‐MB‐231 cells (P < .01), whereas K292R mutant showed a marginal increase. These results strongly suggest that K292‐acetylated Hsp90α promotes the development of breast cancer.
Figure 2.
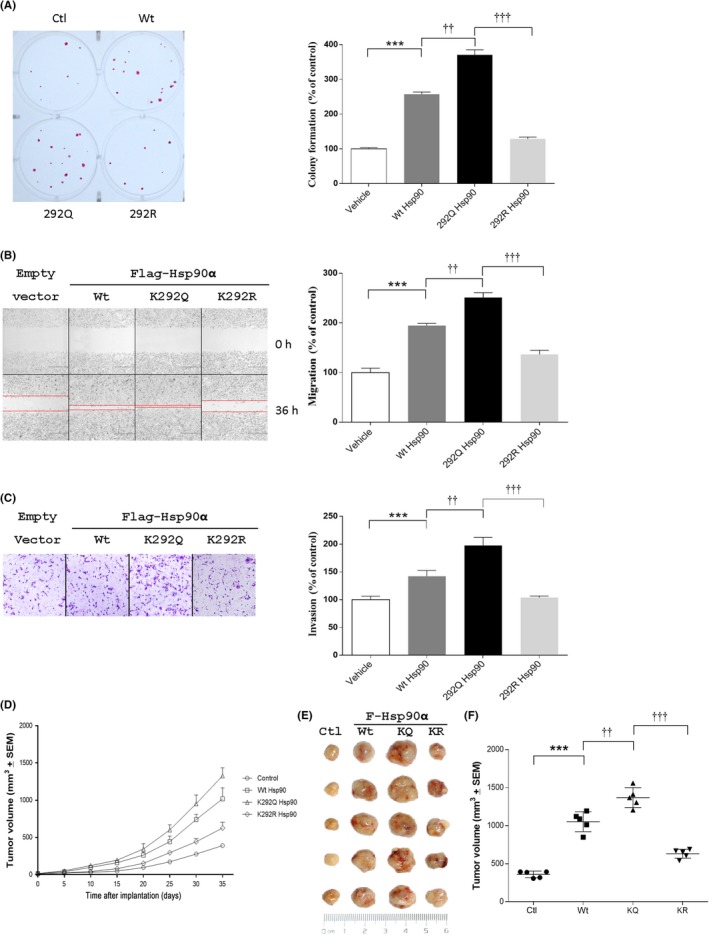
Acetylation of heat shock protein 90α (Hsp90α) at K292 enhances tumorigenesis. Colony formation (A), migration (B), and invasion (C) were evaluated in MDA‐MB‐231 cells stably expressing Flag‐tagged Wt, K292Q, or K292R mutant of Hsp90α. Images were processed with ImageJ, and values are presented as bar plots (mean ± SEM, n = 5). D, Acetylation levels of Hsp90α at K292 were detected by western blotting in MDA‐MB‐231 cells used for (A–C). E, Tumor growth curves of MDA‐MB‐231 cells stably expressing Wt, K292Q (KQ), or K292R (KR) mutant of Hsp90α in xenografted mice. F, Photographs of sc tumors from indicated cells in xenografted mice. G, Final distribution of tumor volumes. ***P < .001 compared with control group; †† P < .01, ††† P < .001 compared with K292Q Hsp90 group. Ctl, control
3.3. K292‐acetylated Hsp90α acts as a target for the screening of Hsp90 inhibitor
In tumor cells, Hsp90 is present in a high‐affinity conformation in multichaperone complexes.19 Therefore, we analyzed the binding of K292‐acetylated Hsp90 to cochaperones and client proteins. As shown in Figure 3A, K292Q mutant showed increased affinity of Hsp90 with cochaperones Cdc37, HOP, and AHA1, whereas K292R mutant decreased the affinity. Similarly, K292Q mutant remarkably increased whereas K292R mutant compromised the binding of Hsp90 with kinases epidermal growth factor receptor (EGFR), Raf‐1, and CDK4, as well as the kinase downstream substrates p‐HSF1 and p‐eEF2K (Figure 3B). Therefore, the binding of Hsp90 with its client and cochaperone was significantly impacted by the acetylation at K292.
Figure 3.
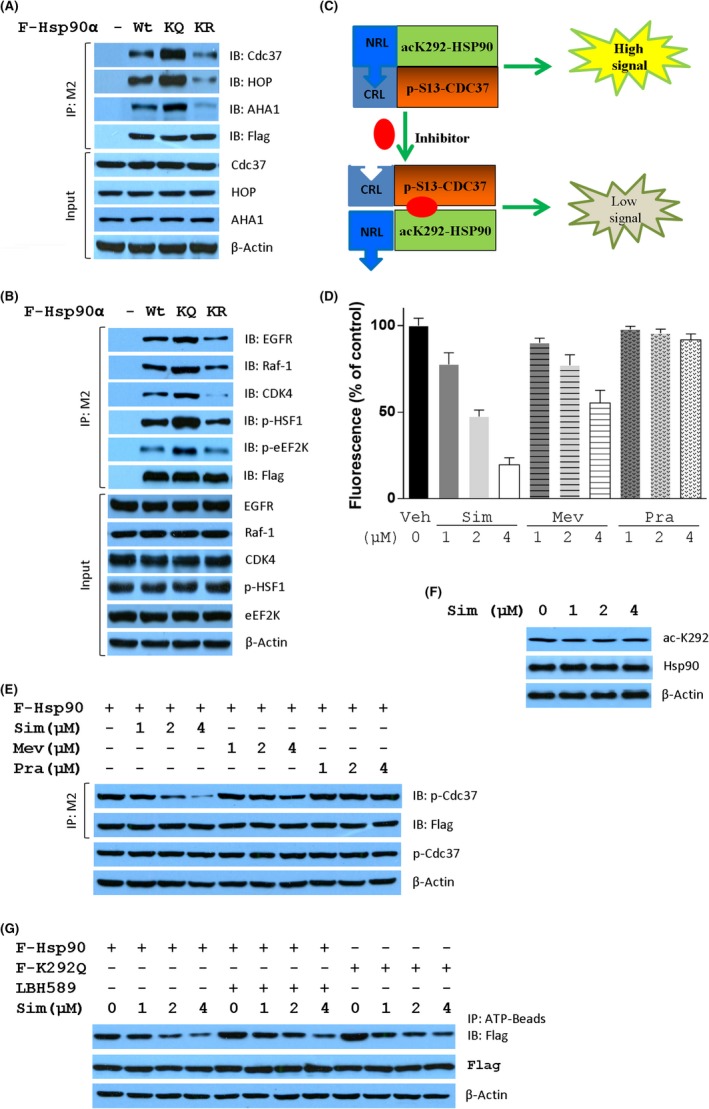
Simvastatin (Sim) is an inhibitor of heat shock protein 90 (Hsp90) acetylated at K292. Acetylation status at K292 influences the formation of Hsp90 chaperone machinery (A) and the interaction between Hsp90 and kinase clients (B). Cell lysates from MDA‐MB‐231 cells stably expressing Flag‐tagged Wt, K292Q (KQ), or K292R (KR) mutant of Hsp90α were immunoprecipitated (IP) with M2 beads followed by immunoblotting (IB) with the indicated antibodies. Protein expression levels in cell lysates were detected by western blotting as indicated. C, Schematic model for high throughput screening of Hsp90 inhibitor disrupting the formation of K292‐acetylated Hsp90/p‐S13 Cdc37 machinery. Complementation‐based bioluminescence assay was used to identify small molecules interrupting the interaction of K292Q Hsp90 with S13D Cdc37. D, Distinct inhibitory activity of three structure‐related statins on the interaction between K292Q Hsp90 and S13D Cdc37. E, Inhibitory effects of three statins on the interaction between Hsp90α and p‐S13 Cdc37 in MDA‐MB‐231 cells stably expressing Flag‐tagged Hsp90α. F, Sim did not affect the acetylation at K292 of Hsp90 in MDA‐MB‐231 cells. G, Inhibitory effect of Sim on the interaction between Wt, LBH589‐treated Wt, or K292Q mutant of Hsp90α and ATP. AHA1, Activator of Hsp90 ATPase protein 1; CDK4, cyclin‐dependent kinase 4; CRL, C‐terminal of Renilla luciferase; EGFR, epidermal growth factor receptor; HOP, Hsp70‐Hsp90 organizing protein 1; Mev, mevastatin; NRL, N‐terminal of Renilla luciferase; p‐eEF2K, S366 phosphorylated Eukaryotic elongation factor 2 kinase; p‐HSF1, S326 phosphorylated Heat shock factor 1; Pra, pravastatin; Veh, vehicle
Among Hsp90 cochaperones, activation of protein kinase clients is mediated by Hsp90‐Cdc37 machinery facilitating the phosphorylation of kinase substrates.20, 21 The phosphorylation at Ser13 is essential for Cdc37 to modulate Hsp90‐mediated kinase folding. This prompted us to establish a monitoring platform to find agents that have potential efficacy in targeting K292‐acetylated Hsp90. Thus, an SRL‐PFAC assay22 was generated (Figure 3C) and 8 hits (Table 1) from the LOPAC library (Sigma‐Aldrich) of 1280 pharmacologically active compounds were identified to disrupt K292‐acetylation mimic Hsp90 (K292Q)/S13 phosphorylation mimic Cdc37 (S13D) interaction in MDA‐MB‐231 cells. Except cisplatin, the most promising was Mev, an HMGCR reductase inhibitor, that shows synergistic anticancer effects with LBH589.16 Interestingly, Sim, an analog of Mev, showed a stronger ability to disrupt the complemented RL activity than Mev in a dose‐dependent manner, whereas pravastatin did not show any effect (Figure 3D). Similar effects have been observed on the interaction between Hsp90 and p‐Cdc37 (Figure 3E), indicating that the disruption of Hsp90/p‐Cdc37 by Sim would be independent of the mevalonate signaling pathway.
Table 1.
Eight hits from 1280 pharmacologically active compounds were found to disrupt the formation of the K292Q‐Hsp90/S13D‐Cdc37 complex in MDA‐MB‐231 cells
| Inhibition (%) | Compound (10 μmol/L) |
|---|---|
| 54 | Bosutinib |
| 97 | Cisplatin |
| 92 | Simvastatin |
| 80 | Carmofur |
| 58 | Nisoldipine |
| 61 | Mevastatin |
| 89 | Phenoxybenzamine HCl |
| 54 | Rolitetracycline |
To explore whether Sim had any effect on Hsp90 binding with ATP, Wt, and LBH589‐treated or K292Q mutant of Hsp90 were analyzed for their affinity with ATP. As shown in Figure 3F, Sim did not attenuate the basal level of acetylation at K292, whereas it dramatically decreased the affinity of Hsp90 to ATP (Figure 3G), indicating that Sim can interfere with Hsp90 binding with ATP. The phenomenon that low concentration of LBH589 increased the binding of Hsp90 with ATP might be ascribed to its effect on Hsp90 acetylation, and was further proven by the observation that K292Q mutant showed increased affinity with ATP. Simvastatin compromised the binding of Hsp90 with ATP but less in the case of either LBH589‐induced acetylation or K292Q mutation in a dose‐dependent manner. Therefore, Sim could be an inhibitor targeting K292‐acetylated Hsp90.
3.4. K292‐acetylated Hsp90 is a direct target of Sim
Owing to the dissociation of Hsp90 from Cdc37 by Sim, we next tested whether Sim could inhibit Hsp90 function and induce the degradation of its client proteins in TNBC cells. As expected, Sim time‐dependently decreased the expression of Raf‐1, Cyclin D1, and CDK4, as well as AKT, HSF1, and their phosphorylated forms p‐AKT and p‐HSF1, produced by protein kinase in both MDA‐MB‐231 (Figure 4A) and MDA‐MB‐468 (Figure 4B) cell lines. The expression of Hsp90 clients, such as AKT, HSF1, ERK, and MEK, as well as their phosphorylated forms, were also measured in both MDA‐MB‐231 (Figure 4C) and MDA‐MB‐468 (Figure 4D) cell lines treated with various concentrations of Sim. Interestingly, Simvastatin slightly decreased the expression of AKT, HSF1, ERK, and MEK, but decreased remarkably their phosphorylated forms as well as Raf‐1, TGFβR2, and PDGFRβ, which are involved in the regulatory pathways of tumor development. However, Sim had neither time‐dependent (Figure S1) nor dose‐dependent (Figure S2) effects on the expression of heat shock proteins, including Hsp27, Hsp40, Hsp70, and Hsp90, nor the cochaperone of Hsp90, Cdc37, or phosphorylated Cdc37. Therefore, Sim might provide a new therapeutic scheme for cancer by downregulating multiple oncogenic proteins simultaneously through proteasomal degradation. These data suggest that Sim stimulates the degradation of Hsp90 clients without heat shock response, indicating that Sim is different from ATP‐competing Hsp90 inhibitors.
Figure 4.
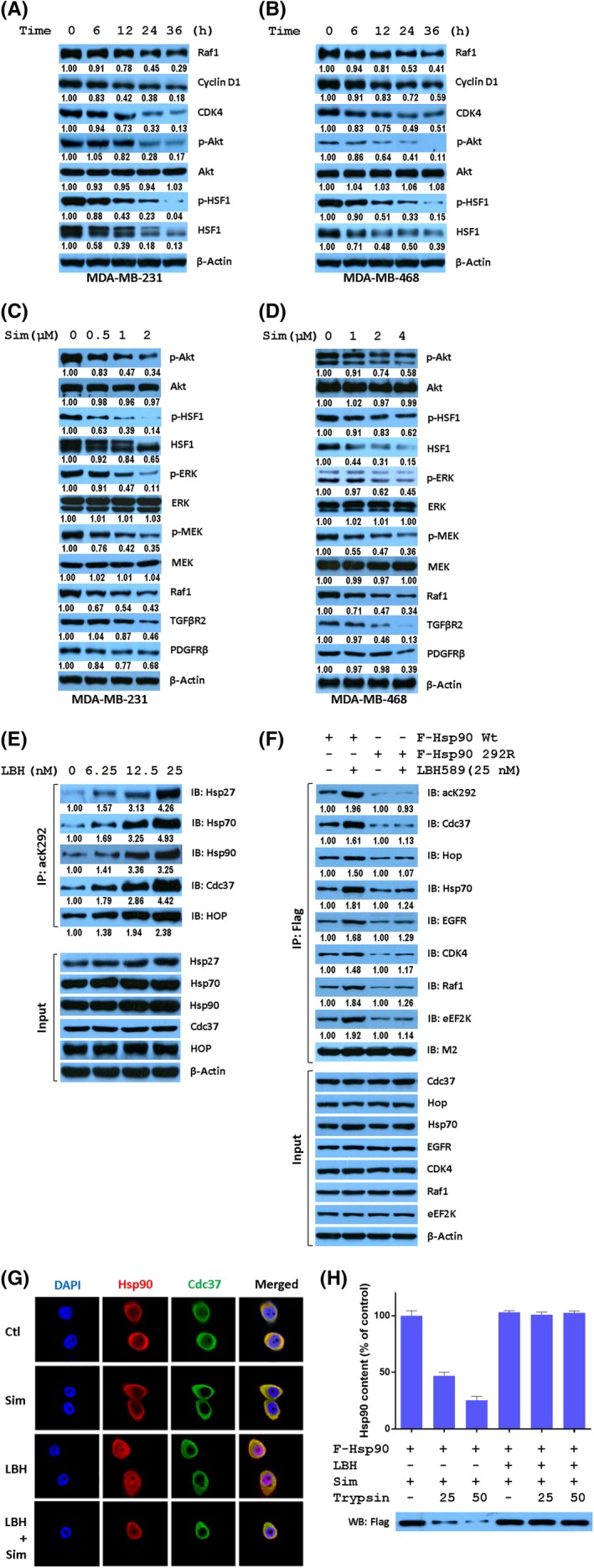
K292‐acetylated heat shock protein 90α (Hsp90α) is a direct target of simvastatin (Sim). Expression of Hsp90 clients was time‐dependently decreased in MDA‐MB‐231 cells treated with 2 μmol/L Sim (A) or MDA‐MB‐468 cells treated with 8 μmol/L Sim (B) for indicated times. Expression of Hsp90 clients in MDA‐MB‐231 (C) or MDA‐MB‐468 (D) cells were dose‐dependently decreased by treatment at indicated concentrations of Sim for 24 hours. E, LBH589 (LBH) enhanced the complex formation of K292‐acetylated Hsp90α with its cochaperones in a dose‐dependent manner. Cell lysates from MDA‐MB‐231 cells treated as indicated for 24 h were immunoprecipitated (IP) using acK292 antibody followed by immunoblotting (IB) with indicated antibodies. F, Acetylation status at K292 is the determinant for the complex formation of acetylated Hsp90 with its cochaperones and kinase clients. MDA‐MB‐231 cells were transfected with Wt or K292R mutant of Hsp90 followed by the treatment as indicated for 24 h. Cell lysates were immunoprecipitated with M2 beads, and the coprecipitated proteins were detected by western blotting. G, Sim interfered with the colocalization of Hsp90 and Cdc37 in MDA‐MB‐468 cells. Cells were incubated with monoclonal anti‐Hsp90 antibody and polyclonal andti‐Cdc37 antibody, and then with fluorescent antibodies. H, Sim binds preferentially with K292‐acetylated Hsp90α. Cell lysates from MDA‐MB‐231 cells stably expressing Flag‐tagged Hsp90α treated with or without 25 nmol/L LBH589 for 24 h were immunoprecipitated with acK292 antibody or M2 beads. Beads were washed, and incubated with 8 μmol/L Sim for 2 hours followed by digestion with indicated concentrations of trypsin for 5 min, and Hsp90α was detected by western blotting (WB). CDK4, cyclin‐dependent kinase 4; Ctl, control; eEF2K, Eukaryotic elongation factor 2 kinase; EGFR, epidermal growth factor receptor; HOP, Hsp70‐Hsp90 organizing protein 1; PDGFRβ, Platelet‐derived growth factor receptor β; p‐HSF1,S326 phosphorylated Heat shock factor 1; TGFβR2, TGF‐β receptor type 2
Previous studies indicated that acetylation of Hsp90 alters the interaction between Hsp90 and its co‐chaperone or clients.6, 7 We observed a progressive increase in K292‐acetylated Hsp90 and its affinity with Hsp27, Hsp70, as well as Hsp90 cochaperone Cdc37 and HOP with increasing concentration of LBH589 in a dose‐dependent manner (Figure 4E). This result indicates the significance of acetylation at K292 for Hsp90 chaperone function. Importantly, LBH589 increased the acetylation at K292 and the affinity of Hsp90 with its cochaperone Cdc37, HOP, Hsp70, and kinase clients EGFR, CDK4, Raf1, and eEF2K, whereas K292 acetylation‐deficient Hsp90 did not respond to LBH589 treatment (Figure 4F), suggesting that the binding of Hsp90 to its cochaperone and clients depends on acetylated K292. Cdc37 is the most noteworthy cochaperone for its role in the maturation and function of oncogenic kinases. To explore the explicit effect of Sim and LBH589 on Hsp90 function, we tested whether these agents disrupt the Hsp90‐Cdc37 interaction in MDA‐MB‐231 cells. Simvastatin was observed by confocal microscopy to ablate the codistribution signal (yellow) of Hsp90 (red) and Cdc37 (green) compared with LBH589 treatment alone (Figure 4G).
To investigate whether K292‐acetylated Hsp90 is a direct target of Sim, K292‐acetylated and nonacetylated Hsp90 were purified with antibodies against acK292 and Hsp90, respectively, from Flag‐tagged Hsp90‐expressing HEK293T cells treated with or without 100 nmol/L LBH589 for 24 hours. Equal amounts of purified Hsp90 were mixed with Sim for 2 hours followed by digestion with indicated amounts of trypsin. K292‐acetylated Hsp90 complex with Sim was more stable against the digestion with trypsin than K292 nonacetylated Hsp90 complex with Sim (Figure 4H), indicating that K292‐acetylated Hsp90 is a direct target of Sim.
3.5. Simvastatin and LBH589 have a synergistic inhibition on tumor cell growth in vitro
Recently, preclinical data from different cancer cell lines and tumor xenograft models indicated that Hsp90 inhibitor shows synergistic effects in killing cancer cells when combined with LBH589.16, 23 We hypothesize that Sim could be an ideal drug in the synergistic inhibition of TNBC with LBH589. The antiproliferative effect of cells treated by increasing amount of Sim without (Figure 5A) or with 25 nmol/L LBH589 (Figure 5B) was determined using the CCK‐8 assay. LBH589 showed significantly high cytotoxicity against all TNBC cells detected, whereas Sim acted as a weak antiproliferative agent when used alone (Table S1). To further evaluate the combinatory effect of Sim and LBH589, the effects of half‐diluted Sim supplemented with 25 nmol/L LBH589 on cell proliferation, were examined. When used together, Sim significantly inhibited cell growth compared with each single treatment. The IC50 of Sim under 25 nmol/L LBH589 was increased by 6‐ to 16‐fold compared with Sim alone (Table S1). The CI for proliferation and viability was also measured by using simultaneously half‐diluted Sim and/or LBH589, showing significant synergistic effects in all TNBC cell lines (Table S2).
Figure 5.
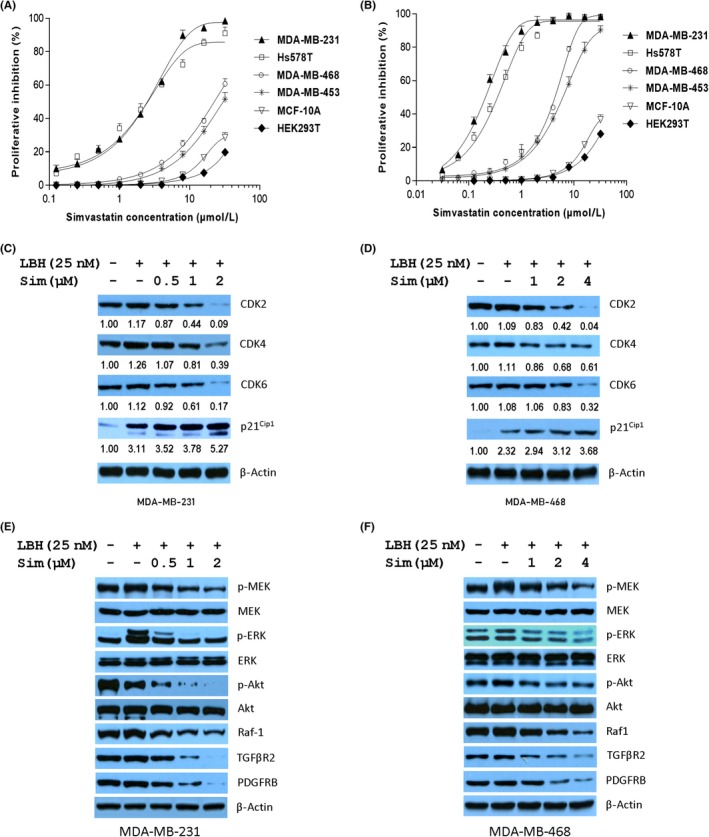
LBH589 (LBH) enhances the inhibitory effects of simvastatin (Sim) on heat shock protein 90 (Hsp90) chaperone activity. Cell proliferation were assessed for triple negative breast cancer cells treated with Sim (A) or Sim plus 25 nmol/L LBH (B) for 72 hours using CCK‐8 reagents. MDA‐MB‐231 cells were treated as indicated for 24 hours followed by western blotting for Hsp90 client proteins associated with cell cycle (C) or protein kinase clients (E), and similarly, Hsp90 client proteins associated with cell cycle (D) or protein kinase clients (F) in MDA‐MB‐468 cells. CDK, cyclin‐dependent kinase; PDGFRβ, Platelet‐derived growth factor receptor β; p‐HSF1, Heat shock factor protein 1; TGFβR2,TGF‐β receptor type 2
To further confirm that the Hsp90 chaperone system could be impaired by Sim in cancer cells, the expression of protein kinase clients was analyzed by western blot. In the presence of LBH589, Sim inhibited the expression of CDK2/4/6, but dose‐dependently increased the expression of cell cycle kinase inhibitor p21cip1 in both cell lines, which could result in G1 cell cycle arrest (Figure 5C,D). Raf1 is one of the components of the MAPK/ERK pathway in cancer cells. Both TGFβR2 and PDGFRβ are members of the Ser/Thr protein kinases involved in the activation of signaling from Raf to MEK to MAPK in the protein kinase cascade. Thus, the combination of LBH589 and Sim could inhibit cellular survival and the MAP/ERK pathway. We observed that Sim induced a defect in phosphorylation of Akt, ERK, and MEK, but their expression levels remained unchanged in both cell lines treated with Sim in the presence of LBH589 (Figure 5E,F). Therefore, the combination of LBH589 and Sim diminishes the chaperone activity of K292‐acetylated Hsp90.
Next, we tested the alteration of acetylation at K292 in Hsp90 and the consequence of Hsp90 binding with its cochaperone and client proteins in both MDA‐MB‐231 and MDA‐MB‐468 cells treated with the combination of LBH589 and Sim. Treatment with LBH589 at 25 nmol/L for 12 hours, which showed no significant cell killing effects, remarkably induced the acetylation of Hsp90 at K292 and cochaperone complexes of Hsp90 with S13‐phosphorylated Cdc37, Cdc37, Hsp70, HOP, and p23, whereas Sim lessened the chaperone complex significantly with or without LBH589. In accordance with western blot results in Figures 3A and 4C, Sim blocked the formation of the K292‐acetylated Hsp90/cochaperone complex and, more strongly, the formation of the K292‐acetylated Hsp90/p‐Cdc37 complex (Figure 6A,B). Although LBH589 elevated the expression of Hsp27, Hsp70, and Hsp90 in a dose‐dependent manner (Figure 4E), neither Sim alone nor Sim with LBH589 had similar effects (Figures S2, S3). However, Sim dose‐dependently decreased the expression of protein kinase clients independent of LBH589 (Figures 4C,D, 5C–F). Interestingly, Sim impaired the binding of K292‐acetylated Hsp90 to kinase clients CDk2/4/6, Raf1, TGFβR2, and p‐HSF1 (Figure 6C,D) without obvious abrogation in the expression of protein kinase clients under combined treatment for 12 hours.
Figure 6.

Simvastatin (Sim) diminishes the chaperone activity of K292‐acetylated heat shock protein 90α (Hsp90α). Sim abrogated the complex formation of K292‐acetylated Hsp90 with Hsp90 cochaperones in MDA‐MB‐231 (A) and MDA‐MB‐468 (B) cells treated as indicated for 12 hours. Cell lysates were immunoprecipitated (IP) using acK292 antibody followed by immunoblotting (IB) with the indicated antibodies. Similarly, Sim abrogated the complex formation of K292‐acetylated Hsp90α with protein kinase clients in MDA‐MB‐231 (C) and MDA‐MB‐468 cells (D). E, Expression of Hsp90 clients regulated by combined treatment with Sim and LBH589 (LBH) is independent of the mevalonate (MEV) pathway. CDK, cyclin‐dependent kinase; Ctl, control; HOP, Hsp70‐Hsp90 organizing protein 1; HSF1, Heat shock factor 1; p‐HSF1, S326 phosphorylated Heat shock factor 1; TGFβR2, TGF‐β receptor type 2
To test whether the expression of Hsp90 clients regulated by the combined treatment is reliant upon the mevalonate pathway,16 mevalonate was used to test the quantitation of Hsp90 clients. As expected, mevalonate did not rescue the expression of Hsp90 clients decreased by Sim treatment for 24 hours (Figure 6E). Unlike after 12 hours of treatment, the expression of Hsp90 clients decreased by treatment with Sim with or without LBH589 for 24 hours. More importantly, STK33, required by mutant Kras‐dependent cancer cells,24 was also attenuated by Sim. These results indicated that Sim preferentially disrupts the K292‐acetylated Hsp90/Cdc37 complex independent of the mevalonate pathway.
3.6. Combination of Sim and LBH589 might be a potential strategy for TNBC therapy
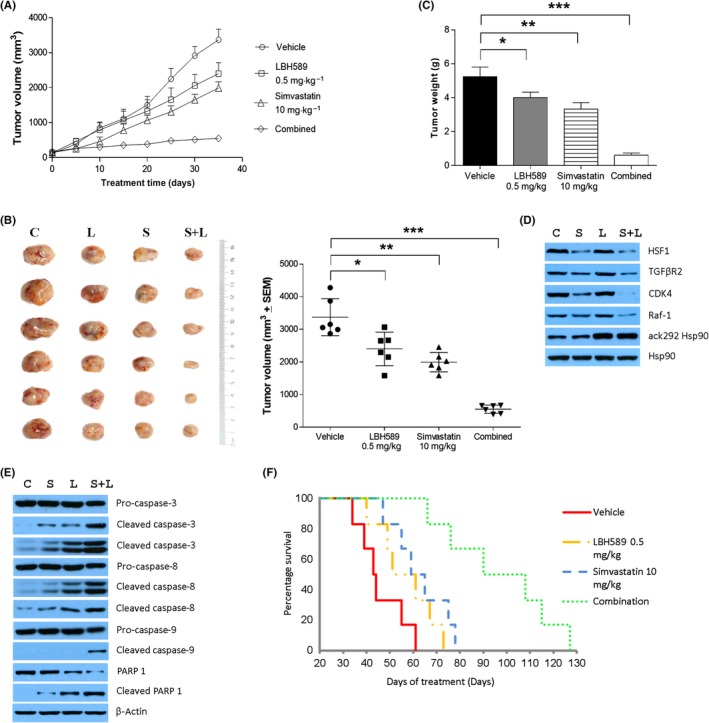
To assess the synergistic effect of Sim with LBH589 on the growth of MDA‐MB‐231 cells in vivo, xenografts of MDA‐MB‐231 cells were established in BALB/c nude mice. Tumor‐bearing animals were treated with either control vehicle, Sim (10 mg/kg, ig qd), LBH589 (0.5 mg/kg, ip qd) or both. The combination of Sim and LBH589 was significantly more efficacious in preventing tumor growth, compared with each treatment alone (Figure 7A). Animals in the combination group showed the slowest tumor growth, with final volume and weight, compared with that of control, LBH589, and Sim groups (Figure 7B,C). Overall, the results showed that the combination was significant superior compared to either individual treatment.
Figure 7.
Simvastatin and LBH589 have a synergistic inhibition on tumor growth in MDA‐MB‐231 cell xenografted mice. A, Tumor growth curves during treatment with vehicle (control; C), LBH589 (L), simvastatin (S), or S+L in xenografted mice. B, Photographs of sc tumors from indicated treatments in xenografted mice and the final distribution of tumor volumes. C, Final weight of tumors. D,E, Immunoblotting analysis of heat shock protein 90 (Hsp90) clients (D) and apoptotic proteins (E) in xenograft tumor tissues. F, Survival of mice was monitored. CDK, cyclin‐dependent kinase; HSF1, Heat shock factor 1; PARP, poly(ADP‐ribose) polymerase; TGFβR2, TGF‐β receptor type 2
Combined treatment with Sim and LBH589 decreased the expression of HSF1, TGFβR2, CDK4, and Raf‐1 (Figure 7D) but enhanced the cleavage of caspase‐3, caspase‐8, caspase‐9, and PARP, compared with vehicle control or either agent alone in tumor samples from xenograft mice (Figure 7E). The longevity of the post‐xenograft mice as an indicator of the tumorigenicity of the MDA‐MB‐231 cells was also examined. The median survival time for each group of mice illustrated that the combined treatment significantly prolonged the survival of MDA‐MB‐231 tumor‐bearing mice (Figure 7F, Table 2). These results strongly suggest that the combined treatment would be a potential strategy for TNBC patients.
Table 2.
Combined treatment with LBH589 and simvastatin (Sim) significantly prolonged the survival of MDA‐MB‐231 tumor‐bearing mice
| Group | Median survival time (MST, days) |
|---|---|
| Vehicle | 43.5 |
| LBH589 (0.5 mg/kg) | 56.0 |
| Sim (10 mg/kg) | 62.0* |
| Combination | 90.0*, *** |
* P < .05 compared with vehicle.
** P < .05 compared with vehicle.
*** P < .05 compared with Sim 10 mg/kg.
4. DISCUSSION
Triple‐negative breast cancer, lacking estrogen receptor, progesterone receptor, or human epidermal growth factor receptor 2 genes, poses a significant clinic challenge with particularly unfavorable prognoses due to the absence of defined molecular targets. Therefore, new therapeutic strategies are needed. In this study, we found that Sim in combination with LBH589 could specifically target TNBC both in vitro and in vivo, via disrupting the formation of K292‐acetylated Hsp90/Cdc37 chaperone machinery.
Hsp90 is required for the maturation and function of oncogenic client proteins in oncogenesis and malignant progression.24 Interruption of Hsp90 chaperone activity would lead to the proteasomal degradation of oncogenic client proteins.25 However, none of Hsp90 inhibitors have been approved for clinic treatment. Thus, a new approach disrupting Hsp90/Cdc37 complex may represent an effective strategy for cancer treatment.22, 26, 27, 28
HDAC inhibitors directly promote the acetylation of Hsp90, with destabilization of the ATP‐dependent active Hsp90‐client protein complex.5, 6 As a consequence, client proteins are driven to poly‐ubiquitination and proteasomal degradation. In this study, we show that Wt and K292Q mutant of Hsp90 stimulate proliferation and other malignant features significantly in MDA‐MB‐231 cells. However, K292R mutant of Hsp90 only obtained mild enhancement of malignant phenotypes, suggesting that Hsp90 is dominantly acetylated at K292 in MDA‐MB‐231 cells. We also validate this by quantifying Hsp90 and K292‐acetylated Hsp90 in paired human breast carcinoma and para‐carcinoma tissues (Figure 1C). Consequently, we conclude that significantly increased Hsp90 expression and the acetylation at K292 meet the stressed intracellular environment of cancer cells.
Unlike acetylation at K294,7 acetylation at K292 alone is sufficient to affect the affinity of Hsp90 to ATP (Figure 3F), co‐chaperone and client proteins (Figures 4F, 6A,B,C,D). Point mutation at K292 significantly impacts the binding of Hsp90 to clients and cochaperones, indicating the importance of acetylation at K292 for the formation of Hsp90/cochaperone or Hsp90/client protein complex. In addition, we verified that Sim is a promising Hsp90 inhibitor by specifically targeting K292‐acetylated Hsp90 to interfere multiple oncogenic pathways. Taken together, we identified a promising anticancer target, the K292‐acetylated Hsp90, and an attractive therapeutic strategy for TNBC, Sim with HDACi.
CONFLICT OF INTERESTS
The authors have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
Authors thank Dr. Joseph McDermott for proofreading of the manuscript. We appreciated assistance from Ms. Ping Yang from the Facility Core and Ms. Linlin Tao from the small animal laboratory, Fudan University School of Pharmacy. Y.Y received funding from the National Natural Science Foundation of China (Nos. 81272391 and 81572721).
Kou X, Jiang X, Liu H, et al. Simvastatin functions as a heat shock protein 90 inhibitor against triple‐negative breast cancer. Cancer Sci. 2018;109:3272–3284. 10.1111/cas.13748
REFERENCES
- 1. Taipale M, Krykbaeva I, Koeva M, et al. Quantitative analysis of HSP90‐client interactions reveals principles of substrate recognition. Cell. 2012;150(5):987‐1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shrestha L, Bolaender A, Patel HJ, Taldone T. Heat shock protein (HSP) drug discovery and development: targeting heat shock proteins in disease. Curr Top Med Chem. 2016;16(25):2753‐2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Karagoz GE, Rudiger SG. Hsp90 interaction with clients. Trends Biochem Sci. 2015;40(2):117‐125. [DOI] [PubMed] [Google Scholar]
- 4. Mollapour M, Neckers L. Post‐translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim Biophys Acta. 2012;1823(3):648‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bali P, Pranpat M, Bradner J, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280(29):26729‐26734. [DOI] [PubMed] [Google Scholar]
- 6. Yang Y, Rao R, Shen J, et al. Role of acetylation and extracellular location of heat shock protein 90alpha in tumor cell invasion. Cancer Res. 2008;68(12):4833‐4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scroggins BT, Robzyk K, Wang D, et al. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol Cell. 2007;25(1):151‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blows FM, Driver KE, Schmidt MK, et al. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: a collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med. 2010;7(5):e1000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zagouri F, Sergentanis TN, Chrysikos D, Papadimitriou CA, Dimopoulos MA, Psaltopoulou T. Hsp90 inhibitors in breast cancer: a systematic review. Breast. 2013;22(5):569‐578. [DOI] [PubMed] [Google Scholar]
- 10. Chiosis G, Dickey CA, Johnson JL. A global view of Hsp90 functions. Nat Struct Mol Biol. 2013;20(1):1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mehta PP, Whalen P, Baxi SM, Kung PP, Yamazaki S, Yin MJ. Effective targeting of triple‐negative breast cancer cells by PF‐4942847, a novel oral inhibitor of Hsp 90. Clin Cancer Res. 2011;17(16):5432‐5442. [DOI] [PubMed] [Google Scholar]
- 12. Buckley NE, D'Costa Z, Kaminska M, Mullan PB. S100A2 is a BRCA1/p63 coregulated tumour suppressor gene with roles in the regulation of mutant p53 stability. Cell Death Dis. 2014;5:e1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124(1):30‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gryder BE, Sodji QH, Oyelere AK. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med Chem. 2012;4(4):505‐524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang J, Zhong Q. Histone deacetylase inhibitors and cell death. Cell Mol Life Sci. 2014;71(20):3885‐3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin Z, Zhang Z, Jiang X, et al. Mevastatin blockade of autolysosome maturation stimulates LBH589‐induced cell death in triple‐negative breast cancer cells. Oncotarget. 2017;8(11):17833‐17848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee HS, Park SB, Kim SA, et al. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci Rep. 2017;7:41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stine JE, Guo H, Sheng X, et al. The HMG‐CoA reductase inhibitor, simvastatin, exhibits anti‐metastatic and anti‐tumorigenic effects in ovarian cancer. Oncotarget. 2016;7(1):946‐960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10(8):537‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Keramisanou D, Aboalroub A, Zhang Z, et al. Molecular mechanism of protein kinase recognition and sorting by the Hsp90 kinome‐specific cochaperone Cdc37. Mol Cell. 2016;62(2):260‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang T, Hamza A, Cao X, et al. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol Cancer Ther. 2008;7(1):162‐170. [DOI] [PubMed] [Google Scholar]
- 22. Zhang T, Li Y, Yu Y, Zou P, Jiang Y, Sun D. Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J Biol Chem. 2009;284(51):35381‐35389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. George P, Bali P, Annavarapu S, et al. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17‐AAG is highly active against human CML‐BC cells and AML cells with activating mutation of FLT‐3. Blood. 2005;105(4):1768‐1776. [DOI] [PubMed] [Google Scholar]
- 24. Azoitei N, Hoffmann CM, Ellegast JM, et al. Targeting of KRAS mutant tumors by HSP90 inhibitors involves degradation of STK33. J Exp Med. 2012;209(4):697‐711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garcia‐Carbonero R, Carnero A, Paz‐Ares L. Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol. 2013;14(9):e358‐e369. [DOI] [PubMed] [Google Scholar]
- 26. Joo JH, Dorsey FC, Joshi A, et al. Hsp90‐Cdc37 chaperone complex regulates Ulk1‐ and Atg13‐mediated mitophagy. Mol Cell. 2011;43(4):572‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li D, Xu T, Cao Y, et al. A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc Natl Acad Sci USA. 2015;112(16):5017‐5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Smith JR, Clarke PA, de Billy E, Workman P. Silencing the cochaperone CDC37 destabilizes kinase clients and sensitizes cancer cells to HSP90 inhibitors. Oncogene. 2009;28(2):157‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials