

The largest outbreak of MARV occurred in Angola in 2004 to 2005 and had a 90% case fatality rate. There are no approved treatments available for MARV. Development of antivirals as therapeutics requires a fundamental understanding of the viral life cycle. Because of the close similarity of MARV to another member of Filoviridae family, EBOV, it was assumed that the two viruses have similar mechanisms of regulation of transcription and replication. Here, characterization of the role of VP30 and its phosphorylation sites in transcription of the MARV genome demonstrated differences from those of EBOV. The identified phosphorylation sites appeared to inhibit transcription and appeared to be involved in interaction with both NP and VP35 ribonucleoproteins. A small molecule targeting PP1 inhibited transcription of the MARV genome, effectively suppressing replication of the viral particles. These data demonstrate the possibility developing antivirals based on compounds targeting PP1.
KEYWORDS: antiviral, filovirus, Marburg virus, phosphatase, protein phosphorylation, replication, transcription
ABSTRACT
The filoviruses Marburg virus (MARV) and Ebola virus (EBOV) cause hemorrhagic fever in humans and nonhuman primates, with high case fatality rates. MARV VP30 is known to be phosphorylated and to interact with nucleoprotein (NP), but its role in regulation of viral transcription is disputed. Here, we analyzed phosphorylation of VP30 by mass spectrometry, which resulted in identification of multiple phosphorylated amino acids. Modeling the full-length three-dimensional structure of VP30 and mapping the identified phosphorylation sites showed that all sites lie in disordered regions, mostly in the N-terminal domain of the protein. Minigenome analysis of the identified phosphorylation sites demonstrated that phosphorylation of a cluster of amino acids at positions 46 through 53 inhibits transcription. To test the effect of VP30 phosphorylation on its interaction with other MARV proteins, coimmunoprecipitation analyses were performed. They demonstrated the involvement of VP30 phosphorylation in interaction with two other proteins of the MARV ribonucleoprotein complex, NP and VP35. To identify the role of protein phosphatase 1 (PP1) in the identified effects, a small molecule, 1E7-03, targeting a noncatalytic site of the enzyme that previously was shown to increase EBOV VP30 phosphorylation was used. Treatment of cells with 1E7-03 increased phosphorylation of VP30 at a cluster of phosphorylated amino acids from Ser-46 to Thr-53, reduced transcription of MARV minigenome, enhanced binding to NP and VP35, and dramatically reduced replication of infectious MARV particles. Thus, MARV VP30 phosphorylation can be targeted for development of future antivirals such as PP1-targeting compounds.
IMPORTANCE The largest outbreak of MARV occurred in Angola in 2004 to 2005 and had a 90% case fatality rate. There are no approved treatments available for MARV. Development of antivirals as therapeutics requires a fundamental understanding of the viral life cycle. Because of the close similarity of MARV to another member of Filoviridae family, EBOV, it was assumed that the two viruses have similar mechanisms of regulation of transcription and replication. Here, characterization of the role of VP30 and its phosphorylation sites in transcription of the MARV genome demonstrated differences from those of EBOV. The identified phosphorylation sites appeared to inhibit transcription and appeared to be involved in interaction with both NP and VP35 ribonucleoproteins. A small molecule targeting PP1 inhibited transcription of the MARV genome, effectively suppressing replication of the viral particles. These data demonstrate the possibility developing antivirals based on compounds targeting PP1.
INTRODUCTION
Marburg virus (MARV) and Ebola virus (EBOV) are members of the family Filoviridae, order Mononegavirales, and cause highly lethal hemorrhagic fevers in human and nonhuman primates (1). Human outbreaks of MARV infection occur regularly in Central Africa; the largest and the most deadly outbreak was reported in Angola in 2004 to 2005 with a 90% case fatality rate (2). Currently, there are no approved vaccine or antiviral therapies for humans infected with MARV.
The MARV genome is a negative-sense single-stranded RNA of approximately 19 kb that encodes seven genes: NP (nucleoprotein), VP35, VP40, GP (glycoprotein), VP30, VP24, and L (RNA-dependent RNA polymerase) (3, 4). MARV VP30 together with NP, VP35, and L form the nucleocapsid complex. However, VP30 is dispensable for nucleocapsid formation (5, 6). Experiments with an EBOV minigenome demonstrated a VP30 requirement for transcriptional activity (7). In contrast, in an MARV minigenome system, NP, VP35, and L were sufficient to achieve viral transcription and replication, rendering VP30 nonessential (8–11). On the other hand, RNA interference (RNAi) knockdown of VP30 in MARV-infected cells strongly suppressed the expression of all viral proteins and reduced the release of progeny virus (12), pointing to an important role of VP30 in the MARV life cycle. Thus, the role of VP30 in the MARV life cycle remains unresolved.
We along with others previously demonstrated that EBOV VP30 is phosphorylated at six N-proximal serine residues at positions 29 to 31,42, 44, and 46 and at threonine residues at positions 143 and 146 (13, 14). Phosphorylation of VP30 blocks transcription of EBOV genes (13, 15). This blockade is accompanied by a shift of polymerase activity from transcription to replication (9, 14). Finally, we identified the role of cellular protein phosphatase 1 (PP1) in dephosphorylation of EBOV VP30 and demonstrated that a small-molecule compound, 1E7-03, targeting PP1 increases VP30 phosphorylation and strongly inhibits replication of EBOV (14).
Investigation of the effects of mutations of putative phosphorylation sites in plasmid-expressed MARV VP30 on its total phosphorylation resulted in identification of several phosphorylation sites, most of which are located in the serine-rich region spanning amino acids 40 to 51. Some of the identified phosphorylation sites were demonstrated to be shared with EBOV VP30 (5). The role of MARV VP30 phosphorylation in viral transcription, however, remained unexplored. In this study, we performed a comprehensive analysis of MARV VP30 phosphorylation using high-resolution mass spectrometry (MS). We investigated the role of VP30 in viral transcription and determined the role of the identified phosphorylation sites. We also tested the PP1-targeting compound 1E7-03 for the inhibition of MARV minigenome, for its effect on MARV VP30 phosphorylation in cultured cells, and for replication of MARV particles. We found that the effect of MARV VP30 phosphorylation on transcription activity of viral polymerase is different from the role of phosphorylation of EBOV VP30. We also found that 1E7-03 effectively inhibits replication of MARV particles.
RESULTS
Dynamic phosphorylation of the N-terminal part of MARV VP30.
We analyzed phosphorylation of vector-expressed VP30 and VP30 in MARV viral particles using mass spectrometry. Flag-tagged MARV VP30 was expressed in 293T cells, immunoprecipitated from cellular lysates, and resolved by 10% SDS-PAGE (Fig. 1A, lane 1). Previously, we observed a strong increase in EBOV VP30 phosphorylation in cells treated with the PP1-targeting compound 1E7-03 or a pan-phosphatase inhibitor, okadaic acid (14). Thus, we also analyzed MARV VP30 phosphorylation in 293T cells treated with 1E7-03 or okadaic acid (Fig. 1A, lanes 2 and 3). Interestingly, either treatment resulted in a reduced level of VP30 that can be associated with downregulation of RNA polymerase II activity (16–18), which drives the expression of VP30 from the cytomegalovirus promoter. High-resolution liquid chromatography-tandem MS (LC-MS/MS) analysis of tryptic peptides extracted from the gel identified 47% of MARV VP30 protein sequence excluding the N-terminal part containing a Flag tag and the C-terminal part (Fig. 1B, green and red indicate peptides identified with high and low confidence, respectively, and the N-terminal Flag tag sequence is italicized). Analysis of posttranslational modifications using Proteome Discover, version 1.4, identified phosphorylation of Ser-46, Ser-47, Ser-50, Ser-51, Thr-53, Ser-63, Thr-64, Ser-68, Thr-102, Thr-172, and Ser-173 (Fig. 1B, indicated with asterisks); the corresponding peptides are listed in Fig. 1C. The MS/MS spectra of peptides containing representative phosphorylated residues Ser-47 and Ser-63 and a nonphosphorylated peptide (residues 186 to 196) are shown in Fig. 1D to F.
FIG 1.

Analysis of MARV VP30 phosphorylation by LC-MS/MS. (A) Purification of Flag-tagged MARV VP30 for LC-MS/MS analysis. Cells were transfected with a VP30-expressing plasmid, untreated (lane 1) or treated with 10 μM 1E7-03 overnight (lane 2) or 0.1 μM okadaic acid (OA) for 2 h (lane 3). Lane 4, molecular weight (MW) markers. The arrow indicates the position of VP30. (B) MS/MS analysis of MARV VP30 conducted with Proteome Discoverer, version 1.4, and the SEQUEST search engine. Peptides were in-gel digested with trypsin, eluted, and subjected to MS analysis on a Thermo linear trap quadrupole (LTQ) Orbitrap XL mass spectrometer. Shown is the sequence of VP30. Peptides identified with high and low probability are shown in green and blue, respectively, and the phosphorylated residues are indicated with asterisks. (C) Identified VP30 phosphopeptides. (D to F) MS/MS spectra of selected phosphorylated and nonphosphorylated VP30 peptides: SsAESSPTNHIPR, ARPPsTFNLSKPPPPPK, and AALSLTcAGIR, as indicated. Lowercase letters indicate phosphorylation (s) or alkylation (c). Color peaks indicate matched MS/MS fragments, where green indicates precursors, and blue and red indicate y and b ions, respectively.
To analyze phosphorylation of VP30 in MARV particles, we propagated the virus in 293T cells and purified samples in a sucrose gradient. Viral preparations were inactivated by heating at 95°C in 4% SDS for 15 min, and MARV proteins were analyzed by 10% SDS-PAGE (Fig. 2A). The protein gel was cut into equal fragments, and peptides were extracted after in-gel trypsinization and analyzed by high-resolution LC-MS/MS as described above. We detected VP30 with 57% coverage (Fig. 2B) and identified 12 phosphorylated serine and threonine residues (Fig. 2C), including Ser-8, Ser-63, Thr-64, Ser-68, Thr-118, Ser-130, Ser-132, Ser-134, Thr-172, Ser-173, Ser-179, and Thr-181. The MS/MS spectra of peptides containing representative phosphorylated residues Ser-8, Ser-63, and Ser-130 are shown in Fig. 2D to F. Interestingly, the identified phosphorylation sites in MARV virion-associated VP30 only partially overlapped those in plasmid-expressed VP30 as a cluster of amino acids from Ser-46 to Thr-53 was not phosphorylated. These data suggested that the cluster is dynamically phosphorylated, as recently described for the cluster of N-terminal amino acids in EBOV VP30 (19).
FIG 2.

Analysis of MARV virion-associated VP30 phosphorylation by LC-MS/MS. (A) Purification of MARV for LC-MS/MS analysis. (B) MS/MS analysis of MARV VP30 conducted with Proteome Discoverer, version 1.4, and the SEQUEST search engine. Peptides were in-gel digested with trypsin, eluted, and subjected to MS analysis on a Thermo LTQ Orbitrap XL mass spectrometer. Shown is the sequence of VP30. Peptides identified with high and median probabilities are shown in green and blue, respectively, and the phosphorylated residues are shown with black asterisks. (C) Identified VP30 phosphopeptides. (D to F) MS/MS spectra of representative phosphorylated peptides from virion-derived MARV VP30: MQQPRHRsR, ARPPsTFNLSKPPPPPK, and SPQDcGsPSLSK, as indicated. Lowercase letters indicate phosphorylation (s) or alkylation (c). Color peaks indicate matched MS/MS fragments, whereby green indicates precursors, and blue and red indicate y and b ions, respectively.
Comparison of VP30 sequences across filoviruses (Fig. 3) demonstrated that except for Ser-50, all the identified phosphorylation residues are conserved in the Ravn virus, which is the only other member, in addition to MARV, of the genus Marburgvirus. However, among the other filoviruses, including EBOV, which belong to the genus Ebolavirus, only one site appeared to be conserved.
FIG 3.

Comparison of MARV VP30 (GenBank accession number P35258) with Ravn virus VP30 (RAVV) (Q1PDC6), EBOV VP30 (EBOV) (Q05323), human respiratory syncytial virus M2-1 (HRSV) (P04545), and human metapneumovirus M2-1 (HMPV) (Q8QN58) proteins. Multiple-sequence alignment was done using Clustal Omega. The blue box in the top group of sequences indicates a region rich in arginines and lysines unique for filoviruses, which supports the role of this protein as a transcription antitermination factor. The red box in the second group indicates a zinc finger motif, and the blue box in this group indicates another region rich in arginines and lysines, followed by a leucine-rich region which indicates its role as an oligomerization domain. Note that two leucines are also conserved in RSV and HMPV. The common core roughly corresponds to residues 132 to 253 of MARV VP30. The symbols indicate various levels of sequence conservation: asterisk, fully conserved; semicolon, conserved between groups of amino acids with strongly similar properties; period, conserved between groups of amino acids with weak similarity. Dashes indicate gaps in sequence alignment. The phosphorylated residues identified in the current study are shown in brown.
Small-molecule targeting of protein phosphatase 1 increases phosphorylation of MARV VP30.
To determine if treatment with PP1 inhibitors, a PP1-targeting compound 1E7-03 and okadaic acid, increases phosphorylation of MARV VP30, we quantified the detected phosphopeptides using SIEVE, version 2.1, software that performs label-free quantification. Nonphosphorylated peptide AALSLTcAGIR (in which the lowercase “c” indicates alkylation) was used for normalization (Fig. 4A) and phosphopeptides ARPPsTFNLSKPPPPPK and SsAESSPTNHIPR (in which the lowercase “s” indicates phosphorylated residues) were quantified (Fig. 4B and C). Treatment of cells with 1E7-03 or okadaic acid significantly increased the levels of SsAESSPTNHIPR and decreased the levels of ARPPsTFNLSKPPPPPK (Fig. 4D). These data demonstrate that small-molecule targeting of PP1 induced phosphorylation in the cluster of phosphorylated amino acids from Ser-46 to Thr-53 of MARV VP30.
FIG 4.
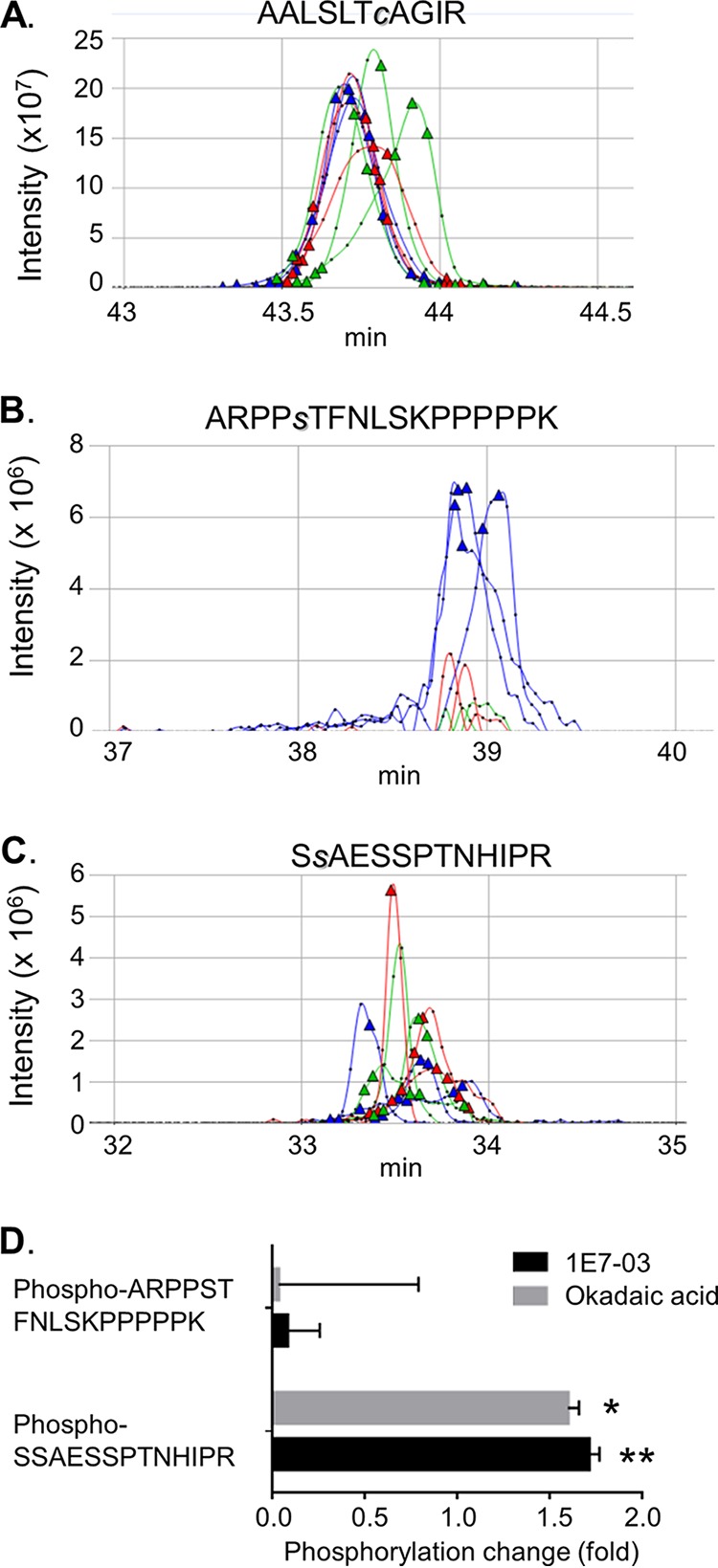
Treatment with phosphatase inhibitors increases phosphorylation of MARV VP30. (A to C) Quantitative analysis of MARV peptide expression using SIEVE, version 2.1. Ion elution profiles are shown in blue for control samples, in red for the cells treated with 1E7-03, and in green for the cells treated with okadaic acid. The AALSLTcAGIR peptide was used for normalization. Triangles indicate the time points at which MS/MS was conducted. Representative data from three independent experiments are shown. (D) Ratios of the ion peaks for ARPPsTFNLSKPPPPPK and SsAESSPTNHIPR peptides in 1E7-03- or okadaic acid-treated cells to that in nontreated cells. Values are means ± standard deviations based on three samples. Peaks were integrated, and a P value was calculated using SIEVE, version 2.1. *, P = 8.7 × 10−15; **, P = 4.3 × 10−11.
Most of the identified VP30 phosphorylation sites are located in disordered regions.
The full-length three-dimensional structure of MARV VP30 has not been experimentally determined. Only the structures of the C-terminal domains of MARV VP30 (PDB accession number 5T3W), EBOV VP30 (residues 142 to 266, PDB accession number 2I8B; residues 142 to 271, PDB 5DVW), and Ebola virus Reston VP30 (residues 142 to 266, PDB accession number 3V7O) (20, 21) have been solved. For structural insight and a fuller understanding of the implication of the identified MARV VP30 phosphorylation sites, we modeled the structure of full-length (281 amino acids) MARV VP30 protein (GenBank accession number YP_001531157) using the Rosetta/Robetta protocols (22). The constructed three-dimensional structure demonstrated that VP30 comprises two distinct domains, residues 1 to 129 and residues 130 to 281 (Fig. 5, gold). Fine-mapping revealed structured domains alternating with unstructured domains. Most of the N terminus, specifically, residues 1 to 18 and residues 26 to 72, was found to be intrinsically disordered. This region was found to be followed by an ordered region (residues 75 to 115). These two subdomains form domain 1. A disordered region at residues 116 to 148 was found to connect domain 1 to the C-terminal domain 2. Domain 2 was found to be mostly structured but with three short disordered fragments at residues 206 to 210, 272 to 274, and 276 to 281 imparting flexibility. Disorder mapping using RONN software (23) confirmed the results from Rosetta/Robetta (data not shown). Next, we superimposed the modeled structure of MARV VP30 on the crystal structure of the EBOV VP30 C-terminal domain (PDB accession number 2I8B) (Fig. 5, blue) for comparison. The superimposition showed a close similarity of the C-terminal domains of EBOV and MARV VP30. This common core is also shared by M2-1 transcription antiterminator proteins of human respiratory syncytial virus (HRSV) (PDB accession number 4C3D) and human metapneumovirus (HMPV) (PDB accession number 4CS7) (24, 25). Similar to findings for HRSV and HMPV, the domain was found to be rich in lysines and arginines to bind RNA. This was further supported by the presence of the zinc finger (ZnF) domain common to these proteins (Fig. 3). The N terminus of MARV VP30, which was found to be disordered, is likely to be engaged in interaction with RNA and is accessible for phosphorylation.
FIG 5.
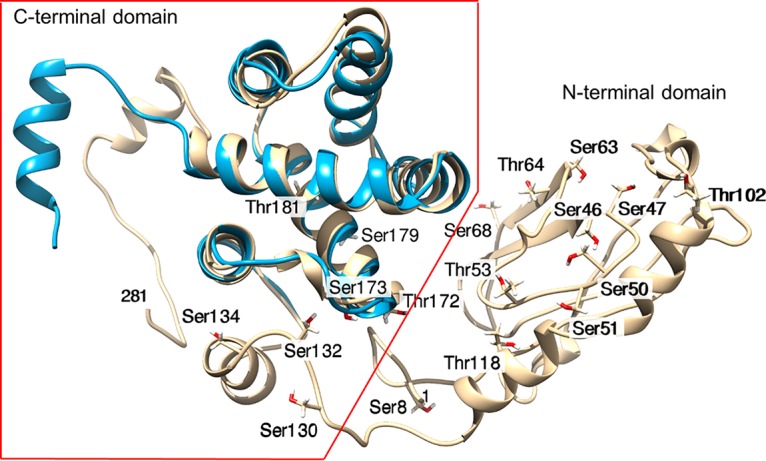
Mapping of the identified phosphorylation sites on the modeled three-dimensional structure of MARV VP30. The modeled full-length structure of MARV VP30 is shown in gold. The molecule comprises the unstructured N-terminal half and the ordered C-terminal half. The indicated phosphorylated residues show oxygen atoms in red. The superimposed structure of the C-terminal half of EBOV VP30 is shown in cyan.
Mapping of the 18 identified phosphorylation sites to the three-dimensional structure showed that nearly all of the phosphorylation sites are located in the unstructured N-terminal domain: Ser-8 is in the flexible N terminus; Ser-46, Ser-47, Ser-50, Ser-51, and Thr-53 are in a random coil; Ser-63 is just before a β-strand; Thr-64 is in the β-strand; Ser-68 is in a coil immediately following the β-strand; and Thr-102 is in a coil (Fig. 5). Residues Thr-118, Ser-130, Ser-132, and Ser-134 lie in the long loop connecting to the ordered C-terminal domain, Thr-172 and Ser-173 are located on a short linker loop in the C-terminal domain, and Ser-179 and Thr-181 lie on ordered helices but are otherwise accessible (Fig. 5). Analysis of the identified phosphorylation sites for the relative solvent accessibility using a previously published approach (26) demonstrated that they are generally moderately to very accessible (Table 1). These values are consistent with the extended and flexible nature of the N terminus of MARV VP30.
TABLE 1.
Relative solvent-accessible surface area values of MARV VP30
| Positiona | Residue | rSASA valueb |
|---|---|---|
| 8 | Ser | 0.57 |
| 46 | Ser | 0.44 |
| 47 | Ser | 0.15 |
| 50 | Ser | 0.30 |
| 51 | Ser | 0.16 |
| 53 | Thr | 0.25 |
| 63 | Ser | 0.18 |
| 64 | Thr | 0.57 |
| 68 | Ser | 0.42 |
| 102 | Thr | 0.67 |
| 118 | Thr | 0.17 |
| 130 | Ser | 0.83 |
| 132 | Ser | 0.41 |
| 134 | Ser | 0.26 |
| 172 | Thr | 0.64 |
| 173 | Ser | 0.19 |
| 179 | Ser | 0.19 |
| 181 | Thr | 0.09 |
The majority (14 out of 18) of the identified phosphorylation sites are located in unstructured and flexible regions. Thr-181, which lies in the structured C-terminal domain inside a helical turn, is poorly exposed. Nevertheless, this, as well as all other phosphorylated residues, is solvent accessible. Ser-130 lies on a highly flexible loop and has a high relative solvent-accessible surface area (rSASA) value.
The average rSASA value based on all 18 phosphorylation sites is 0.36.
VP30 is required for effective transcription of MARV.
Previous studies demonstrated that only NP, VP35, and L are required for MARV transcription and replication whereas VP30 is dispensable (8–11). We explored these findings further by using an improved MARV minigenome that utilized a Renilla luciferase reporter gene together with codon-optimized support plasmids. This system shows a better signal-to-noise ratio and higher expression of the reporter gene than nonoptimized plasmids (27). First, the minigenome system was optimized to achieve the greatest activity of Renilla luciferase in the MARV minigenome by comparing at least three different concentrations for all support plasmids expressing NP, VP35, VP30, and L, as well as MARV minigenome and T7 polymerase. The Renilla luciferase activity was normalized to that of a firefly luciferase expressed from a cotransfected plasmid. As expected, the absence of either L or T7 or minigenome completely abolished MARV minigenome transcription in both Vero-E6 and 293T cells (Fig. 6A and B, graphs). Lack of a VP30 expression vector resulted in significant 27% and 33% reductions in MARV transcription in Vero-E6 and 293T cells, respectively. These data were further confirmed by analysis of Renilla luciferase protein expression by Western blotting, which demonstrated its reduced luciferase expression in the absence of VP30 (Fig. 6A and B, blots). Thus, VP30 is required for effective MARV transcription, but, in contrast to EBOV VP30, lack of MARV VP30 does not completely abolish transcription.
FIG 6.

VP30 is required for effective transcription. Luciferase signal in the absence of VP30 was normalized to that in the presence of VP30 in Vero-E6 (A) and 293T (B) cells. Top panels show minigenome analysis; bottom panels show Western blot analysis of Renilla luciferase expression. Values are means ± standard errors of the means based on triplicate samples. *, P < 0.05 for comparisons of results to those with the full set of plasmids (one-way analysis of variance and Dunnett's test). The experiments in panels A and B were performed three times, resulting in essentially similar data.
VP30 phosphorylation inhibits transcription of MARV genome.
We next determined the role of the identified VP30 phosphorylation sites in MARV polymerase activity. We used plasmid-expressed VP30 with an N-terminal Flag tag to replace the nine phosphorylation sites identified by MS (Ser-46, Ser-47, Ser-50, Ser-51, Thr-53, Ser-63, Thr-64, Ser-68, and Thr-102) with alanines. None of the substitutions affected VP30 expression (Fig. 7, bottom panel). Testing of the constructs in the minigenome system using luciferase signal demonstrated that alanine substitutions of Ser-46, Ser-47, Ser-50, Ser-51, and Thr-53 significantly increased (P < 0.05) MARV transcription by 40 to 70% (Fig. 7, top).
FIG 7.

VP30 dephosphorylation increases MARV transcription. At top, effects of alanine substitutions for Thr and Ser phosphorylation sites on minigenome activity are shown. Luciferase signals of mutated minigenomes were normalized to the signal of nonmutated minigenome (100%). Bottom panels show expression of the mutated VP30 constructs as analyzed by Western blotting. The VP30 Western blot image was spliced from two different gels run in parallel because of the limited number of samples which could be loaded on one gel and to place the samples in the order consistent with the discussion in the rest of the manuscript. The GAPDH Western blot image was spliced from two different gels run in parallel because of the limited number of samples which could be loaded on one gel. Values are means ± standard errors of the means based on triplicate samples. *, P < 0.05 for comparisons of results to those with the nonmutated (wild-type [WT]) VP30 (one-way analysis of variance and Dunnett's test). The experiment was performed at least three times, resulting in essentially similar data.
We next mimicked VP30 phosphorylation by generating VP30 aspartic acid mutants, S46D and S51D. None of the mutations affected VP30 expression or minigenome transcription (Fig. 8A). We also tested the effect of a cluster of alanine and aspartic acid mutations in the residues shown to affect transcription, Ser-46, Ser-47, Ser-50, Ser-51, and Thr-53 (hereafter designated cluster Ser-46–Thr-53), and residues previously shown to be critical for the interaction of VP30 with NP (5) Ser-40 and Ser-42 (cluster Ser-40/Ser-42). Consistently with the data on individual mutants, alanine mutants of both clusters (designated S46-T53A and S40/42A, respectively) resulted in a significant increase in transcription (Fig. 8B). In contrast, aspartic acid substitutions resulted in no effect (cluster Ser-40/Ser-42) or a significant reduction of transcription (cluster Ser-46–Thr-53) (Fig. 8B, S40/42D and S46-T53D, respectively). The minigenome analysis was repeated with increasing concentrations of VP30 wild type (WT) or the mutants (Fig. 8C). We observed a proportionate increase in transcription for the alanine mutants of both clusters but no effect for VP30 WT and no effect or a marginal reduction for the aspartic acid mutants. Taken together, these data suggest that phosphorylation of MARV VP30 inhibits transcription of the viral genome.
FIG 8.
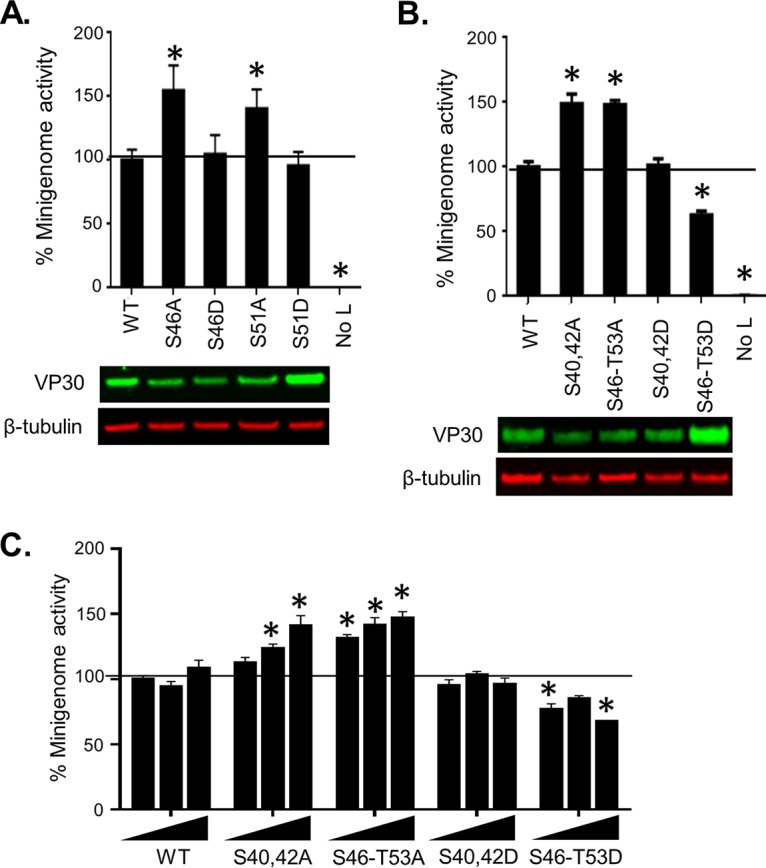
VP30 phosphorylation inhibits transcription. The effects of alanine and aspartic acid single (A) and cluster (B and C) substitutions of phosphorylated serine residues in VP30 on minigenome activity normalized to activity with nonmutated VP30 (100%) are shown in the graphs. For the experiments shown in panels A and B, a single VP30 concentration of 25 ng was used, whereas for the experiment shown in panel C, three different concentrations of VP30 (25, 50 and 75 ng) were used. Expression of the mutated VP30 constructs was analyzed by Western blotting (A and B, bottom panels). Values are means ± standard errors of the means based on triplicate samples. *, P < 0.05 for comparisons of results to those with the nonmutated (wild-type [WT]) VP30 (one-way analysis of variance and Dunnett's test). The experiments were performed three times, resulting in essentially similar data.
VP30 phosphorylation is necessary for its binding to NP and VP35.
Previously, phosphorylation of MARV VP30 Ser-40 and Ser-42 was shown to be critical for interaction of VP30 with NP (5). Here, we determined whether additional phosphorylated residues, Ser-46, Ser-47, Ser-50, Ser-51, and Thr-53, directly identified by MS and shown to be required for the viral polymerase activity in this study are also involved in NP interaction. We cotransfected 293T cells with NP and WT VP30 or the mutants S40/42A, S46-T53A, S40/42D, and S46-T53D and analyzed lysates by coimmunoprecipitation assays (Fig. 9A). Both sets of alanine substitutions decreased binding to NP while aspartic acid substitutions mimicking phosphorylation did not affect the interaction (Fig. 9B). These results suggest that VP30 phosphorylation facilitates its binding to NP. Since EBOV VP30 also binds to VP35 (9), we tested the binding of MARV VP30 mutants to VP35. WT VP30 or the mutants and VP35 bearing an influenza virus hemagglutinin tag (HA-VP35) were expressed in 293T cells, and their interaction was analyzed by coimmunoprecipitation assays (Fig. 9C). In contrast to EBOV where phosphorylated VP30 inhibited interaction with VP35 (9), both MARV VP30 alanine and aspartic acid substitutions significantly reduced VP30 interaction with VP35 (Fig. 9D). The reduction of binding associated with aspartic acid substitutions may suggest that a charged amino acid substitution does not fully mimic phosphorylation in the interaction with VP35. These data suggest that VP30 phosphorylation residues located at the clusters Ser-40/Ser-42 and Ser-46–Thr-53 are critical for VP30 binding to both NP and VP35. These data contrast with findings for EBOV for which phosphorylation of VP30 dissociates it from the polymerase complex due to disrupted interaction with VP35 (9).
FIG 9.
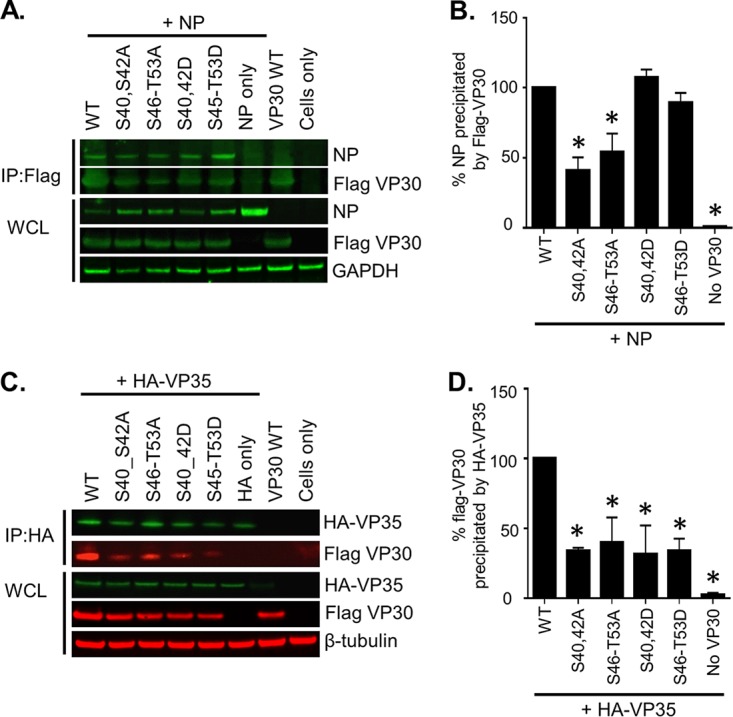
VP30 interacts with NP and VP35. Coimmunoprecipitation was performed of Flag-VP30 phosphorylation mutants with NP (A and B) or HA-VP35 (C and D). 293T cells expressing Flag-tagged VP30 constructs and NP or HA-tagged VP35 were lysed at 48 h posttransfection, and protein complexes were immunoprecipitated (IP) overnight at 4°C using mouse anti-Flag affinity gel or mouse anti-HA agarose. An aliquot for expression control (whole-cell lysate [WCL]) was collected from the cell lysate before precipitation. Western blot analysis performed with anti-NP and anti-Flag antibodies (A, green). (C) Western blotting was performed with anti-HA (green) and anti-Flag (red) antibodies visualized with a LiCor Odyssey Imaging System. Panels B and D show the quantification of immunoprecipitated Flag-VP30 and NP and of Flag-VP30 and HA-VP35, respectively, performed in Image Studio Lite software. Precipitation of VP30 WT by NP/HA-VP35 was set to 100%. Values are means ± standard errors of the means based on duplicate samples. *, P < 0.05 for comparisons of results to those with the nonmutated (wild-type [WT]) VP30 (one-way analysis of variance and Dunnett's test). The experiments were performed at least two times, resulting in essentially similar data.
Small-molecule targeting of phosphatase 1 decreases transcription of MARV genome.
We previously demonstrated that the PP1-targeting compound 1E7-03 increased phosphorylation of EBOV VP30 and shifted EBOV polymerase activity from transcription to replication with subsequent dramatic inhibition of viral replication (14). Here, we tested the effect of 1E7-03 on transcription of MARV minigenome. Treatment of Vero-E6 cells with 3 μM 1E7-03, which was determined to be noncytotoxic (14), resulted in a more than 50% decrease in luciferase signal (Fig. 10A). This effect of 1E7-03 is consistent with the effects of VP30 omission from the minigenome system, which showed a reduction of transcription (Fig. 6).
FIG 10.

Treatment with 1E7-03 reduces transcription by promoting binding of phosphorylated VP30 with NP and VP35. (A) Effects of 1E7-03 treatment on minigenome activity. The luciferase signal was normalized to activity in nontreated cells (100%). (B to E) Coimmunoprecipitation of Flag VP30 WT with NP or HA-VP35, as indicated. Western blot analysis with anti-NP and anti-Flag antibodies (green) (B) and with anti-HA (green) or anti-Flag (red) antibodies (D) was visualized with a Li-Cor Odyssey imaging system. Quantification of immunoprecipitated Flag-VP30 and NP and Flag-VP30 and HA-VP35 from the experiments shown in panels B and D, respectively, was performed in Image Studio Lite software. Precipitation of VP30 WT by NP/HA-VP35 was set to 100%. Values are means ± standard errors of the means based on duplicate samples. *, P < 0.05 for results compared to those with the DMSO control (one-way analysis of variance and Dunnett's test). The experiment shown in panel A was performed three times, and the experiments shown in panels B to E were performed at least two times, resulting in essentially similar data.
We have previously demonstrated that treatment of cells with 1E7-03 or okadaic acid significantly increases the levels of phosphorylated VP30 peptides (Fig. 4D). To confirm whether the interactions of VP30 with NP and VP35 are modulated by phosphorylation, we performed coimmunoprecipitation experiments on lysates transfected with VP30 and NP or VP35 in the presence of the PP1-targeting molecule 1E7-03, the phosphatase inhibitor okadaic acid, or dimethyl sulfoxide (DMSO). Both inhibitors caused a significant increase in VP30-NP (Fig. 10B and C) and VP30-VP35 interactions (Fig. 10D and E). These data support our previous findings, which showed that alanine substitutions reduce interactions with NP and VP35 (Fig. 9). Prior studies with EBOV VP30 demonstrated a similar increase in interaction of phosphorylated VP30 with NP; however, they also demonstrated a decreased interaction with VP35 which results in a switch from transcription to genome replication (9). Thus, phosphorylated VP30 remains bound to the nucleocapsid by strengthening its interactions with both NP and VP35. These data demonstrate that the PP1-targeting molecule 1E7-03 facilitates binding of phosphorylated VP30 to the polymerase complex, further supporting our hypothesis that phosphorylated VP30 acts as a repressor of MARV transcription.
Treatment with 1E7-03 inhibits replication of MARV particles.
To test the effect of 1E7-03 on replication of MARV particles, Vero-E6 cells were infected with the virus at a multiplicity of infection (MOI) of 0.01 PFU/cell and treated daily with 3 μM 1E7-03 starting at the day of infection or 24 h prior to the infection, the regimens found to be the most effective in our previous study with EBOV (14). Treatment with the vehicle (DMSO) had no effect on MARV replication (data not shown). Control infected cells, which did not receive the treatment, showed a prominent and pronounced cytopathic effect starting at day 6 postinfection, but 1E7-03-treated cells did not show any cytopathic effect up to day 8 (Fig. 11A). Titration of MARV in supernatants showed that 1E7-03 treatment that was started at the time of infection significantly reduced viral titers. On day 2, the reduction was 1.6 log10, and on day 8, the reduction was 1.7 log10 (Fig. 11B). Adding the compound starting at 24 h prior to infection resulted in no detectable virus until day 2 postinfection, equating to at least a 1.3-log10 reduction of viral titer, and very low virus titers on the subsequent days. For example, on days 4, 5, and 6 postinfection, the relative reductions in the viral titers were 2.3, 2.7, and 3.2 log10, respectively. Thus, 1E7-03 effectively suppressed MARV replication.
FIG 11.

1E7-03 inhibits replication of MARV. Vero-E6 cells were infected with MARV at an MOI of 0.01 PFU/cell and treated daily with 1E7-03 at 3 μM starting at 24 h prior to the infection or during the infection. (A) Bright-field microscopy of cell monolayers infected with MARV and treated with 1E7-03 starting at the indicated time points. Monolayers on days 1 to 4 are not shown due to the lack of visible cytopathic effect. (B) MARV titers in supernatants of infected monolayers treated with 1E7-03 starting at the indicated time points. Values are means ± standard deviations based on triplicate samples. *, P < 0.05 for results compared to those in the untreated cells (Student's t test). The dotted line indicates the limit of detection of the assay.
DISCUSSION
EBOV replication requires NP, polymerase L, and its cofactor VP35, whereas viral transcription also needs transcription factor VP30 (11). An early study suggested that MARV VP30 is not required for transcription and replication of MARV minigenome that required only NP, L, and VP35 proteins (10). However, a rescue of recombinant MARV from cDNA required VP30 (28). A more recent study showed that the addition of VP30 to NP, L, and VP35 plasmids increased the efficacy of MARV replication (29). Moreover, small interfering RNA (siRNA)-mediated knockdown of MARV VP30 in MARV-infected cells significantly reduced production of all viral proteins and release of viral particles (12). In our study, the absence of an MARV VP30 supporting plasmid reduced but did not completely abrogate the luciferase activity in an MARV minigenome system as we observed 27% and 33% reductions in Vero E6 and 293T cells, respectively (Fig. 6A and B).
Previous studies demonstrated that EBOV VP30 phosphorylation inhibits transcription but enhances replication, suggesting the switch of the polymerase activity from transcription to replication upon VP30 phosphorylation (9, 14). A previous study used an indirect method involving alanine substitutions of phosphorylated serine residues in a plasmid encoding MARV VP30, followed by treatment with phosphatase, to demonstrate that the protein is phosphorylated on serine residues within amino acids 40 to 46 and 47 to 51 (5). Here, we used a direct, label-free quantitative proteomic approach to identify VP30 phosphorylation sites both in cells transfected with a VP30-encoding plasmid, as in all filovirus studies to date (5, 13, 14, 30–32), and, for the first time for any filovirus, in purified viral particles. Analysis of plasmid-expressed VP30 in 293T cells identified phosphorylation of Ser-46, Ser-47, Ser-50, Ser-51, Thr-53, Ser-63, Thr-64, Ser-68, Thr-102, Thr-172, and Ser-173. Treatment with phosphatase inhibitors, okadaic acid and the PP1-targeting 1E7-03 compound, induced phosphorylation of a peptide containing Ser-46, Ser-47, Ser-50, Ser-51, and Thr-53 residues but did not affect phosphorylation of a peptide containing residues Ser-63, Thr-64, and Ser-68 (Fig. 4) or a peptide with residue Thr-102 (data not shown). This observation suggests that MARV VP30 is phosphorylated on its N-terminal serine-rich region and that this phosphorylation is likely to be controlled by cellular PP1, which is similar to our previous observation with EBOV VP30 (14).
To further analyze the functionality of the MARV VP30 phosphorylation site, the phosphorylated residues were replaced with alanines. Consistently with the proteomics observations, replacement of Ser-46, Ser-47, Ser-50, Ser-51, or Thr-53 resulted in a significant increase of transcriptional activity (Fig. 7 and 8). In contrast, alanine substitutions for the additional newly identified phosphorylation residue Ser-63, Thr-64, Ser-68, or Thr-102 had no effect, suggesting that only the N-terminal phosphorylation is important for MARV transcription. Furthermore, analysis of purified MARV particles, while confirming part of the identified phosphorylation sites, demonstrated the lack of phosphorylation in the N-terminal cluster comprising Ser-46, Ser-47, Ser-50, Ser-51, and Thr-53, which inhibit transcription (Fig. 7 and 8) and are involved in interaction with NP and VP35 (Fig. 9). Interestingly, a recent study demonstrated that alanine substitution of the corresponding amino acids in the N terminus of EBOV VP30 increased transcription of EBOV minigenome (33), indirectly supporting our results. Taken together, these data suggest that these amino acids are dynamically phosphorylated, as also recently demonstrated for serine 29 of EBOV VP30 (19).
Interactions of EBOV VP30 with NP and VP35 are critical for stabilization of nucleocapsid (34) and synthesis of RNA (9, 34). A recent study demonstrated that the VP30 binding region of EBOV NP is conserved across filoviruses, including MARV (34). Previous studies demonstrated that EBOV VP30 phosphorylation strengthens VP30 interaction with NP (13) and facilitates transport of the nucleocapsid proteins to the sites of RNA synthesis (9) while its dephosphorylation enhances binding to VP35 (9) and viral leader RNA (35), thus promoting the initiation of transcription (15, 19). Here, we demonstrate that MARV VP30 phosphorylation also enhances VP30 binding to NP, as alanine but not aspartic acid substitutions weaken this interaction (Fig. 9A and B), which is consistent with a previous study (5). Unexpectedly and unlike results with the EBOV VP30, alanine substitutions also weakened the interaction of MARV VP30 with VP35 (Fig. 9C and D), suggesting that dephosphorylation of MARV VP30 diminishes its interaction with VP35. Aspartic acid substitutions also diminished this interaction, suggesting that substitution of charged amino acids does not fully mimic phosphorylation. Moreover, in the presence of the PP1-targeting molecule 1E7-03, which prevents VP30 dephosphorylation, interactions of VP30 WT with both NP and VP35 were strengthened. These data demonstrate that the most effective transcription of MARV occurs when VP30 is dephosphorylated, which is accompanied by its dissociation from NP and VP35. We propose a model in which dissociation of a dephosphorylated form of VP30 from NP and VP35 facilitates efficient transcription by the polymerase complex (Fig. 12). In contrast, phosphorylated VP30 binds to both NP and VP35, reducing transcription. This model can explain the transcriptional activity only if VP30 is dynamically phosphorylated, as was previously demonstrated for the P protein of respiratory syncytial virus (36) and more recently for EBOV VP30 (19). According to this model, the dephosphorylated form of MARV VP30 promotes high-level transcription of genome, while its phosphorylated form works as a repressor of the polymerase activity. The model is further confirmed by comparison of the phosphorylation status of plasmid-expressed VP30 with that in purified viral particles, which demonstrated the lack of phosphorylation of the N-terminal cluster of amino acids 46 to 53 (Fig. 2). As such, VP30 incorporated in viral particles does not interact with NP and VP35 through specific phosphorylation and does not act as a repressor of transcription, allowing the virus to quickly start a new cycle of infection upon entering a cell. Keeping the production of mRNA in infected cells, in which VP30 is phosphorylated, at an optimal level might ultimately be beneficial for the virus as excessive production of viral RNA would induce a strong innate immune response.
FIG 12.
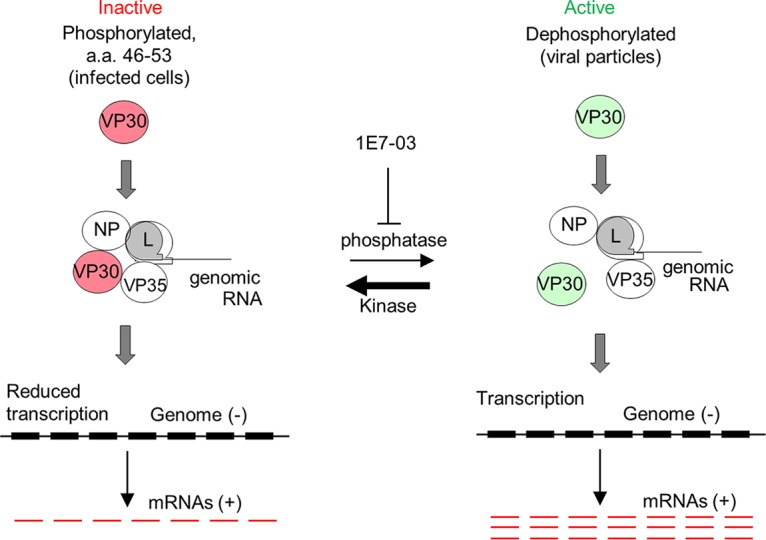
A putative model of the role of VP30 phosphorylation in MARV transcription. The phosphorylated form of VP30 (red, left) binds to NP and VP35 and suppresses transcription. The dephosphorylated form of VP30 (green, right) is dissociated from NP and VP35 and does not inhibit transcription. The phosphorylated form of VP30 may work as a repressor of MARV polymerase by associating with NP and VP35. Treatment of cells with 1E7-03 prevents dephosphorylation of VP30 by PP1 that results in suppression of MARV transcription.
Two members of the Pneumovirinae family of the order Mononegavirales, respiratory syncytial virus (HRSV) and human metapneumovirus (HMPV), have an M2-1 protein, which is considered to be a counterpart of filovirus VP30. HRSV M2-1 was identified as the first transcription elongation factor in the order Mononegavirales and was found to be essential for replication of viral particles (37), but its HMPV analog appeared to be dispensable for virus replication (38). The core domain of RSV M2-1 comprised of residues 58 to 177 (M2-158–177) was demonstrated to be structurally homologous to the C-terminal domain of EBOV VP30 (24). Moreover, the ordered oligomerization and the presence of core domains alternating with disordered segments in our structural model of MARV VP30 are reminiscent of the similar structural features identified in the crystal structure of HMPV M2-1 (25). These common features suggest a common mechanism that uses structural flexibility to allow conformational changes for recruitment of these proteins to the viral transcriptional machinery. The location of the majority of phosphorylation sites of MARV VP30 in the predicted disordered regions is remarkable and indicates the importance of structural flexibility for the phosphorylation and/or regulatory role of the phosphorylation sites.
We previously showed that the PP1-targeting molecule 1E7-03 increases phosphorylation of EBOV VP30, disabling transcription but increasing genome replication and ultimately suppressing replication of EBOV particles (14). This study shows that for MARV, the mechanism of the antiviral effect is different as the compound inhibited transcription by sequestering both NP and VP35 away from the polymerase complex, leading to a dramatic suppression of viral growth. Therefore, targeting the interaction of PP1 with EBOV and MARV VP30 effectively blocks replication of both viruses, despite the differences in VP30-mediated regulation of transcription/replication of the two viruses.
The role of phosphorylation of viral proteins in the viral live cycle has been increasingly recognized, as demonstrated for multiple viruses, including influenza A virus (39), Newcastle disease virus (40), hepatitis C virus (41), dengue virus (42), and Rift valley virus (43), among others. While phosphatases are involved in multiple normal physiological reactions of cells, targeting of their noncatalytic sites, which prevents their interaction with only certain substrates, including viral proteins, may be a viable approach to develop broadly specific antivirals. The 1E7-03 small-molecule compound that was used in this study is nontoxic, presumably because it targets only one PP1 noncatalytic site and probably prevents the interaction of PP1 holoenzyme with EBOV VP30, whereas the host cell PP1 regulatory subunits typically bind PP1 through several interacting sites with a much higher affinity (44). As such, the approach may be viable for development of broad-spectrum antivirals.
In summary, this study demonstrates the following: (i) a direct identification of novel phosphorylation sites of MARV VP30 by MS, some of which are dynamically phosphorylated, (ii) that phosphorylated VP30 works as a repressor of transcription activity, (iii) that the repressor activity of VP30 is a result of its binding to NP and VP35, (iv) that the binding to NP and VP35 occurs through VP30 phosphorylation, and (v) that dephosphorylation of VP30 can be prevented by a small molecule targeting noncatalytic sites of PPs, which results in effective inhibition of viral replication. It should be noted that while identification of phosphorylation sites and testing of the antiviral effects of 1E7-03 were performed with live MARV, investigation of mechanisms of the effects were performed with a minigenome, rather than live virus, which is a potential limitation of the study. Overall, these data suggest that regulation of transcription activity of MARV is different from that of EBOV.
MATERIALS AND METHODS
Viruses and cell lines.
MARV strain Angola was obtained from the Special Pathogens Program, National Microbiology Laboratory, Winnipeg, Canada, and passaged in Vero-E6 cells. All work with MARV was performed in biosafety level 4 (BSL-4) facilities of the Galveston National Laboratory. Vero-E6 and 293T cells were obtained from ATCC (Manassas, VA). 293T cells were maintained in Dulbecco's modified Eagle medium (DMEM)-high glucose and supplemented with 10% fetal bovine serum (FBS) and 0.1% gentamicin sulfate (Corning, Manassas, VA); cells were plated on PureCoat amine flat-bottom multiwell plates (Corning). Vero-E6 cells were maintained in modified Eagle medium (MEM), supplemented with 10% FBS, 1% sodium pyruvate, 1% MEM nonessential amino acids (Sigma, St. Louis, MO), and 0.1% gentamicin sulfate and plated on tissue culture-treated 6- or 24-well plates. All other cell culture reagents were from Thermo Fisher Scientific (Waltham, MA).
Purification of MARV particles.
293T cells were infected with MARV at a multiplicity of infection 0.1 PFU/cell and incubated for 5 days. Supernatants of infected cells were clarified from cell debris by low-speed centrifugation and then subjected to ultracentrifugation through a 25% sucrose cushion (2 h, +4°C, 175,000 × g). Pellets were resuspended in STE buffer (10 mM Tris, 1 mM EDTA, 0.1 M NaCl) and further purified by ultracentrifugation in a 20 to 60% sucrose gradient (1.5 h, +4°C, 288,000 × g). The virus-containing band was collected, and MARV virions were washed from sucrose by dilution in STE buffer, followed by ultracentrifugation (1 h, +4°C, 175,000 × g). The obtained viral particles were suspended in STE buffer with 4% SDS and heat inactivated at 95°C for 15 min. Virus inactivation was confirmed by inoculation of Vero-E6 cell monolayers with an aliquot of inactivated virus preparation or a live virus control and by microscopic observation of cytopathic effect for up to 10 days.
Mass spectrometry and data analysis.
Flag-tagged VP30 was expressed in 293T cells and immunoprecipitated with anti-Flag antibodies (Sigma). Recombinant Flag-tagged VP30 or proteins of the purified virus were resolved by 10% SDS-PAGE. Mass spectra of peptides were obtained with a data-dependent four-event scan method, which included survey Fourier transform (FT)-MS parent scans followed by sequential data-dependent FT-MS/MS scans on the three most abundant peptide ions from the parent scan. Protein identification was carried out with Proteome Discoverer, version 1.4, software using the SEQUEST engine for a protein database search and the International Protein Index (IPI) Human Protein Database (version 1.79). A sequential database search was performed using the human FASTA database. Only peptides with a cross-correlation (XCorr) cutoff of 2.6 for [M + 2H]2+, 3.0 for [M + 3H]3+, and higher charge states were applied. These SEQUEST criteria resulted in a 1 to 2% false discovery rate (FDR). FDR was determined by searching a decoy database. SIEVE, version 2.1, software (Thermo Fisher Scientific) was used for label-free quantitative analysis of the high-resolution MS spectra produced by Orbitrap MS scans.
Modeling MARV VP30 structure.
The three-dimensional structure of the full-length MARV VP30 was obtained using the Rosetta/Robetta suite of protocols (22, 45) and the sequence of MARV strain Musoke (GenBank accession number P35258). The BLAST/PSI-BLAST suite of programs was used to identify boundaries of domains, the units of protein folding. This involved mapping the sequence against the Pfam database; when a domain with a reasonable match was identified, it was further used for template-based structural modeling. Domains with no reasonable match were modeled using Rosetta fragment assembly methodology. In either case, the multiple obtained structures were refined against the novel Rosetta force field (22, 45). The best 10 structures, assessed by energy minimization, were generated by the protocol, and the top five of them were likely to include the accurate structure according to Critical Assessment of Structure Prediction (CASP) competition analyses (46, 47). The final structure was selected by visual inspection and analyzed and validated by the Rosetta/Robetta output and PSIPRED (48) and RAMPAGE (49); the structures were visualized and further analyzed within Chimera (50). Relative solvent-accessible surface area (rSASA) values were calculated using parameter-optimized surfaces (POPS) as previously described (51, 52) (Table 1).
Analysis of virus replication in the presence of 1E7-03.
Triplicate monolayers of Vero-E6 cells were treated with 3 μM 1E7-03 for 24 h before infection or at the time of infection with MARV at an MOI of 0.01 PFU/cell. Both treatment groups were then treated with 3 μM 1E7-03 every 24 h for 10 days. A total of 100 μl of supernatant was collected from each well every 24 h and frozen at −80°C until the titration. To determine the titers of MARV in supernatants, aliquots were titrated using a standard plaque assay procedure using Vero-E6 monolayers and an overlay of MEM containing 2% FBS and 0.5% methylcellulose. Monolayers were incubated at 37°C for 11 days, the overlay was removed, and the cells were fixed and stained with 0.25% crystal violet in 10% buffered formalin (Thermo Fisher Scientific). Plaques were counted and are expressed as the number of PFU/milliliter.
Construction of mutated VP30 plasmids.
DNA fragments containing alanine or aspartic acid substitutions of the phosphorylated serine and threonine residues identified by mass spectrometry, as well as serine residues at positions 40 and 42 and serine/threonine residues at positions 46, 47, 50, 51, and 53, were synthesized by Integrated DNA Technologies (Coralville, IA). The synthesized DNA fragments were digested with NotI and XhoI restriction enzymes and cloned into a pCAGGS-Flag-VP30 plasmid kindly provided by Christopher Basler (Icahn School of Medicine at Mount Sinai). Aspartic acid substitutions of S46 and S51 were introduced by overlap PCR (53) using Pfx polymerase (Thermo Fisher Scientific) with two sets of external primers complementary to the 5′ and 3′ ends of the pCAGGS-Flag-VP30 sequence and two internal primers containing the mismatched bases. After sequence confirmation, the mutated plasmids were digested with NotI and XhoI restriction endonucleases and cloned back into pCAGGS-Flag-VP30 vector. Mutated VP30 plasmids were transfected in 293T cells using TransIT-LT1 transfection reagent (Mirus, Madison, WI) at 3 μl per μg of plasmid DNA, and the cell lysates were harvested at 48 h posttransfection by resuspension in 5× passive lysis buffer (Promega, Madison, WI).
Analysis of VP30 protein by Western blotting.
Cell lysates were denatured at 95°C for 5 min and separated by NuPAGE 4 to 12% Bis-Tris gels along with a Novex sharp prestained protein standard, and proteins were transferred to nitrocellulose membrane using an iBlot Gel transfer system (all items from Thermo Fisher Scientific). Membranes were blocked with 5% milk in phosphate-buffered saline containing 0.1% Tween 20 (PBST) for 1 h and incubated with M2 mouse monoclonal antibody specific for Flag at a 1:1,000 dilution (F1804; Sigma-Aldrich) and secondary anti-mouse IgG antibodies conjugated to horseradish peroxidase at a 1:3,000 dilution (Cell Signaling Technology, Danvers, MA). Protein bands were visualized using chemiluminescent Pierce ECL Western blotting substrate (Thermo Fisher Scientific), scanned using a Li-Cor Odyssey Fc imaging system, and quantified with Image Studio Lite, version 5.2 (Li-Cor Biotechnology, Lincoln, NE).
Minigenome experiments.
The MARV minigenome expressing a Renilla luciferase reporter gene and pCAGGS plasmid expressing firefly luciferase were kindly provided by Christopher Basler. The codon-optimized support plasmids expressing MARV NP pCAGGS-NPco, VP35 pCAGGS-VP35co, VP30 pCAGGS-VP30co, and L pCAGGS-L and the plasmid expressing T7 RNA polymerase pC-T7 were provided by Jonathan S. Towner (Centers for Disease Control and Prevention, Atlanta, GA) and kindly shared by Thomas Ksiazek (University of Texas Medical Branch at Galveston, TX). Vero-E6 cells and 293T cells were seeded to approximately 50% confluence and transfected with the following amounts of plasmids: 250 ng of pCAGGS-NPco, 25 ng of pCAGGS-VP35co, 25 ng of pCAGGS-VP30co or pCAGGS-Flag V30 or its mutants, 250 ng of pCAGGS-Lco, 100 ng of pC-T7co, or 300 ng of MARV minigenome and 2 ng of pCAGGS-firefly luciferase plasmid. In addition, the cells were treated with 3 μM 1E7-03 or DMSO as indicated in the text. The plasmids were transfected with Lipofectamine 3000 reagent (Thermo Fisher Scientific) at a ratio of 1:2 per the manufacturer's protocol. At 48 h posttransfection, cells were lysed with passive lysis buffer to measure transcription by a dual-luciferase reporter assay (Promega). To express activity of the minigenome, Renilla luciferase activity was normalized to firefly luciferase activity, and the resulting value was expressed as percent activity relative to that of the nonmutated VP30 plasmid. Minigenome expression was also confirmed by performing Western blot analysis using primary polyclonal rabbit antibodies specific for Renilla luciferase (PA5-322; Thermo Fisher Scientific) and IRDye 800CW-labeled goat anti-rabbit IgG secondary antibodies (Li-Cor Biotechnology).
Coimmunoprecipitation assays.
Synthetic DNA containing an HA tag at the N terminus of VP35 was generated by Integrated DNA Technologies and cloned into NotI and XhoI restriction sites in pCAGGS vector. 293T cells cotransfected with 2 μg each of pCAGGS-NPco or pCAGGS-HA-VP35 and either pCAGGS-Flag-VP30 or mutants or empty vector were collected 48 h later by lysing cells in Pierce immunoprecipitation (IP) Lysis/Wash Buffer and Halt Protease and Phosphatase Inhibitor Cocktail, EDTA-free (100×) (Thermo Fisher Scientific). For coimmunoprecipitation assays done in the presence of PP1 inhibitors, DMSO or 3 μM 1E7-03 or 0.1 μM okadaic acid was added 24 h prior to harvesting of lysates. Lysates were clarified by low-speed centrifugation, and immunoprecipitation of Flag-VP30 or HA-tagged VP35 proteins was performed using anti-DYKDDDDK (Flag) affinity gel (Bimake.com, Houston, TX) or a Pierce HA tag IP/co-IP kit (Thermo Fisher Scientific) per the manufacturer's protocols. Total protein in lysates was quantified by a Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific). Approximately 400 μg of total protein lysate was incubated with 20 μl of washed affinity Flag gel resin or anti-HA agarose slurry and incubated overnight at 4°C. Immunoprecipitated lysates, as well as whole-cell lysates, were denatured and analyzed by Western blotting using the following primary antibodies diluted at 1:1,000 in StartingBlock T20 (Tris-buffered saline [TBS]) blocking buffer: rabbit anti-Flag polyclonal antibody (catalog number F7425), mouse monoclonal anti-FLAG M2 antibody, and rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) polyclonal antibody (9545) (all, Sigma-Aldridge); rabbit anti-NP polyclonal antibody (IBT Bioservices, Rockville, MD); rabbit HA-probe polyclonal antibody (sc-805; Santa Cruz Biotechnology, Dallas, TX); mouse beta-3 tubulin monoclonal antibody (32-2600; Thermo Fisher Scientific). After three washes with PBST (0.1%), the blots were incubated with either IRDye 680RD-labeled goat anti-mouse IgG or IRDye 800CW-labeled goat anti-rabbit IgG secondary antibodies (Li-Cor Biotechnology) for 1 h, scanned in a Li-Cor Odyssey Fc imaging system, and quantified by Image Studio Lite, version 5.2, software (Li-Cor Biotechnology).
ACKNOWLEDGMENTS
This project was supported by NIH Research grants U19AI109664 (A.B. and S.N.) and 5G12MD007597 (to S.N.).
We thank Christopher Basler (Georgia State University) for providing the plasmids expressing MARV Flag VP30, MARV minigenome, and firefly luciferase, Jonathan S. Towner (Centers for Disease Control and Prevention, Atlanta, GA) for providing codon-optimized support plasmids for MARV NP, VP30, VP35, L, and T7 polymerase, and Benjamin Gelman (University of Texas Medical Branch, Galveston, TX) for providing access to the Li-Cor Odyssey Imaging system.
S.N. and A.B. have a patent on inhibitors of protein phosphatase 1 and their use for the treatment or prevention of filovirus infections.
REFERENCES
- 1.Brauburger K, Hume AJ, Muhlberger E, Olejnik J. 2012. Forty-five years of Marburg virus research. Viruses 4:1878–1927. doi: 10.3390/v4101878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.CDC. 2014. Marburg hemorrhagic fever (Marburg HF). Chronology of Marburg hemorrhagic fever outbreaks. Centers for Disease Control and Prevention, Atlanta, GA: https://www.cdc.gov/vhf/marburg/resources/outbreak-table.html. [Google Scholar]
- 3.Feldmann H, Muhlberger E, Randolf A, Will C, Kiley MP, Sanchez A, Klenk HD. 1992. Marburg virus, a filovirus: messenger RNAs, gene order, and regulatory elements of the replication cycle. Virus Res 24:1–19. doi: 10.1016/0168-1702(92)90027-7. [DOI] [PubMed] [Google Scholar]
- 4.Bukreyev AA, Volchkov VE, Blinov VM, Dryga SA, Netesov SV. 1995. The complete nucleotide sequence of the Popp (1967) strain of Marburg virus: a comparison with the Musoke (1980) strain. Arch Virol 140:1589–1600. doi: 10.1007/BF01322532. [DOI] [PubMed] [Google Scholar]
- 5.Modrof J, Moritz C, Kolesnikova L, Konakova T, Hartlieb B, Randolf A, Muhlberger E, Becker S. 2001. Phosphorylation of Marburg virus VP30 at serines 40 and 42 is critical for its interaction with NP inclusions. Virology 287:171–182. doi: 10.1006/viro.2001.1027. [DOI] [PubMed] [Google Scholar]
- 6.Becker S, Rinne C, Hofsass U, Klenk HD, Muhlberger E. 1998. Interactions of Marburg virus nucleocapsid proteins. Virology 249:406–417. doi: 10.1006/viro.1998.9328. [DOI] [PubMed] [Google Scholar]
- 7.Sanchez A, Kiley MP, Holloway BP, Auperin DD. 1993. Sequence analysis of the Ebola virus genome: organization, genetic elements, and comparison with the genome of Marburg virus. Virus Res 29:215–240. doi: 10.1016/0168-1702(93)90063-S. [DOI] [PubMed] [Google Scholar]
- 8.Martinez MJ, Volchkova VA, Raoul H, Alazard-Dany N, Reynard O, Volchkov VE. 2011. Role of VP30 phosphorylation in the Ebola virus replication cycle. J Infect Dis 204(Suppl 3):S934–940. doi: 10.1093/infdis/jir320. [DOI] [PubMed] [Google Scholar]
- 9.Biedenkopf N, Hartlieb B, Hoenen T, Becker S. 2013. Phosphorylation of Ebola virus VP30 influences the composition of the viral nucleocapsid complex: impact on viral transcription and replication. J Biol Chem 288:11165–11174. doi: 10.1074/jbc.M113.461285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muhlberger E, Lotfering B, Klenk HD, Becker S. 1998. Three of the four nucleocapsid proteins of Marburg virus, NP, VP35, and L, are sufficient to mediate replication and transcription of Marburg virus-specific monocistronic minigenomes. J Virol 72:8756–8764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muhlberger E, Weik M, Volchkov VE, Klenk HD, Becker S. 1999. Comparison of the transcription and replication strategies of Marburg virus and Ebola virus by using artificial replication systems. J Virol 73:2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fowler T, Bamberg S, Moller P, Klenk HD, Meyer TF, Becker S, Rudel T. 2005. Inhibition of Marburg virus protein expression and viral release by RNA interference. J Gen Virol 86:1181–1188. doi: 10.1099/vir.0.80622-0. [DOI] [PubMed] [Google Scholar]
- 13.Modrof J, Muhlberger E, Klenk HD, Becker S. 2002. Phosphorylation of VP30 impairs Ebola virus transcription. J Biol Chem 277:33099–33104. doi: 10.1074/jbc.M203775200. [DOI] [PubMed] [Google Scholar]
- 14.Ilinykh PA, Tigabu B, Ivanov A, Ammosova T, Obukhov Y, Garron T, Kumari N, Kovalskyy D, Platonov MO, Naumchik VS, Freiberg AN, Nekhai S, Bukreyev A. 2014. Role of protein phosphatase 1 in dephosphorylation of Ebola virus VP30 protein and its targeting for the inhibition of viral transcription. J Biol Chem 289:22723–22738. doi: 10.1074/jbc.M114.575050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinez MJ, Biedenkopf N, Volchkova V, Hartlieb B, Alazard-Dany N, Reynard O, Becker S, Volchkov V. 2008. Role of Ebola virus VP30 in transcription reinitiation. J Virol 82:12569–12573. doi: 10.1128/JVI.01395-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Washington K, Ammosova T, Beullens M, Jerebtsova M, Kumar A, Bollen M, Nekhai S. 2002. Protein phosphatase-1 dephosphorylates the C-terminal domain of RNA polymerase-II. J Biol Chem 277:40442–40448. doi: 10.1074/jbc.M205687200. [DOI] [PubMed] [Google Scholar]
- 17.Ammosova T, Yedavalli VR, Niu X, Jerebtsova M, Van Eynde A, Beullens M, Bollen M, Jeang KT, Nekhai S. 2011. Expression of a protein phosphatase 1 inhibitor, cdNIPP1, increases CDK9 threonine 186 phosphorylation and inhibits HIV-1 transcription. J Biol Chem 286:3798–3804. doi: 10.1074/jbc.M110.196493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ammosova T, Obukhov Y, Kotelkin A, Breuer D, Beullens M, Gordeuk VR, Bollen M, Nekhai S. 2011. Protein phosphatase-1 activates CDK9 by dephosphorylating Ser175. PLoS One 6:e18985. doi: 10.1371/journal.pone.0018985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biedenkopf N, Lier C, Becker S. 2016. Dynamic phosphorylation of VP30 is essential for Ebola virus life cycle. J Virol 90:4914–4925. doi: 10.1128/JVI.03257-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartlieb B, Muziol T, Weissenhorn W, Becker S. 2007. Crystal structure of the C-terminal domain of Ebola virus VP30 reveals a role in transcription and nucleocapsid association. Proc Natl Acad Sci U S A 104:624–629. doi: 10.1073/pnas.0606730104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clifton MC, Kirchdoerfer RN, Atkins K, Abendroth J, Raymond A, Grice R, Barnes S, Moen S, Lorimer D, Edwards TE, Myler PJ, Saphire EO. 2014. Structure of the Reston Ebolavirus VP30 C-terminal domain. Acta Crystallogr F Struct Biol Commun 70:457–460. doi: 10.1107/S2053230X14003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raman S, Vernon R, Thompson J, Tyka M, Sadreyev R, Pei J, Kim D, Kellogg E, DiMaio F, Lange O, Kinch L, Sheffler W, Kim BH, Das R, Grishin NV, Baker D. 2009. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins 77(Suppl 9):89–99. doi: 10.1002/prot.22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang ZR, Thomson R, McNeil P, Esnouf RM. 2005. RONN: the bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 21:3369–3376. doi: 10.1093/bioinformatics/bti534. [DOI] [PubMed] [Google Scholar]
- 24.Blondot ML, Dubosclard V, Fix J, Lassoued S, Aumont-Nicaise M, Bontems F, Eleouet JF, Sizun C. 2012. Structure and functional analysis of the RNA- and viral phosphoprotein-binding domain of respiratory syncytial virus M2-1 protein. PLoS Pathog 8:e1002734. doi: 10.1371/journal.ppat.1002734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leyrat C, Renner M, Harlos K, Huiskonen JT, Grimes JM. 2014. Drastic changes in conformational dynamics of the antiterminator M2-1 regulate transcription efficiency in Pneumovirinae. Elife 3:e02674. doi: 10.7554/eLife.02674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Somavarapu AK, Balakrishnan S, Gautam AK, Palmer DS, Venkatraman P. 2014. Structural interrogation of phosphoproteome identified by mass spectrometry reveals allowed and disallowed regions of phosphoconformation. BMC Struct Biol 14:9. doi: 10.1186/1472-6807-14-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Albariño CG, Shoemaker T, Khristova ML, Wamala JF, Muyembe JJ, Balinandi S, Tumusiime A, Campbell S, Cannon D, Gibbons A, Bergeron E, Bird B, Dodd K, Spiropoulou C, Erickson BR, Guerrero L, Knust B, Nichol ST, Rollin PE, Ströher U. 2013. Genomic analysis of filoviruses associated with four viral hemorrhagic fever outbreaks in Uganda and the Democratic Republic of the Congo in 2012. Virology 442:97–100. doi: 10.1016/j.virol.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Enterlein S, Volchkov V, Weik M, Kolesnikova L, Volchkova V, Klenk HD, Muhlberger E. 2006. Rescue of recombinant Marburg virus from cDNA is dependent on nucleocapsid protein VP30. J Virol 80:1038–1043. doi: 10.1128/JVI.80.2.1038-1043.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Albarino CG, Uebelhoer LS, Vincent JP, Khristova ML, Chakrabarti AK, McElroy A, Nichol ST, Towner JS. 2013. Development of a reverse genetics system to generate recombinant Marburg virus derived from a bat isolate. Virology 446:230–237. doi: 10.1016/j.virol.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peyrol J, Thizon C, Gaillard JC, Marchetti C, Armengaud J, Rollin-Genetet F. 2013. Multiple phosphorylable sites in the Zaire Ebolavirus nucleoprotein evidenced by high resolution tandem mass spectrometry. J Virol Methods 187:159–165. doi: 10.1016/j.jviromet.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 31.Sanger C, Muhlberger E, Lotfering B, Klenk HD, Becker S. 2002. The Marburg virus surface protein GP is phosphorylated at its ectodomain. Virology 295:20–29. doi: 10.1006/viro.2002.1374. [DOI] [PubMed] [Google Scholar]
- 32.Kolesnikova L, Mittler E, Schudt G, Shams-Eldin H, Becker S. 2012. Phosphorylation of Marburg virus matrix protein VP40 triggers assembly of nucleocapsids with the viral envelope at the plasma membrane. Cell Microbiol 14:182–197. doi: 10.1111/j.1462-5822.2011.01709.x. [DOI] [PubMed] [Google Scholar]
- 33.Kruse T, Biedenkopf N, Hertz EPT, Dietzel E, Stalmann G, Lopez-Mendez B, Davey NE, Nilsson J, Becker S. 2018. The Ebola virus nucleoprotein recruits the host PP2A-B56 phosphatase to activate transcriptional support activity of VP30. Mol Cell 69:136–145.e6. doi: 10.1016/j.molcel.2017.11.034. [DOI] [PubMed] [Google Scholar]
- 34.Kirchdoerfer RN, Moyer CL, Abelson DM, Saphire EO. 2016. The Ebola virus VP30-NP interaction is a regulator of viral RNA synthesis. PLoS Pathog 12:e1005937. doi: 10.1371/journal.ppat.1005937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biedenkopf N, Schlereth J, Grunweller A, Becker S, Hartmann RK. 2016. RNA binding of Ebola virus VP30 is essential for activating viral transcription. J Virol 90:7481–7496. doi: 10.1128/JVI.00271-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asenjo A, Rodriguez L, Villanueva N. 2005. Determination of phosphorylated residues from human respiratory syncytial virus P protein that are dynamically dephosphorylated by cellular phosphatases: a possible role for serine 54. J Gen Virol 86:1109–1120. doi: 10.1099/vir.0.80692-0. [DOI] [PubMed] [Google Scholar]
- 37.Collins PL, Hill MG, Cristina J, Grosfeld H. 1996. Transcription elongation factor of respiratory syncytial virus, a nonsegmented negative-strand RNA virus. Proc Natl Acad Sci U S A 93:81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buchholz UJ, Biacchesi S, Pham QN, Tran KC, Yang L, Luongo CL, Skiadopoulos MH, Murphy BR, Collins PL. 2005. Deletion of M2 gene open reading frames 1 and 2 of human metapneumovirus: effects on RNA synthesis, attenuation, and immunogenicity. J Virol 79:6588–6597. doi: 10.1128/JVI.79.11.6588-6597.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng W, Li J, Wang S, Cao S, Jiang J, Chen C, Ding C, Qin C, Ye X, Gao GF, Liu W. 2015. Phosphorylation controls the nuclear-cytoplasmic shuttling of influenza A virus nucleoprotein. J Virol 89:5822–5834. doi: 10.1128/JVI.00015-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qiu X, Zhan Y, Meng C, Wang J, Dong L, Sun Y, Tan L, Song C, Yu S, Ding C. 2016. Identification and functional analysis of phosphorylation in Newcastle disease virus phosphoprotein. Arch Virol 161:2103–2116. doi: 10.1007/s00705-016-2884-x. [DOI] [PubMed] [Google Scholar]
- 41.Lee KY, Chen YH, Hsu SC, Yu MJ. 2016. Phosphorylation of serine 235 of the hepatitis C virus non-structural protein NS5A by multiple kinases. PLoS One 11:e0166763. doi: 10.1371/journal.pone.0166763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Noppakunmongkolchai W, Poyomtip T, Jittawuttipoka T, Luplertlop N, Sakuntabhai A, Chimnaronk S, Jirawatnotai S, Tohtong R. 2016. Inhibition of protein kinase C promotes dengue virus replication. Virol J 13:35. doi: 10.1186/s12985-016-0494-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baer A, Shafagati N, Benedict A, Ammosova T, Ivanov A, Hakami RM, Terasaki K, Makino S, Nekhai S, Kehn-Hall K. 2016. Protein phosphatase-1 regulates Rift Valley fever virus replication. Antiviral Res 127:79–89. doi: 10.1016/j.antiviral.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bollen M, Beullens M. 2002. Signaling by protein phosphatases in the nucleus. Trends Cell Biol 12:138–145. doi: 10.1016/S0962-8924(01)02247-4. [DOI] [PubMed] [Google Scholar]
- 45.Rohl CA, Strauss CE, Misura KM, Baker D. 2004. Protein structure prediction using Rosetta. Methods Enzymol 383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 46.Siew N, Elofsson A, Rychlewski L, Fischer D. 2000. MaxSub: an automated measure for the assessment of protein structure prediction quality. Bioinformatics 16:776–785. doi: 10.1093/bioinformatics/16.9.776. [DOI] [PubMed] [Google Scholar]
- 47.Ben-David M, Noivirt-Brik O, Paz A, Prilusky J, Sussman JL, Levy Y. 2009. Assessment of CASP8 structure predictions for template free targets. Proteins 77(Suppl 9):50–65. doi: 10.1002/prot.22591. [DOI] [PubMed] [Google Scholar]
- 48.Jones DT. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 49.Lovell SC, Davis IW, Arendall WB III, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. 2003. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins 50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 50.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 51.Fraternali F, Cavallo L. 2002. Parameter optimized surfaces (POPS): analysis of key interactions and conformational changes in the ribosome. Nucleic Acids Res 30:2950–2960. doi: 10.1093/nar/gkf373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cavallo L, Kleinjung J, Fraternali F. 2003. POPS: A fast algorithm for solvent accessible surface areas at atomic and residue level. Nucleic Acids Res 31:3364–3366. doi: 10.1093/nar/gkg601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]