

Hepatitis B virus (HBV) causes acute and chronic hepatitis. Approximately 260 million people are chronically infected with HBV and under an increased risk of developing cirrhosis and hepatocellular carcinoma. Host immune responses, particularly HBV-specific CD8+ T cell responses, largely determine the outcome of HBV infection. It is widely accepted that antigen inexperienced CD8+ T cells should be initially activated by professional antigen-presenting cells (pAPCs) in lymphoid tissues to differentiate into effector CD8+ T cells. However, this notion has not been tested for HBV-specific CD8+ T cells. In this study, we show that HBV-specific CD8+ T cell responses can be induced in the liver. Surprisingly, antigen presentation by hepatocytes is more important than cross-presentation by hematopoietic cells for the induction of HBV-specific CD8+ T cell responses. These results revealed a previously unappreciated role of antigen presentation by hepatocytes in the induction of HBV-specific CD8+ T cell responses.
KEYWORDS: hepatitis B virus, cross-presentation, endogenous antigen presentation, liver, intrahepatic lymphocytes, T cell priming, tolerance
ABSTRACT
CD8+ T cells are the key cellular effectors mediating the clearance of hepatitis B virus (HBV) infections. However, early immunological events surrounding the priming of HBV-specific CD8+ T cell responses remain poorly understood. This study examined the importance of priming location and the relative contribution of endogenous antigen presentation by hepatocytes versus cross-presentation by bone marrow-derived cells to the induction of functional HBV-specific CD8+ T cell responses using the animal models of acute and chronic HBV infection. Functional HBV-specific CD8+ T cell responses could be induced to intrahepatically expressed HBV even when T cell homing to the lymphoid tissues was severely suppressed, suggesting that functional priming could occur in the liver. The expansion of HBV-specific CD8+ T cells was significantly reduced in the mice whose major histocompatibility complex (MHC) class I expression was mostly restricted to nonhematopoietic cells, suggesting the importance of cross-presentation by hematopoietic cells in the induction of HBV-specific CD8+ T cells. Strikingly, the expansion and cytolytic differentiation of HBV-specific CD8+ T cells were reduced even more severely in the mice whose MHC class I expression was restricted to hematopoietic cells. Collectively, these results indicate that cross-presentation is required but relatively inefficient in terms of inducing the cytolytic differentiation of HBV-specific CD8+ T cells by itself. Instead, the expansion and functional differentiation of HBV-specific CD8+ T cells are primarily dependent on hepatocellular antigen presentation.
IMPORTANCE Hepatitis B virus (HBV) causes acute and chronic hepatitis. Approximately 260 million people are chronically infected with HBV and under an increased risk of developing cirrhosis and hepatocellular carcinoma. Host immune responses, particularly HBV-specific CD8+ T cell responses, largely determine the outcome of HBV infection. It is widely accepted that antigen inexperienced CD8+ T cells should be initially activated by professional antigen-presenting cells (pAPCs) in lymphoid tissues to differentiate into effector CD8+ T cells. However, this notion has not been tested for HBV-specific CD8+ T cells. In this study, we show that HBV-specific CD8+ T cell responses can be induced in the liver. Surprisingly, antigen presentation by hepatocytes is more important than cross-presentation by hematopoietic cells for the induction of HBV-specific CD8+ T cell responses. These results revealed a previously unappreciated role of antigen presentation by hepatocytes in the induction of HBV-specific CD8+ T cell responses.
INTRODUCTION
CD8+ T cells play a central role in eliminating viruses (1). Virus-specific CD8+ T cells recognize their cognate antigenic peptide associated with major histocompatibility complex (MHC) class I molecules to induce cell death and inflammatory cytokine expression, thereby preventing the spread of infection (1). MHC class I molecules normally present antigens that are synthesized in the cytoplasm. This process, termed endogenous antigen presentation, ensures that healthy bystander cells that have merely endocytosed viruses or virus-associated antigens are not targeted by virus-specific CD8+ T cells (2). To induce effective antiviral CD8+ T cell responses, the initial activation of T cells (i.e., T cell priming) should be mediated by bone marrow-derived professional antigen-presenting cells (pAPCs), typically by dendritic cells (DCs) (3). Therefore, against a virus that does not infect pAPCs, functional CD8+ T cell responses should be generated only if pAPCs acquire and present extracellular antigen on their MHC class I molecules. This process, termed cross-priming (4, 5) or cross-presentation, was shown to prime virus-specific CD8+ T cells during virus infection (6–8). However, the specific role of cross-presentation in priming antiviral CD8+ T cell responses is controversial with some arguing that cross-presentation is physiologically irrelevant (9, 10).
Hepatitis B virus (HBV) is a noncytopathic, enveloped, double-stranded DNA virus that causes acute and chronic hepatitis and hepatocellular carcinoma (11–13). Similar to other noncytopathic viruses, the clearance of HBV requires functional virus-specific CD8+ T cell responses (14). Despite the central role that HBV-specific CD8+ T cells play in HBV infection, very little is known about where and how they are generated during HBV infection. By adoptively transferring HBV-specific CD8+ T cell receptor (TCR) transgenic T cells into HBV transgenic mice, we have previously shown that HBV-specific naive T cells are activated in the liver and that intrahepatic antigen presentation induces functionally defective antigen-specific CD8+ T cell responses in a PD-1-dependent manner (15). Importantly, activation of myeloid dendritic cells (mDCs) by an agonistic anti-CD40 antibody induced vigorous expansion and functional differentiation of the adoptively transferred T cells (15). While these results suggest that activated mDCs are required for the maximum expansion and functional differentiation of adoptively transferred HBV-specific naive CD8+ T cells, we do not know whether mDCs are also required for normal T cell precursors in immunologically naive animals to differentiate into effector CD8+ T cells. Nor do we know the exact mechanism by which mDCs induce expansion and functional differentiation of HBV-specific CD8+ T cells after antigen recognition.
In the present study, we examined whether mDCs are required for generating HBV-specific CD8+ T cell responses from normal T cell precursors, as opposed to adoptively transferred TCR transgenic naive T cells, and whether functional HBV-specific CD8+ T responses could be primed in the liver using animal models of acute and chronic HBV infection (16, 17). We also examined the relative contribution of MHC class I expression on nonhematopoietic cells versus hematopoietic cells for the functional differentiation of HBV-specific CD8+ T cell responses that were observed after activation of mDCs by an agonistic anti-CD40 antibody (15). The results suggest that intrahepatic cross-presentation by DCs in the liver augments HBV-specific CD8+ T cell expansion, while concomitant or subsequent hepatocellular presentation of endogenously synthesized antigen is essential for expansion and cytolytic differentiation of HBV-specific CD8+ T cells induced by DC activation.
RESULTS
Myeloid dendritic cells are required for natural HBV-specific CD8+ T cell precursors to differentiate into effector T cells in a mouse model of acute HBV infection.
To examine whether DCs are required for generating HBV-specific T cell responses from natural T cell precursors in immunologically naive animals, we used CD11c-DOG mice that express the human diphtheria toxin (DTX) receptor on CD11c+ cells. The system allows us to deplete DCs by DTX administration with no signs of toxicity (18). Groups of 3 to 4 CD11c-DOG mice were treated with DTX or saline (NaCl), and 1 day later, hydrodynamically injected with a plasmid DNA encoding an HBV1.3 supergenomic DNA (1.3-HBV plasmid) to transduce HBV replication and gene expression in the liver (17). As controls, two groups of four C57BL/6 (B6) mice were also treated with DTX or saline and hydrodynamically injected with 1.3-HBV plasmid. The mice were then sacrificed on day 14 after hydrodynamic injection to analyze intrahepatic and splenic lymphocytes for the total number of CD8+ T cells specific for an epitope located between residues 93 and 100 of the HBV core region (COR93) and the extent to which they coexpress gamma interferon (IFN-γ) after in vitro stimulation by cognate COR93 peptide. As shown in Fig. 1A and B, at the time of hydrodynamic transfection, the frequencies of CD11c+ CD11b+ cells (mostly, myeloid DCs) and CD11c+ CD11b− cells (mostly, lymphoid DCs) were strongly reduced in the liver, lymph nodes, and spleen of CD11c-DOG mice by DTX administration (black bars) compared to NaCl (white). In contrast, DTX treatment of B6 mice did not reduce the frequencies of CD11c+ CD11b+ cells or CD11c+ CD11b− cells (Fig. 1C and D). As expected, COR93-specific CD8+ T cells were not detectable in the DTX-treated CD11c-DOG mice (Fig. 2A and B, black bars) on day 14 after hydrodynamic injection, while saline-treated control CD11c-DOG mice mounted vigorous, IFN-γ-producing COR93-specific CD8+ T cell responses in the liver (Fig. 2A and B, white bars). Importantly, HBV input DNA, as well as replicative intermediates, was still present in the livers of DTX-treated CD11c-DOG mice on day 14, presumably reflecting the absence of intrahepatic COR93-specific CD8+ T cell cells (Fig. 2C). In contrast, HBV input DNA and replication were abolished in the liver of saline-treated CD11c-DOG mice (Fig. 2C). DTX treatment of B6 mice had no impact on COR93-specific CD8+ T cell cells (Fig. 2D and E). Taken together, these results indicate that DCs are required for natural HBV-specific T cell precursors to differentiate into effector T cells in immunologically naive mice and eliminate the virus from the liver after hydrodynamic transduction of HBV.
FIG 1.
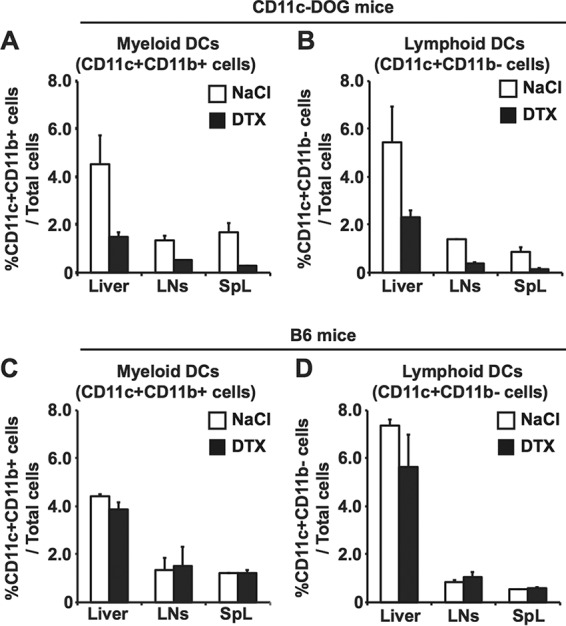
The efficiency of depletion of dendritic cells in CD11c-DOG mice by DTX. The frequencies of myeloid dendritic cells (CD11c+ CD11b+ cells) and lymphoid dendritic cells (CD11c+ CD11b− cells) in the livers, lymph nodes (LNs), and spleens (SpL) of CD11c-DOG mice (A and B) and B6 mice (C and D) were examined on day 1 after DTX (black bars) and saline (white bars) treatment. The data represent means ± the SD for three mice.
FIG 2.
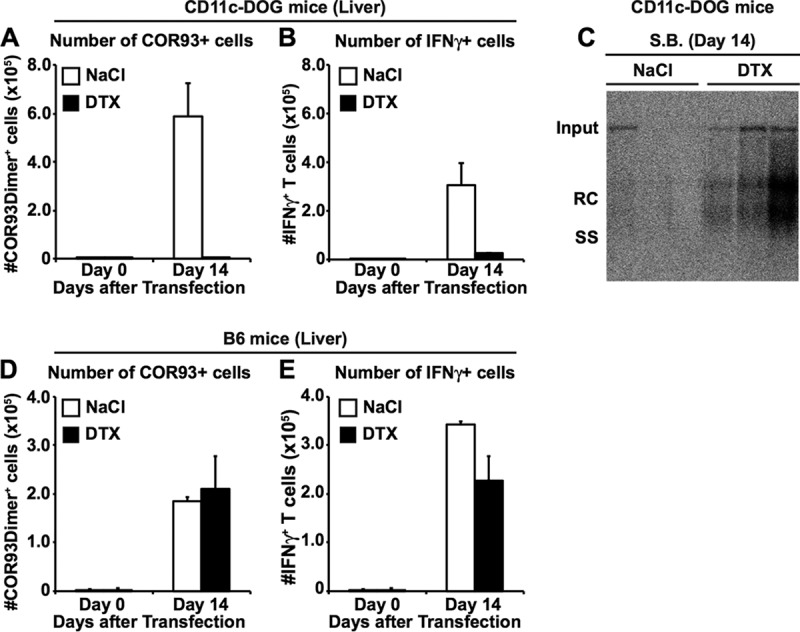
Dendritic cells are required for the induction of HBV-specific CD8+ T cells from natural T cell precursors. Groups of 3 to 4 CD11c-DOG mice and B6 mice were treated with either 200 ng of human DTX or saline and 1 day later hydrodynamically injected with HBV plasmid DNA and treated with same amount of DTX every other day thereafter. (A to C) On day 14 after hydrodynamic transfection of 1.3-HBV plasmid DNA, the CD11c-DOG and B6 mice were sacrificed for HBV-specific CD8+ T cell analyses. (A and B) Numbers of COR93-specific CD8+ T cells (A) and IFN-γ-producing CD8+ T cells (B) in the livers of CD11c-DOG mice that were treated either with saline (n = 3, white bars) or 200 μg of DTX (n = 4, black bars). The data represent means ± the SD for three or four mice. (C) Southern blot analysis (S.B.) of 30 μg of total liver DNA isolated from the mice shown in panel A and B. Bands corresponding to the expected size of the integrated transgene (Tg), relaxed circular (RC), and single-stranded (SS) HBV DNA are indicated. (D and E) The number of COR93-specific CD8+ T cells (D) and IFN-γ-producing CD8+ T cells (E) in the livers of B6 mice that were treated either with saline (n = 3, white bars) or 200 μg of DTX (n = 4, black bars). The data represent means ± the SD for three or four mice.
Induction of functional HBV-specific CD8+ T cell responses is independent of T cell homing to lymphoid organs.
We then examined whether HBV-specific CD8+ T cell responses could be induced in the liver independently of T cell homing to lymphoid organs. Groups of 3 to 4 B6 mice that had received splenectomies were injected with anti-CD62L antibodies (αCD62L) every other day to prevent naive T cell homing to the lymph nodes (19, 20). On the next day after the second αCD62L treatment, mice were hydrodynamically injected with 1.3-HBV plasmid DNA. The mice were sacrificed on day 14 after hydrodynamic transfection, and their intrahepatic lymphocytes were analyzed for the total number of COR93-specific CD8+ T cells (Fig. 3A) and the extent to which they coexpress IFN-γ after in vitro stimulation by cognate COR93 peptide (Fig. 3B). The results were compared to their counterparts in the control animals that received sham surgery and saline. As shown in Fig. 3, the number of multimer positive and IFN-γ-positive COR93-specific CD8+ T cell were comparable between αCD62L-treated (Fig. 3, black bar) and control animals (Fig. 3, white bar), indicating that HBV-specific CD8+ T cell responses can be efficiently induced in the livers of immunologically naive animals independently of T cell homing to the lymphoid tissues.
FIG 3.
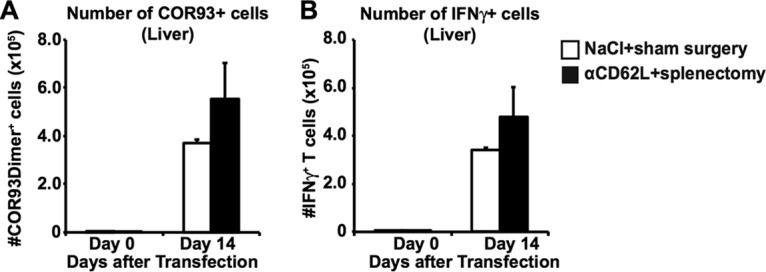
Functional COR93-specific CD8+ T cell responses after acute HBV transduction under reduced T cell homing to the lymphoid organs. The absolute number of COR93-specific CD8+ T cells (A) and the percentages of IFN-γ-producing COR93-specific CD8+ T cells (B) were analyzed in the livers of mice that received saline and sham surgery (white) or anti-CD62L antibodies (αCD62L) and splenectomy (black). Splenectomy was performed at least 1 month before the experiment. αCD62L was administered every other day. The data represent means ± the SD for three mice.
To confirm this notion, a group of 3 to 4 splenectomized B6 mice were adoptively transferred with naive CD8+ T cells isolated from transgenic mice that express a TCR-specific COR93 (15) and then treated with αCD62L every other day. On day 3 after T cell transfer, the mice were hydrodynamically transfected with HBV plasmid DNA, and intrahepatic, lymph nodal, and splenic COR93-specific CD8+ T cells were analyzed for CD69 expression 3 days later. The results were compared with those in control animals that received sham surgery and saline. As shown in Fig. 4A, administration of αCD62L reduced the homing of COR93-specific CD8+ T cells to the lymph nodes by 84.7%. Despite the 84.7% reduction in T cell homing to the lymph nodes, the intrahepatic COR93-specific CD8+ T cells in the αCD62L-treated mice were fully activated to express CD69, and they expanded as vigorously as their counterparts in control animals (Fig. 4B).
FIG 4.
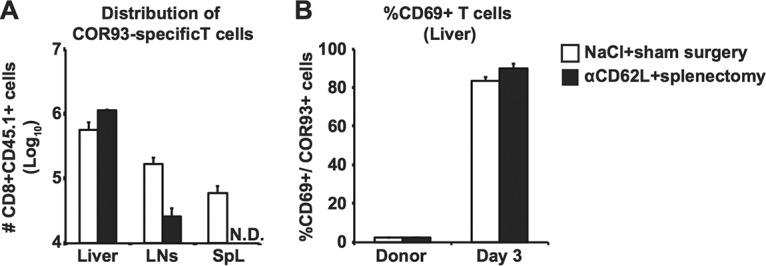
Normal expansion and activation of adoptively transferred TCR transgenic T cells after acute HBV transduction under reduced T cell homing to lymphoid organs. (A) Absolute numbers of adoptively transferred COR93-specific CD8+ T cells in the livers, lymph nodes, and spleens of the B6 mice that received saline plus sham surgery (white) or anti-CD62L antibodies (αCD62L) and splenectomy (black). Splenectomy and sham surgery were performed at least 1 month before the experiment. αCD62L was administered on days 1 and 2 after adoptive transfer and every 3 days thereafter. On the next day after adoptive transfer, the mice were hydrodynamically transfected with 1.3-HBV plasmid DNA. The absolute numbers of COR93-specific CD8+ T cells were monitored on day 3 after the hydrodynamic transfection. The lymph nodal data represent the total number of CD8+ CD45.1 cells in bilateral axillary, inguinal, and mesenteric lymph nodes. (B) Frequency of CD69 expressing cells among the intrahepatic COR93-specific CD8+ T cells shown in Fig. 4A. The data represent means ± the SD of three mice.
To test whether the intrahepatic induction of functional HBV-specific CD8+ T cells occurs irrespective of model systems, we performed a similar experiment with the HBV transgenic mouse lineage 1.3.32 that replicates HBV at a high level in the liver (16). A group of 3 to 4 lineage 1.3.32 transgenic mice received splenectomy or sham surgery. One month later, the mice were treated with αCD62L or control antibody every other day. On the next day after the second treatment with αCD62L or control antibody, the mice were treated with anti-CD40 antibody to activate mDCs and adoptively transferred with naive COR93-specific CD8+ T cells. Again, the homing of COR93-specific CD8+ T cells to the lymph nodes was almost completely blocked by administration of αCD62L on day 1 after adoptive transfer (data not shown). As shown in Fig. 5, HBV-specific CD8+ T cells expanded as vigorously in HBV transgenic mice that received splenectomy and αCD62Lantibody as in control HBV transgenic recipients that received sham surgery and control antibody (Fig. 5A). Furthermore, the fractions of IFN-γ-positive (Fig. 5B) and granzyme B-positive (Fig. 5C) COR93-specific CD8+ T cells were similar between the two groups. Collectively, these results confirm that intrahepatic T cell activation, expansion, and functional differentiation could occur in the liver irrespective of homing to the lymphoid tissues if mDCs are activated by the agonistic anti-CD40 antibody.
FIG 5.

Normal expansion and functional differentiation of HBV-specific T cells in HBV transgenic mice under reduced T cell homing to lymphoid organs. The absolute numbers of adoptively transferred COR93-specific CD8+ T cells (A), and the fractions of IFN-γ (B)- and granzyme B (C)-expressing COR93-specific CD8+ T cells in the livers of the HBV-transgenic mice that received saline plus sham surgery (white) or anti-CD62L antibodies (αCD62L) and splenectomy (black) were analyzed on day 7 after adoptive transfer. The numbers of COR93-specific CD8 T cells in HBV transgenic mice before adoptive transfer (Pre) and the fractions of IFN-γ and granzyme B-expressing donor COR93-specific CD8+ T cells (Donor) are also indicated in the corresponding panels. Splenectomy was performed at least 1 month before the experiment. αCD62L was administered 48 and 24 h before adoptive transfer and every 3 days after the second antibody administration. The data represent means ± the SD for three mice.
Antigen presentation by nonhematopoietic cells plays a dominant role in the robust expansion of HBV-specific CD8 T cells.
Next, we examined whether cross-presentation by hematopoietic cells is required for the HBV-specific T cell priming. Groups of 3 to 4 B6 mice and β2-microglobulin-deficient (β2m−/−) mice were lethally irradiated and reconstituted with bone marrow derived from β2m−/− mice and B6 mice, respectively. This should yield two groups of mice in which antigen presentation is mostly restricted to either nonhematopoietic cells (Fig. 6, red bars) or hematopoietic cells (Fig. 6, blue bars). B6 mice were reconstituted with bone marrow from B6 mice to serve as controls (Fig. 6, white bars). Six to ten weeks after bone marrow transplantation, we examined the reconstitution efficacy by analyzing the total number of lymphocytes, I-A/I-E+ cells (mostly professional antigen-presenting cells [pAPCs]), and CD11b+ cells (mostly myeloid cells) in the liver, as well as H-2Kb (i.e., MHC class I)-expressing cells in the corresponding population. As shown in Fig. 6A to C, the total numbers of lymphocytes, I-A/I-E+ cells, and CD11b+ cells were largely comparable between the three groups, although total number of lymphocytes and I-A/I-E + cells were significantly reduced in B6 mice that received β2m−/− bone marrow. The numbers of H-2Kb-expressing lymphocytes, I-A/I-E+ cells, and CD11b+ cells in B6 mice that received β2m−/− bone marrow (red bars) decreased to 14.1, 2.9, and 14.1% of the corresponding population in B6 mice that received B6 bone marrow (white bars), suggesting that MHC class I expression in B6 mice that received β2m−/− bone marrow (red bars) is mostly restricted to nonhematopoietic cells. In contrast, the numbers of H-2Kb-positive cells of these cell populations were largely comparable between B6 mice that received B6 bone marrow (white bars) and β2m−/− mice that received B6 bone marrow (blue bars), suggesting that MHC class I expression in β2m−/− mice that received B6 bone marrow (blue bars) is restricted to hematopoietic cells.
FIG 6.
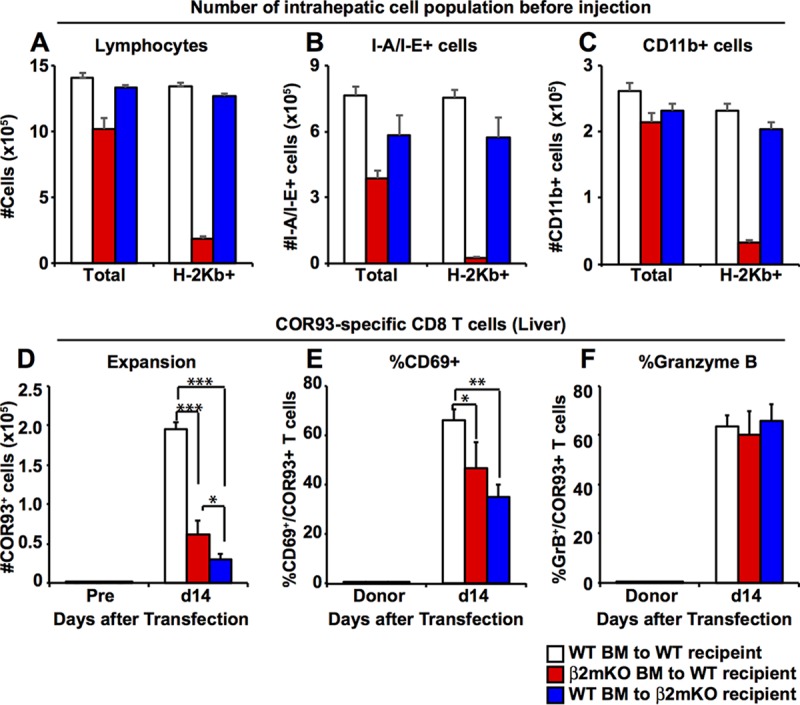
Relative contributions of MHC class I expression on hematopoietic cells versus nonhematopoietic cells to the induction of HBV-specific CD8+ T cells after HBV transduction. (A to C) Six to ten weeks after bone marrow transplantation, the reconstitution efficiency of bone marrow chimeric mice was evaluated by measuring the number of total and H-2Kb-expressing lymphocytes (A), I-A/I-E+ cells (mostly pAPCs) (B), and CD11b+ cells (mostly Kupffer cells, myeloid DCs, and neutrophils) (C). (D) The number of adoptively transferred COR93-specific CD8+ T cells in the liver of B6 mice in which MHC class I expression is mostly restricted to nonhematopoietic cells (red bars) or hematopoietic cells (blue bars) was analyzed on day 7 after hydrodynamic transfection with 1.3-HBV plasmid DNA. The number of COR93-specific T cells transferred to control B6 mice reconstituted with B6 bone marrow is shown in white. The numbers of COR93-specific CD8 T cells in the recipient mice (Pre) were analyzed before adoptive transfer of COR93-specific T cells. (E and F) Percentages of CD69-positive and GrB-positive COR93-specific CD8+ T cells in the liver, respectively. The data represent means ± the SD for three mice. The fractions of CD69+ and granzyme B-expressing donor COR93-specific CD8+ T cells (Donor) are also indicated.
Next, the reconstituted mice were adoptively transferred with naive COR93-specific CD8+ T cells and hydrodynamically injected with HBV plasmid DNA. The mice were sacrificed 2 weeks after hydrodynamic transfection to analyze COR93-specific CD8+ T cells for the expansion and the extent to which they coexpress CD69 and granzyme B (GrB).
As shown in Fig. 6D, intrahepatic COR93-specific naive CD8+ T cells expanded vigorously in the control animals (Fig. 6, white bars). In contrast, intrahepatic COR93-specific CD8+ T cells expanded much less vigorously in the mice whose MHC class I expression was mostly restricted to nonhematopoietic cells (red bar), suggesting that MHC class I expression on hematopoietic cells was required for robust expansion of HBV-specific CD8+ T cells after hydrodynamic transfection. Interestingly, however, MHC class I expression on hematopoietic cells alone was not sufficient to induce robust expansion of COR93-specific CD8+ T cells, since COR93-specific CD8+ T cells expanded even less vigorously in the mice whose MHC class I expression was restricted to hematopoietic cells (Fig. 6D, blue bar) than their counterparts in the mice whose MHC class I expression was mostly restricted to nonhematopoietic cells (Fig. 6D, red bar). As shown in Fig. 6E, approximately 65% of intrahepatic COR93-specific T cells expressed CD69 in the control animals (Fig. 6E, white bar), whereas the expression of CD69 was moderately, but significantly reduced when MHC class I expression was restricted to nonhematopoietic cells (Fig. 6E, red bar) or to hematopoietic cells (Fig. 6E, blue bar). In contrast, however, the abilities of intrahepatic COR93-specific CD8+ T cells to express granzyme B were not reduced in the mice whose MHC class I expression was mostly restricted to either nonhematopoietic cells (red bars) or hematopoietic cells (blue bars) (Fig. 6F). Taken together, these results suggest that antigen presentation by hematopoietic cells and nonhematopoietic cells are both required for the maximum expansion and partial functional activation of HBV-specific CD8+ T cells after hydrodynamic transfection of 1.3 HBV-DNA.
To further examine the importance of MHC class I expression on hematopoietic and nonhematopoietic cells, we reconstituted lineage 1.3.32 transgenic mice with bone marrow derived from β2m−/− mice, yielding 1.3.32 transgenic mice whose MHC class I expression was mostly restricted to nonhematopoietic cells (Fig. 7, red bars). We also crossed 1.3.32 transgenic mice with β2m−/− mice for two generations and then reconstituted them with bone marrow derived from B6 mice to generate 1.3.32 transgenic mice whose MHC class I expression was restricted to hematopoietic cells (Fig. 7, blue bars). Parental 1.3.32 transgenic mice were also reconstituted with bone marrow derived from B6 mice to serve as controls (white bars). Six to ten weeks later, we confirmed that reconstitution efficacy was similar to the hydrodynamic transfection experiment shown in Fig. 6 (data not shown). The reconstituted mice were then injected with the agonistic anti-CD40 antibody to activate pAPCs and adoptively transferred with naive COR93-specific CD8+ T cells. The mice were sacrificed on days 1 and 7 after adoptive transfer to examine COR93-specific CD8+ T cells. The CD8+ T cell responses were correlated with the degree of liver damage and HBV gene expression on day 7 monitored by serum alanine aminotransferase (sALT) activity and Northern blot analyses, respectively.
FIG 7.

Relative contributions of MHC class I expression on hematopoietic cells versus nonhematopoietic cells to the induction of HBV-specific CD8+ T cells in HBV transgenic mice. The numbers of adoptively transferred COR93-specific CD8+ T cells in the livers (A) and spleens (D) and the percentage of the COR93-specific CD8+ T cells in the lymph nodes (G) of HBV-transgenic mice in which MHC class I expression is mostly restricted to nonhematopoietic cells (red bars) or hematopoietic cells (blue bars) were determined. COR93-specific T cells transferred to control HBV-transgenic mice reconstituted with B6 bone marrow are shown in white. The numbers of COR93-specific CD8+ T cells in the recipient transgenic mice before adoptive transfer (Pre) are also indicated. (B, E, and H) Percentages of CD69-positive COR93-specific CD8+ T cells in the recipient livers, spleens, and lymph nodes on days 1 and 7 after adoptive transfer and in the donor spleen (Donor). (C, F, and I) Percentages of GrB-positive COR93-specific T cells in the recipient livers, spleens, and lymph nodes and in the donor spleen (Donor). The data represent means ± the SD for three mice.
As shown in Fig. 7, the number of intrahepatic COR93-specific CD8+ T cells was slightly increased on day 1 in HBV transgenic mice whose MHC class I expression was mostly restricted to nonhematopoietic cells (Fig. 7A, red bar) but were significantly reduced in HBV transgenic mice whose MHC class I expression was restricted to hematopoietic cells (Fig. 7A, blue bar) compared to control mice (Fig. 7A, white bars). Intrahepatic CD69 expression was comparable between control mice (Fig. 7B, white bars) and HBV transgenic mice whose MHC class I expression was mostly restricted to nonhematopoietic cells (Fig. 7B, red bar) but was significantly reduced in HBV transgenic mice whose MHC class I expression was restricted to hematopoietic cells (Fig. 7B, blue bar). These results suggest the importance of MHC class I expression on nonhematopoietic cells in early sequestration and activation of HBV-specific CD8+ T cells in the liver. By day 7, the number of intrahepatic COR93-specific CD8+ T cells was reduced by approximately 25-fold in HBV transgenic mice whose MHC class I expression was mostly restricted to nonhematopoietic cells (Fig. 7A, red bar) compared with control mice (Fig. 7A, white bar), suggesting that MHC class I expression on hematopoietic cells was required for the robust expansion of COR93-specific CD8+ T cells in anti-CD40-treated HBV transgenic mice. Strikingly, the number of intrahepatic COR93-specific CD8+ T cells was reduced by approximately 1,800-fold in HBV transgenic mice whose MHC class I expression was restricted to hematopoietic cells (Fig. 7A, blue bar) compared to controls (Fig. 7C, white bar). The reduced COR93-specific CD8+ T cells in the liver do not reflect their redistribution to or preferential expansion lymphoid organs since the number of COR93-specific CD8+ T cells was also reduced in the spleen (Fig. 7D), and the fraction of their counterparts in the lymphoid organs was also significantly reduced (Fig. 7G). These results suggest that MHC class I expression on nonhematopoietic cells is even more critical for the expansion of COR93-specific CD8+ T cells than MHC class I expression on hematopoietic cells. Similarly, granzyme B expression in COR93-specific CD8+ T cells was not induced in these organs of HBV transgenic mice whose MHC class I expression was restricted to hematopoietic cells (Fig. 7C, F, and I, blue bars), while it was preferentially induced in the liver of the COR93-specific CD8+ T cells in control HBV transgenic mice (Fig. 7C, F, and I, white bars) and HBV transgenic mice whose MHC class I expression was mostly restricted to nonhematopoietic cells (Fig. 7C, F, and I, red bar), suggesting that antigen presentation by hepatocytes was required for granzyme B expression by COR93-specific CD8+ T cells after adoptive transfer into HBV transgenic mice. As expected, the number of intrahepatic COR93-specific CD8+ T cells was directly correlated with the serum ALT activity (Fig. 8A), and inversely correlated with the intrahepatic HBV gene expression level (Fig. 8B), indicating the importance of T cell expansion and recognition of antigen on hepatocytes for causing liver disease and clearing HBV. Collectively, these results suggest that antigen presentation by nonhematopoietic cells is essential for expansion and granzyme B expression of HBV-specific CD8+ T cells after adoptive transfer to HBV transgenic mice. On the other hand, antigen presentation by hematopoietic cells is also required for robust expansion of HBV-specific CD8+ T cells, but its role in the expansion and the induction of cytolytic ability is relatively minor compared to antigen presentation by nonhematopoietic cells.
FIG 8.

Pathogenic and antiviral effect of HBV-specific CD8+ T cells. (A) The serum alanine aminotransferase (sALT) activity in the HBV transgenic mice described in Fig. 7 is expressed as units/liter. The data represent means ± the SD for three to four mice. (B) Northern blot analysis of total liver RNA isolated from the same mice. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used to normalize the amount of RNA bound to membrane.
DISCUSSION
The present study demonstrated that myeloid dendritic cells (mDCs), but not T cell homing to the secondary lymphoid tissues, are required for normal HBV-specific CD8+ T cell precursors to differentiate into effector T cells and clear HBV from the liver. The data also suggest that the maximum expansion of HBV-specific CD8+ T cells requires cooperation between cross-presentation by hematopoietic cells and direct presentation by nonhematopoietic cells (Fig. 9). Collectively, the study sheds new light on the importance of local antigen presentation for the induction of a fully functional HBV-specific CD8+ T cell response.
FIG 9.
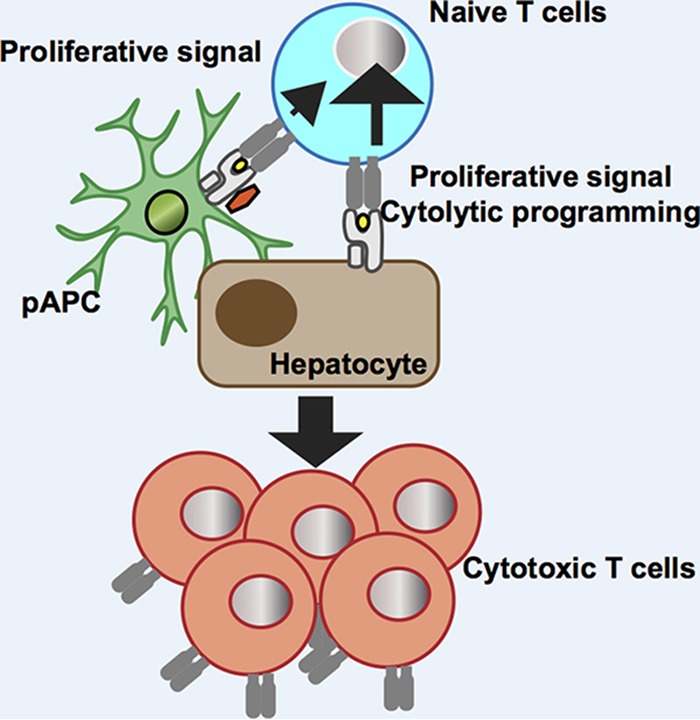
Schematic summary of the study. Presentation of endogenously synthesized antigen by hepatocytes plays a central role in the expansion and differentiation of cytotoxic HBV-specific CD8+ T cells, while cross-presentation by hematopoietic cells is required for maximum expansion of the CD8+ T cells.
The liver is widely regarded as a tolerogenic organ (21). Indeed, we have shown previously that T cell priming in the liver induces PD-1 mediated functional exhaustion of adoptively transferred, COR93-specific transgenic CD8+ T cells (15). Bertolino and colleagues also showed that intrahepatically primed CD8+ T cells were functionally defective and prone to apoptosis, while the same naive CD8+ T cells activated within the lymph nodes were fully cytolytic (20, 22). Therefore, under noninflammatory conditions, intrahepatic T cell priming appears to be clearly immunosuppressive. In contrast, the results of this study suggest that the induction of HBV-specific CD8+ T cell responses can occur independently of T cell homing to the lymphoid organs if intrahepatic antigen-presenting cells are properly activated. Blockade of T cell homing to lymph nodes by anti-CD62L antibody did not prevent natural HBV-specific T cell precursors to differentiate into effector CD8+ T cells (Fig. 3 and 5). These findings challenge the current models of antigen presentation in which pAPCs acquire antigen in the peripheral tissues, mature, and migrate to the draining lymph nodes to prime T cells. The notion that efficient T cell priming could occur in the liver was not without precedent as an OVA-specific CD8+ T cell response was induced in an orthotropic liver transplantation model in which acutely introduced OVA-derived SIINFEKL epitope could be only presented by the transplanted liver (23). In the present study, we showed that efficient T cell priming could occur in the liver to the endogenously synthesized viral antigen. The role of DCs in the induction of virus-specific CD8+ T cells is firmly established. Indeed, DCs were required for normal HBV-specific CD8+ T cell precursors to differentiate into effector CD8+ T cells and eliminate HBV from the liver after hydrodynamic transfection of 1.3-HBV plasmid (Fig. 2). In contrast, the physiological importance of cross-presentation by DCs remains contentious (10). Here, we provided the first in vivo evidence that HBV-specific CD8+ T cells could be activated to intrahepatically expressed HBV in the absence of MHC class I expression on hepatocytes (Fig. 6E, 7B, 7E, and 7H blue bars), presumably reflecting cross-presentation. We also showed that the expansion of adoptively transferred COR93-specific CD8+ T cells was significantly reduced in the near absence of MHC class I expression on hematopoietic cells (Fig. 6D, 7A, 7D, and 7G, red bars). These results suggest that antigen presentation by hematopoietic cells, i.e., cross-presentation, is required to induce maximum expansion of HBV-specific CD8+ T cells. The requirement of cross-presentation for maximum T cell expansion probably indicates the importance of positive costimulation, since pAPCs are the only cell population capable of delivering costimulation. We do not know exactly which pAPC population should express MHC class I, but in light of recent reports, cross-presentation by BATF3 expressing DCs (24) might contribute to the maximum expansion of HBV-specific CD8+ T cells.
Surprisingly, antigen presentation by hepatocytes appears to be far more efficient than that by pAPCs to induce HBV-specific CD8+ T cell responses in the liver. The expansion of COR93-specific CD8+ T cells was strongly reduced in the absence of MHC class I expression on nonhematopoietic cells in both the hydrodynamic transfection and transgenic mouse systems (Fig. 6D, 7A, 7D, and 7G, blue bars), and the cytolytic ability of CD8+ T cells was also absent in the HBV transgenic mice where nonhematopoietic cells do not express MHC class I (Fig. 7C, F, and I, blue bars). These results do not reflect poor reconstitution efficacy in these mice because the number of MHC-class I expressing lymphocyte, pAPCs (I-A/I-E+), and myeloid cells (CD11b+ cells) in these mice (Fig. 6A, B, and C, blue bars) are largely comparable to those in control mice (white bars). These results suggest that antigen presentation by activated pAPCs by itself is insufficient to induce a robust CD8+ T cell response against an intrahepatically expressed antigen. These results probably reflect relative inefficiency of cross-presentation to provide antigenic stimulus, as indicated by relatively low CD69 expression on day 1 in the absence of MHC class I expression on nonhematopoietic cells (Fig. 7B, E, and H). It should be emphasized, however, that activation of DCs is required for the expansion and functional differentiation of HBV-specific CD8+ T cell responses (15). It is therefore possible that activated DCs mainly contribute to the expansion and functional differentiation of HBV-specific CD8+ T cells by producing soluble factors, such as type I interferons and interleukin-15, rather than providing the antigenic stimulus. We postulate that cross-presentation is crucial only in T cell priming phase when the number of naive T cells is still very low. As T cells expand and greatly outnumber DCs, T cells may become more dependent on endogenous antigen presentation by hepatocytes. Experiments are under way to test this hypothesis.
There are several caveats in this study. First, the CD8+ T cell analysis in this study was limited to a single CD8+ T cell epitope (COR93) in a single HBV antigen (HBV Core). While we have shown previously that COR93-specific and the envelope-derived epitope ENV28-specific CD8+ T cells respond in a similar manner to the intrahepatically expressed HBV (15), it is unclear whether the nature of antigen and T cell epitope influences the priming location and the dependency on cross-presentation versus hepatic antigen presentation. Second, we transferred an unphysiological number of naive T cells in selected experiments. In these experiments, the ratio between antigen-presenting cells and naive T cells is probably different from physiological condition. The relative contribution of either direct or cross-presentation to T cell activation and/or expansion is likely dependent on both the amount of antigen (or antigen positive cells) and the number of antigen-specific T cells that are present in the liver at the time of priming. Therefore, it is possible that the cross-presentation plays a more significant role during natural infection than these experimental settings. Third, the study does not address the contribution of cross-presentation by liver sinusoidal endothelial cells or stellate cells to the induction of HBV-specific CD8+ T cells, although we have shown previously that the HBV-expressing hepatocytes were the major cell population that prime HBV-specific CD8+ T cells in the liver (15).
In summary, the present study revealed the importance of local antigen presentation for the induction of HBV-specific CD8+ T cell responses. Antigen presentation by pAPCs is critical for the induction of HBV-specific CD8+ T cell responses, and it does not have to occur in lymphoid organs. Unexpectedly, antigen presentation by pAPCs per se is far from sufficient to induce expansion and functional differentiation of HBV-specific CD8+ T cells. The data presented here are important for understanding the immunological basis for the priming of HBV-specific CD8+ T cell responses and may help in the design of therapeutic vaccine strategies for chronic HBV infection.
MATERIALS AND METHODS
Ethics statement.
All animal studies followed the guidelines in the NIH Guide for the Care and Use of Laboratory Animals, or the Guidelines for Animal Experiments of Nagoya City University Graduate School of Medical Sciences, and were approved by The Scripps Research Institute Animal Care and Use Committee (protocol 08-0159) or by the Nagoya City University Medical School Institutional Animal Care and Use Committee.
Mice.
The HBV transgenic mouse lineage 1.3.32 (inbred B6, H-2b) was described previously (16). Lineage 1.3.32 expresses all of the HBV antigens and replicates HBV in the liver and kidney at high levels without any evidence of cytopathology. The mice were matched for age (8 weeks) and sex (male) in all experiments and (for the 1.3.32 animals) serum HBeAg levels before experimental manipulations. The generation of COR93-specific TCR transgenic mice (BC10.3) was described previously (15). BC10.3 transgenic mice have been backcrossed more than 10 times onto the B6 background and then mated once with CD45.1 mice (H-2b), so that the COR93-specific TCR transgenic T cells could be easily followed by anti-CD45.1 antibody staining. β2-Microglobulin (β2m) knockout mice on B6 backgrounds were purchased from Jackson Laboratory. CD11c-DOG mice (inbred B6, H-2b), kindly provided by Günter Hämmerling, were described previously (18). HBV transgenic mice from lineage 1.3.32 were crossed with β2m knockout mice for two generations to produce β2m-deficient HBV transgenic mice on a B6 background. All experiments were approved by the Scripps Research Institute Animal Care and Use Committee.
The preparation of bone marrow chimeras.
Bone marrow chimeras were prepared as described previously (25) with minor modifications. Briefly, 8- to 10-week-old male HBV transgenic mice on the B6 background were lethally irradiated twice with two 550-cGy doses, for a total dose of 1,100 cGy from a 137Cs source (Gammacell 40 irradiator; Atomic Energy of Canada, Ltd., Ottawa, Canada) and 1 day later injected intravenously with 107 bone marrow cells collected from the femurs and tibias of β2m knockout mice. To receive bone marrow graft from β2m knockout mice, the recipients were administered 100 μg of anti-NK 1.1 PK 136 antibody to deplete their NK cells 48 and 24 h before irradiation to prevent acute bone marrow graft rejection mediated by the recipient's NK cells, and then donor T cells (CD90.2+ cells) were depleted by positive selection using MACS (Miltenyi Biotec), according to the manufacturer's instructions to avoid graft-versus-host disease. Finally, the bone marrow graft was injected into the tail veins of the irradiated recipient mice. In addition, 8- to 10-week-old male β2m-deficient HBV transgenic mice on B6 background were lethally irradiated and reconstituted with bone marrow cells collected from B6 mice. These procedures yielded two groups of mice in which expression of MHC class I molecules is mostly restricted to nonhematopoietic cells or hematopoietic cells. The efficiency of reconstitution was monitored by fluorescence-activated cell sorting analysis to quantitate the numbers of total and H-2Kb (MHC class I)-expressing CD45+ cells (lymphocytes), I-A/I-E+ cells (mostly pAPCs), and CD11b+ cells (myeloid cells) in the peripheral blood. Six to ten weeks later, after reconstitution was complete, the reconstituted mice were used for the experiments.
Splenectomy.
In some experiments, mice were subjected to splenectomy following classical procedures (26). Groups of 3 to 4 control mice received sham surgery. The splenectomized mice and control mice were used at least 1 month after surgery.
Peptide.
A synthetic peptide corresponding to the previously described, HBV core protein (COR)-specific CTL epitope, COR93 (Kb; MGLKFRQL), was purchased from Mimotope (Victoria, Australia).
Adoptive transfer of COR93-specific CD8+ T cells.
More than 98% of the splenic CD8+ T cells in BC10.3 transgenic mice were COR93 specific and exhibit a naive T cell phenotype (15). COR93-specific CD8+ T cells were isolated from the spleen cells of BC10.3 transgenic mice by negative selection using MACS (Miltenyi Biotec), according to the manufacturer's instructions, and 2 × 106 of CD8+ T cells were then injected into the tail veins of mice.
Hydrodynamic injection of plasmid pT-MCS-HBV1.3 and pCMV-SB.
The generation of plasmid pT-MCS-HBV1.3, which contains an HBV 1.3 supergenomic DNA flanked by the inverted-repeat recognition sequences of the Sleeping Beauty transposase, has been described (17). A total of 27.0 μg of pT-MCS-HBV1.3 and 4.5 μg of the Sleeping Beauty transposase expression plasmid pCMV-SB wa injected into the tail veins of mice in a volume of saline equivalent to 8% of the body mass of the mouse (e.g., 1.6 ml for a mouse of 20 g). The total volume was delivered within 5 to 8 s. Cohorts were defined by matching mice on the basis of sALT, HBsAg, and HBeAg parameters on day 1 after injection of HBV1.3 DNA.
DTX treatment.
Diphtheria toxin (DTX) was purchased from Sigma-Aldrich, dissolved in PBS, and administered intraperitoneally (200 ng/mouse) every other day in some experiments.
Anti-CD40 and anti-CD62L antibody treatment.
The FGK45 hybridoma, which produce a rat IgG2a monoclonal antibody against mouse CD40 (αCD40) was provided by A. Rolink (Basel Institute for Immunology, Basel, Switzerland) (27). HBV transgenic mice were injected intravenously with 100 μg of αCD40 16 h before adoptive transfer of HBV-specific naive T cells into HBV transgenic mice. A monoclonal anti-CD62L antibody (αCD62L, clone Mel-14) was purchased from BD Bioscience, and mice were administered 100 μg of αCD62L intraperitoneally in 200 μl of phosphate-buffered saline (PBS) as indicated in Results.
Lymph mononuclear cell preparation.
Spleen cells, lymph node cells, and intrahepatic lymphocytes (IHLs) were prepared as previously described (28, 29). Briefly, spleen cells and lymph node cells were isolated by being pressed through a 70-μm-pore size cell strainer (Becton Dickinson) with the plunger of a 1-ml syringe, washed three times with PBS, and used for further analysis. For IHL isolation, livers were perfused with 10 ml of PBS via the portal vein to remove circulating lymphocytes, and the liver cell suspension was pressed through a 70-μm-pore size cell strainer and digested with 10 ml of RPMI 1640 medium (Life Technologies), containing 0.02% (wt/vol) collagenase IV (Sigma) and 0.002% (wt/vol) DNase I (Sigma), for 40 min at 37°C. Cells were washed with RPMI 1640 and then overlaid on a Percoll/Histopaque solution consisting of 12% Percoll (Pharmacia) and 88% Histopaque-1083 (Sigma-Aldrich). After centrifugation for 20 min at 1,500 × g, the IHLs were isolated at the interface. The lymph mononuclear cells were washed twice with RPMI 1640 medium and used for further analysis.
Flow cytometric T cell analysis.
Lymph mononuclear cells isolated from the liver, spleen and lymph nodes were incubated with a mixture containing Pacific Blue-conjugated anti-mouse CD8 or CD45.1, PerCP-conjugated anti-mouse CD4, PerCP-Cy5.5-conjugated anti-mouse CD8, PE-Cy7-conjugated anti-mouse CD25 or B220, allophycocyanin (APC)-conjugated anti-mouse CD11c, fluorescein isothiocyanate (FITC)-conjugated anti-mouse CD69 or CD62L, and phycoerythrin (PE)-conjugated anti-mouse H-2Kb, CD11b, or PD-1 and Alexa 700-conjugated anti-mouse CD44 or I-A/I-E for 1 h on ice. After washing, the cells were incubated for 30 min with APC-conjugated anti-mouse IgG at 4°C to detect dimer positive cells. Dimer without peptide was used as a control. Intracellular cytokine staining was performed using PE-conjugated anti-mouse IFN-γ, APC-conjugated anti-mouse granzyme B (GrB; Caltag) after incubation for 5 h at 37°C in the presence of brefeldin A, as described previously (15, 28, 29). All antibodies were purchased from BD Bioscience and eBioscience.
Tissue DNA and RNA analyses.
Total liver DNA and RNA were analyzed for HBV replicative intermediates by Southern blotting, and for HBV RNA by Northern blotting, exactly as previously described (28, 30). The relative abundance of specific DNA and RNA molecules was determined by phosphor imaging analysis, using the Optiquant image analysis software (Packard).
Biochemical analyses.
The extent of the hepatocellular injury was monitored by measuring sALT activity at multiple time points after treatment as described previously (28, 30).
Statistical analysis.
A Student t test was performed using Microsoft Excel. The data are depicted as means ± the SD, and P values of <0.05 were considered significant (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
ACKNOWLEDGMENTS
We thank Francis V. Chisari (The Scripps Research Institute) for guidance and encouragement, as well as providing HBV transgenic mice lineage 1.3.32 and TCR transgenic mice lineage BC10.3; Günter Hämmerling (German Cancer Research Center) for providing CD11c-DOG mice; and Christina Whitten Bauer for excellent technical assistance.
This study was partially supported by a grant R01AI79060 from the National Institutes of Health, grants-in-aid from the Ministry of Education, Cultures, Sports, Science, and Technology, Japan, and from Japan Society for the Promotion of Science (KAKENHI grants 26461015 and 17K09436), and the Research Program on Hepatitis from the Japan Agency for Medical Research and Development (AMED project codes 17fk0310106h0001, 16fk0310512h0005, and 17fk0310101h0001).
Y.M. and M.I. designed experiments. Y.M., K.K., K.S., and M.I. executed experiments. Y.M., K.K., K.S., Y.T., and M.I. analyzed and interpreted experiments. Y.M. and M.I. wrote the original and revised manuscripts. K.K. and Y.T. edited the manuscript.
REFERENCES
- 1.Wong P, Pamer EG. 2003. CD8 T cell responses to infectious pathogens. Annu Rev Immunol 21:29–70. doi: 10.1146/annurev.immunol.21.120601.141114. [DOI] [PubMed] [Google Scholar]
- 2.Pamer E, Cresswell P. 1998. Mechanisms of MHC class I-restricted antigen processing. Annu Rev Immunol 16:323–358. doi: 10.1146/annurev.immunol.16.1.323. [DOI] [PubMed] [Google Scholar]
- 3.Banchereau J, Steinman RM. 1998. Dendritic cells and the control of immunity. Nature 392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 4.Bevan MJ. 1976. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J Exp Med 143:1283–1288. doi: 10.1084/jem.143.5.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bevan MJ. 2006. Cross-priming. Nat Immunol 7:363–365. doi: 10.1038/ni0406-363. [DOI] [PubMed] [Google Scholar]
- 6.Sigal LJ, Crotty S, Andino R, Rock KL. 1999. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature 398:77–80. doi: 10.1038/18038. [DOI] [PubMed] [Google Scholar]
- 7.Xu R-H, Remakus S, Ma X, Roscoe F, Sigal LJ. 2010. Direct presentation is sufficient for an efficient anti-viral CD8+ T cell response. PLoS Pathog 6:e1000768. doi: 10.1371/journal.ppat.1000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haan den JM, Bevan MJ. 2001. Antigen presentation to CD8+ T cells: cross-priming in infectious diseases. Curr Opin Immunol 13:437–441. doi: 10.1016/S0952-7915(00)00238-7. [DOI] [PubMed] [Google Scholar]
- 9.Ochsenbein AF, Sierro S, Odermatt B, Pericin M, Karrer U, Hermans J, Hemmi S, Hengartner H, Zinkernagel RM. 2001. Roles of tumour localization, second signals and cross priming in cytotoxic T-cell induction. Nature 411:1058–1064. doi: 10.1038/35082583. [DOI] [PubMed] [Google Scholar]
- 10.Zinkernagel RM. 2002. On cross-priming of MHC class I-specific CTL: rule or exception? Eur J Immunol 32:2385–2392. doi:. [DOI] [PubMed] [Google Scholar]
- 11.Ganem D, Prince AM. 2004. Hepatitis B virus infection–natural history and clinical consequences. N Engl J Med 350:1118–1129. doi: 10.1056/NEJMra031087. [DOI] [PubMed] [Google Scholar]
- 12.Chisari FV. 2000. Rous-Whipple Award Lecture. Viruses, immunity, and cancer: lessons from hepatitis B. Am J Pathol 156:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isogawa M, Tanaka Y. 2014. Immunobiology of hepatitis B virus infection. Hepatol Res 45:179–189. doi: 10.1111/hepr.12439. [DOI] [PubMed] [Google Scholar]
- 14.Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, Chisari FV. 2003. CD8+ T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol 77:68–76. doi: 10.1128/JVI.77.1.68-76.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Isogawa M, Chung J, Murata Y, Kakimi K, Chisari FV. 2013. CD40 activation rescues antiviral CD8+ T cells from PD-1-mediated exhaustion. PLoS Pathog 9:e1003490. doi: 10.1371/journal.ppat.1003490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guidotti LG, Matzke B, Schaller H, Chisari FV. 1995. High-level hepatitis B virus replication in transgenic mice. J Virol 69:6158–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang PL, Althage A, Chung J, Chisari FV. 2002. Hydrodynamic injection of viral DNA: a mouse model of acute hepatitis B virus infection. Proc Natl Acad Sci U S A 99:13825–13830. doi: 10.1073/pnas.202398599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hochweller K, Striegler J, Hämmerling GJ, Garbi N. 2008. A novel CD11c.DTR transgenic mouse for depletion of dendritic cells reveals their requirement for homeostatic proliferation of natural killer cells. Eur J Immunol 38:2776–2783. doi: 10.1002/eji.200838659. [DOI] [PubMed] [Google Scholar]
- 19.Lepault F, Gagnerault MC, Faveeuw C, Boitard C. 1994. Recirculation, phenotype and functions of lymphocytes in mice treated with monoclonal antibody MEL-14. Eur J Immunol 24:3106–3112. doi: 10.1002/eji.1830241229. [DOI] [PubMed] [Google Scholar]
- 20.Bowen DG, Zen M, Holz L, Davis T, McCaughan GW, Bertolino P. 2004. The site of primary T cell activation is a determinant of the balance between intrahepatic tolerance and immunity. J Clin Invest 114:701–712. doi: 10.1172/JCI200421593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crispe I. 2003. Hepatic T cells and liver tolerance. Nat Rev Immunol 3:51–62. doi: 10.1038/nri981. [DOI] [PubMed] [Google Scholar]
- 22.Bertolino P, Bowen DG, McCaughan GW, Fazekas de St Groth B. 2001. Antigen-specific primary activation of CD8+ T cells within the liver. J Immunol 166:5430–5438. doi: 10.4049/jimmunol.166.9.5430. [DOI] [PubMed] [Google Scholar]
- 23.Klein I, Crispe IN. 2006. Complete differentiation of CD8+ T cells activated locally within the transplanted liver. J Exp Med 203:437–447. doi: 10.1084/jem.20051775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM. 2008. Batf3 deficiency reveals a critical role for CD8 dendritic cells in cytotoxic T cell immunity. Science 322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakamoto Y, Guidotti LG, Kuhlen CV, Fowler P, Chisari FV. 1998. Immune pathogenesis of hepatocellular carcinoma. J Exp Med 188:341–350. doi: 10.1084/jem.188.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reeves JP, Reeves PA, Chin LT. 2001. Survival surgery: removal of the spleen or thymus. Curr Protoc Immunol Chapter 1:Unit 1.10. doi: 10.1002/0471142735.im0110s02. [DOI] [PubMed] [Google Scholar]
- 27.Schoenberger SP, Toes REM, van der Voort EIH, Offringa R, Melief CJM. 1998. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 28.Isogawa M, Furuichi Y, Chisari FV. 2005. Oscillating CD8+ T cell effector functions after antigen recognition in the liver. Immunity 23:53–63. doi: 10.1016/j.immuni.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 29.Isogawa M, Kakimi K, Kamamoto H, Protzer U, Chisari FV. 2005. Differential dynamics of the peripheral and intrahepatic cytotoxic T lymphocyte response to hepatitis B surface antigen. Virology 333:293–300. doi: 10.1016/j.virol.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 30.Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. 1996. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity 4:25–36. doi: 10.1016/S1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]