

Abstract
Biphenyl hydrolase-like protein (BPHL) is a novel human serine hydrolase that was originally cloned from a breast carcinoma cDNA library and shown to convert valacyclovir to acyclovir and valganciclovir to ganciclovir. However, the exclusivity of this process has not been determined and, indeed, it is possible that a number of esterases/proteases may mediate the hydrolysis of valacyclovir and similar prodrugs. The objectives of the present study were to evaluate the in situ intestinal permeability and stability of valacyclovir in wildtype (WT) and Bphl knockout (KO) mice, as well as the in vivo oral absorption and intravenous disposition of valacyclovir and acyclovir in the two mouse genotypes. We found that Bphl knockout mice had no obvious phenotype and that Bphl ablation did not alter the jejunal permeability of valacyclovir during in situ perfusions (i.e., 0.54 × 10−4 in WT vs. 0.53 × 10−4 cm/sec in KO). Whereas no meaningful changes occurred between genotypes in the gene expression of proton-coupled oligopeptide transporters (i.e., PepT1, PepT2, PhT1, PhT2), enzymatic upregulation of Cyp3a11, Cyp3a16, Abhd14a and Abhd14b was observed in some tissues of Bphl knockout mice. Most importantly, we found that valacyclovir was rapidly and efficiently hydrolyzed to acyclovir in the absence of BPHL, and that hydrolysis was more extensive after the oral vs. intravenous route of administration (for both genotypes). Taken as a whole, BPHL is not obligatory for the conversion of valacyclovir to acyclovir either presystemically or systemically.
Keywords: Biphenyl hydrolase-like protein, Bphl knockout mice, Valacyclovir, Intestinal stability and permeability, Oral absorption
Graphical abstract

1. Introduction
By targeting transporters and enzymes, prodrugs have proven to be a valuable strategy in overcoming pharmaceutical, pharmacokinetic and pharmacodynamic barriers. Such barriers include poor solubility, permeability and stability, and unacceptable toxicity of the active parent drug [1–7]. Once entering the human body, prodrugs can be activated chemically such as through pH sensitivity [8–10], or enzymatically such as through paraoxonase, serine hydrolase, carboxylesterase, and acetylcholinesterase [11–14]. One notable example is the antiviral ester prodrug valacyclovir, which is actively transported by the proton-coupled oligopeptide transporter PepT1 (SLC15A1) from intestinal lumen. Previous studies have suggested that this prodrug is converted to the active parent drug acyclovir by the serine hydrolase biphenyl hydrolase-like (BPHL) protein [15–18]. As illustrated by this prototypic prodrug, a comprehensive understanding of all possible transporters and activating enzymes involved in valacyclovir’s intestinal oral absorption, activation and systemic availability can aid in the rational design of future drug candidates.
Along with other prodrug targeted transporters, such as the organic anion transporter (OAT) family and the organic cation transporter (OCT) family, the PepT1 transporter is predominately expressed on the apical membrane of enterocytes and its biochemistry, biology, pharmacokinetics and substrate specificity have been well characterized [19–23]. As such, PepT1 is a potential drug design target for improving intestinal absorption and the systemic availability of poorly absorbed drug molecules [4,24,25]. In addition to increasing oral drug absorption, an essential step is the conversion of prodrug to its active therapeutic moiety. Recent advances in biochemistry and biology concerning the structural and functional characteristics of enzymes have provided abundant information to benefit prodrug design. More sophisticated strategies have been developed involving the covalent binding of special promoieties to relevant parent drugs. These prodrugs can then be activated by one or more enzymes presystemically, systemically or at the site of action [12,26,27].
BPHL, also called human valacyclovirase, an is an α-amino acid ester hydrolase that was initially cloned from a human breast carcinoma cDNA library and found to be highly expressed in human liver and kidney [28]. BPHL protein consists of 274 amino acids containing the Gly-X-Ser-X-Gly motif, which is characteristic of serine hydrolases involved in the degradation of aromatic compounds by cleavage of carbon-carbon bonds [29]. In 2008, a study on the crystal structure and biochemical analysis of recombinant BPHL identified the key determinants for substrate recognition [30]. Specifically, these authors reported that the protein had a unique active site defined by a flexible and mostly hydrophobic acyl pocket, a localized negative electrostatic potential, and a large open leaving group-accommodating groove. Most importantly, BPHL protein also had a pivotal acidic residue, Asp-123, located after the nucleophile Ser-122, whose side chain was directed into the substrate binding pocket, thereby, playing a critical role in the substrate discriminating ability of serine hydrolases [30]. Moreover, the recombinant BPHL enzyme (originally from Caco2 cells) was shown to effectively hydrolyze many amino acid ester nucleoside prodrugs, converting compounds such as valacyclovir to acyclovir [18] and valganciclovir to ganciclovir [31]. Physiologically, BPHL was reported as a likely candidate for protecting against homocysteine thiolactone toxicity [32].
Despite the fact that BPHL is a major enzyme for the activation of valine ester prodrugs, the in vivo impact of BPHL on prodrug stability, permeability and activation has yet to be investigated. It is also uncertain as to whether valacyclovir (and similar prodrugs) are hydrolyzed exclusively by BPHL or if other enzymes contribute to the in vivo hydrolysis (activation) of valacyclovir to acyclovir. With this in mind, the objectives of this study were to evaluate the in situ intestinal permeability and stability of valacyclovir in wildtype and Bphl knockout mice, and to determine the in vivo oral absorption and intravenous disposition of valacyclovir and acyclovir in the two mouse genotypes. This is the first time that a phenotypic, biochemical, molecular and functional assessment of BPHL ablation has been reported under both in situ and in vivo study conditions.
2. Materials and methods
2.1. Chemicals
Anti-mouse BPHL antiserum was generated by Lampire Biological Laboratories, Inc (Pipersville, PA USA) using the C-terminal 20 amino acid residues (HNLHLRFADEFNRLVEDFLQ, amino acids 272–291) of the protein. Valacyclovir and acyclovir were purchased from Sigma-Aldrich (St. Louis, MO USA). Valacyclovir-D4 and acyclovir-D4 internal standards were purchased from Toronto Research Chemicals (Toronto, Ontario Canada). Other chemicals and reagents were from standard sources.
2.2. Animals
Bphl knockout mice on a pure C57BL/6N background were generously provided by the Institut Clinique de la Souris (Illkirch Cedex, France). Animals were bred and housed in a temperature-controlled environment with 12-hour light and dark cycles. They received a standard diet and water ad libitum, as provided by the Unit for Laboratory Animal Medicine, University of Michigan, Ann Arbor, MI. Gender-matched mice (8–10 weeks) were used in accordance with the Guide for Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health. All relevant mouse strains were validated by genotyping.
2.3. Initial phenotypic analysis
Wildtype and Bphl knockout mice were evaluated for viability, fertility, serum clinical chemistry and histology, as described previously [23].
2.4. Quantitative real-time PCR (qRT-PCT) and immunoblot analysis
Gene transcripts of Bphl, PepT1, PepT2, PhT1, PhT2, Ces1, Ces2, Cyp3a11, Cyp3a16, Cyp3a41 and Cyp3a44 were evaluated in the small intestine, colon, kidney, spleen and liver of wildtype and Bphl knockout mice using the 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA), as described previously [21]. The primers are listed in Table 1. Briefly, all reactions were performed in triplicate, where each reaction included 30 ng of cDNA that was reverse-transcribed using the Omniscript RT kit (Qiagen, Valencia, CA) and 16-mer random primers from RNA isolated using the RNeasy Plus Mini Kit (Qiagen, Valencia, CA). The qRT-PCR thermal conditions were one cycle at 50°C for 2 min, one cycle at 95°C for 10 min, and forty cycles at 95°C for 15 sec followed by 60°C for 1 min. Relative levels of the target gene transcripts in mice were calculated using the ΔCT method, where the ratio of target gene to Gapdh was equal to 2–ΔCT, ΔCT = CT(gene) – CT (Gapdh). The mouse Gapdh gene was set as an internal control of cDNA quality and quantity.
Table 1.
Primers used in qRT-PCR
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Gapdh (EC1.2.1.12) |
5’-GAGACAGCCGCATCTTCTTGT-3’ | 5’-CACACCGACCTTCACCATTTT-3’ |
| Bphl (EC3.1.-.-) |
5’-CCCTGAAATCCCGGATCTG -3’ | 5’-AAGTTACTGCAGTGCCGAAGGT-3’ |
| PepT1 (Slc15a1) |
5’-CCACGGCCATTTACCATACG-3’ | 5’-TGCGATCAGAGCTCCAAGAA-3’ |
| PepT2 (Slc15a2) |
5’-TGCAGAGGCACGGACTAGATAC-3’ | 5’-GGGTGTGATGAACGTAGAAATCAA-3’ |
| PhT1 (Slc15a4) |
5’-GCTGCCACCTGCATTACTACTTC-3’ | 5’-CGTACTTCACAGACACAATGAGGAA-3’ |
| PhT2 (Slc15a3) |
5’-GCTGAAGCTTGCGTTCCAA-3’ | 5’-AACAGGTGGGCACTTTCAGAGT-3’ |
| Ces1 (EC3.1.1.-) |
5’- GCCCTGGAGCTTCGTGAA -3’ | 5’- TCAATGCTGCCTCTGGGTTT -3’ |
| Ces2 (EC3.1.1.-) |
5’- GCTGGAAGGCTGCAGTTCTG -3’ | 5’- GTGCTTGTCCTGAGAAGCCTTT -3’ |
| Cyp3a11 (EC1.14.99.38) |
5’- AACTGCAGGATGAGATCGATGAG -3’ | 5’- TTCATTAAGCACCATATCCAGGTATT -3’ |
| Cyp3a16 (EC1.14.99.-) |
5’- CTGCAGGAGGAGATCGATGAG -3’ | 5’- CCATCGCCATCACGGTATC -3’ |
| Cyp3a41 (EC1.14.-.-) |
5’- CTGACAGACAAGCAGGGATGAA -3’ | 5’- GTAGAGGAGCACCAGGATGATTG -3’ |
| Cyp3a44 (EC1.14.-.-) |
5’- CAGCTCTCTCACTGGATACATTGG -3’ | 5’- GTACGGGTCCCATATCGGTAGA -3’ |
| Abhd14a (EC3.-.-.-) |
5’- CCAGAGGCAACTCTCGAATCTT-3’ | 5’- CACGACTTCTGCCCTGCAT -3’ |
| Abhd14b (EC3.-.-.-) |
5’- GGCTCCTCCAGCTTTCAACA -3’ | 5’- CCCTGCCCCTTCCATGAC -3’ |
Protein levels of BPHL were determined in the liver, jejunum, spleen and kidney of wildtype and Bphl knockout mice by immunoblot analysis, as described previously [21,33,34]. In brief, after processing the tissues, membrane proteins were denatured at 40°C for 45 min, resolved by 7.5% SDS-PAGE, transferred to a PVDF membrane (Millipore, Billerica, MA), and then blotted for one hour with rabbit anti-mouse BPHL antisera (1:5000). The membrane was washed three times with TBST and then incubated with the secondary antibody, goat anti-rabbit IgG-HRP (1:3000) (Bio-Rad, Hercules, CA). For β-actin, the membrane was blotted with a mouse monoclonal antibody (1:1000) (Santa Cruz Biotechnology, Santa Cruz, CA), followed by the secondary antibody, goat anti-mouse IgG-HRP (1:1000) (Santa Cruz Biotechnology). The membrane was then washed five times with TBST, the UCL substrate (Millipore) added, and the x-ray film exposed and developed.
2.5. In situ single-pass intestinal perfusion studies
Prodrug design can be used to increase the permeability and stability of the parent drug [35]. The effects of Bphl gene deletion on valacyclovir permeability and stability were evaluated using in situ single-pass jejunal perfusion studies, as described previously with wildtype mice as the control group [20,21,23]. In brief, following an overnight fast, wildtype and Bphl knockout mice were anesthetized with sodium pentobarbital (40–60 mg/kg ip) and an 8-cm jejunal segment was isolated. The segment was then rinsed with 0.9% isotonic saline solution and a glass cannula (0.2 mm OD) was inserted at each end of the intestinal segment. Perfusion buffer (135 mM NaCl, 5 mM KCl and 10 mM MES/Tris, pH 6.5) containing 50 μM valacyclovir was perfused through the jejunal segment at a flow rate of 0.1 mL/min and the outlet perfusate was collected every 10 min for 90 min. Water flux during the perfusion was corrected by mass balance using a gravimetric method.
Hydrolysis studies were also performed in which, following jejunal perfusions of 1.0 mM valacyclovir, plasma samples were collected from the portal vein at 5 and 30 min after initiating the perfusion.
2.6. Ultra performance liquid chromatography with photodiode array detector assay conditions for in situ perfusion studies
Following centrifugation of the 100-μL perfusate samples, a 25-µL aliquot was injected into the ultra performance liquid chromatography (UPLC) system, as previously described with minor modification [36]. Specifically, the method employed a UPLC Acquity H-Class System (Waters Corp, Milford, MA) coupled to a Photodiode Array Detector set at 254 nm. A 100-cm Acquity HSS T3 column (1.8 μm) was maintained at 40°C with gradient elution at a flow rate of 0.5 mL/min. The mobile phase started with 100% water (0.1% TFA), and increased linearly to 10% acetonitrile and 90% water (0.1% TFA) over 5 min. This level was maintained for 2 min, then reverted back to 100% water (0.1% TFA) at 7 min, which was maintained until the end of the run (12 min). The lower limits of quantitation for acyclovir and valacyclovir in perfusate buffer were 50 and 10 nM, respectively.
2.7. Oral and intravenous pharmacokinetic studies
The effects of Bphl gene deletion on the oral absorption and disposition of valacyclovir were evaluated, as described previously for valacyclovir in wildtype mice [17]. For oral studies, 25 nmol/g of valacyclovir (in 0.2 mL of water per 20 g body weight) was administered by oral gavage. For intravenous bolus studies, 25 nmol/g of valacyclovir (in 0.1 mL of saline per 20 g body weight) was administered by tail vein injection. Blood samples (~ 20 μL aliquots), after both dosing routes, were obtained by tail biopsy at 2, 5, 15, 30, 60, 90, 120 and 180 min post-injection, with an additional sample taken at 45 min for the oral studies. The blood samples were then mixed with 1 μL EDTA-K3 and centrifuged at 3,000 g for 5 min at 4°C. Following centrifugation, the plasma was harvested and a 10-μL aliquot was mixed with 40 μL of acetonitrile containing 200 nM of the internal standards VACV-d4 and ACV-d4. Samples were then centrifuged at 17,000 g for 10 min at 4°C to precipitate the proteins, the supernatant transferred carefully into a new tube, and frozen at −80°C for subsequent analysis by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
2.8. LC-MS/MS assay conditions for in vivo pharmacokinetic studies
Plasma supernatant (10 μL aliquot), obtained from the in vivo pharmacokinetic studies (see above), was analyzed by LC-MS/MS, as previously described [37]. In brief, the LC–MS/MS analysis consisted of a Shimadzu HPLC system (Shimadzu, Tokyo, Japan) coupled to an API 4000 triple quadrupole/linear ion trap (QTRAP) mass spectrometer (AB Sciex, Foster City, CA, USA). Analytes were separated on a Waters Atlantis T3 C18 column (5 μm, 150 × 2.1 mm, Dublin, Ireland). The mobile phase consisted of water containing 2 mM ammonium acetate and 0.2% formic acid (v/v) (Phase A) and acetonitrile containing 0.2% formic acid (v/v) (Phase B), which was delivered at a flow rate of 0.2 ml/min. Gradient elution was applied for compound separation and the program was timed as follows: Phase B was started at 2% for 2 min, then increased linearly to 4% over the next 2 min, further increased to 50% over the next 2 min, and then returned to 2% at 6.5 min, which was maintained until the end of run (9 min). The mass spectrometer was operated in positive ion mode using electrospray ionization. The following transitions were monitored in a multiple reaction monitoring mode: VACV, 325.2 > 152.1; ACV, 226.2 > 152.1; VACV-d4, 329.2 > 152.1; ACV-d4, 230.2 > 152.1. The lower limit of quantification was 2 nM for both valacyclovir and acyclovir.
2.9. Data analysis
Data were reported as mean ± SE, unless otherwise noted. Statistical differences between two groups were determined by an unpaired t-test using GraphPad Prism 7.0 (GraphPad Software, Inc., La Jolla, CA). A value of p ≤ 0.05 was considered statistically significant. Pharmacokinetic analyses were carried out by noncompartmental analysis (NCA) using Phoenix WinNonlin 8.0 (Certara, St. Louis, MO USA).
3. Results
3.1. Initial phenotypic analysis
Bphl knockout mice appeared normal with no obvious behavioral abnormalities as compared to wildtype mice. They also had normal fertility, litter size, gender distribution, viability, and body weight. As shown in Table 2, no significant differences were observed in serum clinical chemistry between wildtype and Bphl knockout mice, although a statistical difference was noted for sodium (i.e., 147 vs 144 mM, respectively). Histological evaluation, as judged by hematoxylin and eosin staining, demonstrated normal morphology in the small and large intestines, liver, kidney and spleen of wildtype and Bphl knockout mice (Fig. 1).
Table 2.
Serum clinical chemistry of wildtype and Bphl knockout mice
| Wildtype | Bphl Knockout | |
|---|---|---|
| Sodium (mmol/L) | 144 ± 1 (10) | 147 ± 1* (8) |
| Potassium (mmol/L) | 9.1 ± 0.4 (10) | 7.7 ± 0.7 (8) |
| Chloride (mmol/L) | 109 ± 1 (10) | 109 ± 1 (8) |
| Creatine (mg/dL) | 0.32 ± 0.05 (9) | 0.27 ± 0.03 (8) |
| TRIG (mg/dL) | 64 ± 6 (10) | 76 ± 5 (8) |
| CHOL (mg/dL) | 79 ± 10 (10) | 67 ± 3 (8) |
| AST (U/L) | 121 ± 12 (10) | 226 ± 57 (8) |
| Glucose (mg/dL) | 250 ± 79 (9) | 242 ± 13 (8) |
| Albumin (g/dL) | 2.5 ± 0.3 (10) | 2.4 ± 0.3 (8) |
| Protein (g/dL) | 5.1 ± 0.1 (9) | 5.2 ± 0.1 (8) |
| Calcium (mg/dL) | 9.2 ± 0.4 (9) | 7.0 ± 1.0 (8) |
| ALT (U/L) | 79 ± 41 (9) | 132 ± 31 (8) |
| CPK (U/L) | 1629 ± 197 (9) | 1562 ± 344 (6) |
| Bilirubin (mg/dL) | 0.12 ± 0.01 (9) | 0.10 ± 0.00 (7) |
| BUN (mg/dL) | 42 ± 5 (9) | 31 ± 2 (8) |
| ALP (U/L) | 86 ± 11 (9) | 105 ± 5 (8) |
Data are expressed as mean ± SE (number of mice). TRIG, triglycerides; CHOL, Cholesterol; AST, aspartate aminotransferase; ALP, alkaline phosphatase; CPK, creatine phosphokinase; BUN, blood urea nitrogen; ALT, alanine aminotransferase.
p ≤ 0.05, as compared to wildtype mice, using an unpaired t-test.
Figure 1.

Histological evaluation, using hematoxylin and eosin staining, of the duodenum, jejunum, ileum, cecum, colon, liver, kidney and spleen from wildtype and Bphl knockout (KO) mice. One representative example is shown of six individual stains. Magnification 20×.
3.2. Confirmation of BPHL ablation
Even though knockout mice were identified by genotyping, select tissues were further tested by qRT-PCR for Bphl gene expression and by immunoblotting for the presence of BPHL protein. As shown in Fig. 2A, Bphl transcripts were barely detectable in the duodenum, jejunum, ileum, colon, liver, kidney and spleen of Bphl knockout mice. In wildtype mice, Bphl mRNA expression was greatest in the kidney and liver, moderate in the colon and ileum, and low in the small intestine and spleen. This result was confirmed in Fig. 2B, where abundant expression of BPHL protein was observed in the kidney and liver of wildtype mice, but faint expression of protein in the jejunum and spleen. In contrast, no BPHL protein expression was observed in the counterpart tissues of Bphl knockout mice.
Figure 2.
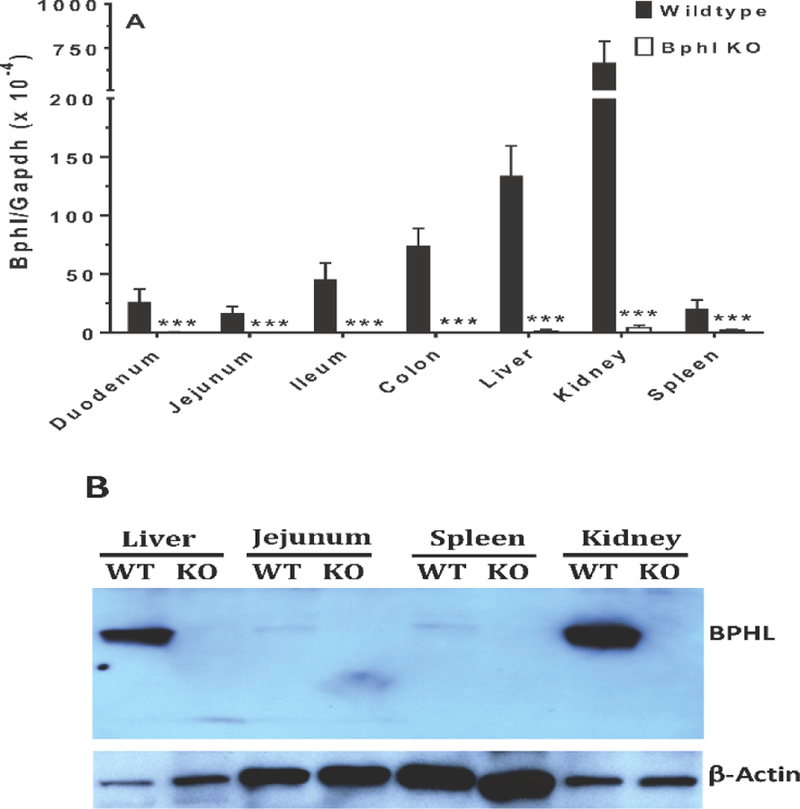
BPHL gene and protein expression in select tissues from wildtype and Bphl knockout mice. (A) Bphl transcripts were measured by quantitative real-time PCR and the data expressed as mean ± SE (n=6, each sample run in triplicate). ***p ≤ 0.001, as compared to wildtype, using an unpaired t-test. (B) BPHL protein was measured by immunoblotting in which 10 μg of protein lysate was loaded for liver and kidney, and 40 μg of protein lysate loaded for jejunum and spleen. One representative example is shown of three individual immunoblots. WT, wildtype mice; KO, Bphl knockout mice.
3.3. Gene expression of select transporters and enzymes in tissues from wildtype and Bphl knockout mice
It is important to know whether Bphl gene ablation resulted in a compensatory response for relevant transporters and/or enzymes of mice. The four mammalian proton-coupled oligopeptide transporters were examined first in several tissues of wildtype and Bphl knockout mice. The intestinal PepT1 transporter was of particular interest because of its strategic value as a target for improving drug (and prodrug) oral absorption. As shown in Fig. 3A, no significant differences were observed between the two genotypes in the gene expression of PepT1 in tissues, with the exception of colon in which PepT1 mRNA was increased. In regard to PepT2, no differences in gene expression were noted between wildtype and Bphl knockout mice in any of the tissues tested (Fig. 3B). In contrast, PhT1 gene expression was increased in the duodenum, ileum, colon, kidney and spleen of Bphl knockout mice (Fig. 3C). Finally, PhT2 gene expression was increased only in the ileum during Bphl ablation (Fig. 3D).
Figure 3.
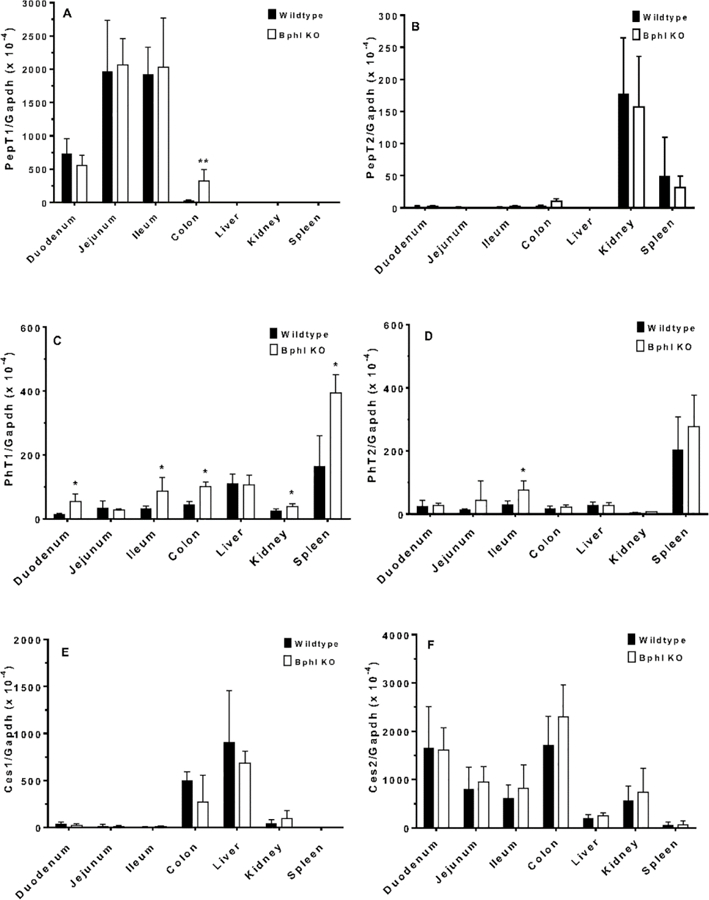
Quantitative real-time PCR analysis of relevant transporters and enzymes in select tissues from wildtype and Bphl knockout (KO) mice. Data are expressed as mean ± SE (n=6, each sample run in triplicate). *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001, as compared to wildtype mice, using an unpaired t-test. Refer to Table 1 for gene identification.
Enzymes that might impact the hydrolysis of valacyclovir were examined next. As shown in Fig. 3E and 3F, no differences were observed between wildtype and Bphl knockout mice in the gene expression of Ces1 or Ces2, respectively. Although Cyp3a11 transcripts were increased in the liver during Bphl ablation (Fig. 3G), as were Cyp3a16 transcripts in the duodenum and liver (Fig. 3H), no differences in Cyp3a41 (Fig. 3I) and Cyp3a44 (Fig. 3J) gene expression were discerned in any of the tissues tested between wildtype and Bphl knockout mice. Nonetheless, significant increases in Abhd14a gene expression were observed in the duodenum, kidney and spleen of Bphl knockout mice (Fig. 3K). Likewise, during Bphl ablation, Abhd14b transcripts were significantly increased in the duodenum, ileum, colon, liver, kidney and spleen of these mice (Fig. 3L).
3.4. Intestinal permeability and stability of valacyclovir during in situ perfusion of prodrug in wildtype and Bphl knockout mice
Single-pass jejunal perfusion studies were performed in order to evaluate the effect of Bphl gene ablation on the intestinal permeability, stability, and presystemic metabolism of valacyclovir. As shown in Fig. 4A, there was no significant difference in the effective permeability of valacyclovir in the jejunum of wildtype and Bphl knockout mice (i.e., 0.54 ± 0.10 and 0.53 ± 0.05 × 10−4 cm/sec, respectively). Moreover, valacyclovir was stable during the jejunal perfusion, as assessed by the very low appearance of acyclovir in outlet perfusate of both genotypes during the experiment, that is about 5% through 80 min and 5–10% at 90 min (Fig. 4B). Presystemic metabolism was determined by sampling the portal vein after 5 and 30 min of jejunal perfusion of valacyclovir. As shown in Fig. 4C, after 5 min of perfusion, prodrug hydrolysis was significantly greater in wildtype mice as compared to Bphl knockout mice (i.e., 8.0 ± 1.0 vs. 23.3 ± 4.4% valacyclovir remaining intact, respectively). However, after 30 min of perfusion, there was no significant difference between genotypes in the proportion of valacyclovir found intact in portal vein blood (6.8 ± 2.3% in wildtype vs. 11.7 ± 6.7% in Bphl knockout mice).
Figure 4.

Intestinal permeability and stability of valacyclovir during in situ jejunal perfusions of prodrug in wildtype and Bphl knockout (KO) mice. (A) Effective permeability of 50 μM valacyclovir in both genotypes, and (B) acyclovir concentrations in the outlet perfusate of both genotypes as a function of time. Data are expressed as mean ± SE (n=3–4). (C) Percent ofvalacyclovir and acyclovir found in the portal vein blood, at 5 and 30 min, following jejunal perfusions of 50 μM valacyclovir. Data are expressed as mean ± SE (n=3). *p ≤ 0.05, as compared to wildtype mice, using an unpaired t-test.
3.5. Oral and intravenous pharmacokinetic studies of valacyclovir in wildtype and Bphl knockout mice
The effect of Bhpl ablation on valacyclovir absorption, disposition and conversion to acyclovir was evaluated in vivo after both oral and intravenous administrations. The pharmacokinetics of valacyclovir (and acyclovir) were considered first after intravenous dosing of prodrug since disposition and conversion could be evaluated in the absence of absorption. As shown in Fig. 5A, similar (but not superimposable) plasma concentration - time profiles were observed between wildtype and Bphl knockout mice for prodrug and active parent drug. On average, the Cmax and AUC0–180 were about 30–40% greater for both drug species, whereas the Tmax for acyclovir was about 2.5-fold greater in Bphl knockout mice (Table 3). The changes in valacyclovir Cmax and AUC0–180, though most were not statistically significant, reflect the reduced conversion of valacyclovir to acyclovir in the absence of BPHL. The log-linear terminal half-life (T½) showed only a modest difference between genotypes (i.e., 10% larger for prodrug and 20% smaller for active parent drug in Bphl knockout mice).
Figure 5.

Plasma concentration-time profile of acyclovir (ACV) and valacyclovir (VACV) after (A) intravenous and (B) oral administrations of 25 nmol/g valacyclovir in wildtype (WT) and Bphl knockout (KO) mice. Data are expressed as mean ± SE (n=3) in which the y-axis is displayed on a linear scale (left-sided panels) and on a logarithmic scale (right-sided panels).
Table 3.
Pharmacokinetic Parameters of Valacyclovir and Acyclovir in Wildtype and Bphl Knockout Mice after the Oral and Intravenous Administrations of 25 nmoL/g Valacyclovir
| Oral | Cmax (μM) |
Tmax (min) |
AUC0–180 (μM·min) |
T1/2 (min) |
|---|---|---|---|---|
| Wildtype | ||||
| VACV | 1.3 ± 0.6 | 5 ± 0 | 22.1 ± 6.5 | 48.9 ± 18.4 |
| ACV | 7.6 ± 0.6 | 15 ± 0 | 402 ± 126 | 47.5 ± 10.1 |
| VACV/ACV (%) | 17 ± 6 | 33 ± 0 | 5.5 ± 2.4 | 103 ± 1 |
| Bphl Knockout | ||||
| VACV | 2.0 ± 0.5 | 5 ± 0 | 38.3 ± 10.3 | 37.2 ± 2.6 |
| ACV | 9.4 ± 0.2* | 12 ± 3 | 419 ± 81 | 37.0 ± 6.6 |
| VACV/ACV(%) | 22 ± 2 | 43 ± 2* | 9.1 ± 3.0 | 100 ± 1 |
| Intravenous |
Cmax (μM) |
Tmax (min) |
AUC0–180 (μM·min) |
T1/2 (min) |
| Wildtype | ||||
| VACV | 9.1 ± 0.4 | 2 ± 0 | 109 ± 8 | 50.2 ± 15.4 |
| ACV | 6.8 ± 0.3 | 5 ± 0 | 259 ± 18 | 45.9 ± 6.0 |
| VACV/ACV(%) | 134 ± 1 | 40 ± 0 | 37.3 ± 3.9 | 109 ± 1 |
| Bphl Knockout | ||||
| VACV | 11.6 ± 0.5* | 2 ± 0 | 145 ± 11 | 53.7 ± 19.8 |
| ACV | 8.8 ± 2.6 | 12 ± 3 | 373 ± 97 | 37.3 ± 6.2 |
| VACV/ACV(%) | 131 ± 3 | 17 ± 2*** | 34.2 ± 9.3 | 144 ± 1*** |
p ≤ 0.05
p ≤ 0.001, as compared to wildtype mice for tde same route of administration, using an unpaired t-test.
After oral dosing of valacyclovir, the plasma concentration - time profiles of acyclovir were quite similar between the two genotypes (Fig. 5B). Accordingly, the Cmax of acyclovir was about 20% greater but the AUC0–180 only 4% greater in Bphl knockout mice (Table 3). Small (20%) reductions were also observed for the Tmax and T½ values of acyclovir during Bphl ablation. On the other hand, larger differences were noted between genotypes for valacyclovir in which the Cmax and AUC0–180 were about 50 and 70% greater, respectively, in Bphl knockout mice as compared to wildtype animals. Although no change was found in the Tmax of valacyclovir, a small 25% reduction was observed in the T½ of prodrug during Bphl ablation. Interestingly, the ratio of AUC0–180 for prodrug to active parent drug (i.e., VACV/ACV) was 70% greater after oral dosing in Bphl knockout mice. In contrast, the AUC ratio differed by only 8% between genotypes after intravenous dosing.
Collectively, these findings demonstrate that, in the absence of BPHL, other enzymes are available to convert valacyclovir to acyclovir in vivo and that the conversion is more efficient after oral than intravenous administration of prodrug.
4. Discussion
Biphenyl hydrolase-like protein (BPHL) is a novel human serine hydrolase that was originally cloned from a breast carcinoma cDNA library [28] and, because of its ability to catalyze the hydrolytic activation of valacyclovir (to acyclovir), referred to as valacyclovirase (VACVase). VACVase, initially purified from rat liver, was reported to be a basic protein that associated intracellularly with mitochondria [38]. This enzyme was further characterized by studying its hydrolysis kinetics for six amino acid esters of acyclovir, including that of valacyclovir. A human valacyclovirase was later identified from the human colon carcinoma cell line, Caco-2 [18], after which the human gene was cloned and a crystal structure characterized [30]. Although several studies investigated the hydrolytic activation for a broad range of amino acid ester nucleoside prodrugs by BPHL [18,31,39], no study has yet evaluated the in vivo exclusivity of this process. Indeed, a number of esterases/proteases may mediate the hydrolysis of valacyclovir and similar prodrugs.
In this study, the in situ intestinal permeability and stability of valacyclovir were evaluated during jejunal perfusions, and the in vivo absorption and disposition of valacyclovir (and acyclovir) evaluated during oral and intravenous bolus dosing of prodrug in wildtype and Bphl knockout mice. The use of transgenic mice, in which the Bphl gene was ablated, offers a unique opportunity to test the relative importance of BPHL in hydrolyzing valacyclovir, as compared to other possible enzymes. Major findings of this study revealed, for the first time, that: 1) no obvious phenotype was observed for Bphl knockout mice; 2) enzymatic upregulation of Cyp3a11, Cyp3a16, Abhd14a and Abhd14b was observed in some tissues of Bphl knockout mice; 3) Bphl ablation had no effect on the jejunal permeability of valacyclovir in mice; 4) valacyclovir was stable during intestinal perfusions and, following absorption, was found mostly as acyclovir (>75%) in portal vein blood; 5) valacyclovir was rapidly and efficiently hydrolyzed to acyclovir in vivo in the absence of BPHL; and 6) hydrolysis was more extensive after the oral vs. intravenous route of administration (for both genotypes), as judged by the 10- to 20-fold higher AUC0–180 values of acyclovir to valacyclovir after oral dosing as compared to the only 2- to 3-fold higher AUC0–180 values of acyclovir to valacyclovir after intravenous dosing. Taken as a whole, BPHL is not obligatory for the in vivo conversion of valacyclovir prodrug to its active parent drug acyclovir.
Assuming that BPHL was solely responsible for valacyclovir to acyclovir hydrolysis, one would have expected substantial differences between wildtype and Bphl knockout mice in the systemic exposure of valacyclovir and acyclovir following oral and intravenous administrations of prodrug. Although some differences in AUC0–180 were noted, none of the changes achieved a level of statistical significance. For example, after oral administration of prodrug, the AUC0–180 of valacyclovir was increased 70%, the AUC0–180 of acyclovir was essentially unchanged, and the VACV/ACV ratio was increased 70% in Bphl knockout mice. In contrast, after intravenous administration of prodrug, the AUC0–180 of valacyclovir was increased 30%, the AUC0–180 of acyclovir was increased 40%, and the VACV/ACV ratio was essentially unchanged in Bphl knockout mice. Thus, during BPHL ablation, the hydrolysis of valacyclovir to acyclovir was less extensive following oral dosing, suggesting that presystemic metabolism of valacyclovir was more important than systemic metabolism of prodrug. This finding is corroborated by our in situ jejunal perfusion results in mice showing that, as early as 5 min, < 10% of valacyclovir remains intact in portal vein blood, and by studies in humans showing that valacyclovir is nearly completely converted to acyclovir by first-pass intestinal and/or hepatic metabolism [40].
Ces1 and Ces2 are mouse carboxylesterases that are expressed in the liver (CES1 predominates in human) and small intestine (contains CES2 alone in human), and hydrolyze ester prodrugs, including the antiviral substrates valacyclovir and oseltamivir [41]. Cyp3a11, Cyp3a16, Cyp3a41 and Cyp3a44 are mouse orthologs of the human CPY3A4/5 genes that are highly expressed in both small intestine and liver, and account for the metabolism in over 50% of clinically used drugs [42]. Abhd14a and Abhd14b are mouse orthologs of the human hydrolases (ABHD14A/B) that are expressed about equally in most human tissues, with lowest expressions in brain, testis and placenta [12]. They are believed to have some role in transcriptional initiation and have been shown to hydrolyze the model substrate p-nitrophenyl butyrate. Although the present study design did not allow us to identify which additional enzymes were involved in the hydrolysis of valacyclovir during BPHL ablation, some compensatory (upregulatory) mechanisms were noted in Bphl knockout mice. For instance, gene expression was significantly increase d for Cyp3a16 in the duodenum and liver, and for Abhd14a in the duodenum, ileum and kidney of mice lacking the Bphl gene. More prevalent changes were observed for Abhd14b in which gene expression was significantly increased in the duodenum, ileum, colon, liver, kidney and spleen of Bphl knockout mice. Although speculative, the increased exposure of ACV in Bphl knockout mice, after intravenous administration of VACV, might be the result of significant upregulation of Abhd14a/b in the liver, kidney and spleen (Figs 3K and 3L).
In regard to the proton-coupled oligopeptide transporters, PepT1 was upregulated in the colon of Bphl knockout mice. However, this change is unlikely to have a significant impact on the in vivo pharmacokinetic results because valacyclovir is almost entirely absorbed in the small intestine [43]. Significant changes in the gene expression of PhT1 and PhT2 are also unlikely to impact the absorption of valacyclovir since these transporters are not expressed at the apical membrane of small intestinal epithelia but are, instead, located intracellularly [44].
In conclusion, the in situ intestinal permeability and stability of valacyclovir, and in vivo absorption and disposition of valacyclovir (and acyclovir) were evaluated after oral and intravenous administrations in wildtype and Bphl knockout mice. Whereas the jejunal permeability of valacyclovir was unchanged between the two genotypes, rapid and extensive hydrolysis of prodrug was still evident in mice even during Bphl gene deletion. Thus, BPHL is not obligatory for the in vivo conversion of valacyclovir to acyclovir either presystemically or systemically. The relevance of the upregulation of mouse Abhd14a and/or Abhd14b hydrolases in small intestinal segments, liver, kidney and spleen is currently unknown but deserves further attention.
Acknowledgments
This work was supported by the National Institutes of Health National Institute of General Medical Sciences grant R01GM115481 (to DES).
Abbreviations:
- ABHD14A
alpha/beta hydrolase domain containing 14a
- ABHD14B
alpha/beta hydrolase domain containing 14b
- ACV
acyclovir
- Bphl
biphenyl hydrolase-like protein
- KO
knockout
- PepT1
peptide transporter 1 (SLC15a1)
- PepT2
peptide transporter 2 (SLC15a2)
- PhT1
peptide/histidine transporter 1 (SLC15a4)
- PhT2
peptide/histidine transporter 2 (SLC15a3)
- POT
proton-couple oligopeptide transporter
- SLC
solute carrier family
- VACV
valacyclovir
- WT
wildtype
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflicts of interest, financial or otherwise.
References
- [1].Muller CE, Prodrug approaches for enhancing the bioavailability of drugs with low solubility, Chem Biodivers 6(11) (2009) 2071–83. [DOI] [PubMed] [Google Scholar]
- [2].Jornada DH, dos Santos Fernandes GF, Chiba DE, de Melo TR, dos Santos JL, Chung MC, The Prodrug Approach: A Successful Tool for Improving Drug Solubility, Molecules 21(1) (2015) 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Song X, Lorenzi PL, Landowski CP, Vig BS, Hilfinger JM, Amidon GL, Amino acid ester prodrugs of the anticancer agent gemcitabine: synthesis, bioconversion, metabolic bioevasion, and hPEPT1-mediated transport, Mol Pharm 2(2) (2005) 157–67. [DOI] [PubMed] [Google Scholar]
- [4].Landowski CP, Vig BS, Song X, Amidon GL, Targeted delivery to PEPT1-overexpressing cells: acidic, basic, and secondary floxuridine amino acid ester prodrugs, Mol Cancer Ther 4(4) (2005) 659–67. [DOI] [PubMed] [Google Scholar]
- [5].Tsume Y, Amidon GL, Selection of suitable prodrug candidates for in vivo studies via in vitro studies; the correlation of prodrug stability in between cell culture homogenates and human tissue homogenates, J Pharm Pharm Sci 15(3) (2012) 433–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tarasenko N, Cutts SM, Phillips DR, Berkovitch-Luria G, Bardugo-Nissim E, Weitman M, Nudelman A, Rephaeli A, A novel valproic acid prodrug as an anticancer agent that enhances doxorubicin anticancer activity and protects normal cells against its toxicity in vitro and in vivo, Biochem Pharmacol 88(2) (2014) 158–68. [DOI] [PubMed] [Google Scholar]
- [7].Gabizon AA, Tzemach D, Horowitz AT, Shmeeda H, Yeh J, Zalipsky S, Reduced toxicity and superior therapeutic activity of a mitomycin C lipid-based prodrug incorporated in pegylated liposomes, Clin Cancer Res 12(6) (2006) 1913–20. [DOI] [PubMed] [Google Scholar]
- [8].Murdter TE, Friedel G, Backman JT, McClellan M, Schick M, Gerken M, Bosslet K, Fritz P, Toomes H, Kroemer HK, Sperker B, Dose optimization of a doxorubicin prodrug (HMR 1826) in isolated perfused human lungs: low tumor pH promotes prodrug activation by beta-glucuronidase, J Pharmacol Exp Ther 301(1) (2002) 223–8. [DOI] [PubMed] [Google Scholar]
- [9].Romanski M, Urbaniak B, Kokot Z, Glowka FK, Activation of Prodrug Treosulfan at pH 7.4 and 37 degrees C Accompanied by Hydrolysis of Its Active Epoxides: Kinetic Studies with Clinical Relevance, J Pharm Sci 104(12) (2015) 4433–4442. [DOI] [PubMed] [Google Scholar]
- [10].Swift LP, Cutts SM, Rephaeli A, Nudelman A, Phillips DR, Activation of adriamycin by the pH-dependent formaldehyde-releasing prodrug hexamethylenetetramine, Mol Cancer Ther 2(2) (2003) 189–98. [PubMed] [Google Scholar]
- [11].Tougou K, Nakamura A, Watanabe S, Okuyama Y, Morino A, Paraoxonase has a major role in the hydrolysis of prulifloxacin (NM441), a prodrug of a new antibacterial agent, Drug Metab Dispos 26(4) (1998) 355–9. [PubMed] [Google Scholar]
- [12].Long JZ, Cravatt BF, The metabolic serine hydrolases and their functions in mammalian physiology and disease, Chem Rev 111(10) (2011) 6022–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Landowski CP, Lorenzi PL, Song X, Amidon GL, Nucleoside ester prodrug substrate specificity of liver carboxylesterase, J Pharmacol Exp Ther 316(2) (2006) 572–80. [DOI] [PubMed] [Google Scholar]
- [14].Peauger L, Azzouz R, Gembus V, Tintas ML, Sopkova-de Oliveira Santos J, Bohn P, Papamicael C, Levacher V, Donepezil-Based Central Acetylcholinesterase Inhibitors by Means of a “Bio-Oxidizable” Prodrug Strategy: Design, Synthesis, and in Vitro Biological Evaluation, J Med Chem 60(13) (2017) 5909–5926. [DOI] [PubMed] [Google Scholar]
- [15].Balimane PV, Tamai I, Guo A, Nakanishi T, Kitada H, Leibach FH, Tsuji A, Sinko PJ, Direct evidence for peptide transporter (PepT1)-mediated uptake of a nonpeptide prodrug, valacyclovir, Biochem Biophys Res Commun 250(2) (1998) 246–51. [DOI] [PubMed] [Google Scholar]
- [16].Ganapathy ME, Huang W, Wang H, Ganapathy V, Leibach FH, Valacyclovir: a substrate for the intestinal and renal peptide transporters PEPT1 and PEPT2, Biochem Biophys Res Commun 246(2) (1998) 470–5. [DOI] [PubMed] [Google Scholar]
- [17].Yang B, Hu Y, Smith DE, Impact of peptide transporter 1 on the intestinal absorption and pharmacokinetics of valacyclovir after oral dose escalation in wild-type and PepT1 knockout mice, Drug Metab Dispos 41(10) (2013) 1867–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kim I, Chu XY, Kim S, Provoda CJ, Lee KD, Amidon GL, Identification of a human valacyclovirase: biphenyl hydrolase-like protein as valacyclovir hydrolase, J Biol Chem 278(28) (2003) 25348–56. [DOI] [PubMed] [Google Scholar]
- [19].Drozdzik M, Groer C, Penski J, Lapczuk J, Ostrowski M, Lai Y, Prasad B, Unadkat JD, Siegmund W, Oswald S, Protein abundance of clinically relevant multidrug transporters along the entire length of the human intestine, Mol Pharm 11(10) (2014) 3547–55. [DOI] [PubMed] [Google Scholar]
- [20].Hu Y, Smith DE, Species differences in the pharmacokinetics of cefadroxil as determined in wildtype and humanized PepT1 mice, Biochem Pharmacol 107 (2016) 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hu Y, Xie Y, Wang Y, Chen X, Smith DE, Development and characterization of a novel mouse line humanized for the intestinal peptide transporter PEPT1, Mol Pharm 11(10) (2014) 3737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hu Y, Chen X, Smith DE, Species-dependent uptake of glycylsarcosine but not oseltamivir in Pichia pastoris expressing the rat, mouse, and human intestinal peptide transporter PEPT1, Drug Metab Dispos 40(7) (2012) 1328–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hu Y, Smith DE, Ma K, Jappar D, Thomas W, Hillgren KM, Targeted disruption of peptide transporter Pept1 gene in mice significantly reduces dipeptide absorption in intestine, Mol Pharm 5(6) (2008) 1122–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dai T, Li N, Zhang L, Zhang Y, Liu Q, A new target ligand Ser-Glu for PEPT1-overexpressing cancer imaging, Int J Nanomedicine 11 (2016) 203–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Foley D, Pieri M, Pettecrew R, Price R, Miles S, Lam HK, Bailey P, Meredith D, The in vitro transport of model thiodipeptide prodrugs designed to target the intestinal oligopeptide transporter, PepT1, Org Biomol Chem 7(18) (2009) 3652–6. [DOI] [PubMed] [Google Scholar]
- [26].Schellmann N, Deckert PM, Bachran D, Fuchs H, Bachran C, Targeted enzyme prodrug therapies, Mini Rev Med Chem 10(10) (2010) 887–904. [DOI] [PubMed] [Google Scholar]
- [27].Walther R, Rautio J, Zelikin AN, Prodrugs in medicinal chemistry and enzyme prodrug therapies, Adv Drug Deliv Rev (2017). [DOI] [PubMed]
- [28].Puente XS, Lopez-Otin C, Cloning and expression analysis of a novel human serine hydrolase with sequence similarity to prokaryotic enzymes involved in the degradation of aromatic compounds, J Biol Chem 270(21) (1995) 12926–32. [DOI] [PubMed] [Google Scholar]
- [29].Lawson WB, Specificity in the alkylation of serine at the active site of alpha-chymotrypsin by aromatic alpha-bromo amides, Biochemistry 19(10) (1980) 2140–4. [DOI] [PubMed] [Google Scholar]
- [30].Lai L, Xu Z, Zhou J, Lee KD, Amidon GL, Molecular basis of prodrug activation by human valacyclovirase, an alpha-amino acid ester hydrolase, J Biol Chem 283(14) (2008) 9318–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kim I, Song X, Vig BS, Mittal S, Shin HC, Lorenzi PJ, Amidon GL, A novel nucleoside prodrug-activating enzyme: substrate specificity of biphenyl hydrolase-like protein, Mol Pharm 1(2) (2004) 117–27. [DOI] [PubMed] [Google Scholar]
- [32].Marsillach J, Suzuki SM, Richter RJ, McDonald MG, Rademacher PM, MacCoss MJ, Hsieh EJ, Rettie AE, Furlong CE, Human valacyclovir hydrolase/biphenyl hydrolase-like protein is a highly efficient homocysteine thiolactonase. PLoS One 9 (10) (2014) e110054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jappar D, Wu SP, Hu Y, Smith DE, Significance and regional dependency of peptide transporter (PEPT) 1 in the intestinal permeability of glycylsarcosine: in situ single-pass perfusion studies in wild-type and Pept1 knockout mice, Drug Metab Dispos 38(10) (2010) 1740–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hu Y, Song F, Jiang H, Nunez G, Smith DE, SLC15A2 and SLC15A4 mediate the transport of bacterially derived di/tripeptides to enhance the nucleotide-binding oligomerization domain-dependent immune response in mouse bone marrow-derived macrophages, J Immunol (2018) [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- [35].Hsieh PW, Hung CF, Fang JY, Current prodrug design for drug discovery, Curr Pharm Des 15(19) (2009) 2236–50. [DOI] [PubMed] [Google Scholar]
- [36].Epling D, Hu Y, Smith DE, Evaluating the intestinal and oral absorption of the prodrug valacyclovir in wildtype and huPepT1 mice, Biochem Pharmacol (2018) [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- [37].Shi J, Hu Y, Smith DE, Zhu HJ, A sensitive liquid chromatographt-tandem mass spectrometry method for the quantification of valacyclovir and ite metabolite acyclovir in mouse and human plasma, J Chromatogr B (2018) [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- [38].Burnette TC, Harrington JA, Reardon JE, Merrill BM, de Miranda P, Purification and characterization of a rat liver enzyme that hydrolyzes valaciclovir, the L-valyl ester prodrug of acyclovir, J Biol Chem 270(26) (1995) 15827–31. [DOI] [PubMed] [Google Scholar]
- [39].Tao W, Zhao D, Sun M, Li M, Zhang X, He Z, Sun Y, Sun J, Enzymatic activation of double-targeted 5’-O-L-valyl-decitabine prodrug by biphenyl hydrolase-like protein and its molecular design basis, Drug Deliv Transl Res 7(2) (2017) 304–311. [DOI] [PubMed] [Google Scholar]
- [40].Valtrex product monograph GlaxoSmithKline Inc, Ontario, Canada, https://ca.gsk.com/media/593038/valtrex.pdf (2015). [Google Scholar]
- [41].Laizure SC, Herring V, Hu Z, Witbrodt K, Parker RB, The role of human carboxylesterases in drug metabolism: have we overlooked their importance?, Pharmacotherapy 33(2) (2013) 210–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gonzalez FJ, Coughtrie M, Tukey RH, Drug metabolism. Goodman and Gilman’s The Pharmacological Basis of Therapeutics, 13 ed., Hill McGraw, New York, 2018. pp. 85–100. [Google Scholar]
- [43].Yang B, Smith DE, In silico absorption analysis of valacyclovir in wildtype and Pept1 knockout mice following oral dose escalation, Pharm Res 34(11) (2017) 2349–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Smith DE, Clemencon B, Hediger MA, Proton-coupled oligopeptide transporter family SLC15: physiological, pharmacological and pathological implications, Mol Aspects Med 34(2–3) (2013) 323–36. [DOI] [PMC free article] [PubMed] [Google Scholar]