

Abstract
Mutations in the ATP1A3 gene (the α‐3 subunit of the Na+/K+ ATPase) are associated with rapid‐onset dystonia‐parkinsonism; alternating hemiplegia of childhood; and cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS syndrome). The authors report 3 cases with pleiotropic movement disorders, including a novel mutation in a patient who presented with ataxia and dysphagia. Case 1 had a history of attention deficit hyperactivity disorder and developed dysphagia, chorea, and limb dystonia after a febrile illness at age 12 years. Case 2 presented with limb dystonia at age 26 years and dysarthia and dysphagia after a febrile illness. Case 3 had a history of learning disability and developed progressive ataxia with cerebellar atrophy at age 20 years. In all cases, deleterious mutations were identified in ATP1A3. They illustrate wide phenotypic variability, including chorea and ataxia. New cases are likely to be diagnosed as knowledge about the phenotypic spectrum expands.
Keywords: rapid‐onset dystonia, parkinsonism, dystonia, ataxia
Mutations in the ATP1A3 gene, which encodes the α‐3 subunit of the Na+/K+ ATPase, are associated with rapid dystonia parkinsonism (RDP), alternating hemiplegia of childhood, early life epilepsy with microcephaly, and CAPOS syndrome (cerebellar ataxia, arreflexia, pes cavus, optic atrophy, and sensorineural hearing loss).1, 2, 3 Extensive phenotypic variation is described, and genotype‐phenotype correlations continue to be refined.4 In addition to the prominent motor symptoms, there is increasing recognition of cognitive and psychiatric manifestations.5, 6 We describe 3 patients who were evaluated in our movement disorders clinic and had distinct phenotypes illustrating this variability.
Case Series
Case 1
A 10 year‐old African‐American boy had a history of neonatal feeding difficulties and attention deficit hyperactivity disorder (ADHD), which was treated with methylphenidate. One day after recovering from a febrile illness, he developed dysarthia and dysphagia. Over the next several weeks, he developed orobuccal and upper extremity dystonia and chorea. Despite negative testing, he was originally treated for Sydenham's chorea with antibiotics, valproic acid, and steroids. No improvement was seen. Investigation with imaging, copper studies, blood, and cerebrospinal fluid autoimmune and infectious studies were negative. Genetic testing with sequencing of ATP1A3 demonstrated a mutation in exon 17 c.2267G>A, p.Arg756His. This mutation has been reported previously in an infant with a distinct phenotype that included spells of hypotonia, dysphagia and developmental delay7 (Video 1).
Case 2
A 26 year‐old African‐American woman with a history of migraines presented with limb dystonia at age 26 years. She first noticed flexion of the fingers on the left hand. Over the course of 2 years, there was involvement of the left wrist and left foot. Investigation with basic blood work, including ceruloplasmin, tests of liver function, and antinuclear antibody were normal. Nerve‐conduction studies and electromyographic examination of the left leg were normal. A magnetic resonance image of the brain and whole spine was unremarkable, and she was treated with trihexyphenidyl and botulinum toxin injections with modest benefit. After a febrile illness and transiently stopping the trihexyphenidyl, she developed severe dysarthia, dysphagia, and worsening of her limb dystonia. Re‐examination disclosed hypomimia and postural imbalance. Genetic testing with sequencing of ATP1A3 demonstrated a mutation in exon 8 c.829G>A, p.Glu277Lys. This case was previously reported8 (Video 2).
Case 3
A 28‐year‐old man with a history of mild learning disability and migraines developed falls at age 21 years. There were no clear triggers. This progressed to prominent gait and appendicular ataxia with dysarthria, associated action tremor, and myoclonus. Oculomotor abnormalities included intrusions and dysmetric saccades with no optic nerve or retinal pathology. Within a few years, he became dependent on a walker and wheelchair for longer distances. Nerve‐conduction and electromyography studies were unremarkable. Neuroimaging demonstrated vermian cerebellar atrophy, which was stable on repeated examinations (Fig. 1). An extensive diagnostic workup was performed, including metabolic, mitochondrial, autoimmune, and genetic causes of cerebellar degeneration. Whole‐exome sequencing demonstrated a de novo mutation in exon 8 c.946G>A, p.Gly316Ser.9 Repeat mutation analysis of the ATP1A3 gene in a CLIA (Clinical Laboratory Improvement Amendments) laboratory confirmed the mutation in the proband. Repeat testing of the parents in a CLIA laboratory was declined. To the best of our knowledge, this the first full, published report of this mutation, as we did not find it in large publically available cohorts.10, 11 Both the G‐to‐A substitution and the predicted Gly‐to‐Ser substitution occur at highly conserved nucleotides and amino acids, respectively. Computational analysis tools (SIFT and Mutation Taster) predict that this mutation will be deleterious and disease causing. Two other well‐known benign variants were also found (homozygous exon 7 c666T>G, p.Thr222Thr [rs2217342] and heterozygous exon 23 c.*39C>G [rs919390]) (Video 3).
Figure 1.
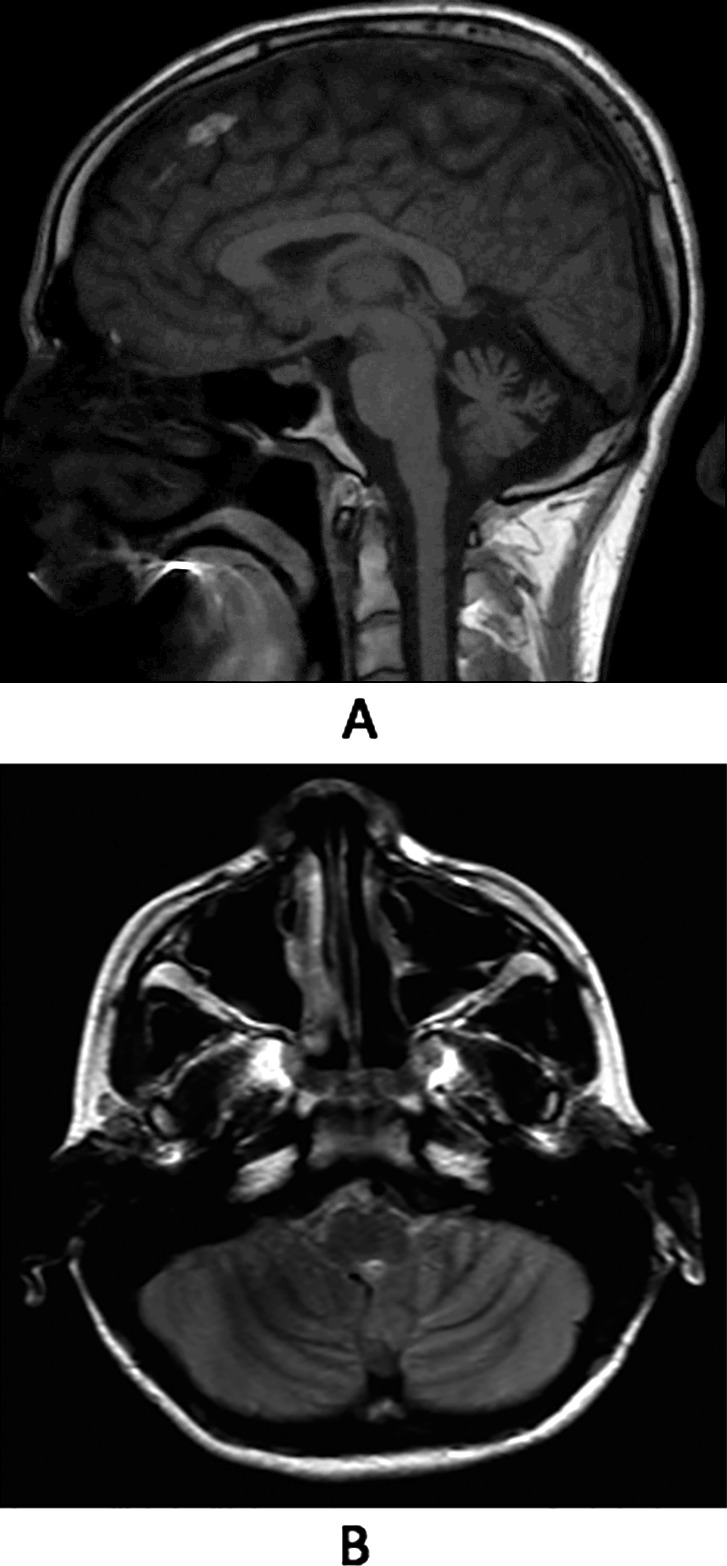
Magnetic resonance images of the patient described in case 3. (A) In a sagittal, fluid‐attenuated inversion recovery image, vermian atrophy is seen. (B) An axial T2 image reveals thinning of cerebellar folia with prominent cerebrospinal fluid spaces.
Discussion
These 3 cases illustrate the phenotypic variability associated with ATP1A3 mutations. Case 1 is remarkable for the typical core features of RDP, including abrupt onset of dystonia after a physiological stressor, predominant bulbar findings, and a rostrocaudal gradient.12 In addition, chorea was part of the movement disorder. The previous history of ADHD may suggest additional neuropsychiatric manifestations of ATP1A3, as recently described.5, 6
Case 2 shares the core features of bulbar symptomatology and rapid worsening after a physiological stressor but is notable for hemidystonia in the absence of structural abnormalities, as previously reported.8 Case 3 illustrates a novel mutation with features of cerebellar ataxia and prominent atrophy on imaging. It expands even further the phenotype of yet another syndrome associated with ATP1A3 mutations that includes ataxia with arreflexia, optic atrophy, pes cavus, and hearing loss.2 Notably, this case lacked the additional findings and presented exclusively with ataxia, suggesting that this gene should be included in the consideration of the myriad etiologies of ataxic disorders.
This case series illustrates how clinical medicine has benefitted from expanding knowledge in genomic science. Notably, there was no parkinsonism in these cases, which lacked the bradykinesia and postural imbalance in the original descriptions of RDP.13 It underscores how an accurate phenotypic description allows the precise determination of different syndromes attributed to the same gene. Recent reports demonstrate overlapping features of alternating hemiplegia of childhood and RDP, which previously were considered distinct phenotypes associated with ATP1A3 mutations.14 Similar considerations may be entertained with ataxia and CAPOS syndrome. As genetic knowledge continues to expand, precise clinical phenotyping is crucial for useful, pragmatic phenotype‐genotype correlations. This series should encourage clinicians to test for mutations in ATP1A3 when the phenomenology includes the core features and to entertain its possibility when associated with chorea or ataxia. New cases are likely to be diagnosed as awareness and diagnostic technologies continue to evolve.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
C.M.d.G: 1A, 1B, 1C, 3A
M.D.: 1C, 3B
N.S.: 3B
Disclosures
Funding Sources and Conflicts of Interest: The authors report no particular sources of funding and no conflicts of interest relevant to this work.
Financial Disclosures for the previous 12 months: Claudio M. de Gusmao received compensation from the Movement Disorders Society to assist in the educational design of an online course for advanced therapies in Parkinson's disease. He was the recipient of the Marshall Wolff Neurology Fellowship and the Silverman Family Bachman‐Strauss Fellowship for Movement Disorders. Nutan Sharma received support from the following funding sources: the National Institute of Neurological Disorders and Stroke (P50 NS037409), the Silverman Family Bachmann‐Strauss Fellowship for Movement Disorders, and The Marshall Wolf Neurology Fellowship. Marisela Dy does not have any relevant disclosures.
Supporting information
Videos accompanying this article are available in the supporting information here.
Video S1: Video clip of the patient described in case 1. The timeline is indicated over months to indicate stability over time. Dysarthia and orobuccal dystonia are seen with drooling and a fixed smile. Upper extremity dystonia is noted, and choreiform intrusions are most prominent during gait.
Video S2: Video clip of the patient described in case 2. Hemidystonia is noted with abnormal posturing of the left arm and leg; jaw dystonia is seen at rest.
Video S3: Video clip of the patient described in case 3. Cerebellar features, such as scanning dysarthria and oculomotor dysmetria, are seen with associated appendicular and axial ataxia.
Supporting information may be found in the online version of this article.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Heinzen EL, Arzimanoglou A, Brashear A, et al. Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol 2014;13:503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Demos MK, van Karnebeek CD, Ross CJ, et al. A novel recurrent mutation in ATP1A3 causes CAPOS syndrome [serial online]. Orphanet J Rare Dis 2014;9:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Paciorkowski AR, McDaniel SS, Jansen LA, et al. Novel mutations in ATP1A3 associated with catastrophic early life epilepsy, episodic prolonged apnea, and postnatal microcephaly. Epilepsia 2015;56:422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brashear A, Ozelius LJ, Sweadner KJ. ATP1A3 mutations: what is the phenotype? Neurology 2014;82:468–469. [DOI] [PubMed] [Google Scholar]
- 5. Cook JF, Hill DF, Snively BM, et al. Cognitive impairment in rapid‐onset dystonia‐parkinsonism. Mov Disord 2014;29:344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brashear A, Cook JF, Hill DF, et al. Psychiatric disorders in rapid‐onset dystonia‐parkinsonism. Neurology 2012;79:1168–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brashear A, Mink JW, Hill DF, et al. ATP1A3 mutations in infants: a new rapid‐onset dystonia‐parkinsonism phenotype characterized by motor delay and ataxia. Dev Med Child Neurol 2012;54:1065–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tarsy D, Sweadner KJ, Song PC. Case 17‐2010: a 29‐year‐old woman with flexion of the left hand and foot and difficulty speaking. N Engl J Med 2010;362:2213–2219. [DOI] [PubMed] [Google Scholar]
- 9. Sweadner KJ, Cook J, Snively B, Ozelius L, Toro C, Brashear A. A case of rapid‐onset dystonia‐parkinsonism (RDP) with prominent ataxia [abstract]. Neurology 2015;84(P2):117. [Google Scholar]
- 10. Exome Aggregation Consortium (ExAC) . ExAC Browser. Cambridge, MA: ExAC; 2015. Available at: http://exac.broadinstitute.org.11/24/2015. [Google Scholar]
- 11. National Heart, Lung, and Blood Institute (NHLBI) . NHLBI GO exome sequencing project (ESP). Exome Variant Server. Seattle, WA: NHLBI; 2010. Available at: http://evs.gs.washington.edu/EVS.11/24/2015. [Google Scholar]
- 12. Brashear A, Dobyns WB, de Carvalho Aguiar P, et al. The phenotypic spectrum of rapid‐onset dystonia‐parkinsonism (RDP) and mutations in the ATP1A3 gene. Brain 2007;130(pt 3):828–835. [DOI] [PubMed] [Google Scholar]
- 13. Dobyns W, Ozelius L, Kramer P, et al. Rapid‐onset dystonia‐parkinsonism. Neurology 1993;43:2596–2602. [DOI] [PubMed] [Google Scholar]
- 14. Termsarasab P, Yang AC, Frucht SJ. Intermediate phenotypes of ATP1A3 mutations: phenotype‐genotype correlations [serial online]. Tremor Other Hyperkinet Mov (N Y) 2015;5:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Videos accompanying this article are available in the supporting information here.
Video S1: Video clip of the patient described in case 1. The timeline is indicated over months to indicate stability over time. Dysarthia and orobuccal dystonia are seen with drooling and a fixed smile. Upper extremity dystonia is noted, and choreiform intrusions are most prominent during gait.
Video S2: Video clip of the patient described in case 2. Hemidystonia is noted with abnormal posturing of the left arm and leg; jaw dystonia is seen at rest.
Video S3: Video clip of the patient described in case 3. Cerebellar features, such as scanning dysarthria and oculomotor dysmetria, are seen with associated appendicular and axial ataxia.