

Abstract
Background
Patients with inflammatory bowel disease have higher incidence of airway hyperresponsiveness compared to the general population. Lung inflammation leading to airway hyperresponsiveness causes illnesses for more than ten percent of the population in USA.
Aims
We investigated the lung response to bacterial endotoxin in colitic mice.
Methods
Rag-1 mice were transplanted with negatively selected splenic T cells. Some mice groups were treated with NSAID to develop colitis. All mice were treated with bacterial endotoxin and necropsied 3 weeks later.
Results
Colitic mice developed intensified lung inflammation on day 21 of treatment with bacterial endotoxin. Pulmonary lymphocytes from colitic mice displayed a proinflammatory cytokine profile, expressed high ICAM1 and low FoxP3. CD11c+, CD8+ cells bound and responded to non-systemic antigens from gut-localized microbiota and had higher expression of TLR4.
Conclusions
Colitic mice developed exacerbated lung inflammation in response to bacterial endotoxin compared to non-colitic mice. Proinflammatory cytokines from pulmonary lymphocytes induced high expression of ICAM1 and suppressed FoxP3 on CD4+ cells. CD11c+, CD8+ cells binding and responding to gut-localized antigens as well as high expression of TLR4 indicate innate and adaptive lung response to bacterial endotoxin. Inflammatory cells from colons of colitic mice homed in the lungs as well as the intestine suggesting recirculation of sensitized immunocompetent cells. These data support our hypothesis that colitis intensifies lung inflammation.
Keywords: Inflammatory bowel disease (IBD), Airway hyperresponsiveness, Lymphocyte recirculation, Effector and regulatory cells, Innate and adaptive immune responses, Bacterial endotoxin
Introduction
Extra-intestinal manifestations occur in inflammatory bowel disease. Patients with inflammatory bowel disease (IBD) have significantly increased prevalence of asthma, bronchitis, arthritis, and psoriasis. IBD-related airway diseases are the second most common chronic inflammatory disease assessed in patients with IBD [1–4].
According to the most recent estimates by the Centers for Disease Control and Prevention, there are more than 25 million people nationwide diagnosed with asthma [5]. Multiple etiologies are known to elicit asthma episodes. Lung inflammation leading to airway hyperresponsiveness in more than 60% of patients is caused by allergic response to a wide range of heterologous substances delivered through respiratory, digestive or even by skin contact [6, 7].
The epithelial lining of the colon and lungs have similarities that may allow the same disease state to affect both sites [1]. Common airway inflammatory changes have been thought to represent the same type of inflammatory changes that occur in the bowel [2, 3].
It has been suggested that skin and eye complications occur as a consequence of the recruitment of activated effector cells released from the gut into the circulation to extra-intestinal sites where they cause acute damage [4].
Expression of homing receptors determines the destination of lymphocytes to the gut, lungs, or skin. Lymphocyte recirculation in colitis may result in localized inflammation of other organs. Salmi et al. [8] showed that gut-derived lymphocytes can bind to synovial vessels, associated with joint inflammation. Defining the atypical parameters of the lymphocytes in colitis leading to exacerbation of lung inflammation is necessary to advance the therapeutic potential of a treatment strategy [9].
Different proinflammatory patterns are expressed in asthma. IL4, IgE, and IL5 prevail in eosinophilic inflammation [10]. On the other hand, IFNγ and IL12 prevail in neutrophilic inflammation. IL17 production associated with lung inflammation has been reported.
Some proinflammatory cytokine patterns can exclude each other. Choy et al. published data indicating that Th2 and Th17 were reciprocally regulated in asthma [11].
Lung inflammation can be identified by immunophenotyping of macrophages. Asthmatic patients show signs of alternative activation of macrophages associated with high IgE and degranulation of mast cells and eosinophils [10].
Multiple types of thymic (natural) and blood (induced) regulatory cells exist in lung inflammation and play a role in controlling the inflammatory response [12]. Helminth treatment used to expand regulatory T cells to control lung inflammation has shown efficacy in improving asthma [13–15].
The current literature contains numerous animal models used in IBD research [16–23]. We selected the Rag-1 reconstitution model of NSAID-induced colitis that gave us the opportunity to identify the immunocompetent entity causing the pathological finding and to answer questions regarding the mechanisms of initiation and resolution. Cell labeling for tracking lymphocytes pathways becomes more feasible in the Rag-1 mice since they lack functional T and B lymphocytes.
In this report, we presented data supporting our hypothesis that intestinal inflammation intensifies lung response to bacterial endotoxin.
Materials and Methods
Mouse Model
Female Rag-1 mice of C57Bl/6 background were reconstituted with negatively selected splenic T cells from the same gender and background from The Jackson Laboratory, Bar Harbor, ME. To induce colitis, three independent groups of five mice each of reconstituted Rag-1 mice were treated with the NSAID, Piroxicam SIGMA P-5654; 65 mg/250 g mixed into their ground feed NIH-31 diet 7013 from Harlan, WI., USA, for the first and second weeks. Mice were then placed on normal rodent chow without piroxicam for two more weeks before experiments. Control (non-colitic mice) groups with equal numbers were given ground feed without piroxicam. To grade intestinal inflammation, colons were removed, Swiss-rolled, fixed, sent to histology to prepare slides and stain them with H&E.
The inflammation was scored from 0 to 4 using criteria described elsewhere [13].
Mice used in our experiments developed grade-4 colitis, lost weight, and had bloody diarrhea resembling the human ulcerative colitis.
Cell Transfer
Negatively selected T cells were isolated from dispersed spleen cells using Pan T cell separation cocktail antibodies to coat then sheep anti-rat IgG-coated micro-beads (DYNAL BIOTECH) to separate non-T cells according to manufacturer’s specifications. Purified T cells adjusted to 5 × 106 cells/ml of PBS were injected intraperitoneally, 1 ml into each Rag-1 recipient mouse. Half the reconstituted mice were treated with NSAID for 2 weeks to induce colitis, and both groups entered the murine asthma model phase to induce lung inflammation.
Induction of Lung Inflammation in Colitic and Non-colitic Mice
Lung inflammation was induced in non-colitic and colitic Rag-1 in sets of five mice by two intraperitoneal (IP) injections followed by four intranasal (IN) instillations of lipopolysaccharide (LPS) extracted from E. coli (SIGMA, L-2637). LPS used for IP injections at 10 µg per mouse in 0.5 ml alum solution (1 mg/ml) on days zero and seven. Intranasal instillation started at second week of NSAID treatment on days 14, 15 and 19, 20 of starting the experiment. LPS was used at 10 µg in 50 µl sterile saline per mouse. Mice were necropsied on day 21 of starting the LPS treatment (Fig. 2a). Lung inflammation in colitic mice was given a score of 3.3 ± 0.2 on scale 4.0 based on lymphocytes infiltration and fibrosis found around the bronchioles.
Fig. 2.

Diagram for the procedure for treating mice with bacterial endotoxin (a). In b, histology sections stained with H&E of lungs from non-colitic mice (upper panel) and colitic mice (middle panel). The lower picture demonstrates the granulocytic infiltration found in the lung tissue of colitic mice as indicated by the black arrows. The figures are representative of three independent experiments. c is a graphical representation of the inflammatory scores in the colons and lungs from non-colitic and colitic mice
Preparation of Intestinal Lamina Propria Mononuclear Cells (LPL)
Intestinal tissues (colons) were washed extensively with calcium- and magnesium-free HBSS and LPL isolated as described before [14]. Cells were counted and viability was 90% as determined by trypan blue exclusion.
Single-Cell Preparations from the Lungs
Following euthanasia, lungs were collected from groups of five mice and cells were dispersed by exposing the sliced lung tissue to suction and expulsion cycles through a 1-ml syringe. Dispersed cells were passed through cell strainer, counted and viability taken by trypan blue exclusion dye.
Cell Cultures
For cytokine analysis, cells were adjusted to 5 × 106 cells/ml in RPMI 1640 containing 10% FCS, 25 mM HEPES buffer, 2 mM l-glutamine, 2β-ME, 1 mM sodium pyruvate, 100 U/ml penicillin, 5 mg/ml gentamicin, and 100 mg/ml streptomycin. Cell suspensions were dispensed at 200 μl into 96-well microtiter plates (Corning Glass). Cultures were incubated at 37 °C for 48 h. Lamina propria lymphocytes were cultured alone or with 1 mg/ml adherent anti-CD3 and anti-CD28 antibodies prepared from 2C11 and PV1 hybridomas purchased from American Type Culture Collection. Some cultures were stimulated with soluble anti-CD3, LPS, and HP antigen extracts.
Murine Exposure to Helminth
Mice were housed and handled according to National Institutes of Health guidelines and as approved by our Institutional Animal Care and Use Committee. Mice were bred on site in specific pathogen-free animal facilities. At 6 weeks of age, mice were colonized with 150 third-stage infective ensheathed larvae (L3) of H. polygyrus (US National Helminthological Collection No. 81930) by oral gavage.
Preparation of H. polygyrus Adult Worm Antigens
Adult H. polygyrus worm antigens were prepared with a modification from Robinson et al. 1994. Worms were removed from the intestines of 2 weeks colonized mice by scrubbing the dissected duodenum and washed thoroughly in PBS with antibiotics. The washed worms were centrifuged gently at 5×g for five minutes, and the supernatant was carefully decanted. The pellet was freeze-dried overnight and kept at − 80 °C until used. At time of use, 1 g of pooled worm pellets was vortexed in 10 ml of PBS for 5 min and the tube was centrifuged at 300×g for 5 min. Supernatant of the extract was sterile-filtered through low protein-binding, 0.2-µm syringe tip filtration units. Protein content was estimated and adjusted to 1 mg/ml by using a protein measurement kit from BIORAD, CA, USA. The filtered supernatant was dispensed into 1 ml aliquots and stored at − 80 °C until used.
Biotinylation of Antigen Extracts
Two tubes of the 1 ml adult worm antigen extract were adjusted to 1 mg/ml of protein and dialyzed in 0.1 M sodium bicarbonate buffer pH 8.4 for 4 h with two changes of buffer. 200 μl of filtered biotinyl-N-hydroxy-succinimide ester (SIGMA # H1759) 1 mg/ml solution in DMSO were added, and tubes rocked on a platform for 4 h at room temperature and then dialyzed extensively with multiple changes against PBS. The biotinylated antigen was dispensed into single-use aliquots and stored at − 80 °C.
Cytokine ELISA’s
IL17 was measured by using a sandwich ELISA described before [18, 19]. For IFNγ ELISA, we used the XMG 1.2 monoclonal antibody to coat the wells and HB170-biotin for detection (both from ATCC) in the same protocol, and for IL4 we used 11B11 to coat and BVD6-biotin to detect. Corresponding antibodies for IL10 and IL12 were determined by kits from R&D systems and used according to manufacturer’s directions.
Flow Cytometry
Monoclonal antibodies used in flow cytometry to stain the surface receptors included CD4 (Clone GK1.5), CD8 (Clone 53-6.7), CD11c (Clone N418), FoxP3 (Clone FJK-16s), CTLA4 (Clone UC10), ICAM-1(Clone YN1), TLR4 (Clone UT41), and α4β7 integrin (Clone DATK32) with the appropriate fluorochrome were purchased from BD-Pharmingen, e-Bioscience, or R&D systems as needed. Intra-cytoplasmic staining of IL17, IL4, IL12(P40), and IFN γ in CD4+ and CD8+ T cells and monocytes was performed according to manufacturer’s instructions. MLN cells were stimulated overnight with phorbol myristate acetate (PMA) at 1:100 dilution of 1 mg/ml DMSO stock solution and ionomycin (1 μg/ml) both from SIGMA, St. Louis, MO. Golgi-Stop is used at 4 μl in 6 ml culture, BD cat # 554715.
Staining of surface receptors and cytokines was done in parallel with irrelevant isotype control antibodies to ensure specificity. Cell acquisition was done by a FACScan flow cytometer, and analysis used Flow-Jo software in the flow cytometry core facility at the University of Iowa.
Statistical Analysis
Data are expressed as mean ± SE of multiple measurements. A minimum of three experiments were done to test every hypothesis. Statistically significant differences between groups were determined by using the Student’s t-test and ANOVA. Values of p < 0.05 or less were considered significant.
Results
Histopathological Examination of Lung Inflammation in Colitic Mice
In our model of colitis, we induced colon inflammation in Rag-1 mice reconstituted with normal T cells, by treatment with NSAID in ground feed as described in Fig. 1a. Mice developed transmural inflammation with massive lymphocyte infiltration and disrupted epithelium allowing a direct contact between the gut immunocytes and intestinal contents, Fig. 1b). The inflamed colons in colitic mice were given a score of 3.6 ± 0.4 on the basis of the transmural inflammation, and thickening of mucosal, submucosal, and muscular layers associated with massive lymphocyte infiltration and depleted goblet cells. The crypts of Lieberkuhn were reduced with remarkable structural disruption of the villi layer [13, 14].
Fig. 1.

Protocol for induction of colitis (a). Histology sections in colons from non-colitic and colitic mice stained with H&E (b). The figures are representative of three independent experiments
Exposure of colitic mice to LPS, as shown in Fig. 2a, intensified the lung inflammation as compared to non-colitic mice with the same treatment. Lymphocytes infiltrated the lung tissue associated with fibrosis around the bronchioles evident in histology slides. Figure 2b shows the pathological findings at different powers of magnification including large areas of the lungs showing massive lymphocytes infiltration in first and second pictures (100×), the tissue inflammation around bronchioles in the third picture (200×), and the granulocytic infiltration in the lower picture (400×). Development of lung inflammation at such a high frequency in colitic mice (Fig. 2c) indicated that the intensified lung inflammation secondary to intestinal inflammation is underestimated.
Cytokine Profile of Colon and Lung Lymphocytes from Colitic and Non-colitic Mice
Lymphocytes from the lungs of colitic mice were skewed toward a proinflammatory response. Lung lymphocytes stimulated in vitro with anti-CD3 expressed a proinflammatory cytokine profile replicating that found in the inflamed intestine shown in Fig. 3a, b, with the exception of IL4 which reflects the lung-specific response.
Fig. 3.

ELISA analysis of cytokines in culture supernatants of LPL from colitic and non-colitic mice stimulated in vitro with anti-CD3, (a) was mimicked by their lung lymphocytes counterparts (b) with the only exception in enhanced production of IL4. c Is a graph representing the distribution of CD4+, CD8+ and CD11c+, CD8+ in lung cell preparations from colitic and non-colitic mice. The figures represent means and standard errors from three independent experiments
Higher CD4+ with lower CD8+ T cells and higher CD11c+, CD8+ subpopulations in the lungs of colitic mice were detected by flow cytometry (Fig. 3c). After LPS exposure, CD4+ lymphocytes from lungs of colitic mice expressed a proinflammatory response with more IFN-γ+ effector cells in colitic mice compared to non-colitic mice and fewer IL10+ regulatory cells in colitic mice compared to non-colitic mice (Table 1).
Table 1.
Flow cytometry analysis of intracellular staining of IFN-γ and IL10 in CD4 populations from lung lymphocytes in colitic and non-colitic mice
| Lung lymphocytes | Percentage of cells | |
|---|---|---|
| Subpopulation | Non-colitic mice | Colitic mice |
| CD4+, IL10+ | 8.4 ± 0.5 | 1.8 ± 0.01 (P < 0.01) |
| CD4+, IFN-γ+ | 0.6 ± 0.01 | 1.1 ± 0.02 (P < 0.05) |
Expression of Regulatory Versus Inflammatory Markers on Lung Lymphocytes
Flow cytometry analysis of lymphocytes isolated from the lung tissue indicated fewer pulmonary lymphocytes expressing the regulatory marker FoxP3 in colitic mice compared to non-colitic mice (1.0 ± 0.2 vs. 4.6 ± 0.4, P < 0.05), associated with the elevated ICAM1 expression in lungs from colitic mice, 5.5 ± 0.4 versus non-colitic mice, 3.0 ± 0.1, P < 0.05 (Fig. 4). ICAM1 is a strong proinflammatory indicator usually associated with IFNγ production at sites of inflammation. Its expression is a strong indicator of inflammation [24].
Fig. 4.
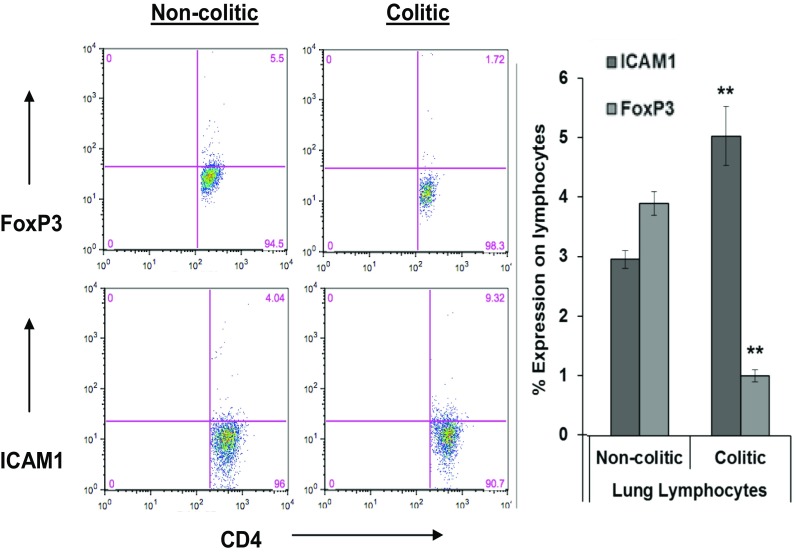
Expression of intercellular adhesion molecule-1 (ICAM1) and FoxP3 by lung lymphocytes from non-colitic and colitic mice. Graph to the right represent means ± S. E. of percentages of positive cells within the CD4 T cells from three independent experiments (** P < 0.05)
Migration of Lymphocytes from the Gut to the Lungs
In order to address the migration of inflammatory lymphocytes from the intestine to the lungs, we had to prove first that they can home in the lungs. We separated LPL from colitic mice and injected them intraperitoneally into untreated Rag-1 mice lacking functional T and B lymphocytes. After inducing colitis, we examined the lungs for the presence of CD4+ and CD8+ cells.
We found that lung cells preparations contained CD4+ and CD8+ lymphocytes (Fig. 5a). These lymphocytes originated from the colon LPL homed in the lungs as well as the colon indicating inflammatory lymphocytes circulation between the peritoneal cavity and the gut as well as the lungs and provide evidence of mishoming of these colon-originated inflammatory lymphocytes into organs other than the colon.
Fig. 5.

Lamina propria lymphocytes from inflamed colons injected into peritoneal cavity of Rag-1 mice, lacking functional T and B lymphocytes. CD4 and CD8 lymphocytes were detected in the lungs as well as the colons (Fig. 6a). Lung lymphocytes from colitic and non-colitic mice were incubated with biotinylated helminth antigens, and flow analysis was done for CD11c+, CD8+ DP gate for binding of the biotinylated antigens (Fig. 6b). In vitro IL17 production by lung lymphocytes and MLN from colitic mice in response to HP antigens (Fig. 6c). The figures are representative of three independent experiments
Next, we examined the lungs of colitic mice for the presence of cells with antigen-presenting capability. We asked if these accessory cells can bind antigens found exclusively in the gut. We prepared biotinylated antigens from the mouse non-systemic, enteric helminth H. polygyrus. We treated lung lymphocytes from colitic and non-colitic mice with the biotinylated antigens and used streptavidin conjugated with a fluorochrome to visualize the cells binding these antigens. CD11c+ cells expressing CD8 are antigen-presenting cells with ability to migrate between different organs. We used flow cytometry analysis of cells from CD11c+, CD8+ gate to see their ability to bind the gut-localized helminth antigens. Comparing lung dendritic cells from colitic and non-colitic mice indicated that lungs from colitic mice had a threefold increase in CD11c+, CD8+ cells capable of binding antigens from the gut microbiome, as shown in Fig. 5b.
Finally, we asked if the lung lymphocytes from colitic mice pre-exposed to HP are capable of responding to the HP antigens in vitro. In Fig. 5c, lymphocytes from the lungs were responsive to HP antigens nearly as much as the MLN cells.
Lung CD11c+ Cells Binding Gut-Localized Antigens Express More TLR4 in Colitic Mice
We analyzed CD11c+ cells from lungs of colitic mice for expression of TLR4 and compared that to their counterparts in non-colitic mice. CD11c+ cells from colitic mice expressed TLR4 twice as much as those from non-colitic mice. This finding proved that these cells can initiate innate immune response by responding to LPS in addition to the adaptive response they can mediate through antigen presentation. This experiment was repeated three times with the same result, as shown in Fig. 6.
Fig. 6.

Flow cytometric analysis of TLR4 expression by CD11c+ dendritic cells from lung cell preparations of colitic and non-colitic Rag-1 mice. Data indicate that colitic mice have a twofold higher expression of TLR4 compared to non-colitic mice. The difference in TLR4 expression is significant at P < 0.05. The data are means ± S. E. from three independent experiments
Discussion
Recent epidemiologic data have indicated extra-intestinal manifestations in inflammatory bowel disease including the lungs. Lung inflammation can be associated with or may occur later than the flare-up times of the intestinal disorder [2]. In this report, we studied lung injury secondary to colitis in mice and revealed novel inflammatory pathways important for developing new strategies to treat this syndrome.
Histopathological examination of the lungs from colitic mice showed extensive lung inflammation with thickening of the airways and massive lymphocyte infiltration as compared to non-colitic mice. Inflammation of the airways induces narrowing of the bronchioles and reduces the airflow that contributes to asthma.
The cytokine profiles in the colons from colitic and non-colitic mice were mimicked by their lung lymphocytes counterparts with the only exception of high level of IL4 which may indicate a Th2 adding to the Th1 cytokine profile that allows switching in antibody isotype to IgE typically seen in asthmatic subjects. Lung inflammation can be identified by immunophenotyping of macrophages. Asthmatic patients show signs of alternative activation of macrophages associated with high IgE and degranulation of mast cells and eosinophils [26]. Our data indicated a robust production of IL4 by lung lymphocytes from colitic mice which may imply a trend of these mice to become asthmatic through switching to the Th2 response.
IL12 (P40), IL17, IFN-γ, and IL10 in cultures from lung lymphocytes reflected a mirror image of cytokines regulation in the LPL in the inflamed colon. The upregulation of the Th1/Th17 cytokines seen in the enhanced IL12 (P40), IL17, and IFN-γ production suggests the presence of granulocytes, as the histology sections demonstrated in Fig. 2b. Production of both Th1 and Th2 cytokines is indicative of heterologous stimulation of classic M1 as well as alternative M2 pathways by macrophages in our model. Heterologous subpopulations of pulmonary macrophages activated through the alternative pathway were shown to contribute to both the induction and resolution of acute lung inflammation in a mouse model of murine pneumonia [31]. In our model of lung injury, macrophages from both classic and alternative activation were expected as we determined that the cytokine profile included Th1, Th2, and Th17.
Similarities in the cytokine profiles in the colon and lungs may hint to migration of inflammatory cells from the colon to the lungs. In order to confirm this, we transplanted LPL from inflamed colons into Rag-1 mice lacking functional T and B lymphocytes.
We could detect CD4+ and CD8+ T cell populations in the lungs from these mice post-colitis development. Lymphocytes from the lungs of colitic mice exposed to H. polygyrus responded to purified worm antigens by producing levels of IL17 comparable to those made by the gut-associated mesenteric lymph nodes (Fig. 5c). These T cells expressed a proinflammatory trend, indicating that they did not change their phenotype during recirculation or by changing the organ of residence.
We analyzed the peripheral blood lymphocytes from mice exposed to the helminth by stimulating them with the same antigens and found no response in cytokine production; however, they responded to stimulation with anti-CD3, indicating the non-systemic nature of H. polygyrus infection (unpublished data).
Flow cytometry data indicated suppressed expression of FoxP3 regulatory marker associated with enhanced expression of ICAM1. We do not know if this is a cause-and-effect relationship. However, the enhanced expression of ICAM1 indicates a proinflammatory trend usually associated with recruiting inflammatory lymphocytes producing TNFα and IFN-γ as well as mast cells and promoting type I hypersensitivity reaction [5, 24].
Studies on the mice early embryonic development showed that the lungs and trachea arise from the anterior foregut endoderm that generates the respiratory system, esophagus, thyroid, and liver, suggesting yet unidentified pathways for circulation between the intestine and the lungs [25]. Because inflammatory lymphocytes recirculating from the gut mucosal tissue of colitic mice (e.g., from the lamina propria) to the lung mucosa are exposed to normal gut flora, we hypothesize that these primed lymphocytes are capable of modifying the lung response to challenge by airborne contaminants like the bacterial endotoxin.
α4β7 is an important gut-homing receptor [25]. Its expression on lymphocytes from the intestine is indicative of binding to MadCAM-1 expressed on endothelium in the intestinal vasculature. Our finding is that in the mouse model of transfer colitis, effector lymphocytes (lacking CTLA4) from the intraepithelial lymphocytes (IEL) have suppressed levels of the gut-homing receptor α4β7 compared to their counterparts from non-colitic mice (unpublished data). This important finding may result in longer circulation time for these lymphocytes which may increase their chance in homing to other organs where they can promote inflammation. This dysfunctional immune cell homing mechanism could be one of the pathways underlying the pulmonary pathology post-colitis development [26, 27].
Meenan et al., in 1997, indicated expression of α4β7 on CD4+, CD45RO+ in patients with colonic inflammation but not activated CD4+, CD25+ cells [28]. Suppressed α4β7 expression on lymphocytes with non-regulatory function implied that these cells are migratory with proinflammatory nature, as data from Figs. 3 and 4 suggest.
Tissue-resident dendritic cells (DC) imprint naïve T cells homing to the tissue of their residence [29]. Accordingly, DC from the gut will make naïve T cells more favorably home to the gut [30], and so it is with the lung DC. However, this may not apply in colitic mice, since we detected inflammatory lymphocytes from LP homing in the lungs.
The disrupted epithelial integrity in colitic mice may result in sensitized lamina propria lymphocytes as they expose to antigens from the intestinal microbiota. We found that CD11c+, CD8+ cells from the lungs of colitic mice are more capable of binding gut-localized antigens compared to non-colitic mice. The presence of antigen-presenting cells sensitized to gut antigens in the lungs makes inflammatory response more favorable. Furthermore, these cells may contribute to mobilization of gut antigens to the lungs when they recirculate. Also, the high expression of TLR4 on CD11c+ cells from the lungs of colitic mice promotes the innate response to LPS. These factors combined raise the risk of exacerbating the lung inflammatory response to antigens localized in the gut and may translate into the higher prevalence of lung inflammation among colitic subjects.
Funding
This work is supported (or supported in part) by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development Grants 1BX002715 and NIH P30 ES005605.
Compliance with ethical standards
Conflict of interest
Dr. Metwali does not have any conflict of interests to disclose
Contributor Information
Ahmed Metwali, Phone: 319-335-6605, Phone: 319-400-2215, Email: ahmed-metwali@uiowa.edu.
David E. Elliott, Phone: 319-335-6605, Email: elliottde@healthcare.uiowa.edu
References
- 1.Bernstein CN, Wajda A, Blanchard JF. The clustering of other chronic inflammatory diseases in inflammatory bowel disease: a population-based study. Gastroenterology. 2005;129:827–836. doi: 10.1053/j.gastro.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 2.Sharma S, Eschun G. Respiratory symptoms may develop years after IBD diagnosis what are the pulmonary manifestations of inflammatory bowel disease? J Respir Dis. 2001;28:774–777. [Google Scholar]
- 3.Marvisi M, Fornasari G. Is the lung a target organ in inflammatory bowel disease? Recent Prog Med. 2001;92:774–777. [PubMed] [Google Scholar]
- 4.Eksteen B, Miles AE, Grant AJ, Adams DH. Lymphocyte homing in the pathogenesis of extra-intestinal manifestations of inflammatory bowel disease. Clin Med. 2004;4:173–180. doi: 10.7861/clinmedicine.4-2-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Center for Disease Control and Prevention. National Surveillance of Asthma: United States, 2001–2010. Vital and Health Statistics, Series 3, Number 35; 2012. [PubMed]
- 6.Maestrelli P, Boschetto P, Fabbri LM, Mapp CE. Mechanisms of occupational asthma. J Allergy Clin Immunol. 2009;123:531–542. doi: 10.1016/j.jaci.2009.01.057. [DOI] [PubMed] [Google Scholar]
- 7.Moldoveanu B, Otmishi P, Jani P, Walker J, Sarmiento X, Guardiola J, Saad M, Yu J. Inflammatory mechanisms in the lung. J Inflamm Res. 2009;2:1–11. [PMC free article] [PubMed] [Google Scholar]
- 8.Salmi M, Andrew DP, Butcher EC, Jalkanen S. Dual binding capacity of mucosal immunoblasts to mucosal and synovial endothelium in humans: dissection of the molecular mechanisms. J Exp Med. 1995;181:137–149. doi: 10.1084/jem.181.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corry DB. Emerging immune targets for the therapy of allergic asthma. Nat Rev. 2002;1:55–62. doi: 10.1038/nrd702. [DOI] [PubMed] [Google Scholar]
- 10.Duan M, Steinfort DP, Smallwood D, et al. CD11b immunophenotyping identifies inflammatory profiles in the mouse and human lungs. Mucosal Immunol. 2016;9:550–563. doi: 10.1038/mi.2015.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choy DF, Hart KM, Borthwick LA, et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med. 2015;7(301):301ra129. doi: 10.1126/scitranslmed.aab3142. [DOI] [PubMed] [Google Scholar]
- 12.Ray A, Khare A, Krishnamoorthy N, Qi Z, Ray P. Regulatory T cells in many flavors control asthma. Nature. 2010;3:216. doi: 10.1038/mi.2010.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Metwali A, Setiawan T, Blum AM, Urban J, Elliott DE, Hang L, Weinstock JV. Induction of CD8+ regulatory T cells in the intestine by Heligmosomoides polygyrus infection. Am J Physiol Gastrointest Liver Physiol. 2006;291:G253–G259. doi: 10.1152/ajpgi.00409.2005. [DOI] [PubMed] [Google Scholar]
- 14.Setiawan T, Metwali A, Blum AM, Ince MN, Urban JF, Jr, Elliott DE, Weinstock JV. Heligmosomoides polygyrus promotes regulatory T-cell cytokine production in the murine normal distal intestine. Infect Immun. 2007;75:4655–4663. doi: 10.1128/IAI.00358-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kitagaki K, Businga TR, Racila D, Elliott DE, Weinstock JV, Kline JN. Intestinal helminths protect in a murine model of asthma. J Immunol. 2006;177:1628–1635. doi: 10.4049/jimmunol.177.3.1628. [DOI] [PubMed] [Google Scholar]
- 16.Strober W, Ludviksson BR, Ivan J, Fuss IJ. The pathogenesis of mucosal inflammation in murine models of inflammatory bowel disease and crohn disease. Ann Intern Med. 1998;128:848–856. doi: 10.7326/0003-4819-128-10-199805150-00009. [DOI] [PubMed] [Google Scholar]
- 17.Hoffmann JC, Pawlowski NN, Kühl AA, Höhne W, Zeitz M. Animal models of inflammatory bowel disease: an overview. Pathobiology. 2002;70(3):121–130. doi: 10.1159/000068143. [DOI] [PubMed] [Google Scholar]
- 18.Jurjusa Abdo R, Khourya Naim N, Reimundb Jean-Marie. Appraisal of state-of-the art animal models of inflammatory bowel disease. J Pharmacol Toxicol Methods. 2004;50:81–92. doi: 10.1016/j.vascn.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 19.Devoss J, Diehl L. Murine models of inflammatory bowel disease (IBD): challenges of modeling human disease. Toxicol Pathol. 2014;42:99–110. doi: 10.1177/0192623313509729. [DOI] [PubMed] [Google Scholar]
- 20.Goyal N, Rana A, Ahlawat A, Bijjem KRV, Kumar P. Animal models of inflammatory bowel disease: a review. Inflammopharmacology. 2014;22:219–233. doi: 10.1007/s10787-014-0207-y. [DOI] [PubMed] [Google Scholar]
- 21.Wirtz S, Neurath MF. Mouse models of inflammatory bowel disease. Adv Drug Deliv Rev. 2007;59:1073–1083. doi: 10.1016/j.addr.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 22.Kiesler P, Fuss IJ, Strober W. Experimental models of inflammatory bowel diseases. Cell Mol Gastroenterol Hepatol. 2015;1:154–170. doi: 10.1016/j.jcmgh.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mizoguchi A. Animal models of inflammatory bowel disease. Prog Mol Biol Trans Sci. 2012;105:263–320. doi: 10.1016/B978-0-12-394596-9.00009-3. [DOI] [PubMed] [Google Scholar]
- 24.Hubbard AK, Rothlein R. Intercellular adhesion molecule-1 (ICAM1) expression and cell signaling cascades. Free Radic Biol Med. 2000;28:1379–1386. doi: 10.1016/S0891-5849(00)00223-9. [DOI] [PubMed] [Google Scholar]
- 25.Herriges M, Morrisey EE. Lung development: orchestrating the generation and regeneration of a complex organ. Development. 2014;141:501–513. doi: 10.1242/dev.098186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mateer SW, Maltby S, Marks E, Foster PS, Horvat JC, Hansbro PM, Keely S. Potential mechanisms regulating pulmonary pathology in inflammatory bowel disease. J Leukoc Biol. 2015;98:727–737. doi: 10.1189/jlb.3RU1114-563R. [DOI] [PubMed] [Google Scholar]
- 27.Adam DH, Eksteen B. Aberrant homing of mucosal T cells and extra-intestinal manifestations of inflammatory bowel disease. Nat Rev. 2006;6:244–251. doi: 10.1038/nri1784. [DOI] [PubMed] [Google Scholar]
- 28.Meenan J, Spaans J, Grool TA, Pals ST, Tytgat GNJ, van Deventer SJH. Altered expression of α4β7, a gut homing integrin, by circulating and mucosal T cells in colonic mucosal inflammation. Gut. 1997;40:241–246. doi: 10.1136/gut.40.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mikhak Z, Strassner JP, Luster AD. Lung dendritic cells imprint T cell lung homing and promote lung immunity through the chemokine receptor CCR4. J Exp Med. 2013;210:1855–1869. doi: 10.1084/jem.20130091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coombes JL, Powrie F. Dendritic cells in intestinal immune regulation. Nat Rev Immunol. 2008;8:435–446. doi: 10.1038/nri2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnston LK, Rims CR, Gill SE, McGuire JK, Manicone AM. Pulmonary macrophage subpopulations in the induction and resolution of acute lung injury. Am J Respir Cell Mol Biol. 2012;47:417–426. doi: 10.1165/rcmb.2012-0090OC. [DOI] [PMC free article] [PubMed] [Google Scholar]