

Summary
Chromatin modifying enzymes, and specifically the protein arginine methyltransferases (PRMTs) have emerged as important targets in cancer. Here, we investigated the role of CARM1 in normal and malignant hematopoiesis. Using conditional knockout mice, we show that loss of CARM1 has little effect on normal hematopoiesis. Strikingly, knockout of Carm1 abrogates both the initiation and maintenance of acute myeloid leukemia (AML) driven by oncogenic transcription factors. We show that CARM1 knockdown impairs cell cycle progression, promotes myeloid differentiation, and ultimately induces apoptosis. Finally, we utilize a selective, small-molecule inhibitor of CARM1 to validate the efficacy of CARM1 inhibition in leukemia cells in vitro and in vivo. Collectively, this work suggests that targeting CARM1 may be an effective therapeutic strategy for AML.
Keywords: AML, arginine methyltransferase, CARM1, AML1-ETO, MLL-AF9, epigenetics, acute myeloid leukemia
IN BRIEF:
Greenblatt et al. show that loss of the protein arginine methyltransfersase CARM1 minimally impacts normal hematopoiesis but strongly impairs leukemogenesis by regulating cell cycle progression, myeloid differentiation, and apoptosis. Targeting CARM1 reduces AML growth in primary patient samples and mouse models.
Introduction:
Protein methyltransferase enzymes play a major role in cancer. While mutations in these enzymes infrequently occur in cancer, when overexpressed, they promote oncogenic transcriptional programs that are critical for generating or maintaining the disease. Accumulating evidence suggests that cancer cells may be uniquely sensitive to inhibition of these enzymes. For example, inhibition of the DOT1L methyltransferase has shown efficacy in MLL-rearranged leukemia, while inhibition of EZH2 methyltransferase activity shows promise for treating patients with B cell lymphoma (Bernt et al., 2011, Daigle et al., 2011, Knutson et al., 2012, McCabe et al., 2012). Inhibition of readers of epigenetic marks such as BRD4 have also shown efficacy in MLL-rearranged leukemia, as have small molecule inhibitors of MENIN and LEDGF, two proteins that bridge the MLL-fusion protein complex to chromatin (Yokoyama et al., 2008, Mereau et al., 2013). Notably, while these epigenetic readers, writers, and cofactors have important roles in normal transcriptional regulation, there appears to be a greater dependence on their activity in leukemia, leading to a therapeutic selectivity.
The enzyme co-activator associated arginine methyltransferase 1 (CARM1), has broad roles in embryonic development and cellular differentiation (Torres-Padilla et al., 2007, Wu et al., 2009). CARM1, also known as protein arginine methyltransferase 4 (PRMT4), is a type I arginine methyltransferase enzyme, that adds asymmetric dimethylation to arginine residues in histones, with specificity for H3R17 and H3R26, as well as an ever increasing number of other protein substrates (Schurter et al., 2001, Shishkova et al., 2017). These substrates include the transcription factor RUNX1, co-activators such as p300, CBP, and AIB1, and chromatin regulatory proteins including members of the SWI/SNF, COMPASS, and Mediator complexes (Xu et al., 2001, Daujat et al., 2002, An et al., 2004, Feng et al., 2006, Naeem et al., 2007, Lee et al., 2011, Wang et al., 2014, Wang et al., 2015, Xu et al., 2017). CARM1 also methylates RNA binding proteins such as PABP1 and multiple splicing factors, including CA150, SAP49, SmB, and U1C (Lee et al., 2002, Cheng et al., 2007). Through these modifications and others, CARM1 regulates critical cellular processes such as RNA splicing and autophagy (Ohkura et al., 2005, Cheng et al., 2007, Shin et al., 2016).
Increased CARM1 expression and/or activity has been reported in prostate, breast, colorectal, lung, and liver cancer (Hong et al., 2004, El Messaoudi et al., 2006, Majumder et al., 2006, Kim et al., 2010, Al-Dhaheri et al., 2011, Osada et al., 2013, Yang et al., 2013). In solid tumors, CARM1 serves as a cofactor for cancer-associated transcription factors such as NF-KB, p53, and steroid hormone receptors, and its expression is correlated with cancer cell proliferation, metastasis, and poor survival outcomes (Covic et al., 2005, Frietze et al., 2008, Jansson et al., 2008, Mann et al., 2013). Emerging evidence also supports the role of the PRMTs in malignant hematopoiesis (Cheung et al., 2007, Zhao et al., 2008, Shia et al., 2012, Liu et al., 2015, Cheung et al., 2016). We previously reported the overexpression of CARM1 in the context of leukemia, showing that 70% of cytogenetically normal acute myeloid leukemia (AML) patients have up-regulation of CARM1 (Vu et al., 2013). Our initial analysis showed that CARM1 levels are highest in undifferentiated human CD34+ cells, with decreased expression as cells undergo cytokine-driven myeloid differentiation in vitro. Furthermore, overexpression of CARM1 blocks the myeloid differentiation of human hematopoietic stem and progenitor cells (HSPCs), while CARM1 knockdown (KD) induces differentiation.
AML1-ETO, which is generated by t(8:21), is the most common translocation in adult de novo AML, occurring in approximately 12% of patients, while translocations involving the MLL gene (on 11q23), occur in 15% of pediatric AML and more than 70% of infant ALL. Evidence exists that CARM1 can regulate the function of the individual components of these oncogenic fusion proteins. AML1 is methylated by CARM1 on R223, leading to the recruitment of a multi-protein complex that regulates the expression of genes critical for myeloid differentiation (Vu et al., 2013). Similarly, a multi-protein complex containing MLL1 is assembled following CARM1-dependent methylation of transcriptional regulatory proteins, which modulates gene expression during differentiation (Kawabe et al., 2012). Although it is unknown if the AML1 and MLL containing fusion proteins are dependent on CARM1 for their function, we hypothesized that CARM1 may play a critical role in transformed hematopoietic cells.
Results
Generation of hematopoietic-specific CARM1 knockout mice
In order to understand the role of CARM1 in the mouse hematopoietic system, we first determined the levels of Carm1 mRNA and protein in different HSPC populations and several mature populations in the mouse bone marrow (BM), purifying each population based on cell surface marker expression. Carm1 mRNA and protein levels were readily detected in HSPCs, and elevated in the common myeloid progenitor (CMP) population, with decreased mRNA and protein expression in mature Mac-1+GR-1+ myeloid cells, megakaryocytic, and erythroid populations (Figure 1A-1B).
Figure 1: Generation of hematopoietic-specific Carm1 knockout mice.
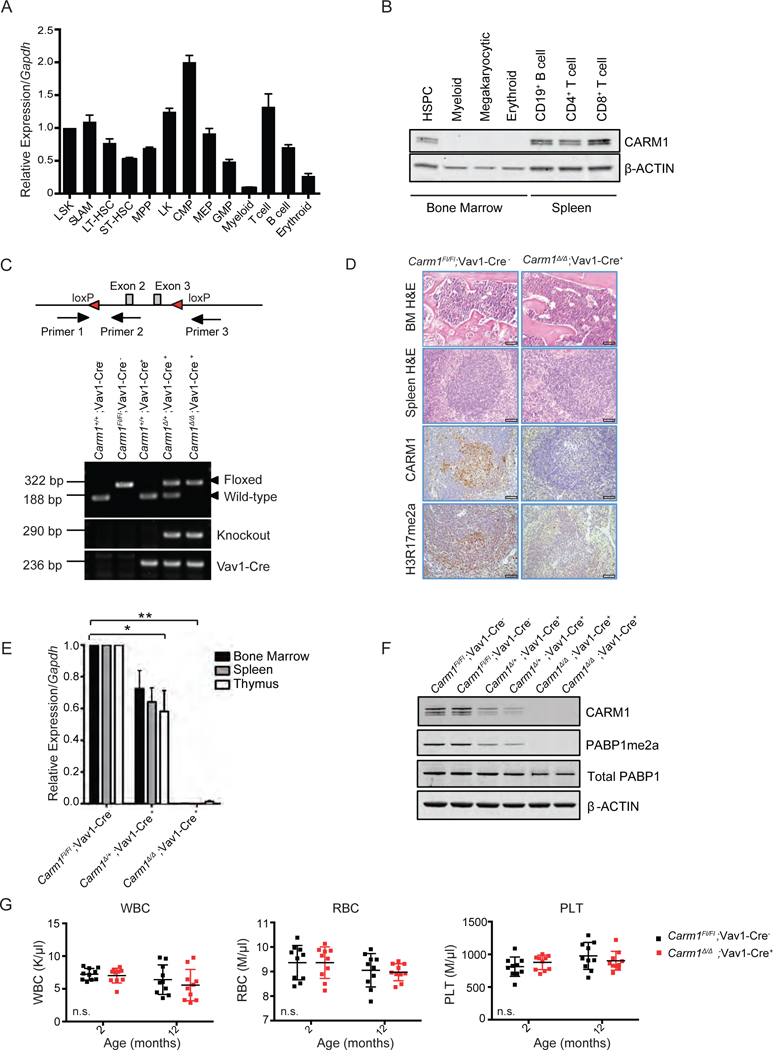
A) Relative Carm1 expression from mRNA isolated from sorted hematopoietic stem and progenitor cells and mature hematopoietic populations analyzed by qRT-PCR. MPPs, multipotent progenitors; CMPs, common myeloid progenitors; GMPs, granulocyte-macrophage progenitors. Carm1 expression is normalized to Gapdh. n=5
B)Total protein isolated from sorted HSPC and mature hematopoietic populations and expression of CARM1 and β-ACTIN analyzed by western blot.
C) Schema of targeting strategy resulting in disruption of transcription at Carm1 exon 2 and location of genotyping primers to confirm the floxed locus (Primer 1 and 2) or knockout of Carm1 (Primer 1 and 3). Representative PCR assessing Carm1 wild-type, floxed, and knockout alleles and Vav1-Cre.
D) Hematoxylin and Eosin (H&E) staining of fixed bone marrow and spleen tissue from 6-week-old Carm1Fl/Fl; Vav1-Cre− and Carm1∆/∆; Vav1-Cre+ mice. Immunohistochemical analysis of CARM1 and H3R17me2a in the spleen of 6-week-old Carm1Fl/Fl and Carm1∆/∆ mice. Scale bar=100 µm.
E) Quantitative RT-PCR analysis for Carm1 expression performed on whole bone marrow, spleen, or thymus cells from Carm1Fl/Fl;Vav1-Cre−, Carm1∆/+;Vav1-Cre+ or Carm1∆/∆;Vav1-Cre+ mice and the expression relative to Gapdh was averaged based on a two-tailed Student’s t-test for samples of unequal variance. n= 5, *p<0.01, **p<0.001
F) Assessment of CARM1 and the asymmetric dimethylation of its specific target PABP1 by western blotting of spleen cells from Carm1Fl/Fl;Vav1-Cre−, Carm1∆/+;Vav1-Cre+ or Carm1∆/∆;Vav1-Cre+ mice. Total PABP1 and β-ACTIN are used as loading controls.
G) Complete blood count analysis of peripheral blood, white blood cells (WBC), red blood cells (RBC), and platelets (PLT) in 2-month-old or 12-month-old Carm1Fl/Fl; Vav1-Cre− and Carm1∆/∆; Vav1-Cre+ mice (n= 10). Data points represent individual mice with mean ± SD. n.s.= no significant differences. All bar graph data represent mean ± SD.
Carm1 knockout mice are born, but they die shortly after birth from defects in the differentiation of the lung parenchyma, adipocytes, and muscle cells (Chen et al., 2002, Kim et al., 2004, Yadav et al., 2008). To evaluate the role of CARM1 in the hematopoietic system, we first generated Carm1 conditional knockout (cKO) mice by crossing Carm1 floxed mice with Vav1-Cre transgenic mice to generate Carm1+/+;Vav1- Cre+, Carm1∆/+;Vav1-Cre+, Carm1∆/∆;Vav1-Cre+ mice, and Vav1-Cre negative littermate controls (Carm1Fl/Fl;Vav1-Cre−). Conditional Carm1 knockout was confirmed by extracting DNA from the BM of each genotype and conducting PCR analysis (Figure 1C). H&E staining of the BM and spleen tissue showed no abnormalities in BM or spleen morphology. Immunohistochemistry confirmed the loss of CARM1 protein and the histone mark H3R17me2a in the spleens of 6-week-old Carm1 cKO mice compared to Carm1Fl/Fl; Vav1-Cre− littermates (Figure 1D). We also documented Carm1 loss at the mRNA level in the bone marrow, spleen, and thymus by quantitative real-time PCR (qRT-PCR) (Figure 1E). Loss of CARM1 activity was confirmed by using an antibody to specific asymmetric methylation sites on a well established CARM1 target, PABP1(R445/R460) (Figure 1F) (Lee et al., 2002, Shishkova et al., 2017). We examined mice at two time points to evaluate the contribution of CARM1 to hematopoietic populations in the peripheral blood (PB). PB samples obtained from Carm1∆/∆;Vav1-Cre+ mice showed no significant changes in the white blood cell, red blood cell, or platelet counts at 2 months or 12 months compared to Carm1Fl/Fl;Vav1-Cre− (Figure 1G).
CARM1 deficiency leads to alterations in HSPC frequency and number
Given the relatively high expression of Carm1 in hematopoietic progenitor populations, compared to mature myeloid cells, we examined how Carm1 loss affects HSPC frequency and cell number. Flow cytometry analysis of 1-year-old Carm1Fl/Fl;Vav1-Cre−, Carm1∆/+;Vav1-Cre+, and Carm1∆/∆;Vav1-Cre+ BM cells revealed a normal frequency of lineage (Lin)-negative cells, but a modest decrease in phenotypic long-term hematopoietic stem cells (LT-HSCs) as measured by SLAM markers or LSK cell markers (Figure 2A-2B). In contrast, the frequency and total cell number of progenitor cells was significantly increased, and within this “LK cell” population, the CMP population was decreased, with an increase in the more mature GMP and MEP populations (Figure 2C-2D).
Figure 2: Evaluation of the role of CARM1 in normal hematopoiesis.
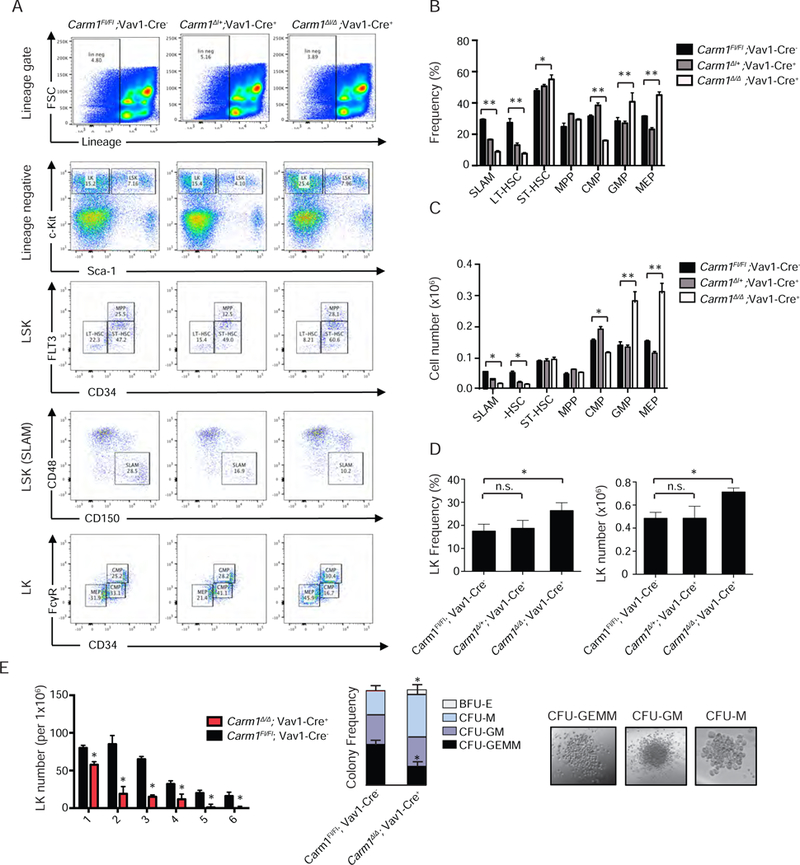
A) Lin–, c-Kit+, and Sca1– progenitor cells isolated from 12-month-old mice and bone marrow cells stained for SLAM markers, as well as FLT3 and CD34 to distinguish the LT-HSC, ST-HSC, and MPP populations. Cells were also stained with CD34 and FcRII/III to distinguish the CMP, GMP, and MEP cell populations. The percentage of each population is indicated.
B) HSPC frequency at 1-year of age. n=3, *p<0.05, **p<0.01
C) Total HSPC cell numbers at 1-year of age. n=5 *p<0.05, **p<0.01
D) LK (Lin−/Kit+/Sca-1−) frequency and total cell n=5, *p<0.05 Data points represent mean ± SD. *p<0.01, n.s. = no significant differences
E) Lin−, c-Kit+ bone marrow cells were isolated from 6–8 week-old mice and plated in M3434 methylcellulose. Colonies were counted 7 days after plating, and serially replated 6 times. n=3, *p< 0.05. Colony differentiation was scored 14 days after plating. Representative colonies are shown. Colonies are defined as burst forming unit eythroid (BFU-E), and colony forming unit monocyte (CFU-M), granulocyte-macrophage (CFU-GM), and granulocyte-eythroid-macrophage-megakaryocyte (CFU-GEMM). Scale bar = 50 µm, *p< 0.05
All bar graph data represent mean ± SD and p values were determined by a two-tailed Student’s t-test for samples of unequal variance. See also Figure S1.
We evaluated the self-renewal and differentiation potential of the LK populations using colony formation (CFU) assays. Sorted Carm1∆/∆ LK cells showed decreased replating potential compared to Carm1Fl/Fl controls, with increased differentiation, based on an increase in the granulocyte/macrophage committed colonies, CFU-M and CFU-GM. (Figure 2E). The effect of CARM1 loss on hematopoietic differentiation appears to be limited to HSPCs, as we failed to identify significant differences in the frequency of mature hematopoietic cell populations, in the BM, spleen, or thymus (Figure S1A-S1D). While we did not observe any changes in T cell frequency in the spleen, a slight but significant decrease in double negative (DN; CD4−CD8−) cells was found in the thymus (Figure S1E). This is consistent with the report of defects in fetal thymic progenitors reported in the non-conditional Carm1 knockout mice (Li et al., 2013). However, we observed no changes in thymic size or cellularity (data not shown), suggesting enhanced thymic T cell differentiation rather than loss of the earlier T cell precursors.
CARM1 is required for AML initiation
To study the role of CARM1 in leukemia initiation, we utilized two mouse models involving the expression of AML1-ETO and MLL-AF9 in hematopoietic cells (Figure 3A). As AML1-ETO (AE) is not fully leukemogenic in retroviral transduction and transplantation (RTT) models, we used an alternatively spliced isoform AML1-ETO exon 9a (AE9a), which lacks the NHR3 and NHR4 domains of ETO but is leukemogenic (Yan el at., 2006). Vav1-Cre− Carm1+/+ and Carm1Fl/Fl, and Vav1-Cre+ Carm1+/+, Carm1∆/+, and Carm1∆/∆ fetal livers were isolated on embryonic day E14.5, and transduced three times with retroviruses expressing AE9a or MLL-AF9 as well as GFP (Figure 3B). Congenic CD45.1+ recipient mice received a lethal dose of 8.5 Gy of irradiation followed by transplantation of 1×105 CD45.2 GFP+ cells together with 5×105 CD45.1+ helper cells by tail vein injection. Peripheral blood from CD45.1+ recipient mice was collected monthly and analyzed for complete blood counts and by flow cytometry to determine the contribution of donor cells (GFP+ CD45.2+) versus helper and recipient cells (CD45.1+) to hematopoiesis (Figure S2A). When we further examined the peripheral blood of the Carm1∆/∆;Vav1-Cre+ recipient cohort, compared to the control groups, we noted that the WBC counts were significantly lower over time, while the GFP+ population was rapidly eliminated from the peripheral blood within three months (Figure S2B-S2E). In both RTT models, mice receiving Carm1∆/∆;Vav1-Cre+ FL cells failed to develop AML, while mice receiving wild-type (Carm1Fl/Fl;Vav1-Cre+) FL cells developed leukemia with a latency of 106 ± 5.5 days for AE9a (Figure 3C) and 153 ± 35 days for MLL-AF9 (Figure 3D). At three months of age, both the AE9a and MLL-AF9 Carm1∆/∆ recipients had significantly lower spleen weights and spleen sizes than the Carm1Fl/Fl controls (Figures S2F-S2I). In the AE9a driven AML model, loss of a single Carm1 allele impaired leukemogenesis, with a significant delay in AML development as well as decreased penetrance from 100% to 33%. The peripheral blood in the Carm1Fl/Fl recipient mice contained numerous circulating blasts, while the bone marrow was replaced by sheets of immature cells, and leukemia infiltrates were present in the spleen (Figure S2J). Bone marrow cytospins from the wild-type recipient mice showed abundant agranular blasts. In contrast, the Carm1∆/∆;Vav1-Cre+ recipients showed normal organ and tissue morphology, and failed to develop leukemia, even one year post-transplant.
Figure 3: Effects of CARM1 knockout on leukemia initiation and maintenance in fusion oncoprotein-driven models of leukemia.
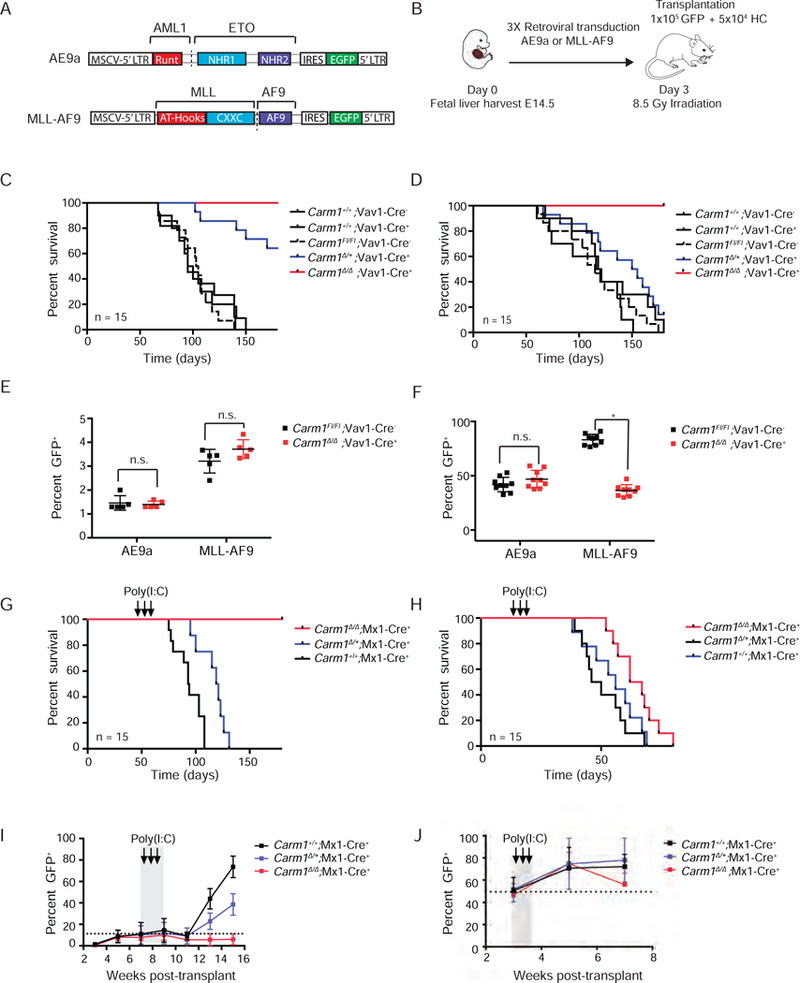
A) Structure of the MIGR1-AE9a and MIGR1-MLL-AF9 retroviral expression plasmids.
B) Schematic of fetal liver retroviral transduction and transplantation (RTT) model.
C) Kaplan-Meier survival analysis of leukemia initiation AE9a Carm1+/+;Vav1-Cre−, Carm1+/+;Vav1-Cre+, Carm1Fl/Fl;Vav1-Cre−, Carm1∆/+;Vav1-Cre+, Carm1∆/ ∆;Vav1-Cre+ recipient mice. n= 15. Log-rank p value for Carm1∆/∆;Vav1-Cre+ or Carm1∆/+;Vav1-Cre+ recipients compared to CARM1Fl/Fl;Vav1-Cre, p<0.0001. Figure represents 2 independent experiments.
D) Kaplan-Meier survival analysis of leukemia initiation MLL-AF9 Carm1+/+;Vav1-Cre−, Carm1+/+;Vav1-Cre+, Carm1Fl/Fl;Vav1-Cre−, Carm1∆/+;Vav1-Cre+, Carm1∆/ ∆;Vav1-Cre+ recipient mice. n=15. Log-rank p value for Carm1∆/∆;Vav1-Cre+ recipients compared to CARM1Fl/Fl;Vav1-Cre− recipients, p<0.001. Log-rank p-value for Carm1∆/+;Vav1-Cre+ recipients compared to CARM1Fl/Fl;Vav1-Cre− recipients, n.s. = no significant difference. Figure represents 2 independent experiments.
E) Homing of AE9a and MLL-AF9 expressing Carm1∆/∆;Vav1-Cre+ or CARM1Fl/Fl;Vav1-Cre− fetal liver cells in the BM of CD45.1 recipient mice, 16 hr post tail vein injection. n=5, *p< 0.01, **p< 0.01, n.s = no significant differences
F) Engraftment of GFP+ cells in the BM of CD45.1 recipient mice, 1-month post-transplant. n=10, n.s = no significant difference, *p<0.01
G) Kaplan-Meier survival analysis of leukemia maintenance AE9a fetal liver recipients post-Poly(I;C) injection. n=15. Log-rank p value of Carm1∆/∆;Mx1-Cre+ or Carm1∆/+;Mx1+ recipients compared to CARM1+/+;Mx1-Cre+ recipients, p<0.001.
H) Kaplan-Meier survival analysis of leukemia maintenance MLL-AF9 fetal liver recipients post-Poly(I;C) injection. n=5. Log-rank p value of Carm1∆/∆;Mx1-Cre+ recipients compared to CARM1+/+;Mx1-Cre+ recipients, p<0.01.
I) Percentage of AE9a Carm1+/+;Mx1-Cre+, Carm1∆/+;Mx1-Cre+ or Carm1∆/ ∆;Mx1+ recipient GFP+ cells in the peripheral blood of AE9a mice over time. Time of poly(I:C) induction of CARM1 deletion is indicated. n=15.
J) Percentage of MLL-AF9 Carm1+/+;Mx1-Cre+, Carm1∆/+;Mx1-Cre+ or Carm1∆/ ∆;Mx1+ recipient GFP+ cells in the peripheral blood of AE9a mice over time. Time of poly(I:C) induction of CARM1 deletion is indicated. n=15. All bar graph data represent mean ± SD and p values were determined by a two-tailed Student’s t-test for samples of unequal variance. See also Figure S2 and S3.
To eliminate the possibility that the impaired leukemogenesis was due to cell engraftment or homing defects, we evaluated five additional mice per group for defects in the homing of GFP+ cells to the BM 16 hr. after transplantation and found no significant differences in the homing capacity of the AE9a or MLL-AF9 expressing Carm1Fl/Fl; Vav1-Cre− or Carm1∆/∆; Vav1-Cre+ cells (Figure 3E). BM engraftment was also monitored, by examining 10 Carm1Fl/Fl;Vav1-Cre− or Carm1∆/∆; Vav1-Cre+ recipient mice 1-month post transplantation. While AE9a Carm1∆/∆;Vav1-Cre+ FL cells showed equal engraftment to the WT CD45.1 helper cells, the MLL-AF9 Carm1∆/∆ cells were out-competed by helper cells in competitive repopulation (Figure 3F), suggesting that Carm1 null MLL-AF9 expressing FL cells have a decreased capacity to compete in the bone marrow or may be lost due to terminal differentiation.
Carm1 is required for the maintenance of AE9a driven leukemia
Next, we studied the consequence of Carm1 loss on established AML leukemia maintenance. For these experiments, Mx1-Cre positive Carm1+/+, Carm1Fl/+, or Carm1Fl/Fl FL cells were transformed by AE9a or MLL-AF9 and transplanted to lethally irradiated CD45.1+ recipients. Recipient mice were monitored every two weeks to evaluate their complete blood count (CBC) and the percentage of GFP+ cells in the peripheral blood. When mice reached a threshold of 10% circulating GFP+ cells, Cre expression and gene excision were induced by three intraperitoneal injections of poly(I:C) given every other day. The deletion of Carm1 post poly(I:C) induction was confirmed by PCR analysis after BM aspiration (Figure S2K). Deletion of Carm1 significantly abrogated leukemia maintenance in the AE9a recipient mice, with none of the mice showing evidence of disease by 150 days post-transplant (p< 0.001) (Figure 3G). CARM1 also plays an important in MLL-AF9 driven leukemia maintenance, as the Carm1∆/∆ recipient mice showed improved survival (Figure 3H). In the AE9a recipient mice, deletion of one allele of Carm1 significantly delayed leukemia mortality in the AE9a recipient mice based on the percentage of GFP+ cells in the peripheral blood (Figure 3I), while we observed no change in survival when only a single allele of Carm1 was deleted in the MLL-AF9 model (Figure 3J). Sorted GFP+ cells were obtained from the MLL-AF9 Carm1∆/∆ recipient mice at the time of death and evaluated for changes in Carm1 mRNA expression. These leukemia cells had Carm1 expression that was 40–90% of the MLL-AF9 Carm1+/+ recipient mice (Figure S2L), suggesting that these cells had incomplete deletion of Carm1 and likely underwent clonal selection in order to emerge as AML. We also noted oncogene specific differences in CARM1 protein expression; fetal cells transduced with AE or AE9a showed higher levels of CARM1, while there were no changes in MLL-AF9 transduced cells (Figure S2M).
Restoration of CARM1 enzymatic activity rescues the impaired differentiation phenotype
Next, we evaluated the self-renewal and differentiation potential of AE9a and MLL-AF9 expressing Carm1Fl/Fl;Vav1-Cre− or Carm1∆/∆;Vav1-Cre+ cells using CFU assays, and assessed the dependence of the observed phenotype on CARM1 enzymatic activity. As expected, AE9a and MLL-AF9 expressing Carm1Fl/Fl;Vav1-Cre− FL cells demonstrated impaired myeloid differentiation. However, the AE9a and MLL-AF9 Carm1∆/∆; Vav1-Cre+ cells displayed increased monocyte/macrophage differentiation in CFU assays compared to AE9a and MLL-AF9 expressing Carm1Fl/Fl; Vav1-Cre− cells (Figure S3A). We also evaluated the AE9a Carm1∆/∆;Vav1-Cre+ cells for the expression of a macrophage specific marker, F4/80, and found a significant increase in its expression in the Carm1∆/∆; Vav1-Cre+ mice compared to the Carm1Fl/Fl; Vav1-Cre− cells, after the second replating (Figure S3B).
We then expressed WT CARM1 (CARM1WT) or the enzymatically dead mutant CARM1-E267Q (CARM1EQ) and RFP in Carm1 null hematopoietic cells, to determine whether the enzymatic activity of CARM1 was needed to rescue the aberrant oncogene-driven CFU differentiation phenotype. The restoration of total Carm1 mRNA and protein levels was documented by qRT-PCR and western blot (Figure S3C and S3D). The SWI/SNF subunit BAF155 is a previously published substrate of CARM1, and methylation of BAF155 R1064 is a predictor of cancer cell migration and metastasis (Wang et al., 2014). We found that CARM1EQ did not restore the asymmetric dimethylation of BAF155 (Figure S3E) and failed to rescue the aberrant differentiation (Figures S3F-S3I). The results demonstrate that the methyltransferase activity of CARM1 is required to manifest the effects of the fusion oncoproteins on differentiation in hematopoietic cells.
Knockdown of CARM1 affects AML cell biology
To further elucidate the role of CARM1 in human AML cells, we profiled the TCGA AML dataset to determine if CARM1 expression correlates with prognosis (Cancer Genome Atlas Research., 2013). Indeed, CARM1 high expressing patients had significantly worse survival outcomes than patients with low CARM1 expression (p= 0.036) (Figure 4A). CARM1 is overexpressed in multiple cancer cell lines and patient samples. However, hematopoietic tissue and leukemia cell lines specifically appear to be dependent on CARM1 based on the analysis of large scale RNAi screens using cancer cell lines (Figure 4B).
Figure 4: AML cell dependence on CARM1.
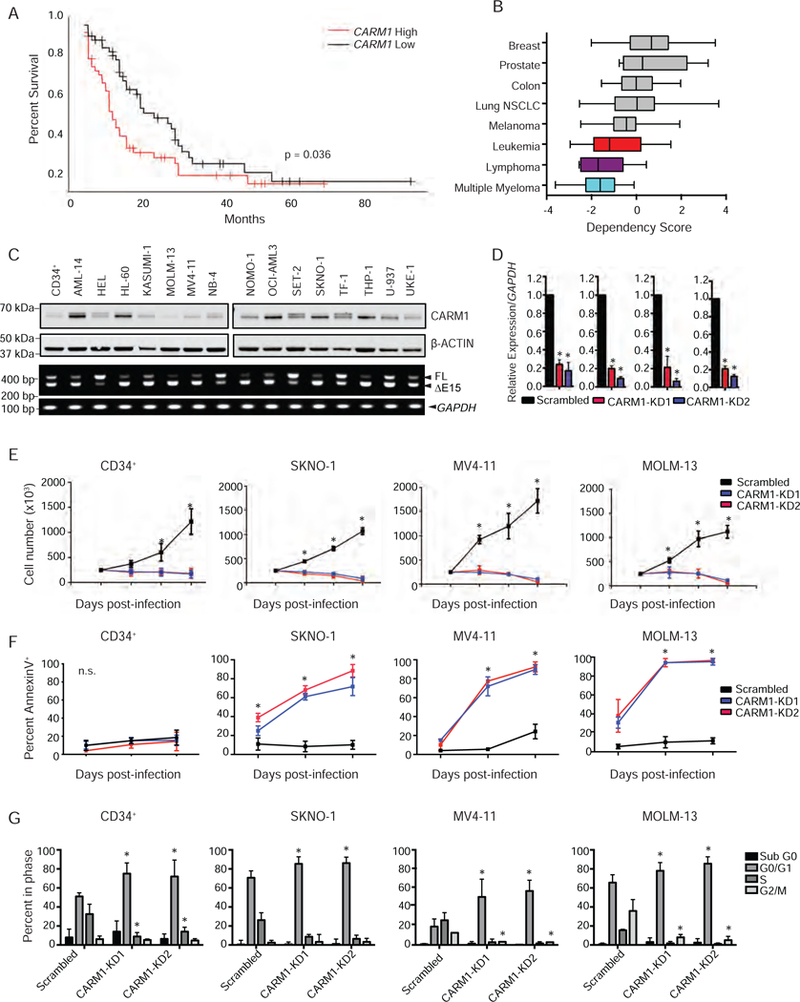
A) Kaplan-Meier survival analysis of TCGA AML cohort with high or low levels of CARM1 mRNA expression. n=162, Log-rank p value = 0.036
B) Cancer cell line dependency scores obtained from http://depmap.org plotted for Breast (n=33), Prostate (n=8), Colon (n=25), NSCLC (n=90), Melanoma (n=36), Leukemia (n=28), Lymphoma (n=7), and Multiple Myeloma (n=12) cell lines screened with shRNA targeting CARM1. Box plots are annoted with the lower quartile, median, and upper quartile for each cancer cell line’s z-scores. Whiskers represent the maximum and minimum values, excluding outliers.
C) CARM1 protein and full length (FL) and exon 15 deletion (∆E15) isoform expression in 15 AML cell lines and CD34+ cells was assessed by immunoblotting and semi-quantitative PCR, respectively.
D) Assessment of CARM1 KD at the mRNA level in RFP+ sorted CD34+, SKNO-1, MV4–11, and MOLM-13 cells by qRT-PCR. n= 3, *p< 0.001
E) Cell counts from RFP+ CD34+, SKNO-1, MV4–11, and MOLM-13 cells infected with a CARM1 KD virus or scrambled control. n=3, *p< 0.01
F) Percentage of Annexin V+ cells analyzed by flow cytometry from CD34+, SKNO-1, MV4–11, and MOLM-13 cells infected with a CARM1 KD virus or scrambled control on days 3, 5, and 7 post-infection. n=3, *p<0.01
G) Percentage of cells in sub-G1, G1, S-phase, or G2/M in CD34+ cells, SKNO-1, MV4–11, and MOLM-13 cells, 3 days after CARM1 KD. n=3, *p= 0.01
All bar graph data represent mean ± SD and p values were determined by a two-tailed Student’s t-test for samples of unequal variance. See also Figure S4.
In order to understand the dependency of AML cells on CARM1, we profiled 16 AML cell lines for basal levels of CARM1 protein expression, mRNA expression, and isoform abundance (Figures 4C). The majority of the 16 cell lines had elevated CARM1 protein and mRNA expression, compared to CD34+ cord-blood (CB) cells (Figure 4C, Figure S4A). We also found an increased level of BAF155 asymmetric demethylation; while it may be possible to use this mark as a readout for functional CARM1 activity, it did not correlate with the level of CARM1 protein expression (Figures S4B and S4C). CARM1 has two alternative splice isoforms, full length CARM1 and an isoform lacking exon 15 (Shlensky et al., 2015). We confirmed the protein expression of both isoforms of CARM1 in hematopoietic cell lines and quantified the relative isoform abundance using semi-quantitative PCR (Figure S4D). Neither CARM1 isoform ratio nor CARM1 mediated-BAF155 methylation predicted sensitivity to CARM1 inhibition (Figures S4E and S4F).
To evaluate the biological basis of CARM1 dependency, we generated vectors that express two different shRNAs that efficiently target CARM1. We examined the effects of CARM1 KD on SKNO-1 cells, and two MLL-rearranged cell lines (MOLM-13 and MV4–11). CD34+ cells and the leukemia cell lines transduced with these vectors showed significantly decreased CARM1 mRNA (Figure 4D). Next we evaluated the effect of CARM1 KD on apoptosis induction in these cells. The AML cell lines displayed reduced total cell counts and rapid apoptosis, as shown by staining for Annexin V, following CARM1 KD (Figure 4E-4F). We examined cell cycle progression and observed decreased cells in S-phase and a G0/G1 arrest for all three AML cell lines following CARM1 KD (Figure 4G). Interestingly, while CARM1 KD CD34+ CB cells showed impaired cell cycle progression and proliferation, increased apoptosis was not observed.
Aberrant transcriptional networks regulated by CARM1 in AML cells
To define the transcriptional functions of CARM1 in AML cells, we performed gene expression analyses by RNA-Sequencing (RNA-Seq) in SKNO-1, MOLM-13, and MV4–11 cells after CARM1 KD. We found 138 genes (105 downregulated and 33 upregulated) that were significantly modulated in all 3 cell lines [log2 fold change > 1 and false discovery rate (FDR) < 0.05] by KD of CARM1. Differentially expressed upregulated and downregulated genes for each cell line are shown in Table S1. Differentially expressed pathways common to all three cell lines included mitotic cell cycle, DNA replication and DNA repair (Figure 5A). A comparison of the top 100 differentially regulated genes in the three leukemia cells lines, confirmed that the CARM1 KD constructs effectively downregulated CARM1 and generated similar gene expression profiles (Figure 5B). To define the molecular pathways regulated by CARM1, we performed Gene Ontology (GO) analysis on the 105 commonly downregulated transcripts and observed that genes involved in cell cycle progression were highly enriched (Figure 5C, Table S2). We also applied gene set enrichment analysis (GSEA) to identify pathways differentially regulated by the loss of CARM1, and found statistically significant downregulation of several GSEA hallmarks common to all three leukemia cell lines. Heat maps of FDR (q < 0.01) values from these GSEA of hallmarks gene set collections are shown in Figure 5D. E2F transcriptional targets had a normalized enrichment score (NES) of −3.78 and a highly significant p value (p<0.00001) (Figure 5E). We independently evaluated 5 E2F target genes, CENPA, CDC25A, MYBL2, TOP2A, and UHRF1, and found that all were significantly down-regulated by CARM1 KD in MV4–11 and MOLM-13 cells (Figure 5F). In order to understand the role of the E2F family in AML cells, we determined the relative abundance of E2F1–8, relative to GAPDH, in our panel of 15 AML cell lines and CD34+ cells. E2F1, E2F4, and E2F8 showed consistently higher relative abundance compared to the other E2F family members in all cell lines examined (Figure 5G).
Figure 5: Transcriptional analysis of CARM1 knockdown in AML cell lines.
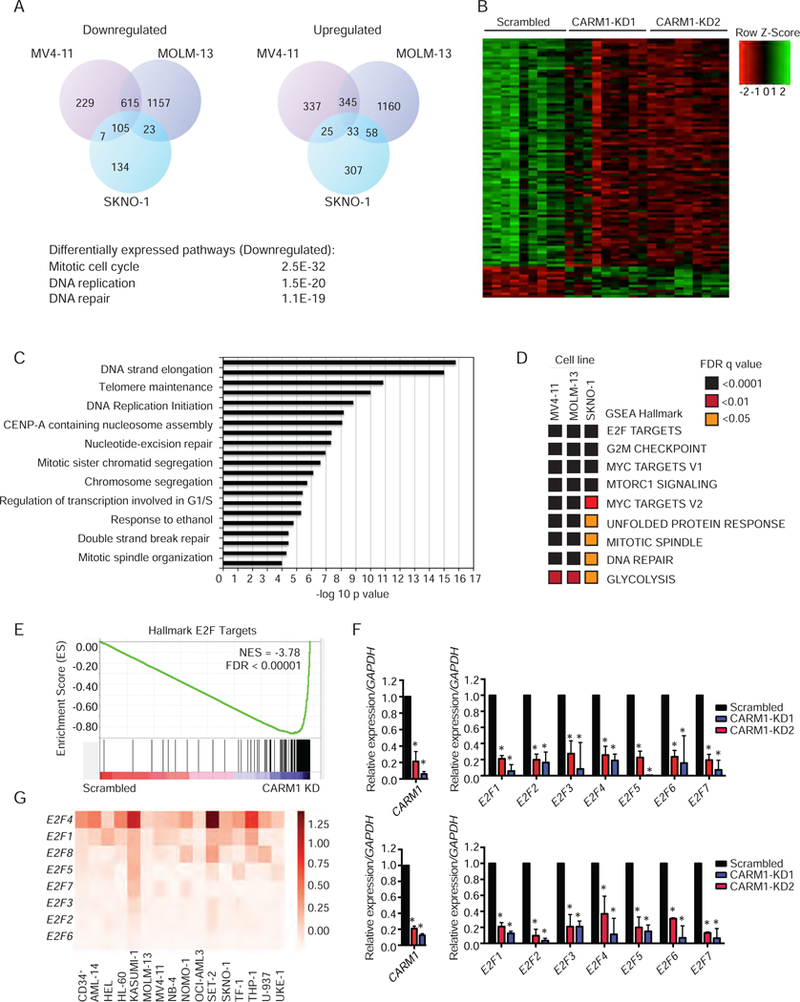
A) Venn diagram representation of the number of significantly upregulated and downregulated genes based on RNA-Seq, 3 days post lentiviral infection with CARM1 shRNA in SKNO-1, MV4–11, and MOLM-13 cell lines. n=3. Data represent genes that were significantly upregulated or downregulated with both shRNAs compared to scrambled control.
B) Heat map of top 100 differentially regulated genes for all three cell lines, representing replicates of cells expressing control (Scrambled) or two independent CARM1 shRNAs (CARM1-KD1 and CARM1-KD2).
C) Gene ontology analysis of significant downregulated genes in all 3 cells lines.
D) Heat maps of FDR (q < 0.01) values from GSEA of hallmarks gene set collections.
E) Representative GSEA plot depicting the down-regulation of E2F targets. NES, normalized enrichment score, FDR, false discovery rate
F) Quantitative RT-PCR analysis, showing expression of E2F1, E2F2 and E2F target genes CENPA, CDC25, MYBL2, TOP2A, and UHRF1 in MV4–11(top) and MOLM-13 (bottom) cells with KD of CARM1. Mean and SD are expressed as a percentage of GAPDH expression. *p< 0.05.
G) Relative transcript levels of E2F family mRNA in leukemia cell lines and CD34+ cells, normalized to GAPDH. Heat map represents the average of three independent experiments. All bar graph data represent mean ± SD and p values were determined by a two-tailed Student’s t-test for samples of unequal variance. See also Figure S5 and Tables S1–S3.
Since CARM1 has been reported to play a role in splicing regulation (Cheng et al., 2007), we also analyzed the RNA-Seq results using multivariate analysis of transcript splicing (MATS), defining the five classes of alternative splicing events. We identified an average of 1080 alternative splicing events per cell line, mainly skipped exons affecting gene products involved in cell cycle regulation, DNA repair and translation (Figure S5A-S5B, Table S3). Events were considered significant if present in both CARM1 KD samples and not the scrambled control for each cell line. We validated an intron retention event that occurs in the PMAIP1 gene (Figure S5C and S5D) and found that retention of an alternative exon resulted in a premature stop codon and decreased mRNA production (Figure S5E).
Pharmacological inhibition of CARM1 suppresses AML in vitro
Although several small molecule inhibitors of CARM1 have been recently reported, many display a lack of selectivity for CARM1 or fail to produce a biological response. The discovery of potent and selective CARM1 inhibitors (Drew et al., 2017), has made it possible to evaluate the implications of pharmacological inhibition of CARM1 in vitro and in vivo (Figure 6A). We first tested the in vitro activity of a selective CARM1 inhibitor, EPZ025654. EPZ025654 reduced methyl-BAF155 levels in a time and concentration-dependent manner, with full reduction requiring at least three days of treatment (Figures 6B and6C).
Figure 6: Effects of pharmacological inhibition of CARM1 on target methylation and AML cells in vitro and in vivo.
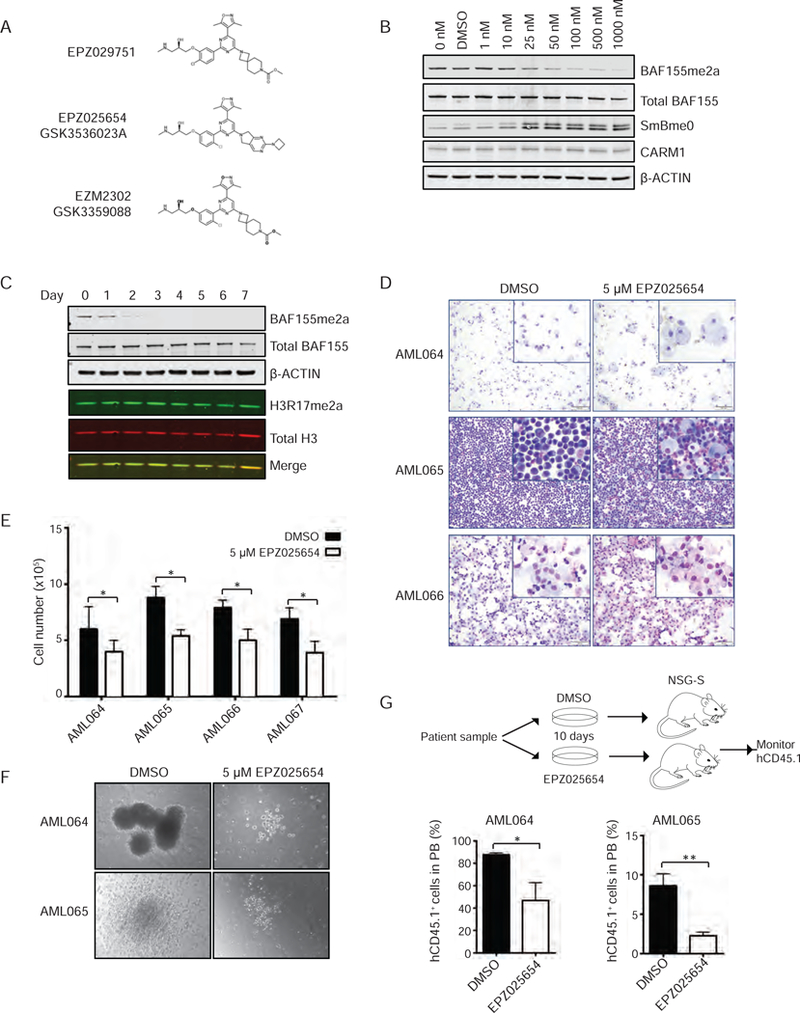
A) Chemical structures of CARM1 inhibitors and control compound utilized in study.
B) Methyl-BAF155 in MV4–11 cells following doses of EPZ025654 ranging from 1 nM to 1000 nM, 48 hr treatment with EPZ025654.
C) Immunoblot analysis of 7-day time course of MV4–11 cells treated with 25 nM of EPZ025654.
D) Cytospins of three patient samples treated with DMSO or 5 µM of EPZ025654 for 10 days. Scale bar= 20 µm
E) Quantification of total cell number of patient samples cultured in methylcellulose with DMSO or 5 µM EPZ025654. n=3, *p< 0.05
F) Images of colony formation of patient samples cultured in methylcellulose with human cytokines and DMSO or 5 µM of EPZ025654 for 10 days. Scale bar= 100 µm
G) Schema of xenotransplantation experiment, indicating ex vivo treatment of patient samples with DMSO or EPZ025654, followed by i.v. transplantation to NSG-S mice. Percentage of human CD45.1+ cells in the peripheral blood, 3 months post-transplant. n=5, *p< 0.05 All bar graph data represent mean ± SD and p values were determined by a two-tailed Student’s t-test for samples of unequal variance. See also Figures S6 and S7.
We also utilized a control compound (EPZ029751), that displays similar chemical properties to the active EPZ025654 inhibitor, but 1000-fold less biochemical potency against CARM1. We incubated SKNO-1 cells with increasing doses of EPZ025654 or EPZ029751 for 48 hr (Figure S6A). Treatment with the inhibitor led to a concentration-dependent decrease in BAF155me2a with an IC50 of 18 nM, while EPZ029751 did not. Similarly, quantification of immunoblot analysis of CD34+, HEL, MV4–11, KASUMI-1, SKNO-1, and an AE9a cell line derived from the leukemic mice indicated dose-dependent inhibition of methyl-BAF155, with an IC50 of 18–200 nM (Figure S6B). While we observed a significant loss in cell viability and decreased total cell numbers in SKNO-1 cells treated with EPZ025654, the cells did not show evidence of apoptosis, as measured either by Annexin V staining or cleavage of PARP (Figure S6C and S6D). In contrast, we found a dose-dependent loss of cell viability (Figure S6E) and expression of senescence-associated β-galactosidase activity (Figure S6F-S6G) that was specific to cells treated with EPZ025654, but not EPZ029751. Finally, we assessed whether EPZ025654 could inhibit MLL-rearranged or AE driven gene expression. Translocation (8;21) AML samples in the Eastern Cooperative Oncology Group cohort, have significantly higher CARM1 expression compared to normal CD34+ controls (Figure S6H). This led us to hypothesize that CARM1 is a direct target of the AML1-ETO fusion protein. We confirmed that two independent shRNA could efficiently knockdown AML1-ETO in Kasumi-1 cells and resulted in a significant downregulation of CARM1 mRNA (Figure S6I). Furthermore, AE specific target genes showed significant changes in expression following EPZ025654 (Figure S6J). In contrast, we found no significant changes in hallmarks of MLL-rearranged leukemia cell transcription in response to CARM1 inhibition (Figure S6K).
We next evaluated the effects of EPZ025654 on primary patient cells treated with EPZ025654 for 10 days and found morphological evidence of myeloid differentiation. We also evaluated the patient cells in methylcellulose supplemented with human cytokines to determine the ability of the cells to form colonies. While some of the patient samples failed to generate colonies, we noted that EPZ025654 treatment decreased overall cell numbers in all four of the samples examined (Figure 6E), and decreased colony size and morphological evidence of myeloid differentiation for the AML064 and AML065 samples (Figure 6F) We also performed xenotransplantation assays, treating two of the patient samples with DMSO or EPZ025654 for 7 days in liquid culture, then transplanting 1×106 cells into NSG-S mice. Mice receiving the AML064 or AML065 cells treated with EPZ025654 showed significantly less engraftment of human CD45.1+ cells in the peripheral blood, compared to mice receiving cells treated with DMSO (Figure 6G). We next evaluated EZM2302, an active compound structurally related to EPZ025654 that is highly orally bioavailable and can be given to mice with little off target effects (Drew et al., 2017). We generated MLL-AF9 primary transplantation mice and treated them with 100 mg/kg of EZM2302 or vehicle twice-daily (BID) (Figure 7A). The inhibitor treated mice showed improved survival (n= 9, p< 0.005) (Figure 7B), as well as significantly fewer GFP+ cells in the PB over time (data not shown).
Figure 7. Effects of chemical inhibition on AML cell line viability.
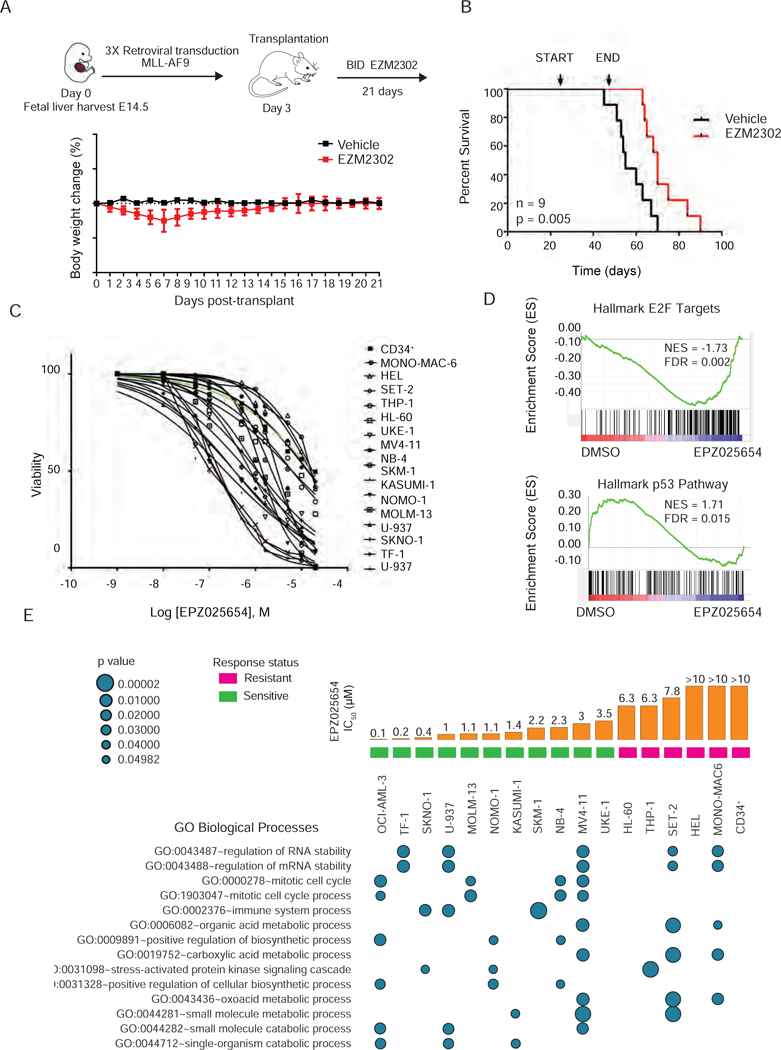
A) Schematic of fetal liver retroviral transduction and in vivo treatment strategy (top). Daily body weight change for mice treated with either vehicle or 100 mg/kg EZM2302 for three weeks. n=9
B) Kaplan-Meier survival analysis of MLL-AF9 fetal liver recipient mice treated with EZM2302 or vehicle for 21 days. Treatment start and end are indicated. n= 9, long-rank p value is 0.005.
C) Dose response for the cellular viability of 16 AML cells lines and CD34+ cells treated in vitro with EPZ025654 for 10 days. IC50 values were calculated using a non-linear regression analysis.
D) Representative GSEA plots depicting the top upregulation of the p53 pathway (NES = 1.71, FDR = 0.015) and down-regulation of E2F targets (NES = −1.73, FDR = 0.002) in SKNO-1 cells treated with EPZ025654 for 7 days. Results are compared to SKNO-1 cells treated with DMSO.
E) Summary of L1000 analysis showing significant gene ontology biological processes enriched in 16 AML cell lines and CD34+ cells treated with 5 µM of EPZ025654 for 6 days. Results represent three biological replicates, and three technical replicates. Bar graphs represent the mean IC50 of three idependent experiments.
To assess the specific effects of EPZ025654 in leukemia cells compared to normal CD34+ cells, we examined cell viability after 10 days of treatment using a panel of 16 leukemia cell lines (Table S4). The AE positive cell lines exhibited an IC50 in the 0.4–3 µM range, while the CD34+ cells were relatively resistant to CARM1 inhibition (Figure 7C). Of these 16 AML cell lines, mutations in leukemia-associated genes or expression of CARM1 isoform mRNA, CARM1 protein levels, or target methylation failed to explain the difference in sensitivity to CARM1 inhibition. This led us to explore other mechanisms for the sensitivity to CARM1 inhibition and loss in viability. We utilized RNA-Seq to evaluate SKNO-1 cells treated with EPZ025654, and L1000 profiling (Subramanian et al., 2017) to simultaneously profile the gene expression of 18 AML cell line and CD34+ cells after 6 days of treatment. We applied GSEA to identify pathways differentially regulated by CARM1 inhibition and found a significant downregulation of E2F targets, similar to what we observed with CARM1 knockdown; we also identified significant upregulation of the p53 pathway (Figure 7D). For the L1000 analysis, DEGs were identified using a log2 fold change greater than 1.2 and GO enrichment analysis was performed using a background list of the 978 L1000 genes. Functional annotations were ranked based on counts in sensitive versus resistant cell lines and the p values for the top networks are shown (Figure 7E and Table S5). While we noted gene expression profiles representing changes in RNA stability, the most significant differences between sensitive and resistant cell lines were genes associated with the regulation of cell cycle progression, including changes in TP53 and CDKN1A expression.
Finally, we evaluated the effects of EZM2302 on normal hematopoiesis, treating five WT CD45.1 mice with 100 mg/kg BID for three weeks. In vivo efficacy was evaluated by immunoblot analysis of the WBCs in the spleen of mice treated with the inhibitor or vehicle control. We observed inhibition of the asymmetric dimethylation of PABP1 and an increase in mono-methyl SmB (Figure S7A). There were no significant changes in body weight over the course of the experiment, indicating that the inhibitor was well tolerated (Figure S7B). Flow cytometry analysis post-treatment revealed a normal frequency of lineage-negative cells, phenotypic LT-HSCs as measured by SLAM markers or CD34−FLT3− LSK cell markers. The frequency of progenitor cells, including the CMP, GMP, and MEP populations were also unchanged (Figure S7C), and so were the mature hematopoietic populations in the bone marrow, spleen, and thymus (Figure S7D). Peripheral blood counts and spleen size and weight also displayed no significant differences in mice treated with EZM2302 or the vehicle control (Figures S7E and S7F).
Discussion:
Understanding the mechanisms by which oncogenic transcriptional regulators redirect gene expression in AML is critical to developing therapeutics. Here, we demonstrated that CARM1 plays a critical role in myeloid leukemogenesis. In the absence of CARM1, both MLL-AF9 and AE9a expressing FL cells showed loss of self-renewal and increased monocyte/macrophage differentiation in vitro. WT CARM1 was able to rescue the oncogene-driven block in differentiation, while an enzymatically dead version of CARM1 was not. This confirms that the methyltransferase activity of CARM1, and not just its scaffolding function, is required for its inhibitory effects on hematopoietic cell differentiation.
In contrast to its role in transformed cells, CARM1 appears to play a more modest role in normal HSPC differentiation and proliferation. Given its role in many cellular processes, and its apparent unique substrates, we imagine that lack of CARM1 is compensated by other Type I PRMT family members. In fact, scavenging by other PRMTs has been noted in systems in which PRMT1 activity is lost, leading to increased global monomethylation and symmetric demethylation (Dhar et al., 2013). The observation that different arginine methyltransferase members can compete for the same substrates, such as the transcription factor RUNX1, also may explain the mild phenotype we observed in normal hematopoietic cells. Taken together, this lack of an effect on normal hematopoiesis may indicate a favorable therapeutic index for CARM1 inhibitors, with the potential for positive outcomes for patients. The dispensability of Carm1 stands in marked contrast to the essential role for Prmt5 in adult hematopoiesis, as cKO of Prmt5 leads to BM aplasia and lethal pancytopenia (Liu 2015).
Methyltransferase knockdown or enzyme inhibition can often induce a complex phenotype including the early inhibition of proliferation, gene expression associated with myeloid differentiation, and ultimately the induction of apoptosis. Consistent with this paradigm, CARM1 knockdown affected pathways associated with proliferation and cell cycle progression, including E2F-, MYC, and mTOR- regulated processes. Indeed, a role for CARM1 as a coactivator of E2F family members has been previously described in breast cancer cells (El Messaoudi et al., 2006, Frietze et al., 2008). E2F proteins have been implicated in embryonic stem cell fate decisions and myeloid differentiation, two developmental stages where CARM1 expression is tightly regulated. In solid tumors, dysregulation of E2F protein is a key difference between tumor and normal tissue, with higher expression correlating with tumor differentiation and metastasis. Thus, CARM1 may “lock in” aberrant transcriptional programs in a cell type specific manner, and its loss may have different effects on transcription in normal vs. malignant cells. While these transcriptional changes occurred early, we also noted a delayed induction of myeloid differentiation and rapid progression to apoptosis.
A number of groups have developed small-molecule inhibitors targeting the methyltransferase activity of the PRMTs, and demonstrated their anti-tumor activity in the context of mantle cell lymphoma and AML (Chan-Penebre et al., 2015, Tarighat et al., 2016). The promising nature of these results have led to the first PRMT-directed targeted therapy in cancer patients (NCT02783300).This first in human, dose-escalation study will assess the pharmacokinetics, pharmacodynamics, and clinical activity of PRMT5 inhibition in subjects with advanced or recurrent solid tumors. EPZ025654 provides a powerful tool compound for understanding the biological effects of acutely inhibiting CARM1 enzymatic activity, as well as exploring the basis for its effects on gene expression, and cell behavior. This inhibitor has potent anti-proliferative activity in multiple myeloma cells in vitro and human multiple myeloma tumor xenografts in vivo. In a leukemia setting, the small molecule inhibitor EPZ025654 triggers senescence and gene expression changes that accompany myeloid differentiation. This inhibitor shares many characteristics with other methyltransferase inhibitors, e.g. small molecule inhibitors of Dot1L and EZH2, which also trigger hematopoietic differentiation followed by apoptosis (Daigle et al., 2011, Beguelin et al., 2013). Although EPZ025654 shows a similar discrepancy between the dose needed to inhibit specific asymmetric arginine demethylation and that needed to elicit a biological response, the sensitivity of the AML cells to CARM1 inhibition, relative to normal HSCs, suggests a promising therapeutic window, which could lead to selective targeting of leukemia cells.
We wish to point out that neither the PRMT5 inhibitors, nor the CARM1 inhibitors reported thus far, affect the level of their respective dimethylated histone targets in cancer cell lines. This suggests that methylated histone substrates are relatively stable in cells, or that our reagents may not be able to identify small changes in histone methylation that may be important. It also implies that the non-histone substrates of these enzymes could be the more critical substrates in cancer cells. The fact that PRMT5 and CARM1 are primarily localized in the cytoplasm in cancer cells supports this latter hypothesis.
Investigating the basis for leukemia cell sensitivity to CARM1 inhibition, we found that the relative level of expression of the two isoforms of CARM1 was highly variable and did not correlate with sensitivity to CARM1 inhibition, consistent with previous reports (Shlensky et al., 2015). CARM1 mRNA or protein levels also failed to predict sensitivity to EPZ025654 in our leukemia cell lines, in multiple myeloma cell lines, or in the larger Cancer Cell Line Encyclopedia database (Drew et al., 2017). This suggests that for many small molecule inhibitors of epigenetic modifying enzymes (e.g. for JQ1), the expression level of the enzyme being targeted may not be the primary predictor of response in cancer cells.
Gene expression analysis of leukemia cells treated with EPZ025654 indicated a complex phenotype, including inhibition of RNA stability, E2F target downregulation, and induction of a p53 response signature. This phenotype may be partially explained by the effects on CARM1 on post-transcriptional RNA processing through the methylation of proteins such as PABP1 and SmB. While CARM1 inhibition did not induce apoptosis, we observed features of senescence and gene expression changes that accompany myeloid differentiation. Therefore, combination therapies including CARM1 inhibition may be required to induce apoptosis in leukemia cells. In fact, defects in pathways such as splicing regulation may further sensitize myeloid cells to alternative splicing induced by CARM1 inhibition. Despite this complex phenotype, patient xenografts and mouse models of AML were highly sensitive to CARM1 inhibition in vivo, showing significantly reduced AML cell growth and improved survival.
Thus, we find that AML is exquisitely sensitive to Carm1 genetic loss or chemical inhibition, while deficiency of CARM1 has little impact on normal hematopoiesis. Collectively, these data suggest that inhibiting CARM1 should be investigated as a potentially effective therapeutic strategy for AML.
STAR Methods
CONTACT FOR REAGENT and RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephen Nimer (s.nimer@med.miami.edu).
GlaxoSmithKline provided EPZ025654 (GSK3536023) and EZM2302 (GSK3359088) for the purposes of this research, while Epizyme Inc. provided EPZ029751. EPZ025654, EPZ029751, and EZM2302 were discovered by Epizyme, Inc. and developed through a Research Collaboration and License Agreement between GlaxoSmithKline and Epizyme, Inc.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice.
The generation of mice with targeted disruption of Carm1 (Carm1∆/∆ mice) have been previously described (Yadav 2003) and were provided by Dr. Tenen. B6.SJL CD45.1 recipient mice and Mx1-Cre (Stock: 003556) and Vav1-Cre (Stock: 008610) mice were purchased from The Jackson Laboratory. Carm1 cKO mice were generated by crossing Carm1-floxed mice with Mx1-Cre or Vav1-Cre transgenic mice. Carm1 gene excision in Mx1-Cre+ Carm1Fl/Fl mice was induced by poly(I:C) HMW (InvivoGen), given by 3 intraperitoneal injections at a dose of 10 mg/kg every other day. Female 6 to 8-week-old CD45.1 mice were used as recipients for all transplantation studies. CD45.1 transplant recipient mice (B6.SJL-Ptprca Pepcb/BoyJ, Stock: 002014) and NSG-S mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3, CSF2, KITLG)1Eav/MloySzJ), Stock: 013062) were purchased from the Jackson Laboratory. All animal studies were conducted in compliance with NIH guidelines for the care and use of laboratory animals and were approved by the IACUC of the University of Miami.
Cell lines.
Leukemia cell lines were purchased from ATCC or DSMZ and cultured according to provider’s instructions. Cell lines were authenticated using short-tandem repeat (STR) assays at the Characterized Cell Line Facility at MD Anderson Cancer Center. All cell lines were grown in the recommended cell culture media at 37°C in 5% CO2, with the exception of patient samples TBCF-AML064 and TBCF-AML065 which were cultured under hypoxic conditions (2% O2). SKNO-1 cells were supplemented with 10 ng/mL GM-CSF (Peprotech), while TF-1 cells were supplemented with 10 ng/ml IL-3 (Peprotech).
Primary cell culture.
Human leukemia tissues were collected from routine medical procedures by the Tissue Banking Core Facility (TBCF) at the University of Miami, according to IRB approved Protocol No: 20060858. The TBCF personnel removes all patient identifying information and labels tissue samples with a bar code before transferring samples for future use and storage at the Live Tumor Culture Core (LTCC). For patient sample colony formation assays, 1×105 cells were cultured for 10 days in H4435 methylcellulose (Stem Cell Technologies) per 10 cm plate. Liquid culture of patient samples was conducted in IMDM supplemented with 20% BIT (Stem Cell Technologies), low density lipoprotein (LDL) human plasma (EMD Millipore, #437644), ß-mercaptoethanol, pen/strep, 20 ng/ml IL-6, 20 ng/ml IL-3, 20 ng/ml GM-CSF, 50 ng/ml FLT3, and 50 ng/ml SCF.
METHODS DETAILS
Purification and culture of CB derived CD34+ cells.
Normal, human CD34+ HSPCs were purified by positive selection using the Midi MACS (magnetic-activated cell sorting) LS+ separation columns and isolation Kit (Miltenyi Biotec # 130–042-401) starting with mononuclear cells that were isolated from CB by Ficoll-Hypaque Plus (GE Healthcare #17–1440-03) density centrifugation. CD34+ cells were cultured in X-vivo 15 medium (Lonza #04–744Q) and 20% BIT 9500 medium (STEMCELL Technologies #09500), supplemented with SCF (100 ng/ml), FLT-3 ligand (10 ng/ml), IL-6 (20 ng/ml), and TPO (100 ng/ml) as the basic culture. All cytokines were purchased from PeproTech.
Fetal liver (FL) transplantation assays.
FL cells isolated from embryonic day 14.5 (E14.5) mice were lysed with RBC lysis buffer and infected 3 times with retroviral vectors using retronectin coated plates (Clontech #T110A) and 4 ng/mL polybrene (Millipore #TR-1003-G). Approximately 5×105 GFP+ FL cells were injected into the tail vein of lethally irradiated (8.5 Gy) B6.SJL recipient mice. Survival of the mice was monitored daily. For the homing assay, BM cells were isolated 16 hr following tail vein injection and the frequency of donor-derived cells was identified by flow cytometry for GFP, simultaneously staining for Lin, Sca-1, and Mac-1 markers.
In vitro rescue experiments.
Carm1∆/∆ FL cells were infected twice with a retrovirus expressing MIGR1-AE9a or MIGR1-MLL-AF9 and GFP, followed by a single lentiviral infection with PCDH vector alone, PCDH-Carm1WT or PCDH-Carm1EQ, expressing RFP. Cells co-expressing both GFP and RFP were sorted and 1×103 were plated in M3434 methylcellulose in triplicate. Cells were counted, harvested, and replated at the same density every 7 days.
Western blotting.
Cells were lysed with cell lysis buffer (Cell Signaling #9803) supplemented with PhosSTOP phosphatase inhibitor (Roche # 4906845001) and complete protease inhibitor cocktail (Sigma #11873580001). 15 ug of total protein were loaded and western blots were run following typical procedures. The following antibodies were used: CARM1 (Millipore #09–818), Anti-dimethyl-BAF155 (Millipore ABE1399), total BAF155 (D7F86) (Cell Signaling #11956), Asymmetric-methyl-PABP1 (Cell Signaling # 3505), total PABP1 (Cell Signaling #49926), Asymmetric-dimethyl H3R17 (Abcam #ab8284), total H3 (C-16)(Santa Cruz #sc-8654), Mono-methyl SmB (Sigma-Aldrich #S0698) and β-Actin (Sigma-Aldrich #A5441). Quantification of protein levels was performed using the Odyssey CLx imaging system (LI-COR Biosciences).
RNAi.
Lentiviral pLKO.1 vectors expressing RFP and containing shRNA targeting CARM1 were obtained from the RNAi Consortium (Moffat 2006). For all figures, CARM1-KD1 refers to sense sequence CTATGGGAACTGGGACACTTT and CARM1-KD2 refers to sense sequence CGATTTCTGTTCCTTCTACAA. Lentiviral pLKO.1 vectors containing shRNA targeting AML1-ETO were obtained from Sigma-Aldrich. AE-KD1 refers to sense sequence GCAAGTTGAAACGCTTTCTTA and AE-KD2 refers to sense sequence GCGCTGAATTGCATTATGGAA.
Immunohistochemisty.
Hematoxylin and Eosin (H&E) staining of fixed bone marrow and spleen tissue was conducted using 6-week-old Carm1Fl/Fl and Carm1∆/∆ mice. Antibodies used for analysis of CARM1 and H3R17me2a in 6-week-old mice were CARM1 (Bethyl labs #A300–420A) and Asymmetric-dimethyl H3R17(Abcam #ab8284), respectively. Senescence staining was conducted using the Senescence β - galactosidase kit (Cell Signaling, #9860). Briefly, cytospins containing 2×105 SKNO-1 cells were incubated with staining solution for 36 hr at 37 degrees in a dry incubator prior to imaging.
Flow cytometry.
Flow cytometric analysis and cell sorting were performed as described previously (Liu 2015). For hematopoietic stem/progenitor cell staining, bone marrow was isolated from 12-month-old Carm1Fl/Fl, Carm1∆/+, and Carm1∆/∆ mice and stained with a modified biotin lineage cocktail of TER-119, B220, Gr-1, CD11b, CD3, CD4, CD5, CD8, and CD127. The following anti-mouse monoclonal antibodies were used for the staining of cell surface markers: anti-mouse CD11b (Mac-1, M1/70), CD41a (MWReg30), Gr-1(RB6–8C5), Ter119 (Ter119), CD45R/B220 (RA3–6B2), CD43 (S7), CD24 (M1/69), CD19 (MB19–1, eBioscience), Sca-1(D7), CD117 (c-KIT, 2B8), CD135 (A2F10.1), CD34 (RAM34). All antibodies except where mentioned were purchased from BD Biosciences. All data were analyzed by FlowJo Version 9.3.3 analysis software (TreeStar).
RNA extraction and quantitative RT-PCR.
Leukemia cell line pellets containing 5×106 cells were collected and resuspended in Trizol (Invitrogen #15596–026). RNA was extracted using an RNAeasy mini kit (Qiagen). Equal amounts of RNA were used for RT reaction according to manufacturer’s instructions (qScript™ cDNA SuperMix, Quanta Biosciences). Quantitative PCR (qPCR) was performed with the SYBR® Green PCR Master Mix or the TaqMan Universal PCR Master Mix (Applied Biosystems) and a “7500 Real-Time PCR System” thermocycler (Applied Biosystems). Gene expression levels were normalized to the reference gene GAPDH. Primer pairs and Taqman probes used for PCR were from Invitrogen and ThermoFisher Scientific respectively, and are listed in the Key Resources Table. For the detection of CARM1 isoform levels by RT-PCR, cDNA was generated from total RNA for a panel of 16 AML cell lines. The primer sequences used were: CARM1, forward primer: Exon 13 5′-CAGCACCTACAACCTCAGCA −3′, reverse: Exon 16 5′-GGCTGTTGACTGCATAGTGG 3′. For human GAPDH rRNA, forward primer 5′ CAATGACCCCTTCATTGACC −3′, reverse: 5′- GATCTCGCTCCTGGAAGATG −3′.
RNA sequencing alignment and analysis.
RNA-Seq library prep was carried out using the Illumina TruSeq Total Stranded kit (RS-122–2201) with Ribo-Zero rRNA reduction following manufacturer’s standard protocol without modification. Paired-end sequencing (150 base pairs) was performed on an Illumina Nextseq 500. RNA sequencing reads were mapped to GRCh38.p7 assembly using STAR aligner version 2.5.2b with Ensembl annotations. Gene level counts were estimated using RSEM version 1.3.0. DESeq2 version 1.12.4 was used for sample normalization and differential expression analysis. Gene Set Enrichment Analysis (GSEA v.2.0) was performed on a preranked list based on the DESeq2 t-statistic using preassembled gene sets from MSigDB v5.2. Significant splicing events based on junction counts and reads on target were determined using rMATs version 3.2.5.
CARM1 inhibitor studies.
For in vitro studies, 100 mM stock solutions of EPZ025654 or EPZ029751 were prepared in DMSO and stored at −20° C. Final DMSO concentrations were kept below 0.1%. Apoptosis was measured using the Annexin V Apoptosis Detection Kit (BD Pharmingen #556547) with DAPI stain for DNA content (Biolegend #422801). Cell viability was assessed by a CellTiter-Glo Luminescent Cell Viability Assay (Promega) on day 10, according to the manufacturer’s protocol. For in vivo studies, EZM2302 or vehicle (0.5% methylcellulose in dH20) was administered orally BID at a dose of 100 mg/kg for 21 days. Body weights were measured daily for the duration of the study.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics.
A student’s t-test (two-sided) was applied, and changes were considered statistically significant when p< 0.05. The data were normally distributed and variation within and between groups was not estimated. The sample size was not pre-selected and no inclusion/exclusion criteria were used. The data shown are the averages and s.d. of at least 3 biological replicates. Statistical analysis used Microsoft Excel or GraphPad Prism software. Prediction of cancer dependencies for CARM1 RNAi in cancer cell lines was based on a nonlinear regression model (ATLANTIS) and obtained from https://depmap.org, which are based on multiple shRNA hairpins and analysis for off-target effects using the computational tool DEMETER (Tsherniak et al., 2017), with negative z-scores representing a higher dependence on CARM1. For the L1000 analysis, the differentially expressed genes (DEGs) were identified using a log2 fold change greater than 1.2. Gene Ontology (GO) enrichment analysis of the DEGs was performed using the Database for Annotation, Visualization, and Integrated Discovery (david.abcc.ncifcrf.gov) using a background list of the 978 L1000 genes.The default thresholds of count greater than 2 and EASE score (a modified Fisher Exact p value) greater than 0.1, as well as a Benjamini-Hochberg multiple test correction were used to compute GO annotations.
Survival analysis.
The level-3 data of The Cancer Genome Atlas (TCGA) Acute Myeloid Leukemia (LAML) cohort were obtained from the Genomic Data Commons (GDC) Data Portal. RNA-Seq raw counts of 173 cases using hg38 (“LAML”, level 3, and platform “IlluminaHiSeq_RNASeqV2”) were normalized using within-lane Loess normalization to adjust for GC-content effect on read counts and between-lane upper-quartile normalization for distributional differences between lanes using EDASeq 2.12.0 in R v3.4.1. We performed survival analysis comparing 33% of patients with the highest CARM1 expression with 33% of patients with the lowest expression. Kaplan–Meier plots were generated using Graphpad Prism and reports the p value of the log-rank test.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PRMT4 (IHC, western blot, chIP) Rabbit | Millipore | 09–818, RRI D AB_1587416 |
| Asymmetric-dimethyl H3R17 (IHC, western blot, chIP) Rabbit |
Abcam | ab8284, RRID:AB_306434 |
| Asymmetric-dimethyl BAF155 Rabbit | Millipore | ABE1399 |
| BAF155 (D7F86) Rabbit | Cell Signaling | 49926 |
| Total Histone H3 (C-16) | Santa Cruz | Sc-8654, RRID:AB_2118303 |
| Mono-methyl SmB | Sigma Aldrich | S0698, RRID:AB_2193867 |
| Asymmetric-Methyl-PABP1 (Arg455/Arg460) (C60A10) | Cell Signaling | #3505, RRID:AB_2298971 |
| Anti-PABP antibody [10E10] | Abcam | Ab6125, RRID:AB_2156878 |
| GAPDH FL-335 Rabbit | Santa Cruz | Sc-25778, RRID:AB_10167668 |
| Donkey Anti-Mouse IgG 680RD | LI-COR- Biosciences | 926–68072 RRID:AB_10953628 |
| Donkey Anti-Rabbit IgG 680RD | LI-COR- Biosciences | 926–68073, RRID:AB_10954442 |
| Donkey Anti-Mouse IgG 800CW | LI-COR- Biosciences | 926–32212, RRID:AB_621847 |
| Donkey Anti-Rabbit IgG 800CW | LI-COR- Biosciences | 926–32213, RRID:AB_621848 |
| Biotin-CD3e | BD Biosciences | 553059, RRID:AB_394592 |
| Biotin-CD4 | BD Biosciences | 553728, RRID:AB_395012 |
| Biotin-CD5 | BD Biosciences | 553019, RRID:AB_394557 |
| Biotin-CD8 | BD Biosciences | 553029, RRID:AB_394567 |
| Biotin-CD127 | BD Biosciences | 555288, RRID:AB_395706 |
| Biotin-Gr-1 | BD Biosciences | 553125, RRID:AB_394641 |
| Biotin-B220 | BD Biosciences | 553086, RRID:AB_394616 |
| Biotin-Ter119 | BD Biosciences | 553672, RRID:AB_394985 |
| APC-c-Kit | BD Biosciences | 553356, RRID:AB_398536 |
| Strep-PerCP-Cy5.5 | BioLegend | 405214, RRID:AB_2716577 |
| PE-CD71 | Biolegend | 113808, RRID:AB_313569 |
| FITC-Ter119 | Biolegend | 116206, RRID:AB_313707 |
| FITC-Annexin-V | Biolegend | 640905, RRID:AB_2561291 |
| PE-cy7-Sca-1 | Thermo Fisher Scientific |
255981, RRID:AB_469668 |
| APC-Mac-1 | Thermo Fisher Scientific |
17–0112, RRID:AB_469343 |
| Biological Samples | ||
| Human: Umbilical cord blood for CD34 purification | NYBB nybloodcenter.org |
N/A |
| Human: AML064 | TCBF-UM | TCBF-AML064 |
| Human: AML065 | TCBF-UM | TCBF-AML065 |
| Human: AML066 | TCBF-UM | TCBF-AML065 |
| Human: AML067 | TCBF-UM | TCBF-AML065 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Poly (I:C) HMW | Invivogen | Tlrl-pic-5 |
| EPZ025654/GSK3536023 CARM1 tool compound | Epizyme | N/A |
| EZM2302/GSK3359088 CARM1 in vivo compound | Epizyme | N/A |
| EPZ029751 control compound | Epizyme | N/A |
| M3434 Methylcellulose, Mouse | STEMCELL technologies |
03434 |
| H4435 Methylcellulose, Human | STEMCELL technologies |
04435 |
| Methyl cellulose viscosity 400 cP (In vivo vehicle) | Sigma-Aldrich | M0262 |
| Critical Commercial Assays | ||
| CellTiter-Glo | Promega | G7572 |
| CD34 Microbead Kit, Human | Miltenyi | 130–046-702 |
| Senescence β-Galactosidase Staining Kit | Cell Signaling | #9860 |
| RNeasy Mini Kit | Qiagen | #74106 |
| Deposited Data | ||
| RNA-sequencing: CARM1 knockdown in AML cell lines | This paper | GEO: GSE103528 |
| Experimental Models: Cell Lines | ||
| Human: AML-14 | OSU | RRID:CVCL_8286 |
| Human: HEL | ATCC | TIB-180, RRID:CVCL_2481 |
| Human: HL-60 | ATCC | CCL-240, RRID:CVCL_A794 |
| Human: KASUMI-1 | ATCC | CRL-2724, RRID:CVCL_0589 |
| Human: MOLM-13 | DSMZ | ACC-554, RRID:CVCL_RS50 |
| Human: MV4–11 | ATCC | CRL-9591, RRID:CVCL_0064 |
| Human: NB-4 | DSMZ | ACC-207, RRID:CVCL_0005 |
| Human: NOMO-1 | DSMZ | ACC-542. RRID:CVCL_1609 |
| Human: OCI-AML3 | DSMZ | ACC-582, RRID:CVCL_1844 |
| Human: SET-2 | DSMZ | ACC-608, RRID:CVCL_2187 |
| Human: SKNO-1 | DSMZ | ACC-690, RRID:CVCL_2196 |
| Human: TF-1 | ATCC | CRL-2003, RRID:CVCL_0559 |
| Human: THP-1 | ATCC | TIB-202, RRID:CVCL_0006 |
| Human: U937 | ATCC | CRL-1593.2, RRID:CVCL_0007 |
| Human: UKE-1 | Coriell | RRID:CVCL_4Z62 |
| Experimental Models: Organisms/Strains | ||
| Mouse: CARM1 conditional knockout | Laboratory of Dan Tenen |
N/A |
| Mouse: B6.SJL-Prprca PEPCb/BoyJ Age: 6–8 weeks |
The Jackson Laboratory |
Stock: 002014 |
| Mouse: Vav1-cre B6.Cg-Tg(Vav1-cre) A2Kio/J | The Jackson Laboratory |
Stock: 008610 |
| Mouse: Mx1-cre B6.Cg-Tg(Mx1-cre) 1Cgn/J | The Jackson Laboratory |
Stock: 003556 |
| Mouse: NOD.Cg-Prkdcscid
Il2rgtm1Wjl Tg(CMV- IL3,CSF2,KITLG)1Eav/MloySzJ |
The Jackson Laboratory |
Stock: 013062 |
| Oligonucleotides | ||
| Human CARM1-KD1- CTATGGGAACTGGGACACTTT | RNAi Core MSKCC Moffat 2006 |
N/A |
| Human CARM1KD2- CGATTTCTGTTCCTTCTACAA | RNAi Core MSKCC Moffat 2006 |
N/A |
| Human AML1-ETO-KD1- GCAAGTTGAAACGCTTTCTTA |
Sigma-Aldrich | TRCN0000013366 |
| Human AML1-ETO-KD2- GCGCTGAATTGCATTATGGAA |
Sigma-Aldrich | TRCN0000013367 |
| See Table S6 for Taqman primer sequences | ||
| Recombinant DNA | ||
| MIGR1 plasmid-GFP | Addgene Pear 1998 | #27490 |
| MIGR1-AE9a-GFP | Addgene Yan 2006 | #12433 |
| MIGR1-MLL-AF9-GFP | Addgene Saito 2015 | #71443 |
| pCDH-PRMT4-WT-RFP | This paper | N/A |
| pCDH-PRMT4-EQ-RFP | This paper | N/A |
| pLKO1.1 TRC Cloning vector | Addgene Moffatt 2006 | #10878 |
| Software and Algorithms | ||
| Dep map |
depmap.org Tsherniak 2017 |
N/A |
| FlowJo Version 9.3.3 | TreeStar | N/A |
| Other | ||
Significance:
Therapeutic approaches are sorely needed for acute leukemia patients, who receive non-specific chemotherapy agents and often fail to achieve sustained remissions. CARM1 is an epigenetic modifying enzyme, which is significantly overexpressed in AML as well as in prostate, breast, lung, liver, and colorectal cancers, and regulates cellular differentiation. We have shown the dependency of AML cells, but not normal hematopoietic stem cells, on CARM1 activity, based on CARM1 knockout, knockdown, and chemical inhibition. These studies show that CARM1 regulates essential processes in leukemia cells, is critical for leukemic transformation, and can be targeted with small molecule inhibitors, providing experimental support for an effective “epigenetically-targeted” strategy for AML.
HIGHLIGHTS:
- Conditional knockout of Carm1 has little effect on normal hematopoiesis
- Loss of Carm1 abrogates the initiation and maintenance of AML in mouse models
- CARM1 knockdown impairs cell cycle progression and induces apoptosis in AML cells
- A potent and selective CARM1 inhibitor impairs AML cell proliferation
Acknowledgements:
We thank the members of the Nimer lab for their assistance and thoughtful input on the manuscript. We thank Nicholas Lyons and Lev Litichevskiy at the Broad Institute for their assistance with project design and data analysis. We also thank the Oncogenomics Shared Resource, Cancer Modeling Shared Resource, and the Flow Cytometry Core Facility of the Sylvester Comprehensive Cancer Center. The Graphical Abstract was designed by Christine Ryan. This study was supported by R01-CA-166835, (SDN). The L1000 analysis was funded by the Data Coordination and Integration Center for LINCs-BD2K (U54HL127624).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests:
Michael T. McCabe and Ryan G. Kruger are employees and shareholders of GSK.
DATA AVAILABILITY
Accession numbers. RNA-sequencing and data sets generated in this study have been deposited in Gene Expression Omnibus database using the accession number GSE103528.
References:
- Al-Dhaheri M, Wu J, Skliris GP, Li J, Higashimato K, Wang Y, White KP, Lambert P, Zhu Y, Murphy L, and Xu W (2011). CARM1 is an important determinant of ERalpha-dependent breast cancer cell differentiation and proliferation in breast cancer cells. Cancer Res 71, 2118–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An W, Kim J, and Roeder RG (2004). Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell 117, 735–748. [DOI] [PubMed] [Google Scholar]
- Beguelin W, Popovic R, Teater M, et al. (2013). EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23, 677–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, et al. (2011). MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 20, 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N., Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr., Laird PW, et al. (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368, 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, Rioux N, Munchhof MJ, Jin L, Jacques SL, et al. (2015). A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol 11, 432–437. [DOI] [PubMed] [Google Scholar]
- Chen SL, Loffler KA, Chen D, Stallcup MR, and Muscat GE (2002). The coactivator-associated arginine methyltransferase is necessary for muscle differentiation: CARM1 coactivates myocyte enhancer factor-2. J Biol Chem 277, 4324–4333. [DOI] [PubMed] [Google Scholar]
- Cheng D, Cote J, Shaaban S, and Bedford MT (2007). The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol Cell 25, 71–83. [DOI] [PubMed] [Google Scholar]
- Cheung N, Chan LC, Thompson A, Cleary ML, and So CW (2007). Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol 9, 1208–1215. [DOI] [PubMed] [Google Scholar]
- Cheung N, Fung TK, Zeisig BB, Holmes K, Rane JK, Mowen KA, Finn MG, Lenhard B, Chan LC, and So CW (2016). Targeting Aberrant Epigenetic Networks Mediated by PRMT1 and KDM4C in Acute Myeloid Leukemia. Cancer Cell 29, 32–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covic M, Hassa PO, Saccani S, Buerki C, Meier NI, Lombardi C, Imhof R, Bedford MT, Natoli G, and Hottiger MO (2005). Arginine methyltransferase CARM1 is a promoter-specific regulator of NF-kappaB-dependent gene expression. EMBO J 24, 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, et al. (2011). Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daujat S, Bauer UM, Shah V, Turner B, Berger S, and Kouzarides T (2002). Crosstalk between CARM1 methylation and CBP acetylation on histone H3. Curr Biol 12, 2090–2097. [DOI] [PubMed] [Google Scholar]
- Dhar S, Vemulapalli V, Patananan AN, Huang GL, Di Lorenzo A, Richard S, Comb MJ, Guo A, Clarke SG, and Bedford MT (2013). Loss of the major Type I arginine methyltransferase PRMT1 causes substrate scavenging by other PRMTs. Sci Rep 3, 1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew AE, Moradei O, Jacques SL, Rioux N, Boriack-Sjodin AP, Allain C, Scott MP, Jin L, Raimondi A, Handler JL, et al. (2017). Identification of a CARM1 Inhibitor with Potent In Vitro and In Vivo Activity in Preclinical Models of Multiple Myeloma. Sci Rep 7, 17993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Messaoudi S, Fabbrizio E, Rodriguez C, Chuchana P, Fauquier L, Cheng D, Theillet C, Vandel L, Bedford MT, and Sardet C (2006). Coactivator-associated arginine methyltransferase 1 (CARM1) is a positive regulator of the Cyclin E1 gene. Proc Natl Acad Sci U S A 103, 13351–13356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Yi P, Wong J, and O’Malley BW (2006). Signaling within a coactivator complex: methylation of SRC-3/AIB1 is a molecular switch for complex disassembly. Mol Cell Biol 26, 7846–7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frietze S, Lupien M, Silver PA, and Brown M (2008). CARM1 regulates estrogen-stimulated breast cancer growth through up-regulation of E2F1. Cancer Res 68, 301–306. [DOI] [PubMed] [Google Scholar]
- Hong H, Kao C, Jeng MH, Eble JN, Koch MO, Gardner TA, Zhang S, Li L, Pan CX, Hu Z, et al. (2004). Aberrant expression of CARM1, a transcriptional coactivator of androgen receptor, in the development of prostate carcinoma and androgen-independent status. Cancer 101, 83–89. [DOI] [PubMed] [Google Scholar]
- Jansson M, Durant ST, Cho EC, Sheahan S, Edelmann M, Kessler B, and La Thangue NB (2008). Arginine methylation regulates the p53 response. Nat Cell Biol 10, 1431–1439. [DOI] [PubMed] [Google Scholar]
- Kawabe Y, Wang YX, McKinnell IW, Bedford MT, and Rudnicki MA (2012). Carm1 regulates Pax7 transcriptional activity through MLL1/2 recruitment during asymmetric satellite stem cell divisions. Cell Stem Cell 11, 333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Lee J, Cheng D, Li J, Carter C, Richie E, and Bedford MT (2010). Enzymatic activity is required for the in vivo functions of CARM1. J Biol Chem 285, 1147–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee J, Yadav N, Wu Q, Carter C, Richard S, Richie E, and Bedford MT (2004). Loss of CARM1 results in hypomethylation of thymocyte cyclic AMP-regulated phosphoprotein and deregulated early T cell development. J Biol Chem 279, 25339–25344. [DOI] [PubMed] [Google Scholar]
- Kim YR, Lee BK, Park RY, Nguyen NT, Bae JA, Kwon DD, and Jung C (2010). Differential CARM1 expression in prostate and colorectal cancers. BMC Cancer 10, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, et al. (2012). A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol 8, 890–896. [DOI] [PubMed] [Google Scholar]
- Lee J, and Bedford MT (2002). PABP1 identified as an arginine methyltransferase substrate using high-density protein arrays. EMBO Rep 3, 268–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Bedford MT, and Stallcup MR (2011). Regulated recruitment of tumor suppressor BRCA1 to the p21 gene by coactivator methylation. Genes Dev 25, 176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhao Z, Carter C, Ehrlich LI, Bedford MT, and Richie ER (2013). Coactivator-associated arginine methyltransferase 1 regulates fetal hematopoiesis and thymocyte development. J Immunol 190, 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Cheng G, Hamard PJ, Greenblatt S, Wang L, Man N, Perna F, Xu H, Tadi M, Luciani L, and Nimer SD (2015). Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis. J Clin Invest 125, 3532–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder S, Liu Y, Ford OH 3rd, Mohler JL, and Whang YE (2006). Involvement of arginine methyltransferase CARM1 in androgen receptor function and prostate cancer cell viability. Prostate 66, 1292–1301. [DOI] [PubMed] [Google Scholar]
- Mann M, Cortez V, and Vadlamudi R (2013). PELP1 oncogenic functions involve CARM1 regulation. Carcinogenesis 34, 1468–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A 3rd, Diaz E, et al. (2012). EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112. [DOI] [PubMed] [Google Scholar]
- Mereau H, De Rijck J, Cermakova K, Kutz A, Juge S, Demeulemeester J, Gijsbers R, Christ F, Debyser Z, and Schwaller J (2013). Impairing MLL-fusion gene-mediated transformation by dissecting critical interactions with the lens epithelium-derived growth factor (LEDGF/p75). Leukemia 27, 1245–1253. [DOI] [PubMed] [Google Scholar]
- Naeem H, Cheng D, Zhao Q, Underhill C, Tini M, Bedford MT, and Torchia J (2007). The activity and stability of the transcriptional coactivator p/CIP/SRC-3 are regulated by CARM1-dependent methylation. Mol Cell Biol 27, 120–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkura N, Takahashi M, Yaguchi H, Nagamura Y, and Tsukada T (2005). Coactivator-associated arginine methyltransferase 1, CARM1, affects pre-mRNA splicing in an isoform-specific manner. J Biol Chem 280, 28927–28935. [DOI] [PubMed] [Google Scholar]
- Osada S, Suzuki S, Yoshimi C, Matsumoto M, Shirai T, Takahashi S, and Imagawa M (2013). Elevated expression of coactivator-associated arginine methyltransferase 1 is associated with early hepatocarcinogenesis. Oncol Rep 30, 1669–1674. [DOI] [PubMed] [Google Scholar]
- Schurter BT, Koh SS, Chen D, Bunick GJ, Harp JM, Hanson BL, Henschen-Edman A, Mackay DR, Stallcup MR, and Aswad DW (2001). Methylation of histone H3 by coactivator-associated arginine methyltransferase 1. Biochemistry 40, 5747–5756. [DOI] [PubMed] [Google Scholar]
- Shia WJ, Okumura AJ, Yan M, Sarkeshik A, Lo MC, Matsuura S, Komeno Y, Zhao X, Nimer SD, Yates JR 3rd, and Zhang DE (2012). PRMT1 interacts with AML1-ETO to promote its transcriptional activation and progenitor cell proliferative potential. Blood 119, 4953–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HJ, Kim H, Oh S, Lee JG, Kee M, Ko HJ, Kweon MN, Won KJ, and Baek SH (2016). AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 534, 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishkova E, Zeng H, Liu F, Kwiecien NW, Hebert AS, Coon JJ, and Xu W (2017). Global mapping of CARM1 substrates defines enzyme specificity and substrate recognition. Nat Commun 8, 15571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlensky D, Mirrielees JA, Zhao Z, Wang L, Mahajan A, Yu M, Sherer NM, Wilke LG, and Xu W (2015). Differential CARM1 Isoform Expression in Subcellular Compartments and among Malignant and Benign Breast Tumors. PLoS One 10, e0128143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, et al. (2017). A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 171, 1437–1452 e1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarighat SS, Santhanam R, Frankhouser D, Radomska HS, Lai H, Anghelina M, Wang H, Huang X, Alinari L, Walker A, et al. (2016). The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia 30, 789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Padilla ME, Parfitt DE, Kouzarides T, and Zernicka-Goetz M (2007). Histone arginine methylation regulates pluripotency in the early mouse embryo. Nature 445, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu LP, Perna F, Wang L, Voza F, Figueroa ME, Tempst P, Erdjument-Bromage H, Gao R, Chen S, Paietta E, et al. (2013). PRMT4 blocks myeloid differentiation by assembling a methyl-RUNX1-dependent repressor complex. Cell Rep 5, 1625–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zeng H, Wang Q, Zhao Z, Boyer TG, Bian X, and Xu W (2015). MED12 methylation by CARM1 sensitizes human breast cancer cells to chemotherapy drugs. Sci Adv 1, e1500463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhao Z, Meyer MB, Saha S, Yu M, Guo A, Wisinski KB, Huang W, Cai W, Pike JW, et al. (2014). CARM1 methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer Cell 25, 21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Bruce AW, Jedrusik A, Ellis PD, Andrews RM, Langford CF, Glover DM, and Zernicka-Goetz M (2009). CARM1 is required in embryonic stem cells to maintain pluripotency and resist differentiation. Stem Cells 27, 2637–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Chen H, Du K, Asahara H, Tini M, Emerson BM, Montminy M, and Evans RM (2001). A transcriptional switch mediated by cofactor methylation. Science 294, 2507–2511. [DOI] [PubMed] [Google Scholar]
- Yadav N, Cheng D, Richard S, Morel M, Iyer VR, Aldaz CM, and Bedford MT (2008). CARM1 promotes adipocyte differentiation by coactivating PPARgamma. EMBO Rep 9, 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M, Kanbe E, Peterson LF, Boyapati A, Miao Y, Wang Y, Chen IM, Chen Z, Rowley JD, Willman CL, and Zhang DE (2006). A previously unidentified alternatively spliced isoform of t(8;21) transcript promotes leukemogenesis. Nat Med 12, 945–949. [DOI] [PubMed] [Google Scholar]
- Yang Y, and Bedford MT (2013). Protein arginine methyltransferases and cancer. Nat Rev Cancer 13, 37–50. [DOI] [PubMed] [Google Scholar]
- Yokoyama A, and Cleary ML (2008). Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 14, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Jankovic V, Gural A, Huang G, Pardanani A, Menendez S, Zhang J, Dunne R, Xiao A, Erdjument-Bromage H, et al. (2008). Methylation of RUNX1 by PRMT1 abrogates SIN3A binding and potentiates its transcriptional activity. Genes Dev 22, 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.