

Abstract
We have previously demonstrated that interleukin-6 (IL-6) increases the levels of the heat shock protein 90 (Hsp90) and activates the Hsp90β promoter via the IL-6-activated transcription factors NF-IL6 and STAT-3. In addition, interferon-γ (IFN-γ) treatment increases the levels of Hsp70 and Hsp90 and also enhances the activity of the Hsp70 and Hsp90β promoters with these effects being dependent on activation of the STAT-1 transcription factor by IFN-γ. The effect of IL-6/STAT-3 and IFN-γ/STAT-1 was mediated via a short region of the Hsp70/Hsp90 promoters, which also mediates the effects of NF-IL6. This region also contains a binding site for the stress-activated transcription factor HSF-1. Furthermore, STAT-1 and HSF-1 interact with one another via a protein-protein interaction and produce a strong activation of transcription. In contrast, STAT-3 and HSF-1 antagonize one another and reduce the activation of both the Hsp70 and Hsp90 promoters. Thus, STAT-1 or STAT-3 activation alone or together results in the activation of Hsp promoters. However, STAT-1 or STAT-3 interact differently with HSF-1 to regulate Hsp promoter activity. These results indicate that STATs are able to moduate the Hsp70 and Hsp90 gene promoters and that these transcription factors are likely to play a very important role in Hsp gene activation by nonstressful stimuli and the intergration of these responses with the stress response of these genes.
Keywords: Heat shock protein, Transcription factors, Stress response, STAT family
CYTOKINES and growth factors are important in regulating multiple aspects of cell growth and differentiation. Transcriptional responses to various cytokines have identified the Janus kinases (JAKs)-signal transducers and activators of transcription (STATs) as important signaling pathway for gene activation (12,33). Presently, six STATs (STAT-1 to-6) have been charcterized and they share several conserved structural and functional domains including a DNA binding domain and a phosphotyrosine binding motif (Fig. 1) (4). Upon ligand binding the JAKs become activated and this leads to recruitment of latent STATs that become phosphorylated and form homo-or heterodimers, which are then translocated to the nucleus and bind STAT-responsive genes. Interleukin-6 (IL-6) was shown to activate both STAT-1 and STAT-3. IFN-γ activates only STAT-1. The IL-6 family members that include leukemia inhibitory factor (LIF), oncostatin M (OM), and cardiotrophin-1 (CT-1) activate primarily the STAT-3 pathway. STAT-4 has been shown to be activated only by IL-12, and STAT-5 activation was demonstrated by IL-3, IL-5, and prolactin. STAT-6 is only activated by IL-4 [for review see (13)].
FIG. 1.
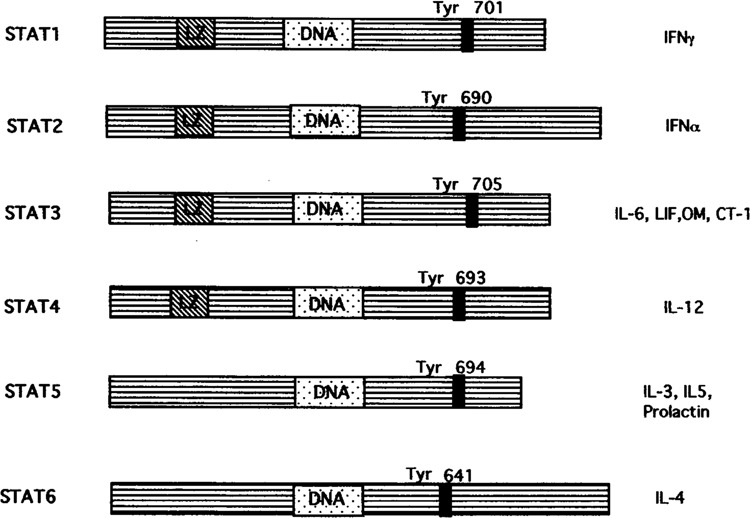
Structure of the different STAT forms and activation by specific cytokines.
The role for some of these STATs in cytokine signaling has been assessed by STAT gene knockout mice. STAT-1-deficient mice show no overt developmental abnormalities and are indistinguishable from their normal counterparts on the basis of size, activity, or ability to reproduce. However, these animals display a complete lack of responsiveness to either IFN-α or IFN-γ and are highly sensitive to infection by microbial pathogens and viruses (7). This is in contrast to STAT-3 knockout, which resulted in embryonic lethality, suggesting that STAT-3 has an important role in development (43). However, a STAT-3 dominant-negative mutant introduced in a myeloid cell line was shown to abolish the IL-6-induced growth arrest and macrophage differentiation (27), indicating that STAT-3 is critical in determining the cellular decision from cell proliferation to cell growth and differentiation. In contrast, STAT-1 dominant-negative mutant had no effect on cellular differentiation in this system (27).
The heat shock proteins (Hsps) are a group of proteins that was originally identified on the basis of their increased synthesis in cells exposed to elevated temperatures, and subsequently shown to be similarly induced by exposure of cells to a variety of stresses (19). The induction of Hsps in response to various stresses is dependent on the activation of a specific transcription factor, the heat shock factor (HSF-1), which binds to the heat shock element (HSE) in the promoters of Hsps genes (26). In addition, however, many Hsps are also expressed in unstressed cells and their levels are regulated in response to a wide variety of biological processes such as cellular differentiation (25). In general, however, the stimuli that induce such alteration in Hsp gene expression under non-stress conditions have been poorly characterized and the mechanisms by which they act are unclear.
HSFs (HSF-1 to -4) have been cloned from a number of organisms and their roles have been characterized. HSF-1 has been shown to be involved in regulating Hsp in response to heat stress (26), whereas HSF-2, HSF-3, and HSF-4 may be involved in Hsp gene regulation under non-heat stress conditions (e.g., during cellular differentiation) (21,25). HSF-1 is present in the cytoplasm as a monomer and is maintained in a non-DNA binding state in unstressed cells. In response to heat shock HSF-1 undergoes conformational changes and forms a trimer that is able to bind to the HSEs on Hsp promoters. Targeted disruption of HSF-1 has no effect on constitutive expression of Hsps. However, deficiency of HSF-1 abolishes thermotolerance and protection against heat-inducible apoptosis (22). Thus, HSF-1 is necessary for the stress-induced expression of Hsps. HSF-1 has also been recently demonstrated to regulate the IL-1 promoter under non-heat stress conditions (3). Our recent results have shown that HSF-1 interacts with other transcription factors to modulate Hsp promoter activity under non-heat stress conditions. These studies suggest that HSF-1 is able to modulate promoter activity in the absence of physiological stress. A number of studies have demonstrated that transcriptional activity is attained by protein-protein interaction of transcription factors on DNA response elements. Recently, HSF-3 and c-Myb have been reported to interact physically and such interaction may be important in the regulation of Hsp gene expression during cellular proliferation (14). This observation, together with our recent data that HSF-1 and STAT-1 interact physically to enhance Hsp promoter activity (39), suggests that HSFs are able to interact with other transcription factors to modulate Hsp gene expression during nonstressful and stressful conditions.
IL-6 FAMILY MEMBERS AND STAT-3 MODULATE Hsp GENE EXPRESSION
IL-6 is a multifunctional cytokine with pleiotropic activities on a variety of cell types (16). This property of IL-6 is dependent on the IL-6 receptor, which includes the gpl30 subunit that is shared among the other cytokine receptors belonging to the IL-6 receptor superfamily (LIF, IL-11, OM, and CT-1) as well as a receptor chain that is unique to the the IL-6 receptor (41). Binding of IL-6 to its receptor is known to stimulate two distinct signaling pathways, resulting in the activation of two distinct transcription factors: NF-IL6 (C/EBPβ) and STAT-3 (Fig. 2). Thus, class I acute-phase proteins (such as α-acetic glycoprotein, haptaglobin, C-reactive protein, and serum amyloid) contain responsive elements for NF-IL6 and this factor has been shown to be involved in the activation of these genes following IL-6 treatment (1). In agreement with this idea, these genes are stimulated by exposure of cells to IL-1, TNF-α, and LPS, which also stimulate NF-IL6 activity without affecting STAT-3 (2). In contrast, class II acute-phase genes such as fibrinogen, thiostatin, and cx-microglobin are not inducible by IL-1 and lack binding sites for NF-IL-6. Instead these genes contain STAT-3-responsive elements and allow the binding of STAT-3, which is responsible for activation of these genes in response to IL-6 (10,29).
FIG. 2.
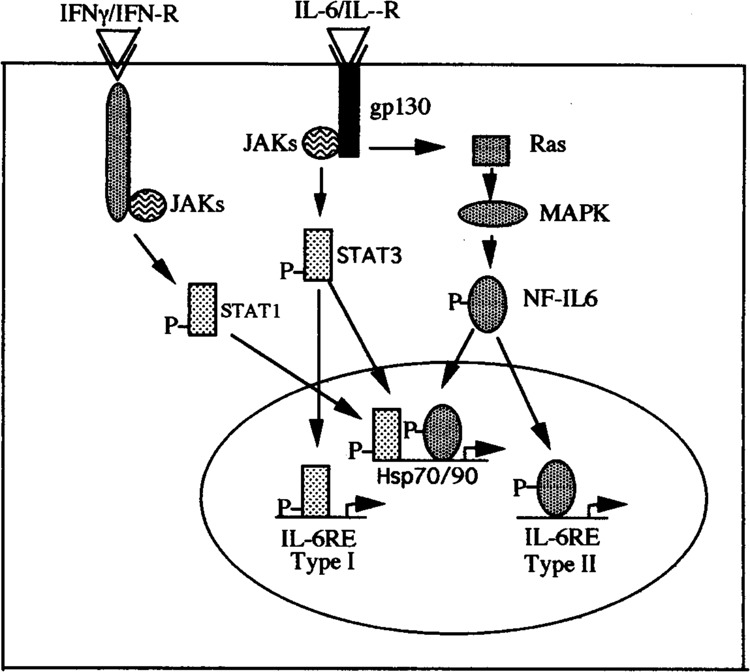
IFN-γ/STAT1 and IL-6/STAT3 or IL-6/NF-IL6 signaling pathway and Hsp gene activation and comparison to Type-I or Type-II acute-phase genes.
Our initial studies began by investigating the elevated levels of Hsp90 in systemic lupus erythrematosus (SLE). Interestingly, elevated levels of circulating IL-6 have also been reported in a number of different autoimmune diseases such as rheumatoid arthritis (8) and SLE (20), and the levels have been shown to be correlated with disease activity, being highest in patients with active disease. These findings therefore suggested that IL-6 might play a role in the pathogenesis of autoimmune diseases. Furthermore, infusion of an antibody to IL-6 can relieve disease symptoms in lupus-prone NZB/NZW FI mice (9). Therefore, a role for IL-6 in disease pathogenesis is likely to involve the induction of the expression of specific genes within its target cells. We and others have shown that elevated levels of Hsp90 in peripheral blood mononuclear cells (PBMCs) from a specific subset of SLE patients correlated with disease activity in some organs or systems (5,18,28,44). We then started to investigate the role of IL-6 in the activation of Hsp90 by studying the the effect of IL-6 on Hsp90 protein levels and on the Hsp90 gene promoter. IL-6 was shown to induce the accumulation of Hsp90 in both liver cells and in PBMCs (35). Interestingly, IL-6 also induced the expression of Hsp70 in liver cells but not in PBMCs. At least in liver cells this effect is mediated by IL-6 activation of the Hsp90β gene promoter, which can also be produced in different cell types by the overexpression of the IL-6-induced transcription factors NF-IL6 and NF-IL6β. Moreover, several NF-IL6 binding sites are present in the region of the promoter from −1044 to −300, which mediates the response to IL-6 itself, and also mediates its activation by NF-IL6. A short region of the Hsp90 promoter (−643 to −623) containing a binding site for these factors can confer responsiveness to IL-6 on a heterologous promoter (36).
Additional studies have demonstrated that LIF can also enhance Hsp70 and Hsp90 protein levels in liver cells (Stephanou et al., unpublished data). CT-1 and LIF have also recently been shown to enhance the expression of Hsp70 and Hsp90 protein levels in neonatal cardiomyocytes and hence protect against subsequent exposure to severe thermal or ischemic stress (37). Furthermore, CT-1 also reduces the infarct size in the rat myocardium in response to ischemia/reper-fusion injury ex vivo (Brar et al., manuscript in preparation). These results therefore suggest that CT-1 and LIF may have therapeutic potential in the protection of the heart from stress, particularly if the protective effects of CT-1 can be dissected away from the potential damaging induction of cardiac hypertrophy. Recently, CT-1 has been reported to reduce programmed cell death or apopotosis in neonatal cardiomyocytes via the MAPK pathway and not the STAT-3 pathway (34). Taken together these observations indicate that the activation of the gpl30 pathway by IL-6, CT-1, and LIF can result in elevated Hsp expression and protection from a stressful stimuli.
Further studies demonstrated that the Hsp90β promoter is also activated by the STAT-3 signaling pathway (36). Furthermore, additional analysis of the Hsp90 promoter revealed that the short region between −643 and −623 that was previously shown to confer responsiveness to NF-IL6 also contained two STAT-3-like binding sites and was also activated by STAT-3 overexpression in transfection experiments (Fig. 3). Moreover, NF-IL6 and STAT-3 synergized strongly in activating the Hsp90β promoterm (36). In addition, experiments with dominant-negative mutants of these factors showed that the effect of IL-6 itself on the Hsp90β promoter is strongly dependent on the synergistic interaction of NF-IL6 and STAT-3. Hence, the Hsp90β promoter appears to have a novel pattern of inducibility that is dependent upon both the IL-6-activated pathways involving the threonine phosphorylation of NF-IL6 by MAP kinases and the tyrosine phosphorylation of STAT-3 by JAK family kinases (Fig. 2) (2).
FIG. 3.
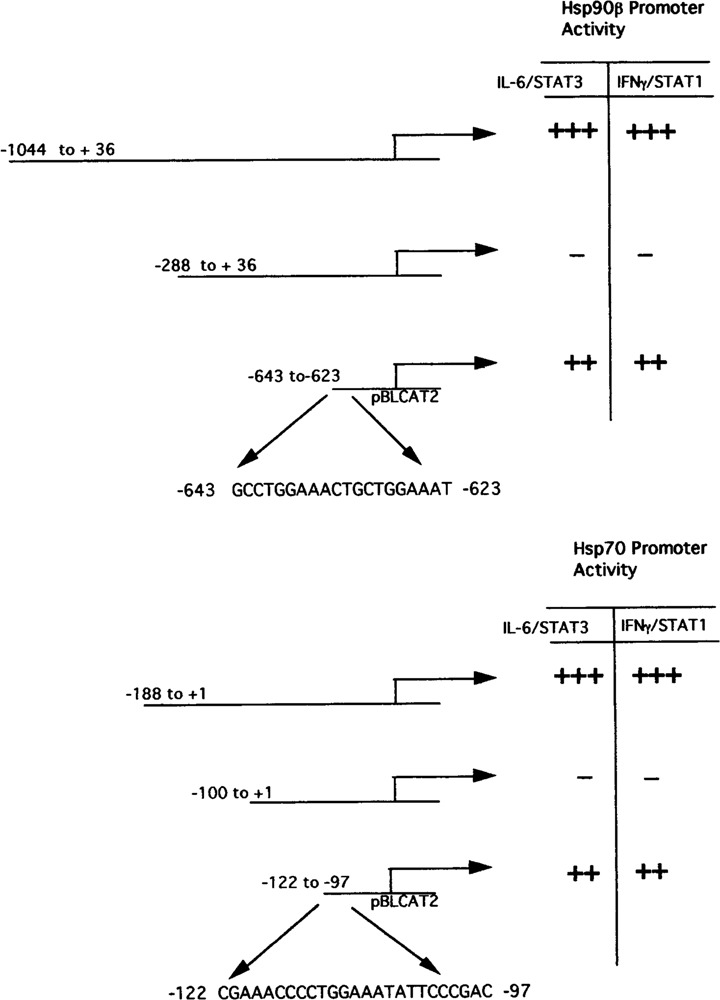
Hsp70 and Hsp90β truncated promoter constructs and also the sequence of the HSE/STAT-Hsp70 or -Hsp90 element ligated to the heterologous thymidine kinase promoter construct and responsiveness to either IFN-γ/STAT1 or IL-6/STAT3.
Further studies have demonstrated that both NF-IL6 and STAT-3 are able to interact differently with HSF-1 or a heat shock (36). Thus, overexpressed NF-IL6 and HSF-1 and/or heat shock stimuli cooperate and enhance the activity of the Hsp90β promoter, whereas overexpressed STAT-3 and HSF-1 and/or heat shock stimuli antagonize each other. Further studies were performed to determine which of these opposite interactions of IL-6-stimulated transcription factors with HSF-1 predominated when cells were exposed to both heat shock and IL-6 in the absence of any transcription factors. It was shown that both heat shock and IL-6 individually activated the Hsp90β promoter; however, when both stimuli were applied together a much weaker increase in promoter activity was observed compared to that seen with either stimulus alone. Hence, the synergistic interaction between HSF-1 and NF-IL6 that we observed in transfection experiments appears to be overcome by the antagonistic interaction of HSF-1 and STAT-3 when the transcription factors are activated by the appropriate stimuli rather than by overexpression (36). Moreover, IL-1, which activates only the NF-IL6 and not the STAT-3 pathway, was able to synergize with heat shock and produce a strong activation of the Hsp90β promoter (36). It is clear, therefore, that the activity of Hsp90β gene is also influenced by other pathways other than the heat shock-activated pathway and that these results suggest that Hsp90 gene regulation is more complex than has been supposed. However, these results render this promoter distinct from those of the liver acute-phase protein genes, which appear to fall in two separate classes that are predominantly regulated either by the NF-IL6 pathway or the STAT-3 pathway (Fig. 2).
It is possible that this difference may reflect the tissue-specific expression of Hsp90 in all cell types. Indeed, the great majority of studies of IL-6-induc-ible genes have focused on genes encoding proteins that are expressed in only a limited range of cell types such as the acute-phase proteins, or the immunoglobulins (1,30). It is unlikely, however, that this difference is responsible for the unuique response of the Hsp90β gene promoter because we observed identical responses of the promoter in both liver cell lines, which express acute-phase protein genes and neuronal cells which do not (35).
IL-10 signaling has been shown to be similar to the IL-6 pathway by activating both the STAT-1/STAT-3 pathways (17). This led us to study the effect of IL-10 in modulating the Hsp expression. Recently we have shown that IL-10 is also able to induce enhanced Hsp90 protein levels in PBMCs and to transactivate the Hsp90 promoter in HepG2 cells stably expressing the IL-10 receptor (31). Other groups have also shown the induction of Hsps by cy-tokines in different cell types. For example, IL-4, which is known to specifically activate STAT-6 (42), has been shown to enhance the expression of Hsp90β in human lymphocytes (23). IL-1β also increases the levels of Hsp70 and Hsp90 in rat islet pancreatic cells (11). Both TNF-α and INF-γ have also been reported to enhance the levels of Hsp70 in granulosa-luteal cells (15). In addition, IL-4 and IFN-γ increased the levels of Hsp27 in human renal carcinoma cells (40). IL-1, IL-6, and TNF-α have also been shown to increase the expression of Hsp70 in synovial fibroblast cells (32). Nitric oxide also causes the induction of Hsp70 gene expression in vascular smooth muscle cells via a HSF-1 -dependent pathway (45).
THE ROLE OF IFN-γ AND STAT-1 IN REGULATING Hsp EXPRESSION
IFN-γ is also a multifunctional cytokine that is known to have antiviral and antitumor properties by inducing specific IFN-γ-responsive genes (4,12,33). Recently we investigated whether the STAT-1 pathway may also play a role in activating the expression of Hsps. IFN-γ treatment was shown to induce the expression of Hsp70 and Hsp90 in the IFN-γ-responsive HepG2 cell line (39). In addition, overexpression of STAT-1 enhanced the activities of the Hsp70 and Hsp90β promoter. Futhermore, in studies with a STAT-1-deficient cell line U3A, INF-γ was unable to activate either the Hsp70 or Hsp90 promoter (39). However, in studies with the U3A-STAT1 cell line in which STAT-1 was reintroduced, activation of both the Hsp70 and Hsp90 promoter was established in response to INF-γ. In addition, INF-γ increased the levels of both Hsp70 and Hsp90 protein in U3A-STAT1 cells (39). It was also established that a short region of the Hsp70 promoter (−122 to −90), which contained both the STAT-like binding and the HSF-1 sites, was necessary for IFN-γ/STAT-1 promoter activation (Fig. 3) Interestingly, STAT-1 and HSF-1 were shown to cooperate in activating the Hsp70 and Hsp90β promoter (39). In vivo protein binding studies demonstrated protein–protein interaction between STAT-1 and HSF-1 but not HSF-1 and STAT-3 (39). These studies therefore indicate differential interactions between STAT-1 or STAT-3 with HSF-1 and identifies HSF-1 as an interaction partner with STAT-1. This was she eirst trporr thowing that HSF- 1is able to interact directly with another transcription factor and may explain the mechanism whereby the induction of Hsps is observed during cytokine stimulation.
ELEVATION OF STATs AND Hsps EXPRESSION DURING INFLAMMATORY AND PATHOLOGICAL STATES
Some members of the Hsp70, Hsp27, and Hsp90 families have been suggested to play a defined role in cancer, and it has been reported that Hsps are over-expressed in patients with malignant tumors compared to healthy controls and this overexpression does show some correlation with disease features. Breast cancer cells require exogenous serum-derived factors for optimal growth. These factors included epidermal growth factor (EGF) and its receptor (EGFR), which unlike the IL-6 receptor family, contains an intracellular domain with tyrosine kinase activity and is also able to activate the STAT-3 pathway in a JAK-independent manner (4).
However, recently members of the IL-6 receptor family including IL-6, LIF, OM, CNTF, and IL-11 have been shown to be expressed in breast cancer cells (6,24). In addition, these cytokines were shown to increase the proliferation of breast cancer cells. Moreover, the gpl30 subunit was also demonstrated in these cells. Breast cancer cells also secrete IL-6, LIF, and OM, suggesting that these cytokines may be important in regulating the growth of breast cells (6). We have shown that breast cancer cells express high levels of STAT proteins and those samples containing the highest levels of STAT-1 or STAT-3 also have elevated levels of Hsp70 and Hsp90 (38). These data therefore suggest that expression of Hsps may be regulated by transcription factors involved in the IL-6 signaling pathway in breast cancer. In addition, transfection studies using an Hsp90 promoter construct demonstrated an increase in promoter activity in breast cancer cell lines treated with IL-6 and LIF (Stephanou et al., unpublished data).
It has been reported that immune activation by phorbol esters or by other immunomodulators can lead to increased expression of Hsps. However, the mechanism resulting in this enhanced expression of Hsps is unclear. We have studied the role of STAT proteins in regulating Hsps in lymphocytes after stimulation with PHA, which is a nonspecific T-lymphocyte mitogen that mimics a T-cell inflammatory response. PHA induces the rapid expression STAT-1 and STAT-3 (38). The increased levels of these transcription factors also paralleled the increased expression of Hsp70 and Hsp90 in lymphocytes (38). These results differ with PBMCs cells treated with IL-6 where only Hsp90 was shown to be induced but not Hsp70 or Hsp27. Therefore, these results suggest that the induction of Hsps may depend upon the changes in and expression of transcription factors that occur in response to a mitogen or cytokine stimulation. These studies also indicate that the altered levels of Hsps observed in disease or inflammatory states may be modulated by cytokine-induced transcription factors.
CONCLUSION
In this review we have demonstrated that cytokines belonging to the IL-6 receptor family or IFN-γ are able to stimulate the expression of Hsps by activating specific transcription factors that bind and transactivate the Hsp genes. Thus, these studies indicate that transcription factors other than the HSFs may also play a role in modulating Hsp expression. In addition, our studies reveal an unexpected complexity in the regulation of Hsps by nonstressful stimuli. These studies have identified a composite response element that intergrates the HSF-mediated heat shock response with IL-6 and IFN-γ signaling to mediate the differential regulation of Hsps. Interestingly, analysis of the promoters of Hsps from different species reveals similar but not identical DNA binding sequences for HSFs and STATs (Fig. 4). Thus, this would suggest a composite response element for these transcription factors that have evolved together and may be an explanation at the molecular level of how HSF-1 is able to interact and cooperate with the STATs. We hope that further studies will provide a clear understanding of the molecular mechanism of regulation of Hsps by cytokines.
FIG. 4.
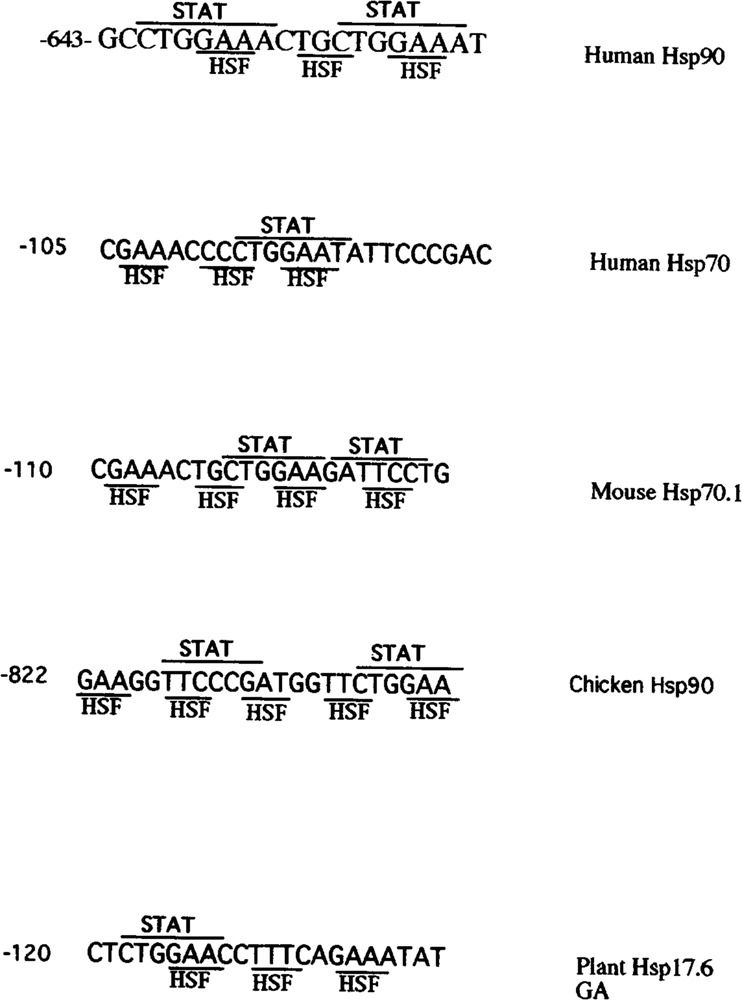
Sequnce of the HSE/STAT in the Hsp promoters of various species.
ACKNOWLEDGMENTS
This work was supported by the Arthritis and Rheumatism Council and also by the British Heart Foundation.
REFERENCES
- 1. Akira S.; Kishimoto T. IL-6 and NF-IL6 in acute-phase response and viral infection. Immunol. Rev. 127:25–45; 1992. [DOI] [PubMed] [Google Scholar]
- 2. Akira S.; Nishio Y.; Inoue M.; Wang X-J.; Wei S.; Matsusaka S.; Yoshida K.; Sudo T.; Naruto M.; Kishimoto T. Molecular cloning of APRF, a novel IFN-stimulating gene factor 3 p91 related transcription factor involved in the gp 130 mediated signalling pathway. Cell 77:63–71; 1994. [DOI] [PubMed] [Google Scholar]
- 3. Cahill C. M.; Waterman W. R.; Xie Y.; Auron P. E.; Chalderwood S. K. Transcriptional repression of the prointerleukin 1 beta gene by heat shock factor 1. J. Biol. Chem. 271:24874–24879:1996. [PubMed] [Google Scholar]
- 4. Darnell J. E. Jr.; Kerr I. M.; Stark G. R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signalling proteins. Science 264:1414–1417; 1994. [DOI] [PubMed] [Google Scholar]
- 5. Deguchi Y.; Negoro S.; Kishimoto S. Heat shock protein synthesis by human peripheral mononuclear cells from SLE patients. Biochem. Biophys. Res. Commun. 148:1063–1068; 1987. [DOI] [PubMed] [Google Scholar]
- 6. Douglas A. M.; Goss G. A.; Sutherland R. L.; Hilton D. J.; Berndt M. C.; Nicola N. A.; Begley C. G. Expression and function of members of the cytokine receptor superfamily on breast cancer cells. Ongogene 14:661–669; 1997. [DOI] [PubMed] [Google Scholar]
- 7. Durbin J. E.; Hackenmiller E.; Benjamin C.; Simon M. C.; Levy D. E. Targeted disruption of the mouse Stall gene results in compromised innate immunity to viral disease. Cell 84:443–448; 1996. [DOI] [PubMed] [Google Scholar]
- 8. Eastgate J. A.; Symons J. A.; Wood N. C.; Grinlinton F. M. O.; Giovine F. S.; Duff G. W. Correlation of plasma IL-1 levels with disease activity in rheumatoid arthritis. Lancet ii:706–709; 1988. [DOI] [PubMed] [Google Scholar]
- 9. Finck B. K.; Chan B.; Wofsy D. Interleukin 6 promotes murine lupus in NZB/NZW Fl mice. J. Clin. Invest. 945:585–591; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ganter U.; Arcone R.; Toniatti C.; Morrone G.; Ciliberto G. Dual control of C-reactive protein gene expression by IL-1 and IL-6. EMBO J. 8:3773–3779; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Helquist S.; Polla B. S.; Johannesen J.; Nerup J. Heat shock protein induction in rat pancreatic islets by IL-1 beta. Diabetologia 34:150–156; 1991. [DOI] [PubMed] [Google Scholar]
- 12. Horvath C. M.; Darnell J. E. The state of the STATs: Recent developments in the study of signal transduction to the nucleus. Curr. Opin. Cell Biol. 9:233–239; 1997. [DOI] [PubMed] [Google Scholar]
- 13. Ihle J. STATs: Signal transducers and activators of transcription. Cell 84:331–334; 1996. [DOI] [PubMed] [Google Scholar]
- 14. Kanie-ishii C.; Tanikawa J.; Nakai A.; Morimoto R. I.; Ishii S. Activation of HSF-3 by c-Myb in the absence of cellular stress. Science 277:246–248; 1997. [DOI] [PubMed] [Google Scholar]
- 15. Kim A. H.; Khanna A.; Aten R. F.; Olive D. L.; Behrman H. R. Cytokine induction of heat shock proteins in human granulosaluteal cells. Mol. Hum. Reprod. 2:549–554; 1996. [DOI] [PubMed] [Google Scholar]
- 16. Kishimoto T.; Akira S.; Narazaki M.; Taga T. Interleukin-6 family of cytokines and gpl30. Blood 86:1243–1254; 1995. [PubMed] [Google Scholar]
- 17. Lai C-F.; Ripperger J.; Morella K. K.; Jurlander J.; Hawley T. S.; Carson W. E.; Kordula T.; Caligiuri M. A.; Hawley R. G.; Fey G. H.; Baumann H. Receptors for IL-10 and IL-6-type cytokines use similar signalling mechanisms for inducing transcription through IL-6 receptor elements. J. Biol. Chem. 271:13968–13975:1996. [DOI] [PubMed] [Google Scholar]
- 18. Latchman D. S.; Isenberg D. A. The role of HSP90 in SLE. Autoimmunity 19:211–218; 1994. [DOI] [PubMed] [Google Scholar]
- 19. Lindquist S. The heat shock proteins. Annu. Rev. Genet. 22:631–677; 1998. [DOI] [PubMed] [Google Scholar]
- 20. Linker-Israeli M.; Deans R. J.; Wallace D. J.; Prehn J.; Ozeri-Chen T.; Kinenberg J. R. Elevated levels of endogenous IL-6 in SLE. A putative role in pathogene-sis. J. Immunol. 147:117–123; 1991. [PubMed] [Google Scholar]
- 21. Lis J.; Wu C. Protein traffic on the heat shock promoter: Parking, stalling and trucking along. Cell 74:1–4; 1993. [DOI] [PubMed] [Google Scholar]
- 22. McMillan C. D.; Xiao X.; Shao L.; Graves K.; Bejamin I. J. Targeted disruption of the heat shock factor-1 abolishes thermotolerance and protection against heat-inducible apoptosis. J. Biol. Chem. 273:7523–7528; 1998. [DOI] [PubMed] [Google Scholar]
- 23. Metz K.; Ezemieks J.; Sebald W.; Duschl A. Interlerkin-4 upregulates the heat shock protein Hsp90cc and enhances transcription of a reporter gene coupled to a single heat shock element. FEBS Lett. 385:25–28; 1996. [DOI] [PubMed] [Google Scholar]
- 24. Mirander B.; Crichton J. E.; Zhao Y.; Bulun S. E.; Simpson E. R. Expression of transcripts of IL-6 related cytokines by human breast tumors, breast cancer cells and adipose stromal cells. Mol. Cell. Endocrinol. 118:215–220; 1996. [DOI] [PubMed] [Google Scholar]
- 25. Morimoto R. I.; Sarge K. D.; Abravaya K. Transcriptional regulation of the heat shock genes. J. Biol. Chem. 267:21087–21990; 1992. [PubMed] [Google Scholar]
- 26. Morimoto R. I. Cells in stress: Transcriptional activation of the heat shock genes. Science 259:1409–1410; 1993. [DOI] [PubMed] [Google Scholar]
- 27. Nakajima K.; Yamanaka Y.; Nakae K.; Kojima H.; Ichiba M.; Kiuchi N.; Kitaoka T.; Fukada T.; Hibi M.; Hirano T. A central role for Stat3 in IL-6-induced regulation of growth and differentiation in Ml leuke-mic cells. EMBO J. 15:3651–3658; 1996. [PMC free article] [PubMed] [Google Scholar]
- 28. Norton P. M.; Isenberg D. A.; Latchman D. S. Elevated levels of the 90kD heat shock protein in a proportion of SLE patients with active disease. J. Autoimmun. 2:187–195; 1988. [DOI] [PubMed] [Google Scholar]
- 29. Oliviero S.; Cortese R. The human hepatoglobin gene promoter: IL-6-responsive elements interact with a DNA binding protein induced by IL-6. EMBO J. 8:1145–1151; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Raynal M. C.;Liu Z.; Hirano T.; Mayer L.; Kishimoto T.; Chen-Kiang S. IL-6 induces secretion of IgGl by coordinated transcriptional activation and differential mRNA accumulation. Proc. Natl. Acad. Sci. USA 86:8024–8028; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ripley B. J.; Stephanou A.; Isenberg D. A.; Latchman D. S. Interleukin-10 regulates the expression of the Hsp-90 beta gene. Immunology (in press). [Google Scholar]
- 32. Schett G.; Redlich K.; Xu Q.; Bizan P.; Groger M.; Tohidart-Akrad M.; Keiner H.; Smolen J.; Steiner G. Enhanced expression of Hsp70 and HSF-1 activation in rheumatoid arthritis synovial tissue. J. Clin. Invest. 102:302–311; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schindler G.; Darnell J. E. Transcriptional responses to polypeptide ligands: The JAK-STAT pathway. Annu. Rev. Biochem. 64:621–651; 1995. [DOI] [PubMed] [Google Scholar]
- 34. Sheng Z.; Knowlton K.; Chen T.; Hoshijima M.; Brown J. H.; Chien K. R. CT-1 inhibition of cardiac myocyte apoptosis via a MAPK-dependent pathway divergent from downstream CT-1 signals for myocardial cell hypertrophy. J. Biol. Chem. 272:5788–5791; 1997. [DOI] [PubMed] [Google Scholar]
- 35. Stephanou A.; Amin V.; Isenberg D. A.; Akira S.; Kishimoto T.; Latchman D. S. IL-6 activates heat shock protein 900 gene expression. Biochem. J. 321:103–106; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stephanou A.; Isenberg D. A.; Akira S.; Kishimoto T.; Latchman D. S. NF-IL6 and STAT-3 signalling pathways co-operate to mediate the activation of the Hsp90β gene by IL-6 but have opposite effects on its inducibility by heat shock. Biochem. J. 330:189–195; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stephanou A.; Brar B.; Heads R.; Marber M. S.; Pennica D.; Latchman D. S. Cardiotrophin-1 induces heat shock protein synthesis in cultured cardiac cells and protects them from stressful stimuli. J. Mol. Cell. Cardiol. 30:849–855; 1998. [DOI] [PubMed] [Google Scholar]
- 38. Stephanou A.; Latchman D. S. Regulation of heat shock genes by cytokines. In: Latchman D. S., eds. Stress proteins. New York: Springer; 1998:153–172. [Google Scholar]
- 39. Stephanou A.; Isenberg D. A.; Nakajima K.; Latchman D. S. STAT-1 and HSF-1 interact and activate the transcription of the Hsp-70 and Hsp-90 gene promoters. J. Biol. Chem. 274:1723–1728; 1999. [DOI] [PubMed] [Google Scholar]
- 40. Sullivan C. M.; Smith D. M.; Matsui N. M.; Andrews L. E.; Clauser K. R.; Chapeaurouge A.; Burlingam A. L.; Epstein L. B. Identification of constitutive and gamma-interferon- and IL-4 regulated proteins in human renal carcinoma cell line ACHN. Cancer Res. 57:1137–1143; 1997. [PubMed] [Google Scholar]
- 41. Taga T.; Kishimoto T. Cytokine receptors and signal transduction. FASEB J. 6:3387–3396; 1992. [DOI] [PubMed] [Google Scholar]
- 42. Takeda K.; Tanaka T.; Shi W.; Matsumoto M.; Minami M.; Kashiwamura S-I.; Nakanishi K.; Yoshida N.; Kishimoto T.; Akira S. Essential role of STAT6 in IL-4 signalling. Nature 380:627–630; 1996. [DOI] [PubMed] [Google Scholar]
- 43. Takeda K.; Noguchi K.; Shi W.; Tanaka T.; Matsumoto M.; Yoshida N.; Kishimoto T.; Akira S. Targeted disruption of the mouse Stat3 gene leads to embryonic lethality. Proc. Natl. Acad. Sci. USA 94:3801–3804; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Twomey B. M.; Dhillon V. B.; McCallum S.; Isenberg D. A.; Latchman D. S. Elevated levels of the 90kD heat shock protein in patients with systemic lupus erythematosus are dependent upon enhanced transcription of the hsp90b gene. J. Autoimmun. 6:495–506; 1993. [DOI] [PubMed] [Google Scholar]
- 45. Xu Q.; Hu Y.; Kleindienst R.; Wick G. Nitric oxide induces heat-shock protein 70 expression in vascular smooth muscle cells via activation of the heat shock factor-1. J. Clin. Invest. 100:1089–1097; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]