

Abstract
Coordinated production of IFN-γ by innate and adaptive immune cells is central for host defense, but can also trigger immunopathology. The investigation of the lymphoid cell-specific contribution to the IFN-γ-mediated intestinal pathology during Toxoplasma gondii infection identified CD4+ T cells as a key cell population responsible for IFN-γ-dependent intestinal inflammation and Paneth cell loss, where T-bet-dependent ILC1 cells play a minor role in driving the parasite-induced immunopathology. This was evident from the analysis of T-bet deficiency that did not prevent the intestinal inflammation and instead revealed that T-bet-deficient CD4+ Th1 cells are sufficient for T. gondii-triggered acute ileitis and Paneth cell loss. These results revealed that T-bet independent Th1 effector cells are major functional mediators of the type I immunopathological response during acute gastrointestinal infection.
Introduction
The pro-inflammatory cytokine IFN-γ plays a major role in host resistance to large and diverse groups of intracellular pathogens. Coordinated IFN-γ production by innate and adaptive immune cells throughout all stages of infections is indispensable for controlling pathogen burden. Research has shown that IFN-γ plays a crucial role in host resistance to numerous intracellular pathogens, including Salmonella, Mycobacterium tuberculosis, Leishmania major and Toxoplasma gondii1, 2, 3, 4, 5, 6, 7, 8, 9, 10. Although much of the attention has been focused on T cell-derived IFN-γ in determining host resistance to pathogens, recent reports have shown a contribution of NK cells, group 1 innate lymphoid cells (ILC1s), and neutrophils in mediating type I immunity against intracellular pathogens11, 12, 13, 14, 15, 16, 17. Failure to produce IFN-γ in a time and cell-type specific manner results in acute susceptibility to all groups of intracellular pathogens, including viruses, bacteria, and protozoan parasites18, 19, 20, 21.
There is also a growing body of literature indicating that a type I immune response to mucosal pathogens is associated with intestinal inflammation17, 22, 23, 24. While the precise mechanism connecting IFN-γ production with intestinal pathology remains elusive, recent studies suggest that IFN-γ mediates its pathological effects in part via selective elimination of Paneth cells, an epithelial cell type that is essential for mucosal barrier function25, 26, 27. The work from our and other laboratories demonstrated that IFN-γ-mediate loss of Paneth cell antimicrobial peptides (AMPs) and the uncontrolled expansion of Gram-negative bacteria of the Enterobacteriaceae family25, 28, 29. The expanded Enterobacteriaceae bacteria in the absence of Paneth cell AMPs contributes to severe intestinal inflammation25. Importantly, preservation of Paneth cells by blocking IFN-γ completely prevented intestinal dysbiosis and intestinal inflammation25. However, it is not entirely clear how IFN-γ initiates Paneth cell loss. It is also unknown how multiple IFN-γ-producing cells cooperate in type I intestinal inflammation.
Among several factors regulating IFN-γ expression, the T-box transcription factor T-bet, encoded by Tbx21, is known as the master-regulator of IFN-γ expression in the CD4+ T helper type I lineage30, 31. This transcriptional factor also plays a role in the functional maturation of intraepithelial lymphocytes (IELs) required for their host protective and pathological effector functions32, 33. Recently it was revealed that T-bet regulates the development of ILC1s and functions as the lineage-defining transcription factor for ILC1s11, 34. These cells are uniquely positioned for host protection against mucosal pathogens35, but their involvement in IFN-γ-mediated pathological response is incompletely understood. We sought to dissect the relative contribution of IFN-γ-producing lymphoid cells in T. gondii-mediated intestinal immunopathology. To address this question we orally infected T-bet-deficient mice (Tbx21−/−) with the parasite and assessed the immunopathological response in the small intestine. Unexpectedly, T. gondii-infected Tbx21−/− mice exhibited substantial intestinal inflammation, loss of Paneth cells and their AMPs. Similar to WT controls, T-bet-deficient mice developed intestinal dysbiosis in the small intestine characterized by the expansion of Enterobacteriaceae bacteria, indicating a limited role for ILC1s in the parasite-mediated immunopathological response. Further analysis of T. gondii-infected WT and Tbx21−/− mice revealed that T-bet, while being highly expressed in WT Th1 cells, is not essential for the CD4+ T cell differentiation into Th1 cells in vivo. Additional adoptive transfer experiments formally dismissed a T-cell-intrinsic requirement for T-bet in the Th1 effector choice, since T-bet-deficient CD4+ T cells were capable of IFN-γ production, although with a delayed kinetics when compared to WT controls. Most importantly, we revealed that T-bet-deficient Th1 cells are fully capable to induce IFN-γ-mediated intestinal pathology and sufficient for the parasite-mediated Paneth cell loss. Thus, our findings formally established the ability of CD4+ T cells to acquire Th1 effector phenotype and mediate immunopathological responses in the absence of T-bet.
Results
T. gondii induce intestinal inflammation in the absence of T-bet
T. gondii infection results in a complex interplay between multiple cell types that are required for IFN-γ-mediated host resistance to the parasite and are responsible for severe intestinal inflammation36, 37, 38. Recent experiments revealed particular roles for neutrophils, ILC1s, and CD4+ T cells as the major mediators of the IFN-γ-dependent immunopathological response to the parasite39. To further dissect how IFN-γ-producing cells contribute to intestinal pathology, we focused on the consequences of T-bet deficiency on the outcome of the T. gondii-driven gastrointestinal pathology. T-bet, considered being the master regulator of IFN-γ production is also essential for the development of ILC1s and maturation of IELs. Therefore, we investigated if T-bet deficiency alleviates the parasite-triggered intestinal immunopathology.
Analysis of the small intestines from T. gondii-infected mice revealed that the parasite-induced severe intestinal inflammation in both WT and T-bet-deficient mice (Fig. 1A–1C). The intestinal inflammation observed in the Tbx21−/− mice was unforeseen, since the described essential IFN-γ-producing cell types, CD4+ Th1s and ILC1s are impaired and absent, respectively, in animals lacking T-bet11. Moreover, IFN-γ is a major mediator of parasite-induced intestinal inflammation25, 40, 41, and it is largely accepted that loss of T-bet results in abrogation of IFN-γ expression31. Because selective Paneth cell loss is a key characteristic of T. gondii-mediated intestinal inflammation25, we next evaluated a role for T-bet in the disappearance of Paneth cells. Our experiments revealed that T. gondii-infected Tbx21−/− mice demonstrated a significant loss of Paneth cells by day 7 post-infection (Fig. 1D–1F).
Figure 1. T. gondii infection leads to intestinal immunopathology in Tbx21-deficient mice.
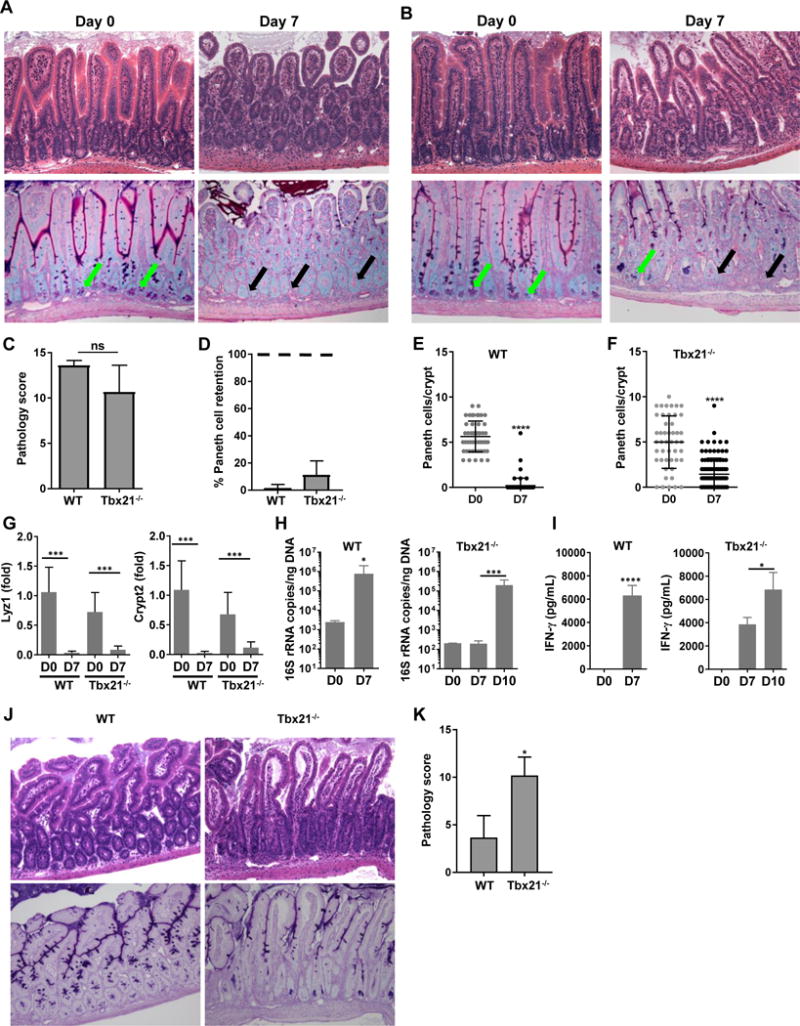
Histological analysis of the small intestines and visualization of Paneth cells in (A) WT and (B) Tbx21−/− mice orally infected with 20 cysts of the ME49 strain were performed on day 7 post-infection. Green arrows indicate Paneth cells; black arrows point toward crypts lacking Paneth cells. The results shown are representative of three independent experiments. (C) Histological changes in the small intestine were analyzed on day 7 post-infection based on an additive scoring system. (D-F) Paneth cell quantification analysis of the small intestines and visualization of Paneth cells in WT and Tbx21−/− were performed on days 0 and 7 post-infection. In (D), quantification of number of Paneth cells in naïve mice (100%) is marked by the dotted line. (G) qRT-PCR analysis of relative Lyz1 and Crypt2 expression measured in the small intestine of naïve WT and Tbx21−/− mice (n=4) or mice infected with 20 cysts of ME49 on day 7 post-infection (n=12). (H) qRT-PCR analysis of Enterobacteriaceae from the lumens of the small intestines of naïve mice, or mice infected with 20 cysts of ME49 on day 7 (WT) and days 7 and 10 (Tbx21−/−) following infection. (I) Analysis of serum IFN-γ in WT and Tbx21−/− mice infected with T. gondii was performed on day 7 and 10 post-infection. (J, K) Histological changes in the small intestine of WT and Tbx21−/− mice were analyzed on day 10 post-infection based on an additive scoring system. The data shown are representative of three (G-K) and four independent experiments (A-F; n = 3-5 mice per group for each independent experiment). Statistical analyses were done using Mann-Whitney analysis of individual groups, *P<0.05, ***P<0.001, ****P<0.0001.
We next analyzed Paneth cell-specific transcripts in the small intestines from T. gondii-infected WT and Tbx21−/− mice. A quantitative analysis of the examined Paneth cell-specific AMP transcripts revealed that T. gondii infection resulted in complete or nearly complete loss of AMP signal in both WT and T-bet-deficient mice (Fig. 1G).
We and others have previously observed that T. gondii infection triggers dramatic and selective Enterobacteriaceae expansion, a large family of Gram-negative bacteria frequently associated with intestinal inflammation25, 28. To assess a role for T-bet in the parasite-induced intestinal dysbiosis, we quantified Enterobacteriaceae in the small intestines of infected Tbx21−/− mice on days 7 and 10 post-infection. We detected a similar, even though slightly delayed expansion of Enterobacteriaceae in Tbx21−/− mice when compared to infected WT animals (Fig. 1H), which correlated with delayed systemic induction of IFN-γ observed in T. gondii-infected Tbx21−/− mice (Fig. 1I). Furthermore, day 10 expansion of Enterobacteriaceae and IFN-γ induction in T-bet-deficient mice coincided with significantly increased immunopathology compared to WT animals (Fig. 1J, 1K). When combined, our results establish that T. gondii infection triggers intestinal inflammation in the absence of T-bet.
Th1 CD4+ T cells mediate intestinal pathology in the absence of T-bet
Detailed analyses of T. gondii-infected mice revealed a major role for polarized Th1 cells in mediating intestinal inflammation25, 38, 42, 43, 44. CD4+ T cell-derived IFN-γ is required and sufficient for T. gondii-triggered intestinal inflammation and Paneth cell loss in WT mice25. To examine the possibility that lack of T-bet results in a morphologically similar gastrointestinal pathology, but is driven by a distinct mechanism, we examined the roles for both CD4+ T cells and IFN-γ in parasite-induced T-bet independent immunopathology. We revealed that similar to WT controls, blocking IFN-γ in Tbx21−/− mice prevented intestinal inflammation and Paneth cell death (Fig. 2). Furthermore, depletion of CD4+ T cells during infection in Tbx21−/− mice prevented Paneth cell loss (Fig. 2). These experiments demonstrate that both CD4+ T cells and IFN-γ are essential for parasite-induced intestinal inflammation, while T-bet is largely dispensable for the parasite-induced inflammation and Paneth cell loss.
Figure 2. T-bet-independent immunopathological response to T. gondii is mediated by CD4+ T cells and IFN-γ.
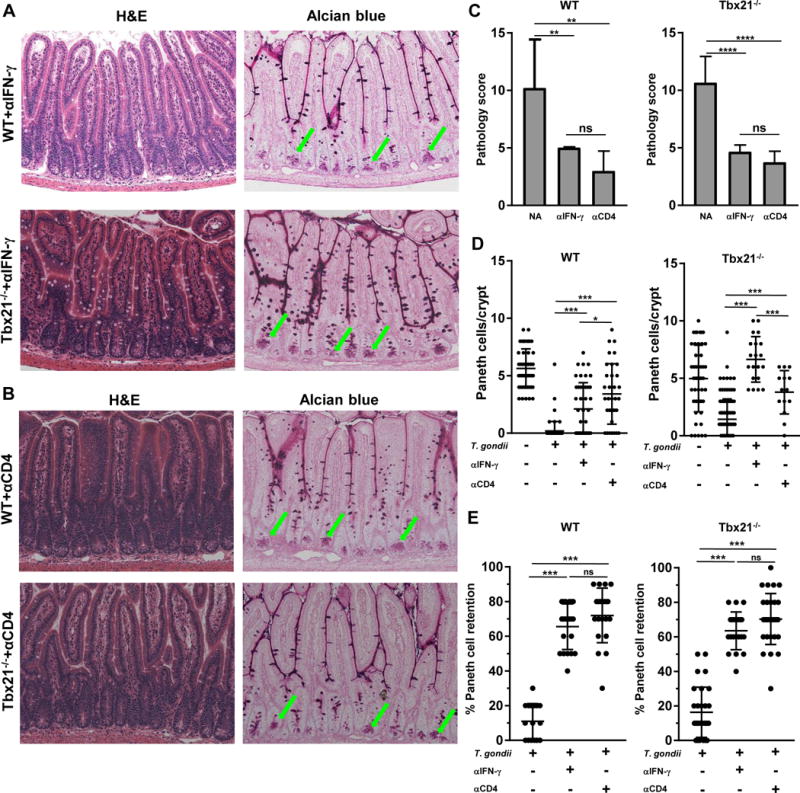
WT and Tbx21−/− mice were treated with (A) anti-IFN-γ or (B) anti-CD4 antibodies and then orally infected with T. gondii. Green arrows indicate Paneth cells. (C) Histological changes in the small intestine of infected WT and Tbx21−/− mice treated with anti-IFN-γ or anti-CD4 antibodies were analyzed on day 7 post-infection based on an additive scoring system. (D) Paneth cell quantification of the small intestines and visualization of Paneth cells in WT and Tbx21−/− mice were performed on day 7 post-infection. (E) Quantification of the retention of Paneth cells per individual crypt in infected WT and Tbx21−/− mice treated with anti-IFN-γ or anti-CD4 antibodies on day 7 post-infection. Data shown are representative of three independent experiments (n = 3-5 mice per group for each independent experiment). Statistical analyses were done using Mann-Whitney analysis of individual groups, *P<0.05, ***P<0.001.
Critically, analysis of CD4+ T cells of T. gondii-infected mice revealed that while in WT mice IFN-γ-producing CD4+ T cells co-expressed T-bet, CD4+ T cells isolated from Tbx21−/− mice remained capable of producing IFN-γ (Fig. 3A-F and Sup. Fig. 1), even though at the modestly reduce level (Fig 3E). Lack of T-bet did not result in induction of Th2 or Th17 transcription factors or the expression of their effector cytokines (Sup. Fig. 2). Thus, the transcription factor T-bet is not essential for IFN-γ expression by CD4+ T cells in response to T. gondii infection. We next addressed ILC1s’ contribution to T. gondii-induced intestinal immunopathology, since it was recently reported that T-bet-dependent ILC1s are a primary source of IFN-γ during T. gondii infection11. As expected, ILC1s are present in the lamina propria of the small intestines of infected WT, but not Tbx21−/− mice (Fig. 3G-K). However, the intestinal inflammation and Paneth cell loss observed in Tbx21−/− animals established that ILC1s are not essential for T. gondii-triggered intestinal immunopathology.
Figure 3. T-bet-independent Th1 response to T. gondii.
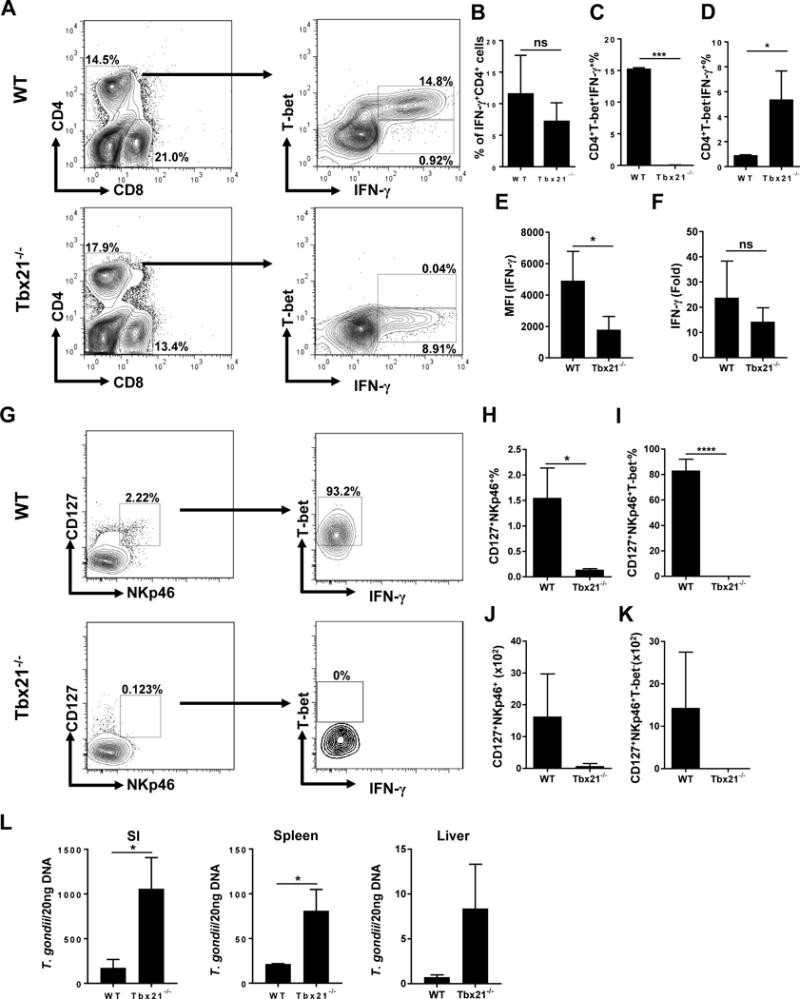
(A) WT and Tbx21−/− mice were infected with 20 cysts of the ME49 strain of T. gondii. Mesenteric lymph nodes (mLNs) were harvested and T-bet expression and IFN-γ production by CD4+ T cells were analyzed by flow cytometry. Average frequencies of (B) CD4+IFN-γ+, (C) CD4+T bet+IFN-γ+, (D) CD4+T bet−IFN-γ+ and (E) MFI of CD4+ T cell IFN-γ were analyzed on day 7 post-infection. The results are representative of three independent experiments. (F) Quantitative analysis of IFN-γ expression in the small intestines of WT and Tbx21−/− mice infected with the parasite on day7 post-infection by qRT-PCR. (G) Small intestinal lamina propria of WT and Tbx21−/− were harvested on day 7 post-infection and T-bet expression and IFN-γ production by ILC1s were analyzed by flow cytometry. Average frequencies of (H) Lin-CD127+NKp46+ and (I) Lin-CD127+NKp46+T-bet+, and absolute quantification of (J) Lin-CD127+NKp46+ and (K) Lin-CD127+NKp46+T-bet+ cells analyzed from the lamina propria of mice orally infected with T. gondii. (L) Quantitative analysis of T. gondii burden in the small intestines (SI), spleen, and livers of WT and Tbx21−/− mice infected with the parasite. Data are representative from three (G-K) and four (A-F, L; n = 3-5 mice per group for each independent experiment) independent experiments. Statistical analyses were done using Mann-Whitney analysis of individual groups: *P<0.05, *** P<0.001. NS=not significant.
To examine if the absence of the transcription factor T-bet plays a role in parasite burden during T. gondii-driven intestinal inflammation, we analyzed T. gondii levels in intestinal and systemic tissues. Tbx21−/− mice had significantly elevated parasite burden and were more susceptible to acute T. gondii infection when compared with their WT controls (Fig. 3L). Thus, our results reveal that T-bet is dispensable for CD4+ T cell-derived IFN-γ, but remains critical for parasite restriction.
T-bet-deficient CD4+ T cells are sufficient for IFN-γ-mediated Paneth cell loss and intestinal pathology
Host protective and pathological functions of CD4+ Th1 effector cells are regulated by the cooperation of multiple T cell-intrinsic and extrinsic factors45. To investigate the intrinsic pathobiological functions of T-bet-deficient CD4+ T cells, we took advantage of the previous observation that Paneth cell development remains intact in RAG1−/− and RAG2−/− mice, similar to IFN-γ deficient animals (Fig. 4A, 4B)25. Furthermore, we have previously demonstrated that T. gondii-infected RAG1−/− mice retain Paneth cells in response to the parasitic infection, unless reconstituted with IFN-γ-producing CD4+ T cells25. In agreement with our previous results, we observed that reconstitution of RAG1−/− mice with WT splenocytes was sufficient for induction of intestinal inflammation and Paneth cell death in response to T. gondii infection (Fig. 4C, 4E-G). Similar to adoptively transferred WT splenocytes, transferring Tbx21−/− splenocytes was sufficient for Paneth cell loss in T. gondii-infected RAG1−/− mice (Fig. 4D, 4E 4G). As expected, depletion of CD4+ cells or blocking IFN-γ in reconstituted mice alleviated the intestinal inflammation seen during infection with the parasite (Fig. 4C, 4D). Additionally, quantification of the intestinal histology revealed that blocking IFN-γ in mice reconstituted with either WT or T-bet-deficient splenocytes was sufficient to ameliorate the T. gondii-mediated pathology (Fig. 4E). Moreover, when the Paneth cell retention per individual crypt was quantified in mice reconstituted with WT or Tbx21−/− splenocytes treated with anti-CD4+ cells or anti-IFN-γ we observed a significant increase in the presence of Paneth cells maintained within the intestinal crypts (Fig. 4F, 4G).
Figure 4. CD4+ T cell-intrinsic T-bet expression is dispensable for IFN-γ-mediated intestinal pathology during T. gondii infection.
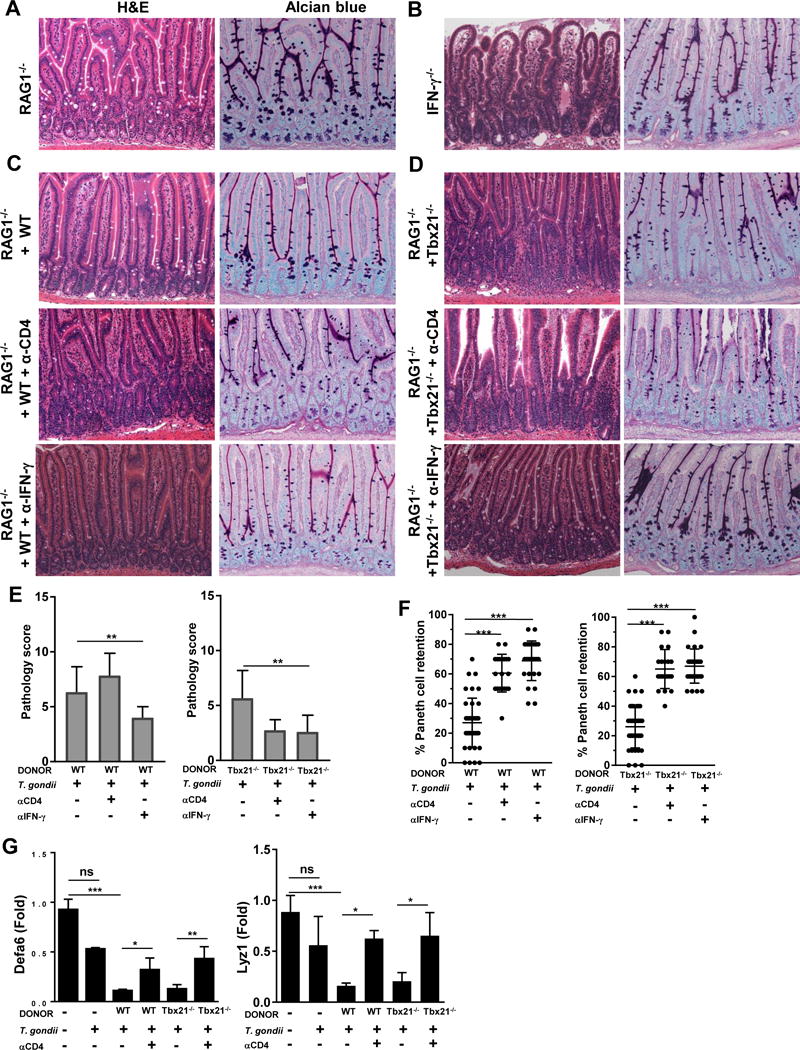
(A) Histological analysis of the small intestines and visualization of Paneth cells in RAG1−/− and (B) IFN-γ−/− mice orally infected with 20 cysts of the ME49 strain were performed on day 7 post-infection. RAG1−/− mice were reconstituted with (C) WT or (D) Tbx21−/− splenocytes, then infected with T. gondii and additionally treated with anti-CD4 or anti-IFN-γ antibodies. (E) Histological changes in the small intestine of reconstituted RAG1−/− mice treated with anti-CD4 or anti-IFN-γ antibodies were analyzed on day 7 post-infection based on an additive scoring system. (F) Quantification of the retention of Paneth cells per individual crypt in infected reconstituted RAG1−/− animals treated with anti-CD4 or anti-IFN-γ antibodies on day 7. (G) qRT-PCR analysis of relative Defa6 and Lyz1 expression measured in the small intestines of RAG1−/− mice reconstituted with WT or Tbx21−/− splenocytes and additionally treated with anti-CD4 antibodies on day 7 post-infection. Data are representative from three independent experiments (n = 4-5 mice per group). Statistical analyses were done using Mann-Whitney analysis of individual groups: *P<0.05, ** P<0.01, *** P<0.001. NS=not significant.
Next, to identify if the T-bet-deficient CD4+ T cells were capable of expressing IFN-γ, we examined their cytokine production by flow cytometry. Similar to RAG2−/− mice reconstituted with WT splenocytes, we observed no significant differences in the frequency of IFN-γ+CD4+ T cells during T. gondii infection (Fig. 5A, 5B). However, there was a partial reduction in the amount of IFN-γ produced by T-bet-deficient CD4+ T cells when compared to WT controls (Fig. 5C). As anticipated, ILC1s were retained in RAG1−/− mice (Sup. Fig. 3) but no immunopathology was observed in response to T. gondii infection (Fig. 4A), suggesting that ILC1s alone are insufficient to drive intestinal inflammation in the absence of CD4+ T cells.
Figure 5. CD4+ T cell-intrinsic T-bet expression is dispensable for IFN-γ production in vivo.
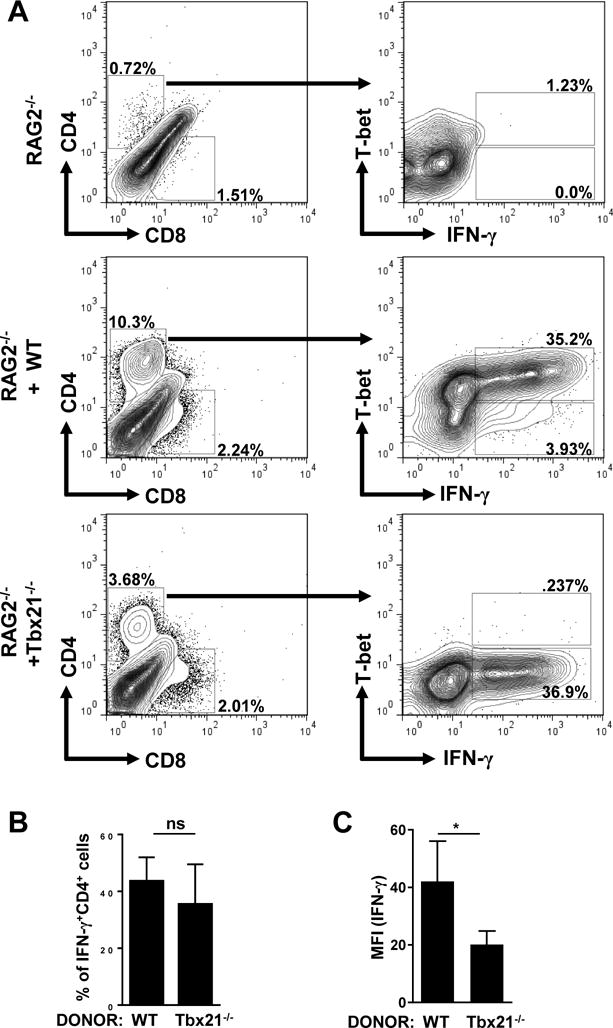
(A) RAG2−/− mice were untreated or reconstituted with WT or Tbx21−/− splenocytes, and then infected with 20 cysts of the ME49 strain of T. gondii. Spleens were harvested on day 7 post-infection and T-bet expression and IFN-γ production by CD4+ T cells were analyzed by flow cytometry. (B) Average frequencies of CD4+IFN-γ+ were analyzed from the spleens of RAG2−/− mice reconstituted with WT or Tbx21−/− splenocytes and infected with T. gondii on day 7 post-infection. (C) MFI of CD4+ T cell IFN-γ (A). Data shown are representative of three independent experiments (n = 4-5 mice per group). Statistical analyses were done using Mann-Whitney analysis of individual groups: *P<0.05. NS=not significant.
Overall, the combined analysis of the RAG1−/−, IFN-γ−/−, and RAG1−/− or RAG2−/− mice reconstituted with WT or Tbx21−/− splenocytes formally demonstrate that parasite-mediated intestinal inflammation is primarily driven by the Th1 cells independent of the transcription factor T-bet.
Discussion
In this study, we revealed that T-bet is largely dispensable for the induction of IFN-γ secretion by CD4+ T cells and for the Th1-dependent immunopathological response during the acute parasitic infection. The analysis of T. gondii-infected Tbx21−/− mice and T-bet-deficient cells adoptively transferred into RAG1−/− and RAG2−/− mice revealed that T-bet-deficient CD4+ T cells are fully capable of differentiating into Th1 effector cells that were both required and sufficient for parasite-triggered intestinal inflammation. The observed intestinal inflammation and bacterial dysbiosis along with Paneth cell loss in T. gondii-infected Tbx21−/− mice also established that a limited role for ILC1s in mediating intestinal pathology during parasitic infection.
It is well established that in addition to host defense, IFN-γ is a central mediator of the immunopathological response observed during T. gondii infection. T. gondii-infected IFN-γ-deficient mice, while having an uncontrolled parasitic burden, do not develop intestinal inflammation in response to the infection. IFN-γ is also a major cytokine required for both Paneth cell loss and severe intestinal dysbiosis observed during acute responses to the parasite. The ability of multiple cell types to produce IFN-γ in response to T. gondii infection raised a question regarding the relative contribution of IFN-γ-producing innate and adaptive immune cells in mediating intestinal inflammation39. In this regard, T-bet is a unique molecule involved in the regulation of IFN-γ by CD4+ T cells and ILC1s. While T-bet controls IFN-γ production in CD4+ T cells via regulating cytokine expression, it is also the master regulator of ILC1 development and therefore lack of T-bet abolishes the ILC1 contribution to host defense. Thus, we anticipated that T-bet, similar to IFN-γ, is essential for the induction of a type I immunopathological response. Contrariwise, we observed that T-bet-deficient mice develop severe intestinal inflammation comparable to that seen in parasite infected WT mice. These results, combined with lack of pathology in T. gondii-infected RAG1−/− mice, unexpectedly establish that ILC1s alone cannot induce the IFN-γ-dependent intestinal immunopathological response observed during T. gondii infection. Further investigation of mechanisms responsible for the Paneth cell loss detected in T. gondii-infected Tbx21−/− mice revealed that T-bet is not required for IFN-γ production by CD4+ T cells. Instead, it appeared that while in WT mice all IFN-γ-producing CD4+ T cells expressed T-bet, this was a result of co expression of the cytokine and the transcriptional factor rather than T-bet-dependent regulation of IFN-γ expression. This conclusion was further confirmed in the adoptive transfer studies with WT and T-bet-deficient CD4+ T cells.
Our results extend several studies demonstrating the presence of Th1 cells in the absence of T-bet. Using experimental intraperitoneal infection with the same pathogen it was previously observed that the lack of T-bet results in impaired CD4+ T cell migration rather than IFN-γ expression by T. gondii-specific CD4+ T cells46. A requirement for T-bet in regulation of CD4+ T cell migration and IEL functional maturation further suggests that T-bet regulates the effector functions of CD4+ T cells rather than their Th1 effector choice in vivo32. In this regard it is important to note that the requirement for T-bet in mediating IFN-γ-dependent immunopathological responses in the brain during malaria infection47 or in the pancreas during autoaggressive response was previously documented48. While it is tempting to speculate that immune activation driven by the intestinal microbiota may explain why T-bet is dispensable for T. gondii-initiated intestinal damage, induction of intestinal inflammation in a murine model of transfer colitis argues against this simplistic explanation since CD4+ T cell driven colitis can be induced in the presence or absence of T bet32, 49. Whether there are additional tissue and activation-specific requirements for mediating IFN-γ-dependent immunopathological responses at present is unknown. Overall, the analysis of the natural oral route of T. gondii infection in Tbx21−/− mice formally established that T-bet is dispensable for the pathogen-driven pathological response, an observation that has several important implications. Our experiments ruled out ILC1s as the primary mediators of the IFN-γ-dependent immunopathological response. Taking into consideration a limited role for ILC3s in the pathogen-driven intestinal pathology, and the preserved intestinal tissues in T cell deficient mice infected with the parasite, we conclude that CD4+ T cell-derived IFN-γ rather than innate sources of this cytokine is responsible for the intestinal pathology. We also demonstrated that T-bet-deficient CD4+ T cells are not only capable of producing IFN-γ, but retain their pathological potential and can drive intestinal damage, demonstrating that T-bet is dispensable for the pathogen-induced type I immunopathological disorder.
Methods
Animals
C57BL/6, Tbx21−/−, IFN-γ−/−, and RAG1−/− and RAG2−/− mice were obtained from Jackson Laboratory. Control and experimental mice were age-matched within all individual experiments. All mice were maintained at the American Association of Laboratory Animal Care-accredited animal facility at the University of Rochester Medical Center.
Toxoplasma gondii infections, histopathology, and RT-PCR
All mice were infected orally with an average of 20 T. gondii cysts (ME49 strain). At days 7 and 10 post-infection, the animals were necropsied, and portions of the small intestines were fixed in Carnoy’s fixative, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin or Alcian blue. Quantification of Paneth cells per intestinal crypt was performed in a double blinded manner. The severity of intestinal pathology was analyzed based on the following additive scoring system50. For crypt and villi integrity: 0, normal; 1, irregular villi and crypts; 2, mildly inflamed; 3, severe villi and crypt loss; 4, complete villi and crypt loss with an intact epithelial cell layer; 5, complete loss of villi and crypts and surface epithelium. For infiltration of inflammatory cells into the mucosa: 0, normal; 1, mild; 2, modest; and 3, severe. For infiltration of the submucosa: 0, normal; 1, mild; 2, modest; and 3, severe. For infiltration of the muscle: 0, normal; 1, mild; 2, modest; and 3, severe. These scores were added, resulting in a total scoring range of 0 to 14.
Total RNA was isolated from the intestines of naïve or T. gondii-infected mice using Trizol with subsequent purification using a PureLink RNA Mini Kit (Invitrogen). cDNA was prepared using a iScript cDNA Synthesis Kit (Bio-Rad). Optimized primers targeting each gene were designed using Primer 3 software46. These primers included the following: Lyz1, 5′-gccaaggtctacaatcgttgtgagttg-3′, 5′-cagtcagccagcttgacaccacg-3′; Cryptdin 2 (Crypt2), 5′-ccaggctgatcctatccaaa-3′, Defensin 6 (Defa6) 5′-ccttccaggtccaggctgat-3′ and 5′-tgagaagtggtcatcaggcac-3′, 5′-gtcccattcatgcgttctct-3′; and Hprt, 5′-gcccttgactataatgagtacttcagg-3′, 5′-ttcaacttgcgctcatcttagg-3′46. cDNA was amplified using SsoFast Eva Green Supermix (BioRad). A MyiQ Real-Time PCR Detection System (BioRad) was used to obtain Ct values. The relative expression of each sample was determined after normalization to HPRT using the ddCt method.
The relative abundances of Proteobacteria (family Enterobacteriaceae) were measured by qPCR using a MyiQ Real-Time PCR Detection System with taxon-specific or universal 16S rRNA gene primers as previously described25.
To determine T. gondii pathogen loads, total genomic DNA from animal tissue was isolated by using the DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer’s instructions. PCR were performed by using SsoFast EvaGreen Supermix (Bio-Rad). Samples were measured by qPCR using a MyiQ Real-Time PCR Detection System (Bio-Rad), and data from genomic DNA was compared with a defined copy number standard of the T. gondii gene B1.
Measurements of CD4+ T Cell Responses
To assay the responses of mice infected with T. gondii, the lamina propria, mesenteric lymph nodes (mLNs), and spleens were harvested from WT, Tbx21−/−, and RAG1−/− mice on day 7 post-infection. Single cell suspensions of mLNs and spleens were restimulated with 1 μg/ml αCD3 (BD Biosciences) for 5 hr in the presence of GolgiPlug (Brefeldin A, BD Biosciences). After isolation or in vitro restimulation, the cells were washed once in PBS + 1% FBS and stained with fluorochrome-conjugated antibodies. Cell fluorescence was measured using an LSRII flow cytometer, and data were analyzed using FlowJo software (Tree Star).
For the adoptive transfer experiments, splenocytes were isolated from WT or Tbx21−/− mice and were transferred (15×106 cells) to RAG1−/− or RAG2−/− mice 4 weeks prior to T. gondii infection. The control and reconstituted RAG-deficient mice were infected orally with 20 cysts per mouse of the ME49 strain and were additionally treated with αIFN-γ or αCD4 antibodies during the course of T. gondii infection as previously described25.
Statistical analysis
All data were analyzed with Prism (Version 7; GraphPad). These data were considered statistically significant when P values were less than 0.05.
Supplementary Material
Supplementary Figure 1. T-bet independent Th1 responses to T. gondii in the lamina propria. (A) WT and Tbx21−/− mice were infected with 20 cysts of the ME49 strain of T. gondii. Lamina propria cells were harvested on day 7 post-infection and IFN-γ production and T-bet expression by CD3+CD4+ T cells were analyzed by flow cytometry. Average frequencies of (B) CD3+CD4+, (C) CD3+CD4+IFN-γ+, and (D) CD3+CD4+T bet+ and absolute cell numbers of (E) CD3+CD4+T bet+ cells were analyzed on day 7 post-infection. Data are representative of three independent experiments (n = 3-5 mice per group for each independent experiment). Statistical analyses were done using Mann-Whitney analysis of individual groups: * P<0.05. NS=not significant.
Supplementary Figure 2. T. gondii infection does not result in Th2 or Th17 immune responses in the absence of T-bet. (A) WT and Tbx21−/− mice were infected with 20 cysts of the ME49 strain of T. gondii. mLNs were harvested on day 7 post-infection and RORγt and IL-17 expression by CD4+ T cells were analyzed by flow cytometry. Average frequencies of (B) CD4+RORγt+ and (C) CD4+IL-17+ were analyzed from the mLNs of mice orally infected with T. gondii. (D) qRT-PCR analysis of relative RORγt, IL-23p19, IL-17, IL-22, and Gata3 expression measured in the small intestine of infected WT and Tbx21−/− mice on day 7 post-infection. Data are representative of three (A-C) or four (D) independent experiments; n = 3-5 mice per group for each independent experiment. Statistical analyses were done using Mann-Whitney analysis of individual groups: NS=not significant.
Supplementary Figure 3. T-cell reconstitution of RAG2−/− mice leads to reduced frequency of ILC1s. (A) RAG2−/− mice were untreated or reconstituted with WT or Tbx21−/− splenocytes, and then infected with 20 cysts of the ME49 strain of T. gondii. (A) Small intestinal lamina propria cells were harvested on day 7 of infection and T-bet expression and IFN-γ production by ILC1s were analyzed by flow cytometry. Average frequencies of (B) CD127+NKp46+ and (C) CD127+NKp46+T bet+, and absolute quantification of (D) CD127+NKp46+ and (E) CD127+NKp46+T bet+ cells analyzed from the lamina propria of mice orally infected with T. gondii. Data are representative of three independent experiments (n = 4-5 mice per group for each independent experiment). Statistical analyses were done using Mann-Whitney analysis of individual groups: ** P<0.01. NS=not significant.
Acknowledgments
This work was supported by NIAID Grants R56AI085263, R01AI121090, and the Burroughs Wellcome Foundation.
References
- 1.Sacks D, Noben-Trauth N. The immunology of susceptibility and resistance to Leishmania major in mice. Nat Rev Immunol. 2002;2(11):845–858. doi: 10.1038/nri933. [DOI] [PubMed] [Google Scholar]
- 2.Scott P. IFN-gamma modulates the early development of Th1 and Th2 responses in a murine model of cutaneous leishmaniasis. J Immunol. 1991;147(9):3149–3155. [PubMed] [Google Scholar]
- 3.Sakai S, Kauffman KD, Sallin MA, Sharpe AH, Young HA, Ganusov VV, et al. CD4 T cell-derived IFN-gamma Plays a Minimal Role in Control of Pulmonary Mycobacterium tuberculosis Infection and Must Be Actively Repressed by PD-1 to Prevent Lethal Disease. PLoS Pathog. 2016;12(5):e1005667. doi: 10.1371/journal.ppat.1005667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hess J, Ladel C, Miko D, Kaufmann SH. Salmonella typhimurium aroA-infection in gene-targeted immunodeficient mice: major role of CD4+ TCR-alpha beta cells and IFN-gamma in bacterial clearance independent of intracellular location. J Immunol. 1996;156(9):3321–3326. [PubMed] [Google Scholar]
- 5.Ravindran R, Foley J, Stoklasek T, Glimcher LH, McSorley SJ. Expression of T-bet by CD4 T cells is essential for resistance to Salmonella infection. J Immunol. 2005;175(7):4603–4610. doi: 10.4049/jimmunol.175.7.4603. [DOI] [PubMed] [Google Scholar]
- 6.Mittrucker HW, Kaufmann SH. Immune response to infection with Salmonella typhimurium in mice. J Leukoc Biol. 2000;67(4):457–463. doi: 10.1002/jlb.67.4.457. [DOI] [PubMed] [Google Scholar]
- 7.Svensson A, Nordström I, Sun JB. Protective immunity to genital herpes simpex virus type 2 infection is mediated by T-bet. The Journal of …. 2005 doi: 10.4049/jimmunol.174.10.6266. [DOI] [PubMed] [Google Scholar]
- 8.Sullivan BM, Jobe O, Lazarevic V. Increased susceptibility of mice lacking T-bet to infection with Mycobacterium tuberculosis correlates with increased IL-10 and decreased IFN-γ production. The Journal of …. 2005 doi: 10.4049/jimmunol.175.7.4593. [DOI] [PubMed] [Google Scholar]
- 9.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR. Distinct effects of T-bet in TH1 lineage commitment and IFN-γ production in CD4 and CD8 T cells. Science. 2002 doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 10.Ravindran R, Foley J, Stoklasek T. Expression of T-bet by CD4 T cells is essential for resistance to Salmonella infection. The Journal of …. 2005 doi: 10.4049/jimmunol.175.7.4603. [DOI] [PubMed] [Google Scholar]
- 11.Klose CS, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell. 2014;157(2):340–356. doi: 10.1016/j.cell.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 12.Askenase MH, Han SJ, Byrd AL, Morais da Fonseca D, Bouladoux N, Wilhelm C, et al. Bone-Marrow-Resident NK Cells Prime Monocytes for Regulatory Function during Infection. Immunity. 2015;42(6):1130–1142. doi: 10.1016/j.immuni.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldszmid RS, Caspar P, Rivollier A, White S, Dzutsev A, Hieny S, et al. NK cell-derived interferon-gamma orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity. 2012;36(6):1047–1059. doi: 10.1016/j.immuni.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sturge CR, Burger E, Raetz M, Hooper LV, Yarovinsky F. Cutting Edge: Developmental Regulation of IFN-gamma Production by Mouse NeutrophIL-Precursor Cells. Journal of immunology. 2015;195(1):36–40. doi: 10.4049/jimmunol.1500366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spees AM, Kingsbury DD, Wangdi T, Xavier MN, Tsolis RM, Baumler AJ. Neutrophils are a source of gamma interferon during acute Salmonella enterica serovar Typhimurium colitis. Infect Immun. 2014;82(4):1692–1697. doi: 10.1128/IAI.01508-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hou B, Benson A, Kuzmich L, DeFranco AL, Yarovinsky F. Critical coordination of innate immune defense against Toxoplasma gondii by dendritic cells responding via their Toll-like receptors. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(1):278–283. doi: 10.1073/pnas.1011549108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity. 2013;38(4):769–781. doi: 10.1016/j.immuni.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim BH, Shenoy AR, Kumar P, Das R, Tiwari S, MacMicking JD. A family of IFN-gamma-inducible 65-kD GTPases protects against bacterial infection. Science. 2011;332(6030):717–721. doi: 10.1126/science.1201711. [DOI] [PubMed] [Google Scholar]
- 19.MacMicking JD. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nature reviews Immunology. 2012;12(5):367–382. doi: 10.1038/nri3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang SY, Boisson-Dupuis S, Chapgier A, Yang K, Bustamante J, Puel A, et al. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol Rev. 2008;226:29–40. doi: 10.1111/j.1600-065X.2008.00698.x. [DOI] [PubMed] [Google Scholar]
- 21.Hunn JP, Feng CG, Sher A, Howard JC. The immunity-related GTPases in mammals: a fast-evolving cell-autonomous resistance system against intracellular pathogens. Mamm Genome. 2011;22(1–2):43–54. doi: 10.1007/s00335-010-9293-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nature immunology. 2013;14(3):221–229. doi: 10.1038/ni.2534. [DOI] [PubMed] [Google Scholar]
- 23.Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, et al. Interleukin-12 and -23 Control Plasticity of CD127(+) Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria. Immunity. 2015;43(1):146–160. doi: 10.1016/j.immuni.2015.06.019. [DOI] [PubMed] [Google Scholar]
- 24.Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol. 2003;3521(7):533. doi: 10.1038/nri1132. [DOI] [PubMed] [Google Scholar]
- 25.Raetz M, Hwang SH, Wilhelm CL, Kirkland D, Benson A, Sturge CR, et al. Parasite-induced TH1 cells and intestinal dysbiosis cooperate in IFN-gamma-dependent elimination of Paneth cells. Nat Immunol. 2013;14(2):136–142. doi: 10.1038/ni.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farin HF, Karthaus WR, Kujala P, Rakhshandehroo M, Schwank G, Vries RG, et al. Paneth cell extrusion and release of antimicrobial products is directly controlled by immune cell-derived IFN-gamma. The Journal of experimental medicine. 2014;211(7):1393–1405. doi: 10.1084/jem.20130753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villeret B, Brault L, Couturier-Maillard A, Robinet P, Vasseur V, Secher T, et al. Blockade of IL-1R signaling diminishes Paneth cell depletion and Toxoplasma gondii induced ileitis in mice. Am J Clin Exp Immunol. 2013;2(1):107–116. [PMC free article] [PubMed] [Google Scholar]
- 28.Molloy MJ, Grainger JR, Bouladoux N, Hand TW, Koo LY, Naik S, et al. Intraluminal containment of commensal outgrowth in the gut during infection-induced dysbiosis. Cell Host Microbe. 2013;14(3):318–328. doi: 10.1016/j.chom.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heimesaat MM, Bereswill S, Fischer A, Fuchs D, Struck D, Niebergall J, et al. Gram-negative bacteria aggravate murine small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii. J Immunol. 2006;177(12):8785–8795. doi: 10.4049/jimmunol.177.12.8785. [DOI] [PubMed] [Google Scholar]
- 30.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100(6):655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 31.Lazarevic V, Glimcher LH, Lord GM. T-bet: a bridge between innate and adaptive immunity. Nature reviews Immunology. 2013;13(11):777–789. doi: 10.1038/nri3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reis BS, Hoytema van Konijnenburg DP, Grivennikov SI, Mucida D. Transcription factor T-bet regulates intraepithelial lymphocyte functional maturation. Immunity. 2014;41(2):244–256. doi: 10.1016/j.immuni.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klose CS, Blatz K, d’Hargues Y, Hernandez PP, Kofoed-Nielsen M, Ripka JF, et al. The transcription factor T-bet is induced by IL-15 and thymic agonist selection and controls CD8alphaalpha(+) intraepithelial lymphocyte development. Immunity. 2014;41(2):230–243. doi: 10.1016/j.immuni.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 34.Sciume G, Hirahara K, Takahashi H, Laurence A, Villarino AV, Singleton KL, et al. Distinct requirements for T-bet in gut innate lymphoid cells. J Exp Med. 2012;209(13):2331–2338. doi: 10.1084/jem.20122097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sonnenberg GF, Artis D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med. 2015;21(7):698–708. doi: 10.1038/nm.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yarovinsky F. Innate immunity to Toxoplasma gondii infection. Nat Rev Immunol. 2014;14(2):109–121. doi: 10.1038/nri3598. [DOI] [PubMed] [Google Scholar]
- 37.Hunter CA, Sibley LD. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat Rev Microbiol. 2012;10(11):766–778. doi: 10.1038/nrmicro2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cohen SB, Denkers EY. The gut mucosal immune response to Toxoplasma gondii. Parasite immunology. 2015;37(3):108–117. doi: 10.1111/pim.12164. [DOI] [PubMed] [Google Scholar]
- 39.Sturge CR, Yarovinsky F. Complex immune cell interplay in the gamma interferon response during Toxoplasma gondii infection. Infection and immunity. 2014;82(8):3090–3097. doi: 10.1128/IAI.01722-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liesenfeld O, Kang H, Park D, Nguyen TA, Parkhe CV, Watanabe H, et al. TNF-alpha, nitric oxide and IFN-gamma are all critical for development of necrosis in the small intestine and early mortality in genetically susceptible mice infected perorally with Toxoplasma gondii. Parasite immunology. 1999;21(7):365–376. doi: 10.1046/j.1365-3024.1999.00237.x. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki Y, Sher A, Yap G, Park D, Neyer LE, Liesenfeld O, et al. IL-10 is required for prevention of necrosis in the small intestine and mortality in both genetically resistant BALB/c and susceptible C57BL/6 mice following peroral infection with Toxoplasma gondii. J Immunol. 2000;164(10):5375–5382. doi: 10.4049/jimmunol.164.10.5375. [DOI] [PubMed] [Google Scholar]
- 42.Mennechet FJ, Kasper LH, Rachinel N, Li W, Vandewalle A, Buzoni-Gatel D. Lamina propria CD4+ T lymphocytes synergize with murine intestinal epithelial cells to enhance proinflammatory response against an intracellular pathogen. J Immunol. 2002;168(6):2988–2996. doi: 10.4049/jimmunol.168.6.2988. [DOI] [PubMed] [Google Scholar]
- 43.Egan CE, Craven MD, Leng J, Mack M, Simpson KW, Denkers EY. CCR2 dependent intraepithelial lymphocytes mediate inflammatory gut pathology during Toxoplasma gondii infection. Mucosal Immunol. 2009;2(6):527–535. doi: 10.1038/mi.2009.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hand TW, Dos Santos LM, Bouladoux N, Molloy MJ, Pagan AJ, Pepper M, et al. Acute gastrointestinal infection induces long-lived microbiota-specific T cell responses. Science. 2012;337(6101):1553–1556. doi: 10.1126/science.1220961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DuPage M, Bluestone JA. Harnessing the plasticity of CD4 T cells to treat immune mediated disease. Nat Rev Immunol. 2016 doi: 10.1038/nri.2015.18. [DOI] [PubMed] [Google Scholar]
- 46.Harms Pritchard G, Hall AO, Christian DA, Wagage S, Fang Q, Muallem G, et al. Diverse roles for T-bet in the effector responses required for resistance to infection. J Immunol. 2015;194(3):1131–1140. doi: 10.4049/jimmunol.1401617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oakley MS, Sahu BR, Lotspeich-Cole L, Solanki NR, Majam V, Pham PT, et al. The transcription factor T-bet regulates parasitemia and promotes pathogenesis during Plasmodium berghei ANKA murine malaria. J Immunol. 2013;191(9):4699–4708. doi: 10.4049/jimmunol.1300396. [DOI] [PubMed] [Google Scholar]
- 48.Esensten JH, Lee MR, Glimcher LH, Bluestone JA. T-bet-deficient NOD mice are protected from diabetes due to defects in both T cell and innate immune system function. J Immunol. 2009;183(1):75–82. doi: 10.4049/jimmunol.0804154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zimmermann J, Kuhl AA, Weber M, Grun JR, Loffler J, Haftmann C, et al. T-bet expression by Th cells promotes type 1 inflammation but is dispensable for colitis. Mucosal Immunol. 2016 doi: 10.1038/mi.2016.5. [DOI] [PubMed] [Google Scholar]
- 50.Kirkland D, Benson A, Mirpuri J, Pifer R, Hou B, DeFranco AL, et al. B cell-intrinsic MyD88 signaling prevents the lethal dissemination of commensal bacteria during colonic damage. Immunity. 2012;36(2):228–238. doi: 10.1016/j.immuni.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. T-bet independent Th1 responses to T. gondii in the lamina propria. (A) WT and Tbx21−/− mice were infected with 20 cysts of the ME49 strain of T. gondii. Lamina propria cells were harvested on day 7 post-infection and IFN-γ production and T-bet expression by CD3+CD4+ T cells were analyzed by flow cytometry. Average frequencies of (B) CD3+CD4+, (C) CD3+CD4+IFN-γ+, and (D) CD3+CD4+T bet+ and absolute cell numbers of (E) CD3+CD4+T bet+ cells were analyzed on day 7 post-infection. Data are representative of three independent experiments (n = 3-5 mice per group for each independent experiment). Statistical analyses were done using Mann-Whitney analysis of individual groups: * P<0.05. NS=not significant.
Supplementary Figure 2. T. gondii infection does not result in Th2 or Th17 immune responses in the absence of T-bet. (A) WT and Tbx21−/− mice were infected with 20 cysts of the ME49 strain of T. gondii. mLNs were harvested on day 7 post-infection and RORγt and IL-17 expression by CD4+ T cells were analyzed by flow cytometry. Average frequencies of (B) CD4+RORγt+ and (C) CD4+IL-17+ were analyzed from the mLNs of mice orally infected with T. gondii. (D) qRT-PCR analysis of relative RORγt, IL-23p19, IL-17, IL-22, and Gata3 expression measured in the small intestine of infected WT and Tbx21−/− mice on day 7 post-infection. Data are representative of three (A-C) or four (D) independent experiments; n = 3-5 mice per group for each independent experiment. Statistical analyses were done using Mann-Whitney analysis of individual groups: NS=not significant.
Supplementary Figure 3. T-cell reconstitution of RAG2−/− mice leads to reduced frequency of ILC1s. (A) RAG2−/− mice were untreated or reconstituted with WT or Tbx21−/− splenocytes, and then infected with 20 cysts of the ME49 strain of T. gondii. (A) Small intestinal lamina propria cells were harvested on day 7 of infection and T-bet expression and IFN-γ production by ILC1s were analyzed by flow cytometry. Average frequencies of (B) CD127+NKp46+ and (C) CD127+NKp46+T bet+, and absolute quantification of (D) CD127+NKp46+ and (E) CD127+NKp46+T bet+ cells analyzed from the lamina propria of mice orally infected with T. gondii. Data are representative of three independent experiments (n = 4-5 mice per group for each independent experiment). Statistical analyses were done using Mann-Whitney analysis of individual groups: ** P<0.01. NS=not significant.