

Abstract
A crucial step in the understanding of any phenotype is the correct identification of the signaling pathways that are significantly impacted in that phenotype. However, most current pathway analysis methods produce both false positives as well as false negatives in certain circumstances. We hypothesized that such incorrect results are due to the fact that the existing methods fail to distinguish between the primary dis-regulation of a given gene itself and the effects of signaling coming from upstream. Furthermore, a modern whole-genome experiment performed with a next-generation technology spends a great deal of effort to measure the entire set of 30,000–100,000 transcripts in the genome. This is followed by the selection of a few hundreds differentially expressed genes, step that literally discards more than 99% of the collected data. We also hypothesized that such a drastic filtering could discard many genes that play crucial roles in the phenotype. We propose a novel topology-based pathway analysis method that identifies significantly impacted pathways using the entire set of measurements, thus allowing the full use of the data provided by NGS techniques. The results obtained on 24 real data sets involving 12 different human diseases, as well as on 8 yeast knock-out data sets show that the proposed method yields significant improvements with respect to the state-of-the-art methods: SPIA, GSEA and GSA.
Availability
Primary dis-regulation analysis is implemented in R and included in ROntoTools Bioconductor package (versions ≥ 2.0.0). https://www.bioconductor.org/packages/release/bioc/html/ROntoTools.html
Index Terms: Pathway analysis, gene expression, primary disregulation, target pathway
I. Introduction
The goal of pathway analysis methods is to identify the most perturbed pathways in a given condition. Pathways are divided in two main categories: i) signaling pathways, that are defined as graphs in which nodes represent genes/proteins and edges are interactions between them, and ii) metabolic pathways in which the nodes represent biochemical compounds and the edges represent reactions, carried out by enzymes which are coded by genes [32]. Such pathways describe all known phenomena involved in a biological process (e.g. cell cycle), disease (e.g. Alzheimer’s disease), etc. In this paper, we focus on signaling pathways to be able to map the measured expression level of the genes to the corresponding nodes in those pathways. Intuitively, the impact of a given phenotype on a given pathway should be determined by the number of differentially expressed (DE) genes on the given pathway, the magnitude of the changes in the expression level of the genes, and the type, direction and strength of the interactions between the genes in that pathway.
The simplest pathway analysis approach is the over-representation analysis (ORA) [23]. This approach considers only the number of DE genes that are present in a given pathway. ORA techniques calculate the probability of finding a certain number of DE genes among all the genes in a pathway just by chance. Another approach to pathway analysis is the functional class scoring (FCS) [24], [32]. This approach takes into consideration all measured expression changes, as well as the correlation between the expression change of the genes and the phenotype. The most popular techniques in the FCS category are Gene Set Enrichment Analysis (GSEA) [45] and Gene Set Analysis (GSA) [10]. These two techniques rank the genes based on the correlation between their expression and a given phenotype, and calculate a score that reflects the degree to which a given pathway is represented at extremes of the ranked list. Neither of these two approaches considers the interactions between genes, their direction, type, strength, etc. In essence, all these methods treat the pathways as simple sets of genes.
However, databases such as KEGG [34], BioCarta [5] and Reactome [21] provide pathways that consist of much more than just sets of genes. These databases provide complex graphs for each signaling pathways in which each node is a gene/protein and each edge is an interaction between two such genes or proteins. Ignoring the wealth of knowledge captured in the topology of the pathway is clearly sub-optimal. Even though these databases provide more detailed information about the topology of the pathways, there are thousands of genes that have not been annotated yet. Furthermore, many of the existing annotations may be inaccurate [24]. However, we believe that accuracy and reliability of pathways annotation is growing and using this type of information can only help the interpretation of high-throughput experiments.
One of the first methods to exploit the information about the interactions among genes tried to analyze the entire set of known interactions, in order to find circuits, or subnetworks, that are affected by the phenotype in analysis. The interactions are obtained by combining different sources of information, such as pathways, interaction databases and literature [18].
More recently, more sophisticated methods that are able to fully take into consideration all the interactions between genes in signaling pathways to find which pathway is most impacted by a given phenotype have been proposed [9]. These are sometimes referred to as “topology-aware” or “third generation” pathway analysis methods [24], [32]. The method proposed in this paper belongs to this latest generation of pathway analysis methods, inasmuch it considers the topology of the pathways, as well as the changes in expression level of the genes.
However, even the most sophisticated current pathway analysis methods still produce both false positives as well as false negatives in certain circumstances. We hypothesized that such incorrect results are due to the fact that the existing methods fail to distinguish between the primary dis-regulation of a given gene itself and the effects of signals coming from upstream. We hypothesize that better results could be achieved if one distinguishes between genes that are true sources of perturbation, e.g. due to mutations, copy number variations, epigenetic changes, etc. and genes that merely respond to perturbation signals coming from upstream. Intuitively, a pathway should be more significantly impacted if it hosts more genes that are such true sources of perturbation. The method proposed here is an attempt at capturing these differences by calculating a “primary dis-regulation” for every gene and using them to compute a total pathway perturbation and subsequent significance.
Another issue related to the traditional topological data analysis approaches involves the need for a selection of differentially expressed (DE) genes. Traditionally, the pathway analysis step is performed after a set of DE genes has been selected using some thresholds on some criteria such as fold-change and/or p-values. Typically, a set of a few hundred genes are selected as DE. However, a modern whole-genome experiment performed with a next-generation technology (NGS) provides measurements for the entire set of transcripts in the genome, albeit for a non-trivial cost in computation necessary for the assembly and quantification of millions of short reads. In addition to the high computational cost, other drawbacks are related to the large amount of storage space, and the need to specialized bioinformatics expertise to set-up and run the environment necessary for the analysis. Given that this great deal of effort is spent in order to measure over 30,000 transcripts, it makes little sense to discard approximately 99% of these measurements in order to focus on 300 or so genes that are declared to be differentially expressed. Subsequently, the pathway analysis step aims to identify system-level changes based on only these 1% of the original data collected. More recently, approaches that are able to identify significantly impacted pathways based on the entire set of measurements have been proposed [54]. Henceforth, we will refer to the original approach based on DE genes as the cut-off -based approach, and to the threshold-free approach as the all genes approach. We assessed the novel method proposed here with both types of input.
In the methods section, we describe our new proposed method in details. In the discussion section, we evaluate our method using 24 data sets involving 12 conditions from different experiments comparing human diseased versus normal tissues. The results of the proposed method using the cut-off -based approach are compared with SPIA (cut-off) [48], which also uses a pre-selected list of DE genes as input. The results of the proposed method using the all genes approach are compared with GSEA [45], GSA [10] and SPIA (all genes) [54] which use entire set of genes as input. These existing methods have been selected as the reference in our comparisons because they are among the most cited and widely used methods in the literature [32]. We also evaluate our method using eight yeast knock-out data sets from different experiments comparing samples with knock-out gene versus normal samples. The comparisons show that the proposed method is able to perform better than the most widely used pathway analysis methods, in identifying the target pathways as statistically significant.
II. Methods
The measured expression change of a gene in a given phenotype can be seen as the result of influences from upstream genes superposed on the dis-regulation incurred by that particular gene itself. We will refer to this later quantity as the primary dis-regulation (pDis). The diffusion of signals between genes in regulatory networks, called “network propagation”, can be used to find the active genes and subnetworks as well as the function of the genes in different conditions [19]. Widely used methods in this field are introduced in [57] and [52]. Here, we are using a similar approach that uses propagation between genes to calculate pDis in order to find the most impacted pathways. We propose a pathway analysis method that focuses on this primary dis-regulation.
The change in the expression level of a gene i, ΔE(gi), can be seen as a sum of the primary dis-regulation (pDis) and the secondary dis-regulation (sDis):
| (1) |
The secondary dis-regulation of the gene gi is the term that is meant to capture the perturbation reaching this particular gene from upstream. This can be calculated by adding the expression change of upstream genes normalized by the number of their downstream genes:
| (2) |
In the equation above, ΔE(gj) is the measured fold change of the gene gj that is somewhere directly upstream of gi, U is the set of all such genes directly upstream of gi, and Nds(gj) is the number of genes immediately downstream of gj (see Fig. 1). The quantity βi,j represents efficiency of the interaction between gene i and gene j. It captures a specific value if an interaction is available between two genes. We used +1 if the interaction type is activation or expression and −1 if it is inhibition or repression as default values. This is the same approach used by the impact analysis [9].
Fig. 1.
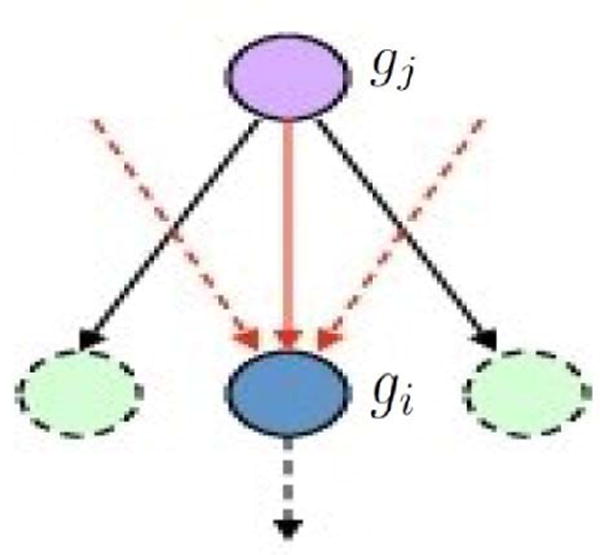
An example of one upstream gene and its three downstream genes. pDis(gi) is calculated using its measured fold change of ΔE(gi) and measured fold change of upstream genes (e.g. ΔE(gj)). In this example, the number of downstream genes for gj is Nds(gj) = 3.
The primary dis-regulation, which gives the change in a gene expression inherent to the gene itself, can then be derived as follows:
| (3) |
The primary dis-regulation is meant to capture information about the genes that are sources of perturbation in a given phenotype, rather than those genes that change as a result of upstream changes. For instance, a mutation that induce expression changes would be captured by the gene’s primary dis-regulation, while expression changes due to upstream signaling would be captured by the secondary dis-regulation. A mutation is an example that is sufficient but not necessary to create primary dis-regulation. Other potential cause could be copy number variations, epigenetic changes such as methylation, etc. The intuition motivating the computation of the primary dis-regulation is that pathways that have more genes that are sources of perturbation are more likely to be truly involved in the phenotype.
The process of calculating all values of the primary dis-regulation for all genes in a given pathway can be summarized using the matrix equation:
| (4) |
In this equation, the matrix B represents the adjacency matrix of each signaling pathway normalized by the number of downstream genes of each gene.
In equation 4, I is an identity matrix with dimensions equal to the number of genes in a pathway, and ΔE is the vector of measured expression changes of the genes in that pathway:
The score for pathway k is calculated as the sum of the absolute values of primary dis-regulation of all the genes in the pathway, totalpDis:
| (5) |
The quantity totalpDis of a pathway represents the amount of primary dis-regulation of the whole pathway in the condition under study.
The significance of each pathway is assessed by computing the probability of obtaining just by chance a totalpDis value more extreme than the one observed. This probability is estimated using a bootstrap approach where the null distribution for totalpDis for each pathway is generated by sampling random gene expression changes from the original set of expression changes. The number of bootstraps used was 2,000. This process is repeated for all pathways and yields a p-value for each pathway. Subsequently, the set of p-values for all pathways are corrected for multiple comparisons using the false discovery rate (FDR). The average running time for a data set is 6.3 minutes on an architecture using a single Intel Xeon core @ 2.66GHz with 1TB of RAM.
Cut-off dependent versus cut-off free analysis
Pathway analysis techniques often take a subset of statistically significant genes as input, based on cut-offs for expression change and/or p-value. It has been shown in [35] that small variations of the threshold used to select the subset of differentially expressed (DE) genes has dramatic effects on the outcome of the methods. Hence, the accuracy of any pathway analysis methods using a subset of DE genes will also be very dependent on the threshold(s) used. Furthermore, when using a cut-off, some genes that play an important biological role may fail to meet the selection criteria and thus, not included in the set of DE genes. This can potentially impede the identification of the biologically meaningful pathways.
Recently, it has also been shown that the accuracy of a pathway analysis method can be improved by using the entire set of measurement from an experiment rather than a subset of DE genes [54]. This means that a selection of a set of DE genes may no longer be needed in many situations.
With respect to the method proposed in this paper, the use of a subset of DE genes will affect the values of the pDis of other genes in a pathway. The pDis of a gene is simply equal to the expression change when there are no upstream DE genes. However, when such upstream genes do exist, pDis is calculated using the expression changes of upstream genes as well. The inclusion of all genes in the calculation will have a strong impact on the result, even if the expression changes are small. This allows the analysis to retain all of the information in the data, avoiding arbitrary threshold choices.
We refer to this method as pDis analysis (all genes), as opposed to pDis analysis (cut-off) for cut-off based. Here, we show the results from both types of input sets applied to our new method proposed in this paper.
III. Discussion and Results
Ranks and p-values for targeted data sets
To date there is no universally accepted technique for the validation of the results of pathway analysis methods. The assessment of the results of different pathway analysis methods usually involves the selection of a few data sets, and then the interpretation of the results either with the help of biologists in the field, or by searching the published literature. This approach is very limited because it can only be applied to a small number of data sets. Furthermore, it is subjective, and may lead to bias in the results since most of the time the expert who performs the assessment is also a co-author of the paper. Finally, the biological phenomena are so complex that with enough literature search, a large number of pathways can be implicated in almost any condition. In this work we follow two validation approaches. The first one is the validation approach introduced in [47]. We like this evaluation approach because it is objective, reproducible, based on multiple data sets, and it does not require an unavoidably biased “expert” human evaluation of the results [47]. This approach requires testing on a large number (at least 10 but preferably more) of different data sets coming from a variety of different conditions, tissues, and laboratories. The data sets are selected such that there are specific pathways in the target pathway databases that model each of the given diseases. For each data set, the pathway corresponding to the phenotype is considered to be the target pathway (e.g. the colorectal cancer pathway will be the target pathway in a colorectal cancer data set). The evaluation focuses on the ability of each method to identify these true positive pathways as significant, and rank them as high as possible. In this paper we validated the proposed method using 24 data sets involving 12 different human diseases. These data sets are shown in Table I.
TABLE I.
The twenty-four data sets from the GEO database used to evaluate the pathway analysis methods compared in this paper. Each data set corresponds to a disease for which there is a target pathway in KEGG.
| GEO ID | Pubmed | Reference | Disease | Target Pathway | |
|---|---|---|---|---|---|
| 1 | GSE1297 | 14769913 | [6] | Alzheimer’s Disease | hsa05010 |
| 2 | GSE5281 | 17077275 | [27] | Alzheimer’s Disease | hsa05010 |
| 3 | GSE5281 | 17077275 | [27] | Alzheimer’s Disease | hsa05010 |
| 4 | GSE5281 | 17077275 | [27] | Alzheimer’s Disease | hsa05010 |
| 5 | GSE20153 | 20926834 | [61] | Parkinson’s disease | hsa05012 |
| 6 | GSE20291 | 15965975 | [60] | Parkinson’s disease | hsa05012 |
| 7 | GSE8762 | 17724341 | [40] | Huntington’s disease | hsa05016 |
| 8 | GSE4107 | 17317818 | [16] | Colorectal Cancer | hsa05210 |
| 9 | GSE8671 | 18171984 | [41] | Colorectal Cancer | hsa05210 |
| 10 | GSE9348 | 20143136 | [15] | Colorectal Cancer | hsa05210 |
| 11 | GSE14762 | 19252501 | [58] | Renal Cancer | hsa05211 |
| 12 | GSE781 | 14641932 | [26] | Renal Cancer | hsa05211 |
| 13 | GSE15471 | 19260470 | [1] | Pancreatic Cancer | hsa05212 |
| 14 | GSE16515 | 19732725 | [36] | Pancreatic Cancer | hsa05212 |
| 15 | GSE19728 | NA | NA | Glioma | hsa05214 |
| 16 | GSE21354 | NA | NA | Glioma | hsa05214 |
| 17 | GSE6956 | 18245496 | [56] | Prostate Cancer | hsa05215 |
| 18 | GSE6956 | 18245496 | [56] | Prostate Cancer | hsa05215 |
| 19 | GSE3467 | 16365291 | [13] | Thyroid Cancer | hsa05216 |
| 20 | GSE3678 | NA | NA | Thyroid Cancer | hsa05216 |
| 21 | GSE9476 | 17910043 | [44] | Acute myeloid leukemia | hsa05221 |
| 22 | GSE18842 | 20878980 | [42] | Non-Small Cell Lung Cancer | hsa05223 |
| 23 | GSE19188 | 20421987 | [17] | Non-Small Cell Lung Cancer | hsa05223 |
| 24 | GSE3585 | 17045896 | [3] | Dilated cardiomyopathy | hsa05414 |
The second approach uses knock-out data sets. In this case, the exact source of perturbation is known: the specific gene being knock-out. Thus the pathways that include this gene will be truly relevant to the phenotype, since they contain the very source of the perturbation that created the phenotype. In other words, these pathways are true positives and are also considered the target pathways in our validation.
The p-values (representing the probability of observing the given perturbations just by chance) are used to assign significance to each pathway. The list of pathways is then ranked based on these p-values.
In order to formalize and quantify the assessment, we define an “improvement factor” that will be used to compare the performance of two pathway analysis methods. If the target pathway for a given data set goes from not significant in the results of method 1 to significant in the results of method 2, the improvement factor for this data set will be 1 (see Fig. 2). If the target pathway goes from significant to not significant, the improvement factor will be −1. If the significance of the target pathway does not change but the ranking improves, the improvement factor will be +0.5. Finally, if the significance does not change but the ranking worsens, the improvement will be −0.5. If the ranking remains the same, the improvement is zero for that data set. The improvement of method 2 compared to method 1 is the average of improvement factors associated to each target pathway over the set of 24 different data sets. If the overall improvement is positive, then method 2 is considered to perform better than method 1 based on this validation method.
Fig. 2.
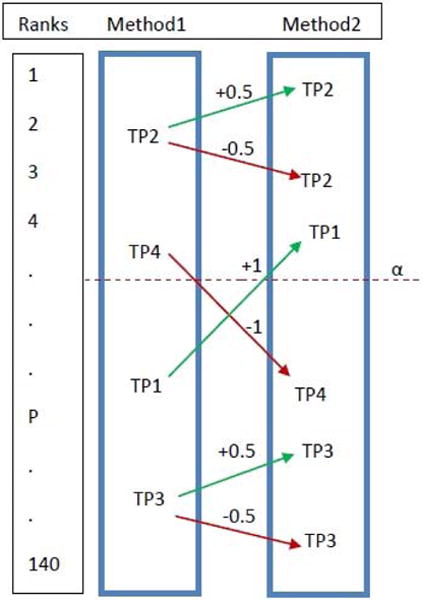
The criteria used to assess the results. Alpha (α) represents the chosen significance threshold. The green and red arrows denote situations in which method 2 is better or worse than method 1, respectively. The number on each arrow represents the value the improvement factor in each case. If a target pathway becomes significant in the results of method 2, the improvement factor for that target pathway will be +1 (e.g. target pathway TP1); if the pathway becomes not significant, the improvement factor is considered −1 (e.g. TP4). If the target remains on the same side of the significance threshold, the improvement factor is considered +0.5 or −0.5 based on the improvement or deterioration of the rank, respectively (e.g. TP2 and TP3).
The proposed method was implemented using the R statistical programming environment [50]. The code is currently available by request from the authors. We are also planning to make the code available as a Bioconductor R package. We used KEGG signaling pathways as input pathways. The pathways were obtained from the “SPIA” R package version 2.14.0 [49] as included in Bioconductor version 2.13, released on October 15th, 2013. We selected all pathways that have at least one interaction with the type of activation, inhibition, repression or expression between their genes. This resulted in a set of 139 pathways. The results of pDis analysis (all genes) are compared to GSA, GSEA and SPIA (all genes) and the results of pDis analysis (cut-off) are compared to SPIA (cut-off). SPIA (cut-off) combines two different p-values. One is the perturbation p-value (pPERT) of a pathway. The perturbation p-value is computed based on the perturbation accumulation of the pathway, which is the sum of the perturbation factors of its genes. The other p-value of SPIA is the hypergeometric p-value, based on the number of DE genes in the pathway in a given data set. Since the number of DE genes in each pathway does not depend on the analysis method, the hypergeometric p-value is the same in SPIA (cut-off) and the method proposed in this paper.
Each data set was normalized by the “mar” normalization method available in the “affy” R package (version 1.38.1) [20] from Bioconductor version 2.12, release on April 4th, 2013. For each gene, the probe id was mapped to gene Entrez ID. The fold change between normal and disease conditions for each probe was calculated by using the “limma” package (version 3.16.8) [43] from Bioconductor version 2.12, release on April 4th, 2013. We used the log2-transform of the fold changes for each gene in our analysis. The moderated t-test was performed on each probe to compute the significance of the changes between two phenotypes. For the methods that use cut-off approach, we used a 5% threshold to select the DE genes.
Ranks and p-values of target pathways for 24 disease data sets
The ranks and p-values of target pathways in all human disease data sets are shown in Fig. 3. The details of the results for the proposed and reference methods are provided in Table III (SPIA and pDis analysis (cut-off) and Table IV, V and VI (GSEA, GSA, SPIA (all genes) and pDis analysis (all genes)). The distributions of the ranks and the p-values obtained for the target pathways in four methods are shown as boxplots in Fig. 3.
Fig. 3.
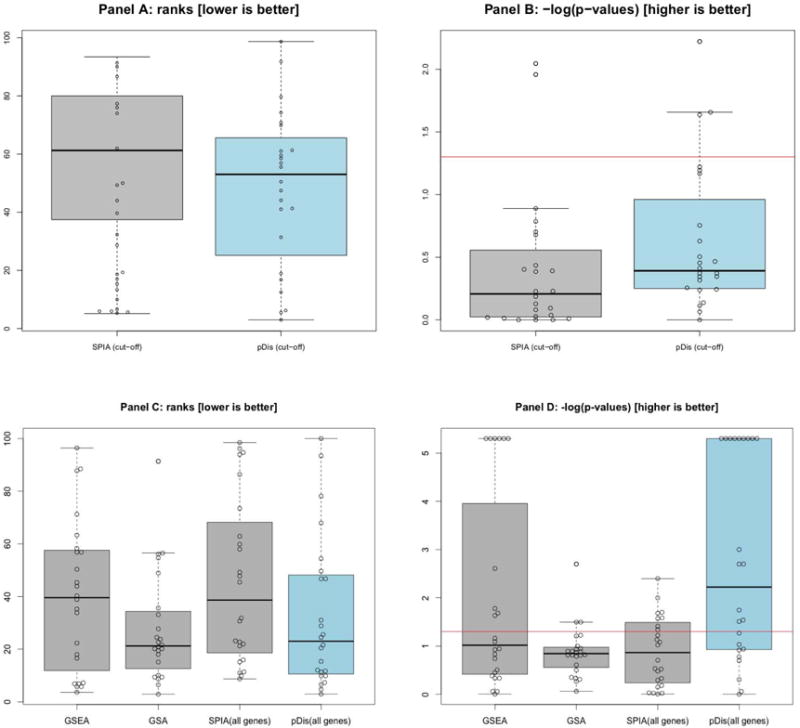
The ranks (in the left column, lower is better) and negative log of p-values of the target pathways (in the right column, higher is better) in the proposed and reference methods. The first row (panel A and panel B) shows the comparison between methods using a set of DE genes: pDis (cut-off) and SPIA (cut-off). The second row (panel C and panel D) shows the comparison between methods using all genes: GSEA, GSA and SPIA (all genes), pDis (all genes). For SPIA, the comparisons are based on the perturbation p-value (pPERT). All human signaling pathways from KEGG (139 pathways) were used in the comparisons. The data show the results obtained for the target pathways in the 24 data sets shown in Table I. The bold line in the boxplots represents the median of the distribution. These distributions show that the proposed method pDis analysis (in blue) is never significantly worse than any of the existing methods, while it yields a statistically significant improvement in 5 out of the 8 comparisons (see Table II).
TABLE III.
The ranks and the p-values of the 24 target pathways for SPIA (cut-off) and pDis analysis (cut-off). The improvement factor based on Fig. 2 is calculated for each data set considering the 5% significance threshold using FDR-corrected p-values. The average improvement factor shows that pDis analysis (cut-off) improves 12.5% compared to SPIA. As shown, the average p-value and rank for the target pathways are lower (i.e. better) in pDis analysis (cut-off) than in SPIA.
| GEO ID | Target pathway | SPIA (pPERT) | pDis analysis (cut-off) | Improvement Compared to SPIA | |||||
|---|---|---|---|---|---|---|---|---|---|
| p-values | FDR | ranks | p-values | FDR | ranks | ||||
| 1 | GSE1297 | Alzheimer’s Disease | 0.916 | 1.00 | 78.79 | 0.729 | 0.987 | 70.83 | +0.5 |
| 2 | GSE5281 | Alzheimer’s Disease | 0.807 | 1.00 | 71.53 | 0.022 | 0.328 | 6.20 | +0.5 |
| 3 | GSE5281 | Alzheimer’s Disease | 0.956 | 1.00 | 92.54 | 0.006 | 0.201 | 2.99 | +0.5 |
| 4 | GSE5281 | Alzheimer’s Disease | 0.831 | 0.985 | 82.20 | 0.068 | 0.359 | 18.94 | +0.5 |
| 5 | GSE20153 | Parkinson’s disease | 1 | 1.00 | 62.82 | 1 | 1.00 | 98.72 | −0.5 |
| 6 | GSE20291 | Parkinson’s disease | 0.129 | 0.712 | 18.10 | 0.425 | 0.803 | 50.48 | −0.5 |
| 7 | GSE8762 | Huntington’s disease | 1 | 1.00 | 69.49 | 0.425 | 0.524 | 79.66 | −0.5 |
| 8 | GSE4107 | Colorectal Cancer | 0.011 | 0.213 | 5.15 | 0.023 | 0.184 | 12.50 | −0.5 |
| 9 | GSE8671 | Colorectal Cancer | 0.406 | 0.778 | 50.74 | 0.351 | 0.772 | 44.12 | +0.5 |
| 10 | GSE9348 | Colorectal Cancer | 0.198 | 0.503 | 37.96 | 0.387 | 0.679 | 56.93 | −0.5 |
| 11 | GSE14762 | Renal Cancer | 0.009 | 0.07 | 12.04 | 0.482 | 0.786 | 61.31 | −0.5 |
| 12 | GSE781 | Renal Cancer | 0.412 | 1.00 | 36.94 | 0.859 | 0.935 | 91.79 | −0.5 |
| 13 | GSE15471 | Pancreatic Cancer | 0.651 | 0.843 | 76.47 | 0.451 | 0.757 | 59.56 | +0.5 |
| 14 | GSE16515 | Pancreatic Cancer | 0.94 | 1.00 | 88.15 | 0.452 | 0.796 | 55.56 | +0.5 |
| 15 | GSE19728 | Glioma | 0.979 | 1.00 | 91.24 | 0.235 | 0.485 | 47.45 | +0.5 |
| 16 | GSE21354 | Glioma | 0.367 | 0.744 | 49.26 | 0.342 | 0.560 | 61.03 | −0.5 |
| 17 | GSE6956 | Prostate Cancer | 0.21 | 1.00 | 17.28 | 0.771 | 0.981 | 74.26 | −0.5 |
| 18 | GSE6956 | Prostate Cancer | 0.592 | 1.00 | 54.13 | 0.555 | 0.993 | 41.32 | +0.5 |
| 19 | GSE3467 | Thyroid Cancer | 0.745 | 0.957 | 77.78 | 0.57 | 0.925 | 58.52 | +0.5 |
| 20 | GSE3678 | Thyroid Cancer | 0.59 | 0.987 | 59.70 | 0.313 | 0.706 | 41.04 | +0.5 |
| 21 | GSE9476 | Acute myeloid leukemia | 0.164 | 0.841 | 18.11 | 0.064 | 0.825 | 5.51 | +0.5 |
| 22 | GSE18842 | Non-Small Cell Lung Cancer | 0.395 | 0.857 | 44.53 | 0.06 | 0.348 | 16.79 | +0.5 |
| 23 | GSE19188 | Non-Small Cell Lung Cancer | 0.97 | 1.00 | 93.43 | 0.176 | 0.560 | 31.39 | +0.5 |
| 24 | GSE3585 | Dilated cardiomyopathy | 1 | 1.00 | 81.18 | 0.577 | 0.825 | 69.89 | +0.5 |
| Average | 0.595 | 0.854 | 57.06 | 0.389 | 0.682 | 48.19 | +3/24=12.5% | ||
TABLE IV.
The ranks and the p-values of the 24 target pathways for GSA and pDis analysis (all genes). The improvement factor based on Fig. 2 is calculated for each data set considering 5% significance threshold using FDR-corrected p-values. The average improvement factor shows that pDis analysis (all genes) improves the results 33.3% compared to GSA. Twelve target pathways were found to be significant in pDis analysis (all genes) while non of the target pathways have significant FDR-corrected p-values in GSA. As shown, the average p-value for the target pathways are lower (i.e. better) in the pDis analysis (all genes) than in GSA.
| GEO ID | Target pathway | GSA | pDis analysis (all genes) | Improvement Compared to GSA | |||||
|---|---|---|---|---|---|---|---|---|---|
| p-values | FDR | ranks | p-values | FDR | ranks | ||||
| 1 | GSE1297 | Alzheimer’s Disease | 0.100 | 0.514 | 19.42 | 5e-06 | 5.7e-05 | 4.74 | +1.0 |
| 2 | GSE5281 | Alzheimer’s Disease | 0.316 | 0.872 | 33.09 | 5e-06 | 3.6e-05 | 7.29 | +1.0 |
| 3 | GSE5281 | Alzheimer’s Disease | 0.116 | 0.488 | 23.74 | 5e-06 | 2.6e-05 | 9.85 | +1.0 |
| 4 | GSE5281 | Alzheimer’s Disease | 0.164 | 0.537 | 27.69 | 5e-06 | 2.6e-05 | 9.85 | +1.0 |
| 5 | GSE20153 | Parkinson’s disease | 0.542 | 0.885 | 54.67 | 0.002 | 0.008 | 21.53 | +1.0 |
| 6 | GSE20291 | Parkinson’s disease | 0.246 | 0.629 | 35.61 | 5e-06 | 2.2e-05 | 11.67 | +1.0 |
| 7 | GSE8762 | Huntington’s disease | 0.154 | 0.876 | 15.10 | 5e-06 | 1.6e-05 | 15.32 | +1.0 |
| 8 | GSE4107 | Colorectal Cancer | 0.154 | 0.764 | 20.14 | 0.002 | 0.009 | 20.43 | +1.0 |
| 9 | GSE8671 | Colorectal Cancer | 0.002 | 0.069 | 2.87 | 0.116 | 0.248 | 46.71 | −0.5 |
| 10 | GSE9348 | Colorectal Cancer | 0.032 | 0.342 | 9.35 | 0.054 | 0.172 | 31.02 | −0.5 |
| 11 | GSE14762 | Renal Cancer | 0.132 | 0.600 | 21.58 | 0.029 | 0.062 | 46.71 | −0.5 |
| 12 | GSE781 | Renal Cancer | 0.492 | 0.865 | 56.47 | 0.998 | 0.998 | 100 | −0.5 |
| 13 | GSE15471 | Pancreatic Cancer | 0.112 | 0.622 | 17.98 | 0.168 | 0.247 | 67.88 | −0.5 |
| 14 | GSE16515 | Pancreatic Cancer | 0.062 | 0.625 | 8.63 | 0.121 | 0.242 | 49.63 | −0.5 |
| 15 | GSE19728 | Glioma | 0.136 | 0.548 | 24.46 | 5e-06 | 2.2e-05 | 11.31 | 1.0 |
| 16 | GSE21354 | Glioma | 0.128 | 0.547 | 22.66 | 5e-06 | 2.1e-05 | 12.04 | +1.0 |
| 17 | GSE6956 | Prostate Cancer | 0.060 | 0.440 | 9.35 | 0.031 | 0.124 | 24.45 | −0.5 |
| 18 | GSE6956 | Prostate Cancer | 0.032 | 0.451 | 6.47 | 0.001 | 0.013 | 6.56 | +1.0 |
| 19 | GSE3467 | Thyroid Cancer | 0.152 | 0.687 | 20.14 | 0.018 | 0.061 | 28.83 | −0.5 |
| 20 | GSE3678 | Thyroid Cancer | 0.464 | 0.814 | 56.11 | 0.201 | 0.364 | 54.38 | + 0.5 |
| 21 | GSE9476 | Acute myeloid leukemia | 0.128 | 0.902 | 10.07 | 5e-06 | 9.7e-05 | 2.92 | +1.0 |
| 22 | GSE18842 | Non-Small Cell Lung Cancer | 0.156 | 0.6992 | 20.86 | 0.496 | 0.635 | 78.10 | −0.5 |
| 23 | GSE19188 | Non-Small Cell Lung Cancer | 0.446 | 0.844 | 48.92 | 0.874 | 0.925 | 93.43 | −0.5 |
| 24 | GSE3585 | Dilated cardiomyopathy | 0.866 | 0.919 | 91.36 | 0.093 | 0.364 | 25.54 | +0.5 |
| Average | 0.216 | 0.647 | 27.368 | 0.133 | 0.186 | 32.51 | +8/24=33.3% | ||
TABLE V.
The ranks and the p-values of the 24 target pathways for GSEA and pDis analysis (all genes). The improvement factor based on Fig. 2 is calculated for each data set considering 5% significance threshold using FDR-corrected p-values. The average improvement factor shows that pDis analysis (all genes) improves the results 37.5% compared to GSEA. Twelve target pathways were found to be significant in pDis analysis (all genes) while only 2 target pathways have significant FDR-corrected p-values in GSEA. As shown, the average p-value and rank for the target pathways are lower (i.e. better) in the pDis analysis (all genes) than in GSEA.
| GEO ID | Target pathway | GSEA | pDis analysis (all genes) | Improvement Compared to GSEA | |||||
|---|---|---|---|---|---|---|---|---|---|
| p-values | FDR | ranks | p-values | FDR | ranks | ||||
| 1 | GSE1297 | Alzheimer’s Disease | 5e-06 | 4e-05 | 5.75 | 5e-06 | 5.7e-05 | 4.74 | +0.5 |
| 2 | GSE5281 | Alzheimer’s Disease | 5e-06 | 4e-05 | 3.59 | 5e-06 | 3.6e-05 | 7.29 | −0.5 |
| 3 | GSE5281 | Alzheimer’s Disease | 5e-06 | 4e-04 | 5.75 | 5e-06 | 2.6e-05 | 9.85 | −0.5 |
| 4 | GSE5281 | Alzheimer’s Disease | 5e-06 | 4e-05 | 7.19 | 5e-06 | 2.6e-05 | 9.85 | −0.5 |
| 5 | GSE20153 | Parkinson’s disease | 0.995 | 1 | 96.40 | 0.002 | 0.008 | 21.53 | +1 |
| 6 | GSE20291 | Parkinson’s disease | 5e-06 | 4e-05 | 6.83 | 5e-06 | 2.2e-05 | 11.67 | −0.5 |
| 7 | GSE8762 | Huntington’s disease | 5e-06 | 0.08 | 6.83 | 5e-06 | 1.6e-05 | 15.32 | +1 |
| 8 | GSE4107 | Colorectal Cancer | 0.081 | 0.171 | 35.25 | 0.002 | 0.009 | 20.43 | +1 |
| 9 | GSE8671 | Colorectal Cancer | 0.312 | 0.625 | 56.83 | 0.116 | 0.248 | 46.71 | −0.5 |
| 10 | GSE9348 | Colorectal Cancer | 0.118 | 0.283 | 33.81 | 0.054 | 0.172 | 31.02 | +0.5 |
| 11 | GSE14762 | Renal Cancer | 0.148 | 0.261 | 45.32 | 0.029 | 0.062 | 46.71 | −0.5 |
| 12 | GSE781 | Renal Cancer | 0.356 | 0.584 | 58.27 | 0.998 | 0.998 | 100 | −0.5 |
| 13 | GSE15471 | Pancreatic Cancer | 0.020 | 0.038 | 38.84 | 0.168 | 0.247 | 67.88 | −1 |
| 14 | GSE16515 | Pancreatic Cancer | 0.002 | 0.019 | 16.54 | 0.121 | 0.242 | 49.63 | −1 |
| 15 | GSE19728 | Glioma | 0.069 | 0.121 | 50.35 | 5e-06 | 2.2e-05 | 11.31 | +1 |
| 16 | GSE21354 | Glioma | 0.114 | 0.248 | 43.88 | 5e-06 | 2.1e-05 | 12.04 | +1 |
| 17 | GSE6956 | Prostate Cancer | 0.023 | 0.170 | 22.30 | 0.031 | 0.124 | 24.45 | −0.5 |
| 18 | GSE6956 | Prostate Cancer | 0.016 | 0.068 | 17.98 | 0.001 | 0.013 | 6.56 | +1 |
| 19 | GSE3467 | Thyroid Cancer | 0.463 | 0.682 | 71.22 | 0.018 | 0.061 | 28.83 | +0.5 |
| 20 | GSE3678 | Thyroid Cancer | 0.182 | 0.353 | 40.28 | 0.201 | 0.364 | 54.38 | −0.5 |
| 21 | GSE9476 | Acute myeloid leukemia | 0.4662 | 0.808 | 56.83 | 5e-06 | 9.7e-05 | 2.92 | +1 |
| 22 | GSE18842 | Non-Small Cell Lung Cancer | 0.414 | 0.727 | 63.30 | 0.496 | 0.635 | 78.10 | −0.5 |
| 23 | GSE19188 | Non-Small Cell Lung Cancer | 0.870 | 0.995 | 87.76 | 0.874 | 0.925 | 93.43 | −0.5 |
| 24 | GSE3585 | Dilated cardiomyopathy | 0.874 | 1 | 88.48 | 0.093 | 0.364 | 25.54 | +0.5 |
| Average | 0.23 | 0.34 | 39.98 | 0.133 | 0.186 | 32.51 | +2.5/24=10% | ||
TABLE VI.
The ranks and the p-values of the 24 target pathways for SPIA (all genes) and pDis analysis (all genes). The improvement factor based on Fig. 2 is calculated for each data set considering 5% significance threshold using FDR-corrected p-values. The average improvement factor shows that pDis analysis (all genes) improves the results 43.75% compared to SPIA (all genes). Twelve target pathways were found to be significant in pDis analysis (all genes) while only one target pathways have significant FDR-corrected p-values in SPIA (all genes). As shown, the average p-value and rank for the target pathways are lower (i.e. better) in the pDis analysis (all genes) than in SPIA (all genes).
| GEO ID | Target pathway | SPIA (all genes) | pDis analysis (all genes) | Improvement Compared to SPIA (all genes) | |||||
|---|---|---|---|---|---|---|---|---|---|
| p-values | FDR | ranks | p-values | FDR | ranks | ||||
| 1 | GSE1297 | Alzheimer’s Disease | 0.095 | 0.414 | 22.72 | 5e-06 | 5.7e-05 | 4.74 | +1 |
| 2 | GSE5281 | Alzheimer’s Disease | 0.661 | 0.880 | 73.48 | 5e-06 | 3.6e-05 | 7.29 | +1 |
| 3 | GSE5281 | Alzheimer’s Disease | 0.060 | 0.255 | 23.10 | 5e-06 | 2.6e-05 | 9.85 | +1 |
| 4 | GSE5281 | Alzheimer’s Disease | 0.332 | 0.695 | 47.72 | 5e-06 | 2.6e-05 | 9.85 | +1 |
| 5 | GSE20153 | Parkinson’s disease | 0.021 | 0.170 | 11.36 | 0.002 | 0.008 | 21.53 | +1 |
| 6 | GSE20291 | Parkinson’s disease | 0.020 | 0.174 | 10.98 | 5e-06 | 2.2e-05 | 11.67 | +1 |
| 7 | GSE8762 | Huntington’s disease | 0.955 | 0.992 | 96.21 | 5e-06 | 1.6e-05 | 15.32 | +1 |
| 8 | GSE4107 | Colorectal Cancer | 0.010 | 0.101 | 9.84 | 0.002 | 0.009 | 20.43 | +1 |
| 9 | GSE8671 | Colorectal Cancer | 0.991 | 1.00 | 98.48 | 0.116 | 0.248 | 46.71 | +0.5 |
| 10 | GSE9348 | Colorectal Cancer | 0.197 | 0.433 | 45.45 | 0.054 | 0.172 | 31.02 | +0.5 |
| 11 | GSE14762 | Renal Cancer | 0.004 | 0.025 | 15.90 | 0.029 | 0.062 | 46.71 | −1 |
| 12 | GSE781 | Renal Cancer | 0.074 | 0.338 | 21.21 | 0.998 | 0.998 | 100 | −0.5 |
| 13 | GSE15471 | Pancreatic Cancer | 0.039 | 0.125 | 30.68 | 0.168 | 0.247 | 67.88 | −0.5 |
| 14 | GSE16515 | Pancreatic Cancer | 0.046 | 0.144 | 31.81 | 0.121 | 0.242 | 49.63 | −0.5 |
| 15 | GSE19728 | Glioma | 0.301 | 0.502 | 59.84 | 5e-06 | 2.2e-05 | 11.31 | +1 |
| 16 | GSE21354 | Glioma | 0.026 | 0.118 | 21.96 | 5e-06 | 2.1e-05 | 12.04 | +1 |
| 17 | GSE6956 | Prostate Cancer | 0.083 | 0.543 | 15.15 | 0.031 | 0.124 | 24.45 | −0.5 |
| 18 | GSE6956 | Prostate Cancer | 0.027 | 0.294 | 8.71 | 0.001 | 0.013 | 6.56 | +1 |
| 19 | GSE3467 | Thyroid Cancer | 0.936 | 0.990 | 93.93 | 0.018 | 0.061 | 28.83 | +0.5 |
| 20 | GSE3678 | Thyroid Cancer | 0.951 | 0.977 | 94.69 | 0.201 | 0.364 | 54.38 | +0.5 |
| 21 | GSE9476 | Acute myeloid leukemia | 0.512 | 0.793 | 62.87 | 5e-06 | 9.7e-05 | 2.92 | +1 |
| 22 | GSE18842 | Non-Small Cell Lung Cancer | 0.294 | 0.591 | 49.24 | 0.496 | 0.635 | 78.10 | −0.5 |
| 23 | GSE19188 | Non-Small Cell Lung Cancer | 0.712 | 0.824 | 86.36 | 0.874 | 0.925 | 93.43 | −0.5 |
| 24 | GSE3585 | Dilated cardiomyopathy | 0.471 | 0.801 | 57.95 | 0.093 | 0.364 | 25.54 | +0.5 |
| Average | 0.325 | 0.507 | 45.40 | 0.133 | 0.186 | 32.51 | +11/24=43.75% | ||
The paired t-test and the paired Wilcoxon test were performed to compare the distribution of the ranks and p-values of target pathways in each method. The results are shown in table II. The statistical tests are performed as one-tail tests in order to test whether the ranks and p-values of target pathways in proposed methods are significantly lower than the reference methods. The results show that the p-values of the target pathways in pDis analysis (cut-off) are significantly lower than SPIA. Furthermore, the ranks and the p-values of the target pathways in pDis analysis (all genes) is significantly lower than GSEA. The p-values of pDis analysis (all genes) are also lower than those yielded by GSA but not significantly so (at 5%).
TABLE II.
Results of the statistical tests that were performed to compare the results of the various methods. pDis analysis (cut-off) was compared to SPIA (cut-off). pDis analysis (all genes) was compared to GSEA, GSA and SPIA (all genes). Each p-value shows whether the ranks and the p-values of the target pathways in proposed method are significantly lower than the reference methods (at 5% significance threshold). The results show that pDis analysis (cut-off) yields significantly better p-values than SPIA (cut-off) for the target pathways. Also, pDis analysis (all genes) yields lower p-values as well as lower ranks compared to GSEA and SPIA (all genes).
| P-value (paired t.test p-value) | SPIA (cut-off) | GSA | GSEA | SPIA (all genes) |
|---|---|---|---|---|
| pDis analysis (cut-off) | 0.01 | – | – | – |
| pDis analysis (all genes) | – | 0.07 | 0.074 | 0.01 |
| Ranks (paired Wilcoxon.test p-value) | SPIA (cut-off) | GSA | GSEA | SPIA (all genes) |
|---|---|---|---|---|
| pDis analysis (cut-off) | 0.13 | – | – | – |
| pDis analysis (all genes) | – | 0.75 | 0.29 | 0.05 |
The pDis analysis (all genes) yields better results compared to GSEA, in term of both ranks (panel C in Fig. 3, Wilcoxon test p-value = 0.29), as well as p-values of the target pathways (panel D in Fig. 3, t-test p-value = 0.074). The proposed method yields significantly better results compared to SPIA (all genes) in terms of both ranks (panel C in Fig. 3, Wilcoxon test p-value = 0.05), as well as p-values of the target pathways (panel D in Fig. 3, t-test p-value =0.01). The results also show that the proposed method provides more significant p-values compared to GSA, even thought the differences are not statistically significant (see Table II). There is not significant difference between the ranks yielded by pDis (all genes) and GSA. The figure also shows the comparison between pDis analysis (cut-off) and SPIA (cut-off). The proposed method yields significantly better results compared to SPIA (cut-off) in terms of p-values (panel B in Fig. 3, t-test p=0.01). The results are also better in terms of ranks, even though the difference is not statistically significant (panel A in Fig 3, Wilcoxon test p-value =0.13).
As some diseases are complex phenotypes involving fundamental biochemical pathways, other pathways might be significantly impacted in addition to the target pathway. Therefore, we studied the detailed results of pDis analysis (all genes) on a data set, in order to show that our method is not limited to identifying the target pathway as significantly impacted, but it is also able to correctly report relevant fundamental biochemical mechanisms in the condition under study. We chose to perform detailed analysis of the first neurodegenerative disease as it appears in table I. We provide the information about the p-values of all the analyzed pathways with FDR-corrected p-value lower that 5% for the data set studying alzheimer disease [6] (see table VII). The pathways with bold font in each table indicate the pathways that are known to be related to that disease based on existing literature. We can see that most of the significant pathways are biologically meaningful in the condition in analysis, showing high precision in the results. These results indicate that the proposed method is able to report the target pathways as more significant and ranked higher, compared to the state-of-the-art methods for pathway analysis, as well as it is able to report as significant the pathways that are known to be associated to a given disease.
TABLE VII.
The resulting ranks and p-values for all the pathways from analyzing the data set that studies alzheimer disease [6] that have FDR-corrected p-value lower than 5%. We studied the association of these top pathways to alzheimer disease. The pathway shown in red is the target pathway with the name corresponding to the disease under study. The bold pathways are the ones with known association with alzheimer disease based on existing literature. The number of bold and red pathways represents the number of true positives found by the method. Here we can see 16 true positives with FDR-corrected p-value lower than 5%.
| Name | ID | p-values | FDR | ranks | references | |
|---|---|---|---|---|---|---|
| 1 | Alzheimer’s disease | 05010 | 5e-06 | 5e-05 | 4.74 | |
| 2 | Cytokine-cytokine receptor interaction | 04060 | 5e-06 | 5e-05 | 4.74 | [39] |
| 3 | Glutamatergic synapse | 04724 | 5e-06 | 5e-05 | 4.74 | [11] |
| 4 | GABAergic synapse | 04727 | 5e-06 | 5e-05 | 4.74 | [28] |
| 5 | Dopaminergic synapse | 04728 | 5e-06 | 5e-05 | 4.74 | [30] |
| 6 | Long-term depression | 04730 | 5e-06 | 5e-05 | 4.74 | [37] |
| 7 | Endocrine and other factor-regulated calcium reabsorption | 04961 | 5e-06 | 5e-05 | 4.74 | |
| 8 | Parkinson’s disease | 05012 | 5e-06 | 5e-05 | 4.74 | [2], [8] |
| 9 | Amyotrophic lateral sclerosis (ALS) | 05014 | 5e-06 | 5e-05 | 4.74 | [31] |
| 10 | Huntington’s disease | 05016 | 5e-06 | 5e-05 | 4.74 | [12], [8] |
| 11 | Vibrio cholerae infection | 05110 | 5e-06 | 5e-05 | 4.74 | |
| 12 | Pathogenic Escherichia coli infection | 05130 | 5e-06 | 5e-05 | 4.74 | |
| 13 | Oocyte meiosis | 04114 | 1e-03 | 0.01 | 10.95 | |
| 14 | Long-term potentiation | 04720 | 1e-03 | 0.01 | 10.95 | [51], [25] |
| 15 | Retrograde endocannabinoid signaling | 04723 | 1e-03 | 0.01 | 10.95 | [33] |
| 16 | Gastric acid secretion | 04971 | 1e-03 | 0.01 | 10.95 | |
| 17 | Pancreatic secretion | 04972 | 1e-03 | 0.01 | 10.95 | |
| 18 | VEGF signaling pathway | 04370 | 2e-03 | 0.01 | 13.87 | [38], [14] |
| 19 | Epithelial cell signaling in Helicobacter pylori infection | 05120 | 2e-03 | 0.01 | 13.87 | |
| 20 | Systemic lupus erythematosus | 05322 | 2e-03 | 0.01 | 13.87 | |
| 21 | Salmonella infection | 05132 | 3e-03 | 0.02 | 15.33 | |
| 22 | Calcium signaling pathway | 04020 | 0.01 | 0.03 | 16.79 | [59], [7] |
| 23 | Salivary secretion | 04970 | 0.01 | 0.03 | 16.79 | |
| 24 | Arrhythmogenic right ventricular cardiomyopathy (ARVC) | 05412 | 0.01 | 0.03 | 16.79 | |
| 25 | Gap junction | 04540 | 0.01 | 0.03 | 18.25 | [29], [46] |
| 26 | Phosphatidylinositol signaling system | 04070 | 0.01 | 0.04 | 18.98 | [62], [4] |
| 27 | Morphine addiction | 05032 | 0.01 | 0.04 | 19.71 | |
| 28 | Protein processing in endoplasmic reticulum | 04141 | 0.01 | 0.04 | 20.80 | [53] |
| 29 | Shigellosis | 05131 | 0.01 | 0.04 | 20.80 | |
| 30 | Renal cell carcinoma | 05211 | 0.01 | 0.05 | 21.90 |
Ranks and p-values for the target pathways for eight yeast knock-out data sets
We also validate our approach using eight data sets that comes from experiment studying eight yeast knock-out genes. We obtained the KEGG signaling pathways for yeast from the “ROntoTools” R package version 1.2.0 [55] as included in Bioconductor version 2.12 released on April 4th, 2013. We used all pathways that have at least one interaction of type activation, inhibition, expression, or repression. There are nine such yeast pathways in KEGG. We used the data provided by [22] as our wild type and knock-out sample. These are contained in the in the data sets GSE42215 [22] and GSE42527 [22], respectively. We selected eight knock-out data sets whose knock-out genes belong to at least one pathway considered in the analysis. The log2-fold changes for each knock-out sample were calculated by comparing expression levels of that sample with the wild type samples. Each data set was processed as described in section III. We performed the pDis analysis (all genes), SPIA (all genes) and GSA, for each of the eight knock-out sample using the calculated log2-fold changes. The target pathways for each knock-out data are the pathways that include the knock-out genes. The ranks and p-values of the target pathways for eight yeast knockout data sets are shown in the tables VIII and IX. The data show an improvement of about 50% with respect to SPIA (all genes) and an improvement of about 20% with respect to GSA. The GSEA results were not included in the comparison on the knock-out data sets because all data sets involve yeast and GSEA is not available for yeast pathways. The statistical tests are performed as one-tail in order to test whether the ranks and p-values of target pathways in proposed methods are significantly lower than the reference methods. The proposed method yields significantly better results compared to SPIA (all genes) in terms of both p-values (t-test p-value = 0.002) as well as ranks of the target pathways (Wilcoxon test p-value = 0.01). The result show that pDis (all genes) provides lower p-values (t-test p-value = 0.09) and lower ranks for the target pathways, although not significantly (Wilcoxon test p-value = 0.36) when compared to GSA.
TABLE VIII.
The ranks and the p-values of the target pathways for SPIA (all genes) and pDis analysis (all genes) using 8 yeast knock-out gene data sets. The improvement factor based on Fig. 2 is calculated for each data set considering 5% significance threshold using FDR-corrected p-values. The average improvement factor shows that pDis analysis (all genes) improves the results 50% compared to SPIA (all genes). Three target pathways were found to be significant after FDR-correction in pDis analysis (all genes) while only one target pathways have significant FDR-corrected p-values in SPIA (all genes). As shown, the average p-value and rank for the target pathways are lower (i.e. better) in the pDis analysis (all genes) than in SPIA (all genes). The results show that pDis analysis (all genes) yields significantly better p-values than SPIA (all genes) for the target pathways (p-value from t.test = 0.002) as well as it has significantly lower ranks for the target pathways compared to SPIA (all genes) (p-value from Wilcoxon test = 0.01).
| knock-out gens | target pathway | SPIA (all genes) | pDis analysis (all genes) | improvement factors compared to SPIA | |||||
|---|---|---|---|---|---|---|---|---|---|
| p-values | FDR | ranks | p-values | FDR | ranks | ||||
| 1 | APC9 | Cell cycle Meiosis - yeast |
0.422 0.917 |
1 1 |
3 5 |
0.133 0.184 |
0.411 0.414 |
2 4 |
+0.5 +0.5 |
| 2 | TPK3 | Meiosis- yeast | 5e-06 | 4.5e-05 | 1 | 5e-06 | 4e-05 | 1 | – |
| 3 | RGT2 | Meiosis- yeast | 0.075 | 0.225 | 3 | 0.001 | 0.009 | 1 | +1 |
| 4 | USA1 | Protein processing in endoplasmic reticulum | 1 | 1 | 7.5 | 0.092 | 0.27 | 3 | +0.5 |
| 5 | TIF4631 | RNA transport | 1 | 1 | 7.5 | 0.084 | 0.756 | 1 | – |
| 6 | URM1 | Sulfur relay system | 0.048 | 0.306 | 1 | 0.040 | 0.36 | 1 | +1 |
| 7 | SSM4 | Protein processing in endoplasmic reticulum | 1 | 1 | 7.5 | 0.004 | 0.018 | 2 | +0.5 |
| 8 | CUE1 | Protein processing in endoplasmic reticulum | 1 | 1 | 7.5 | 0.208 | 0.624 | 3 | +0.5 |
| Average | 0.606 | 0.725 | 4.77 | 0.082 | 0.318 | 2 | 4.5/9=50% | ||
TABLE IX.
The ranks and the p-values of the target pathways for GSA and pDis analysis (all genes) using 8 yeast knock-out gene data sets. The improvement factor based on Fig. 2 is calculated for each data set considering 5% significance threshold using FDR-corrected p-values. The average improvement factor shows that pDis analysis (all genes) improves the results 22.2% compared to GSA. Three target pathways were found to be significant after FDR-corrected-correction in pDis analysis (all genes) while no target pathways have significant FDR-corrected p-values in GSA. As shown, the average p-value and rank for the target pathways are lower (i.e. better) in the pDis analysis (all genes) than in GSA. The results show that pDis analysis (all genes) yields better p-values than GSA for the target pathways (p-value from t.test = 0.09) as well as it has lower ranks for the target pathways compared to GSA (p-value from Wilcoxon test = 0.36).
| knock-out gens | target pathway | GSA | pDis analysis (all genes) | improvement factors compared to GSA | |||||
|---|---|---|---|---|---|---|---|---|---|
| p-values | FDR | ranks | p-values | FDR | ranks | ||||
| 1 | APC9 | Cell cycle Meiosis - yeast |
0.05 0.06 |
0.28 0.28 |
1 2 |
0.133 0.184 |
0.411 0.414 |
2 4 |
−0.5 −0.5 |
| 2 | TPK3 | Meiosis- yeast | 0.40 | 0.63 | 5 | 5e-06 | 4e-05 | 1 | +1 |
| 3 | RGT2 | Meiosis- yeast | 0.98 | 0.98 | 9 | 0.001 | 0.009 | 1 | +1 |
| 4 | USA1 | Protein processing in endoplasmic reticulum | 0.09 | 0.78 | 1 | 0.092 | 0.27 | 3 | −0.5 |
| 5 | TIF4631 | RNA transport | 0.78 | 0.88 | 6 | 0.084 | 0.756 | 1 | +0.5 |
| 6 | URM1 | Sulfur relay system | 0.02 | 0.09 | 2 | 0.040 | 0.36 | 1 | +0.5 |
| 7 | SSM4 | Protein processing in endoplasmic reticulum | 0.04 | 0.43 | 1 | 0.004 | 0.018 | 2 | +1 |
| 8 | CUE1 | Protein processing in endoplasmic reticulum | 0.04 | 0.31 | 1 | 0.208 | 0.624 | 3 | −0.5 |
| Average | 0.27 | 0.51 | 3.1 | 0.082 | 0.318 | 2 | 2/9=22.2% | ||
False positives under the null hypothesis
As we have demonstrated, the proposed method produces significantly lower p-values for the target pathways compared with the existing methods, across the set of 24 data sets used in the validation. However, lower p-values for the target pathways could be produced if the new method indiscriminately lowered the p-values for all pathways, thus introducing many false positives.
In order to show that this is not the case, we ran a number of experiments with completely random data. In each of these experiments, a set of expression changes are assigned to the genes from a random normal distribution with mean of zero and standard deviation of 1. This was repeated 1,000 times and p-values for the pathways were computed in each iteration. The pathway p-values for these random data sets, produced the distribution for the p-values under the null hypothesis. Null-hypothesis distributions were also calculated for each target pathway and showed no abnormal tendencies. The distribution of the pooled p-values for all pathways over the 1,000 iterations is shown in Fig. 4. Both the distribution of the pooled p-values, as well as all null distributions associated with each individual target pathway were uniform, demonstrating that our method does not yield more significant p-values for the target pathways by lowering all p-values. These distributions demonstrate that the proposed method does not produce any more false positives than appropriate for any significance threshold.
Fig. 4.
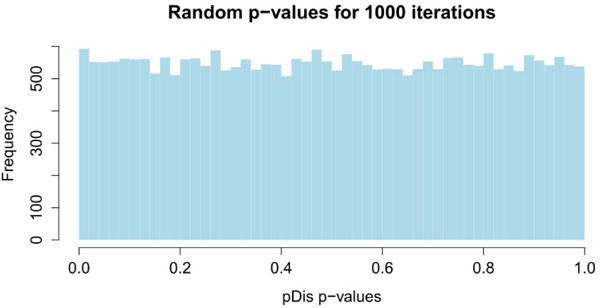
The null distribution of the p-values obtained from pDis analysis for all KEGG signaling pathway (139 pathways) in 1,000 iterations. The input gene expression values were chosen from a random normal distribution with mean of 0 and standard deviation of 1. The histogram shows the null distribution of the pooled p-values. Uniform distributions were also obtained for each individual target pathway (data not shown). The uniform distributions prove that pDis analysis does not produce any more false positives than expected.
IV. Conclusion
Here we proposed a new topological pathway analysis method based on the amount of perturbation associated with each individual gene. The proposed pDis analysis considers the dis-regulation of each gene in every pathway to calculate a p-value with respect to the distribution of the dis-regulation under the null hypothesis. The proposed method is able to use either i) a pre-selected number of DE genes, pDis analysis (cut-off), or ii) the entire list of measured expression levels, pDis analysis (all genes). The results showed that the proposed method yields significant improvements with respect to the state-of-the-art methods: SPIA, GSEA and GSA. The comparisons have been performed with a validation method that used 24 different data sets involving 12 different human diseases and eight different data sets involving eight knocked out genes in yeast.
Acknowledgments
This research was supported in part by the following grants: NIH R01 DK089167, R42 GM087013 and NSF DBI-0965741, and by the Robert J. Sokol Endowment in Systems Biology. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of any of the funding agencies.
Biographies

Sahar Ansari is a Ph.D. candidate in the department of Computer Science, Wayne State University, Detroit, MI, USA. She received his B.Sc. in Electrical Engineering in 2010 from Sharif University of Technology, Tehran, Iran. Her research interests include systems biology, data mining and biostatistics.

Calin Voichita earned his Ph.D. in computer science from Wayne State University, Detroit, MI, USA. His research efforts are focused on ways to detect abnormalities in the behavior of biological networks. He designed and implemented methods to propagate measured expression changes through gene signaling pathways without the need to select differentially express genes. He is the author of the ROntoTools package available as part of Bioconductor that make these methods available in the R language. These methods allow life scientists to take advantage of the wealth of information available with the recent RNAseq technologies. Furthermore, he applies his computational intelligence experience to develop methods that are able to identify patients that will not respond to general treatments and may require personalized solutions.

Michele Donato received his master’s degree in computer engineering from Universit di Pisa in 2006 (Italy). He received a second master’e degree in computer science from the department of computer science at Wayne State University in 2015 (USA). He also successfully defended his Ph.D. thesis to the department of computer science at Wayne State University in 2015. Currently he is a postdoctoral research fellow at Stanford University (USA).

Rebecca Tagett has a Bachelors in Physics, a Masters in Molecular Biology, and 10 years R&D experience in industry as a Computational Biologist. A PhD Candidate and a member of the Intelligent Systems and Bioinformatics Laboratory (ISBL) in the Department of Computer Science at Wayne State University, her research focuses on phenotypic prediction using multi-omics. Her interests are Functional Genomics, Scientific Writing, Bioinformatics and Biostatistics. She is a member of the International Society for Computational Biology (ISCB).

Sorin Draghici earned a Ph.D. in computer science from the University of St. Andrews, United Kingdom. He holds the Robert J. Sokol MD Endowed Chair in Systems Biology in the Department of Obstetrics and Gynecology, and is a professor in the Department of Clinical and Translational Science and the Department of Computer Science, as well as the head of the Intelligent Systems and Bioinformatics Laboratory at Wayne State University. He is also the chief of the Bioinformatics and Data Analysis Section in the Perinatology Research Branch of the National Institute for Child Health and Development. A senior member of IEEE, Dr. Draghici is an editor of IEEE/ACM Transactions on Computational Biology and Bioinformatics, Journal of Biomedicine and Biotechnology, and International Journal of Functional Informatics and Personalized Medicine. He has published two books on microarray analysis entitled Data Analysis Tools for Microarrays (Chapman and Hall/CRC Press, 2003) and Statistics and Data Analysis for Microarrays Using R and Bioconductor, Second Edition (Chapman & Hall/CRC Mathematical & Computational Biology, 2011), eight book chapters and more than 100 peer-reviewed journal and conference papers.
Contributor Information
Sahar Ansari, Department of Computer Science, Wayne State University, Detroit, MI, USA.
Calin Voichita, Department of Computer Science, Wayne State University, Detroit, MI, USA.
Michele Donato, Department of Computer Science, Wayne State University, Detroit, MI, USA.
Rebecca Tagett, Department of Computer Science, Wayne State University, Detroit, MI, USA.
Sorin Draghici, Department of Computer Science, Wayne State University, Detroit, MI, USA.
References
- 1.Badea Liviu, Herlea Vlad, Dima Simona Olimpia, Dumitrascu Traian, Popescu Irinel, et al. Combined Gene Expression Analysis of Whole-Tissue and Microdissected Pancreatic Ductal Adenocarcinoma identifies Genes Specifically Overexpressed in Tumor Epithelia. Hepatogastroenterology. 2008;55(88):2016. [PubMed] [Google Scholar]
- 2.Bailey Rachel M, Covy Jason P, Melrose Heather L, Rousseau Linda, Watkinson Ruth, Knight Joshua, Miles Sarah, Farrer Matthew J, Dickson Dennis W, Giasson Benoit I, et al. Lrrk2 phosphorylates novel tau epitopes and promotes tauopathy. Acta Neuropathologica. 2013;126(6):809–827. doi: 10.1007/s00401-013-1188-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barth Andreas S, Kuner Ruprecht, Buness Andreas, Ruschhaupt Markus, Merk Sylvia, Zwermann Ludwig, Kääb Stefan, Kreuzer Eckart, Steinbeck Gerhard, Mansmann Ulrich, et al. Identification of a common gene expression signature in dilated cardiomyopathy across independent microarray studies. Journal of the American College of Cardiology. 2006;48(8):1610–1617. doi: 10.1016/j.jacc.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 4.Bhaskar Kiran, Miller Megan, Chludzinski Alexandra, Herrup Karl, Zagorski Michael, Lamb Bruce T. The pi3k-akt-mtor pathway regulates abeta oligomer induced neuronal cell cycle events. Mol Neurodegener. 2009;4:14. doi: 10.1186/1750-1326-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.BioCarta. BioCarta - Charting Pathways of Life. http://www.biocarta.com.
- 6.Blalock Eric M, Geddes James W, Chen Kuey Chu, Porter Nada M, Markesbery William R, Landfield Philip W. Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(7):2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheung King-Ho, Shineman Diana, Müller Marioly, Cardenas Cesar, Mei Lijuan, Yang Jun, Tomita Taisuke, Iwatsubo Takeshi, Lee Virginia M-Y, Foskett J Kevin. Mechanism of ca 2+ disruption in alzheimer’s disease by presenilin regulation of insp 3 receptor channel gating. Neuron. 2008;58(6):871–883. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Correia Sónia C, Santos Renato X, Perry George, Zhu Xiongwei, Moreira Paula I, Smith Mark A. Neurodegenerative Diseases. Springer; 2012. Mitochondrial importance in alzheimer’s, huntington’s and parkinson’s diseases; pp. 205–221. [DOI] [PubMed] [Google Scholar]
- 9.Drăghici Sorin, Khatri Purvesh, Tarca Adi Laurentiu, Amin Kashayp, Done Arina, Voichiţa Călin, Georgescu Constantin, Romero Roberto. A systems biology approach for pathway level analysis. Genome Research. 2007;17(10):1537–1545. doi: 10.1101/gr.6202607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Efron Bradley, Tibshirani Robert. On testing the significance of sets of genes. The Annals of Applied Statistics. 2007;1(1):107–129. [Google Scholar]
- 11.Francis Paul T. Glutamatergic systems in alzheimer’s disease. International Journal of Geriatric Psychiatry. 2003;18(S1):S15–S21. doi: 10.1002/gps.934. [DOI] [PubMed] [Google Scholar]
- 12.Haim Lucile Ben, Ceyzériat Kelly, Sauvage Maria Angeles Carrillo-de, Aubry Fabien, Auregan Gwennaëlle, Guillermier Martine, Ruiz Marta, Petit Fanny, Houitte Diane, Faivre Emilie, et al. The jak/stat3 pathway is a common inducer of astrocyte reactivity in alzheimer’s and huntington’s diseases. The Journal of Neuroscience. 2015;35(6):2817–2829. doi: 10.1523/JNEUROSCI.3516-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He Huiling, Jazdzewski Krystian, Li Wei, Liyanarachchi Sandya, Nagy Rebecca, Volinia Stefano, Calin George A, Liu Chang-gong, Franssila Kaarle, Suster Saul, et al. The role of microRNA genes in papillary thyroid carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(52):19075–19080. doi: 10.1073/pnas.0509603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hohman Timothy J, Bell Susan P, Jefferson Angela L. The role of vascular endothelial growth factor in neurodegeneration and cognitive decline: Exploring interactions with biomarkers of alzheimer disease. JAMA Neurology. 2015 doi: 10.1001/jamaneurol.2014.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong Yi, Downey Thomas, Eu Kong Weng, Koh Poh Koon, Cheah Peh Yean. A ‘metastasis-prone’signature for early-stage mismatch-repair proficient sporadic colorectal cancer patients and its implications for possible therapeutics. Clinical & Experimental Metastasis. 2010;27(2):83–90. doi: 10.1007/s10585-010-9305-4. [DOI] [PubMed] [Google Scholar]
- 16.Hong Yi, Ho Kok Sun, Eu Kong Weng, Cheah Peh Yean. A susceptibility gene set for early onset colorectal cancer that integrates diverse signaling pathways: implication for tumorigenesis. Clinical Cancer Research. 2007;13(4):1107–1114. doi: 10.1158/1078-0432.CCR-06-1633. [DOI] [PubMed] [Google Scholar]
- 17.Hou Jun, Aerts Joachim, Den Hamer Bianca, Van Ijcken Wilfred, Bakker Michael Den, Riegman Peter, van der Leest Cor, van der Spek Peter, Foekens John A, Hoogsteden Henk C, Grosveld Frank, Philipsen Sjaak. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS ONE. 2010;5(4):e10312. doi: 10.1371/journal.pone.0010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ideker Trey, Ozier Owen, Schwikowski Benno, Siegel Andrew F. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 2002;18(suppl 1):S233–S240. doi: 10.1093/bioinformatics/18.suppl_1.s233. [DOI] [PubMed] [Google Scholar]
- 19.Ideker Trey, Sharan Roded. Protein networks in disease. Genome Research. 2008;18(4):644–652. doi: 10.1101/gr.071852.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Irizarry Rafael A, Gautier Laurent, Bolstad Benjamin Milo, Miller Crispin, Astrand Magnus, Cope Leslie M, Gentleman Robert, Gentry Jeff, Halling Conrad, Huber Wolfgang, MacDonald James, Rubinstein Benjamin IP, Workman Christopher, Zhang John. affy: Methods for Affymetrix Oligonucleotide Arrays. 2005 R package version 1.6.7. [Google Scholar]
- 21.Joshi-Tope G, Gillespie Marc, Vastrik Imre, D’Eustachio Peter, Schmidt Esther, Bono Bernard de, Jassal Bijay, Gopinath GR, Wu GR, Matthews Lisa, Lewis Suzanna, Birney Ewan, Stein Lincoln. REACTOME: a knowledgebase of biological pathways. Nucleic Acids Research. 2005;33:D428–432. doi: 10.1093/nar/gki072. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kemmeren Patrick, Sameith Katrin, van de Pasch Loes AL, Benschop Joris J, Lenstra Tineke L, Margaritis Thanasis, O’Duibhir Eoghan, Apweiler Eva, Wageningen Sake van, Ko Cheuk W, et al. Large-scale genetic perturbations reveal regulatory networks and an abundance of gene-specific repressors. Cell. 2014;157(3):740–752. doi: 10.1016/j.cell.2014.02.054. [DOI] [PubMed] [Google Scholar]
- 23.Khatri Purvesh, Drăghici Sorin. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 2005;21(18):3587–3595. doi: 10.1093/bioinformatics/bti565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khatri Purvesh, Sirota Marina, Butte Atul J. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Computational Biology. 2012;8(2):e1002375. doi: 10.1371/journal.pcbi.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar Ashok. Long-term potentiation at ca3–ca1 hippocampal synapses with special emphasis on aging, disease, and stress. Frontiers in Aging Neuroscience. 2011;3 doi: 10.3389/fnagi.2011.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lenburg Marc E, Liou Louis S, Gerry Norman P, Frampton Garrett M, Cohen Herbert T, Christman Michael F. Previously unidentified changes in renal cell carcinoma gene expression identified by parametric analysis of microarray data. BMC Cancer. 2003;3(1):31. doi: 10.1186/1471-2407-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang Winnie S, Dunckley Travis, Beach Thomas G, Grover Andrew, Mastroeni Diego, Walker Douglas G, Caselli Richard J, Kukull Walter A, McKeel Daniel, Morris John C, et al. Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain. Physiological Genomics. 2007;28(3):311–322. doi: 10.1152/physiolgenomics.00208.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Limon Agenor, Reyes-Ruiz Jorge Mauricio, Miledi Ricardo. Loss of functional gabaa receptors in the alzheimer diseased brain. Proceedings of the National Academy of Sciences. 2012;109(25):10071–10076. doi: 10.1073/pnas.1204606109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maragakis Nicholas J, Rothstein Jeffrey D. Mechanisms of disease: astrocytes in neurodegenerative disease. Nature Clinical Practice Neurology. 2006;2(12):679–689. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- 30.Martorana Alessandro, Koch Giacomo. Is dopamine involved in alzheimer’s disease? Frontiers in Aging Neuroscience. 2014;6 doi: 10.3389/fnagi.2014.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mhatre Molina, Floyd Robert A, Hensley Kenneth. Oxidative stress and neuroinflammation in alzheimer’s disease and amyotrophic lateral sclerosis: common links and potential therapeutic targets. Journal of Alzheimer’s Disease. 2004;6(2):147–157. doi: 10.3233/jad-2004-6206. [DOI] [PubMed] [Google Scholar]
- 32.Mitrea Cristina, Taghavi Zeinab, Bokanizad Behzad, Hanoudi Samer, Tagett Rebecca, Donato Michele, Voichiţa Călin, Drăghici Sorin. Methods and approaches in the topology-based analysis of biological pathways. Frontiers in Physiology. 2013;4:278. doi: 10.3389/fphys.2013.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mulder Jan, Zilberter Misha, Pasquaré Susana J, Alpár Alán, Schulte Gunnar, Ferreira Samira G, Köfalvi Attila, Martín-Moreno Ana M, Keimpema Erik, Tanila Heikki, et al. Molecular reorganization of endocannabinoid signalling in alzheimer’s disease. Brain. 2011;134(4):1041–1060. doi: 10.1093/brain/awr046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogata Hiroyuki, Goto Susumu, Sato Kazushige, Fujibuchi Wataru, Bono Hidemasa, Kanehisa Minoru. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research. 1999;27(1):29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pan Kuang-Hung, Lih Chih-Jian, Cohen Stanley N. Effects of threshold choice on biological conclusions reached during analysis of gene expression by DNA microarrays. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(25):8961–8965. doi: 10.1073/pnas.0502674102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pei Huadong, Li Liang, Fridley Brooke L, Jenkins Gregory D, Kalari Krishna R, Lingle Wilma, Petersen Gloria, Lou Zhenkun, Wang Liewei. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell. 2009;16(3):259–266. doi: 10.1016/j.ccr.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pellegrino Laurel D, Peters Matthew E, Lyketsos Constantine G, Marano Christopher M. Depression in cognitive impairment. Current Psychiatry Reports. 2013;15(9):1–8. doi: 10.1007/s11920-013-0384-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Religa Piotr, Cao Renhai, Religa Dorota, Xue Yuan, Bogdanovic Nenad, Westaway David, Marti Hugo H, Winblad Bengt, Cao Yihai. Vegf significantly restores impaired memory behavior in alzheimer’s mice by improvement of vascular survival. Scientific Reports. 2013;3 doi: 10.1038/srep02053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rubio-Perez Jose Miguel, Morillas-Ruiz Juana Maria. A review: inflammatory process in alzheimer’s disease, role of cytokines. The Scientific World Journal. 2012:2012. doi: 10.1100/2012/756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Runne Heike, Kuhn Alexandre, Wild Edward J, Pratyaksha Wirahpati, Kristiansen Mark, Isaacs Jeremy D, Régulier Etienne, Delorenzi Mauro, Tabrizi Sarah J, Luthi-Carter Ruth. Analysis of potential transcriptomic biomarkers for Huntington’s disease in peripheral blood. Proceedings of the National Academy of Sciences. 2007;104(36):14424–14429. doi: 10.1073/pnas.0703652104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sabates-Bellver Jacob, Van der Flier Laurens G, de Palo Mariagrazia, Cattaneo Elisa, Maake Caroline, Rehrauer Hubert, Laczko Endre, Kurowski Michal A, Bujnicki Janusz M, Menigatti Mirco, et al. Transcriptome profile of human colorectal adenomas. Molecular Cancer Research. 2007;5(12):1263–1275. doi: 10.1158/1541-7786.MCR-07-0267. [DOI] [PubMed] [Google Scholar]
- 42.Sanchez-Palencia Abel, Gomez-Morales Mercedes, Gomez-Capilla Jose Antonio, Pedraza Vicente, Boyero Laura, Rosell Rafael, Fárez-Vidal Ma Esther. Gene expression profiling reveals novel biomarkers in nonsmall cell lung cancer. International Journal of Cancer. 2011;129(2):355–364. doi: 10.1002/ijc.25704. [DOI] [PubMed] [Google Scholar]
- 43.Smyth Gordon K. Limma: linear models for microarray data. Springer; New York: 2005. pp. 397–420. [Google Scholar]
- 44.Stirewalt Derek L, Meshinchi Soheil, Kopecky Kenneth J, Fan Wenhong, Pogosova-Agadjanyan Era L, Engel Julia H, Cronk Michelle R, Dorcy Kathleen Shannon, McQuary Amy R, Hockenbery David, et al. Identification of genes with abnormal expression changes in acute myeloid leukemia. Genes, Chromosomes and Cancer. 2008;47(1):8–20. doi: 10.1002/gcc.20500. [DOI] [PubMed] [Google Scholar]
- 45.Subramanian Aravind, Tamayo Pablo, Mootha Vamsi K, Mukherjee Sayan, Ebert Benjamin L, Gillette Michael A, Paulovich Amanda, Pomeroy Scott L, Golub Todd R, Lander Eric S, Mesirov Jill P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceeding of The National Academy of Sciences of the Unites States of America. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takeuchi Hideyuki, Mizoguchi Hiroyuki, Doi Yukiko, Jin Shijie, Noda Mariko, Liang Jianfeng, Li Hua, Zhou Yan, Mori Rarami, Yasuoka Satoko, et al. Blockade of gap junction hemichannel suppresses disease progression in mouse models of amyotrophic lateral sclerosis and alzheimer’s disease. PloS One. 2011;6(6):e21108. doi: 10.1371/journal.pone.0021108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tarca Adi L, Drǎghici Sorin, Bhatti Gaurav, Romero Roberto. Down-weighting overlapping genes improves gene set analysis. BMC Bioinformatics. 2012;13(1):136. doi: 10.1186/1471-2105-13-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tarca Adi L, Drǎghici Sorin, Khatri Purvesh, Hassan Sonia S, Mittal Pooja, Kim Jung-Sun, Kim Chong J, Kusanovic Juan P, Romero Roberto. A novel signaling pathway impact analysis (SPIA) Bioinformatics. 2009;25(1):75–82. doi: 10.1093/bioinformatics/btn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tarca Adi Laurentiu, Kathri Purvesh, Draghici Sorin. SPIA: Signaling Pathway Impact Analysis (SPIA) using combined evidence of pathway over-representation and unusual signaling perturbations. 2013 R package version 2.14.0. [Google Scholar]
- 50.R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2005. [Google Scholar]
- 51.Tu Shichun, Okamoto Shu-ichi, Lipton Stuart A, Xu Huaxi. Oligomeric a β-induced synaptic dysfunction in alzheimer’s disease. Molecular Neurodegeneration. 2014;9(1):48. doi: 10.1186/1750-1326-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vanunu Oron, Magger Oded, Ruppin Eytan, Shlomi Tomer, Sharan Roded. Associating genes and protein complexes with disease via network propagation. PLoS Computational Biology. 2010;6(1):e1000641. doi: 10.1371/journal.pcbi.1000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Viana Ricardo JS, Nunes Ana F, Rodrigues Cecília MP. Endoplasmic reticulum enrollment in alzheimer’s disease. Molecular Neurobiology. 2012;46(2):522–534. doi: 10.1007/s12035-012-8301-x. [DOI] [PubMed] [Google Scholar]
- 54.Voichiţa Călin, Donato Michele, Drǎghici Sorin. Machine Learning and Applications (ICMLA), 2012 11th International Conference on. Vol. 1. Boca Raton, FL, USA: IEEE; Dec 12–15, 2012. Incorporating gene significance in the impact analysis of signaling pathways; pp. 126–131. [Google Scholar]
- 55.Voichita Calin, Draghici Sorin. ROntoTools: R Onto-Tools suite. R package version 1.2.0. [Google Scholar]
- 56.Wallace Tiffany A, Prueitt Robyn L, Yi Ming, Howe Tiffany M, Gillespie John W, Yfantis Harris G, Stephens Robert M, Caporaso Neil E, Loffredo Christopher A, Ambs Stefan. Tumor immunobiological differences in prostate cancer between African-American and European-American men. Cancer Research. 2008;68(3):927–936. doi: 10.1158/0008-5472.CAN-07-2608. [DOI] [PubMed] [Google Scholar]
- 57.Wang Peggy I, Marcotte Edward M. It’s the machine that matters: predicting gene function and phenotype from protein networks. Journal of Proteomics. 2010;73(11):2277–2289. doi: 10.1016/j.jprot.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Yi, Roche Olga, Yan Mathew S, Finak Greg, Evans Andrew J, Metcalf Julie L, Hast Bridgid E, Hanna Sara C, Wondergem Bill, Furge Kyle A, et al. Regulation of endocytosis via the oxygen-sensing pathway. Nature Medicine. 2009;15(3):319–324. doi: 10.1038/nm.1922. [DOI] [PubMed] [Google Scholar]
- 59.Woods Neha Kabra, Padmanabhan Jaya. Calcium Signaling. Springer; 2012. Neuronal calcium signaling and alzheimer’s disease; pp. 1193–1217. [DOI] [PubMed] [Google Scholar]
- 60.Zhang Yanli, James Michael, Middleton Frank A, Davis Richard L. Transcriptional analysis of multiple brain regions in Parkinson’s disease supports the involvement of specific protein processing, energy metabolism, and signaling pathways, and suggests novel disease mechanisms. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2005;137(1):5–16. doi: 10.1002/ajmg.b.30195. [DOI] [PubMed] [Google Scholar]
- 61.Zheng Bin, Liao Zhixiang, Locascio Joseph J, Lesniak Kristen A, Roderick Sarah S, Watt Marla L, Eklund Aron C, Zhang-James Yanli, Kim Peter D, Hauser Michael A, et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Science Translational Medicine. 2010;2(52):52ra73. doi: 10.1126/scitranslmed.3001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zubenko George S, Stiffler J Scott, Hughes Hugh B, Martinez A Julio. Reductions in brain phosphatidylinositol kinase activities in alzheimer’s disease. Biological Psychiatry. 1999;45(6):731–736. doi: 10.1016/s0006-3223(98)00073-0. [DOI] [PubMed] [Google Scholar]