

Papillomaviruses encode the DNA-binding E1 and E2 proteins, which form a complex and are essential for genome replication. Both proteins are targeted to the nucleus via nuclear localization signals. Our studies have uncovered that cytoplasmic mutant E1 or E2 proteins can be localized to the nucleus when E1 or E2 is also present. An interaction between E1 and E2 is necessary to target cytoplasmic E1 mutant proteins to the nucleus, but cytoplasmic E2 mutant proteins can be targeted to the nucleus without a direct interaction, which points to a novel function of E1.
KEYWORDS: E1, E2, papillomavirus, nuclear localization, replication
ABSTRACT
The papillomavirus (PV) E2 protein is a nuclear, sequence-specific DNA-binding protein that regulates transcription and nuclear retention of viral genomes. E2 also interacts with the viral E1 protein to replicate the viral genome. E2 residue K111 is highly conserved among PV and has been implicated in contributing to nuclear transport, transcription, and replication. Cottontail rabbit (Sylvilagus floridanus) PV (CRPV or SfPV1) E2 K111R, A, or Q mutations are transcription deficient and localized to the cytoplasm, comparable to other PV types. The addition of a nuclear localization signal (NLS) resulted in nuclear E2 K111 mutant proteins but did not restore transcriptional activation, and this is most likely due to an impaired binding to the cellular Brd4 protein. Surprisingly, coexpression of E1 with E2 K111 mutations resulted in their nuclear localization and, for K111A and R mutations, the activation of an E1/E2-dependent reporter construct. Interestingly, the nuclear localization of E2 K111Q mutant protein was independent from the presence of the conserved bipartite NLS in E1 and the direct interaction between E1 and E2. On the other hand, the cytoplasmic E1 NLS mutation could be targeted to the nucleus by wild-type E2, and this was dependent upon an interaction between E1 and E2. In summary, our studies have uncovered that E1 and E2 control each other's subcellular localization: direct binding of E2 to E1 can direct E1 to the nucleus independently from the E1 NLS, and E1 can direct E2 to the nucleus without an intact NLS or direct binding to E2.
IMPORTANCE Papillomaviruses encode the DNA-binding E1 and E2 proteins, which form a complex and are essential for genome replication. Both proteins are targeted to the nucleus via nuclear localization signals. Our studies have uncovered that cytoplasmic mutant E1 or E2 proteins can be localized to the nucleus when E1 or E2 is also present. An interaction between E1 and E2 is necessary to target cytoplasmic E1 mutant proteins to the nucleus, but cytoplasmic E2 mutant proteins can be targeted to the nucleus without a direct interaction, which points to a novel function of E1.
INTRODUCTION
Infections with high-risk human papillomaviruses (HPV) such as HPV16 or HPV18 can give rise to anal, cervical, oropharyngeal, penile, and vaginal cancer (1). HPV are small viruses with a double-stranded circular DNA genome which replicate as nuclear plasmids. Papillomaviruses encode the E1 and E2 proteins, which are required for the replication of the viral genome. E1 is the replicative helicase that unwinds the viral genome and recruits host cell factors to initiate replication of the viral genome (2). E2 consists of a conserved 200-amino-acid (aa) amino-terminal domain responsible for interaction with viral E1 and host cell factors, a nonconserved, unstructured hinge region, and a conserved carboxy-terminal DNA-binding and dimerization domain of 90 aa (3). The interaction of E2 with E1 directs E1 to the viral replication origin (2). E2 functions in addition as a regulator of viral transcription, which is mediated by interactions with host cell proteins such as Brd4 (4–6). The E2-Brd4 interaction is highly conserved, as key residues in E2, such as K37 and I73, involved in the interaction with Brd4, are present in a large number of PV (7). K37 and I73 E2 mutations that decrease binding to Brd4 are greatly impaired in stimulating transcription (8). Studies with cottontail rabbit (Sylvilagus floridanus) PV (CRPV or SfPV1) have shown that viral mutant genomes encoding BRD4-binding-deficient E2 R37K, R37A, or I73L no longer induce tumors in domestic rabbits (9). This is most likely due to both reduced viral transcription and expression of certain host genes, such as c-fos (10).
Both E1 and E2 have to be in the nucleus to bind to the viral genome and control its replication and transcription. In line with this, a highly conserved bipartite nuclear localization signal (NLS) has been identified in E1 (2). E1 also harbors a conserved nuclear export sequence (NES) that is embedded in the bipartite NLS (2). The NES of the HPV11 and HPV31 E1 proteins has been shown to be negatively regulated by CDK-dependent phosphorylation of conserved serines (11, 12). This suggests that nuclear accumulation of at least some E1 proteins is tightly regulated.
E2 proteins also harbor NLS sequences. In bovine PV1 (BPV1) and HPV16 the highly conserved DNA recognition helix in the DNA binding domain acts as a transferable NLS (13, 14). For HPV11 E2, a conserved patch of basic residues in the hinge region acts as an NLS (15). In addition, mutation of highly conserved K111 (Fig. 1A) in BPV1 E2 resulted in cytoplasmic protein localization, but the sequence encompassing K111 does not act as a transferable NLS (14, 16, 17). In line with K111 being important for nuclear localization of E2, a BPV1 E2 K111R mutant genome no longer induces focus formation, and HPV11 E2 K111R and K111A mutations lose their abilities to activate transcription and E1/E2-dependent replication of the viral origin (16–19). Quinlan et al. demonstrated that BPV1 E2 K111 can be modified by acetylation and that this is important for transcriptional activation and subcellular localization (17). A recent analysis of HPV31 E2 confirmed that K111R and K111A mutations do not support E1/E2-dependent replication, whereas K111Q, which is regarded as mimicking acetylated lysine, does (20). This suggested that modification of K111 by acetylation is important for E2 functions (20).
FIG 1.
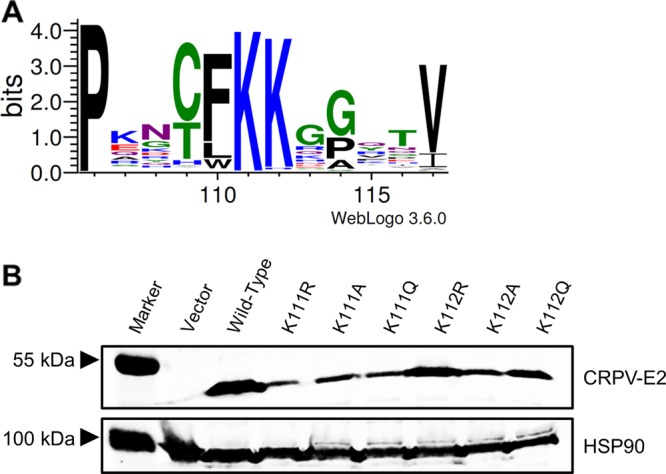
Expression levels of CRPV E2 K111 and K112 mutant proteins. (A) Partial sequence alignment of the region between amino acids 106 and 117 of all 353 annotated E2 proteins. The sequences were obtained from https://pave.niaid.nih.gov/ and adjusted to each other. A sequence logo was generated using WebLogo 3 (http://weblogo.threeplusone.com/) (33). (B) C33a cells were transiently transfected with expression vectors for E2 wt or E2 mt proteins. An empty expression vector (pSG5) was used as a control. At 48 h after transfection, whole-cell extracts were analyzed in immunoblot analysis using the indicated antibodies.
Taken together, these data indicate that conserved lysine K111 is important for subcellular localization, transcriptional activation, and replication functions of E2 and can be modified by acetylation.
RESULTS
Characterization of CRPV E2 K111 and K112 mutant proteins.
To address the role of K111 or K112 for the activities of CRPV E2, K111 and K112 were mutated to R, A, and Q. Immunoblot analyses of whole-cell extracts obtained after expression of vectors for the different CRPV E2 proteins in C33a cells indicated that mutation of K111 or K112 reduced protein levels compared to those of the wild-type (wt) protein (Fig. 1B). Immunofluorescence analyses revealed that the wt and K112 mutant (mt) proteins were localized to the nucleus, whereas K111 mutant proteins were retained in the cytoplasm (Fig. 2). Functional assays using the E2-dependent pC18-Sp1-luc reporter construct or a CRPV early promoter construct (pGL CRPV [PL-PE7]) demonstrated a greatly reduced activation of both reporter constructs by K111R, K111A, K111Q, and K112A, whereas K112R and K112Q displayed wt activation levels (Fig. 3). The loss of transcriptional activation by K111 mutant proteins could be explained by their cytoplasmic localization, since activation by E2 occurs mainly upon recognition of E2 binding sites in the upstream region of viral and synthetic promoters. To test whether the lack of transcriptional activation is due to subcellular mislocalization, the simian virus 40 (SV40) T antigen nuclear localization signal (NLS) was attached to the amino terminus of wt E2 and K111 mutations. Immunofluorescence analysis of transfected cells revealed that the addition of the NLS did not change the nuclear localization of wt E2 but directed K111A, R, and Q mutant proteins into the nucleus (Fig. 4A). However, functional assays demonstrated that NLS-E2 K111R, A, and Q mutations did not gain the ability to activate the pC18-Sp1-luc reporter plasmid, in contrast to NLS-E2 wt (Fig. 4B). This suggested that residue K111 is important not only for the nuclear localization of CRPV E2 but also for its nuclear transcription activation function. To test whether mutation of K111 modulates the interaction with the cellular coactivator Brd4, an in vivo protein-protein interaction assay was applied in which a FRET (Förster resonance energy transfer) signal, which is generated when blue fluorescent protein (BFP)- and super yellow fluorescent protein (SYFP)-labeled fusion proteins interact in vivo, is detected by flow cytometry (Fig. 5A) (21). When we established the FACS-FRET assay for the E2-Brd4 interaction, we found that E2 needs to be tagged with BFP at the amino terminus, whereas it is crucial to link SYFP to the carboxy terminus of Brd4 to gain a FRET signal (data not shown). BFP-NLS-E2 or BFP-NLS-E2 K111Q next was cotransfected with SYFP-Brd4 or SYFP-Brd4 ΔCTM (aa 1 to 1223), a mutant protein lacking the E2 interaction domain (22), and the fraction of FRET-positive cells was determined (Fig. 5B and C). As a positive control, a SYFP-BFP fusion protein gained a nearly 100% FRET signal, whereas the cotransfection of only the expression vectors for BFP and SYFP resulted in a FRET signal below 1%. When we tested the different combinations, we found that wt NLS-E2 resulted in a 50% FRET signal with Brd4. In contrast, the signal was completely absent when Brd4 ΔCTM was used. Interestingly, NLS-E2 K111Q displayed a significantly reduced FRET signal of 25% with bromodomain-containing protein 4 (Brd4) full length (fl) and no signal with Brd4 ΔCTM (Fig. 5C). This suggests that the lack of promoter stimulation by E2 K111Q is due to both mislocalization to the cytoplasm and a reduced interaction with Brd4 in the nucleus.
FIG 2.
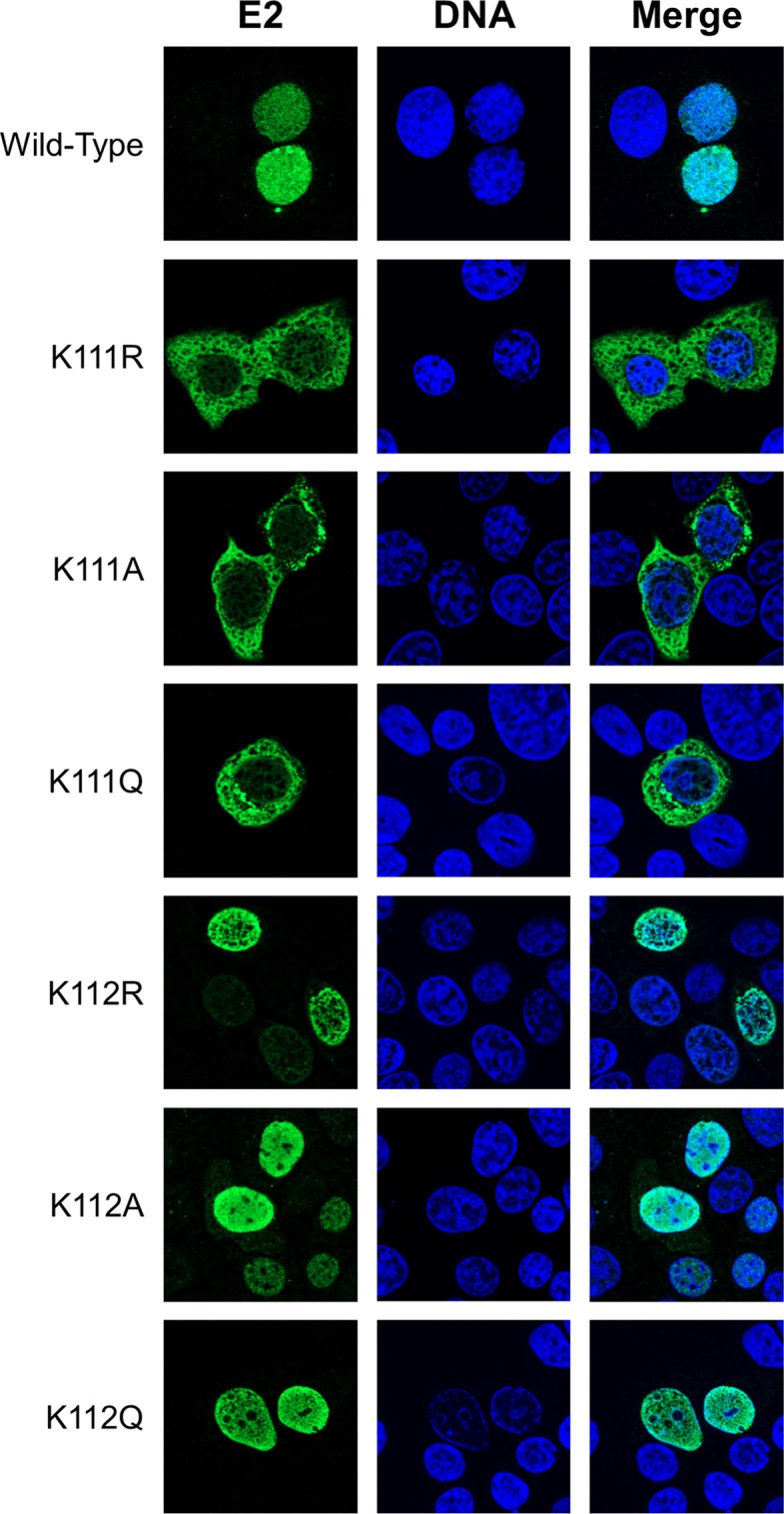
Mutation of K111 affects subcellular localization of the E2 protein. C33a cells were transfected with 500 ng expression vector for each E2 protein (as indicated on the left side). Forty-eight hours after transfection, cells were stained with an anti-CRPV E2 antibody to detect E2 proteins (green) and analyzed by immunofluorescence microscopy. DNA was stained with DAPI (blue).
FIG 3.

K111 mutations do not enable transcriptional activity. C33a cells were either transiently transfected with 50 ng pC18-Sp1-luc (A) or 50 ng pGL CRPV (PL-PE7) firefly reporter construct (B), 10 ng of the empty expression vector (pSG5), or the expression vectors for the different CRPV E2 proteins. A schematic presentation of the reporter constructs is shown above the respective graphs. The values represent the relative luciferase activities of the E2 proteins (wild type or mutants) to the basal activity of the used firefly reporter construct. Error bars indicate the standard error of the mean (SEM) from at least six independent experiments.
FIG 4.
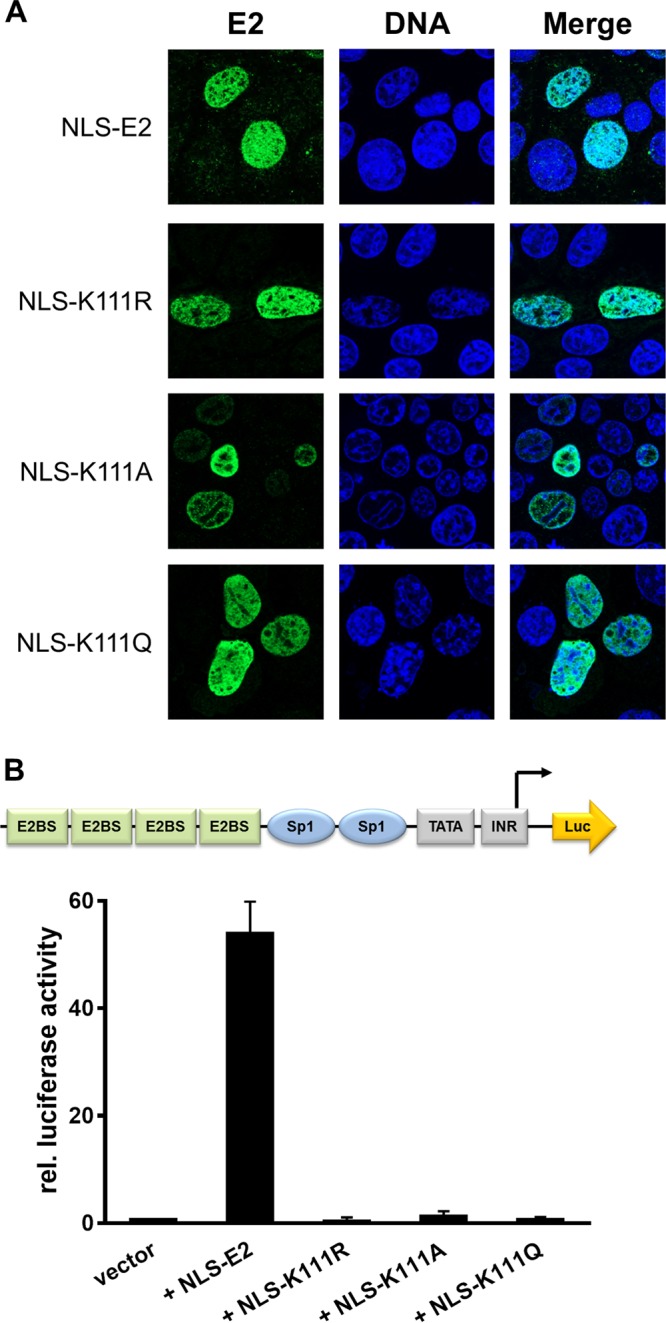
Relocalization of cytoplasmic K111 mutant proteins does not lead to a gain of transcriptional activity. (A) C33a cells were transfected with 500 ng expression vector, and 48 h after transfection cells were stained with an anti-CRPV E2 antibody to detect E2 proteins (green) and analyzed by immunofluorescence microscopy. DNA was stained with DAPI (blue). (B) C33a cells were transfected with 50 ng pC18-Sp1-luc and 10 ng of the empty expression vector (pSG5) or expression vectors for the different CRPV E2 proteins. The values represent the luciferase activities of the E2 proteins relative to the basal activity of the used firefly reporter construct. Error bars indicate the standard errors of the means (SEM) from at least three independent experiments.
FIG 5.

Flow cytometry-FRET analyses indicate a reduced interaction of K111Q with Brd4 in the nucleus. (A) Gating strategy to determine E2 and Brd4 interactions in a flow cytometry-based FRET assay. Untransfected C33a cells were used as a mock control or transfected with 500 ng expression vectors for BFP, SYFP, BFP, and SYFP as well as a BFP-SYFP fusion protein. As a first step, the BFP/SYFP-double positive cells were gated (row 1). Cells giving a false-positive FRET signal resulting from YFP excitation at 405 nm were excluded (row 2). The remaining cells were further analyzed for a positive FRET signal by setting a gate incorporating cells which are cotransfected with the expression vectors for BFP and SYFP, consequently generating no FRET signal (row 3). (B) FACS plots showing FRET-positive cells in living C33a cells cotransfected with the indicated BFP and SYFP fusion proteins. SYFP-BFP fusion protein was used as a positive control, and cotransfected BFP and SYFP expression vectors were negative controls. (C) Mean values of FRET-positive cells from at least three independent experiments that were analyzed as shown in panel B. Error bars indicate the standard errors of the means (SEM). A paired two-tailed t test was used to determine statistical significance (**, P < 0.01).
Coexpression of CRPV E1 relocalizes E2 K111 mutant proteins to the nucleus.
BPV1, HPV11, and HPV31 E2 K111 mutant proteins display a greatly reduced ability to stimulate the E1/E2-dependent replication of the viral origin. We therefore analyzed the properties of CRPV E2 K111 mutations in the presence of the CRPV E1 protein. Surprisingly, coexpression of a hemagglutinin (HA)-tagged version of CRPV E1 induced nuclear localization of all K111 mutations (Fig. 6A). To analyze the functional consequences for transient replication, the pGL CRPV (PL-PE7) reporter construct was used that contains, in addition to CRPV early promoters, the viral origin of replication and thus replicates in the presence of E1 and E2. Interestingly, the coexpression of E1 with E2 K111R and A mutant proteins resulted in stimulation of the CRPV promoter reporter plasmid similar to that of wt E2 (Fig. 6B). This suggested that the coexpression of E1 not only changes the subcellular localization of these mutant proteins but also renders them functionally active. In contrast, the K111Q mutation, despite being localized to the nucleus, showed significantly impaired activity. To investigate whether the reduced activity is due to an impaired interaction between E2 K111Q and Brd4 fl in the presence of E1, we carried out FACS-FRET measurements as described above. This revealed that the addition of E1 did not significantly restore the interaction between E2 K111Q and Brd4 fl (Fig. 6C).
FIG 6.

E2 K111 mutant proteins relocalize to nucleus in the presence of CRPV E1. (A) C33a cells were cotransfected with 500 ng E1 expression vector (pSG 3×HA:CRPV E1) and 500 ng E2 expression vectors (as indicated on the left). Forty-eight hours after transfection, cells were stained with an anti-HA antibody to detect the E1 protein (red) and an anti-CRPV E2 antibody to detect E2 proteins (green) and analyzed by immunofluorescence microscopy. DNA was stained with DAPI (blue). (B) C33a cells were transiently transfected with 50 ng pGL CRPV (PL-PE7) firefly reporter construct, 100 ng E1 expression vector (pSG CRPV E1), and 10 ng of the different E2 expression vectors. Empty expression vector (pSG5) was used to adjust differences in DNA amounts. The values represent the relative luciferase activities of E1/E2 (E2 wild-type or E2 K111 mutant proteins) to the basal activity of the used firefly reporter construct. Error bars indicate the standard errors of the means (SEM) from at least five independent experiments. A paired two-tailed t test was used to determine statistical significance (*, P < 0.05). ns, nonsignificant. (C) C33a cells were cotransfected with expression vectors for BFP and SYFP fusion proteins and, as indicated, increasing DNA amounts of CRPV E1 expression vector (pSG CRPV E1). SYFP-BFP fusion protein was used as a positive control, and cotransfected BFP and SYFP expression vectors were negative controls. Shown are the mean values of FRET-positive cells from at least three independent experiments. Error bars indicate the SEM. A paired two-tailed t test was used to determine statistical significance (**, P < 0.01).
Relocalization of E1 mt by E2 is dependent upon binding of E1 to E2 but not vice versa.
This result indicated that E1 can relocalize E2 K111 mutant proteins to the nucleus. Therefore, we investigated whether this is dependent upon the conserved bipartite NLS and/or the NES in the amino terminus of E1 described for BPV1, HPV11, and HPV31 (2). An alignment of CRPV E1 with HPV11 and HPV31 revealed that the bipartite NLS, the NES, the cyclin binding motif, and regulatory serines are highly conserved between these PV types (Fig. 7A). Respective mutations in E1, namely, CRPV E1 NLS mt, NES mt, and NLS mt/NES mt, were generated, and subcellular localization of these E1 proteins was tested by immunofluorescence analysis upon transfection of C33a cells (Fig. 7B). In addition, the subcellular distributions of the different E1 and E2 wild-type and mutant proteins were quantified in two independent experiments by a combinatorial flow cytometry-microscopy assay using an ImageStreamX Mark II imaging flow cytometer. Cells were stained in solution with 4′,6-diamidino-2-phenylindole (DAPI) to mark nuclei, and anti-E2 and anti-HA (to detect E1) antibodies and images of at least 247 E2-positive or 638 E1-positive cells per condition were acquired. A modified Pearson correlation coefficient (similarity score) for the E1 and DAPI and E2 and DAPI signals was determined for individual cells using IDEAS image analysis software (Fig. 8C). A decrease in the mean similarity score (mss) for the E1/DAPI or E2/DAPI signals indicates a more cytoplasmic distribution and an increase a more nuclear distribution for the corresponding E1 or E2 protein (Fig. 8C). In line with previously published data for HPV11 and HPV31, CRPV E1 NLS mt was localized to the cytoplasm and displayed an mss of −0.11 compared to an mss of 0.85 for wt E1 (Fig. 7B and 8C). The E1 NES mt showed a predominant nuclear localization and an increased mss of 1.62 (Fig. 7B and 8C). The combined E1 NLS mt/NES mt was evenly distributed between the nucleus and cytoplasm and, in line with this, displayed an mss of 0.65, which is reduced compared to that of wt E1 (Fig. 7B and 8C). Consistent with HPV11 or HPV31 E1 NLS mt (12, 23), coexpression of wt E2 resulted in a predominant nuclear localization of E1 NLS mt and increased the mss to 1.55 (Fig. 8). This was also the case for E1 NLS mt/NES mt, for which wt E2 increased the nuclear distribution (mss of 1.64) (Fig. 8). The nuclear localization of wt E1 was also increased in the presence of wt E2 (mss of 1.59), whereas the coexpression of wt E2 with the E1 NES mt only slightly increased its nuclear distribution (mss of 1.82) (Fig. 8). The coexpression of the different E1 proteins did not alter the wt E2 mss of 1.38 (Fig. 8C). In contrast, the cytoplasmic distribution of E2 K111Q (mss of 0.49) was shifted dramatically to the nucleus by all coexpressed E1 proteins (Fig. 8). Accordingly, the mss increased to 1.62 for wt E1, 1.59 for E1 NLS mt, 1.66 for E1 NES mt, and 1.63 for E1 NLSmt/NES mt, which is similar to values for wt E2 and indicates that nuclear redistribution of E2 K111Q is not dependent upon E1's bipartite NLS (Fig. 8). Surprisingly, E1 NLS mt and the NLS mt/NES mt retained their cytoplasmic and cytoplasmic/nuclear localization (Fig. 8A). Consistent with this, E2 K111Q increased the mss of E1 NLS mt and E1 NLS mt/NES mt to much lower levels than those of wt E2 (Fig. 8C).
FIG 7.

Generation and subcellular localization of CRPV E1 NLS, NES, and NLS/NES mutant proteins. (A) Partial sequence alignment of HPV11, HPV31, and CRPV E1 proteins. Sequences were obtained from https://pave.niaid.nih.gov/. Highlighted are the bipartite nuclear localization signal (NLS), the embedded nuclear export signal (NES), and the cyclin binding motif (CBM). Shown below are the sequences of the generated CRPV E1 mutant proteins. Mutated residues are shown in boldface and are underlined. (B) C33a cells were transfected with 500 ng E1 expression vectors (pSG 3×HA:CRPV E1, pSG 3×HA:CRPV E1 NLS mt, pSG 3×HA:CRPV E1 NES mt, or pSG 3×HA:CRPV E1 NLS mt/NES mt). Forty-eight hours after transfection, cells were stained with an anti-HA antibody to detect E1 proteins (red) and analyzed by immunofluorescence microscopy. DNA was stained with DAPI (blue).
FIG 8.

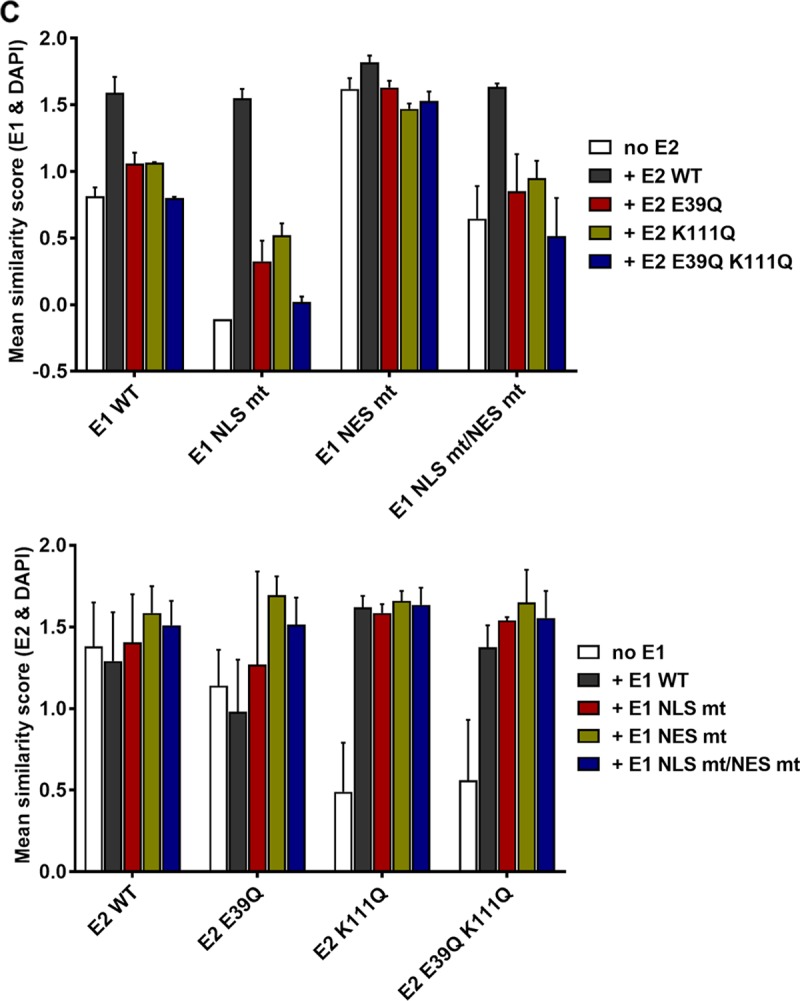
CRPV E1 does not recruit CRPV E2 into the nucleus by direct binding. (A) C33a cells were cotransfected with 500 ng E1 expression vector (pSG 3×HA:CRPV E1, pSG 3×HA:CRPV E1 NLS mt, pSG 3×HA:CRPV E1 NES mt, or pSG 3×HA:CRPV E1 NLS mt/NES mt) and 500 ng E2 expression vector (pSG CRPV E2, pSG CRPV E2 E39Q, pSG CRPV E2 K111Q, or pSG CRPV E2 E39Q/K111Q). Forty-eight hours after transfection, cells were stained with an anti-HA antibody to detect E1 proteins (red) and an anti-CRPV-E2 antibody to detect E2 proteins (green). (B) C33a cells were transfected with 500 ng E2 expression vectors (pSG CRPV E2 E39Q or pSG CRPV E2 E39Q/K111Q). After 48 h, cells were stained with an anti-CRPV-E2 antibody to detect E2 proteins. Cells were analyzed by immunofluorescence microscopy. DNA was stained with DAPI (blue). (C) Subcellular distribution of E1 and E2 wt and mt proteins. C33a cells were transfected with wt and mt E1 and E2 alone (no E1, no E2) or in combination as indicated, and mean similarity scores for E1 and DAPI (upper) and E2 and DAPI (lower) were obtained after staining for DAPI, anti-E1 (HA), and anti-E2. Single-cell images in the focus plane were recorded and analyzed using IDEAS image analysis software. The data shown are from two independent experiments and are derived from at least 247 E2-positive and 637 E1-positive cells per experiment.
In addition, we analyzed the functional consequences of the different E1 and E2 wild-type and mutant proteins to stimulate transient replication activity. Reporter assays revealed that wt E1 and E1 NES mt proteins alone activated the reporter 8-fold, whereas E1 NLS mt and E1 NLS mt/NES mt were less active, indicating that the activation is caused by nuclear E1. This finding is consistent with observations that the amino-terminal domain of different HPV E1 proteins acts as a transcriptional activator in yeast and C33a cells (24). wt E1 in combination with wt E2 activated the reporter ∼200-fold (Fig. 9A). Similar activation levels were observed for the combination of wt E2 with the different E1 mutant proteins, consistent with their E2-induced nuclear localization. The combination of the different E1 mutant proteins with E2 K111Q always resulted in activation levels that were comparable to those of the respective E1 proteins alone, suggesting that E2 K111Q, despite being localized to the nucleus, is severely deficient in supporting activation of the reporter. Consistent with the cytoplasmic localization of the E1 NLS mt, the combination of this mutation with E2 K111Q resulted in the lowest levels of reporter gene activation. To unlink E2 transcription activity and reporter replication, we measured the amounts of newly replicated pGL CRPV (PL-PE7) reporter constructs by quantitative PCR (qPCR) after digestion with DpnI as previously described (25). wt E1, wt E2, or E2 E39Q alone did not induce replication of the reporter construct (Fig. 9B). wt E1 in combination with wt E2 induced a 15.4-fold increase of newly replicated CRPV reporter construct, whereas wt E1 combined with E2 K111Q gave rise to only a 2.6-fold increase (Fig. 9B). This indicated that the E2 K111Q mutation is greatly impaired not only in transcriptional activation but also in supporting origin replication.
FIG 9.

Functional analysis of E1 and E2 mutant proteins. C33a cells were transiently transfected with 50 ng pGL CRPV(PL-PE7) firefly reporter construct, 100 ng E1 expression vectors, and 10 ng E2 expression vectors (as indicated). Empty expression vector (pSG5) was used to adjust differences in DNA amounts. (A) The values represent the relative luciferase activities of either E1, E2, or E1/E2 to the basal activity of the used firefly reporter construct. Error bars indicate the standard errors of the means (SEM) from at least six independent experiments. (B) Total DNA was extracted and digested with DpnI. The extent of DNA replication of the pGL CRPV (PL-PE7) plasmid was determined by qPCR. Data are shown relative to the extent of replication in the presence of the empty vector (vector). Error bars indicate the SEM.
To test whether the localization of E2 K111Q into the nucleus by E1 was mediated by a direct interaction between E1 and E2, we made use of the E2 E39Q mutation, which impairs binding to E1 (26). When expressed alone, E2 E39Q displayed a nuclear localization with an mss of 1.14 (Fig. 8). However, in contrast to wt E2, it only weakly induced nuclear localization of E1 NLS mt (mss of 0.33 versus 1.55 for wt E2) or NLS mt/NES mt (mss of 0.85 versus 1.64 for wt E2) (Fig. 8). This is similar to findings for HPV31 (12) and strongly suggests that a direct interaction between E1 and E2 is required for the nuclear localization of E1 NLS mt by E2. Combining E39Q with K111Q resulted in a cytoplasmic E2 protein when expressed alone with an mss of 0.56 (Fig. 8). In combination with wt or mt E1, E2 E39Q/K111Q was relocalized to the nucleus, comparable to E2 K111Q, and the mss increased to 1.38 (wt E1), 1.54 (E1 NLS mt), 1.65 (E1 NES mt), and 1.56 (E1 NLS mt/NES mt) (Fig. 8). This surprisingly suggested that E39Q/K111Q, despite being impaired in the direct E1-E2 interaction, is localized to the nucleus only when E1 is present. Reporter assays indicated that E2 E39Q alone is able to activate the reporter as well as E2 wt, which is consistent with previous observations (Fig. 9A) (9). In combination with wt E1, a weaker activation of transcription and DNA replication by E2 E39Q than by wt E2 was observed (Fig. 9A), which indicates that reporter gene expression is a combination of activation of transcription by E1 and E2 and replication induced by E1 and E2. When combined with E1 NLS mt, NES mt, or NLS mt/NES mt, E2 E39Q displayed reduced activity compared to that of E2 wt, which was especially pronounced for the E1 NLS mt (Fig. 9A). Surprisingly, E2 E39Q/K111Q diminished activation by all E1 proteins to lower levels than those with E2 K111Q alone (Fig. 9A). This suggests that the loss of interaction between E1 and the E2 K111Q mutant protein creates a transcriptional inhibitor that blocks E1's ability to activate transcription. Furthermore, E2 E39Q/K111Q did not support plasmid replication (Fig. 9B).
DISCUSSION
The PV E2 protein is a multifunctional protein which is involved in viral genome replication, viral transcription, and nuclear retention of viral genomes after cell division. Residues K111 and K112 are highly conserved among PV E2 proteins, and several studies have shown that the conservative mutation of K111 to R results in proteins that lose their ability to stimulate transcription and to activate replication in conjunction with E1 (17–20). A comparable phenotype has been observed for the K111A mutation in HPV11, BPV1, and HPV31 E2 (16, 19, 20). This can be explained by the observation that BPV1 E2 K111A and R mutant proteins are predominantly cytoplasmic instead of nuclear (16, 17). Recent studies have shown that K111 is also subject to acetylation in BPV1 and HPV31 E2 (17, 20). A HPV31 K111Q mutation, which mimics acetylated lysine, retained all activities and displayed nuclear localization.
Using CRPV E2 as a model, we can confirm that mutation of K111 to A or R gives rise to proteins which no longer activate transcription and are mislocalized to the cytoplasm. However, we also find that the CRPV E2 K111Q mt behaves identically to the A and R mutations, indicating substantial differences between HPV31 and CRPV. The lack of transcriptional activation is not simply due to mislocalization, as the recruitment of E2 K111 mt into the nucleus by addition of an SV40 NLS does not restore transcriptional activation. In line with this, an in vivo protein-protein interaction analysis indicated that E2 K111Q has reduced binding to Brd4. Thus, mutation of CRPV E2 K111 changes not only subcellular localization but also the interaction with Brd4. Residue K111 is not involved in the direct interaction between HPV16 E2 aa 1 to 201 and Brd4 aa 1343 to 1362 (7). The reduced interaction could therefore be due to the induction of structural changes by K111Q, which might affect the identified E2-Brd4 interaction surface, or that the full-length Brd4 protein has an extended interaction surface which includes K111.
BPV1 and HPV11 E2 K111 mutations also fail to stimulate E1/E2-dependent origin replication (16, 18, 19), which could be explained by their cytoplasmic localization. However, our data reveal that E2 K111 mutant proteins are localized to the nucleus in the presence of E1, which indicates that E1 can modulate the nuclear localization of E2. Nevertheless, nuclear E2 K111Q failed to stimulate the activity and the replication of the CRPV reporter plasmid in the presence of E1, which is in line with the lack of transcriptional activity observed for NLS-E2 K111Q. E2 K111Q might be transported into the nucleus in complex with E1 and use the bipartite NLS of E1. Inactivation of the conserved NLS in CRPV E1 resulted in a protein that was cytoplasmic, consistent with findings for BPV1, HPV11, and HPV31 proteins (12, 23, 27). Interestingly, the E1 NLS mt became nuclear in the presence of wt E2, confirming previous studies with HPV11 and HPV31 (12, 23). This provides further evidence that E1 and E2 influence each other's nuclear localization. Surprisingly, the coexpression of E2 K111Q with E1 NLS mt induced a nuclear localization of E2 K111Q but not of the E1 NLS mt. This indicates that it is not the bipartite NLS in E1 that directs the E2 K111Q mt to the nucleus. The analysis of the E1 NLS mt/NES mt revealed that this protein is, in contrast to the E1 NLS mt, evenly distributed between the cytoplasm and nucleus, indicating that the bipartite NLS is not completely inactivated by the introduced mutations, which we think is unlikely, as KRK was changed to GGK and KAVK to AAVG, removing 4 of the 5 basic residues within the bipartite NLS. Alternatively, a second NLS exists in E1. Therefore, we cannot fully exclude that E2 K111Q is recruited to the nucleus by an unknown NLS in E1. In the CRPV E2 E39Q mt, a crucial residue for the interaction with E1 is mutated, and in line with this, the protein is greatly impaired in supporting the E1/E2-dependent replication of the CRPV origin (9, 26). Coexpression of E2 E39Q did not direct the E1 NLS mt into the nucleus, confirming previous data obtained with HPV31 (12) and strongly suggesting that an interaction between E1 and E2 is required to direct E1, in the absence of its own NLS, to the nucleus. However, the E2 E39Q/K111Q mt showed a cytoplasmic localization that became nuclear in the presence of the E1 NLS mt, but the cytoplasmic location of E1 NLS mt was only slightly decreased. This does not support the idea that a direct interaction between E1 and E2 K111Q mt is responsible to direct E2 K111Q to the nucleus. Taken together, the findings that neither the conserved NLS in E1 nor the interaction between E1 and E2 are required to direct E2 K111Q to the nucleus strongly suggest that E1 does not recruit E2 into the nucleus by direct binding and providing a nuclear import function but rather by a different mechanism. The observation that cytoplasmic E1 is able to direct E2 K111 mutant proteins to the nucleus argues strongly that this is not mediated by nuclear activity of E1, such as the initiation of DNA replication or the induction of DNA damage (2). Currently, no functions of cytoplasmic E1 have been described, and further studies are required to elucidate the mechanism.
It is currently unclear why E2 K111 mutant proteins from diverse PV do not enter the nucleus. K111 could be a crucial residue within an NLS for E2. However, a corresponding peptide from BPV1 E2 does not act as a transferable NLS (14). Abroi et al. proposed that BPV1 E2 K111 forms aggregates which may prevent nuclear entry (16). The addition of an NLS resulted in nuclear CRPV E2 K111 mt proteins, making it more likely that the defect is specific for nuclear transport, as one would expect that large E2 aggregates would not be able to enter the nucleus. K111 is located in the fulcrum region of the protein, and mutations of K111 have been proposed to induce structural changes to the N-terminal domain of E2 (28, 29). In line with this, BPV1 E2 K111R shows a temperature-dependent transcriptional phenotype (29). The NLS of CRPV E2 have not yet been defined. The alpha helix important for DNA recognition that acts as an NLS in BPV1 and HPV16 is present in CRPV and thus might act as an NLS. Furthermore, prediction tools (NLStradamus; SeqNLS) indicate that CRPV E2 also has an NLS in the hinge region comparable to HPV11 (15). One or both of these putative NLS might be functional, but this still raises the question of how inactivation of K111 in the amino-terminal domain prevents the putative NLS in the hinge and DNA-binding domain from being active. Along these lines, it will be interesting to learn how expression of E1 reverses this phenotype without direct binding to E2.
In summary, our studies have uncovered novel links between E1 and E2 that control their nuclear localization. Direct binding of E2 to E1 can direct E1 to the nucleus independently from the E1 NLS; on the other hand, E1 can direct E2 to the nucleus without direct binding and an intact NLS. Future studies are required to investigate the underlying mechanisms and their relevance for papillomavirus infections in vitro and in vivo.
MATERIALS AND METHODS
Recombinant plasmids.
Luciferase reporter plasmids pC18-Sp1-luc and pGL CRPV (PL-PE7), containing the CRPV nucleotides [nt] 7346 to 1074 and thus placing the E7 ATG in frame with firefly luciferase, have been previously described (10, 30). The expression plasmids pSG CRPV E2, pSG CRPV E2 E39Q, and pSG CRPV E1 have been previously described (9). To generate CRPV, E2 mutant plasmids pSG CRPV E2 and pSG CRPV E2 E39Q were used as a template, and site-specific mutagenesis PCRs were conducted using one of the associated oligonucleotides (available on request), in combination with the upstream primer E2 225 F (5′-GGTGCTGTACATTGAAAGCCTACTCAGG-3′) and the downstream primer E2 713 R (5′-AGTGTACGCGTGGTTTTGGGCCCTGT-3′). The resulting amplicons were cloned into BsrGI/MluI-digested pSG CRPV E2. To attach the SV40 T antigen NLS to the N terminus of CRPV E2 or CRPV E2 K111 mutations, a corresponding fragment was synthesized (Life Technologies) containing an MfeI restriction site at the 5′ end and a BsrGI restriction site at the 3′ end and cloned into MfeI/BsrGI-digested pSG CRPV E2 or pSG CRPV E2 K111 mt. To obtain plasmid pSG 3×HA:CRPV E1, the coding region of CRPV E1 (CRPV nucleotides [nt] 1365 to 3170) was amplified by PCR using plasmid pLAII CRPV (31) as a template, with an upstream primer containing a BamHI restriction site (5′-TATATAGGATCCGCTGAAGGTACAGAC-3′) and a downstream primer with a BamHI restriction site (5′-CCAAACTCGTGCTCGGATCCTCATAGAG-3′) (9). The amplicon was cloned into BamHI-digested pSG 3×HA vector. To generate pSG 3×HA:CRPV E1 NLS mt, pSG 3×HA:CRPV E1 NES mt, and pSG 3×HA:CRPV E1 NLS mt/NES mt or pSG CRPV E1 NLS mt, pSG CRPV E1 NES mt and pSG CRPV E1 NLS mt/NES mt fragments containing the respective mutations were synthesized by Life Technologies, containing a BsgI restriction site at the 5′ end and a SacI restriction site at the 3′ end, and cloned into BsgI/SacI-digested pSG 3×HA:CRPV E1 or pSG CRPV E1. The pmTagBFP-C1 and pSYFP2-N1 plasmids (kindly provided by M. Schindler) have been used to generate BFP- and YFP-labeled fusion proteins. To obtain the BFP-SYFP fusion protein, the coding region for SYFP was amplified by PCR using plasmid pSYFP2-N1 as a template, with an upstream primer containing an XhoI restriction site (5′-TACTCGAGCTATGGTGAGCAAGGGC-3′) and a downstream primer containing an HpaI restriction site (5′-GCTGCAATAAACAAGTTAACAACA-3′). The amplicon was cloned into XhoI/HpaI-digested pmTagBFP-C1. For the generation of pmTagBFP-C1-NLS:CRPV E2 and pmTagBFP-C1-NLS:CRPV E2 K111Q, the coding region for each CRPV E2 protein was amplified by PCR using the corresponding constructs, with an upstream primer containing an EcoRI restriction site (5′-TAGGGCGAATTCCATGCCCAAGAA-3′) and a downstream primer containing a BamHI restriction site (5′-TCCGGTGGATCCTAAAGCCCA-3′). The PCR products were cloned into EcoRI/BamHI-digested pmTagBFP-C1. To obtain pSYFP2-N1-Brd4 and pSYFP2-N1-Brd4ΔCTM, each coding region was amplified by PCR using pcDNA3-F:hBrd4 or pcDNA3-F:hBrd4(1-1223), respectively (22). For both amplicons, an upstream primer containing an XhoI restriction site (5′-TATATCTCGAGCTATGTCTGCGGAGAGCGG-3′) and two individual downstream primers containing a BamHI restriction site (Brd4, 5′-GCGCGGATCCGCGAAAAGATTTTCT-3′; Brd4ΔCTM, 5′-ATATGGATCCAAGCTGTCGCTGGATGAC-3′) were used. Each amplicon was cloned into XhoI/BamHI-digested pSYFP2-N1. The newly generated constructs were verified by DNA sequencing (GATC, Constance, Germany).
Cell culture.
C33a cells were cultivated in Dulbecco's modified Eagle medium (DMEM) (Life Technologies) supplemented with 10% fetal bovine serum (FBS).
Luciferase-based reporter assays.
For the measurement of the transactivation or transient viral DNA replication activity, 7 × 104 C33a cells were seeded into the wells of a 24-well tissue culture plate the day before transfection. To determine transactivation activity, 50 ng reporter plasmid and 10 ng E2 expression plasmid were diluted in 25 μl Opti-MEM (Invitrogen). To determine transient viral DNA replication activities, 50 ng reporter plasmid, 100 ng E1 expression plasmid, and 10 ng E2 expression plasmid were cotransfected using Opti-MEM (Invitrogen) and FuGENE HD transfection reagent (Promega). If necessary, DNA amounts were adjusted using the respective empty vector.
Forty-eight hours after DNA transfection, luciferase-based reporter assays were carried out as previously described (30).
Quantification of transiently replicating luciferase-based reporter plasmid.
To quantify the transient replication activity, cells were harvested 48 h after DNA transfection. Whole-cell extracts were prepared and used to extract total DNA using the EZ1 DNA tissue kit and the EZ1 work station according to the manufacturer's protocol (Qiagen). As previously described, a quantitative real-time PCR was conducted to quantify the amount of replicated DNA (25).
Immunoblot analysis.
Approximately 1 × 106 C33a cells were seeded into 60-mm culture dishes. The following day, cells were transfected with 1 μg of expression vector DNA, and 48 h posttransfection cells were harvested. The cells were lysed in 100 μl 4× Roti-Load 1 (Carl Roth) and heated to 95°C for 5 min. Equal aliquots were separated in 10% SDS-PAGE, and proteins were transferred to a nitrocellulose membrane (Amersham Protran; GE Healthcare) in a buffer containing 10 mM 3-(cyclohexylamino)-1-propanesulfonic acid (CAPS) (pH 10.3) and 10% methanol at 90 V for 90 min. The membrane was blocked with 5% nonfat dry milk in phosphate-buffered saline (PBS) for 60 min at room temperature. Membranes were incubated with 1:1,000-diluted primary antibodies anti-CRPV E2 (recombinant CRPV E2193–390 protein was used to generate a chicken polyclonal antibody; Davids Biotechnology GmbH, Regensburg, Germany) and anti-heat shock protein 90 (HSP90; sc-69703; Santa Cruz Biotechnology). Bound antibodies were detected with 1:15,000-diluted fluorescence-labeled antibodies (IRDye 680RD goat anti-mouse IgG and IRDye 800CW donkey anti-chicken IgG; LI-COR Biosciences) and an Odyssey FC infrared imaging system (LI-COR Biosciences).
Immunofluorescence microscopy.
For immunofluorescence microscopy, approximately 1 × 105 C33a cells were seeded on coverslips in 6-well culture dishes the day before transfection. Cells were transfected with the according expression vector DNA as indicated in the figures. Forty-eight hours later, the cells were washed twice with PBS and fixed and permeabilized with methanol-acetone (1:1) for 2 min. Cells were incubated with primary antibodies (anti-CRPV E2 [in-house production], 1:50; anti-HA tag, 1:1,600; no. 3724; Cell Signaling) diluted in PBS–3% bovine serum albumin (BSA) in a humidified chamber for 16 h at 4°C. Bound antibodies were detected with anti-rabbit Alexa Fluor 555 (A-21428; 1:2,000; Life Technologies) or anti-chicken Alexa Fluor 488 (A-11039; 1:2,000; Life Technologies) conjugated to a fluorescence dye. Samples were mounted and counterstained with Roti-Mount FluorCare DAPI (Carl Roth). A Zeiss Axio Observer microscope and the appropriate filter sets in combination with a Zeiss ApoTome was used to record immunofluorescence signals as previously described (32).
Flow cytometry-based FRET analysis.
For the flow cytometry-based FRET analysis of protein-protein interactions, approximately 1.5 × 105 C33a cells were seeded into the wells of a 12-well tissue culture plate the day before transfection. The next day, cells were transfected with 500 ng BFP fusion protein and 500 ng of SYFP fusion protein expression vector. After 48 h, cells were washed with cold PBS and harvested using 0.05% trypsin-EDTA (Life Technologies). The cell suspension was diluted in 1 ml 1% fetal calf serum (FCS) in PBS and used for further steps. Flow cytometry-FRET measurements were performed using a MACSQuant VYB (Miltenyi Biotec) equipped with 405-nm, 488-nm, and 561-nm lasers. The detailed gating strategy for FRET-positive cells is depicted in Fig. 5A and has been previously described (21). For the evaluation of the data, MACSQuant software (Miltenyi Biotec) was used.
Subcellular distribution analysis via imaging flow cytometry.
Approximately 1.5 × 105 C33a cells were seeded into the wells of a 12-well tissue culture plate the day before transfection. Cells were transfected with a total amount of 1 μg E1 and/or E2 expression vector DNA. After 48 h cells were harvested as described for the flow cytometry-based FRET analysis. Cell suspensions were transferred into wells of a 96-well tissue culture plate and centrifuged (500 × g; 5 min) to collect the cells. Cells were fixed and permeabilized with methanol-acetone (1:1) for 2 min, washed twice with PBS–1% FCS, and incubated with primary antibodies (anti-CRPV E2 [in-house production], 1:50; anti-HA tag, no. 3724; 1:1,600; Cell Signaling) diluted in PBS–3% BSA for 16 h at 4°C. The next day, cells were collected via centrifugation and washed three times with PBS–1% FCS. Cells were resuspended, and primary antibodies were detected with anti-rabbit Alexa Fluor 555 (A-21428; 1:2,000; Life Technologies) or anti-chicken Alexa Fluor 488 (A-11039; 1:2,000; Life Technologies) conjugated to a fluorescence dye. After 1 h of incubation at room temperature, the cells were again collected and washed three times. DAPI was used for counterstaining.
At least 1 × 104 C33a cells were acquired with the INSPIRE instrument controller software (version 200.1.620.0; Merck-Millipore/Amnis, USA) on an ImageStreamX Mark II imaging flow cytometer (Amnis, USA) at ×40 magnification, with lasers of 405 nm (45.00 mW), 488 nm (5.00 mW), and 561 nm (150.00 mW) and side scatter of 782 nm (2.26 mW). Data were analyzed with IDEAS image analysis software (version 6.2.187.0). All samples were gated on single cells in focus. For quantification of subcellular distribution of the CRPV replication proteins E1 and E2, the similarity feature was applied by using the Nuclear Location wizard. The similarity feature is a measure of the degree to which two images are linearly correlated within a masked region.
ACKNOWLEDGMENTS
We thank S. Pöschel and K. Schenke-Layland from the Core Facility Imagestream, Tübingen, for excellent technical support.
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (SFB773 B4) to T.I. and by grants from the Ministry of Science, Research and Arts of Baden Württemberg (Az. SI-BW01222-91) and the Deutsche Forschungsgemeinschaft DFG (German Research Foundation) (Az. INST 2388/33-1).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Parkin DM, Bray F. 2006. Chapter 2: the burden of HPV-related cancers. Vaccine 24(Suppl 3):S3/11–S3/25. doi: 10.1016/j.vaccine.2005.01.101. [DOI] [PubMed] [Google Scholar]
- 2.Bergvall M, Melendy T, Archambault J. 2013. The E1 proteins. Virology 445:35–56. doi: 10.1016/j.virol.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McBride AA. 2013. The papillomavirus E2 proteins. Virology 445:57–79. doi: 10.1016/j.virol.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McPhillips MG, Oliveira JG, Spindler JE, Mitra R, McBride AA. 2006. Brd4 is required for e2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J Virol 80:9530–9543. doi: 10.1128/JVI.01105-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schweiger MR, You J, Howley PM. 2006. Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function. J Virol 80:4276–4285. doi: 10.1128/JVI.80.9.4276-4285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu SY, Lee AY, Hou SY, Kemper JK, Erdjument-Bromage H, Tempst P, Chiang CM. 2006. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev 20:2383–2396. doi: 10.1101/gad.1448206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abbate EA, Voitenleitner C, Botchan MR. 2006. Structure of the papillomavirus DNA-tethering complex E2:Brd4 and a peptide that ablates HPV chromosomal association. Mol Cell 24:877–889. doi: 10.1016/j.molcel.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Senechal H, Poirier GG, Coulombe B, Laimins LA, Archambault J. 2007. Amino acid substitutions that specifically impair the transcriptional activity of papillomavirus E2 affect binding to the long isoform of Brd4. Virology 358:10–17. doi: 10.1016/j.virol.2006.08.035. [DOI] [PubMed] [Google Scholar]
- 9.Jeckel S, Huber E, Stubenrauch F, Iftner T. 2002. A transactivator function of cottontail rabbit papillomavirus e2 is essential for tumor induction in rabbits. J Virol 76:11209–11215. doi: 10.1128/JVI.76.22.11209-11215.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delcuratolo M, Fertey J, Schneider M, Schuetz J, Leiprecht N, Hudjetz B, Brodbeck S, Corall S, Dreer M, Schwab RM, Grimm M, Wu SY, Stubenrauch F, Chiang CM, Iftner T. 2016. Papillomavirus-associated tumor formation critically depends on c-Fos expression induced by viral protein E2 and bromodomain protein Brd4. PLoS Pathog 12:e1005366. doi: 10.1371/journal.ppat.1005366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng W, Lin BY, Jin G, Wheeler CG, Ma T, Harper JW, Broker TR, Chow LT. 2004. Cyclin/CDK regulates the nucleocytoplasmic localization of the human papillomavirus E1 DNA helicase. J Virol 78:13954–13965. doi: 10.1128/JVI.78.24.13954-13965.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fradet-Turcotte A, Moody C, Laimins LA, Archambault J. 2010. Nuclear export of human papillomavirus type 31 E1 is regulated by Cdk2 phosphorylation and required for viral genome maintenance. J Virol 84:11747–11760. doi: 10.1128/JVI.01445-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klucevsek K, Wertz M, Lucchi J, Leszczynski A, Moroianu J. 2007. Characterization of the nuclear localization signal of high risk HPV16 E2 protein. Virology 360:191–198. doi: 10.1016/j.virol.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skiadopoulos MH, McBride AA. 1996. The bovine papillomavirus type 1 E2 transactivator and repressor proteins use different nuclear localization signals. J Virol 70:1117–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zou N, Lin BY, Duan F, Lee KY, Jin G, Guan R, Yao G, Lefkowitz EJ, Broker TR, Chow LT. 2000. The hinge of the human papillomavirus type 11 E2 protein contains major determinants for nuclear localization and nuclear matrix association. J Virol 74:3761–3770. doi: 10.1128/JVI.74.8.3761-3770.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abroi A, Kurg R, Ustav M. 1996. Transcriptional and replicational activation functions in the bovine papillomavirus type 1 E2 protein are encoded by different structural determinants. J Virol 70:6169–6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quinlan EJ, Culleton SP, Wu SY, Chiang CM, Androphy EJ. 2013. Acetylation of conserved lysines in bovine papillomavirus E2 by p300. J Virol 87:1497–1507. doi: 10.1128/JVI.02771-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brokaw JL, Blanco M, McBride AA. 1996. Amino acids critical for the functions of the bovine papillomavirus type 1 E2 transactivator. J Virol 70:23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooper CS, Upmeyer SN, Winokur PL. 1998. Identification of single amino acids in the human papillomavirus 11 E2 protein critical for the transactivation or replication functions. Virology 241:312–322. doi: 10.1006/viro.1997.8941. [DOI] [PubMed] [Google Scholar]
- 20.Thomas Y, Androphy EJ. 2018. Human papillomavirus replication regulation by acetylation of a conserved lysine in the E2 protein. J Virol 92:e01912-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banning C, Votteler J, Hoffmann D, Koppensteiner H, Warmer M, Reimer R, Kirchhoff F, Schubert U, Hauber J, Schindler M. 2010. A flow cytometry-based FRET assay to identify and analyse protein-protein interactions in living cells. PLoS One 5:e9344. doi: 10.1371/journal.pone.0009344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee AY, Chiang CM. 2009. Chromatin adaptor Brd4 modulates E2 transcription activity and protein stability. J Biol Chem 284:2778–2786. doi: 10.1074/jbc.M805835200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu JH, Lin BY, Deng W, Broker TR, Chow LT. 2007. Mitogen-activated protein kinases activate the nuclear localization sequence of human papillomavirus type 11 E1 DNA helicase to promote efficient nuclear import. J Virol 81:5066–5078. doi: 10.1128/JVI.02480-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morin G, Fradet-Turcotte A, Di Lello P, Bergeron-Labrecque F, Omichinski JG, Archambault J. 2011. A conserved amphipathic helix in the N-terminal regulatory region of the papillomavirus E1 helicase is required for efficient viral DNA replication. J Virol 85:5287–5300. doi: 10.1128/JVI.01829-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Straub E, Fertey J, Dreer M, Iftner T, Stubenrauch F. 2015. Characterization of the human papillomavirus 16 E8 promoter. J Virol 89:7304–7313. doi: 10.1128/JVI.00616-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abbate EA, Berger JM, Botchan MR. 2004. The X-ray structure of the papillomavirus helicase in complex with its molecular matchmaker E2. Genes Dev 18:1981–1996. doi: 10.1101/gad.1220104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lentz MR, Pak D, Mohr I, Botchan MR. 1993. The E1 replication protein of bovine papillomavirus type 1 contains an extended nuclear localization signal that includes a p34cdc2 phosphorylation site. J Virol 67:1414–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antson AA, Burns JE, Moroz OV, Scott DJ, Sanders CM, Bronstein IB, Dodson GG, Wilson KS, Maitland NJ. 2000. Structure of the intact transactivation domain of the human papillomavirus E2 protein. Nature 403:805–809. doi: 10.1038/35001638. [DOI] [PubMed] [Google Scholar]
- 29.Zheng PS, Brokaw J, McBride AA. 2005. Conditional mutations in the mitotic chromosome binding function of the bovine papillomavirus type 1 E2 protein. J Virol 79:1500–1509. doi: 10.1128/JVI.79.3.1500-1509.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stubenrauch F, Zobel T, Iftner T. 2001. The E8 domain confers a novel long-distance transcriptional repression activity on the E8E2C protein of high-risk human papillomavirus type 31. J Virol 75:4139–4149. doi: 10.1128/JVI.75.9.4139-4149.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nasseri M, Meyers C, Wettstein FO. 1989. Genetic analysis of CRPV pathogenesis: the L1 open reading frame is dispensable for cellular transformation but is required for papilloma formation. Virology 170:321–325. doi: 10.1016/0042-6822(89)90388-7. [DOI] [PubMed] [Google Scholar]
- 32.van de Poel S, Dreer M, Velic A, Macek B, Baskaran P, Iftner T, Stubenrauch F. 2018. Identification and functional characterization of phosphorylation sites of the human papillomavirus 31 E8^E2 protein. J Virol 92:e01743-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]