

Abstract
Background
The phenotypic diagnosis of von Willebrand disease (VWD) is a multistep process with classification dependent on the quantification of von Willebrand factor (VWF) multimeric structure. VWF multimer analysis is a technically challenging, lengthy and non‐standardised assay, usually performed in specialist laboratories. Recently, a new semi‐automated multimer assay, the Hydragel 5 von Willebrand multimers (H5VWM) has become available.
Objectives
This study, performed in two European centres, compared existing in‐house multimer assays to the H5VWM in individuals with and without VWD.
Results
Overall agreement of 91.1% was observed in 74 individuals with normal VWF levels, 57 patients grouped as type 1 VWD, 33 type 2A, 16 type 2B, 28 type 2M, 11 type 2N. Patients tested following Desmopressin or VWF concentrate, with thrombotic thrombocytopenic purpura and acquired von Willebrand syndrome were also evaluated. Many of the discrepancies between methods were in patients with genetic mutations linked to more than one type of VWD including p.R1374C/H and p.R1315C. Quantifiable multimer results were available within one working day. Densitometry improved the interpretation of the multimers with slight structural variations that were not apparent by visual inspection of the in‐house method.
Conclusions
5VWM was a rapid, sensitive, standardised assay which used existing technology and could be included as an initial screen of VWF multimers in a VWD diagnostic algorithm in conjunction with traditional multimer analysis.
Keywords: densitometry, HMWM, Hydragel, multimer, VWD, VWF
Essentials.
The diagnosis of von Willebrand disease requires assessment of VWF multimer distribution.
Hydragel 5 VW multimers and in‐house multimer methods were compared in patients with VWD.
91% agreement was observed between methods.
Hydragel 5 VW multimer densitometry improved diagnosis of VWD.
1. INTRODUCTION
Von Willebrand disease (VWD) is a hemorrhagic disorder that is caused by a reduction, dysfunction, or absence of von Willebrand factor (VWF).1 VWF is a large multimeric glycoprotein that binds platelets to the damaged subendothelium and also circulates in complex with factor VIII (FVIII). VWF comprises subunits of variable size from the low molecular weight multimers (LMWM) to subunits of more than 50 in size, the high molecular weight multimers (HMWM). VWD is sub‐typed by the ratio of the activity (VWF Ac) to the concentration of protein (VWF antigen, VWF:Ag).2 A VWF Ac/VWF:Ag ratio of >0.6 indicates a quantitative type 1 disorder. A ratio of <0.6 is indicative of qualitative type 2 disorders, which generally have a disproportionate reduction of VWF activity compared to VWF:Ag.3 Type 2 VWD is further divided into four subtypes; type 2A which is generally ineffective multimer assembly or secretion, type 2M which is linked to reduced binding to platelet GPIb, type 2B which is an increased binding to GPIb, and type 2N which is a decrease or absence in binding to FVIII with preservation of the ability of VWF to bind platelets.4, 5 VWF is absent in type 3.
The diagnosis of type 2 VWD requires a number of specialized assays including analysis of multimer distribution.3 Multimer analysis is not widely undertaken by routine laboratories6 due to lack of method standardization, length of test, requirement of specialist equipment, and often variable and subjective results. In‐house VWF multimers (IHVWM) may be performed using sodium dodecyl sulphate (SDS) agarose gel electrophoresis, often followed by Western blotting, then visualization by methods including via radioactive Iodine (I125) autoradiography, chemiluminescence,7 infrared fluorescent imaging,8 or Alkaline Phosphatase (AP).9 Densitometry of the agarose gels may be performed to aid interpretation of the multimer pattern.10 Interpretation of densitometry is also not standardized between laboratories; some report peaks 1‐3 to represent the LMWM, 4‐5 the intermediate molecular weight multimers (IMWM), and peaks 6 and above represent the HMWM,11 while others report peaks 1‐5 as LMWM, 6‐10 as IMWM, and peaks 10 and above as HMWM.9
A new semi‐automated VWF multimer assay kit has recently been launched. The Hydragel 5 von Willebrand multimers test (H5VWM) is performed with existing Hydrasys 2 Technology (Sebia, France) and the kit includes ready to use buffers, agarose gels, and antibodies.
In this study, two centers compared H5VWM with an existing in‐house SDS agarose gel electrophoresis method in healthy donors and patients with different types of VWD or acquired von Willebrand syndrome (AVWS). Subgroups of patients were also tested following treatment with endogenous or exogenous VWF or in the presence of interferring substances.
2. METHODS
2.1. Sample collection
Two sites participated in this study; site 1, Sheffield Haemophilia and Thrombosis Centre, UK; and site 2, Centrum für Blutgerinnungsstörungen und Transfusionsmedizin (CBT), Bonn, Germany.
Residual 3.2% citrated plasma from normal donors or individuals previously tested for VWD were stored frozen in secondary tubes (Sarstedt, Nümbrecht, Germany) at either −80°C for a maximum of 24 months at site 1 or at −35°C for a maximum of 3 months at site 2. Some samples included in the study had been referred to site 1 from other centers within the UK or Italy and were received frozen on dry ice. Samples were thawed for 5 minutes in a water bath at 37°C and discarded after 2 hours.
Site 1 had ethics approval from the South Sheffield Research Ethics Committee for the use of normal donor plasma and residual patient plasma for service evaluation. Written informed consent was provided for samples referred from Italy. For site 2, the study was approved by the Ethical Committee of the Ärztekammer Nordrhein, Dusseldorf, Germany.
2.1.1. Sample groups
Fifty‐five healthy donors were sourced from either laboratory staff (n = 25) or using commercial frozen donors (n = 30, Cryocheck; Precision Biologic, Halifax, NS, Canada). These comprised 28 males and 27 females with a mean age at donation of 40 years and a range of 19‐66 years.
Patients referred for VWD diagnosis or previously diagnosed with VWD were grouped according to VWF:Ag, VWF activity, and ratio of VWF activity/VWF:Ag. A ratio of >0.6 was used as a cut off for a quantitatively normal distribution. Where VWF Ac was not available, Collagen binding (VWF:CB) results were used. Type 2A or 2M patients were then subclassified by the in‐house multimer results. Type 2B patients were classified by genetic mutation or historical diagnosis of type 2B VWD by low‐dose RIPA12 (see Figure 1). Type 2N VWD were included by the presence of genetic mutation linked to 2N VWD, reduced or absent FVIII/VWF binding.13
Figure 1.
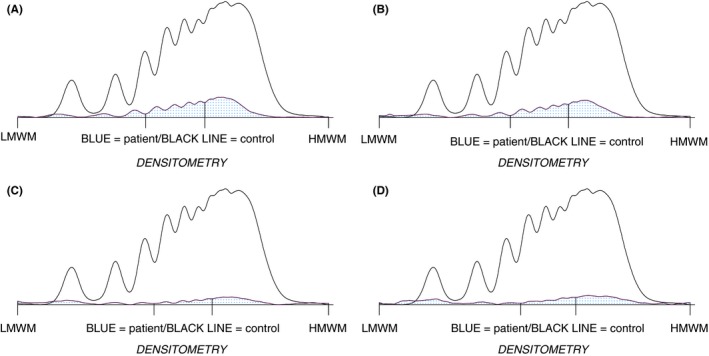
(A‐D) Limit of detection of Hydragel 5 VW Multimers (H5VWM) using diluted normal plasma. H5VWM densitometry from left low molecular weight multimers (LMWM) to right high molecular weight multimers (HMWM) with peaks 1‐3 LMWM, 4‐7 intermediate molecular weight multimers (IMWM), and above 7 HMWM. Normal quality control sample is indicated by a solid black line and test samples in blue. The multimer image is depicted in the top right of each (A), von Willebrand factor antigen (VWF):Ag of 20 IU/dL densitometry LMWM 15.8%, IMWM 38%, HMWM 46.2%. (B) VWF:Ag of 15 IU/dL densitometry LMWM 19%, IMWM 35.8%, HMWM 45.2%. (C) VWF:Ag of 10 IU/dL densitometry LMWM 32%, IMWM 23.4%, HMWM 44.6%. (D) VWF:Ag of 5 IU/dL densitometry LMWM 30.8%, IMWM 23.4%, HMWM 45.8%
Seventy‐four patients had normal VWF Ac, 57 were categorized as type 1, 33 as type 2A, 28 as type 2M, 16 type 2B, 11 type 2N, 3 had type 3 VWD. Thirty patients with AVWS were also included.
A cohort of these patients was also tested following administration of Octim (Desmopressin, Ferring, Parsippany, NJ, USA), Wilfact (LFB, Courtaboeuf, France), Wilate (Octapharma, Lachen, Switzerland), Haemate P or Voncentro (both CSL Behring, King of Prussia, PA, USA).
Samples from four patients with thrombotic thrombocytopenic purpura (TTP) and reduced ADAMTS13 activity (Technozym ADAMTS‐13 Activity ELISA, Pathway, UK) were included.
Possible interference in the H5VWM method by high rheumatoid factor was assessed in eight samples provided by Sebia. Rheumatoid factor levels were determined by both rate Nephelometry (Beckman Coulter, Villepinte, France) with levels ranging from 1000 to 8750 IU/mL (reference interval <20 IU/mL) and by immunoturbidimetry (Randox, Crumlin, UK) with levels ranging from 932 to 4834 IU/mL (reference interval <12.5 IU/mL). The influence of anticoagulant was assessed with three patients whose blood was taken into both citrate and EDTA.
2.2. VWF complex assays
At site 1, VWF assays were performed by Siemens VWF antigen and Innovance VWF Ac (Marburg, Germany), a GP1bM VWF activity assay.2 All assays were performed on Sysmex CS5100i instrumentation (Kobe, Japan). VWF:CB was performed by ELISA using Technozym CBA (Technoclone, Vienna, Austria).
At site 2, VWF assays were performed by VWF Ac or VWF:RCo, and VWF:Ag using ACL TOP750CT instrumentation (all Werfen, Barcelona, Spain). VWF:CB was performed by Euroanalyser one (Euro Diagnostic Systems, Tamil Nadu, India) using Zymutest VWF:CBA kit (Hyphen Biomed, Neuville‐sur‐Oise, France).
2.3. VWF multimer analysis
IHVWM at site 1 was by discontinuous SDS agarose gel electrophoresis, a modification of two methods by omission of acrylamide.14, 15 This was routinely performed at 1.6% agarose (SeaKem HGT [P] agarose; Lonza Biologicals, Slough, UK) but 1.0% agarose was also used in patients that demonstrated a loss of HMWM. Samples were diluted according to their VWF:Ag in buffer containing 10 mMol/L Tris, 1 mMol/l EDTA, 8M urea, and 2% SDS pH 8.0. Pooled normal plasma was included at three positions in the gel as a normal control. Gels were incubated with rabbit anti‐human VWF polyclonal antibody and swine anti‐rabbit antibody conjugated with alkaline phosphatase (both Dako UK Ltd, Ely, UK). Alkaline phosphatase conjugate kit (Biorad Laboratories Ltd, Hercules, CA, USA) was used to visualize the multimers. Multimer patterns were visually inspected by three independent operators since densitometry could not be performed locally. Triplet bands 1‐5 were considered LMWM, 6‐10 as IMWM, and 11 or more as HMWM. Multimer patterns were either considered qualitatively normal, reduction of HWM, gross reduction of HWM (and IMWM), or absence of multimers in comparison to the pooled normal plasma control.
Multimer analysis from site 2 was performed by the laboratory of Professor Budde in Hamburg using a previously described method.16
The Hydragel 5 VWF multimers kit (H5VWM) was used with the Hydrasys 2 Scan instrumentation to perform agarose gel electrophoresis using preformed 2% agarose gels, direct immunofixation, visualization with peroxidase‐labelled antibody, and densitometry. H5VWM Kits were provided by Sebia for this evaluation. Samples were initially tested at a 1:6 dilution of plasma with sample diluent. During this study, individuals with VWF:Ag levels below 20 IU/dL or greater than 150 IU/dL were also tested at 1:4 or 1:12 dilutions of plasma, respectively, according to the manufacturer's recommendations at the commencement of the study. The manufacturer has since revised the methodology to dilute samples greater than 150 IU/dL at 1:10. Densitometry was performed by Hydrasys 2 Scan. The percentage of LMWM, IMWM, and HMWM were assessed using Phoresis software (Sebia) with peaks 1‐3 designated as LMWM, peaks 4‐7 as IMWM, and peaks >7 as HMWM according to the manufacturer's recommendations. A normal level quality control (QC) plasma, Chronolog VWF reference plasma (Havertown, PA, USA) was included on each gel.
Lower limit of detection for the densitometry was evaluated using SSC lot 4 plasma (NIBSC, Potters Bar, UK) diluted to 20, 15, 10, and 5 IU/dL in FVIII and VWF deficient plasma (Werfen).
Historical mutation analysis in Sheffield and for those samples referred from Italy was initially undertaken using Sanger sequencing. Exons 17‐25 and exon 28 were initially analyzed for point mutations in types 2N and 2A, 2B, and 2M. Subsequently, the remainder of the VWF gene was analyzed if necessary. More recently, next generation sequencing was introduced and the entire coding region of VWF analyzed simultaneously. The gene panel comprises 13 genes analyzed using Illumina HiSeq 2500 with SureSelect, followed by bioinformatic analysis to identify sequence to identify sequence variants.
Mutation analysis was available for 66 patients at site 1 only which is a limitation to this study. Of these, five patients had mutations linked with type 1 VWD, 10 patients had p.R1205H Vicenza VWD, seven patients with type 2A VWD, 12 patients with type 2M VWD, and 11 patients had p.R1374H/C amino acid change which is variably linked to types 2A and 2M. Seventeen patients had mutations associated with 2B VWD. Four patients had mutations linked to type 2N VWD.
3. RESULTS
Inter‐assay coefficient of variation (CV), calculated using the normal QC data from 59 gels performed at site 1, was 17% for LMWM, 7.9% for IMMW, and 8.9% for HMWM. Intra‐assay variability was assessed with the normal QC tested in five wells on a single gel. CV was 6.6% for LMWM (mean 19.5%, SD 1.29), 3.6% for IMWM (mean 42.8%, SD 1.56), and 6.0% for HMWM (mean 37.5%, SD 2.26).
A quantitatively normal pattern with both multimer methods was observed in 51 of 55 healthy normal donors. Gaussian distribution was not observed with H5VWM densitometry for LWMW or HMWM so reference ranges were established from 2.5% and 97.5% percentiles for LMWM (11.8%‐23.6%), IMWM (24.6%‐42%), and HMWM (35%‐58.5%).
The effect of low VWF:Ag levels were assessed by dilution of SSC reference plasma to result in VWF:Ag ranging from 20 to 5 IU/dL. At decreasing VWF:Ag, an increase to LMWM was observed (15.8%‐30.8% at 20 and 5 IU/dL, respectively) with loss of IMWM (38%‐23.4%) but little change to HMWM (46.2%‐45.8%). Below 10 IU/dL, a loss of definition to the densitometry peaks was observed (see Figure 1A‐D).
Figure 2 presents the results of the patient cohort excluding those with type 2N, 3, and AVWS for ease of interpretation. The mean VWF and H5VWM densitometry results are depicted in Table 1. Individuals with discrepant IHVWM and H5VWM are detailed in Table 2. Seventy‐four individuals were classified as normal VWF, 72 had a quantitatively normal IHVWM. Two patients, B7, a 52 year old male, and B11, a pregnant female of 30 weeks gestation, had a loss of HMWM by IHVWM but normal densitometry with H5VWM, 38.7% and 37%, respectively.
Figure 2.
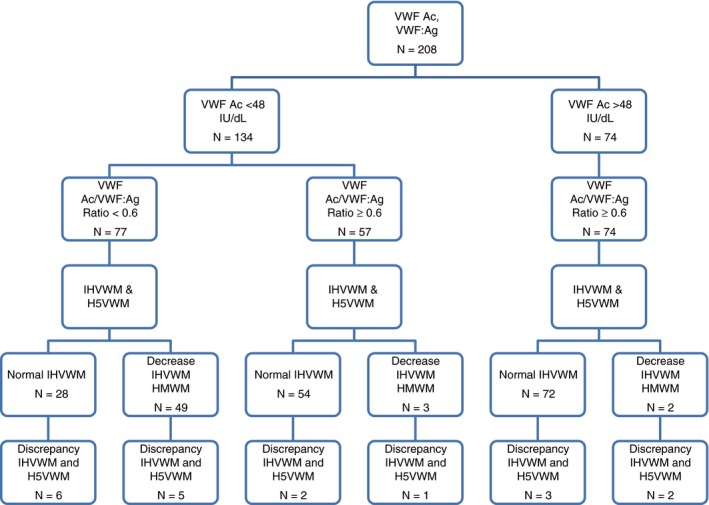
Rationalization of patient results according to the VWF activity, VWF Ac, ratio of VWF Ac/VWF:antigen (VWF:Ag) and in‐house multimer, IHVWM, result. Patients were group according to VWF Ac and then ratio of VWF activity and VWF:Ag. IHVWM pattern was assessed as normal or reduced high molecular weight multimers (HMWM) when less than 11 multimer triplets were visible. The number of patients with discrepancy between IHVWM and Hydragel 5 multimer method, H5VWM, is indicated. Results from patients with type 2N, type 3 and acquired von Willebrand syndrome (AVWS) were excluded
Table 1.
Mean (and range) of VWF parameters and H5VWM densitometry of patient cohorts included in this study
| Group, n | VWF Parameters | Hydrasys 5 densitometry | ||||
|---|---|---|---|---|---|---|
| VWF:Ag (IU/dL) | VWF Ac (IU/dL) | Ratio VWF Act/Ag | LMWM (%) | IMWM (%) | HMWM (%) | |
| Normal donors, 51 | 102 (73‐157) | 94 (58‐167) | 0.92 (0.64‐1.29) | 17.3 (10.8‐25.9) | 33.0 (23.8‐43.4) | 49.9 (34.7‐59.3) |
| Normal VWF, 74 | 99.5 (52‐305) | 89.8 (52‐277) | 0.93 (0.70‐1.28) | 17.1 (9.1‐39.5) | 30.3 (21.7‐39.6) | 52.2 (21.9‐67.8) |
| Type 1 VWD, 47 | 37.7 (12‐66) | 33.2 (8‐58a) | 0.88 (0.65‐1.32) | 20.5 (8.4‐49.3) | 28 (16.2‐41.4) | 51.6 (20.2‐70.9) |
| Type 1 Vicenza, 10 | 10.2 (6‐21) | 9.3 (5‐18) | 0.96 (0.67‐1.43) | 33.6 (21.3‐49.1) | 15.2 (5‐38.6) | 51.2 (37.8‐63.1) |
| Type 2A, 33 | 40.2 (13‐132) | 13.1 (1‐44) | 0.34 (0.04‐0.60) | 60.7 (26.7‐90.6) | 21.7 (3.5‐40.0) | 17.6 (1.1‐40.3) |
| Type 2B, 16 | 43.8 (19‐80) | 14.6 (6‐31) | 0.34 (0.25‐0.60) | 55.4 (24.5‐66.9) | 34.5 (23‐45.2) | 10.2 (3.1‐32.5) |
| Type 2M, 28 | 35.5 (9‐99) | 13.4 (4‐29) | 0.39 (0.16‐0.60) | 32.8 (9.8‐67.7) | 23.4 (5.5‐36.6) | 43.9 (20.2‐71.4) |
| Type 2N, 11 | 68 (34‐138) | 83b (33‐187) | 1.1b (0.9‐1.4) | 21.4 (14.4‐30.4) | 30.8 (6.2‐40.1) | 47.9 (31.7‐64.9) |
| Type 3, 3 | <5 | <4 | — | No peaks | ||
| AVWS, 30 | 79.4 (7‐254) | 58.6 (2‐211) | 0.72 (0.22‐1.38) | 38.3 (11‐85.9) | 25.0 (2.8‐42.2) | 36.7 (0.9‐72.9) |
AVWS, acquired von Willebrand syndrome; HMWM, high molecular weight multimers; IMWM, intermediate molecular weight multimers; LMWM, low molecular weight multimers; VWF Ac, VWF activity; VWF:Ag, VWF antigen.
VWF:CB reduced below normal range.
VWF Ac not available in 5 of 11 patients at site 1.
Table 2.
Patients with discrepant IHVWM and H5VWM
| ID | PROPOSED CLASSIFICATION | VWF:Ag | VWF Ac | Ratio | IHVWM | H5VWM | LMWM | IMWM | HMWM | MUTATION |
|---|---|---|---|---|---|---|---|---|---|---|
| P5 | Normal VWF | 72 | 57 | 0.79 | QNORM | RHWMW | 30.8 | 35.6 | 33.6 | UNK |
| P10 | Normal VWF | 115 | 87 | 0.76 | QNORM | RHMWM | 39.5 | 31.4 | 29.1 | UNK |
| P119 | Normal VWF | 102 | 82 | 0.80 | QNORM | RHWMW | 38.5 | 39.6 | 21.9 | p.P1337L |
| B7 | Normal VWF | 305 | 277 | 0.91 | RHMWM | QNORM | 30.9 | 30.4 | 38.7 | UNK |
| B11 | Normal VWF | 154 | 108 | 0.70 | RHMWM | QNORM | 28.9 | 34.1 | 37.0 | UNK |
| P64 | 1 | 20 | 13 | 0.65 | QNORM | RHMWM | 36.4 | 30.9 | 32.7 | p.C1130F |
| P65 | 1 | 20 | 18 | 0.90 | QNORM | RHMWM | 49.3 | 25.4 | 25.3 | p.C1130F |
| P70 | 1 | 21 | 16 | 0.76 | RHMWM | QNORM | 27.9 | 35.2 | 36.9 | p.P1266Q/p.F1501S |
| P205 | 1 | 45 | 35 | 0.78 | RHMWM | RHMWM | 43.2 | 36.6 | 20.2 | p.P1337L |
| P213 | 1 | 62 | 40 | 0.65 | RHMWM | RHMWM | 43.3 | 35.8 | 20.9 | p.P1337L |
| P94 | 2A | 20 | 11 | 0.55 | RHMWM | QNORM | 27.1 | 35.4 | 37.5 | p.R1374C |
| P96 | 2A | 24 | 10 | 0.42 | RHMWM | QNORM | 32.1 | 28.1 | 39.8 | p.R1374H |
| P111 | 2A | 23 | 10 | 0.43 | RHMWM | QNORM | 45.6 | 16.1 | 38.3 | p.R1374C |
| B8 | 2A | 27 | 16 | 0.59 | RHMWM | QNORM | 26.7 | 33.0 | 40.3 | UNK |
| B9 | 2A | 21 | 8 | 0.38 | RHMWM | QNORM | 30.9 | 33.4 | 35.7 | UNK |
| P121 | 2M | 31 | 13 | 0.42 | QNORM | RHMWM | 50.6 | 24.8 | 24.6 | UNK |
| P124 | 2M | 39 | 19 | 0.49 | QNORM | RHMWM | 54.1 | 25.7 | 20.2 | No mutation in exon 28 |
| P125 | 2M | 24 | 6 | 0.25 | QNORM | RHMWM | 45.1 | 21.5 | 33.4 | p.R1374C |
| P214 | 2M | 25 | 11 | 0.44 | QNORM | RHMWM | 59.9 | 18 | 22.1 | p.D1277E_L1278delinsE |
| P216 | 2M | 25 | 6 | 0.24 | QNORM | RHMWM | 67.7 | 11.1 | 21.1 | p.R1315C |
| P251 | 2M | 25 | 4 | 0.16 | QNORM | RHMWM | 60.6 | 13 | 26.4 | p.R1374C |
| P148 | 2N | 80 | 78 | 0.98 | QNORM | RHMWM | 30.4 | 37.9 | 31.7 | p.Q1053H homozygous |
Patient identifier, classification based on the in‐house VW multimer (IHVWM) result, VWF, multimer, densitometry results, and genetic mutations shown if known. Multimer results were either quantitatively normal, QNORM or demonstrated a reduction of high molecular weight multimers (RHMWM). UNK denotes unknown genetic mutation.
H5VWM densitometry observed a reduction to HMWM below 35% in three further patients. P5 and P10, with HMWM 33.6% and 29.1%, respectively, had no genetic analysis performed (see Figure 3A). P119, an 80‐year‐old male, had 21.9% HWMW and subsequent mutation analysis demonstrated a p.P1337L change in exon 28 linked to 2B VWD (see Figure 3B and Table 2).
Figure 3.
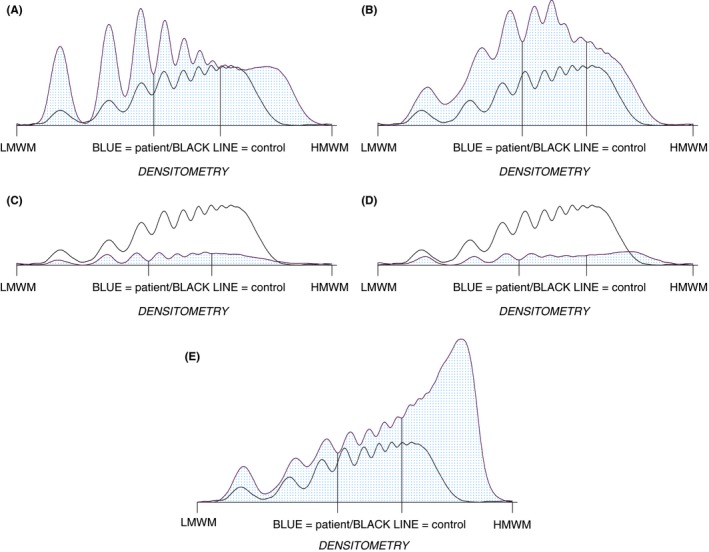
(A‐E) Example Hydragel 5 VW multimers (H5VWM) densitometry of patients from the study. (A) Patient 10, normal VWF and 29% high molecular weight multimers (HMWM). (B) Patient 119 normal VWF, 22% HMWM and p.P1337L mutation linked to type 2B VWD. (C) Patient 70, compound heterozygous for p.F1501S and p.P1266E mutations but categorised as type 1 VWF with loss of IHVWM and normal H5VWM. (D) Patient 125 categorised as type 2M VWD but flattened HMWM peak and p.R1374C mutation. (E) an example of ultra‐large MWM in P165 with TTP and reduced ADAMTS13
Fifty‐seven patients were grouped as type 1 VWD. Fifty‐two of 57 individuals had a qualitatively normal multimer pattern with both methods. Two unrelated patients (P205 and P213) had a loss of HMWM multimers with both methods. HMWM were reduced with H5VWM (20.2% and 20.9%, respectively). Both were known to have p.P1337L mutation linked to type 2B VWD (see Table 2). P213 is the daughter of P119 who was grouped as normal VWF.
P70 (VWF:Ag 21 IU/dL and VWF Ac 16 IU/dL) had a reduction of IHVWM but normal H5VWM (HMWM 36.9%) (see Figure 3C and Table 2) and was found to be compound heterozygous for type 2A and type 2B VWD with p.P1266E and p.Phe1501Ser amino acid substitutions in exon 28. Two related patients (P64 and 65) with a p.C1130F change in exon 26, had visually normal IHVWM, but H5VWM observed an increase in LMWM (49.3 and 36.4%) and decrease in HMWM (25.3 and 32.7%) (see Table 2). VWF:Ag was 20 IU/dL in both patients.
Ten patients had p.R1205H amino acid change (Vicenza VWD), VWF Ac/VWF:Ag ratios greater than 0.6 and qualitatively normal IHVWM. VWF:Ag was less than 15 IU/dL in eight patients. Densitometry of H5VWM demonstrated that all had normal HMWM but six patients had an increase in the lowest LMWM, peak 1 (23.6%‐49.1%).
Thirty‐three patients were categorized as type 2A with a loss of IHVWM. Visible triplet bands ranged from 5 to 10 present. Five (P94, 96, 111, B8, B9) had a normal H5VWM (HMWM 35.7%‐40.3%) and of these, three (P94, P96, and P111), had a point mutation at the same amino acid (p.R1374H/C), two (B8 and B9) did not have mutation analysis available (see Table 2).
A reduction of HMWM by both methods was observed in 16 individuals known to have VWD type 2B. Six to 10 triplet bands were visible by IHVWM whilst H5VWM HMWM ranged from 6.5% to 32.5%.
Twenty‐eight individuals classified as type 2M had normal IHVWM on visual inspection. 6 (P121, P124, P125, P214, P216, and P251) had reduction to HMWM (20.2%‐33.4%) with H5VWM densitometry exhibiting a flattened appearance of the HMWM peak (see Figure 3D and Table 2). An increase in LMWM peak 1 (28.5%‐67.7%) was observed in 16 of 28 patients but only P216 had a low VWF:Ag of 14 IU/dL. Three samples were from patients with a p.R1315C mutation and each had an increase of LMWM, 31.8%‐67.7%, a loss of IMWM, 9.7%‐11.1%, and one (P216) had a reduced HMWM 21.2%. Four individuals, three related, in this group had p.R1374C mutation with two (P125 and P251) having reduced HMWM by H5VWM densitometry.
Eleven patients had type 2N VWD, all had normal IHVWM multimer distribution, and 10 were normal by H5VWM. Densitometry demonstrated some loss of HMWM in P148 (HMWM 31.7%) who was homozygous for p.Q1053H. An absence of all multimers was observed by both methods in type 3 VWD. It was not possible to perform densitometry in this group.
The multimers of 18 patients, at least one patient from each type of VWD, were evaluated prior to and following treatment with Desmopressin (n = 9) or Haemate P (n = 9). The mean densitometry pre Desmopressin was LMWM 16.8%, IMWM 26.3%, and HMWM 56.9%. Post‐Desmopressin means were 16.9%, 27.2%, and 55.9%, respectively, with an increase above the top of the reference range for HMWM observed in five. Paired t test observed no statistically significant difference (P > 0.05) between pre‐ and post‐Desmopressin. Pre‐Haemate P, the mean H5VWM densitometry was LMWM 31.5%, IMWM 25.6%, and HMWM 42.9%. Post‐Haemate P, the means were 33.3%, 38.5%, and 28.2%, respectively, and only a single patient (type 1 VWD) had normal HMWM following treatment. Paired t test observed no statistically significant difference (P > 0.05) between pre‐ and post‐Haemate P for LMWM, conversely, a statistically significant difference was observed for both IMWM (P = 0.002) and HMWM (P = 0.016). Three type 2A VWD patients were treated with Wilfact and one type 3 VWD patient with Voncentro. Post‐concentrate IHVWM were visually normal in two of four, however, H5VWM demonstrated a loss of HMWM ranging from 9.3% to 20.9% in all four individuals.
Thirty previously diagnosed AVWS patients were tested but since the VWF:Ag of five was below 15 IU/dL so densitometry results were excluded. Five of 25 remaining patients demonstrated a loss of HMWM by IHVWM with no triplet bands visible above 10, and normal densitometry was observed with H5VWM. Six patients that had a loss of HWMW by both methods had normal VWF Ac/VWF:Ag ratios of 0.64‐0.76.
The presence of ultra large MWM (ULMWM) with H5VWM (see Figure 3E) was observed in the four individuals with TTP. The ULMWM using the IHVWM were less obvious, particularly at higher concentrations of ADAMTS13. Agreement in multimer results was observed in eight patients with elevated rheumatoid factor and three patients whose blood was drawn in citrate or EDTA.
4. DISCUSSION
Overall agreement of 91.1% was observed between methods in patients with normal VWF or different types of VWD. Where discrepancies occurred, this was generally due to subtle differences detected by densitometry on the Hydragel 5 system which could not be observed by visual inspection of the IHVWM. Losses of HMWM with H5VWM were observed in patients classified as normal VWF or type 1 VWD that had genetic mutations in VWF linked to type 2 VWD. Often these groups of patients may not have multimers routinely analyzed and diagnoses could be missed. Such findings suggest that VWF multimer assays should be performed in all patients with reduced VWF activity and VWF:Ag but testing should also be considered in individuals with normal VWF:Ag and VWF Ac who experience significant bleeding of unknown cause.
The reclassification of type 2 VWD into four subgroups in 1994 has led to many conflicting reports linked to particular mutations in VWF.5 Several mutations present in our cohort, including p.C1130F, p.R1205H, p.R1315C, and p.R1374C/H, have been linked to more than one type or subtype of VWD in the literature. Patients with p.C1130F have previously been classified as both type 2A(IIE)17 and type 1 VWD18 with some authors reporting an aberrant IHVWM pattern,17, 19 however, we observed a normal multimer pattern by IHVWM and a reduction of HMWM with H5VWM in three patients. Ten patients with p.R1205H (Vicenza VWD) were classified as type 1 VWD in this study although this mutation has also been classified as type 2M VWD.20 Three patients with p.R1315C were classified as type 2M but this mutation has also been linked with type 2A VWD.19, 21, 22 Eleven individuals, from four families, had p.R1374C/H, associated with both type 2A19 and 2M23 VWD. Of the seven related patients, four were grouped as type 2A and three as type 2M. This present study confirms that the presence of a particular genetic variant does not always correlate with phenotypic assays. Additional studies are required to compare tradition multimers with the H5VWM assay in patients with these ambiguous mutations.
Post DDAVP samples, eight of which were type 1 VWD and one (P70) with combined type 2A and 2B, had normal multimers with both methods. Following treatment with the VWF concentrates used in this study, only a single type 1 VWD patient, had HMWM within normal limits. Not all high purity human VWF concentrates fully restore HMWM to normal levels,10 although Haemate P has previously been demonstrated to produce a significant increase to HMWM,24 this was not observed in our cohort. Further studies are needed to investigate post treatment samples using H5VWM.
AVWS is caused by a number of mechanisms as reviewed by Mohri.25 There was no clear relationship between VWF Ac/VWF:Ag ratio and multimer profile in our patients however, agreement between multimer methods was observed in 77% of patients. The reasons for the differences are unknown but may be linked to the underlying mechanism of the disorder; further investigation of these patients is therefore warranted.
Plasma from 55 normal donors was initially included to establish adult reference ranges. Inter‐quartile ranges (IQR) were calculated; however, four donors, one female and three males, had LMWM (28.5%‐33.5%) which exceeded the upper quartile +1.5 IQR value of 26.2%. These donors were considered statistical outliers and excluded from further reference range calculations. The results of limited groups of patients suggested that H5VWM was not affected by the presence of rheumatoid factor or use of EDTA in place of sodium citrate as anticoagulant. CV of less than 10% were observed for IMWM and HMWM with the normal QC sample, but LWMW was unexpectedly slightly more variable than the other parameters. Ideally, a pathological QC sample with reduced HMWM would also be included on each gel. This would not be practical with the five‐well H5VWM gel used for this present study, however, an eleven‐well gel (Hydragel 11 VW) is available which offers more scope for the inclusion of two levels of QC plasma.
The sensitivity of VWF activity assays to different VWF mutations clearly impacts on the diagnosis and classification of VWD. Innovance VWF Ac, performed at site 1, is a VWF:GP1bM type assay2 that utilizes two gain of function mutations to glycoprotein 1b (GP1b) in place of Ristocetin and platelets. The VWF:Ag and VWF Ac of multimer discrepant patients were confirmed on repeat. It is possible that the VWF Ac assay was less sensitive to certain mutations than a traditional VWF:RCo. A previous study reported some slight differences in particular in type 2A (IIE), Vicenza VWD, and several type 1 VWD patients.26 Laboratories which use one of the new generation of VWF activity assays should contemplate including a second activity method during the initial diagnosis of VWD. We propose an algorithm for the inclusion of H5VWM as a screening tool in the diagnosis of VWD in the laboratory (see Figure 4). This algorithm uses a VWF Ac/VWF:Ag ratio cut‐off of 0.8 to determine whether VWF multimers should be performed. This increased ratio was chosen to try to ensure that those few patients that had a loss of HMWM but were classified by ratio as normal VWF or type 1 VWD, would be further investigated. Subsequent studies would determine whether a higher ratio cut‐off value is beneficial in the diagnosis of such patients.
Figure 4.
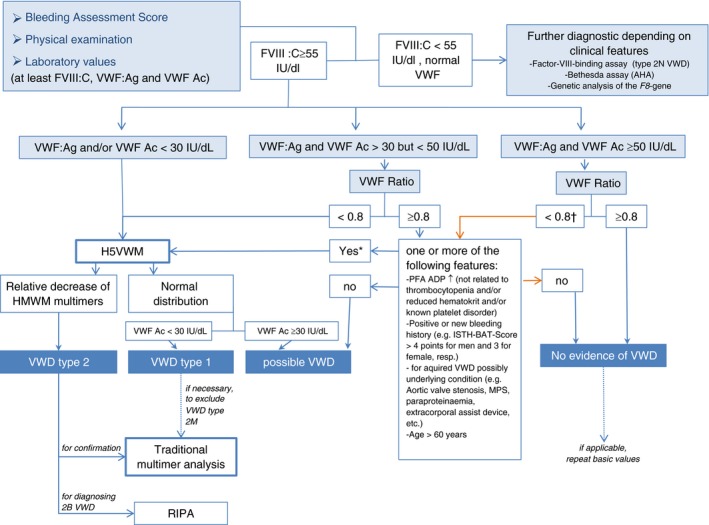
Proposed VWD diagnostic algorithm incorporating H5VWM. *The more of these features the stronger the recommendation for H5VWM and vice versa. †Follow orange line for VWF ≥50 IU/dL in absence of the mentioned features. AHA, acquired hemophilia A; FVIII, factor VIII activity; H5VWM, Hydragel 5 von Willebrand Multimer; MPS, myeloproliferative syndrome; PFA, platelet function analyzer; RIPA, Ristocetin‐Induced Platelet Aggregation; VWF, von Willebrand factor; VWD, von Willebrand disease; VWF ratio, ratio of VWF Act/VWF:Ag
The H5VWM method has recently been reviewed in a letter.27 Interpretable results were available within one working day using minimal plasma, though no triplet patterns could be observed and there is no option to vary the agarose concentration to focus on different multimers. A loss of definition of the multimers was observed at VWF:Ag below 10 and at 5 IU/dL the densitometry was difficult to interpret. At decreasing VWF:Ag the proportion of LMWM doubled, with a corresponding loss of IMWM, although, the percentage of HMWM was unchanged. Twenty‐one patients had VWF:Ag below 15 IU/dL this included eight patients with p.R1205H mutation, three patients with type 3 VWD, and five patients with AVWS. It must be recognized that the densitometry results in these patients should be interpreted with caution.
The accurate phenotypic diagnosis and classification of VWD requires the assessment of VWF multimer distribution. Current, IHVWM methods are laborious, nonstandardized, multi‐step assays which require a degree of expertise to perform and interpret. The difficulty in diagnosis is compounded by reports of different multimer patterns in patients who have identical mutations and similar VWF levels. Sample referral is common practice for many laboratories nevertheless, this incurs transportation costs, and a delay to provision of the final result which may deter clinicians from routinely requesting VWF multimer analysis. The introduction of a rapid, semi‐automated, ready‐to‐use kit which can be used to reduce the workload of traditional multimer methods by rapidly screening out patients with normal multimer patterns and is performed on existing technology is to be welcomed, especially by less specialized laboratories. In the current format, using a single agarose concentration and without the sensitivity to visualize the multimer triplet structure, H5VWM is unlikely to completely replace in‐house multimer analysis but as demonstrated in our proposed algorithm, it would be a useful addition to VWD diagnosis in the routine laboratory. Multimer abnormalities could then be further explored in more detail using in‐house methods with larger gels and at different agarose concentrations as required.
RELATIONSHIP DISCLOSURE
AB has received investigator, speaker fees, and travel funding from Sebia, France.
AUTHOR CONTRIBUTIONS
A. Bowyer designed the research, performed the experiments, and wrote the manuscript. K. Goodfellow and H. Seidel designed the research, performed the experiments, and edited the manuscript. P. Westhofen performed the research and collected subject data. A. Goodeve performed the research, collected subject data, and edited the manuscript. F. Stufano and M. Makris collected subject data and edited the manuscript. S. Kitchen designed the research and edited the manuscript.
ACKNOWLEDGMENTS
We are grateful to Barbara Sampson for assistance with visualization of the in‐house multimer gels and the staff in Coagulation at Leeds St James Hospital for the provision of additional plasma and test results for their patients. We would like to thank Ghislaine Beaulieu, Hector Bautista, and Georges Nouadje from Sebia, France for provision of the Hydrasys analyzer, H5VWM kits, rheumatoid factor samples, training, and advice throughout this study.
Bowyer AE, Goodfellow KJ, Seidel H, et al. Evaluation of a semi‐automated von Willebrand factor multimer assay, the Hydragel 5 von Willebrand multimer, by two European Centers. Res Pract Thromb Haemost. 2018;2:790–799. 10.1002/rth2.12141
Contributor Information
Annette E. Bowyer, Email: annette.bowyer@sth.nhs.uk.
Michael Makris, https://twitter.com/profmakris.
REFERENCES
- 1. Lillicrap D. The basic science, diagnosis and clinical management of von Willebrand disease. 2nd edn Montreal, QC: World Federation of Haemophilia; 2008. [Google Scholar]
- 2. Bodo I, Eikenboom J, Montgomery RR, Patzke J, Schneppenheim R, Di Paola J. Platelet‐dependent von Willebrand factor activity. Nomenclature and methodology: communication from the SSC of the ISTH. JTH.. 2015;13:1345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Laffan MA, Lester W, O'Donnell JS, et al. The diagnosis and management of von Willebrand disease: a United Kingdom Haemophilia Centre Doctors Organization guideline approved by the British Committee for Standards in Haematology. Br J Haematol. 2014;167:453–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sadler JE, Budde U, Eikenboom JCJ, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4:2103–14. [DOI] [PubMed] [Google Scholar]
- 5. Sadler JE. A revised classification of von Willebrand disease. Thromb Haemost. 1994;71:520–5. [PubMed] [Google Scholar]
- 6. Ruggeri ZM, Zimmerman TS. von Willebrand factor and von Willebrand disease. Blood. 1987;70:895–904. [PubMed] [Google Scholar]
- 7. Cumming AM, Wensley RT. Analysis of von Willebrand factor multimers using a commercially available enhanced chemiluminescence kit. J Clin Pathol. 1993;46:470–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pruthi RK, Daniels TM, Heit JA, Chen D, Owen WG, Nichols WL. Plasma von Willebrand factor multimer quantitative analysis by in‐gel immunostaining and infrared fluorescent imaging. Thromb Res. 2010;126:543–9. [DOI] [PubMed] [Google Scholar]
- 9. Ledford‐Kraemer MR. Analysis of von Willebrand factor structure by multimer analysis. Am J Hematol. 2010;85:510–4. [DOI] [PubMed] [Google Scholar]
- 10. Studt JD, Budde U, Schneppenheim R, et al. Quantification and facilitated comparison of von Willebrand factor multimer patterns by densitometry. Am J Clin Pathol. 2001;116:567–74. [DOI] [PubMed] [Google Scholar]
- 11. Mazurier C, Parquet‐Gernez A, Goudemand J, Taillefer MF, Goudemand M. Investigation of a large kindred with type IIB von Willebrand's disease, dominant inheritance and age dependent thrombocytopenia. Br J Haematol. 1988;69:499–505. [DOI] [PubMed] [Google Scholar]
- 12. Favaloro EJ, Patterson D, Denholm A, et al. Differential identification of a rare form of platelet‐type (pseudo‐) von Willebrand disease (VWD) from Type 2B VWD using a simplified ristocetin‐induced‐platelet‐agglutination mixing assay and confirmed by genetic analysis. Br J Haematol. 2007;139:623–6. [DOI] [PubMed] [Google Scholar]
- 13. Nesbitt IM, Goodeve AC, Guilliatt A, Makris M, Preston FE, Peake IR. Characterisation of von Willebrand disease using phenotypic and molecular techniques. Thromb Haemost. 1996;75:959–64. [PubMed] [Google Scholar]
- 14. Ruggeri ZM, Zimmerman TS. The complex multimeric composition of factor VIII/von Willebrand factor. Blood. 1981;57:1140–3. [PubMed] [Google Scholar]
- 15. Enayat MS, Hill FG. Analysis of the complexity of the multimeric structure of factor VIII related antigen/von Willebrand protein using a modified electrophoretic technique. J Clin Pathol. 1983;36:915–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Budde U, Schneppenheim R, Plendl H, Dent JA, Ruggeri ZM, Zimmerman TS. Luminographic detection of von Willebrand factor multimers in agarose gels and on nitrocellulose membranes. Thromb Haemost. 1990;63:312–5. [PubMed] [Google Scholar]
- 17. Schneppenheim R, Michiels JJ, Obser T, et al. A cluster of mutations in the D3 domain of von Willebrand factor correlates with a distinct subgroup of von Willebrand disease: type 2A/IIE. Blood. 2010;115:4894–901. [DOI] [PubMed] [Google Scholar]
- 18. James PD, Notley C, Hegadorn C, et al. The mutational spectrum of type 1 von Willebrand disease: results from a Canadian cohort study. Blood. 2007;109:145–53. [DOI] [PubMed] [Google Scholar]
- 19. Budde U, Schneppenheim R, Eikenboom JCJ, et al. Detailed von Willebrand factor multimer analysis in patients with von Willebrand disease in the European study, molecular and clinical markers for the diagnosis and management of type 1 von Willebrand disease (MCMDM‐1VWD). J Thromb Haemost. 2008;6:762–71. [DOI] [PubMed] [Google Scholar]
- 20. Casonato A, Pontara E, Sartorello F, et al. Identifying type Vicenza von Willebrand disease. J Lab Clin Med. 2006;147:96–102. [DOI] [PubMed] [Google Scholar]
- 21. Ribba A, Hilbert L, Lavergne J, et al. The arginine‐552‐cysteine (R1315C) mutation within the A1 loop of von Willebrand factor induces an abnormal folding with a loss of function resulting in type 2A–like phenotype of von Willebrand disease: study of 10 patients and mutated recombinant von Willebrand factor. Blood. 2001;97:952–9. [DOI] [PubMed] [Google Scholar]
- 22. Casonato A, Pontara E, Sartorello F, Bertomoro A, Durante C, Girolami A. Type 2M von Willebrand disease variant characterized by abnormal von Willebrand factor multimerization. J Clin Lab Med. 2001;137:70–6. [DOI] [PubMed] [Google Scholar]
- 23. James PD, Notley C, Hegadorn C, et al. Challenges in defining type 2M von Willebrand disease: results from a Canadian cohort sudy. J Thromb Haemost. 2007;5:1914–22. [DOI] [PubMed] [Google Scholar]
- 24. Auerswald G, Kreuz W. Haemate P/Humate‐P for the treatment of von Willebrand disease: considerations for use and clinical experience. Haemophilia. 2008;14(Suppl 5):39–46. [DOI] [PubMed] [Google Scholar]
- 25. Mohri H. Acquired von Willebrand Syndrome: features and management. Am J Hematol. 2006;81:616–23. [DOI] [PubMed] [Google Scholar]
- 26. Lawrie AS, Stufano F, Canciani MT, Mackie IJ, Machin SJ, Peyvandi F. A compartive evaluation of a new automated assay for von Willebrand factor activity. Haemophilia. 2013;19:338–42. [DOI] [PubMed] [Google Scholar]
- 27. Favaloro EJ, Oliver S. Evaluation of a new commercial von Willebrand factor multimer assay. Haemophilia. 2017;23:e373–7. [DOI] [PubMed] [Google Scholar]