
Figure 1.
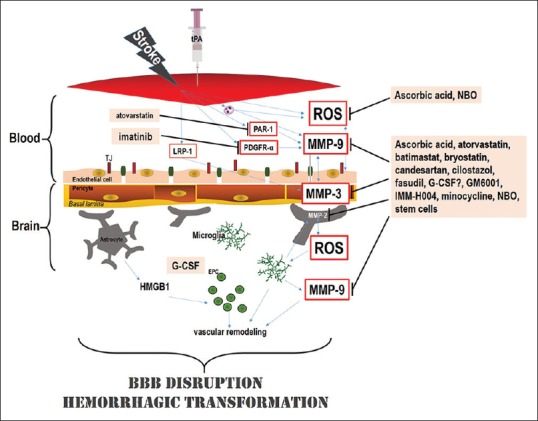
Putative mechanism(s) of action of conjunctive treatments to enhance thrombolytic therapy for acute ischemic stroke. Acute stroke potentially injures endothelial cells which lead to the formation of reactive oxygen species and pro-inflammatory cytokines. Moreover, delayed tissue plasminogen activator therapy could also damage the neurovascular unit and increase blood–brain barrier (BBB) leakage, neurovascular cell death, and hemorrhagic transformation. The hemorrhagic transformation that ensues after delayed Tissue plasminogen activator therapy has also been ascribed to enhanced reperfusion and the influence of tissue plasminogen activator on metalloproteinase activity and other signaling pathways, such as LRP, PAR1, and PDGRF-α signaling. In preclinical stroke modes, adjunctive treatment of ascorbic acid, and NBO has been shown to counteract delayed Tissue plasminogen activator -induced complications through dampening of ROS production and BBB breakdown. Administering atorvastatin, minocycline, cilostazol, GM6001, fasudil, candesartan, bryostatin, and IMM-H004 also reduced delayed tissue plasminogen activator-induced hemorrhagic transformation by preserving the BBB through their effects on MMPs and tight junction proteins. Granulocyte-colony stimulating factor and IMM-H004 have also been demonstrated to reduce the hemorrhagic transformation by enhancing neurovascularization and preserving the integrity of the BBB. Imatinib administration also reduced hemorrhagic transformation by acting through the PDGRF-α receptor, while atorvastatin decreased hemorrhagic transformation by inhibiting PAR1. Stem cells have also been shown to affect multiple targets including BBB and MMPs. EPC: Endothelial progenitor cell; G-CSF: Granulocyte-colony stimulating factor; HMGB1: High-mobility-group-box-1; ROS: Reactive oxygen species; LRP: Lipoprotein receptor-related protein; PAR1: Protease-activated receptor; PDGFR-α: Platelet-derived growth factor α-receptor; NBO: Normobaric oxygen