

Summary
When DNA is unwound during replication, it becomes overtwisted and forms positive supercoils in front of the translocating DNA polymerase. Unless removed or dissipated, this superhelical tension can impede replication elongation. Topoisomerases, including gyrase and topoisomerase IV in bacteria, are required to relax positive supercoils ahead of DNA polymerase, but may not be sufficient for replication. Here, we find that GapR, a chromosome structuring protein in Caulobacter crescentus, is required to complete DNA replication. GapR associates in vivo with positively supercoiled chromosomal DNA, and our biochemical and structural studies demonstrate that GapR forms a dimer-of-dimers that fully encircles overtwisted DNA. Further, we show that GapR stimulates gyrase and topo IV to relax positive supercoils, thereby enabling DNA replication. Analogous chromosome structuring proteins that locate to the overtwisted DNA in front of replication forks may be present in other organisms, similarly helping to recruit and stimulate topoisomerases during DNA replication.
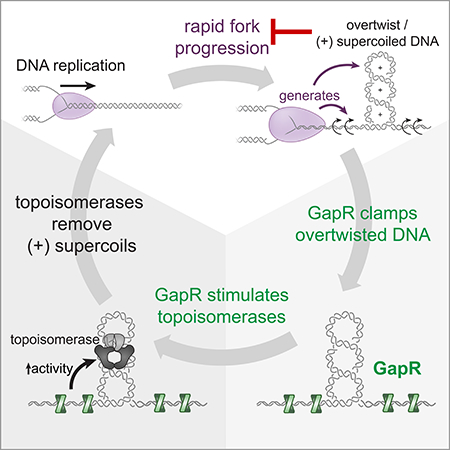
Graphical abstract
eTOC blurb
A bacterial protein that specifically recognizes and encircles overtwisted DNA is required to stimulate the activity of type II topoisomerases and enable DNA replication.
Introduction
In all organisms, chromosomes must be compacted ~1000-fold to fit within the confines of a cell. However, DNA cannot be haphazardly packed, and instead must be organized to facilitate a range of cellular processes, including DNA replication, transcription, and chromosome segregation. Chromosome compaction is achieved in large part by the supercoiling of DNA, forming a series of plectonemic loops in bacteria (Higgins, 2016). DNA can be either positively (+) or negatively (−) supercoiled, depending on whether the DNA duplex winds around a superhelical axis in a left- or right-handed fashion, respectively. The supercoiling of DNA is tightly controlled by topoisomerases, enzymes that can break and rejoin DNA segments to introduce or relieve superhelical strain (Vos et al., 2011). Despite major advances in our understanding of topoisomerases, the molecular mechanisms that regulate when, where, and how these enzymes act in vivo remain poorly understood.
Supercoils are also introduced by cellular processes that unwind the double helix, such as transcription and DNA replication. The ‘twin supercoiled domain’ model posits that, as RNA or DNA polymerase translocates along and unwinds the DNA, it produces overtwisted and (+) supercoiled DNA ahead of it, with underwound, (−) supercoiled DNA arising in its wake (Liu and Wang, 1978; Wu et al., 1988). These supercoils must be dissipated by topoisomerases to prevent buildup of torsional stress that would inhibit unwinding of the duplex and prevent translocation of RNA or DNA polymerase along the DNA (Postow et al., 2001). The action of topoisomerases is especially critical during DNA replication as ~50–100 (+) supercoils per second are generated ahead of the translocating replisome (Postow et al., 2001). Despite the importance of topoisomerases to replication, how the activities of these enzymes are controlled to promote replication elongation remains largely unknown.
Topoisomerases fall into two classes, type I and type II, which respectively cleave either one or two strands of DNA (Vos et al., 2011). Bacteria, which maintain their chromosomes with net-negative supercoil, typically have a type I topoisomerase, topo I, that can relieve excess (−) supercoils, and two type II topoisomerases, DNA gyrase and topo IV, that can relax (+) supercoils. Gyrase also introduces (−) supercoils into relaxed DNA, while topo IV helps decatenate replicated chromosomes. Because gyrase, and to a lesser extent topo IV, are found in nearly all bacteria but are absent from eukaryotes, these enzymes are targeted by a number of clinically-relevant antibiotics, such as the coumerins and quinolones (Vos et al., 2011). Bacteria treated with such drugs cannot replicate their DNA, underscoring the importance of topoisomerases to DNA replication (Khodursky et al., 2000).
Although gyrase, perhaps with topo IV, is often assumed to be sufficient to manage the (+) supercoils that arise during replication (Khodursky et al., 2000), a role for other factors cannot be ruled out. In particular, bacteria, which do not encode histones, harbor a bevy of so-called nucleoid-associated proteins (NAPs) that may influence topoisomerases. NAPs are a functionally heterogeneous set of proteins that are not conserved at the sequence level, but share many similarities such as small size, high basic amino-acid content, and DNA-binding ability (Badrinarayanan et al., 2015). NAPs typically bind DNA sequence non-specifically by recognizing DNA shape, e.g., narrow minor grooves, and many structure the chromosome by bending, wrapping, or bridging DNA, activities that can introduce or constrain supercoils. As NAPs are often highly abundant, their loss can lead to global changes in supercoiling (Lal et al., 2016; Weitao et al., 2000). Several NAPs in Escherichia coli, including SeqA and the larger chromosome structuring protein MukB, can bind to and stimulate topo IV to relax supercoils or decatenate interlinked circular DNA (Hayama and Marians, 2010; Kang et al., 2003; Li et al., 2010). However, to the best of our knowledge, no NAPs have been shown to stimulate gyrase to relax (+) supercoils and a direct role for NAPs in replication elongation has not been established.
Here, we demonstrate that a highly-conserved NAP from the α-proteobacterium Caulobacter crescentus called GapR is essential for DNA replication in fast growth conditions by promoting the ability of gyrase and topo IV to relieve the (+) supercoils that accumulate ahead of the replication fork. GapR was identified as an essential DNA-binding protein that binds AT-rich DNA and has pleiotropic effects when depleted (Arias-Cartin et al., 2017; Ricci et al., 2016; Taylor et al., 2017), but its molecular function and precise roles in the bacterial cell cycle have not been elucidated. Our studies show that a loss of GapR leads to global changes in superhelicity and an inability to complete DNA replication. Intriguingly, we find that, in addition to AT-rich DNA, GapR globally associates with the 3’ ends of highly-expressed genes, but only when they are actively transcribed. These results suggest that GapR recognizes and helps dissipate the (+) supercoils that arise during unwinding of the DNA duplex. Consistent with this model, we demonstrate that GapR preferentially binds overtwisted DNA and can stimulate gyrase and topo IV to remove (+) supercoils. Crystal structures of GapR reveal a unique fold with dimeric GapR that transitions, upon DNA binding, to form a dimer-ofdimers that completely encircles DNA. Strikingly, the structure shows that the GapR clamp specifically recognizes the shape of overtwisted DNA, and cannot accommodate B-form DNA, supporting a model in which GapR specifically localizes to the overtwisted DNA ahead of replication forks where it can stimulate type II topoisomerases.
Results
Identification of the nucleoid-associated protein, GapR
Despite their importance in chromosome organization, most NAPs are not broadly conserved. To identify new NAPs in Caulobacter, we isolated intact nucleoids using sucrose gradient centrifugation and performed mass spectrometry on the proteins that remained tightly associated (Fig. S1A). After sedimentation, fractions were DAPI stained to identify the nucleoid fraction and the slower migrating cytoplasmic fraction, which contains RNA and highly sheared DNA (Fig. S1A). We verified by immunoblotting that a known NAP, integration host factor (IHF), was stably associated with the nucleoid, while DNA binding proteins with higher dissociation rates, such as the transcription factor CtrA, were found in the cytoplasmic fraction (Fig. 1A). Mass spectrometry identified multiple, independent peptides of known NAPs, including HU and IHF, and other tight DNA-binding proteins, including subunits of RNA polymerase (RNAP) and single-stranded DNA binding protein; no known canonical transcription factors were recovered (Table S1).
Figure 1. Identification of GapR and phenotypic characterization of GapR-depleted cells.

(A) Immunoblots of fractions from a nucleoid isolation experiment with antibodies for IHF, CtrA, FLAG, and RpoA.
(B) Schematic of predicted GapR domains.
(C) Growth of the ΔgapR Pxyl-gapR depletion strain assessed by 10-fold serial dilutions on plates containing xylose (gapR induced) or glucose (gapR repressed).
(D) Immunoblots of GapR in wild-type and depletion strains grown in xylose or in glucose for 6 hr. * = non-specific band used as a loading control.
(E) Phase-contrast images and flow cytometry analysis of chromosome content of wild-type and ΔgapR Pxyl-gapR cells in xylose and at the indicated times in glucose. Arrows = sites of incomplete constriction.
(F) Gene expression in GapR-depleted (+glu 6 hr) versus wild-type cells in glucose (+glu). Dashes indicate 2-fold changes in expression.
(G) GapR-depleted cells induce a DNA damage response. Expression changes of LexA regulated genes (left), including recA (right) in GapR-reduced (+xyl) and GapR-depleted (+glu) cells compared to cells treated with novobiocin, DNA damaging agents, the toxin SocB, ethanol stress, or cell-cycle arrested by SciP depletion (left).
(H) Supercoiling-sensitive genes change in GapR-depleted cells. As in (G), except for genes affected > 4-fold by novobiocin treatment. Pearson correlation coefficient (r) and p-value for comparison to novobiocin treatment.
To identify NAPs, we examined our mass spectrometry data for proteins with no annotated function but with properties common to many NAPs: (i) small (< 25 kDa), (ii) a basic region to promote tight DNA binding, (iii) high expression during exponential growth and (iv) conservation across α-proteobacteria. One candidate was an 89 amino-acid protein encoded by the highly-conserved gene gapR (CCNA_03428, Fig. 1B, S1B). We confirmed that GapR was nucleoid-associated by repeating the nucleoid purification on cells expressing GapR-3×FLAG from its native locus. Epitope-tagged GapR was highly enriched in the nucleoid fraction and not detectable in the cytoplasmic fraction, in contrast to α-subunit of RNAP which was present in both fractions (Fig. 1A), suggesting that GapR binds the nucleoid stably and with high affinity.
GapR was also identified in searches for essential Caulobacter DNA-binding proteins (Arias-Cartin et al., 2017; Ricci et al., 2016) and origin-binding proteins (Taylor et al., 2017). These studies suggested that GapR is an essential and highly expressed protein that has pleiotropic effects when depleted, but the molecular function of GapR was not determined.
Depleting GapR leads to cellular filamentation and DNA replication defects
To understand the essential function of GapR, we constructed a depletion strain in which gapR is driven by a glucose-repressible, xylose-inducible promoter on a low-copy plasmid (ΔgapR Pxyl-gapR). Even in the presence of xylose, this strain has reduced levels (~15-fold) of GapR relative to the wild type. The strain remains viable in xylose, but has a significant growth defect (Fig. 1C-D, S1C-D). Further inhibiting gapR expression by shifting cells to a medium with glucose, led to undetectable levels of GapR protein and a five-log defect in plating viability. The GapR depletion strain was not able to grow in glucose and exhibited cell-division defects, becoming highly filamentous, often with multiple, incomplete constriction sites after a shift to a rich medium containing glucose (Fig. 1E). Flow cytometry analysis of chromosome content indicated that most GapR-depleted cells did not accumulate chromosomes despite becoming filamentous (Fig. 1E). Instead, the culture was enriched for G1 cells suggesting that DNA replication is disrupted at the level of initiation and/or elongation.
We used RNA-seq to analyze changes in gene expression after 6 hr of GapR depletion in glucose relative to wild-type cells grown in glucose. We identified 1245 genes that changed expression at least 2-fold (~32% of all genes; 996 genes upregulated and 249 genes downregulated; Fig. 1F) indicating that GapR has broad, pervasive effects on gene expression (Arias-Cartin et al., 2017; Ricci et al., 2016), including a general stress response, indicated by a correlation with the expression changes seen in cells treated with ethanol (Pearson’s r=0.72, p=1.2×10−67; Fig. S1E). We observed similar, but more modest, changes when GapR levels were reduced in xylose (Fig. S1E, Table S2). The GapR-depletion strain in glucose also exhibited a DNA damage response (Arias-Cartin et al., 2017), with expression changes similar to cells treated with DNA damaging agents or expressing the toxin SocB, which leads to replication fork collapse (Pearson’s r=0.57, p=1.6×10−6, Fig. 1G). A DNA damage response was not observed when cells were challenged by ethanol or subjected to cell-cycle disruption by depleting SciP.
GapR binding is not correlated with GapR-dependent changes in gene expression
To gain more insight into GapR function, we analyzed its global DNA binding profile by ChIP-seq using a strain expressing GapR-3×FLAG as the sole copy of GapR from its native locus (Fig. S1F). This profile revealed widespread GapR-3×FLAG binding across the genome (Fig. 2A). Using the 34 highest peaks, we identified two highly enriched motifs, each consisting almost exclusively of A and T, whereas the Caulobacter genome overall is 68% GC, suggesting that GapR preferentially binds AT-rich DNA (Fig. 2B-D). These data agree with previous studies indicating that AT-content of the DNA is one determinant of GapR binding (Arias-Cartin et al., 2017; Ricci et al., 2016). However, there are important exceptions to this trend not previously noted that are discussed below.
Figure 2. GapR binds AT-rich genomic regions and the 3’ ends of highly-expressed genes.

(A) ChIP-seq profiles of GapR-3×FLAG (orange) and wild-type cells (black) using an anti-FLAG antibody.
(B) Motifs enriched in GapR-bound regions.
(C) GapR ChIP-seq versus AT content across the genome calculated using a 100 bp window centered at each position. Mean enrichment at a given % AT (red dots).
(D) AT content (top) and ChIP-seq (bottom) of untreated (orange) and rifampicin-treated (pink) GapR-3×FLAG cells. AT content below the genomic average (32%) is plotted in reverse.
(E) Average GapR ChIP-seq at a given ORF versus change in gene expression in GapR-depleted cells. Shaded area indicates < 2-fold change upon GapR depletion.
(F) GapR accumulates at the end of highly-expressed operons. GapR-3×FLAG (orange), GapR3×FLAG post rifampicin treatment (pink), and RNAP (black) ChIP-seq profiles are shown for a region containing two highly-expressed tRNA. AT content (top) and positions of annotated genes (bottom, tRNA genes in solid black).
(G) GapR accumulates at the 3’ end of highly-expressed operons. Normalized change in GapR ChIP signal was computed from the difference in enrichment (untreated minus rifampicin-treated cells in a 1-kb window) before and after the 3’ ends (left) or 5’ ends (right) of transcription units.
See also Figure S2.
To assess whether GapR directly affects gene expression, we computed the average GapR enrichment within each open reading frame (Fig. 2E) or at each promoter (Fig. S2A) in the genome and plotted these values against the expression changes in GapR-depleted cells. There was no correlation, suggesting that GapR does not directly affect gene expression.
GapR may associate with (+) supercoils at the 3’ end of highly-expressed genes and operons
How, then, does GapR affect global patterns of transcription? Because some NAPs influence the supercoiling state of cells and consequently alter the expression of supercoiling-sensitive genes, we compared the expression profile of cells depleted of GapR in glucose to that of cells treated with novobiocin, an inhibitor of gyrase and, to a reduced extent, topo IV (Table S2). For the 289 genes that changed at least 4-fold in novobiocin-treated cells, there was a strong correlation with the changes observed in GapR-depleted cells (Pearson’s r=0.61, p=5.0×10−31), although the magnitudes of the changes due to GapR-depletion were typically smaller (Fig. 1H). The expression profiles of cells expressing SocB toxin, treated with ethanol, or cell-cycle arrested by SciP depletion did not similarly correlate with the novobiocin response, suggesting that the changes in supercoiling-sensitive genes following GapR depletion are not just a result of general stress or a disruption of the cell cycle (Fig. 1H). The change in supercoiling-sensitive genes suggested to us that GapR-depleted cells may have reduced (−) supercoiling. Consistent with this conclusion, we found that the net (−) superhelicity of chromosomes from GapR-depleted cells was reduced relative to that of wild-type cells, but higher than that of novobiocin-treated cells (Fig. S1G).
Interestingly, we noticed that GapR accumulated in genomic regions that were not AT-rich, but instead corresponded to the 3’ ends of highly-expressed transcription units such as tRNAs (Fig. 2F; S2B). At these locations, the binding of GapR was broad (spanning several kb) and lower in magnitude compared to the AT-rich locations noted above (Fig. 2B-D). GapR binding in these regions was dependent on active transcription, as it was reduced by the addition of the transcriptional inhibitor rifampicin (Fig. 2F; S2B). This transcription-dependent accumulation of GapR was strongly dependent on the expression level of the transcription unit (two-sided t-test: p=1.8×10−33) and was specific to the 3’ ends of transcription units (Fig. 2G, S2D-E). In contrast, the binding of GapR at ATrich genomic regions was not affected by rifampicin (Fig. 2D, S2C). Although it is possible that GapR helps terminate transcription, like E. coli H-NS (Ray-Soni et al., 2016), our RNA-seq analysis of GapR-depleted cells did not reveal any transcriptional read-through (Fig. S2B).
Why would GapR associate with the 3’ ends of highly-expressed operons? Unwinding of the DNA duplex during transcription leads to the overtwisting of DNA and the accumulation of (+) supercoils ahead of RNAP. Thus, GapR may associate preferentially with the DNA structures that arise at the ends of highly-expressed transcription units. Further, we hypothesized that GapR may help eliminate or dissipate (+) supercoils as GapR-depleted cells exhibited decreased (−) superhelicity (Fig. 1H, S1G). If GapR indeed helps relieve (+) supercoil stress, then depleting GapR should disrupt DNA replication, as the unwinding of DNA during replication leads to a significant accumulation of (+) supercoils ahead of replication forks.
GapR is required for proper DNA replication initiation and elongation
To assess the role of GapR in DNA replication, we used flow cytometry to measure DNA content in synchronized, GapR-depleted cells over time (Fig. 3A). The GapR depletion strain was grown in glucose for 2 hr to deplete GapR, synchronized to isolate cells in G1, and released into fresh media with glucose to continue repressing gapR expression. After release, DNA content was monitored by flow cytometry (Fig. 3B). For the wild type, nearly all cells initiated replication by ~40 min postsynchronization, with cell division occurring within 100 min, indicating that replication of the entire chromosome took < 60 min (Fig. 3B-C). In contrast, GapR-depleted cells showed significant defects in both initiation and elongation. Only 20% of cells had initiated within 40 min post-synchronization and only ~60% had initiated after 120 min (Fig. 3C). The rate of replication elongation was also slowed significantly (Fig. 3D), with few cells achieving 2N content (Fig. 3B). We also examined the GapR depletion strain under xylose-inducing conditions, which showed more modest but significant defects in replication initiation and elongation rate (Fig. S3A-D).
Figure 3. Replication initiation and elongation are strongly impaired in GapR-depleted cells.

(A) Assay to assess DNA replication. Wild-type and ΔgapR Pxyl-gapR cells were grown for 2 hr in glucose, synchronized in G1, and released into medium containing glucose. DNA content was measured by SYTOX staining and flow cytometry or DNA sequencing.
(B) Flow cytometry time courses for wild-type and GapR-depleted cells. Blue lines represent 1N and 2N DNA content.
(C) Fraction of cells from (B) that initiated replication, with DNA content > 1N used as a proxy for initiation. Data are mean ± SEM, n ≥ 2.
(D) Replication rates for cells in (B), calculated from a linear fit of the DNA content of cells, normalized to the wild type. Data are mean ± SEM, n ≥ 2.
(E) DNA sequencing of wild-type cells grown in glucose and ΔgapR Pxyl-gapR cells grown for 2 hr in glucose. DNA content, normalized to synchronized G1 cells, is shown as a function of chromosome position using LOWESS smoothing. Blue lines = replication origin and terminus. Cartoon of wildtype fork progression is shown (right).
(F) Replication initiation, based on origin DNA content over time, using DNA sequencing data in (E).
(G) Rates of replication calculated from a linear fit of fork position (from E) in the indicated time ranges.
We then used DNA sequencing to more precisely assess the replication defects of synchronized wildtype and GapR-depleted cells. Replicated regions of the chromosome are easily detected and tracked over time as they have ~2-fold more reads than unreplicated regions (Fig. 3E). Using this assay, we found that nearly all wild-type cells (~90%) initiated replication ~20 min post-synchronization (Fig. 3F), with replication progressing steadily toward the terminus at a rate of ~32–34 kb/min (Fig. 3G), consistent with prior estimates (Jensen, 2006). In contrast, for GapR-depleted cells, <20% of cells had initiated after 20 min, and only ~35% had initiated by 60 min post-release (Fig. 3F). Additionally, the replication forks of GapR-depleted cells showed a progressive slowdown over time, with a rate of only 12 kb/min between 60 and 80 min, and with no cells having finished replication by the 80 min time point when all wild-type cells had completed. We also performed this experiment on the GapR depletion strain shifted to glucose only upon synchronization rather than for 2 hr before synchronization, again observing delays in replication initiation and a progressive decrease in elongation rate (Fig. 3F-G, S3E).
Together, our flow cytometry and DNA-sequencing data suggested that GapR-depleted cells are not competent to finish replication. We directly tested this model by: (i) Monitoring replication by flow cytometry over an extended (4 hr) period. Whereas wild-type cells replicated twice during this period, GapR-depleted cells accumulated intermediate (<2N) chromosome content (Fig. S4A). (ii) If any GapR-depleted cells finish replication, they should constrict and divide; while wild-type cells divided by 120 min, GapR-depleted cells did not divide and became progressively more filamentous over time (Fig. S4B). (iii) The origin-terminus ratio in GapR-depleted cells remained constant after ~60 min (Fig. S4C), indicating that replication initiated but did not complete. Taken together, our results demonstrate that GapR is required for the timely and successful completion of DNA replication.
GapR depleted cells are sensitized to inhibitors of type II topoisomerases
Because our ChIP experiments suggested that GapR associates with (+) supercoiled DNA, we hypothesized that GapR promotes DNA replication by helping to alleviate the (+) supercoils that accumulate ahead of replication forks. This model predicts that cells depleted of GapR should be sensitive to sub-lethal doses of antibiotics that inhibit gyrase and topo IV, the enzymes that remove (+) supercoils. To test this, we measured the plating viability of the GapR depletion strain grown in xylose to reduce GapR levels (Fig. 1C) after treatment with sub-lethal levels of novobiocin, an antibiotic that inhibits gyrase and topo IV. Notably, the GapR depletion strain exhibited nearly 5-logs lower plating viability than wild type in novobiocin (Fig. 4A). The GapR depletion strain was also sensitized to rifampicin and hydroxyurea (Fig. S4D), which also inhibit DNA replication. However, the GapR depletion strain was not more sensitive than the wild type to low doses of mitomycin C, which directly damages DNA, or chloramphenicol, which inhibits translation (Fig. 4A). The antibiotic sensitivities of the GapR depletion strain contrast with that of ΔrecA cells, which had nearly identical plating viability as the wild type on novobiocin and chloramphenicol, but was highly sensitized to mitomycin C. Thus, GapR is likely not required to repair DNA damage or for a general stress response. Instead, these results support a model in which GapR promotes DNA replication by relieving (+) supercoils in front of the elongating replisome.
Figure 4. GapR-depleted cells are sensitized to inhibition of type II topoisomerase activity.

(A) 10-fold serial dilutions of wild-type, ΔgapR Pxyl-gapR + xylose, and ΔrecA cells on plates containing xylose and 0.1 μg/mL novobiocin, mitomycin C, or chloramphenicol.
(B) Wild-type and ΔgapR Pxyl-gapR cells were grown in xylose, synchronized in G1, and released into medium containing xylose. After 20 min, 2.5 μg/mL novobiocin was added (red *). DNA content was measured with flow cytometry. Blue lines represent 1N and 2N DNA content.
(C) DNA content of cells from (B). * = time of novobiocin addition. The fraction of cells with DNA content > 1N was used as a proxy for initiation. Data are mean ± SEM, n ≥ 2.
(D) Relative replication rate of cells from (B), calculated from a linear fit of the DNA content of cells, normalized to the wild type. Data are mean ± SEM, n ≥ 2.
See also Figure S4.
To test whether the synergistic effects of reduced GapR and novobiocin treatment on viability result from defects in DNA replication elongation, we used flow cytometry to monitor DNA content in synchronous populations of wild-type and GapR-depleted cells in the presence of novobiocin. Whereas wild-type cells continued to replicate at a slightly reduced rate in novobiocin, cells with reduced levels of GapR showed very little accumulation of DNA and never reached a 2N state when treated with novobiocin (Fig. 4B-C), having a rate of replication elongation ~20% that of untreated wild-type cells (Fig. 4D). These data confirm that without sufficient GapR, the replication fork becomes highly sensitized to topoisomerase inhibition, suggesting that GapR may affect topoisomerase activity.
GapR binds DNA and can alter DNA topology
Collectively, our in vivo results suggested that GapR promotes DNA replication by attenuating (+) supercoils, thereby relieving the topological stress associated with fork progression. To directly test whether GapR binds and affects DNA topology, we expressed and purified untagged GapR. Gel filtration of GapR produced a single peak that eluted faster than expected for a ~10 kDa protein (Fig. S5A). Size-exclusion chromatography coupled to multi-angle light scattering (SEC-MALS) revealed a single, monodisperse (Mw/Mn = 1.000) peak, with an observed molecular weight of 26.4 kDa (Fig. S5A).
To investigate how GapR binds DNA, we performed electrophoretic mobility shift assays (EMSAs) in the presence of 300 bp, linear double-stranded DNA derived from a GapR ChIP peak (Fig. 5A). We observed the formation of GapR-DNA complexes, with multiple species observed at subsaturating levels of GapR, with an apparent KD of ~220 nM (Fig. S5C). As we progressively reduced the probe length, the number of shifted species also decreased, with two species for a 20 bp probe and a single shift for a 16 bp probe (Fig. 5B, S5B), suggesting that GapR binds to an ~10 bp stretch of DNA. For probes >16 bp, the multiple bands seen could reflect multiple binding events or GapRmediated bridging of different probes. To test the latter possibility, we performed an EMSA on a mixture of probes of two different lengths (Fig. 5C). The resulting EMSA pattern was a union of the individual EMSAs, indicating that GapR does not bridge DNA in vitro. The apparent KD of GapR on DNA was only slightly (~3-fold) lower on a 20 bp probe compared to a 300 bp probe or a ~3 kb plasmid (Fig. S5C), suggesting that GapR does not spread along DNA.
Figure 5. GapR binds DNA and alters DNA topology.

(A-C) EMSAs of GapR binding to linear dsDNA. Purified GapR was incubated with a 300 (A) or 16 bp probe (B), or with 210 and 150 bp probes individually (left and middle, C) and together (right, C). </p/> (D) GapR alters DNA topology. GapR was incubated with relaxed plasmid and then treated with calf thymus topoisomerase I (far left) before reactions were quenched, deproteinized with Proteinase K, electrophoresed, and imaged. Agarose gel analysis of plasmid topology (right), with supercoiled and relaxed standards (left).
(E) GapR alters DNA topology. GapR was incubated with nicked plasmid, treated with T4 DNA ligase (far left) before reactions were quenched and analyzed as in (D) (right). Supercoiled, relaxed, and nicked standards are shown (left).
(F) GapR constrains positive supercoils. Ligation products from (E) were analyzed by 2Dchloroquine electrophoresis (bottom, see Methods). Migration of different plasmid forms are diagrammed (left) with standards shown (top): N=nicked, R=relaxed, L=linear, (−)=negatively supercoiled, (+)=positively supercoiled).
See also Figure S5.
To test whether GapR affects DNA topology, we incubated GapR with relaxed plasmid DNA. If GapR introduces or stabilizes any (+) supercoils that arise, then compensatory (−) supercoils will accumulate elsewhere in the plasmid to maintain a net relaxed state. Addition of calf-thymus topoisomerase I (topo I) can then be added to remove the (−) supercoils; subsequent Proteinase K treatment will remove topo I and GapR, yielding plasmid DNA harboring any supercoils introduced or stabilized by GapR. Changes in supercoiling were assessed by agarose electrophoresis, with each band representing a unique topoisomer. Notably, when both GapR and topo I were added, we observed conversion of relaxed plasmids into supercoiled forms (Fig. 5D), but only when the concentration of GapR was >2.5 μM. At the highest concentration of GapR (20 μM), we saw reduced changes in DNA topology as GapR may coat the plasmid, preventing topo I from accessing the DNA (Fig. S5D). Indeed, we found that such concentrations of GapR also protected plasmid DNA from DNase digestion (Fig. S5F).
We also tested whether GapR could affect DNA topology using a similar assay in which a circular, nicked plasmid was first incubated with GapR and then treated with T4 DNA ligase to trap any supercoils introduced or constrained by GapR; any compensatory supercoils induced by GapR binding would dissipate because of the nick present before ligase is added (Fig. 5E). As above, bound proteins were then degraded and plasmid topology examined by agarose electrophoresis. Ligating nicked plasmid in the absence of GapR yielded relaxed plasmid, as expected. However, in the presence of GapR, we observed a clear increase in plasmid migration rates, again at concentrations of GapR >2.5 μM. To determine if these GapR-constrained topoisomers were (+) or (−) supercoils, we performed two-dimensional chloroquine gel electrophoresis. In the presence of >2.5 μM GapR, the ligated DNA migrated as expected for a (+) supercoiled plasmid, indicating that GapR can constrain or stabilize (+) supercoils (Fig. 5F).
GapR-mediated changes in (+) supercoiling could arise from a propensity for DNA with (+) writhe or for overtwisted DNA, which can convert into (+) writhe. We favored the latter since the increases in (+) writhe measured by gel electrophoresis (Fig. 5D-F) required GapR concentrations >2.5 μM. As GapR binds DNA with a KD of ~200–300 nM, many GapR binding events are likely required to generate the abrupt change in plasmid writhe observed (Fig. 5D-F, S5C). The simplest explanation is that each GapR binding event introduces or stabilizes a small amount of twist in the DNA, with twist from multiple binding events inducing a change in writhe. Importantly however, GapR probably does not directly introduce substantial numbers of supercoils in vivo as immunoblots estimated only ~3,000 GapR molecules per cell (Fig. S1F). Instead, our findings collectively suggest that GapR preferentially recognizes and associates with overtwisted, (+) supercoiled DNA, as occurs in front of active replication forks and at the 3’ ends of highly expressed genes.
GapR forms a dimer-of-dimer clamp that encircles overly twisted DNA
GapR shows no homology to any structurally characterized DNA binding protein and, in fact, contains no known DNA binding motif. To determine how GapR binds DNA and recognizes overtwisted DNA, we solved a crystal structure of C. crescentus GapR in complex with DNA. We obtained this structure by single wavelength anomalous diffraction (SAD) using crystals obtained with selenomethionine substituted GapR in complex with an 11-mer, AT-rich site and refined the structure to final Rwork/Rfree values of 24.1%/26.8% to 2.3 Å resolution (Fig. 6A-B, Table S4). Strikingly, the structure shows that GapR oligomerizes to form a dimer-of-dimers, which completely encircles the 11-mer DNA inside a large, central cavity (Fig. 6A; Movie S1), consistent with our EMSA indicating a binding site of ~10 bp (Fig. 5B). Each GapR subunit consists of three helices. An N-terminal helix (residues 14–51), α1, forms an antiparallel coiled coil dimer with α1 of a second GapR subunit. The α1-α1´ dimer (where ´ indicates the other subunit in a dimer) buries 1340 Å2 of protein surface from solvent. α2 (residues 55–66) and α3 (residues 69–86) are positioned at opposite ends of the α1-α1´ antiparallel coiled coil dimer. The α2-α3 helical pairs from each GapR subunit interdigitate with a α2-α3 helical pair from a second GapR dimer to form the dimer-of-dimers. These cross α2-α3 zipper interactions seal the ends of the dimer-of-dimer, creating the large central DNA binding cavity (Fig. 6A). An electrostatic surface representation of the GapR dimer-of-dimers shows that it forms a DNA-binding clamp with a remarkably electropositive inner DNA binding surface (Fig. 6C; Movie S1). DALI searches revealed no structures with similarity to the GapR monomer, dimer, or dimer-of-dimers.
Figure 6. Crystal structures of apo GapR and GapR-DNA complex.

(A) Structure of the C. crescentus GapR-DNA complex. Diagram of GapR helices (top left). GapR subunits are colored red and cyan (dimer 1), green and yellow (dimer 2). DNA is shown as a grey spheres model. Views are down the axis of the DNA (bottom left) and from the side (right).
(B) Cut away view of the GapR-DNA complex with a sigma A weighted electron density 2Fo-Fc map (blue mesh) contoured at 1.1 σ around DNA.
(C) Electrostatic surface representation of the GapR dimer-of-dimers, with the centrally bound DNA shown as a cartoon. Blue and red represent electropositive and electronegative regions, respectively.
(D) Packing of the GapR dimer-of-dimers in the crystal showing how multiple GapR oligomers could bind a long overtwisted region of DNA.
(E) Comparison of GapR-bound DNA with B-DNA. The widths of the major and minor grooves and the twist are indicated on GapR-bound DNA and B-DNA.
(F) Comparison of DNA-bound and apo GapR dimers. Structures of the DNA-bound GapR and apo GapR α1-α1´ dimers with one subunit colored cyan and the other red (top). Also labeled is Asp68, located between α2 and α3. Close up of the Asp68 side chain, solvent-exposed as part of a continuous helix in the apo structure, while forming N-cap interactions with the amide nitrogens of α3 when GapR binds DNA (bottom).
See also Figure S6.
The electron density for the GapR-bound DNA is clearly resolved (Fig. 6B) and forms a pseudocontinous ladder in the crystal, resulting in multiple dimer-of-dimer molecules along the DNA (Fig. 6D). There are no significant contacts between any of the dimers-of-dimers, supporting the notion that GapR does not cooperatively coat or bridge DNA. Consistent with the idea that GapR does not bind DNA sequence specifically, GapR makes no base contacts and only interacts with the DNA phosphate backbone. Residues from both α1 and α2 helices make key contacts with the DNA. Specifically, Lys34, Lys42 and Lys49 from each α1 helix and Lys56, Lys59, Arg63 and Lys66 from the basic regions of the four α2 helices provide a total of 28 phosphate contacts to DNA (Fig. S6A; Movie S1). The α1 helices (Fig. 6A) and these basic residues track along the phosphate backbone suggesting that they “read” a specific DNA conformation. Indeed, analysis of GapR-bound DNA revealed a significantly narrowed minor groove of 9.8 Å (compared to 12 Å for B-DNA) and widened major groove of 21 Å (compared to 17.2 Å for B-DNA) (Fig. 6E). However, most strikingly, GapR-bound DNA is significantly overtwisted in the structure, with the helical DNA twist being 44.5º compared to 36.0º for B-DNA (Fig. 6E). Importantly, attempts to fit B-DNA, which has a greater diameter than overtwisted DNA, within the GapR DNA binding cavity resulted in clash. Thus, the GapR DNA binding clamp appears to recognize an overtwisted DNA conformation that features narrow minor grooves and widened major grooves, not a specific DNA sequence. Notably, the oligonucleotide used in the crystal structure had high AT-content, which is known to more readily adopt an overtwisted state with narrow minor grooves. GapR does not bind as well to oligonucleotides with reduced AT-content (Fig. S6D).
To investigate whether apo GapR adopts a different conformation, we sought the apo GapR structure. Although apo C. crescentus GapR failed to produce data quality crystals, we obtained the structure of apo Bosea sp. Root381 GapR (herein termed Bosea GapR), which shares 60% sequence identity with the C. crescentus protein (Fig. S6B). We confirmed that the Bosea GapR ortholog also adopts a dimer-of-dimer conformation with DNA in the central cavity (see Methods, Table S4, and Fig. S6C). The apo Bosea GapR structure was solved by SAD phasing and refined to final Rwork/Rfree values of 22.2%/25.9% to 2.4 Å resolution (Table S4). Interestingly, the apo Bosea GapR forms a dimer rather than a dimer-of-dimers (Fig. 6F). The apo structure harbors the same α1-α1´ antiparallel dimer interaction observed in the DNA-bound Caulobacter GapR structure, but the α2 and α3 helices form a continuous helix (Fig. 6F). Although we did not obtain the apo Caulobacter GapR structure, our SEC-MALS analysis was consistent with Caulobacter GapR forming an elongated dimer in solution (Fig. S5A). The finding that GapR is dimeric in the absence of DNA indicates that DNA is not threaded through the dimer-of-dimer clamp, which is implausible, but rather that it adopts the dimerof-dimer state upon binding target DNA.
GapR stimulates the activities of topo IV and gyrase
Collectively, our results indicate that GapR recognizes overtwisted DNA such as that arising in front of replication forks. At these locations GapR may stimulate gyrase and/or topo IV to relieve superhelical stress and promote replication elongation. To test whether GapR promotes the activity of either gyrase or topo IV, we purified the two subunits of both Caulobacter gyrase and topo IV, and reconstituted each enzyme.
We first tested whether GapR can stimulate topo IV to remove (+) supercoils, deliberately selecting reaction conditions that yield relatively low topoisomerase activity to maximize our ability to observe any stimulation by GapR. We found that GapR at 0.4 μM stimulated the rate of (+) supercoil relaxation by topo IV ~4-fold (Fig. 7A). After 60 min, a reaction lacking GapR still had ~60% of the initial supercoiled substrate remaining, whereas the reaction containing GapR had almost no starting substrate left. Notably, the concentrations of GapR that stimulated topo IV activity (Fig. S7A) are insufficient to introduce (+) supercoils alone (Fig. 5D-E, S5E). We also tested whether GapR stimulates topo IV to relax (−) supercoils, and observed a modest, but reproducible ~1.3-fold increase in the rate of this reaction (Fig. S7B). The stimulatory effects of GapR on topo IV were specific as other proteins, including glucose-6-phosphate dehydrogenase (G6PD) and the DNA-binding protein GcrA, did not stimulate topo IV activity (Fig. S7C). We also tested whether GapR can stimulate other bacterial type II topoisomerases, by repeating relaxation assays with E. coli topo IV. We found that 0.4 μM GapR was unable to stimulate (+) supercoil relaxation by E. coli topo IV, but produced a modest increase in (−) supercoil relaxation (Fig. S7D).
Figure 7. GapR stimulates gyrase and topo IV activity.

(A) GapR stimulates relaxation of (+) supercoils by topo IV. Topo IV was incubated with GapR (0.4 μM) before adding (+) supercoiled pUC19 and ATP. Reactions were quenched at the times indicated and analyzed by gel electrophoresis (right). Slower migration in the gel indicates relaxation of (+) supercoils. Relaxed and (+) supercoiled standards are shown (left of the time course). Plot quantifies the % initial supercoiled plasmid (asterisk) at each time point (left). Data indicate mean ± SEM, n ≥ 2.
(B) GapR stimulates relaxation of (+) supercoils by gyrase. Assay performed as in (A) except using gyrase. Plot quantifies % initial supercoiled plasmid (asterisk) at each time point (left).
(C) Model of GapR and topoisomerases in replication. Replication fork progression generates overtwisted DNA, which can convert to (+) writhe and block fork movement. GapR binds to overtwisted DNA and stimulates gyrase and/or topo IV to relieve (+) writhe, allowing replication to resume.
See also Figure S7.
We then tested whether GapR stimulates Caulobacter gyrase. As with topo IV, we observed a clear and reproducible, GapR-dependent increase of ~2-fold for the rates at which gyrase relaxes (+) supercoiled DNA and introduces (−) supercoils into relaxed DNA (Fig. 7B, S7E-F). The stimulatory effects on gyrase were specific as neither G6PD nor GcrA stimulated gyrase (Fig. S7G). GapR had only a modest, but reproducible, stimulatory effect on (+) supercoil relaxation and (−) supercoil introduction by E. coli gyrase (Fig. S7H). These results demonstrate that GapR promotes the activities of both type II topoisomerases from Caulobacter, gyrase and topo IV, including their removal of (+) supercoils. Taken together with our in vivo studies, we conclude that the association of GapR with overtwisted DNA ahead of replication forks helps stimulate the type II topoisomerases, promoting replication fork progression, the timely removal of (+) supercoils ahead of replisomes, and, consequently, the completion of DNA replication in Caulobacter.
Discussion
The successful completion of DNA replication in any organism requires the resolution of complex topological structures that build up around the fork. Although the importance of topoisomerases in this process is well established, how topoisomerases are recruited to, and how their activity is controlled at, the replication forks is still poorly understood. Here, we identified GapR as a crucial regulator of topoisomerases and replisome progression in α-proteobacteria. Loss of GapR sensitizes cells to the inhibition of type II topoisomerases and leads to chromosomal relaxation, significantly slowed replication, and eventual cell death. We propose that the essential function of GapR is to promote the activities of type II topoisomerases, both to maintain global supercoiling levels and, importantly, to attenuate (+) supercoils that would otherwise impede replication forks (Fig. 7C).
Type II topoisomerases have long been known to enable DNA replication by alleviating the torsional stress that accumulates near replication forks. Indeed, the efficacy of many front-line antibiotics, e.g. coumerins and quinolones, is based on their ability to inhibit gyrase, and thereby block bacterial DNA replication (Vos et al., 2011). However, although type II topoisomerases such as gyrase or topo IV are required for DNA replication in vivo, whether these enzymes are sufficient has not been established. Our results indicate that they may not be, and instead may require activators or co-factors, such as GapR.
Recognition of highly twisted DNA by GapR
GapR specifically binds to overtwisted DNA (Fig. 5E-F, 6E), which is precisely the form of DNA that initially arises in front of replication forks. Overtwisted DNA also arises in front of translocating RNA polymerase and our ChIP-seq results demonstrated that GapR binds such regions in a sequenceindependent, but transcription-dependent manner. Consistent with these results, the structure of GapR revealed that the protein makes no base-specific contacts with DNA, but instead recognizes the shape of the DNA. When bound to DNA, GapR adopts a structure with an internal cavity lined with basic residues that contact the negatively charged backbone of the encircled DNA. Importantly, our results indicate that GapR is only able to recognize overtwisted DNA, as the larger diameter of B-DNA cannot be accommodated within the DNA-binding cavity of GapR. Thus, the structure of GapR in complex with DNA reveals a simple, but compelling mechanism for its localization to regions of the genome that are more highly twisted by the action of translocating DNA or RNA polymerase.
Our ChIP-seq analyses and prior studies of GapR revealed that GapR shows a preference for binding AT-rich DNA (Arias-Cartin et al., 2017; Ricci et al., 2016). Notably, such DNA often contain Atracts that can adopt intrinsically bent structures, with narrow minor grooves and increased superhelicity (Haran and Mohanty, 2009). Thus, AT-rich regions and the DNA ahead of replisomes or RNA polymerase may be the primary binding sites for GapR by virtue of a common, overtwisted shape. Our biochemical studies showed that GapR can also associate with relaxed or (−) supercoiled DNA, likely via local regions of overtwisted DNA within these substrates, arising from sequences with intrinsically narrow minor grooves or from thermodynamic fluctuations in twist.
GapR forms a snug clamp around overtwisted DNA raising the question of how it is loaded. The apo structure revealed that GapR is a dimer in the absence of DNA, with α2 and α3 forming a continuous helix (Fig. 6F), suggesting that the region between α2 and α3 is kinked upon DNA binding. A specific residue, Asp68, appears to function as the pivot point between α2 and α3. Among GapR homologs, an aspartic acid is highly conserved at this position, although glutamic acid, glutamine, and aspargine are also found. Notably, these residues are all known to function as N-cap residues and helix breakers. Indeed, while Asp68 points into the solvent in the apo GapR structure as part of the continuous α2 helix, it forms stabilizing N-cap contacts with α3 in the DNA-bound structures (Fig. 6F). Combined, these data suggest a unique mechanism for GapR clamp formation and loading in which the protein uses its α1-α1´ helices to track along the DNA as a caliper to search for sites with narrow minor and expanded major grooves, which then results in stable dimer-of-dimer formation through kinking to form α2-α3 and subsequent interactions between α2-α3 pairs around the DNA. Thus, unlike other DNA binding clamps, GapR likely does not require a clamp loader to dock onto its target DNA.
The preferential binding of GapR to highly twisted DNA explains why many GapR binding events were required to generate increases in plasmid writhe (Fig. 5D-E). Because supercoiling is a combination of twist and writhe, each GapR binding event introduces a small amount of twist into DNA, such that multiple binding events are necessary before inducing a change in writhe. Assuming GapR binds a ~10 bp site with half the available sites bound, the change of +3 writhe we observed implies an overtwisting of the DNA by ~8° per GapR bound, similar to the 8.5° change measured in the structure. However, GapR likely does not directly introduce large numbers of supercoils in vivo as there are only ~3,000 GapR molecules present per cell (Fig. S1F), significantly less than NAPs such as HU, which is present at ~60,000 copies per cell (Rouviere-Yaniv et al., 1979). Additionally, we found that GapR stimulates gyrase and topo IV at lower concentrations than is required to introduce topological changes. These observations, along with the structure of GapR bound to DNA, indicate that the primary role of GapR is not to directly alter DNA topology, but rather to associate with regions of (+) supercoiling, promoting the activity of type II topoisomerases precisely where their activities are needed the most.
Role of GapR in modulating topoisomerase activity
How, mechanistically, might GapR promote gyrase and topo IV activity? The simplest model is that GapR directly binds gyrase and topo IV to promote their binding to DNA or catalytic activity (Fig. S7I, left). By recognizing overtwisted DNA that arises in front of replication forks, GapR may recruit or stimulate gyrase and topo IV to act on the (+) supercoiled DNA that arises in front of forks and that must be dissipated for replication to progress. Notably, the structure of GapR revealed two acidic patches that are solvent-exposed in the DNA-bound, dimer-of-dimers configuration. Most of these acidic residues are highly conserved (Fig. S6B) and they may mediate a direct interaction with the topoisomerases. Alternatively, GapR could stimulate gyrase and topo IV without directly binding these enzymes, instead stabilizing DNA in a way that promotes topoisomerase binding and/or processivity (Fig. S7I, right). For instance, (+) writhed DNA that arises in front of replication forks may normally diffuse away from the fork. GapR binding to overtwisted DNA could ‘pin’ a plectonemic region of DNA, effectively trapping DNA for relaxation by topoisomerases. Further biochemical and enzymatic studies are needed to fully dissect the detailed mechanisms by which GapR stimulates gyrase and topo IV.
Regulation and control of DNA replication elongation
Whether the loss of viability in GapR-depleted cells arises from the defects in replication or the broad changes in transcription, or both, is unclear. The transcriptional changes are, however, likely an indirect effect of GapR depletion as the GapR ChIP profile did not correlate with the global changes in gene expression. We favor the notion, supported by our biochemical and structural studies, that the primary function of GapR is to promote type II topoisomerase activity during DNA replication. Consistent with this model, GapR-venus was suggested to localize to the DNA ahead of the replisome (Arias-Cartin et al., 2017).
Other NAPs have been implicated in controlling bacterial DNA replication. For instance, E. coli IHF and Fis are both critical for DNA replication initiation (Magnan and Bates, 2015). There are also several proteins in E. coli, including MukB and SeqA, that interact with topo IV to stimulate (−) supercoil removal and chromosome decatenation, although neither is required for replication elongation (Hayama and Marians, 2010; Kang et al., 2003; Li et al., 2010). E. coli HU was suggested to stimulate gyrase’s decatenation activity in vitro, but whether this function of HU promotes DNA replication in vivo is unknown (Marians, 1987). E. coli YejK stimulates topo IV to relax (+) supercoils in vitro, but YejK inhibits gyrase and ΔyejK strains have only modest, possibly indirect, effects on DNA replication and exhibit no growth defect (Lee and Marians, 2013). Thus, to our knowledge, GapR is the first NAP required for replication elongation and the first characterized activator of gyrase.
In sum, we suggest that GapR plays a critical role in resolving the superhelical stress associated with DNA replication. GapR may also be required to alleviate the torsional stress that arises in regions of the genome where (+) supercoils accumulate and cannot diffuse, e.g. when the replication forks converge near the terminus or, more frequently, when the transcription and replication machineries converge. At such sites, (+) supercoils must be resolved for replication to continue.
GapR is highly conserved throughout the α-proteobacteria where it likely plays a similar role in activating type II topoisomerases. GapR homologs are not found in other classes of bacteria, but a role for NAPs in replication elongation has not been well explored. There also may be as yet unidentified NAPs that share no homology to GapR, but recognize highly twisted DNA to similarly promote replication. In eukaryotes, there may also be analogous proteins. For example, high mobility group (HMG) proteins bind DNA relatively non-specifically, contributing to chromosome architecture in poorly defined ways. Intriguingly, yeast Hmo1 and the type II topoisomerase Top2 were recently shown to bind together near genes transcribed during S phase to somehow suppress chromosome fragility (Bermejo et al., 2009), and human HMGB1 interacts with and stimulates Top2α decatenation activity (Stros et al., 2007). We anticipate that interactions between stablybound, chromosome architecture proteins and topoisomerases, like that described here for GapR, will ultimately prove essential to the maintenance of genome integrity and the successful execution of DNA replication in most organisms.
STAR Methods
Contact for Reagent and Resource Sharing
Questions about or requests for methods, strains, and resources generated in this study can be directed to the Lead Contact, Michael T. Laub (laub@mit.edu).
Experimental Model and Subject Details
Growth conditions and chemical treatments
Caulobacter strains were grown in PYE (2 g/L bactopeptone, 1g/L yeast extract, 0.3 g/L MgSO4, 0.5 mM 0.5M CaCl2) at 30 °C unless noted. The Pxyl promoter was induced by supplementing media with xylose (0.3%) and repressed with glucose (0.2%). Antibiotics were used at the following concentrations unless noted (liquid/plates): oxytretracycline (1 μg mL−1 / 2 μg mL−1), spectinomycin (25 μg mL−1 / 200 μg mL−1), kanamycin (5 μg mL−1 / 25 μg mL−1), gentamycin (NA / 5 μg mL−1). E. coli strains were grown in LB (10 g/L NaCl, 10 g/L tryptone, 5 g/L yeast extract) and supplemented with antibiotics at the following concentrations (liquid/plates): carbenicillin (50 μg mL−1 / 100 μg mL−1), oxytretracycline (12 μg mL−1 / 12 μg mL−1), spectinomycin (50 μg mL−1 / 50 μg mL−1), kanamycin (30 μg mL−1 / 50 μg mL−1), gentamycin (15 μg mL−1 / 20 μg mL−1). When required, 400 mM IPTG was used to induce gene expression unless noted. Optical density was measured at 600 nm using a Genesys 10 Bio Spectrophotometer.
Strain construction
All Caulobacter strains were derivatives of ML76, an isolate of strain CB15N/NA1000. The insertion of a 3×FLAG at the C-terminus of GapR was constructed via a two-step recombination method using sacB as a counterselection marker. Strain ML2794 was constructed by electroporating plasmid pNPTS138-UP-gapR-3×FLAG-DW into ML76 and first integrants were selected on kanamycin plates. A second recombination step was performed by growing first integrants in PYE overnight and then plating on sucrose. Sucrose resistant clones were restreaked to verify they were kan sensitive and colonies were verified by PCR with primer pair CCNA_3428–3×flag-2-R and CCNA_03428-UP-F-less-hindiii.
The GapR depletion strain, ML2795, was constructed by electroporating plasmid pMT4427-CCNA_03428 into ML76 and selecting on kanamycin plates. As a second step, plasmid pNTPSSPEC-CCNA_03428-TET was electroporated and cells were selected on kanamycin, spectinomycin and tetracycline. First integrants were then grown overnight with kanamycin, tetracycline, and xylose and then plated on kanamycin, tetracycline, xylose + sucrose plates for sacB counterselection. Sucrose resistant plates were restreaked to verify sensitivity to spectinomycin. ML2954 was constructed by electroporating plasmid pMT4260-PgapR-gapR into ML76 and selecting on kanamycin plates. GapR was subsequently deleted by integrating pNTPS-SPEC-CCNA_03428-TET as described previously.
Plasmid construction
Integration plasmids:
pNPTS138-UP-gapR-3×FLAG-DW was constructed by amplifying a fragment containing the upstream region of gapR with primers CCNA_03428-UP-F-less-hindiii and CCNA_03428–3×flag-1-R to which the coding region for 3×-FLAG was added by a second round of PCR with primers CCNA_3428–3×flag-2-R and CCNA_03428-UP-F-less-hindiii. This upstream fragment was fused by splice-overlap-extension (SOE) PCR to a fragment containing the region downstream of gapR amplified with primers 3428dw-fuse-3×flag-F and CCNA_03428-DW-R. The resulting PCR product was digested with NheI and HindIII restriction enzymes and ligated into pNPTS-138 digested with the same enzymes.
The pNTPS-SPEC-CCNA_03428-TET plasmid was constructed by multiple SOE PCR reactions. First, a fragment containing 703 bp upstream of CCNA_03428 was amplified with the primer pair CC3319TETDELUP-R and FOR_UP3319PNTPSPSTI, the tetracycline (tet) resistance cassette was amplified from template pKO3 using primer pair OL7_tet_F and OL8_tet_R, and a fragment containing 703 bp downstream of CCNA_03428 was amplified using primers CC3319TETDELDWF and CC3319TETDELDW_SPEI-R. Subsequent SOE PCRs were performed to fuse the three fragments together. The resulting product was digested with PstI and SpeI and ligated into pNTPSSPEC digested with the same restriction enzymes.
The pMT4260-PgapR-gapR plasmid was constructed by amplifying a fragment containing the promoter and coding region of GapR with GapR_up_KpnI and GapR_down_NheI, digesting with KpnI and NheI, and ligated pMT4260 digested with the same enzymes.
Replicative plasmids:
pMT4427-CCNA_03428 was constructed by ligation of a PCR fragment amplified with primers CCNA_03428-F-NdeI and CCNA_03428-R-SacI and digested with NdeI and SacI.
Expression plasmids:
pET28-CCNA_03428 was constructed by ligation of a PCR fragment amplified with primers CCNA_3428-F-NdeI and CCNA_3428_R_SacI and digested with NdeI and SacI. pKS22b-hSUMO-GapR was constructed by ligation of a PCR fragment amplified with primers GapR-F-BamHI and GapR-R-NotI and digested with BamHI and NotI.
pKS22b-hSUMO-GyrA was constructed by Gibson assembly cloning with a PCR fragment amplified with primers GyraseA-F-BamHI-homology and GyraseA-R-NotI-homology and the pKS22b vector digested with BamHI and NotI. pKS22b-hSUMO-GyrB was constructed by Gibson assembly cloning with a PCR fragment amplified with primers GyrB-F-BamHI-homology and GyrB-R-NotI-homology and the pKS22b vector digested with BamHI and NotI. pKS22b-hSUMO-ParC was constructed by Gibson assembly cloning with a PCR fragment amplified with primers ParC-F-BamHI-homology and ParC-R-NotI-homology and the pKS22b vector digested with BamHI and NotI. pKS22b-hSUMOParE was constructed by Gibson assembly cloning with a PCR fragment amplified with primers ParE-F-BamHI-homology and ParE-R-NotI-homology and the pKS22b vector digested with BamHI and NotI. For pET15b-GapR(Cres) and pET15b-(Bosea) and their variants, the full length Bosea sp. Root 381 GapR protein and C. crescentus GapR residues 11–89 were purchased as E. coli codon optimized genes from Genscript with NdeI and BamHI sites and subcloned into pET15b using restriction cloning. This resulted in the placement of a cleavable hexa-histidine tag at the N-terminus of the expressed proteins. C. crescentus GapR(11–89) was used for structural studies as the N-terminal 10 residues of the C. crescentus protein are not conserved among GapR homologs (and not present in several homologs including the Bosea sp. Root 381 GapR protein) and are predicted to be disordered.
Method Details
Nucleoid purification and mass spectrometry analysis
The nucleoid purification protocol was performed as follows. A 25 mL culture of exponentially growing ML1241 was centrifuged at 8,500 rpm at 4 °C for 5 min. Cell pellets were resuspended in 0.5 mL of ice-cold buffer A (10 mM Tris-HCl [pH 8.2], 100 mM NaCl, and 20% sucrose) followed by the addition of 0.1 mL of ice-cold buffer B (100 mM Tris-HCl [pH 8.2], 50 mM EDTA, 0.6 mg/mL lysozyme) and incubated for 1 minute on ice. Then, 0.5 mL of Buffer C (10 mM Tris-HCl [pH 8.2], 10 mM EDTA, 10 mM spermidine, 1% Brij-58, and 0.4% deoxycholate) was added and the mixture incubated for 4 min at 5 °C. The lysed cells were loaded onto linear sucrose density gradients (15–50%) containing 10 mM Tris-HCl (pH 8.2) and 100 mM NaCl, and centrifuged for 20 minutes at 10,000 rpm in a Beckmann SW 41 Ti rotor. Fractions (~1 mL each) were collected by piercing a hole at the bottom of the ultracentrifugation tube and DNA content quantified by DAPI (Invitrogen) fluorescence using a plate reader (Spectramax). The fractions were then incubated with 20 U of benzonase at room temperature for 30 min. Fractions were run on a 15% Tris-HCl gel (BioRad) and submitted for tandem mass spectrometry analysis at the Koch Institute Biopolymers and Proteomics core facility. The region of the gel corresponding to proteins in the range of 5–25 kDa was isolated and cut into two fragments for mass spectrometry analysis. Mass spectra were analyzed using Mascot (Matrix Science, London, UK), searching against the Sprot_extra_022412 database assuming digestion with trypsin.
To compare chromosomal supercoiling, nucleoids were isolated as above and loaded on to 10–30% linear sucrose gradients containing varying amounts of ethidium bromide. For novobiocin treated nucleoids, cells were treated with 25 μg/mL novobiocin (Sigma) for 15 min before cell harvest. Intercalation of ethidium bromide into DNA first causes relaxation of the chromosome and decreased sedimentation and then at higher concentrations of ethidium increased sedimentation due to introduction of (+) supercoils. The amount of ethidium bromide at the point of slowest migration is reflective of the concentration of negative supercoils in the chromosome (Worcel and Burgi, 1972). Gradients were centrifuged at 10,000 rpm in a Beckman SW41 for 15 min. The distance sedimented by the nucleoid from the top of the gradient was measured. Sedimentation data from multiple runs with different ethidium bromide concentrations were pooled for analysis. The concentration of ethidium required at the point of slowest migration was estimated by fitting the data with a LOWESS fit using Prism 7 software.
Expression profiling with microarrays and RNA-sequencing
RNA was extracted using hot trizol lysis and the Direct-zol RNA MiniPrep (Zymo). rRNAs were removed using the Ribo-Zero Kit for Gram-negative bacteria (Illumina). The rRNA-depleted RNA was then fragmented using the RNA fragmentation reagents (Ambion) at 70 °C for 8 min. Fragmented RNA was recovered by ethanol precipitation and resuspended in 6 μL water. RNA-seq libraries were prepared based on the previously described dUTP protocol as follows. First-strand cDNA synthesis was performed by addition of 1 μL of 3 μg/μL random primers (Invitrogen) followed by incubation at 5 min at 65 °C. Samples were then placed on ice for 1 min and the following reagents were added: 4 μL first-strand synthesis buffer, 2 μL 100 mM DTT, 1 μL 10 mM dNTPs, 1 μL Superase-in (ThermoFisher) and 4 μL water. Reactions were incubated at room temperature for 2 min and 1 μL Superscript III (ThermoFisher) was added and the following thermocycler program was used: 10 min at 25 °C, 1hr at 50 °C and 15 min at 70 °C. The reaction volumes were brought to 200 μL and a phenol:chloroform:isoamyl alcohol (25:24:1) (Sigma) extraction was performed. cDNA was resuspended in 104 μL water and second-strand synthesis reactions were set up with 30 μL of 5X second-strand synthesis buffer (ThermoFisher), 4 μL of 100 mM dNTPs with dUTP (Promega) instead of dTTP, 4 μL of 5X first-strand synthesis buffer and 2 μL 100 mM DTT. Reactions were incubated on ice for 5 min and 1 μL RNase H (NEB), 1 μL E.coli DNA ligase (NEB) and 4 μL DNA Pol I (NEB) were added, followed by an incubation at 16 °C for 2.5 hrs. cDNA was recovered by Ampure XP (Beckman Coulter) bead purification with 100 μL beads in 450 μL 20% PEG/NaCl solution.
Sequencing libraries were built by first end repairing cDNA with 5 μL T4 DNA polymerase (NEB), 5 μL T4 PNK (NEB), and 1 μL Klenow large fragment (NEB) in 100 μL T4 DNA ligase buffer with 0.25 mM dNTPs for 30 min at room temperature. Repaired DNA was recovered by Ampure XP (Beckman Coulter) bead purification, using 100 μL beads in 300 μL 20% PEG/NaCl solution. Beads were washed twice with 80% ethanol, dried, and resuspended in 32 μL EB. The bead slurry was directly treated with 3 μL Klenow (3’→5’ exo-) (NEB) in 50 μL NEB Buffer #2 with 0.2 mM ATP at 37 °C for 30 min to add 3’ overhangs to DNA. The reaction was cleaned up by addition of 150 μL 20% PEG/NaCl and capture of the Ampure XP beads. After two washes with 80% ethanol, the beads were resuspended in 23 μL EB and the supernatant was transferred to a clean tube. Y-shaped Illumina adapters were ligated onto the DNA in 50 μL total volume in Quick Ligase buffer with 3 μM Y-shaped adaptors and 1 μL Quick Ligase (NEB) for 15 min at room temperature. Y-shaped adaptors were prepared by annealing Illumina PE adapter 1 and Illumina PE adapter 2. 75 μL 20% PEG/NaCl was added, and the DNA then recovered by addition of Ampure beads followed by two ethanol washes. After resuspension in 23 μL water, the Ampure beads were captured and the DNA in the supernatant was transferred to a clean tube. Digestion of the second strand was performed by addition of 6 μL of 5× HF Phusion buffer and 1 μL USER (NEB) and incubation at 37 °C for 15 min, followed by 5 min at 95 °C. DNA libraries were amplified in 80 μL final volume with Phusion DNA polymerase (NEB) in Phusion High GC buffer supplemented with Betaine (0.4 M final) (Sigma). The total number of cycles was optimized for each sample such that 10–14 cycles of PCR was used for each sequencing library. Libraries were purified by addition of 240 μL 20% PEG/NaCl and capture of the Ampure XP beads. After two washes with 80% ethanol, the beads were resuspended in 20 μL EB. Elutions were then run on a 8% TBE polyacrylamide gel (ThermoFisher) for 30 min at 180 V and a 230–500 bps region was gel extracted using a Spin-X 0.22 μm cellulose acetate column (Costar). Single-end sequencing was performed on an Illumina Hi-Seq 2000 at the MIT Bio Micro Center.
For DNA microarray experiments, 10 mL cultures of ML76 were grown in PYE + 0.3% xylose, M2X or M2G to mid-exponential phase and treated with 25 μg/mL novobiocin (Sigma) for 15 min or left untreated to use as a reference for the microarray. RNA was extracted using the RNeasy Mini Kit (Qiagen). The generation of labeled cDNA was performed using 20 μg of RNA in 14 μL RNase-free water to which 1 μL of random primers (Invitrogen) was added. The samples were incubated at 65 °C for 10 min, followed by 2 min incubation on ice. 9.6 μL of trimix (6 μL 5X first-strand buffer, 3 μL 0.1 M DTT, and 0.6 μL dNTP mix [25 mM A,G,T, 10 mM C]), 1 μL of Superscript II (ThermoFisher), and 3 μL of labeled dCTP (Cy5 dye for experimental and Cy3 dye for reference) (Sigma) were added and incubated covered with foil for 10 min at room temperature. Samples were then incubated at 42 °C for 1 hr, after which an additional 1 μL of Superscript II was added for another hour. RNA was degraded by addition of 1.5 μL of 0.5 N NaOH and cDNA purified by addition of 1.5 μL of 0.5 N HCl followed by purification with a PCR Purification Kit (Qiagen). After a wash with PE buffer, cDNA was eluted in 50 μL EB. 1 μL of 20 μg/μL yeast tRNA (Sigma) was added and the total volume was adjusted to 55 μL. 55 μL of 2X-HiRPM hybridization buffer (Agilent) as added to the samples and incubated for 3 min at 95 °C followed by 1 min at 42 °C. 100 μL was loaded onto a custom Agilent array placed in a hybridization chamber and incubated overnight rotating at 65 °C. Arrays were washed for 5 min in Oligo aCGH/Chip-on-chip wash buffer 1 (Agilent), washed for 5 min in Oligo aCGH/Chip-on-chip wash buffer 2 (Agilent) at 30 °C, and washed with acetonitrile for 2 min at room temperature. Arrays were dried Stabilization and Drying Solution (Agilent) and imaged using an Agilent scanner at the MIT Bio Micro Center.
Chromatin Immunoprecipitation Sequencing (ChIP-seq)
ChIP was performed using strain ML2794 (gapR::gapR-3×FLAG) or wild-type CB15N grown in PYE. The ChIP procedure was as follws. Briefly, cell cultures (20 mL) were grown to OD600 ~0.3 and fixed by the addition of 10 mM sodium phosphate [pH 7.6] and 1% formaldehyde (final concentrations) (Sigma). When appropriate, 25 μg/mL of rifampicin (Sigma) was added to cells for 20 minutes prior to fixation. Fixed cells were incubated at room temperature for 10 minutes and then quenched with 0.1 M glycine (Sigma) for 5 min at room temperature followed by 15 min on ice. Cells were washed three times with 1X PBS [pH 7.4] and resuspended in 500 μL of TES buffer (10 mM Tris-HCl [pH 7.5], 1 mM EDTA, 100 mM NaCl) to which 35,000 U of Ready-Lyse (Epicentre) was added. Following 15 min incubation at RT, 500 μL of ChIP buffer (16.7 mM Tris-HCl [pH 8.1], 167 mM NaCl, 1.1% Triton X-100, 1.2 mM EDTA) containing protease inhibitors (Roche cOmplete EDTA-free tablets) was added. After 10 min at 37 °C, the lysates were sonicated on ice and cell debris cleared by centrifugation. Supernatant protein concentration was measured by Bradford assay (Thermo Scientific) and 500 μg of proteins were diluted into 1 mL of ChIP buffer + 0.01% SDS. The diluted supernatants were pre-cleared for 1 hr at 4 °C on a rotator with 50 μL of Protein-A Dynabeads (Life Technologies) pre-blocked overnight in ChIP buffer + 0.01% SDS and 100 μg ultrapure BSA (Ambion). Beads were pelleted and 90 μL of the supernatant was removed as input DNA and stored at −80 °C, the remaining pre-cleared supernatant was incubated rotating at 4 °C overnight with 1 μL of FLAG antibody (Sigma). The immune complexes were captured for 2 hr at 4 °C with 50 μL of pre-blocked Protein-A Dynabeads. Beads were then washed consecutively at 4 °C for 15 min with 1 mL of the following buffers: low salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl [pH 8.1], 150 mM NaCl), high salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl [pH 8.1], 500 mM NaCl), LiCl wash buffer (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl [pH 8.1] and twice with TE buffer (10 mM Tris-HCl [pH 8.1], 1 mM EDTA). Complexes were then eluted twice from the beads with 250 μL of freshly prepared elution buffer (1% SDS, 0.1 M NaHCO3). To reverse crosslinking, 300 mM of NaCl and 2 μL of RNase A (0.5 mg / mL) (Qiagen) were added to the collective eluates which were incubated at 65 °C overnight. Samples were then incubated at 45 °C for 2 hr with 5 μL of Proteinase K (NEB) in the presence of 40 mM EDTA (pH 8.0) and 40 mM Tris-HCl [pH 6.8]. DNA from the samples was then extracted twice with phenol:chloroform:isoamyl alcohol (25:24:1) (Sigma) and subsequently precipitated by adding sodium acetate (pH 5.2), 100 μg glycogen (ThermoFisher) and 1 volume of ice cold isopropanol, and stored at −20 °C overnight. DNA was pelleted and washed with 75% ethanol and resuspended in TE buffer [pH 8.0].
Sequencing libraries were built by first end repairing cDNA with 5 μL T4 DNA polymerase (NEB), 5 μL T4 PNK (NEB), and 1 μL Klenow large fragment (NEB) in 100 μL T4 DNA ligase buffer with 0.25 mM dNTPs for 30 min at room temperature. Repaired DNA was recovered by Ampure XP (Beckman Coulter) bead purification, using 100 μL beads in 300 μL 20% PEG/NaCl solution. Beads were washed twice with 80% ethanol, dried, and resuspended in 32 μL EB. The bead slurry was directly treated with 3 μL Klenow (3’→5’ exo-) (NEB) in 50 μL NEB Buffer #2 with 0.2 mM ATP at 37 °C for 30 min to add 3’ overhangs to DNA. The reaction was cleaned up by addition of 150 μL 20% PEG/NaCl and capture of the Ampure XP beads. After two washes with 80% ethanol, the beads were resuspended in 23 μL EB and the supernatant was transferred to a clean tube. Y-shaped Illumina adapters were ligated onto the DNA in 50 μL total volume in Quick Ligase buffer with 3 μM Y-shaped adaptors and 1 μL Quick Ligase (NEB) for 15 min at room temperature. Y-shaped adaptors were prepared by annealing Illumina PE adapter 1 and Illumina PE adapter 2. 75 μL 20% PEG/NaCl was added, and the DNA then recovered by addition of Ampure beads followed by two ethanol washes. After resuspension in 30 μL water, the Ampure beads were captured and the DNA in the supernatant was transferred to a clean tube. DNA libraries were amplified in 80 μL final volume with Phusion DNA polymerase (NEB) in Phusion High GC buffer supplemented with Betaine (0.4 M final) (Sigma). The total number of cycles was optimized for each sample such that 10–14 cycles of PCR was used for each sequencing library. Libraries were purified by addition of 240 μL 20% PEG/NaCl and capture of the Ampure XP beads. After two washes with 80% ethanol, the beads were resuspended in 20 μL EB. Elutions were then run on a 8% TBE polyacrylamide gel (ThermoFisher) for 30 min at 180 V and the 250–500 bps region was gel extracted using a Spin-X 0.22 μm cellulose acetate column (Costar). Single-end sequencing was performed on an Illumina Hi-Seq 2000 at the MIT Bio Micro Center. RpoC-3×FLAG ChIP profiles were from (GSE73925).
Caulobacter synchronizations
For synchronizations, Caulobacter strains were grown to mid-exponential phase and G1/swarmer cells were isolated using Percoll (GE Healthcare) density gradient centrifugation. Cells were pelleted at 8000 × g for 5 min and resuspended in M2 salts (0.87 g/L Na2HPO4, 0.53 g/L KH2PO4, 0.5 g/L NH4Cl) with an equal volume of Percoll. The suspension was centrifuged at 10000 × g for 20 min and G1 cells (bottom layer) were isolated. The G1 cells were then washed with 5 × 1 mL M2 salts before resuspension and release into appropriate medium.
Immunoblotting
Nucleoid fractions were electrophoresed with 4–20% Tris-HCl gels (Bio-Rad) and either stained with Coomassie or transferred onto PVDF and immunoblotted. For whole cell immunoblots, cells were harvested and their proteins precipitated with 20% TCA, washed with acetone, and normalized by OD for resuspension. Proteins were electrophoresed on 12% Bis-Tris gels in MES buffer (Invitrogen) with at least 2 replicates for each condition. For estimates of GapR copy per cell, a standard curve was generated with purified GapR protein. Antibodies were used at the following concentrations: anti-RpoA (1:7500, Neoclone), anti-FLAG (1:7500, Sigma), anti-IHF (1:1000) (generous gift of S. Goodman), anti-CtrA (1:5000), and anti-GapR (1:1000). Rabbit polyclonal anti-GapR antibody (Covance) was generated from purified His6-Thrombin-GapR and was used without purification at 1:1000. HRP-conjugated secondary antibodies (ThermoFisher) were used at the following concentrations: goat anti-rabbit, 1:4000; goat anti-mouse, 1:7500. The membranes were developed with SuperSignal West Femto Maximum Sensitivity Substrate (ThermoFisher) and visualized with a FluorChem R Imager (ProteinSimple).
Microscopy
Phase contrast images were taken on a Zeiss Observer Z1 microscope using a 100×/1.4 oil immersion objective and an LED-based Colibri illumination system using software Metamorph (Universal Imaging, PA). Cells were diluted to maintain OD600 < 0.4. For phase contrast images, cells were fixed with 0.5% paraformaldehyde (Sigma), pelleted, and resuspended in PBS. Fixed cells were spotted onto PBS 1.5% agarose pads and imaged.
Flow cytometry
Flow cytometry was performed as follows. 300 μL cells were fixed with 700 μL 70% ethanol, spun at 5000 × g for 2.5 min, resuspended in 50 mM Na2CO3 with 5 μg/mL RNase A (Qiagen), and incubated at 50 °C for at least 4 hr. Cells were diluted 1:10 into Na2CO3 buffer containing 0.5 μL/mL SYTOX Green nucleic acid stain (Invitrogen) and analyzed on a BD Accuri C6 flow cytometer. For time course experiments, synchronized swarmer cells were released into fresh media and sampled every 20 min after release. For novobiocin experiments, novobiocin (Sigma) was added to 2.5 μg / mL 20 min after release.
DNA sequencing
DNA sequencing was performed on 500 mL of each Caulobacter strain grown to mid-exponential phase (OD600 ~ 0.2–0.4) in PYE + 0.3% xylose. For GapR depletion, cells were grown in 0.3% xylose and either synchronized or harvested, washed 4 × with PYE, and resuspended in 0.2% glucose for 2 hr before synchronization. The wild-type control was grown in 0.2% glucose before synchronization. After synchronization, cells were released into 25 mL of PYE + glucose at ~OD600 ~ 0.2. At each time-point after release (0, 20, 40, 60, 80 min), 2 mL of cells were centrifuged at 14,000 rpm for 1 min, aspirated, and frozen in liquid nitrogen.
DNA was isolated by CTAB purification. Pellets were resuspended in TE (10 mM Tris-HCl [pH 8.0], 1 mM EDTA) and normalized so that 1 mL of culture with OD600 ~0.1 was present per 10 μL resuspension. 400U of Ready-Lyse (Epicentre) at a final concentration of 0.125% was added to 50 μL of resuspension, and incubated for 5 min at room temperature. The volume was brought up to 800 μL in TE supplemented with 10 mM EDTA, 0.75% Triton X-100, and 2.5 μL of 20 mg/mL Proteinase K (NEB), and incubated at 50–65 °C overnight. 100 μL 5 M NaCl and 100 μL of CTAB (Sigma) was added, and samples then incubated at 65 °C for 10 min. The DNA in each sample was then extracted by phenol:chloroform:isoamyl alcohol (25:24:1) (Sigma), and precipitated with isopropanol. DNA was resuspended in 10 mM Tris-HCl [pH 8.0], and sheared to 300–800 bp fragments using a Bioruptor Plus sonicator (Diagenode). RNase A (Qiagen) was added and the fragments were electrophoresed, and 300–600 bp fragments were size selected, purified with Gel Extraction Kit (Qiagen), and resuspended in 50 μL EB.
Sequencing libraries were built by first end repairing fragmented DNA with 4 μL T4 DNA polymerase (NEB), 4 μL T4 PNK (NEB), and 0.75 μL Klenow large fragment (NEB) in 100 μL T4 DNA ligase buffer with 0.25 mM dNTPs for 30 min at room temperature. Repaired DNA was recovered by Ampure XP (Beckman Coulter) bead purification, using 100 μL beads in 300 μL 20% PEG/NaCl solution. Beads were washed twice with 80% ethanol, dried, and resuspended in 30 μL EB. The bead slurry was directly treated with 3 μL Klenow (3’→5’ exo-) (NEB) in 40 μL NEB Buffer #2 with 0.2 mM ATP at 37 °C for 45 min to add 3’ overhangs to DNA. The reaction was cleaned up by addition of 120 μL 20% PEG/NaCl and capture of the Ampure XP beads. After two washes with 80% ethanol, the beads were resuspended in 30 μL EB and Y-shaped Illumina adapters were ligated onto the DNA in 50 μL total volume in T4 Ligase buffer with 3 μM Y-shaped adaptors and 2 μL T4 DNA ligase (NEB) for 30 min at room temperature. Y-shaped adaptors were prepared by annealing Illumina PE adapter 1 and Illumina PE adapter 2. 150 μL 20% PEG/NaCl was added, and the DNA then recovered by capture of the Ampure beads followed by two ethanol washes. After resuspension in 20 μL water, the Ampure beads were captured and the DNA in the supernatant was transferred to a clean tube. DNA libraries were amplified with Kapa HiFi DNA polymerase (Kapa Biosystems) and multiplexed with Illumina PE PCR primer 1 and Illumina PE PCR primer 2 (multiplexing) in Kapa High GC buffer supplemented with DMSO. The total number of cycles was optimized for each sample such that 10–14 cycles of PCR was used for each sequencing library. DNA was paired-end sequenced on a NextSeq500 at the MIT Bio Micro Center.
qPCR analysis of origin-terminus ratio
Cells were grown in PYE with xylose, washed, and resuspended in PYE with glucose for 2 hr before synchronization and release into fresh glucose. 1 mL samples were harvested in 30 min increments and flash frozen. DNA was extracted by resuspending pellets in 600 μL Cell Lysis Solution (Qiagen) and incubating at 80 °C for 5 min to lyse cells. RNAs were removed by treatment with 50 μg RNase A (Qiagen) at 37 °C for 30 min. 200 μL Protein Precipitation Solution (Qiagen) was added, the sample vortexed, and left on ice for 30 min to precipitate proteins. After spinning at 14,000 rpm for 5 min, the supernatant was transferred to a tube containing 600 μL isopropanol and mixed by inversion. DNA was harvested by spinning at 14,000 rpm for 1 min followed by a wash with 600 μL of 70% ethanol. The DNA pellet was resuspended in 20 μL H2O. For qPCR, DNAs were diluted 1:10 or 1:20 and DNAs was mixed with either Cori_2 or CCNA_01869 forward/reverse primer mix and 2X qPCR Master Mix. All experimental samples and standard curves were loaded onto a 384-well plate in triplicate for qPCR. qPCR was conducted in a LightCycler 480 system (Roche) using the following thermocycler program: 95 °C/10 minutes, 95 °C/15 seconds, 60 °C/30 seconds, 72°C/30 seconds with 40 cycles of steps 2–4. Cp values were calculated from LightCycler 480 software at the second derivative maximum. Technical replicates were averaged to yield a final Cp value for each sample and standard curve point.
Plating viability and drug sensitivity
Strains were grown to mid-exponential phase and diluted in PYE to OD600 ~ 0.2. Strains were then 10-fold serially diluted and 2 μL of each dilution spotted onto plates containing 0.3% xylose and one of the following compounds when appropriate: novobiocin (0.1 μg/mL) (Sigma), mitomycin C (0.1 μg/mL) (Sigma), chloramphenicol (0.1 μg/mL) (Sigma), hydroxyurea (0.3 mg/mL) (Sigma), or rifampicin (0.03 μg/mL) (Sigma). Plates were incubated at 30 °C for two or three days and imaged with a scanner. For experiments with minimal medium, strains were first grown in PYE to OD600 ~ 0.2, diluted into M2 + 0.2% glucose for wild-type cells or M2 + 0.3% xylose for the GapR depletion strain and grown to OD600 ~ 0.2, and then spotted onto M2 agar supplemented with 0.2% glucose or 0.3% xylose and grown for three days before imaging.
Phylogeny analysis
Phylogeny analysis was performed using the STRING database. Alignment logo was generated using WebLogo Version 2.8. Protein alignments were generated with JalView using Muscle with default settings. Genomes shown: Caulobacter crescentus (NA1000), Caulobacter K31, Brevundimonas subvibrioides, Caulobacter phage Cr30, Agrobacterium tumefaciens str. Cherry 2E-2–2, Rhodopseudomonas palustris, Sinorhizobium meliloti (strain SM11), Sphingomonas sp. (strain SKA58), Azospirillum phage Cd, Acetobacter pasteurianus, Wolbachia endosymbiont of Muscidifurax uniraptor, Bosea sp. Root 381, and Rickettsia monacensis.
Protein expression and purification
Purified GcrA and glucose-6-phosphate dehydrogenase (G6PD) were part of the lab collection and a gift from B. Wang in the Laub lab, respectively. Protein expressions was performed in BL21(DE3) cells grown in 2xYT to OD600 ~ 0.4–0.5 at 37 °C and induced with 0.4 mM IPTG for 18–20 hr at 18 °C unless otherwise noted. When appropriate, SUMO protease was added and the cleavage reaction adjusted to 400 mM NaCl. Cleavage proceeded for 2 hr or overnight at 4 °C.
His6-GapR and His7-SUMO-GapR: 1 L of cells was harvested by centrifugation and resuspended in 20 mL buffer A (50 mM sodium phosphate [pH 7.5], 0.5 M NaCl, 20 mM imidazole) supplemented with a SIGMAFAST Protease Inhibitor Tablet (Sigma). The cell resuspension was treated with 20 mg lysozyme (Sigma), 1 mM PMSF (Sigma), and 300 U benzonase (Novagen) at room temperature with gentle agitation for 20 min and then lysed by sonication. The cell debris was cleared by centrifugation for 30 min at 10,000 × g and passed over Ni-NTA agarose resin (Qiagen) pre-equilibrated with buffer A at 4 °C. Resin was washed with buffer A and proteins were eluted stepwise with 2.5 column volumes of buffer A containing increasing amounts of imidazole (50–100-150–250-500 mM imidazole). Fractions containing GapR (150–500 mM) were combined. For His7-SUMO-GapR, SUMO cleavage proceeded overnight at 4 °C, supplemented with 300 U of Benzonase (Novagen) and 10 mM final concentration of MgCl2 to ensure removal of DNA during cleavage. Eluates were diluted or buffer exchanged with PD-10 columns (GE Healthcare) to have < 100 mM NaCl and run on a HiTrap Heparin HP (5mL) (GE Healthcare) column, pre-equilibrated in buffer B (50 mM sodium phosphate [pH 7.5], 10 mM NaCl). GapR and His6-GapR were eluted with a 2-step elution (buffer B + 0.5 M NaCl and buffer B + 1.0 M NaCl), each step being 5 column volumes. High salt fractions containing GapR were concentrated on an Amicon 3K Centrifugal Filter Unit (Millipore) and buffer exchanged into storage buffer (50 mM sodium phosphate pH 7.5, 200 mM NaCl, 10% (v/v) glycerol) by gel filtration using a Superdex 200 Increase 10/300 GL column (GE Healthcare). Fractions containing GapR were identified by SDS-PAGE/Coomassie staining.
Purification of His7-SUMO-ParC and His7-SUMO-ParE was performed as follows. 1 L of cells was induced with 0.4 mM IPTG for 4 hr at 37 °C and harvested and resuspended in 20 mL of buffer A800 (20 mM Tris-HCl [pH 8.0], 30 mM imidazole, 800 mM NaCl, 2 mM ßME, 10% (v/v) glycerol) supplemented with a SIGMAFAST Protease Inhibitor Tablet (Sigma). After lysis and sonication, the lysate was applied to Ni-NTA agarose resin (Qiagen), washed with 10 column volumes of buffer A800, and then washed with 20 column volumes with buffer A400 (buffer A800 with 400 mM NaCl). Proteins were eluted with B400 (buffer A400 with 500 mM imidazole), and cleaved with SUMO protease. After validating SUMO cleavage by SDS-PAGE/Coomassie staining, the mixture was concentrated on an Amicon 50K Centrifugal Filter Unit (Millipore) and applied to a Superdex 200 Increase 10/300 GL column (GE Healthcare) equilibrated in 20 mM Tris-HCl [pH 8.0], 400 mM KCl, 2 mM ßME, and 10% (v/v) glycerol. Fractions containing ParC or ParE identified by SDSPAGE/Coomassie staining were concentrated with a Amicon 50K Centrifugal Filter Unit. Glycerol was added and the buffer adjusted to make a final storage buffer of 20 mM Tris-HCl [pH 8.0], 400 mM KCl, 2 mM ßME, 30% (v/v) glycerol and protein aliquots were stored at −80 °C.
His7-SUMO-GyrA and His7-SUMO-GyrB were purified as follows. For GyrA, 1 L of cells was grown in 2xYT at 37 °C to OD600 ~ 0.5 and induced with 0.4 mM IPTG for 4 hr at 37 °C and resuspended in 20 mL of buffer A1000 (50 mM HEPES, [pH 7.5], 20 mM imidazole, 1000 mM NaCl, 2 mM ßME, 10% (v/v) glycerol) supplemented with a SIGMAFAST Protease Inhibitor Tablets (Sigma). After lysis, sonication, and purification on Ni-NTA agarose resin (Qiagen), His7-SUMO was cleaved off using SUMO protease. After validating SUMO cleavage by SDS-PAGE/Coomassie staining, the mixture was concentrated on an Amicon 50K Centrifugal Filter Unit (Millipore) and applied to a Superdex 200 Increase 10/300 GL column (GE Healthcare) equilibrated in 50 mM HEPES [pH 7.5], 400 mM KCl, 1 mM EDTA, 2 mM ßME, and 10% (v/v) glycerol. Fractions containing GyrA identified by SDS-PAGE/Coomassie staining were pooled and concentrated on a Amicon 50K Centrifugal Filter Unit. Glycerol was added to generate a final storage buffer of 45 mM HEPES [pH 7.5], 360 mM KCl, 0.9 mM EDTA, 1.8 mM ßME, 19% (v/v) glycerol and protein aliquots were stored at −80 °C. For GyrB, 1 L of induced cells was resuspended in 20 mL of buffer A800 (20 mM HEPES [pH 7.5], 30 mM imidazole, 800 mM NaCl, 2 mM ßME, 10% (v/v) glycerol) supplemented with a SIGMAFAST Protease Inhibitor Tablets (Sigma). After lysis, sonication, and purification on Ni-NTA agarose resin (Qiagen), His7-SUMO was cleaved off using SUMO protease. The mixture was concentrated on an Amicon 50K Centrifugal Filter Unit (Millipore) and applied to a Superdex 200 Increase 10/300 GL column (GE Healthcare) equilibrated in 50 mM HEPES [pH 7.5], 500 mM KCl, 1 mM EDTA, 2 mM ßME, and 10% (v/v) glycerol. Fractions containing GyrB identified by SDS-PAGE/Coomassie staining were pooled, concentrated on a Amicon 50K Centrifugal Filter Unit. Glycerol was added and the buffer adjusted to make a final storage buffer of 50 mM HEPES [pH 7.5], 500 mM KCl, 1 mM EDTA, 2 mM ßME, 30% (v/v) glycerol and protein aliquots were stored at −80 °C.
E. coli GyrA and GyrB were purified from pET28-GyrA and pET28-GyrB (generous gift from J. Berger) as follows. Proteins were grown in 2xYT at 37°C to OD600 ~ 0.5 and induced with 0.25 mM IPTG overnight. Pellets were resuspended in buffer A800 (20 mM Tris-HCl, [pH 8.0], 800 mM NaCl, 30 mM imidazole, 10% (v/v) glycerol) supplemented with a SIGMAFAST Protease Inhibitor Tablet (Sigma) and lysed, sonicated, and loaded on Ni-NTA agarose resin (Qiagen). GyrA was washed with buffer B (buffer A1000 except with 100 mM NaCl), and eluted in 20 mM Tris-HCl, [pH 8.0], 100 mM NaCl, 500 mM imidazole, 2 mM ßME, and 10% (v/v) glycerol. Proteins were then washed back into buffer B with 2 mM ßME with a Amicon 50K Centrifugal Filter Unit (Millipore). 1:50 (w/w) TEV protease (Sigma) was added and proteins were incubated overnight at 4°C with shaking. Cleaved GyrA was recovered by passage through Ni-NTA agarose resin (Qiagen), concentrated, and applied to a Superdex 200 Increase 10/300 GL column (GE Healthcare) equilibrated in 50 mM TrisHCl, [pH 8.0], 500 mM KCl, 2 mM ßME, and 10% (v/v) glycerol. Fractions containing GyrA identified by SDS-PAGE/Coomassie staining were pooled, concentrated, and snap frozen. His-TEVGyrB was eluted from Ni-NTA resin with buffer C (buffer A1000 except with 400 mM NaCl), concentrated, and directly applied to a Superdex 200 Increase 10/300 GL column (GE Healthcare) equilibrated in 50 mM HEPES [pH 7.5], 500 mM KCl, 1 mM EDTA, 2 mM ßME, and 10% (v/v) glycerol. Fractions containing His-TEV-GyrB identified by SDS-PAGE/Coomassie staining were pooled and concentrated. Glycerol was added and the buffer adjusted to make a final storage buffer of 50 mM HEPES [pH 7.5], 500 mM KCl, 1 mM EDTA, 2 mM ßME, 30% (v/v) glycerol and protein aliquots were stored at −80 °C.
Archaeoglobus fulgidus reverse gyrase (Rgy) was purified as follows: pSG483 (generous gift from J. Berger) was transformed into E. coli C41(DE3). Cells were grown in 2xYT to A600 = 0.5 at 37 °C and induced with 0.5 mM IPTG for 18–20 h at 18 °C. 1 L of cells was resuspended in 20 mL of buffer A (50 mM Tris-Cl [pH 8.0], 300 mM NaCl) supplemented with a SIGMAFAST Protease Inhibitor Tablet (Sigma). The cell resuspension was treated with 20 mg lysozyme and 1 mM PMSF at room temperature with gentle agitation for 20 min and then lysed by sonication. Lysates were spun for 30 min at 40,000 rpm in a Beckman Ti-45 rotor and the supernatant was passed over Ni-NTA agarose resin (Qiagen) pre-equilibrated with buffer A + 10mM imidazole. The resin was washed with buffer A + 20 mM imidazole and eluted in buffer A + 150 mM imidazole. The eluate was concentrated on Amicon 50K Centrifugal filter unit (Millipore) and supplemented with MgCl2 to a final concentration of 10 mM. The concentrate was applied to a Superdex 200 Increase 10/300 GL column equilibrated in buffer B (20mM Tris-Cl [pH 8.0], 200 mM NaCl, 10 mM MgCl2). Fractions containing Rgy as determined by SDS-PAGE and Coomassie staining were combined and concentrated on Amicon 50K Centrifugal filter unit (Millipore), aliquoted, and stored at −80 °C.
For C. crescentus and Bosea sp. GapR structural studies, pET15b expression plasmids were transformed into C41(DE3) cells. For protein expression, cells containing the plasmids were grown to an OD600 of 0.5 at 37 ºC and induced with 0.5 mM IPTG overnight at 15 ºC. Both proteins were purified using the same protocol. Briefly, the cells containing expressed GapR protein were pelleted and then resuspended in Buffer A (50 mM Tris-Cl [pH 7.5], 300 mM NaCl, 5% glycerol) with 1.0 mM β-mercaptoethanol (BME) and 600 U benzonase. The reconstituted cells were lysed using a micro-fluidizer and then centrifuged at 16000 rpm for 40 min at 4 ºC. The lysate was loaded onto a cobalt-NTA column and washed overnight with Buffer A. The following day, the protein was eluted stepwise, with increasing concentrations of imidazole. Both GapR proteins eluted within the 100–500 mM imidazole range and were >95% pure at this stage. The proteins were further purified via a size exclusion chromatography step.
To produce selenomethionine-substituted proteins for phasing, a C. crescentus GapR(L83M) mutant was generated as the WT protein has only one methionine. The Bosea sp. protein has no methionines except the N-terminal methionine so a Bosea sp. GapR(L48M-I54M-L73M) mutant was constructed. The selenomethionine proteins were purified as per the WT proteins except that 5 mM BME was used in the purification buffers. After purification, the proteins were concentrated to 1 mg/mL, buffer exchanged in buffer A without imidazole and the his-tag was cleaved overnight at RT using a thrombin capture cleavage kit. The cleaved tag was removed by buffer exchanging the protein into buffer A using a 10 kDa MW cutoff centricon.
SEC-MALS
SEC-MALS was performed with assistance from B. Brown on equipment owned by the T. Baker Lab at MIT. Size-exclusion chromatography was performed on a Wyatt WTC-030S5 size-exclusion column with an Agilent HPLC. Multi-angle light scattering was measured with a Wyatt DAWNHELEOS-2 instrument and protein concentration was determined using a Wyatt Optilab rEX.
The system was equilibrated in GapR storage buffer (50 mM sodium phosphate [pH 7.5], 200 mM NaCl, 10% (v/v) glycerol). 100 μL of 1 mg/mL GapR was injected and run at 0.5 mL/min in GapR storage buffer at room temperature. The signals were aligned and normalized internally with the ASTRA software 5.3.4 (Wyatt Technology). Bovine serum albumin (Sigma) was used as a standard. Absorbance at 280 nm was used to calculate protein molecular weight.
Preparation of DNA substrates
For 50, 20, and 16 bp DNA substrates, oligos were denatured at 95 °C for 2 min, and allowed to cool slowly to room temperature. Larger DNA substrates were generated by PCR and purified with PCR Purification Kits (Qiagen).
Negatively supercoiled pUC19 was harvested from E. coli using a Midiprep Kit (Qiagen) or purchased from NEB. Relaxed pUC19 was generated with E. coli Topoisomerase I (NEB). Nicked pUC19 was generated with Nt.BspQI (NEB). Linear pUC19 was generated with SmaI-HF (NEB). Positively supercoiled pUC19 was made using reverse gyrase: a 50 μL reaction containing 16.33 μg of negatively supercoiled pUC19 and ~500 nM reverse gyrase was incubated in 50 mM Tris-HCl [pH 8.0], 50 mM NaCl, 10 mM MgCl2, 1 mM ATP, and 10% PEG 8000 at 80 °C for 30 min. All enzymatic reactions were stopped by adding 1% SDS and 10 mM EDTA (final concentration) and plasmid DNA recovered by phenol/chloroform extraction and isopropanol precipitation.
DNA electrophoresis and visualization
Short (< 300 bp) fragments were electrophoresed in 6% DNA Retardation gels (Invitrogen) at 100 V for 60–90 min at 4 °C. pUC19 plasmids were electrophoresed in 1% TBE agarose gels either at 30 V for 16 hr at 4 °C (binding assays) or at 130 V for 90 min (topology assays). Gels were stained in SYBR Gold (Invitrogen) and imaged with a Typhoon FLA 9500 imager (GE Lifesciences).
For two-dimensional chloroquine gels, electrophoresis was performed by first running reactions on a 1% TBE agarose gel at 130 V for 90 min, then soaking the gel for 2 hr with shaking in 1X TBE supplemented with 4.5 μg/mL chloroquine phosphate (Santa Cruz Biotech). The gel was then turned and electrophoresed in the orthogonal direction at 130 V for 60 min in 1X TBE supplemented with 4.5 μg/mL chloroquine phosphate. Chloroquine is a DNA intercalator that introduces (+) supercoils. In chloroquine, (−) supercoiled plasmids will become more relaxed, and migrate more slowly, whereas (+) supercoiled DNA will be further compacted, increasing its migration speed. The gel was washed 4 × 20 min in distilled water to remove chloroquine, stained with SYBR Gold (Invitrogen), and imaged with a Typhoon FLA 9500 imager (GE Lifesciences).
Electrophoretic mobility shift assays
Reactions (15 μL) with indicated amounts of GapR and linear dsDNA or pUC19 DNA (40 ng) in binding buffer (40 mM sodium phosphate [pH 7.5], 100 mM NaCl, 50 μg/mL ultrapure BSA, 0.5 mM DTT) were incubated at 30 °C for 30 min and then placed on ice. DNA loading buffer was added and 10 μL of the reactions were electrophoresed. For experiments with multiple DNA fragments, 40 ng total of DNA was used and the fragments were mixed at equal weight.
DNA topology assays
For topoisomerase I treatment, mixtures of GapR and relaxed pUC19 DNA (40 ng) in binding buffer (40 mM sodium phosphate [pH 7.5], 100 mM NaCl, 50 μg/mL ultrapure BSA, 0.5 mM DTT) were incubated at 30°C for 30 min. Calf Thymus Topoisomerase I (Invitrogen) was diluted to 1 U/μL in Topoisomerase Dilution Buffer (30 mM potassium phosphate [pH 7.0], 5 mM DTT, 0.1 mM EDTA, 0.2 mg/mL BSA, 50% (v/v) glycerol, 0.1% (w/v) Triton X-100) and 1 U was added to reactions and incubated for an additional 30 min at 30 °C. Reactions were stopped by addition of 1% SDS and 10 mM EDTA (final concentration), and digested with Proteinase K (NEB) for 1 hr at 37°C. DNA loading buffer was then added and samples electrophoresed.
For ligase assays, mixtures of GapR and nicked pUC19 DNA (40 ng) in 1X T4 DNA ligase buffer were incubated at 30 °C for 30 min. T4 DNA ligase (NEB) was diluted to 1 U/μL in 1X T4 DNA ligase buffer and 1 U was added to reactions and incubated for an additional 2 hr at room temperature. Reactions were stopped as with topoisomerase I assays. DNA loading buffer was added and the reactions electrophoresed. GapR protein preparations additionally contained trace nuclease activity, which converted plasmids into their linear forms during the assay. The linear forms are observed as an intermediate band (see Fig. 5F, S5E).
DNase I protection assay
Mixtures of GapR and negatively supercoiled pUC19 DNA (40 ng) in binding buffer (40 mM sodium phosphate [pH 7.5], 100 mM NaCl, 50 μg/mL ultrapure BSA, 0.5 mM DTT) were incubated at 30 °C for 30 min. Turbo DNase (Invitrogen) was diluted to 1 U/μL in 1X DNase buffer and 1 μL was added to reactions and incubated for an additional 5 min at 30°C. Reactions were quenched by addition of 1 μL 10% SDS and incubation at 75°C for 10 min. Samples were subsequently analyzed by electrophoresis.
Crystallization of C. crescentus GapR-DNA complex, Bosea sp. apo and Bosea sp. GapR-DNAbound complex.
To obtain crystals of the C. crescentus GapR-DNA complex, the protein was first concentrated to 20 mg/mL and 1 mM DNA (top strand 5´-TTAAAATTAAA-3´), final concentration, was added. Excess DNA was removed by concentrating the sample with a 30 kDa MW cutoff microcon. Crystals were obtained at RT using the hanging drop vapour diffusion method by mixing the GapR-DNA complex 1:1 with a crystallization solution consisting of 28% PEG 400, 0.1 M HEPES pH 7.5 and 0.2 M CaCl2 and suspending the drop over a reservoir containing the crystallization solution. The crystals grew overnight and reached their final size after 2 weeks. The crystals were cryo-preserved by direct transfer from the drop to the cryo-stream. WT GapR and selenomethionine GapR(L83M) proteins in complex with the DN produced the same crystals. The apo Bosea sp. GapR was crystallized by hanging drop vapor diffusion at RT using protein at 40 mg/mL and mixing the protein 1:1 with a solution of 15% MPD, 10 mM MgCl2, 0.1 M HEPES pH 7.5 and 0.001 M spermine. The crystals grew overnight and could be cryo-preserved straight from the drop. The same crystals were produced with the selenomethionine-substituted Bosea sp. GapR(L48M-I54M-L73M) protein. Crystals of the WT Bosea sp. GapR bound to DNA (5´-TTAAAATTAAA-3´) were grown via hanging drop vapor diffusion at RT by mixing the GapR protein (at 20 mg/mL) at a ratio of 1:1 (one GapR dimer-ofdimer to one DNA duplex) and combining the mixture with a crystallization solution consisting of 1.7 M ammonium sulphate and 0.1 M MES pH 6.5. The crystals reached their final size after 1–2 weeks and were cryo-preserved by looping a crystal into a 2 uL drop consisting of the crystallization reagent supplemented with 15% glycerol for 1–2 s before direct placement of the crystal in the cryostream.
Structure determination and refinement of the C. crescentus GapR-DNA complex, Bosea sp. apo and DNA-bound complex.
All X-ray intensity data were collected at the advanced light source (ALS) beamline 8.3.1. The selenomethionine C. crescentus GapR(L83M)-DNA complex was solved by single wavelength anomalous diffraction (SAD) using data collected from a crystal at the selenium peak. Heavy atom sites were located and density modification performed in Phenix Autosol. The density modified map from Autosol was used to construct the model in O (Jones et al., 1991). The crystal takes the space group C2 (Table S4) and there are four GapR subunits and one DNA duplex in the crystallographic asymmetric unit (ASU). Once the model was fit, it was subjected to refinement in Phenix. After several rounds of refinement, refitting and analysis with Molprobity (Chen et al., 2010), the model converged to Rwork/Rfree values of 24.1%/26.8% to 2.3 Å resolution. The final model contains residues 12–88 of three GapR subunits, 12–89 of the fourth GapR subunit, the entire DNA duplex and 104 water molecules. See Table S4 for final refinement statistics. Parameters of GapR-bound DNA was analyzed using the w3DNA program (Zheng et al., 2009).
The apo Bosea sp. GapR(L48M-I54M-L73M structure) was solved by SAD using data collected from a crystal grown with selenomethionine protein. Autosol was used to locate the selenium sites and produce a density modified, electron density map. The crystal (P41212) contains a single GapR subunit in the ASU; the dimer is generated by crystallographic symmetry. The model was constructed in O (Jones et al., 1991). and refined using phenix-refine. The final structure has Rwork/Rfree values of 22.2%/25.9% to 2.4 Å resolution and contains residues 9–78 of the GapR subunit and 3 solvent molecules. The final refinement statistics are provided in Table S4.
The Bosea sp. GapR-DNA crystals were produced with the WT protein (non-selenomethionine) and the structure was solved by molecular replacement (MR). The Bosea sp. apo structure did not produce a solution in MR however, the C. crescentus GapR dimer-of-dimer with the DNA removed produced an MR solution using the program MolRep. The MR solution was first subjected to rigid body refinement and then x,y,z,b refinement using Phenix-refine to 2.0 Å resolution. After the first round of Phenix-refine (3 cycles) the electron density maps were used to fix the model and replace the C. crescentus GapR residues with those in the Bosea sp. GapR sequence. Molprobity was used throughout to assess the model during refinement (Chen et al., 2010). Density for the DNA was evident after initial refinement rounds however it could not be unambiguously traced. Ethidium bromide staining of the crystals also supported the presence of DNA. The final model includes 349 solvent molecules and four GapR subunits, with residues 6–78, 5–78, 3–78 and 3–80 and has final Rwork/Rfree values of 19.8%/22.2% to 2.0 Å resolution.
Fluorescence polarization-based DNA binding experiments
Fluorescence polarization (FP) experiments were performed using a PanVera Beacon 2000 FP system at 25 °C. Samples were excited at 490 nM and fluorescence emission was measured at 530 nM. 5′Fluoresceinated oligonucleotides used for DNA binding experiments were the site used for crystallization (AT-rich, top strand: 5´-F-TTAAAATTAAA-3´), a GC-rich site (5´-FTGGGCGGGCGCCCGCCCA-3´) and a mixed bp 12mer (5´-F-GTGAGTACTCAC-3´). For each FP experiment, WT C. crescentus GapR protein was titrated into 0.995 mL of reaction buffer (25 mM Tris, pH 7.5, 120 mM NaCl) containing 1 nM fluoresceinated oligonucleotide and points taken. All data were fit using Kaleigraph. The analyses were performed in triplicate (technical duplicates).
Topo IV relaxation assays
Caulobacter topo IV was reconstituted on ice and supercoiling assays were performed as follows. Briefly, equimolar purified ParC and ParE were mixed together on ice for 10 min to form 8.63 μM tetramer. Reconstituted topo IV was serially diluted in topo IV dilution buffer (50 mM Tris-HCl [pH 8.0], 150 mM potassium glutamate, 6 mM MgCl2, 10% (v/v) glycerol). 1.5 μL of 400 nM GapR and 1 μL of diluted topo IV tetramer were incubated together on ice for 30 min, then at 30 °C for 5 min. 40 ng pUC19 DNA in topo IV reaction buffer (50 mM Tris-HCl [pH 8.0], 30 mM potassium glutamate, 6 mM MgCl2, 0.5% (v/v) glycerol, 12.5 μg/mL ultrapure BSA, 12.5 mM DTT, 2 mM ATP) was added to generate 16 μL final volume (final concentration of topo IV was 17.55 nM for (−) supercoil relaxation, 2.34 nM for (+) supercoil relaxation). Topoisomerase concentrations and conditions were selected to deliberately yield relatively low topoisomerase activities. At the indicated times, reactions were quenched by addition of 1% SDS and 10 mM EDTA (final concentration), and treated with Proteinase K (NEB) for 1 hr at 37 °C. Topoisomers were analyzed by electrophoresis. For reactions with G6PD or GcrA, assays proceeded with identical conditions as with GapR, using 400 nM (monomer) to maintain equimolar protein. For reactions with E. coli topo IV, topo IV (TopoGEN) was diluted in topo IV buffer and treated identically as with Caulobacter topo IV. 0.5 U total topo IV (0.03125 U per μL) was used for (−) supercoil relaxation and 0.25 U (0.0156 U per μL) was used for (+) supercoil relaxation.
Gyrase supercoiling assays
Caulobacter gyrase was reconstituted on ice and supercoiling assays performed as follows. Briefly, equimolar GyrA and GyrB were mixed on ice for 10 min to form 9.5 μM tetramer. Reconstituted tetramer was serially diluted in gyrase dilution buffer (25 mM Tris-HCl [pH 8.0], 150 mM potassium glutamate, 2.5 mM MgCl2, 10% (v/v) glycerol), and incubated for 5 min on ice after each dilution. 1.5 μL of 400 nM GapR and 1 μL of diluted gyrase tetramer were incubated on ice for 30 min, then at 30 °C for 5 min. 40 ng of pUC19 DNA in gyrase reaction buffer (30 mM Tris-HCl [pH 8.0], 120 mM potassium glutamate, 5.5 mM MgCl2, 0.1 mg/mL ultrapure BSA, 1 mM DTT, 2 mM ATP, 10% (v/v) glycerol) was added to generate 16 μL final volume (final concentration of gyrase was 12.5 nM for introduction of (−) supercoils, 2.81 nM for (+) supercoil relaxation). Topoisomerase concentrations and conditions were selected to deliberately yield relatively low topoisomerase activities. At the indicated times, reactions were quenched as with topo IV assays. Topoisomers were analyzed by electrophoresis. For reactions with G6PD or GcrA, assays proceeded with identical conditions as with GapR, using 400 nM (monomer) to maintain equimolar protein. For reactions with E. coli gyrase, purified GyrA and His-TEV-GyrB were reconstituted and diluted as with Caulobacter gyrase. Gyrase was used at 2.3 nM for (+) supercoil relaxation and 6.8 nM for (−) supercoil introduction.
Quantification and Statistical Analysis
Calculation of growth rates
For wild-type cells the GapR depletion strain in xylose (Fig. S1C), doubling time was calculated from a linear fit of the log-transformed OD600 values.
Quantification of protein levels by immunoblotting
Image quantification and analysis was done with Fiji/ImageJ. GapR band intensities were normalized against an internal standard. Estimated copy number of GapR per cell was calculated from a linear regression of a standard curve generated from purified GapR. The mean and SEM of at least two replicates for each condition are reported.
Analysis of RNA sequencing
Data analysis was performed using custom scripts written in Python 2.7.6. Single-end sequencing reads were aligned to Caulobacter NC011916.1 with bowtie using the following parameters: bowtie v 1 -M 1 --trim3 30 (trimming the last 30 bp of each read, allowing for 1 mismatch, and if a read has more than 1 reportable alignments, report 1 of them at random). The read coverage was mapped to the Caulobacter genome by assigning each mapped base a value of 1/N where N equals the length of the read. To calculate mRNA abundance, the number of reads mapped to a gene was divided by the length of the gene to yield the mean number of reads per kilobase per million sequencing reads (RPKM). The fold change in gene expression for the GapR depletion was computed by taking the RPKM ratio for each gene from GapR-depleted cells to wild-type cells grown in glucose.
We compared the gene expression of the GapR-depletion strain (GSE100657, Table S2) and cells treated with novobiocin (Table S2) to the following datasets: UV and MMC microarrays, which were merged and averaged to form a consensus “DNA damage” dataset (Modell et al., 2011); a microarray of the induction of the toxin SocB (ArrayExpress E-MEXP-3990); RNA-seq after treatment with 4% ethanol (GSE90030); and a microarray of a SciP depletion strain (GSE22062). We computed the Pearson correlation for GapR depletion and genes that were changed at least 2-fold in at least 2 of 3 conditions (UV, MMC, and SocB).
LexA regulated genes were defined based on the presence of a consensus LexA-binding site (Modell et al., 2011). Ethanol-responsive genes were defined as those that were differentially expressed at least 4-fold in ethanol treated cells (432 genes). Supercoiling-responsive genes were defined as those differentially expressed at least 4-fold in novobiocin treated cells (289 genes total). We computed the Pearson correlation for the supercoiling regulon genes between the each dataset and novobiocin treatement data.
ChIP analysis
For GapR ChIP, reads were mapped to the Caulobacter crescentus NA1000 reference genome (NC011916.1) using BWA. Read extension and pile-up was performed using the MACS software package 1.4.2 (with d=200 and keeping all tags at the same location). The data were further process using custom scripts written in Python 2.7.3. The data were smoothed by convolution with a Gaussian (μ=0, σ=50bp, x=(−200 bp, +200 bp)). The data were then normalized in reads per million (rpm). ChIP of RNA polymerase subunit rpoC (rpoC::rpoC-3×FLAG) in PYE was analyzed similarly.
To identify DNA sequence motifs enriched in GapR bound sequences, we isolated regions with GapR ChIP signal above 1.5 rpm (regions less than 150bps apart were merged). A 200 bp window centered at the maximum signal intensity of each of these regions was retrieved and submitted to DREME for sequence motif analysis. In Fig. 2C-D,F, the AT content at each base pair was computed using a centered 100 bp sliding window. In Fig. 2E, GapR ChIP enrichment for each gene was measured by taking the sum of the GapR signal over the coding region and normalizing by the length of the gene. In Fig. S2A, promoters were defined as the 300 bp region upstream of transcription start sites identified from RNAP ChIP-seq. For Fig. S2A, GapR ChIP signal at promoters was computed by taking the sum of the GapR signal over the defined promoter region divided by 300 (the length of the promoter region).
To analyze the GapR ChIP signal at the ends of transcription units, we first identified transcription units in the Caulobacter genome based on the microbes online operon annotation and we included single genes when not annotated in the microbes online operon database if they were located in between two genes found on the opposite DNA strand. We then looked for a change in enrichment over the rifampicin treated signal associated with the ends of transcription units. To do this, we computed the average enrichment (untreated-rifampicin treated) over 1000 bp following and preceding the end of a transcription unit (last base of the last gene in the transcription unit), we then computed the difference of these two enrichments as a measure of GapR signal (see Fig. S2D). As a control, we applied the same metric to the starts of transcription units (first base of the first gene of the transcription unit). In a separate analysis, we computed the average ChIP signal enrichment (untreated-rifampicin treated) at the ends and starts of high or low expression transcription units for each base pair over a 2000 bp window (Fig. S2E). To do so, we took the difference of these two ChIP signals (untreated-rifampicin treated) at each base pair over a 2000 bp window centered at the ends or starts of transcription units and computed the average over all high or low expression transcription units. In both analysis, we limited our analysis to transcription units at least 1000 bp long and we used RNA-seq data of the GapR-3×FLAG strain to categorize transcription units as high (>150 RPKM) or low (<50 RPKM) expression. When a transcription unit contained more than one gene, the expression value of the last gene in the operon was used.
Analysis of replisome progression by flow cytometry
Flow cytometry was analyzed with FlowJo from 50,000 total events for each experiment. For each experiment, 1N was calculated from the mean of the distribution of G1 cells, and 2N was assumed to be twice the value of 1N. To estimate the fraction of cells that initiated, the fraction of cells with DNA content greater than the 95th percentile of 1N cells was calculated. To estimate replication rate, a linear regression of the mean staining of the 70–71st percentile of cells versus time was determined while accumulation of DNA content was in the linear range. The slope of this regression line was used as the replication rate. At least two independent experiments were used for each analysis, which are reported as mean ± SEM.
For experiments examining replication progression after treatment with novobiocin, average DNA content was calculated using the mode of the distribution, as replisomes in GapR-depleted cells displayed heterogeneous behavior. The replication rate was calculated as a linear fit of the mode of the DNA content versus time. At least two independent experiments were used for each analysis, which are reported as mean ± SEM.
Analysis of DNA replication from DNA sequencing
Data analysis was performed using custom scripts written in Python 2.7.6. Paired-end sequencing reads were aligned to Caulobacter NC011916.1 with bowtie2 using the following parameters: bowtie2 –N 1 (mismatches in seed set to 1). The genome was shifted 1 MB to the right (3016947 to end, 0 to 3016947) to ensure coverage of the origin region. Duplicate reads were then removed using samtools rmdup –S. The central nucleotide of each pair of reads was then mapped to the Caulobacter genome.
The Caulobacter genome was divided into 40,001 bins of 1000 bp each, and the total number of reads in each bin was determined for every sample. Samples were then normalized by dividing by the binned data from the corresponding 0 min (1N chromosome content) control, to reduce sequencing biases. The normalized data was then divided by the median of the 20 kb surrounding ter region to set the copy number of ter as 1. The normalized data was then smoothed by the nonparametric LOWESS method in Python, with a smoothing variable (frac) of 0.04 and plotted against the genomic position.
LOWESS smoothed data was analyzed to measure initiation and replication rate. To measure replication initiation at each time-point, the maximum read enrichment within the 200 kb region surrounding the origin was determined from smoothed data and divided by the mean value of a 20 kb unreplicated region near the ter. The initiated value decreases in wild-type over time as replication forks reach ter since the number of reads at ter is set to 1. Replication rate was determined as follows. The fork position was first defined the position with DNA content halfway between the maximal and minimal DNA content found at 20 min. For each 20 min increment (i.e., 20–40 min, 40–60 min, 60–80 min), the replication rate was then calculated by estimating the distance travelled by the replication fork. This analysis was then repeated six times for positions with DNA content evenly-spaced between the maximal and minimal DNA content. These seven measurements were averaged and reported as mean ± SEM.
Quantification of cell division
Image analysis was performed with the MicrobeJ suite in Fiji/ImageJ. Cells were recognized with built-in algorithms with threshold offset set to 20. Non-cells were excluded based on size thresholds. Cell segmentation was performed with the built-in suite with tolerance set to 0.11. Over 250 cells for each time point was counted to determine the fraction of pre-divisional cells.
Quantification of origin-terminus ratio by qPCR
On each plate, relative quantities of cDNA in a given sample were calculated by comparison to a least-square fit on a 2-fold dilution standard curve (Cp vs. log-transformed standard fold dilution). Terminus region quantities were normalized to origin region quantities for each time point, and normalized by the origin abundance after synchrony (0 min). At least two independent experiments were used for this analysis.
Quantification of DNA binding and topoisomerase assays
Image quantification was done with Fiji/ImageJ. To calculate binding affinities and hill coefficients, the amount of bound and free oligonucleotide was determined as a fraction of the total intensity present in the lane. Curves were fitted using the Prism 7 software, using the Specific binding with Hill slope algorithm. At least two independent experiments were used for each analysis, and are reported as mean ± SEM.
For topoisomerase assays, band intensities were measured and normalized based on the intensity of the input. For relaxation of (+) and (−) supercoils, the intensity of the most supercoiled band was measured and normalized by the total intensity. For relaxed plasmids, the fraction of the total intensity with < 3 topoisomers was measured and normalized by the total intensity. For Fig. S7F and other negative supercoiling assays with gyrase, the fraction of the total intensity with > 4 apparent topoisomers was measured and normalized by the total intensity. At least two independent experiments were used for each analysis, which are displayed as mean ± SEM at each time point. The fold-stimulation due to GapR was calculated from the difference in the slope of the change in supercoiled fractions over time.
Data availability
RNA-seq, ChIP-seq, and DNA-seq data are available in the Gene Expression Omnibus (GEO): GSE100657 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE100657). Coordinates and structure factor amplitudes for the C. crescentus GapR-DNA, apo Bosea sp. GapR and Bosea sp. GapR-DNA complex have been deposited with the Protein Data Bank under the Accession codes 6CG8, 6CFY and 6CFX, respectively.
Supplementary Material
Figure S1. Identification of GapR, a highly conserved α-proteobacterial protein, related to Figure 1.
(A) Schematic of nucleoid purification mass spectrometry assay, with a representative DAPI staining profile. For proteins identified in mass spectrometry, see Table S1.
(B) GapR is highly conserved in α-proteobacteria. The number of GapR homologs identified in and the total number of genomes searched for each class of proteobacteria (left). The occurrence (reported as a percentage of genomes searched) and conservation (reported as average of BLAST bit scores) of GapR homologs is shown for each genome in the individual clades of α-proteobacteria (right). $$PARABREAKHERE$$(C) Growth of wild-type and ΔgapR Pxyl-gapR cells assessed by 10-fold serial dilutions onto PYE (rich medium) plates containing either glucose or xylose (top). Plates were imaged after the indicated number of days. Growth in liquid rich medium containing xylose (middle). Growth of wild-type and ΔgapR Pxyl-gapR cells on M2 minimal medium plates containing either glucose or xylose (bottom). $$PARABREAKHERE$$(D) Growth of wild-type, ΔgapR Pxyl-gapR, and a strain where ΔgapR is complemented by gapR expressed from its native promoter integrated at the xyl locus (ΔgapR xyl::PgapR-gapR).
(E) GapR-depleted cells exhibit a general stress response. Expression changes of ΔgapR Pxyl-gapR cells in xylose or glucose, cells treated with novobiocin, DNA damaging agents, the toxin SocB, ethanol stress, or cell-cycle arrest by SciP depletion. Genes that change >2-fold in ethanol are shown.
(F) Immunoblots of GapR in wild-type and the gapR::gapR-3×FLAG strain (top). Asterisk (*) indicates a non-specific band used as a loading control. GapR-3×FLAG levels are 2.2 ± 0.47 fold higher than wild-type GapR levels, mean ± SEM, n = 2. Immunoblots quantifying GapR protein levels in wild-type cells, using purified GapR standards (bottom). Mean ± SEM, n = 4.
(G) Ethidium bromide (EtBr) nucleoid sedimentation analysis to determine relative levels of (−) supercoiling in wild-type, novobiocin-treated, and GapR-depleted cells grown for 2.5 hr in glucose. LOWESS fit of data is shown with solid lines. Approximate concentration of EtBr required to relax the chromosomes is shown with dashed lines.
Figure S2. GapR ChIP-seq, related to Figure 2.
(A) GapR binding and gene expression changes are not correlated. Scatterplot of gene expression changes in GapR-depleted cells versus average GapR ChIP-seq enrichment at promoters. Blue shaded area encompasses genes that change < 2-fold upon GapR depletion.
(B) GapR binding and gene expression changes at a ribosomal protein locus. ChIP signal (middle) and AT content (top) are plotted as in Fig. 2D, RNA-seq data show reads per million (rpm), and the positions of annotated genes are indicated (bottom, with ribosomal operon genes in solid black).
(C) GapR ChIP signal is unchanged at AT rich peaks in untreated versus rifampicin-treated cells.
(D) Flow chart of the ChIP analysis performed to identify transcription-dependent GapR occupancy at the end of transcription units.
(E) Meta-analysis of GapR occupancy at the 5’ and 3’ ends of genes after rifampicin treatment. Normalized change in GapR ChIP occupancy is calculated from the difference in enrichment (untreated minus rifampicin treated cells) over a 1 kb window before and after the 3’ or 5’ ends of transcription units (TUs). High expression TUs have RPKM > 150, low expression TUs have RPKM < 50. Only TUs > 1000 bp were analyzed.
Figure S3. Replication initiation and elongation is impaired in GapR-reduced cells, related to Figure 3.
(A) Schematic of assay to assess DNA replication. Wild-type and ΔgapR Pxyl-gapR cells were grown in xylose, synchronized in G1, and released into medium with xylose (+ xyl) or glucose (+ glu). DNA content was measured with SYTOX staining and flow cytometry or DNA sequencing at various times post release.
(B) DNA replication time courses for wild type and GapR-reduced strains. Blue lines represent 1N and 2N DNA content.
(C) Fraction of cells from (B) that initiated replication over time, with DNA content > 1N used as a proxy for initiation. Data are mean ± SEM, n ≥ 2.
(D) Replication rate for cells in (B), calculated from a linear fit of the DNA content of cells over time and normalized to wild type. Data are mean ± SEM, n ≥ 2.
(E) DNA sequencing of wild-type cells grown in glucose, ΔgapR Pxyl-gapR cells grown in xylose and released into glucose after synchrony as in (B), and ΔgapR Pxyl-gapR cells depleted for 2 hr in glucose before synchrony (from Fig. 4E). DNA content, normalized to that of synchronized G1 cells, is shown as a function of chromosome position using LOWESS smoothing with 10 kb windows. Blue lines represent locations of the replication origin and terminus. Schematics indicate progression of wild-type replication forks.
Figure S4. Replication elongation defects of GapR-depleted cells, related to Figure 3 and Figure 4.
(A) Extended DNA replication timecourse for wild-type and GapR-depleted cells. Cells were grown in xylose, washed into glucose for 2 hr, synchronized in G1, and released into medium with glucose. DNA content was measured with SYTOX staining and flow cytometry for 4 hr after release.
(B) GapR-depleted cells do not successfully divide. Cells treated as in (A) were fixed and imaged to assess cell division by quantifying the percent of predivisional cells (n > 250 cells per timepoint, left). Representative images of wild-type and GapR-depleted cells at 90, 120, and 210 min post release into glucose (right).
(C) Origin-terminus ratio of wild-type and GapR-depleted cells. DNA was isolated from cells treated as in (A) and the relative copy number of the origin and terminus was determined by qPCR. One representative wild type dataset is shown, and the mean ± SEM is reported for GapR-depleted cells, n ≥ 2.
(D) Sensitivity of GapR-depleted cells to 0.3 mg/mL hydroxyurea (HU) and 0.3 μg/mL rifampicin (rif).
Figure S5. In vitro characterization of GapR binding to DNA, related to Figure 5.
(A) Gel filtration trace of purified GapR (70 μM monomer) (top). Absorbance at 280 nm (blue) and at 260 nm (red) are shown. Arrow indicates approximate elution peak for lysozyme monomer (14.3 kD). SEC-MALS analysis of purified GapR (1 mg/mL = 100 μM monomer) (bottom). Apparent molar mass calculated from MALS (red) and absorbance at 280 nm (grey) are shown. GapR is monodisperse (Mw/Mn = 1.000), with an observed mass of 26.4 kDa (expected 20.7 kDa for homodimer).
(B) EMSAs of GapR binding to linear dsDNA. Purified GapR was incubated with 20 (left) and 50 bp DNA (right).
(C) GapR binding curves for various substrates (left). Apparent KD for each substrate is: 16 bp, 4605 nM GapR; 20 bp, 745 nM; 50 bp, 583 nM; 150 bp, 406 nM; 210 bp, 314 nM; 300 bp, 220 nM; and pUC19 589 nM. Data are mean ± SEM, n ≥ 2. Hill coefficients ± SE (right).
(D) EMSA of GapR binding to (−) supercoiled pUC19.
(E) Trace nuclease contamination in GapR preparation. Relaxed, supercoiled, and linear standards compared to relaxed or (+) supercoiled pUC19 plasmid incubated with 0.4 μM GapR for 1 hr at 30°C, followed by reaction quenching, protein removal with Proteinase K, and electrophoresis.
(F) DNase protection by GapR. GapR was incubated with pUC19 and treated with DNase. Reactions were processed as in (E).
Figure S6. GapR DNA structure and contacts, related to Figure 6 and Figure 7.
(A) GapR-DNA contacts in stereoview. Shown are the side chains from the GapR dimer-of-dimers that interact with the DNA.
(B) Sequence logo of GapR computed from select homologs (top). Conserved acidic surface residues that are not involved in structural contacts or DNA binding (*). Sequence alignment of selected GapR homologs, organized by distance from C. crescentus GapR, with residues numbered based on C. crescentus GapR. Helices of GapR are shown for both the dimer and dimer-of-dimers.
(C) Superimposition of the DNA bound forms of the C. crescentus GapR (magenta) and the Bosea GapR (green), showing that they form the same dimer-of-dimers structure.
(D) Fluorescence polarization assay of C. crescentus GapR binding to AT-rich DNA (blue circles), GC-rich DNA (red squares), and a DNA template without AT stretches (green triangles).
Figure S7. Gyrase and topo IV are specifically stimulated by GapR, related to Figure 7.
(A) GapR stimulates topo IV. Topo IV (2.34 nM) was incubated with the indicated amounts of GapR before adding (+) supercoiled pUC19 and ATP. After incubating for the times indicated, reactions were quenched and analyzed by gel electrophoresis. Slower migration in the gel indicates relaxation of (+) supercoils.
(B) GapR modestly stimulates relaxation of (−) supercoils by topo IV. Reactions are the same as in (A) except with 17.55 nM topo IV, 0.4 μM GapR, and (−) supercoiled pUC19. Relaxed and (−) supercoiled standards are shown (left). Plot quantifies the % initial supercoiled plasmid (asterisk) at each time point (right). Data indicate mean ± SEM, n ≥ 2.
(C) Relaxation of (−) supercoils by topo IV is not stimulated by non-specific proteins. Assay is the same as in (B) except with 0.4 μM E. coli glucose-6-phosphate dehydrogenase (G6PD) or C. crescentus GcrA. A representative gel from at least two experiments is shown.
(D) GapR does not strongly stimulate E. coli Topo IV. Assay is the same as in (B) except with E. coli topo IV.
(E) GapR stimulates gyrase. Assay is the same as in (A) except with 6.25 nM gyrase. Relaxed and (+) supercoiled controls (left). Gyrase generates slower migration as (+) supercoils are relaxed and then induces faster migration as (−) supercoils are introduced.
(F) GapR stimulates introduction of (−) supercoils by gyrase. Assay is the same as in (E) except with 12.5 nM gyrase, 0.4 μM GapR, and relaxed pUC19. Faster migration in the gel indicates introduction of (−) supercoils. Plot quantifies % initial relaxed plasmid (asterisk) at each time point (bottom). Data indicate mean ± SEM, n ≥ 2.
(G) Introduction of (−) supercoils by gyrase is not stimulated by non-specific proteins. Assay is the same as in (F) except with 0.4 μM G6PD or GcrA.
(H) GapR stimulates E. coli gyrase. Assay is the same as in (F) except with E. coli gyrase (2.3 nM, (+) relaxation; 6.8 nM, (−) introduction). Plot quantifies % initial supercoiled plasmid (asterisk) at each time point (bottom). Data indicate mean ± SEM, n ≥ 2.
(I) Models for stimulation of topoisomerase activity by GapR. (Left) GapR may bind overtwisted DNA in front of replication forks, directly recruiting or activating type II topoisomerases to relax positively supercoiled DNA. (Right) GapR could bind overtwisted DNA flanking plectonemic DNA, thereby trapping the substrate of type II topoisomerases.
Table S2. Microarray data from wild-type cells in novobiocin and GapR-depleted cells in xylose, related to Figure 1.
Movie S1. Movie of Caulobacter GapR bound to DNA, related to Figure 6.
Highlights.
GapR is a DNA structuring protein conserved throughout α-proteobacteria
GapR promotes DNA replication initiation and elongation
GapR encircles overtwisted DNA as a dimer-of-dimers
GapR stimulates the relaxation of positive supercoils by gyrase and topo IV
Acknowledgements
We thank J. Berger for expression constructs, S. Goodman for antibodies, and M. LeRoux, M. Guzzo, and S. Bell for comments on the manuscript.
This work supported by NIH grants to M.A.S (R01GM115563), and to M.T.L (R01GM082899), who is also an Investigator of the Howard Hughes Medical Institute (HHMI), an HHMI International Predoctoral Fellowship to D.L.H., and a Jane Coffin Childs Memorial Fellowship to M.S.G.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arias-Cartin R, Dobihal GS, Campos M, Surovtsev IV, Parry B, and Jacobs-Wagner C (2017). Replication fork passage drives asymmetric dynamics of a critical nucleoid-associated protein in Caulobacter. EMBO J. 36, 301–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badrinarayanan A, Le TBK, and Laub MT (2015). Bacterial chromosome organization and segregation. Annu. Rev. Cell Dev. Biol 31, 171–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermejo R, Capra T, González-Huici V, Fachinetti D, Cocito A, Natoli G, Katou Y, Mori H, Kurokawa K, Shirahige K, et al. (2009). Genome-organizing factors Top2 and Hmo1 prevent chromosome fragility at sites of S phase transcription. Cell 138, 870–884. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haran TE, and Mohanty U (2009). The unique structure of A-tracts and intrinsic DNA bending. Q. Rev. Biophys 42, 41–81. [DOI] [PubMed] [Google Scholar]
- Hayama R, and Marians KJ (2010). Physical and functional interaction between the condensin MukB and the decatenase topoisomerase IV in Escherichia coli. Proc. Natl. Acad. Sci. USA 107, 18826–18831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins NP (2016). Species-specific supercoil dynamics of the bacterial nucleoid. Biophysical Reviews 8, 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RB (2006). Coordination between chromosome replication, segregation, and cell division in Caulobacter crescentus. J. Bacteriol 188, 2244–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, and Kjeldgaard M (1991). Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr., a, Found. Crystallogr 47 ( Pt 2), 110–119. [DOI] [PubMed] [Google Scholar]
- Kang S, Han JS, Park JH, Skarstad K, and Hwang DS (2003). SeqA protein stimulates the relaxing and decatenating activities of topoisomerase IV. J. Biol. Chem 278, 48779–48785. [DOI] [PubMed] [Google Scholar]
- Khodursky AB, Peter BJ, Schmid MB, DeRisi J, Botstein D, Brown PO, and Cozzarelli NR (2000). Analysis of topoisomerase function in bacterial replication fork movement: use of DNA microarrays. Proc. Natl. Acad. Sci. USA 97, 9419–9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal A, Dhar A, Trostel A, Kouzine F, Seshasayee ASN, and Adhya S (2016). Genome scale patterns of supercoiling in a bacterial chromosome. Nat Commun 7, 11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, and Marians KJ (2013). Characterization of the nucleoid-associated protein YejK. J. Biol. Chem 288, 31503–31516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Stewart NK, Berger AJ, Vos S, Schoeffler AJ, Berger JM, Chait BT, and Oakley MG (2010). Escherichia coli condensin MukB stimulates topoisomerase IV activity by a direct physical interaction. Proc. Natl. Acad. Sci. USA 107, 18832–18837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LF, and Wang JC (1987). Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 84, 7024–7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnan D, and Bates D (2015). Regulation of DNA Replication Initiation by Chromosome Structure. J. Bacteriol 197, 3370–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marians KJ (1987). DNA gyrase-catalyzed decatenation of multiply linked DNA dimers. J. Biol. Chem 262, 10362–10368. [PubMed] [Google Scholar]
- Modell JW, Hopkins AC, and Laub MT (2011). A DNA damage checkpoint in Caulobacter crescentus inhibits cell division through a direct interaction with FtsW. Genes Dev. 25, 1328–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow L, Crisona NJ, Peter BJ, Hardy CD, and Cozzarelli NR (2001). Topological challenges to DNA replication: conformations at the fork. Proc. Natl. Acad. Sci. USA 98, 8219–8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray-Soni A, Bellecourt MJ, and Landick R (2016). Mechanisms of Bacterial Transcription Termination: All Good Things Must End. Annu. Rev. Biochem 85, 319–347. [DOI] [PubMed] [Google Scholar]
- Ricci DP, Melfi MD, Lasker K, Dill DL, McAdams HH, and Shapiro L (2016). Cell cycle progression in Caulobacter requires a nucleoid-associated protein with high AT sequence recognition. Proc. Natl. Acad. Sci. USA 113, E5952–E5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouviere-Yaniv J, Yaniv M, and Germond JE (1979). E. coli DNA binding protein HU forms nucleosomelike structure with circular double-stranded DNA. Cell 17, 265–274. [DOI] [PubMed] [Google Scholar]
- Stros M, Bacíková A, Polanská E, Stokrová J, and Strauss F (2007). HMGB1 interacts with human topoisomerase IIalpha and stimulates its catalytic activity. Nucleic Acids Res. 35, 5001–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JA, Panis G, Viollier PH, and Marczynski GT (2017). A novel nucleoid-associated protein coordinates chromosome replication and chromosome partition. Nucleic Acids Res. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos SM, Tretter EM, Schmidt BH, and Berger JM (2011). All tangled up: how cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol 12, 827–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitao T, Nordström K, and Dasgupta S (2000). Escherichia coli cell cycle control genes affect chromosome superhelicity. EMBO Rep. 1, 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worcel A, and Burgi E (1972). On the structure of the folded chromosome of Escherichia coli. J. Mol. Biol 71, 127–147. [DOI] [PubMed] [Google Scholar]
- Wu HY, Shyy SH, Wang JC, and Liu LF (1988). Transcription generates positively and negatively supercoiled domains in the template. Cell 53, 433–440. [DOI] [PubMed] [Google Scholar]
- Zheng G, Lu X-J, and Olson WK (2009). Web 3DNA--a web server for the analysis, reconstruction, and visualization of three-dimensional nucleic-acid structures. Nucleic Acids Res. 37, W240–W246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Identification of GapR, a highly conserved α-proteobacterial protein, related to Figure 1.
(A) Schematic of nucleoid purification mass spectrometry assay, with a representative DAPI staining profile. For proteins identified in mass spectrometry, see Table S1.
(B) GapR is highly conserved in α-proteobacteria. The number of GapR homologs identified in and the total number of genomes searched for each class of proteobacteria (left). The occurrence (reported as a percentage of genomes searched) and conservation (reported as average of BLAST bit scores) of GapR homologs is shown for each genome in the individual clades of α-proteobacteria (right). $$PARABREAKHERE$$(C) Growth of wild-type and ΔgapR Pxyl-gapR cells assessed by 10-fold serial dilutions onto PYE (rich medium) plates containing either glucose or xylose (top). Plates were imaged after the indicated number of days. Growth in liquid rich medium containing xylose (middle). Growth of wild-type and ΔgapR Pxyl-gapR cells on M2 minimal medium plates containing either glucose or xylose (bottom). $$PARABREAKHERE$$(D) Growth of wild-type, ΔgapR Pxyl-gapR, and a strain where ΔgapR is complemented by gapR expressed from its native promoter integrated at the xyl locus (ΔgapR xyl::PgapR-gapR).
(E) GapR-depleted cells exhibit a general stress response. Expression changes of ΔgapR Pxyl-gapR cells in xylose or glucose, cells treated with novobiocin, DNA damaging agents, the toxin SocB, ethanol stress, or cell-cycle arrest by SciP depletion. Genes that change >2-fold in ethanol are shown.
(F) Immunoblots of GapR in wild-type and the gapR::gapR-3×FLAG strain (top). Asterisk (*) indicates a non-specific band used as a loading control. GapR-3×FLAG levels are 2.2 ± 0.47 fold higher than wild-type GapR levels, mean ± SEM, n = 2. Immunoblots quantifying GapR protein levels in wild-type cells, using purified GapR standards (bottom). Mean ± SEM, n = 4.
(G) Ethidium bromide (EtBr) nucleoid sedimentation analysis to determine relative levels of (−) supercoiling in wild-type, novobiocin-treated, and GapR-depleted cells grown for 2.5 hr in glucose. LOWESS fit of data is shown with solid lines. Approximate concentration of EtBr required to relax the chromosomes is shown with dashed lines.
Figure S2. GapR ChIP-seq, related to Figure 2.
(A) GapR binding and gene expression changes are not correlated. Scatterplot of gene expression changes in GapR-depleted cells versus average GapR ChIP-seq enrichment at promoters. Blue shaded area encompasses genes that change < 2-fold upon GapR depletion.
(B) GapR binding and gene expression changes at a ribosomal protein locus. ChIP signal (middle) and AT content (top) are plotted as in Fig. 2D, RNA-seq data show reads per million (rpm), and the positions of annotated genes are indicated (bottom, with ribosomal operon genes in solid black).
(C) GapR ChIP signal is unchanged at AT rich peaks in untreated versus rifampicin-treated cells.
(D) Flow chart of the ChIP analysis performed to identify transcription-dependent GapR occupancy at the end of transcription units.
(E) Meta-analysis of GapR occupancy at the 5’ and 3’ ends of genes after rifampicin treatment. Normalized change in GapR ChIP occupancy is calculated from the difference in enrichment (untreated minus rifampicin treated cells) over a 1 kb window before and after the 3’ or 5’ ends of transcription units (TUs). High expression TUs have RPKM > 150, low expression TUs have RPKM < 50. Only TUs > 1000 bp were analyzed.
Figure S3. Replication initiation and elongation is impaired in GapR-reduced cells, related to Figure 3.
(A) Schematic of assay to assess DNA replication. Wild-type and ΔgapR Pxyl-gapR cells were grown in xylose, synchronized in G1, and released into medium with xylose (+ xyl) or glucose (+ glu). DNA content was measured with SYTOX staining and flow cytometry or DNA sequencing at various times post release.
(B) DNA replication time courses for wild type and GapR-reduced strains. Blue lines represent 1N and 2N DNA content.
(C) Fraction of cells from (B) that initiated replication over time, with DNA content > 1N used as a proxy for initiation. Data are mean ± SEM, n ≥ 2.
(D) Replication rate for cells in (B), calculated from a linear fit of the DNA content of cells over time and normalized to wild type. Data are mean ± SEM, n ≥ 2.
(E) DNA sequencing of wild-type cells grown in glucose, ΔgapR Pxyl-gapR cells grown in xylose and released into glucose after synchrony as in (B), and ΔgapR Pxyl-gapR cells depleted for 2 hr in glucose before synchrony (from Fig. 4E). DNA content, normalized to that of synchronized G1 cells, is shown as a function of chromosome position using LOWESS smoothing with 10 kb windows. Blue lines represent locations of the replication origin and terminus. Schematics indicate progression of wild-type replication forks.
Figure S4. Replication elongation defects of GapR-depleted cells, related to Figure 3 and Figure 4.
(A) Extended DNA replication timecourse for wild-type and GapR-depleted cells. Cells were grown in xylose, washed into glucose for 2 hr, synchronized in G1, and released into medium with glucose. DNA content was measured with SYTOX staining and flow cytometry for 4 hr after release.
(B) GapR-depleted cells do not successfully divide. Cells treated as in (A) were fixed and imaged to assess cell division by quantifying the percent of predivisional cells (n > 250 cells per timepoint, left). Representative images of wild-type and GapR-depleted cells at 90, 120, and 210 min post release into glucose (right).
(C) Origin-terminus ratio of wild-type and GapR-depleted cells. DNA was isolated from cells treated as in (A) and the relative copy number of the origin and terminus was determined by qPCR. One representative wild type dataset is shown, and the mean ± SEM is reported for GapR-depleted cells, n ≥ 2.
(D) Sensitivity of GapR-depleted cells to 0.3 mg/mL hydroxyurea (HU) and 0.3 μg/mL rifampicin (rif).
Figure S5. In vitro characterization of GapR binding to DNA, related to Figure 5.
(A) Gel filtration trace of purified GapR (70 μM monomer) (top). Absorbance at 280 nm (blue) and at 260 nm (red) are shown. Arrow indicates approximate elution peak for lysozyme monomer (14.3 kD). SEC-MALS analysis of purified GapR (1 mg/mL = 100 μM monomer) (bottom). Apparent molar mass calculated from MALS (red) and absorbance at 280 nm (grey) are shown. GapR is monodisperse (Mw/Mn = 1.000), with an observed mass of 26.4 kDa (expected 20.7 kDa for homodimer).
(B) EMSAs of GapR binding to linear dsDNA. Purified GapR was incubated with 20 (left) and 50 bp DNA (right).
(C) GapR binding curves for various substrates (left). Apparent KD for each substrate is: 16 bp, 4605 nM GapR; 20 bp, 745 nM; 50 bp, 583 nM; 150 bp, 406 nM; 210 bp, 314 nM; 300 bp, 220 nM; and pUC19 589 nM. Data are mean ± SEM, n ≥ 2. Hill coefficients ± SE (right).
(D) EMSA of GapR binding to (−) supercoiled pUC19.
(E) Trace nuclease contamination in GapR preparation. Relaxed, supercoiled, and linear standards compared to relaxed or (+) supercoiled pUC19 plasmid incubated with 0.4 μM GapR for 1 hr at 30°C, followed by reaction quenching, protein removal with Proteinase K, and electrophoresis.
(F) DNase protection by GapR. GapR was incubated with pUC19 and treated with DNase. Reactions were processed as in (E).
Figure S6. GapR DNA structure and contacts, related to Figure 6 and Figure 7.
(A) GapR-DNA contacts in stereoview. Shown are the side chains from the GapR dimer-of-dimers that interact with the DNA.
(B) Sequence logo of GapR computed from select homologs (top). Conserved acidic surface residues that are not involved in structural contacts or DNA binding (*). Sequence alignment of selected GapR homologs, organized by distance from C. crescentus GapR, with residues numbered based on C. crescentus GapR. Helices of GapR are shown for both the dimer and dimer-of-dimers.
(C) Superimposition of the DNA bound forms of the C. crescentus GapR (magenta) and the Bosea GapR (green), showing that they form the same dimer-of-dimers structure.
(D) Fluorescence polarization assay of C. crescentus GapR binding to AT-rich DNA (blue circles), GC-rich DNA (red squares), and a DNA template without AT stretches (green triangles).
Figure S7. Gyrase and topo IV are specifically stimulated by GapR, related to Figure 7.
(A) GapR stimulates topo IV. Topo IV (2.34 nM) was incubated with the indicated amounts of GapR before adding (+) supercoiled pUC19 and ATP. After incubating for the times indicated, reactions were quenched and analyzed by gel electrophoresis. Slower migration in the gel indicates relaxation of (+) supercoils.
(B) GapR modestly stimulates relaxation of (−) supercoils by topo IV. Reactions are the same as in (A) except with 17.55 nM topo IV, 0.4 μM GapR, and (−) supercoiled pUC19. Relaxed and (−) supercoiled standards are shown (left). Plot quantifies the % initial supercoiled plasmid (asterisk) at each time point (right). Data indicate mean ± SEM, n ≥ 2.
(C) Relaxation of (−) supercoils by topo IV is not stimulated by non-specific proteins. Assay is the same as in (B) except with 0.4 μM E. coli glucose-6-phosphate dehydrogenase (G6PD) or C. crescentus GcrA. A representative gel from at least two experiments is shown.
(D) GapR does not strongly stimulate E. coli Topo IV. Assay is the same as in (B) except with E. coli topo IV.
(E) GapR stimulates gyrase. Assay is the same as in (A) except with 6.25 nM gyrase. Relaxed and (+) supercoiled controls (left). Gyrase generates slower migration as (+) supercoils are relaxed and then induces faster migration as (−) supercoils are introduced.
(F) GapR stimulates introduction of (−) supercoils by gyrase. Assay is the same as in (E) except with 12.5 nM gyrase, 0.4 μM GapR, and relaxed pUC19. Faster migration in the gel indicates introduction of (−) supercoils. Plot quantifies % initial relaxed plasmid (asterisk) at each time point (bottom). Data indicate mean ± SEM, n ≥ 2.
(G) Introduction of (−) supercoils by gyrase is not stimulated by non-specific proteins. Assay is the same as in (F) except with 0.4 μM G6PD or GcrA.
(H) GapR stimulates E. coli gyrase. Assay is the same as in (F) except with E. coli gyrase (2.3 nM, (+) relaxation; 6.8 nM, (−) introduction). Plot quantifies % initial supercoiled plasmid (asterisk) at each time point (bottom). Data indicate mean ± SEM, n ≥ 2.
(I) Models for stimulation of topoisomerase activity by GapR. (Left) GapR may bind overtwisted DNA in front of replication forks, directly recruiting or activating type II topoisomerases to relax positively supercoiled DNA. (Right) GapR could bind overtwisted DNA flanking plectonemic DNA, thereby trapping the substrate of type II topoisomerases.
Table S2. Microarray data from wild-type cells in novobiocin and GapR-depleted cells in xylose, related to Figure 1.
Movie S1. Movie of Caulobacter GapR bound to DNA, related to Figure 6.
Data Availability Statement
RNA-seq, ChIP-seq, and DNA-seq data are available in the Gene Expression Omnibus (GEO): GSE100657 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE100657). Coordinates and structure factor amplitudes for the C. crescentus GapR-DNA, apo Bosea sp. GapR and Bosea sp. GapR-DNA complex have been deposited with the Protein Data Bank under the Accession codes 6CG8, 6CFY and 6CFX, respectively.