

Abstract
Although advances in medical technology and health care have improved the early diagnosis and management for cardiorenal metabolic disorders, the prevalence of obesity, insulin resistance, diabetes, hypertension, dyslipidemia, and kidney disease remains high. Findings from numerous population-based studies, clinical trials, and experimental evidence have consolidated a number of theories for the pathogenesis of cardiorenal metabolic anomalies including resistance to the metabolic action of insulin, abnormal glucose and lipid metabolism, oxidative and nitrosative stress, endoplasmic reticulum (ER) stress, apoptosis, mitochondrial damage, and inflammation. Accumulating evidence has recently suggested a pivotal role for proteotoxicity, the unfavorable effects of poor protein quality control, in the pathophysiology of metabolic dysregulation and related cardiovascular complications. The ubiquitin-proteasome system (UPS) and autophagy-lysosomal pathways, two major although distinct cellular clearance machineries, govern protein quality control by degradation and clearance of long-lived or damaged proteins and organelles. Ample evidence has depicted an important role for protein quality control, particularly autophagy, in the maintenance of metabolic homeostasis. To this end, autophagy offers promising targets for novel strategies to prevent and treat cardiorenal metabolic diseases. Targeting autophagy using pharmacological or natural agents exhibits exciting new strategies for the growing problem of cardiorenal metabolic disorders.
Keywords: Cardiorenal metabolic syndrome, adipose tissue, liver, cardiovascular, autophagy
I. Introduction - Definitions and Differential Diagnosis
a. Nomenclature (metabolic syndrome, cardiometabolic syndrome, cardiorenal metabolic syndrome), NIH/ADA diagnostic criteria
Ample evidence has suggested that risk factors for cardiovascular disease (CVD) often cluster together, in the form of obesity, insulin resistance, type 2 diabetes mellitus, dyslipidemia and hypertension (Alberti, et al., 2009; Alberti, Zimmet, & Shaw, 2005). The initial definition of “Syndrome X” encompassed the presence of insulin resistance/hyperinsulinemia, hypertension, glucose intolerance, and metabolic dyslipidemia [e.g., increased very-low-density lipoprotein (VLDL) and decreased HDL cholesterol (HDL-c)] (Reaven, 1988, 1993). Hyperuricemia, angina and elevated plasminogen activator inhibitor 1 (PAI-1) were later included as additional features of this syndrome (Reaven, 1993), referred to as the “metabolic syndrome” or “cardiometabolic syndrome” (Invitti, Gilardini, & Viberti, 2004; Ren, Pulakat, Whaley-Connell, & Sowers, 2010; Shulman & Mangelsdorf, 2005; Whaley-Connell & Sowers, 2014). Since its first appearance in the medical vernacular three decades ago, the general health impact of this constellation of metabolic disorders is increasingly recognized (Govindarajan, Whaley-Connell, Mugo, Stump, & Sowers, 2005). In 2005, the American Heart Association (AHA)/National Heart, Lung and Blood Institute (NHLBI) revised the definition of metabolic syndrome and required presence of three out of the five following criteria: elevated waist circumference (≥ 102 cm in males and ≥ 88 cm in females), triglycerides ≥ 150 mg/dl, and HDL-c < 40 mg/dl in men and < 50 mg/dl in women, elevated blood pressure ≥ 130/85 mmHg and elevated fasting glucose >100 mg/dl (Grundy, et al, 2006). Here, ethnic-specific waist circumferences should be taken into account when using this criterion in addition to adjusting impaired fasting glucose > 100 mg/dl, in compliance with the International Diabetes Federation (IDF) guidelines. These efforts allow us to handle the cardiorenal metabolic syndrome as a set of easily obtainable measures to identify individuals with a high risk for CVD and type 2 diabetes mellitus. The growing prevalence of obesity and diabetes continues to undermine the management of CVD (Alberti, et al, 2005; Rask Larsen, Dima, Correll, & Manu, 2018). A more comprehensive view of obesity is judged based on waist circumference as opposed to body mass index (BMI) in the Harmonization definition of cardiorenal metabolic syndrome (Alberti, et al., 2005; Smith & Ryckman, 2015). Additional evidence suggests that the waist-to-height ratio may be superior to both waist circumference and BMI (Ashwell, Gunn, & Gibson, 2012). Despite wide acknowledgement that intensive clinical management improves CVD outcomes, many patients with cardiorenal metabolic syndrome still face suboptimal risk factor management (Rask Larsen, et al., 2018).
b. Epidemiology and the exclusion of other endocrine disorders
The National Health and Nutrition Examination Survey (NHANES) survey suggested an age-adjusted prevalence of 22.9% of cardiorenal metabolic syndrome between 2009 and 2010, among those 20 years or older in the United States (Beltran-Sanchez, Harhay, Harhay, & McElligott, 2013). Data from the International Diabetes Federation (IDF) suggested that 25% of worldwide adult population suffer from the syndrome with 5% in those exhibiting normal weight, 22% being overweight, and 60% being obese (J. Li & Pfeffer, 2016). The prevalence of cardiorenal metabolic syndrome is much higher in developed countries compared with developing countries, although westernization of developing countries (high calorie diet and low physical activity) saw a quick rise in cardiorenal metabolic syndrome recently (Naidu, Ponnampalvanar, Kamaruzzaman, & Kamarulzaman, 2017). Moreover, a higher rate was found in urban compared with rural populations (Pradeepa, et al., 2016). A positive correlation is also present with aging. The Lifestyle Interventions and Independence for Elders Study reported a prevalence of cardio renal metabolic syndrome of nearly 50% in community-dwelling sedentary adults aged 70 to 89 years (Botoseneanu, et al., 2015).
c. Evaluation of clinical features of cardiorenal metabolic syndrome
While the actual diagnosis of the cardiorenal metabolic syndrome has little therapeutic relevance other than managing the individual components, attenuating CVD risk is a main drive in clinical practice. Individual components of cardiorenal metabolic syndrome serve as independent risk factors for the overall prevalence of cardiovascular disease (Fig. 1). Presence of concurrent risk factors drastically elevates rates and severity of cardiovascular diseases (Z. L. Li, et al., 2012; Neeland, Poirier, & Despres, 2018). The optimal management for individual components such as blood pressure, lipid, or HA1c goals differ in individuals based on age, ethnicity and the presence of confounding comorbidities (Vaughan & Mattison, 2016). For example, weight control lowers the overall risk of insulin resistance, type 2 diabetes, hypertension, dyslipidemia and albuminuria (Bassuk & Manson, 2005) although control of dyslipidemia has little impact on body weight, blood pressure or glycemic control. An additional consideration is the impact that cardiorenal metabolic abnormalities may predispose patients to other anomalies such as cognitive decline (C. Li & Lumey, 2016; Vaughan & Mattison, 2016). Thus, it is imperative to elucidate the precise mechanism behind the CVD complications of cardiorenal metabolic syndrome including cardiac remodeling, microvascular dysfunction, heart failure, albuminuria, coronary atherosclerosis, calcification and premature senescence (Di Lullo, Bellasi, Russo, Cozzolino, & Ronco, 2017; Ludwig, 2016). Earlier conceptualizations of cardiorenal metabolic syndrome mainly focused on obesity with insulin resistance as a core feature. A large volume of clinical and experimental evidence depicted the pathophysiological mechanisms of cardiorenal metabolic syndrome including identification of genes responsible for syndromic or non-syndromic monogenic oligogenic or polygenic anomalies and interplays among genetic and epigenetic factors (Whaley-Connell & Sowers, 2014; Y. Zhang & Ren, 2016). More recent findings have depicted an essential role for autophagy, a cellular process of degrading long-lived, injured proteins and organelles (Mizushima, 2018; S. Yan, Huda, Khambu, & Yin, 2017), in the pathogenesis of cardiorenal metabolic syndrome. Dysregulated autophagy is present in multiple metabolic anomalies including obesity, insulin resistance, diabetes mellitus and dyslipidemia (K. H. Kim & Lee, 2014; Lamb, Dooley, & Tooze, 2013; Marasco & Linnemann, 2018; Y. Zhang, Sowers, & Ren, 2018). While advances in the understanding of the etiology of this complex disorder have been made (Rask Larsen, et al., 2018; Whaley-Connell & Sowers, 2014; Y. Zhang & Ren, 2016; Y. Zhang, et al., 2018), risk reduction remains suboptimal for those high-risk populations, warranting novel therapeutic strategies.
Figure 1:
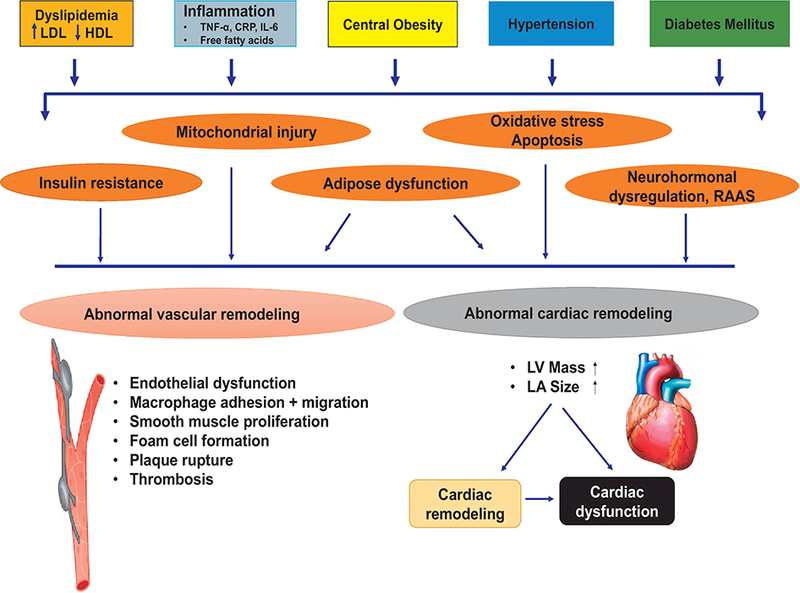
Schematic diagram displaying various contributing factors for cardiorenal metabolic syndrome in the complex pathophysiology of cardiovascular complications. RAAS: renin angiotensin aldosterone system; CRP: C reactive peptide; TNF-α: tumor necrosis factor α; IL-6: interleukin 6 [adapted from (Y. Zhang & Ren, 2016) with modifications].
II. Autophagy: Biochemistry, Cell Biology and Regulation of Autophagy
a. Biochemistry, molecular overview, regulation and functionality of autophagy
The ubiquitin-proteasome system (UPS) and autophagy-lysosome systems are the two main proteolytic pathways in eukaryotic cells to govern protein quality. Unlike the UPS pathway, the autophagy-lysosome mechanism participates in the bulk degradation of cytoplasmic organelles and protein aggregates which cannot be degraded by the UPS system (Levine & Klionsky, 2004, 2017; Mizushima, 2018). To-date, three types of autophagy are reported to remove long-lived, damaged and aggregated cellular components including macroautophagy, chaperone-mediated autophagy (CMA) and microautophagy (Levine & Klionsky, 2004, 2017; Mizushima, 2018). Microautophagy involves invagination of lysosomal or endosomal membranes to engulf cargo content. CMA employs a pentapeptide KFERQ motif and heat shock chaperon to transport cargos into lysosome. Macroautophagy (or autophagy) involves formation of double-membraned autophagosomes to sequester cargo contents and fusion with autolysosomes for degradation (Fig. 2). In autophagy, the double-membrane autophagosomes are capable of identifying and sequestering cellular cargo tagged by autophagy adaptors (e.g., sequestosome 1 [p62/SQSTM1], neighbor of Brca 1 [NBR1] and optineurin) (Stolz, Ernst, & Dikic, 2014). Cargo recognition depends on ubiquitination although non-ubiquitinated cargo may also be cleared under certain circumstances (Levine & Klionsky, 2004). Four steps have been identified in the autophagic process including initiation, engulfment of cargo by a double-membrane structure, formation of autophagolysosomes via the fusion of autophagosome and lysosome, and the final process of lysosomal degradation by acid hydrolases resulting in the recycling of lysosomal contents (Fig. 2) (Levine & Klionsky, 2004). Specific types of autophagy exist mainly depending on the selectivity of cargo delivery modality including glyophagy (glycogen), lipophagy (lipids) and mitophagy (mitochondria) (Evans, Sergin, Zhang, & Razani, 2017; Gatica, Lahiri, & Klionsky, 2018). The formation of isolation membrane begins at phagophore assembly sites including the Golgi, ER, endosomes, and mitochondria. Following the formation of isolated membranes, ubiquitin-like conjugation cascades Atg5-Atg12-Atg16L and Atg4-Atg3-LC3-II catalyze membrane elongation to generate autophagosomes. Atg12 becomes conjugated to Atg5 by an ubiquitination-like reaction through Atg7 and Atg10 prior to the association of Atg16L to the Atg12-Atg5 complex, producing the Atg5-Atg12- Atg16L complex (Nakatogawa & Ohsumi, 2014). The microtubule-associated protein 1 light chain 3 (LC3) is then cleaved at C-terminal by Atg4. The cytosolic LC3-I and phosphatidylethanolamine (PE) are connected by Atg3 and Atg7 to yield the lipidated LC3-II on autophagosomal membranes (Medvedev, Hildt, & Ploen, 2017). Then p62 directs ubiquitinated cargo to autophagosomal compartments (Taniguchi, Yamachika, He, & Karin, 2016), prior to the conjugation with the lysosome for the autophagolysosomes for degradation and recycling (Medvedev, et al., 2017).
Figure 2:
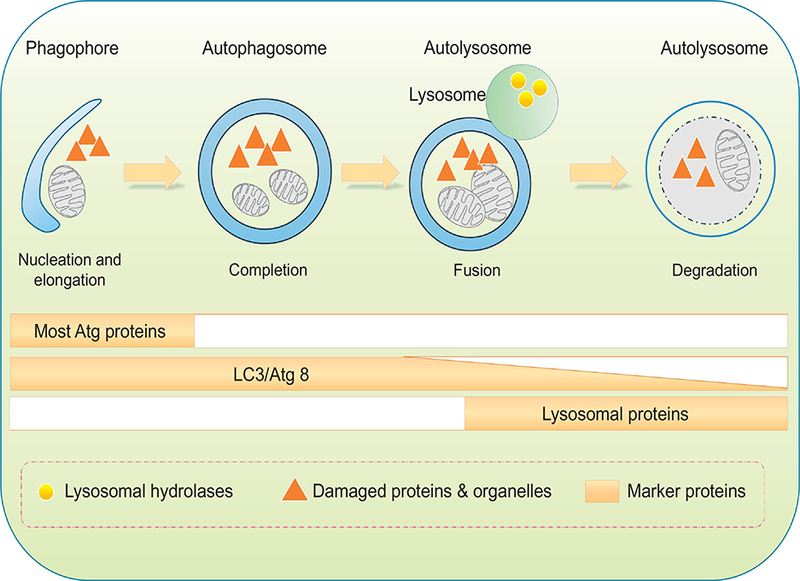
Schematic diagram of sequential autophagy events in a step-wise manner. Phagophore forms with initial sequestration of aged or damaged proteins and organelles; phagophores then undergo a series of further membrane expansion and elongation events to yie ld a completed double-membrane sequestering vesicle named autophagosome. During formation of autophagosome, various substrates (cytosolic proteins, lipids, nucleic acids, glycogen, damaged organelles, and invasive microbes, also known as cargo) of autophagy are encapsulated within the autophagosomal vesicle. Autophagosomes fuse with lysosomes to form autophagolysosmes, where cargos are digested by lysosomal hydrolases. Protein markers are identified throughout each individual steps. Most ATG proteins are visible during early stages of autophagy initiation while the elevated levels of lipidated LC3/Atg8 can sustain for a much longer period of time.
Autophagy is essential to cellular homeostasis and survival through quality control of amino acid pools and energy metabolism (Cheng, Ren, Hait, & Yang, 2013; Ren & Anversa, 2015; Rybstein, Bravo-San Pedro, Kroemer, & Galluzzi, 2018). With stress stimuli including nutrient starvation, hypoxia, oxidative stress, DNA damage, protein aggregates, and intracellular pathogens, autophagy is induced to preserve cellular integrity and physiological homeostasis (C. He & Klionsky, 2009). Autophagy is induced under starvation by posttranslational modifications and protein-protein interactions to release amino acids and fatty acids favoring cell survival (K. H. Kim & Lee, 2014; X. Xu, Pacheco, Leng, Bucala, & Ren, 2013). Mammalian target of rapamycin (mTOR) is the most predominant regulator of autophagy involving 2 complexes - MTORC1 and MTORC2. MTORC1, consisting of mTOR, raptor and mLST8, is rapamycin-sensitive. MTORC2, containing mTOR, rictor, mLST8, PRAS40, and DEPTOR, is rapamycin-insensitive (C. H. Jung, Ro, Cao, Otto, & Kim, 2010; Sciarretta, Forte, Frati, & Sadoshima, 2018). By phosphorylating unc-51 like kinase-1 (ULK1, the orthologue of mammalian Atg1), mTOR forms a complex with ULK1, Atg13 and FIP200, thus inhibiting autophagy (C. H. Jung, et al., 2010). MTORC1 complex (referred to as mTOR) may suppress autophagy through phosphorylation and thus inactivation of ULK/Atg1 and ATG13/Atg13, essential steps in the activation of autophagy. On the other hand, MTORC2 comp lex regulates cell shape, metabolism and autophagy. MTOR functions as a hub to integrate metabolic signals including insulin, growth factors, and cellular energy. One of the well-defined pathways intersecting with MTOR in the regulation of metabolism, growth and longevity is the insulin- insulin like growth factor 1 (IGF1) pathway (Kenyon, 2010). Upon activation of the insulin-IGF-1 cascade, class I phosphatidylinositide-3-kinase (PI3K) is activated to turn on PIP2, to recruit Akt to plasma membranes, where it is phosphorylated by 3-phosphoinositide dependent protein kinase 1/2 (PDPK1/2). Akt next regulates the heterodimeric tuberous sclerosis complex (TSC), which inhibits MTOR through suppression of Rheb (Y. Zhang, et al., 2003). Therefore, the TSC-Rheb-MTOR cascade represents a major signaling axis for growth and autophagy.
Autophagy is turned on by the energy sensor AMP-dependent protein kinase (AMPK), and activation of the ULK/Atg1 initiation complex. Beclin 1 then forms a complex with the PI3K-complex (class III phosphatidylinositol-3-kinase, Vps34, p150, Atg14) along with ULK1/2-complex to generate a phosphatidylinositol-3-phosphate (PI3P)-enriched environment, for nucléation of the isolation membrane. AMPK is sensitive to AMP levels, and may be activated by upstream kinases such as STK11/LKB1 and CaMKK2/CaMKKß (Ca2+/calmodulin-dependent protein kinase kinase 2ß) (Burkewitz, Zhang, & Mair, 2014). Among these AMPK-upstream kinases, STK11/LKB1 is constitutively active while CaMKK2 is responsive to changes in intracellular Ca2+ levels. AMPK also promotes autophagy via phosphorylation of the MTORC1 subunit RPTOR/raptor or dephosphorylation of TSC1/2, resulting in mTOR inhibition. Moreover, AMPK and MTOR may be phosphorylated by ULK/Atg1, offering in a feedback control mechanism of autophagy (Lapierre, Kumsta, Sandri, Ballabio, & Hansen, 2015). A simplified scheme illustrates these autophagy regulatory processes (Fig. 3)
Figure 3:
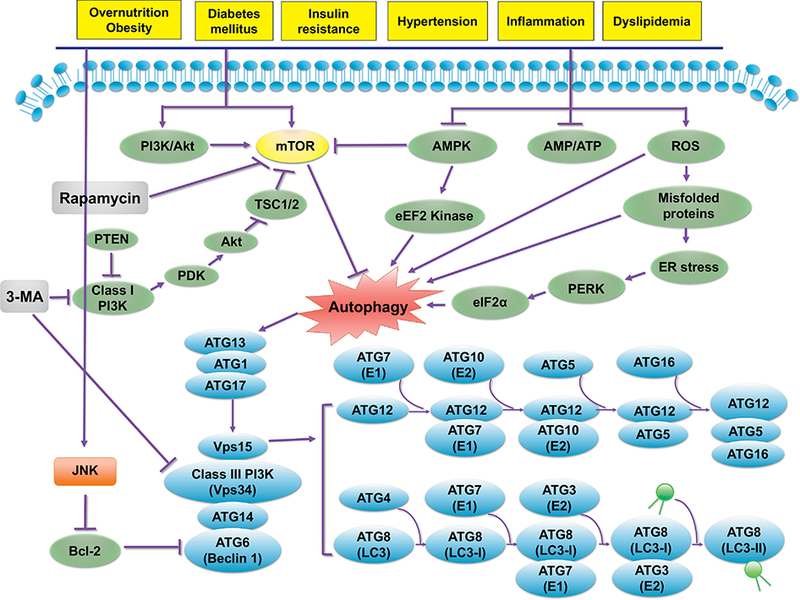
Autophagy regulatory mechanisms in response to various cardiorenal metabolic stimuli (e.g., overnutrition/obesity, hyperlipidemia, insulin resistance, diabetes mellitus, inflammation and hypertension). The PI3K-Akt and AMPK signaling cascades represent the two major cell signaling pathways in response to cardiorenal metabolic stress. Typically, mTOR (with PI3K-Akt being the upstream activator) suppresses autophagy through inhibition of the ULK1 complex (ULK1-ATG13-FIP200) that is required for autophagy induction. AMPK activates the ULK1 complex or indirectly suppress mTOR to initiate autophagy. ROS directly or indirectly (through ER stress) turns on autophagy. The mTOR- independent autophagy regulating autophagy are less clear. One example is c-Jun N-terminal kinase (JNK)-regulated phosphphorylation (or inhibition) of Bcl-2, leading to relieve of Bcl-2-Beclin 1 coupling and its inhibition on autophagy induction. The ULK1 complex consisting of ATG1 (ULK1)-ATG13 and ATG17 recruits Vps34, Beclin-1, and Vpsl5 for autophagosome synthesis, possibly through mTOR-independent pathway(s). Two ubiquitin-like conjugation systems involving ATGs govern the elongation of phagophores. The ATG5-ATG12 conjugation involves ATG7 (El-like) and ATG10 (E2-like), whereas the light chain 3 (LC3, also commonly known as ATG8)-LC3-I- phosphatidylethanolamine (PE) conjugation involves ATG4 (a cysteine protease), ATG7 (El-like), and ATG3 (E2-like). The ATG5-ATG12 conjugate generates a complex with ATG16 to control LC3-I-PE conjugation (resulting in LC3-II). Arrowheads and “T” ended line lines represent activation and inhibition, respectively. AMPK, AMP-activated protein kinase; Akt: also known as Protein Kinase B (PKB); mTOR, mammalian target of rapamycin; Rheb, Ras homology enriched in brain; TSC, tuberous sclerosis complex. Part of this cartoon is modified from (Y. Zhang, et al., 2018).
b. Autophagy in human diseases
Autophagy serves as an indispensable process for cellular homeostasis involved in immunity, inflammation, and metabolism (Choi, Ryter, & Levine, 2013). Either excessive or defective autophagy may be associated with human metabolic diseases (Sinha-Hikim, Sinha-Hikim, & Friedman, 2017), indicating the unique role of autophagy in the regulation of metabolic homeostasis.
Cardiovascular Diseases:
Dysregulated autophagy is commonly seen in cardiovascular diseases including cardiomyopathies, cardiac hypertrophy, ischemia-reperfusion injury, atherosclerosis, diabetes, and drug-induced cardiotoxicity (Bartlett, Trivedi, & Pulinilkunnil, 2017; Evans, Jeong, Zhang, & Razani, 2018; Evans, et al., 2017; X. Xu, Bucala, & Ren, 2013). In ischemia-reperfusion, autophagy is protective during early ischemia, while excessive autophagy is detrimental during reperfusion (H. Ma, Guo, Yu, Zhang, & Ren, 2011; W. J. Yan, Dong, & Xiong, 2013). Basal autophagy is deemed beneficial for cardiac hypertrophy whereas excessive autophagy may promote pathological cardiac remodeling and myopathies (J. Li & Cai, 2015; Nishida & Otsu, 2016). Thus understanding the functional role of autophagy in various cardiovascular diseases and if manipulation of autophagy offers therapeutic benefits are a major focus of research in recent years (Lampert & Gustafsson, 2018; Ren & Taegtmeyer, 2015). In addition, X-linked deficiency in autophagosome-lysosome fusion protein - lysosome-associated membrane protein 2 (LAMP2), leads to Danon’s cardiomyopathy with mitochondrial dysfunction and elevated autophagosomes (Rowland, Sweet, Mestroni, & Taylor, 2016). Along the same line, facilitated autophagy is evident in macrophages from atherosclerotic plaques where autophagy is capa ble of stabilizing atherosclerotic plaques by retarding apoptosis and necrosis (De Meyer, et al., 2015; Evans, et al., 2018; Grootaert, et al., 2018). Although consensus exists that autophagy is an essential cellular quality control pathway in cardiovascular homeostasis, its precise role in disease pathology remains controversial.
Cancer:
Autophagy participates in the initiation, progression, therapeutic efficacy and prognosis of cancer, demonstrating a dual regulation of tumor development (Choi, et al., 2013; Rybstein, et al., 2018). Autophagy might promote cancer cell survival or cancer cell death (Russo & Russo, 2018). Altered autophagy and chromosomal autophagy genes are found in various forms of cancer (S. He & Liang, 2015; Rybstein, et al., 2018). For example, monoallelic disruption of Beclin 1 on chromosome 17q21 is found in human breast, ovarian, and prostate tumors (Negri, et al., 2010). Homozygous deletion of Becn1 in mice leads to embryonic lethality, whereas the monoallelic deletion (Beclin1+/−) promotes spontaneous tumorigenesis, favoring a role for Beclin 1 as a haploinsufficient tumor-suppressor (Yue, Jin, Yang, Levine, & Heintz, 2003). The tumor-suppressing function has been confirmed for multiple autophagy-associated proteins (e.g., Uvrag, Bif1, Atg4C, Atg5, and Atg7) (J. Liu & Debnath, 2016). Consistently, classical tumor suppressing proteins including tensin homolog deleted in chromosome 10 (PTEN), TSC1/2, liver kinase B1 (LKB1), and p53 directly promote autophagy (Choi, et al., 2013). Autophagy is speculated to provide an anti-carcinogenic action by safeguarding against metabolic stress through homeostatic turnover of mitochondria and protein aggregates (J. Liu & Debnath, 2016). However, the apparent pro-survival role of autophagy for cancer cells suggests the necessity for a thorough understanding of the dichotomy roles of autophagy in cancer biology in order to develop anti-cancer drugs capable of inducing or blocking autophagy in a tissue- and time-dependent manner (Russo & Russo, 2018).
Metabolic Diseases:
Autophagy plays an essential role in metabolic diseases including obesity, insulin resistance, diabetes mellitus, and steatohepatitis through not only protein quality control but also release of amino acids, lipids, and other metabolic precursors (Madrigal-Matute & Cuervo, 2016; Sinha, Singh, & Yen, 2017; Y. Zhang, et al., 2018). Dysregulated autophagy promotes metabolic anomalies as evidenced by increased adiposity, insulin resistance, and hepatic storage of triglycerides with genetic deletion of autophagy genes, depic ting a role for autophagy in lipid metabolism (C. He, et al., 2013; Jaber, et al., 2012; Lim, et al., 2014; Y. Liu, et al., 2016; Madrigal-Matute & Cuervo, 2016; Settembre, De Cegli, et al., 2013). Genetic deletion of the autophagy gene Atg7 in the brain decreases food intake (Kaushik, et al., 2011). It is suggested that deletion of autophagy gene may affect metabolic phenotype in an organ specific-manner, as adipose-specific deletion of autophagy promotes insulin sensitivity and metabolic homeostasis (R. Singh, et al., 2009). On the other side of the coin, aberrant (increased or decreased) autophagy was found in various organs (such as liver, adipose tissue, heart and pancreas) in obesity, insulin resistance and diabetes (E. Chang, et al., 2015; H. H. Chang, et al., 2017; Ebato, et al., 2008; C. He, et al., 2012; Hsu, et al., 2016), the effect of which might be alleviated by lipid lowering agents and life style modification such as physical exercise or intermittent fasting (E. Chang, et al., 2015; C. He, et al., 2012; H. Liu, et al., 2017).
Aging:
Aging leads to accumulation of lipofuscin pigments and ubiquitinated protein aggregates along with defective autophagy (Cheng, et al., 2013; Nair & Ren, 2012). Evidence from mice and roundworm Caenorhabditis elegans suggests that autophagy induction prolongs lifespan (Rubinsztein, Marino, & Kroemer, 2011). Furthermore, analysis of gene expression in the brain revealed loss of autophagy with aging. Dietary restriction, a classical way to facilitate autophagy, is perhaps the most robust manipulation to prolong healthy lifespan (Kapahi, Kaeberlein, & Hansen, 2016). More evidence suggests that lysosomal defect is associated with aging-associated pathologies including Parkinson’s and Alzheimer’s disease, as well as shortened lifespan (Shi, Guberman, & Kirshenbaum, 2017). Likewise, targeting lysosomal function has shown promises in promoting longevity (Carmona-Gutierrez, Hughes, Madeo, & Ruckenstuhl, 2016).
III. Pathophysiology of the Cardiorenal Metabolic Syndrome
To better decipher the role of autophagy regulation in cardiorenal metabolic syndrome, it is pertinent to understand the pathophysiology of this medical problem. To date, a number of theories have been proposed describing the onset and progression of cardiorenal metabolic syndrome including excessive caloric intake, oxidative stress, inflammation, insulin resistance, hypertension, dyslipidemia, as well as genetic and epigenetic factors (as depicted in Fig. 1) (da Silva, Rodrigues, Rebelo, Miranda, & Soveral, 2018; Dommermuth & Ewing, 2018). Recent findings suggest that extracellular vesicles from various origins, including endothelial, smooth muscle, skeletal muscle, hepatocytes, adipocytes, macrophages, and microbiota may also serve as bioeffectors for metabolic disorders (Martinez & Andriantsitohaina, 2017; van Greevenbroek, Schalkwijk, & Stehouwer, 2016). Nonetheless, little is known as to why this constellation of metabolic disorders contributes to CVD beyond the sum of its individual components.
a. Insulin resistance and hypertension
Despite the predominant role for obesity and type 2 diabetes in the pathogenesis of cardiorenal metabolic syndrome, insulin resistance, a nexus among components of the syndrome with coexistence of hyperinsulinemia, obesity, dyslipidemia, albuminuria and hypertension, is considered the cardinal feature of this syndrome (Fig. 1) (Alberti, et al., 2005; Reaven, 1993). Insulin resistance triggers inappropriate activation of renin-angiotensin aldosterone system (RAAS) and oxidative stress, and serves as a common denominator for various components of the syndrome (J. A. Kim, Wei, & Sowers, 2008). Ample experimental evidence has depicted a pivotal role for insulin and insulin signaling in metabolism, cell growth, and survival through activation of mitogen-activated protein kinases (MAPKs) and PI3K as well as their downstream signaling cascades (Fu, Wang, & Xiang, 2017; Nielsen, 2017). Resistance to insulin compromises insulin-driven glucose and lipid metabolism in insulin-sensitive organs such as the liver, adipose tissues, skeletal muscles, heart and kidneys, prompting local tissue and organ injury and pathogenesis of albuminuria and hypertension (Fu, et al., 2017; Rask Larsen, et al., 2018). In addition, hyperinsulinemia in insulin resistance contributes to metabolic dyslipidemia manifested as elevated triglycerides, small and dense low-density lipoprotein (LDL) particles and decreased HDL-c, leading to endothelial dysfunction, sympathetic nervous system hyperactivity and hypertension (Fu, et al., 2017). Insulin resistance occurs at multiple levels from the cell surface to the nucleus, including desensitization of the insulin receptor, downregulated insulin receptor substrate (IRS), suppression of PI3K and Akt signals, and lack of inhibition on Foxo1-activated gene transcription (Fu, et al., 2017). Onset of insulin resistance has multiple origins, although obesity and sedentary lifestyle along with unknown genetic factors interact seem to predominate (Templeman, Skovso, Page, Lim, & Johnson, 2017). A key pathological change associated with insulin resistance is elevated blood pressure. It is estimated that over 30% of hypertensive patients display features of cardiorenal metabolic syndrome (Seravalle & Grassi, 2016). In this context, insulin resistance stimulates RAAS to promote volume expansion and endothelial dysfunction. Functional and structural abnormalities in microvasculature such as endothelial dysfunction, poor capillary recruitment, low capillary density and microvascular remodeling may also contribute to insulin resistance in cardiorenal metabolic syndrome (Moreira, et al., 2015). Activation of PI-3 kinase by insulin stimulates nitric oxide synthase (NOS) although overt oxidative stress, particularly from O2, in cardiorenal metabolic syndrome converts NO to ONOO- , resulting in protein damage, loss of NO bioavailability and impaired vasodilation in insulin resistance (Moreira, et al, 2015). To this end, patients usually receive targeted cardiorenal metabolic syndrome therapy only after insulin resistance/prediabetes or diabetes, hypertension and/or hyperlipidemia is confirmed (Rask Larsen, et al., 2018). Dietary and pharmacological manipulations to reduce insulin levels or restore insulin sensitivity are capable of eliciting weight loss and improve abnormalities associated with the cardiorenal metabolic syndrome (Rask Larsen, et al., 2018; Templeman, et al., 2017).
b. Obesity, abnormal fat distribution and adipose function
Obesity places a considerable burden on health care and the global economy, and contributes to the expansion of type 2 diabetes. Excess caloric intake and obesity, particularly visceral fat distribution, are critical to insulin resistance, diabetes, hypertension, dyslipidemia, and reduced life expectancy (Aune, et al., 2016; Perrone-Filardi, et al, 2015). Obese individuals are usually abundant in dysfunctional adipose tissues, triggering metabolic dyslipidemia including elevated triglycerides, apolipoprotein B, LDL-c, low HDL-c, low-grade inflammation, hypertension, and elevated blood glucose (Maiano, Hue, Morin, & Moullec, 2016; Saltiel & Olefsky, 2017; Zylke & Bauchner, 2016). Dysfunctional adipose tissues prompt low-grade inflammation, program adipose expansion and visceral adiposity, skewing the immune system to a pro-inflammatory phenotype favor the development of insulin resistance and hypertension (Alberti et al, 2009; Saltiel & Olefsky, 2017). It is noteworthy that obesity may be associated with a paradox, as findings from epidemiological studies suggest there is improved survival and better prognosis for CVD and kidney disease outcomes in those who are overweight or obese (Lavie, McAuley, Church, Milani, & Blair, 2014). There is still a fair amount of controversy that exists regarding the mechanisms of the paradox as the survival advantage is lost in those with extreme obesity (Oga & Eseyin, 2016) and raises the issue whether the normal range of BMI should be redefined in those afflicted with chronic diseases (Egom, et al, 2017).
c. Neurohormonal dysrégulation
Neuroendocrine function is closely regulated by genetic background and external factors including psychosocial stress, food intake, use of alcohol and other drugs. Dysregulation of these complex neuroendocrine networks promotes the onset and development of metabolic disorders (Seravalle & Grassi, 2016). Increased activation of sympathetic nervous system manifested as increased spillover of norepinephrine is common in obesity (Seravalle & Grassi, 2016), as evidenced by the positive correlation between urinary norepinephrine metabolites and BMI. Altered neural or hormonal outflow contributes to derangements in substrate and energy metabolism. For example, sympathetic activation directly compromises insulin sensitivity while elevated circulating noradrenaline levels promote lipolysis in adipose tissues, but not in muscles. However, local administration of catecholamines induces muscle lipolysis. The lipolytic action of catecholamines is more pronounced in visceral adipocytes along with dampened antilipolytic effect of insulin, resulting in higher free fatty acid (FFA) release from visceral adipocytes (Menzies, et al., 2017). The autonomic innervations of fat tissue may also alter insulin sensitivity through adipose tissue hormones such as resistin and leptin, both with an essential role in metabolic regulation and adiposity (Perez-Perez, et al, 2017). Elevated plasma leptin levels may drive sympathetic activation and dampen insulin sensitivity in obesity (Seravalle & Grassi, 2016). Other components of cardiorenal metabolic syndrome may also contribute to increased sympathetic tone as evidenced by sympathetic activation with hypertension (Seravalle & Grassi, 2016; Tuomikoski & Savolainen-Peltonen, 2017). Not surprisingly, heightened sympathetic tone is responsible for a number of unfavorable cardiorenal metabolic responses (Mahu & Domingos, 2017; Seravalle & Grassi, 2016; Tuomikoski & Savolainen-Peltonen, 2017).
d. Aberrant gene expression and epigenetic disorder
Cardiorenal metabolic syndrome may be consequence of complex genetic and environmental (such as diet) interactions (Park, Kim, Lee, & Kim, 2017). A number of genes such as peroxisome proliferator-activated receptor (PPAR)-α, -ß and -γ are involved in the regulation of insulin sensitivity, adipogenesis, lipid metabolism and blood pressure. Polymorphism in PPAR-α [Leu(162)Val, Val(227)Ala], PPAR-β/δ [+294T > C], or PPAR-γ [Pro(12)Ala and C1431T] can contribute to cardiorenal metabolic syndrome (Seravalle & Grassi, 2016). Moreover, levels of PPAR-γ gene were overtly elevated in oxidative stress (Hatami, et al, 2016). PPARs govern transcriptional regulation of uncoupling proteins (UCPs), members of the mitochondrial anion carrier superfamily regulating body temperature and energy balance (Castrejon-Tellez, et al., 2016). Genetic polymorphism of the promotor region (−866G/A) of UCP-2, is associated with higher prevalence of cardiorenal metabolic syndrome (Park, et al., 2017). UCP-2 gene has been shown to be significantly upregulated in the cardiorenal metabolic syndrome along with PPAR-α and PPAR-γ (Castrejon-Tellez, et al., 2016). Cardiorenal metabolic syndrome is also closely associated with genetic mutation of the adiponectin encoding gene ADIPOQ, which triggers decreased adiponectin levels, insulin resistance and hyperinsulinemia (Perez-Perez, et al, 2017). Recent data from our own group revealed that deficiency of adiponectin may predispose to the onset of obesity cardiomyopathy (R. Guo, Zhang, Turdi, & Ren, 2013) although adiponectin deficiency unexpectedly alleviated high fat diet-induced hepatic injury without affecting hepatic steatosis (R. Guo, Nair, Zhang, & Ren, 2017). Given the role of sympathetic activation in the cardiorenal metabolic syndrome, polymorphism of the ß-adrenergic receptor ADRB1 and ADRB3 genes (Arg389Gly and Trp64Arg) is deemed a susceptible gene for type 2 diabetes and lipid disorder (Burguete-Garcia, et al., 2014; Castrejon-Tellez, et al., 2016). Variant Arg389Gly of ADRB1 is associated with high LDL levels and the variant ADRB3 Trp64Arg is tied with increased levels of homeostatic model assessment-insulin resistance (HOMA-IR) and insulin (Burguete-Garcia, et al., 2014). Recent data have depicted a culprit role for the SLC35D3 gene located at or close to the D6S1009 site on the human chromosome 6 for obesity. Defective SLC35D3 gene disrupts delivery of dopamine in the central nervous system, prompting reduced exercise capacity, energy expenditure, metabolic control, and central obesity (Z. Zhang, et al., 2014). In fact, mutation of SLC35D3 gene occurs in 0.5% in obese population (Burguete-Garcia, et al., 2014). Moreover, exon sequencing in inheritable premature coronary disease, hypertension and diabetes revealed a high frequency of inheritable genetic mutation in Dyrk1B, a regulatory enzyme for muscle-adipose balance and blood glucose. Mutation of Dyrk1B disturbs blood glucose homeostasis (Z. Zhang, et al., 2014) and promotes metabolic syndrome (Keramati, et al, 2014). In addition to the list of genes mentioned above, genetic polymorphism of the lamin A/C (LMNA) (Ji, et al, 2016), insulin receptor substrate 1 (IRS1) (Kloting & Bluher, 2014), melanocortin receptor 4 (MC4R) (Ruiz-Ramirez, Lopez-Acosta, Barrios-Maya, & El-Hafidi, 2016), TCF7L2 (transcription factor 7-like 2) (Palizban, Rezaei, Khanahmad, & Fazilati, 2017), fat mass and obesity associated gene (FTO) (Elouej, et al, 2016; Rotter, et al., 2016) have also been documented to be associated with increased prevalence of cardiorenal metabolic syndrome.
In addition to the “metabolic susceptible genes”, dietary and environmental factors also play an essential role in the prevalence of cardiorenal metabolic syndrome (Gosadi, 2016; Park, et al, 2017). Dietary factor-dependent epigenetic modifications may regulate genome stability, the expression of mRNA and proteins to exert significant health effects (Ji, et al, 2016; Kunes, et al, 2015). Several epigenetic risk markers may be initiated or reversed by dietary and environmental factors such as the inverse correlation between méthylation of suppressors of cytokine signaling-3 (SOCS-3) and the prevalence of metabolic disease (Hohn, Konig, & Jung, 2016). More importantly, the epigenetic DNA modifications (i.e. DNA méthylation, histone modifications) from dietary and environmental factors may predispose offspring to unfavorable metabolic changes in their early life (fetal and perinatal periods) and subsequent progeny for several generations, a phenomenon termed transgenerational inheritance (Smith & Ryckman, 2015).
e. Mitochondrial dysfunction, oxidative stress, inflammation and apoptosis
Ample evidence favors a key role for mitochondrial injury, oxidative stress, and apoptosis in cardiorenal metabolic syndrome (Bhatti, Bhatti, & Reddy, 2016). Mitochondria govern essential cellular function such as ATP production, reactive oxygen species (ROS) production and removal, intracellular Ca2+regulation, and apoptosis (Bhatti, et al, 2016). ROS production occurs within and outside mitochondria through enzymatic and nonenzymatic machineries such as mitochondrial oxidative phosphorylation, nicotinamide adenine dinucleotide phosphate oxidases (NADPH oxidase or NOX), uncoupled NO synthase, xanthine oxidase, D-aminooxidase, p450 cytochromes, and proline hydroxylases. A major shift has been noted in the source of ROS with the progression of metabolic anomalies, from the early (NOX4-dependent), to the intermediate (NOX2-dependent) and late (mitochondria-dependent) stages (C. Y. Han, 2016). Oxidants including ROS and reactive nitrogen species (RNS) promote oxidation of lipids [lipid peroxidation - malondialdehyde, glyoxal, acrolein, and 4-hydroxy-nonenal (4-HNE)] and glucose (glycation - glyoxal and methyl glyoxal), resulting in reactive advanced glycation end-products (AGEs) and advanced lipid oxidation end-products (ALEs) to predispose the onset of cardiorenal metabolic syndrome. Elevated ROS production and dampened mitochondrial biogenesis as well as genes required for mitochondrial oxidative phosphorylation were reported in insulin resistance and type 2 diabetes (F. Dong, Li, Sreejayan, Nunn, & Ren, 2007). Therapeutic strategies to reduce mitochondrial and oxidative injury have been implicated in cardiorenal metabolic syndrome including lifestyle modifications (such as diet and exercise), pharmacological and mitochondria-targeted approaches (Bhatti, et al, 2016). Data from our laboratory revealed that the heavy metal scavenger metallothionein alleviates high fat diet intake and eNOS uncoupling-induced cardiac anomalies, mitochondrial injury, loss of mitochondrial DNA and mitochondrial biogenesis evidenced by peroxisome proliferator-activated receptor γ coactivator 1α (PGC-la) as well as the PGC-lα downstream nuclear factors (Ceylan-Isik, et al., 2009; F. Dong, ét al., 2007).
Inflammation, which initiates in adipose tissues eventually reaching circulation and other parts of body, perpetuates cardiorenal metabolic syndrome (Kloting & Bluher, 2014). Insulin resistance is one of the first consequences of sustained inflammation, since pro-inflammatory cytokines such as interleukins, and TNF-α abrogate insulin receptor phosphorylation and post-receptor action, leading to altered lipid storage, lipid spillover and dyslipidemia (Garbossa & Folli, 2017; Paniagua, 2016). Increased inflammatory infiltration also promotes oxidative stress and apoptosis to disturb metabolic homeostasis.
f. Impaired protein quality control and autophagy
Protein quality control mechanisms including UPS are essential to normal function of various cells, tissues, and organs through regulating synthesis, folding, trafficking, post-translational modification, assembly, disassembly, localization, and degradation of proteins (Parry & Willis, 2016). The occurrence of the pro-oxidative and proinflammatory states in cardiorenal metabolic syndrome, which include low-grade inflammation, oxidative stress, glycosylation, AGE formation, lipid peroxidation, protein oxidation, endoplasmic reticulum (ER) stress, and cytokine release, perturb the highly redox-sensitive UPS to compromise protein quality control, triggering proteotoxicity and cell injury (Hohn, et al., 2016). Proteases, including calpain, matrix metalloproteinase (MMP), cathepsin and caspase, serve as the major proteases governing the breakdown of proteins and participate in the pathogenesis of cardiorenal metabolic syndrome. MMPs and cathepsins alter metabolic processes via modification of extracellular matrix and intracellular proteins. On the other hand, calpain and caspases affect signaling cascade including activation of NF-κΒ and apoptosis (Hua & Nair, 2015). Proteases are gaining attention as biomarkers and therapeutic targets for metabolic diseases. Autophagy, another protein quality control mechanism to remove large injured or long-lived proteins or organelles, may be either up- or down-regulated in various metabolic diseases, exhibiting organ and tissue specificity (Andres, et al., 2016; Giricz, et at, 2017; Z. L. Li, et al., 2012; X. Xu, Hua, Nair, Zhang, & Ren, 2013; X. Xu & Ren, 2015). A reciprocal regulatory mechanism exists between lysosomal autophagy and key metabolic elements such as glucose and lipids (L. Liu, Liao, He, & Li, 2017; Ren & Taegtmeyer, 2015). For example, lipotoxicity in metabolic anomalies impairs lysosomal function and autophagy, further exacerbating lipid accumulation and ultimately cell injury (Taniguchi, et al., 2016). A more in-depth understanding of the role of autophagy in metabolic diseases should yield potential therapeutic strategies for better management of cardiorenal metabolic syndrome. Here, we will discuss dysrégulation and target therapy of autophagy in the pathogenesis and management of cardiorenal metabolic syndrome.
IV. Autophagy Regulation in Cardiorenal Metabolic Function (Short- and Long-term)
Autophagy plays a pivotal role in the maintenance of the body’s metabolism. Clinical and experimental evidence has depicted a link between autophagy and metabolic risk factors such as obesity, dyslipidemia, alcoholism, insulin resistance, hypertension, diabetes mellitus, sepsis, and inflammation (Choi, et al., 2013; Levine, Packer, & Codogno, 2015; H. Ma, et al., 2011; Sinha, et al., 2017; S. Wang & Ren, 2018; Y. Zhang, et al., 2018). Bioengineered models of autophagy also support an essential role for autophagy in systemic metabolic regulation. Global deletion of p62 promotes insulin resistance along with increased body fat mass (Rodriguez, et al., 2006). Heterozygous deletion of Atg7 (homozygous embryonic lethality) or Beclin 2 did not exhibit metabolic abnormalities although it accentuates ob/ob or high fat diet-induced insulin resistance, lipid accumulation and inflammation (Lim, et al., 2014). Atg5-deficient mice present poor removal of apoptotic bodies in development while inhibition of autophagy by Beclin-1 or Atg7 knockdown suppresses apoptosis (Burkewitz, et al., 2014), denoting a tie between autophagy and these biological processes. Monoallelic loss of Beclin 2, a newly identified member in the beclin family, triggers metabolic derangement (increased food intake, body weight and insulin resistance), due to increased brain G protein-coupled receptor (GPCR) cannabinoid 1 receptor governing food intake and energy storage (C. He, et al., 2013). Interestingly, chromosome 1q43, the genetic locus that contains beclin 2, is associated with obesity and diabetes traits in human (Shmulewitz, et al., 2006). It is perceived that Beclin 2 regulates autophagy through binding to PI3K complex, or lysosomal degradation of GPCRs through an adaptor protein GASP1 (GPCR-associated sorting protein 1) (Rocchi & He, 2015). Fig. 4 summarizes autophagy derangement and accumulation of cytotoxic aggregates in response to metabolic stress.
Figure 4:
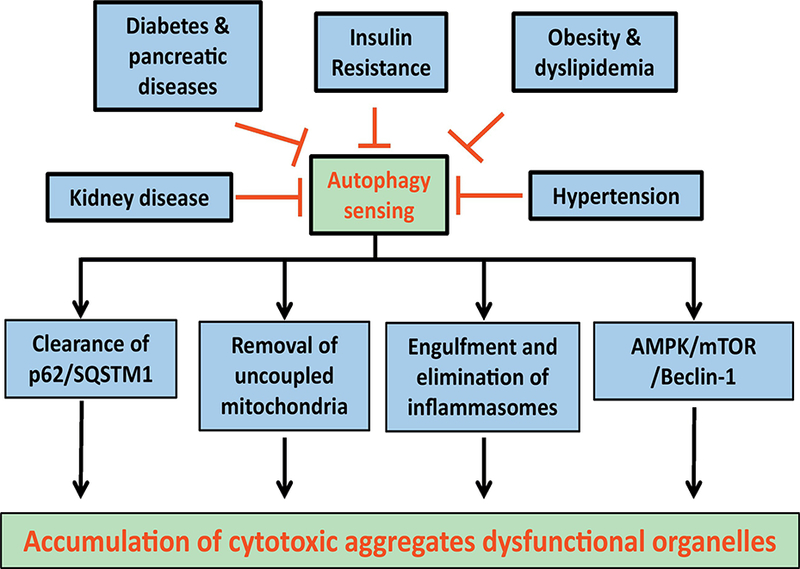
Various components of the cardiorenal metabolic syndrome may compromise autophagy sensing mechanisms leading to compromised autophagy regulatory machineries such as lysosomal degradation, selective removal of mitochondria and other organelles, engulfment of inflammasome and a number of cell signalling molecules governing autophagy, leading to accumulation of cytotoxic aggregates and dysfunctional organelles.
Not only does change in autophagy affect metabolic homeostasis, however metabolic stress also affects autophagy status. Examples of altered autophagy in various individual components of cardiorenal metabolic syndrome are presented in Table 1. Autophagy is suppressed in genetic or diet-induced models of obesity in various tissues, including liver, skeletal muscle and cardiac muscle (E. Chang, et al., 2015; H. H. Chang, et al., 2017; C. He, et al., 2012; Hsu, et al., 2016; H. Liu, et al., 2017; X. Xu, Hua, et al., 2013; X. Xu & Ren, 2015; L. Yang, Li, Fu, Calay, & Hotamisligil, 2010). Elevated circulating insulin (an autophagy-inhibitory hormone) is believed be responsible for changes in autophagy genes, including Vps34, Atg12, Atg8, Atg5, Atg7 and Beclin 1 in hepatic cells from high fat diet or ob/ob mice, the effect of which can be reversed by Atg7 adenoviral transfection (Settembre, De Cegli, et al., 2013). A number of signalling pathways including FoxO1 are known to play a key role in the suppressed autophagy present in hyperinsulinemia (K. H. Kim & Lee, 2014; Sinha, et al., 2017). Given the pivotal role of mitochondria in metabolic homeostasis, mitophagy may serve as a mitochondrial clearance mechanism and a target for metabolic stress (Dorn, 2011; Pickles, Vigie, & Youle, 2018). Nix/BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3L)-Atg8-LC3-II complex, phosphatase and PTEN-induced putative kinase 1 (PINK1) and the E3 ligase Parkinson protein-2 (Parkin) and FUN14 domain containing 1 (FundC1) serve as the main effective mitochondrial clearance machineries (Saito & Sadoshima, 2015). For example, phosphorylation of mitochondrial fusion 2 protein (Mfn2) by PINK recruits Parkin to mitochondria, resulting in the ubiquitination and degradation of Mfn2 activity (Saito & Sadoshima, 2015). Increased mitochondrial fission due to Mfn2 degradation is essential to packaging and digestion of autophagosome containing mitochondria. However, the role of mitophagy and other forms of selective autophagy in cardiorenal metabolic syndrome is less clear. We will discuss the regulatory role of autophagy first in cell metabolism, food intake and energy expenditure before dissecting the pathological role of dysregulated autophagy in individual component of the cardiorenal metabolic syndrome.
Table 1:
Examples of autophagy change in individual compoments of the cardiorenal metabolic syndrome
| Change of Autophagy in Insulin Resistance, Diabetes Mellitus and Pancreatic Defect | |||
|---|---|---|---|
| Model and Organ | Autophagy | Autophagy parameters | Reference |
| Dehydroepiandrosterone-indued insulin resistance; Skeletal muslces | Suppressed | Activated mTOR, decreased LC3- II/LC3-I, and increased p62 | X. Song, et al., 2018 |
| Hepatic steatosis in ob/ob mice; Liver | Suppressed | Increased GFP-LC3 puncta, LC3- II/LC3-I and p62, decreased proteolysis | Inami, et al., 2011 |
| High fat diet-induced insulin resistance; Liver |
Suppressed | Decreased RFP-GFP-LC3 puncta,LC3- II/LC3-I and increased p62 | Qian, et al., 2018 |
| High-fat diet-induced insulin resistance; Hypothalamus | Suppressed | Decreased Atg7, Atg5, andLC3-II | (Meng & Cai, 2011) |
| Palmitic acid-induced insulin resistance; Cardiomyocytes | Enhanced | Increased GFP-LC3 puncta, LC3-II, decreased Atg12-Atg5, Atgl6Ll, Atg3 | (S. Li, H. Li, et al., 2017) |
| Insulin resistance and diabetes in high-fat diet fed mice and db/dbmice; Pancreatic ß cells | Enhanced | Increased autophagosome formation by TEM and LC3-II | Ebato, et al., 2008 |
| Type 1 diabetes in streptozotocin-injected mice; Hearts | Suppressed | Decreased LC3-II, At g 12, and the Atg12-Atg5 complex | (Z. Xie, et al., 2011; X. Xu, Kobayashi, et al., 2013) |
| Insulin resistance in fructose-fed mice; Heart |
Enhanced | Increased LC3-II, Beclin 1, andp62 | Mellor, et al., 2011 |
| Type 2 diabetes in human; Heart | Enhanced | Increased LC3-II, Beclin 1, and decreased p62 | Munasinghe,et al., 2016 |
| Type 2 diabetes in high fat-fed and streptozotocin-treated mice; Pancreatic ß cells | Decreased | Decreased Atg5 andLC3-II | (M. Fan, et al., 2018) |
| Type 1 diabetes in streptozotocin-treated mice; Heart | Decreased | Decreased Beclin 1, Atg7, Atg12, LC3- II, and increased p62 | (Xiao, et al., 2018) |
| Change of Autophagy in Obesity and Dyslipidemia | |||
| Obesity in human; Visceral fat | Increased | Increased Atg5 and LC3-II | (Haim, et al., 2015) |
| Obesity, GFP-LC3+-ob/ob mice; Pancreatic islets | Suppressed | Increased GFP-LC3 puncta, LC3- II/LC3-I and p62, decreased proteolysis | (Quan, et al., 2012) |
| Obesity, both ob/ob mice and high-fat diet; Liver | Suppressed | Decreased Atg7, Atg5, Beclinl, LC3-II, and increased p62 | (Inami, et al., 2011; Qian, et al., 2018; L. Yang, et al., 2010) |
| Obesity in human: Omental and subcutaneous adipose tissue | Increased | increased Atg5, LC3-II, and Atg12-Atg5 | (Kovsan, et al., 2011) |
| Change of Autophagy in Hypertension | |||
| Rat PASMCs, Pulmonary arterial hypertension treated with Chloroquine | Suppressed | increased expression of LC3B-ÏÏ and ATG5 mRNA , increased p62 and BMPR-II protein levels | (Long, et al., 2013) |
| Mice, cardiac hypertrophy induced by TAC, heart failure |
Early suppressed, Late upregulated | decreased LC3-II levels, increased LC3- II levels | (A. Nakai, et al., 2007) |
| Hypertension in obese ZDF rat; Mesenteric arterioles | Increased | Increased Atg5, Atg7, Beclin 1, p62 and LC3-II | (Q. Dong, et al., 2017) |
| Stress-induced hypertension in rats; neurons of the rostral ventrolateral medulla | Impaired | Increased LC3-II and p62, and blocked autophagic flux | (Du, et al., 2017) |
a. Autophagy in cell metabolism, food intake and energy expenditure
Autophagy is highly sensitive to changes in micronutrient environment and participates in the regulation of nutrient uptake, cell metabolism and recycling of essential nutrient components (Evans, et al., 2017; Kimmelman & White, 2017; Saxton & Sabatini, 2017). Growth factors, essential amino acids, lipid and glucose promote multiple cell signalling pathways pivotal for metabolic homeostasis, and the alteration of which directly regulates cellular autophagy status (Fig. 3). For example, starvation and reduced energy in the form of ATP/AMP are the best-characterized inducers for autophagy (Lapierre, et al., 2015). In cells relying on glycolysis, glucose deprivation promotes autophagy through AMPK activation (Hardie, 2014). Interrupted glucose metabolism lowers the levels of ATP and certain precursor molecules for biosynthesis of nucleic acids and fatty acids. In metabolic stress with disturbed glucose metabolism, cells rely on mitochondrial oxidation of fatty acids and amino acids as the main energy source, at the expense of production of dangerous ROS species. These changes in lipid and glucose metabolism affect autophagy as deficiency in amino acid turns on autophagy reminiscent of growth factor withdrawal, leading to clearance of protein aggregates and damaged organelles, and recycling of nutrients to promote cell survival (Altman & Rathmell, 2012). The metabolic by-products including free fatty acids, ß-hydroxybutyrate and ammonia may also participate in the regulation of autophagy in the face of cardiorenal metabolic syndrome (Youm, et al, 2015).
Fatty Acids:
Levels of fatty acids are heavily influenced by food intake and energy expenditure. Autophagy dysrégulation is common in dyslipidemia such as fatty liver disease (S. Yan, et al, 2017), suggesting a tie between autophagy and lipid disorders (M. Yang Zhang & Ren, 2018). Saturated and unsaturated fatty acids including palmitate and oleate stimulate autophagy, via distinct signaling mechanisms such as EIF2AK2 (or PKR) and MAPK8 (or JNK1 ) (Shen, et al, 2012). Stearoyl-CoA desaturase (SCD), which converts saturated lipids into monounsaturated lipids, promotes autophagy with a high content of unsaturated fatty acids in autophagy membranes (Paiardi, et al, 2017). SCD1 is required for early autophagosome formation and supplementation of oleate may rescue SCD1 inhibition-induced autophagic anomalies (Ogasawara, et al, 2014). The essential role of lipids in autophagosome formation also received support by the fact that patatin-like phospholipase-domain-containing 5 (PNPLA5) found in lipid droplets serves as autophagy initiation substrates, encompassing degradation of autophagic adaptors, bulk proteolysis, mitochondrial control and microbial clearance (Dupont, et al, 2014). Therefore, lipid droplets may induce autophagy despite being nutrients (Galluzzi, Pietrocola, Levine, & Kroemer, 2014; Khaldoun, et al, 2014). Autophagy induced by fatty acids may capture and deliver lipid droplets into lysosomes for degradation, representing a rather unique way to combat lipotoxicity in lipid derangement. For example, ω−3 fatty acids may provide protective effects against hepatic lipotoxicity through induction of autophagy (Y. Chen, Xu, Yan, Yu, & Li, 2015).
Amino acids:
In the presence of amino acids (e.g, arginine, leucine, and glutamine), mTORC1 is activated by way of an amino acid sensor (e.g, a leucine sensor), amino acyl-tRNA synthetase (e.g, leucyl-tRNA synthetase). With amino acid shortage, mTORC1 is inhibited to promote ULK1 and autophagy. mTORC1 indirectly regulates autophagy through phosphorylation of transcription factor EB (TFEB), which governs lysosome- and autophagy-specific genes transcriptionally. With insufficiency of amino acids, at least five distinct, non-mutually exclusive mechanisms participate in the regulation of autophagy. First, loss of amino acids lead to the buildup of uncharged tRNA species, and activation of eukaryotic translation initiation factor 2α kinase 4 (eIF2AK4) and transcription factor 4 (ATF4) to facilitate autophagy (Galluzzi, et al, 2014). ATF4 is upregulated in response to microenvironmental stresses including amino acid depletion, oxidative and ER stress to favor autophagy (S. Song Tan, Miao, Li, & Zhang 2017). Second, amino acid shortage in lysosomes facilitates the recruitment of mTORC1 to lysosomal surface and subsequently mTORC1 activation (Galluzzi, et al., 2014). Third, the loss of amino acids especially leucine, glutamate and glutamine suppresses intracellular acetyl-CoA (Galluzzi, et al., 2014), a central metabolite for global protein acetylation and autophagy (Webster, Scott, Traba, Han, & Sack, 2014). Forth, loss of amino acids can decrease the essential intermediate metabolite a-ketoglutarate to stimulate autophagy (Galluzzi, et al.,2014). Last but not least, loss of amino acids may turn on the AMPK-TSC2-mTORC1 cascade to promote autophagy. Findings in C. elegans and mammalian cells suggested that AMPK associates and phosphorylâtes ULK1 to engage autophagy in response to deprivation of multinutrient. It is noteworthy that the amino acid catabolic product ammonia may serve as a potent inducer of autophagy, in contrary to the inhib itory effect of autophagy induced by amino acids (Galluzzi, et al, 2014). Such ammonia-induced autophagy seems to be mediated via an AMPK-dependent and ULKl/ULK2/mTORC1-independent manner (Galluzzi, et al., 2014; Harder, Bunkenborg & Andersen, 2014). These amino acid-driven cellular mechanisms orchestra autophagic responses in the face of amino acid shortage.
Acetyl-CoA and NAD+:
As an essential metabolic intermediate denoting energetic state, acetyl-CoA affects enzymatic specificity, metabolism, mitosis (Pietrocola, Galluzzi, Bravo-San Pedro, Madeo, & Kroemer, 2015) and the balance between cellular catabolism and anabolism (Marino, et al, 2014). Pharmacological or genetic interventions to inhibit acetyl-CoA synthesis facilitate autophagy, while replenishment of acetyl-CoA suppresses starvation-induced autophagy (Marino, et al, 2014; Pietrocola, et al., 2015). Activation of acetyl-CoA-carboxylase leads to lipogenesis and obesogenesis (Sinha-Hikim, et al, 2017), coinciding with suppressed autophagy in fatty liver disease and obesity (Madrigal-Matute & Cuervo, 2016; S. Yan, et al, 2017). As the sole donor of acetyl groups for acetyl transferases, acetyl-CoA influences the acetylation profiles of several proteins including histones and regulates autophagy at the post-translational level via acetylation (Duran, et al, 2013; Pietrocola, et al, 2015). On the other hands, nutrient deprivation promotes buildup of NAD+ (oxidized form) at the expense of NADH (reduced form), both essential substrates for metabolic circuitries such as glycolysis, the Krebs cycle, and oxidative phosphorylation. NAD+ stimulates autophagy through histone deacetylases of the sirtuin family (Houtkooper, Pirinen, & Auwerx, 2012). Levels of relative (NAD+/NADH ratio) and absolute NAD+ may be regulated through inhibition of these enzymes or usage of precursors (e.g, nicotinamide, nicotinamide riboside), resulting in autophagy induction via the NAD+-dependent sirtuins (Houtkooper, et al, 2012).
A thorough understanding of organismal homeostasis through balance between food intake and energy expenditure is essential to the development of strategies to combat cardiorenal metabolic diseases. Other than the classical metabolic regulation of glucose, fatty acid and other nutrients, autophagy also participates in the regulation of hematopoietic stem-cell and T cell metabolism to influence overall metabolic state. Autophagy protects hematopoietic stem cells against metabolic stress, which is pivotal to preserve the regenerative capacity of old hematopoietic stem cells (Ho, et al., 2017). Loss of autophagy in hematopoietic stem cells such as in aging and other disease states leads to build-ups of mitochondria and metabolic stress, resulting in accelerated myeloid differentiation and impaired hematopoietic stem-cell renewal and regenerative ability.
b. Autophagy and insulin resistance
Insulin resistance is a cardinal feature of cardiorenal metabolic syndrome with physical inactivity, inflammation, type 2 diabetes mellitus, obesity, hypertension, and cardiovascular disease. Binding of insulin to its receptor stimulates the intrinsic tyrosine kinase activity of the receptor, and phosphorylâtes target proteins (insulin receptor substrates), to turn on two major kinases namely PI3K (metabolic) and MAPK (growth) (Muscogiuri, et al., 2017). Insulin resistance denotes loss of insulin sensitivity at the receptor or post-receptor levels leading to elevated plasma insulin levels (Muscogiuri, et al, 2017). Given the essential role of insulin as the major anabolic hormone in growth, development, glucose, fat, and protein metabolism, insulin resistance is believed to contribute to the early adverse cardiovascular and metabolic events in cardiorenal metabolic syndrome (Jia, Whaley-Connell, & Sowers, 2018; Ren & Anversa, 2015). Insulin resistance is shown to be distinctly governed by autophagy in insulin responsive tissues such as adipose tissues, skeletal muscles, pancreas, liver, and brain (James, O’Neill, & Nair, 2017; R. Singh, et al., 2009). Levels of insulin are controlled by crinophagy - a lysosomal degradation process of secretory granules. Crinophagy is stimulated at low glucose to promote degradation of insulin secretory granules, while the process is reversed at high glucose levels (Muscogiuri, et al., 2017). Crinophagy helps to maintain the balance between glucose and insulin production. On the other side of the coin, insulin levels and insulin sensitivity inhibit autophagy. Hyperinsulinemia may suppress autophagy, while dampened autophagy further inhibits insulin signaling, denoting a reciprocal regulation between the two (K. H. Kim & Lee, 2014; Sinha, et al., 2017). For example, although Atg7 haploinsufficiency does not exhibit metabolic defect, it promotes insulin resistance, lipid overload, and diabetes when crossed with ob/ob mice (Lim, et al., 2014). Findings from ob/ob mice revealed compromised autophagy in association with insulin resistance and ER stress, the effects of which were rescued by Atg7 (L. Yang, et al, 2010). As indicated in Table 1, compromised autophagy was also noted in livers, skeletal muscle and adipose tissues from diet- and dehydroepiandrosterone-induced insulin resistance (X. Li, et al., 2017; Qian, et al., 2018; X. Song et al., 2018). Insulin sensitization through pharmacological intervention or life style modification may improve insulin signaling and metabolic profiles in association with restored autophagy (H. Liu, et at, 2017; Wei, An, Jin, Li, & Xu, 2018; A. Xu & Sweeney, 2015). Although autophagy may protect ß-cell function (Marsh, et al, 2007), excessive autophagy leads to apoptosis and insulin resistance in cardiomyocytes and ß-cells in stress conditions (Fujitani, Ueno, & Watada, 2010; S. Li, H. Li, et al., 2017). It was indicated recently that inactivation of Vps34 PI 3-kinase, an integral autophagy initiating component, is capable of enhancing insulin sensitivity and metabolism through reprogramming of mitochondrial metabolism (Bilanges, et al., 2017). These findings collectively suggest a rather complicated role of autophagy in the regulation of insulin sensitivity.
c. Autophagy, diabetes mellitus and pancreatic defect
Diabetes mellitus is featured by hyperglycemia, oxidative stress and mitochondrial injury, all of which are under the tight regulation of autophagy (Barlow & Thomas, 2015; James, et al, 2017). Autophagy regulates normal function of pancreatic ß cells and insulin-target tissues, such as liver, skeletal muscle, and adipose tissue. With loss of autophagy homeostasis, damaged mitochondria accumulate, lead ing to an overt rise in ROS. Mice deficient in autophagy gene in ß-cells exhibited accumulation of damaged organelles, injured mitochondria, increases in ROS and reduction in glucose-stimulated insulin secretion (Ichimura, Kominami, Tanaka, & Komatsu, 2008; J. J. Wu, et al., 2009). The autophagy deficient mice were prone to the development of diabetes with marked ß-cell loss upon high fat diet challenge (Ebato, et al., 2008). In particular, pancreatic Atg7 knockout triggered impaired glucose tolerance, reduced insulin secretion, hyperglycemia and diabetes with normal or high fat diet intake (Ebato, et al., 2008) (H. S. Jung et al., 2008) or when cross-bred with ob/ob mice (Quan, et al., 2012). As stated above, pancreatic deficiency in Atg4b exhibited limited autophagic competence, robust increase in body weight in response to distinct metabolic challenges, reduced glucose tolerance and attenuated insulin responses. These mice are more vulnerable to experimentally induced diabetes. Interestingly, stimulation of autophagy by spermidine alleviated metabolic derangement (Fernandez, et al., 2017). These findings suggest that autophagy acts as an important protective mechanism for oxidative stress in insulin-target tissues and influences the resilience of the organism to develop diabetes. Likewise, altered autophagy has been implicated in the progression of diabetes through impaired ß-cell function and onset of insulin resistance (James, et al., 2017). Dysregulation of autophagy has been reported in animal models and human subjects of diabetes, likely due to lipid accumulation and inflammasome activation (Barlow & Thomas, 2015; J. Kim, Lim, & Lee, 2018). Although a causative role for ROS is confirmed in diabetes with proven benefit of antioxidants against diabetic complications in animal studies, clinical studies failed to consolidate the efficacy of antioxidant therapy, suggesting that antagonizing existing ROS by antioxidants may not be sufficient to reduce diabetic injury. Thus it is essential to develop a more effective therapeutic strategy for diabetic complications to eliminate defective mitochondria, which would otherwise generate ROS and release pro-death factors continuously. Enhanced autophagy may function as a unique protective mechanism against oxidative stress in diabetes, as novel anti-diabetic agents such as glucagon-like peptide-1 (GLP-1) agonist are shown to specifically target autophagy in diabetes (Arden, 2018). Other agents capable of enhancing autophagy such as imatinib and trehalose have also been shown to improve metabolic profile and ameliorate inflammation in diabetes with systematic autophagy insufficiency (Castillo, et al, 2013; J. Kim, et al., 2018; Lim, et al., 2014; Q. Xie, Lin, Li, & Chen, 2017). It is noteworthy that autophagy induction may also influence diabetes through its regulation of lipid accumulation in adipose tissues, liver and muscles to affect insulin resistance and pancreatic function (Barlow & Thomas, 2015; Sinha, et al, 2017).
d. Autophagy, obesogenesis, adiposity, adipose development and differentiation
Obesity is a global health issue attributed to the interplay between genetics and environment. Adipose tissues play a central role in obesogenesis in paracrine or endocrine fashion through pro-inflammatory adipokines including leptin, resistin, TNF-α and adiponectin (Mandviwala, Khalid, & Deswal, 2016). These adipokines elicit unfavourable metabolic responses such as cardiac depression, vascular reactivity, proteinuria, inflammation (e.g., interleukins and monocyte chemotactic protein 1) and coagulation (e.g., PAI), contributing to cardiovascular dysfunctions (Mandviwala, et al, 2016). When energy supplies exceed the storage capacity of adipocytes under metabolic stress, adipocyte hyperplasia and hypertrophy occur. Adiposity or obesity promotes cardiovascular anomalies including cardiac remodeling, heart failure, hypertension and a spectrum of non-communicable diseases (Oga & Eseyin, 2016). The relative risk for a 10-cm rise in waist circumference and a 0.1-unit increase in waist-to-hip ratio are both 1.29 (Aune, et al., 2016). Recent evidence has depicted a rather important role for autophagy and lysosomal function in adiposity and organ complications (Jacob, et al., 2017; Sinha-Hikim, et al., 2017; Sinha, et al., 2017), coinciding with the close tie between autophagy and metabolic disorders (Maixner, et al., 2016). Genetic deletion of Atg7 presented increased lipid content when crossed with ob/ob mice (Lim, et al, 2014). Other studies noted the phenotype of adiposity, obesity and insulin resistance with deletion of Bec lin 2 and Atg4b (Fernandez, et al., 2017; C. He, et al., 2013). This is supported by the important regulatory role for autophagy in adipose tissue differentiation, or adipogenesis, which demands cytoplasmic reorganization in mitochondria (Jialal & Devaraj, 2018). Pharmacological inhibition of autophagy in human adipose tissue expiants promoted pro-inflammatory cytokine secretion (Maixner, et al., 2016). Along the same line, upregulation of autophagy in obese Wistar Ottawa Karlsburg W (RT1u) rats contributes to the regulation of visceral adipose tissue function and the balance between pro-inflammatory and protective adipokines (Kosacka, et al, 2018). These data indicate a retraining role for adipose autophagy in adipose tissue inflammation. Nonetheless, activated adipose autophagy contributes to pathogenesis of obesity complication, reminiscent of its role in chronic lung disease (Maixner, et al., 2016). Very recent evidence suggested that suppression of autophagy may induce brown-like adipocyte formation in adipocytes and adipose tissues, a process deemed beneficial for obesity treatment (Leu, et al., 2018). Intriguingly, siRNA-mediated Atg7 knockdown and pharmacological inhibition of autophagy in adipocytes or obese adipose tissues displayed beneficial effects in adiponectin secretion (Slutsky, et al., 2016). Thus, activated adipose autophagy noted in obesity might contribute to the endocrine defect of adipose tissue, linking obesity to its comorbidities. These observations are in line with the clinical findings that elevated adipose tissue autophagy is associated with a high-risk obesity phenotype (visceral fat, insulin resistance) (Maixner, et al., 2016). Recent evidence has supported a key role for transcriptional factors in autophagy-associated adiposity regulation. Transcriptional regulation of E2F1 in visceral (omental) adipose tissue are considered possible route for cardiorenal metabolic risks, including higher circulating free fatty acids, interleukin 6 (IL-6), insulin resistance, and low adiponectin levels. This association disappeared following adjustment to autophagy, indicating a compelling role for autophagy in adipose tissue E2F1-regulated cardiorenal metabolic morbidities. More importantly, E2F1-null adipocytes were less lipolytic, tended to respond better to insulin, secreted less leptin and more adiponectin, and, importantly, were more resilient to pro-inflammatory cytokines (Haim, et al., 2015). Transactivation of Atg4b by CCAAT/enhancer-binding protein ß (C/EBPß) was also reported to promote autophagy, leading to facilitated adipogenesis (L. Guo, et al., 2013).
The role of autophagy alterations in adipose tissue non-adipocyte cells is less clear. Bone marrow-derived macrophages from high fat-fed mice exhibited attenuated autophagy (K. Liu, et al, 2015) although its correlation with lipid handling remains unknown. Lysosomal biogenesis was suggested to participate in lipid mobilization in adipose tissue macrophages in obesity (X. Xu, Grijalva, et al, 2013). In plaque, where macrophages accumulate lipid largely by engulfing modified LDL, autophagy may be required for reverse cholesterol transport (Ouimet, et al., 2011) and is therefore anti-atherogenic. This would be consistent with lipid droplet autophagy (lipophagy) as a mechanism for esterified lipid degradation (Rambold, Cohen, & Lippincott-Schwartz, 2015). The functional role of autophagy in adipose tissue macrophages, which accumulate lipids by either phagocytosis of adipocyte remnants or de novo lipogenesis, remains unclear. In addition, given the potential bidirectional links between autophagy and inflammation, it is intriguing to explore the cell-autonomous immune-metabolic contribution to adipose anomalies once defective autophagy is confirmed in obese adipose tissue immune cells. One of the most commonly considered dogma in immune cells is the link between attenuated autophagy and activated inflammation. It is intriguing to assess such concept in in adipose tissue immune cells or, conversely, that activated autophagy in adipose tissue cells acts to restrain “excessive” inflammatory response (Jansen, et al., 2012). Furthermore, it would be interesting to assess whether this involves transcriptional mechanisms and/or altered immune cell metabolism.
e. Autophagy and NAFLD (non-alcoholic fatty liver disease)
NAFLD is characterized by hepatic steatosis and is a manifestation associated with obesity, insulin resistance and cardiorenal metabolic syndrome, with no perceived cause for secondary hepatic fat accumulation such as alcohol intake, use of steatogenic medication, or hereditary disorders (De Meyer, et al., 2015; Shimano & Sato, 2017). Recent evidence has depicted a role for autophagy in the molecular mechanisms behind the etiology of NAFLD (Evans, et al., 2017; S. Li, X. Dou, et al., 2017; Sinha, Singh, & Yen, 2018). Deletion of autophagy genes, including Atg7, Vps34 and TFEB, exhibited increased hepatic lipid content, liver and body weight while overexpression of Atg7 improved hepatic insulin sensitivity (Jaber, et al, 2012; Settembre, De Cegli, et al., 2013; R. Singh, et al., 2009). Autophagy stimulates lipid metabolism and degradation of lipid stores (lipophagy), offering a new avenue for lipid degradation in addition to the classical cytosolic lipases (J. Liu & Debnath, 2016). This is evidenced by the beneficial effect of autophagy inducers on hepatic and circulating triglycerides or vice versa, lipid reducing agents such as ezetimibe with autophagy inducing properties (S. H. Kim, et al, 2017). The beneficial role for autophagy in NAFLD also received convincing support from the beneficial effect of thyroid hormones on hepatic lipid metabolism. Thyroid hormones are important regulators for lipid metabolism and promote fatty acid oxidation through hepatic autophagy (Mao, Yu, Wang, Guo, & Fan, 2016; Sinha, et al, 2018). The therapeutic promise autophagy is also implicated by its utility in other hepatic diseases including alcoholic liver disease, hepatocellular carcinoma, α1-antitrypsin deficiency, and viral hepatitis (Madrigal-Matute & Cuervo, 2016; Sinha, et al, 2018). Last but not least, it should be noted that controversy exists with regards to the precise role of autophagy in hepatic lipid metabolism and NAFLD, as improved lipid metabolism (reduced lipid content and improved insulin sensitivity) were also reported with hepatic deletion of autophagy genes including FIP200 and Atg7 (K. H. Kim, et al, 2013; D. Ma, et al, 2013; Shibata, et al, 2009). A number of scenarios may be responsible for the apparent discrepancies between autophagy and hepatic lipid metabolism including the necessity for autophagy in lipid droplet formation and induction of protective mitokine (such as Fgf21) with deficiency of certain autophagy gene.
f. Autophagy and hypertension
Hypertension is an independent risk factor for cardiovascular and metabolic disease events (Go, et al,, 2014; Nwankwo, Yoon, Burt, & Gu, 2013). Most major guideline committees recommend that hypertension be defined as a systolic blood pressure is ≥ 140 mm Hg or diastolic blood pressure is ≥ 90 mmHg, or both, on repeated examination. Autophagy, as a catabolic mechanism, maintains endothelial cell alignment, vessel wall biology and atheroprotection, the dysrégulation of which may be a common mechanism for vascular injury and associated pathologies (Nussenzweig, Verma, & Finkel, 2015; Vion, et al, 2017). Evidence support that autophagic vacuoles are significantly reduced quickly after low-dosed isoproterenol administration (Pfeifer, Föhr, Wilhelm, & Dämmrich, 1987). Endothelial autophagy helps to limit atherosclerotic plaque formation by retarding apoptosis, senescence, and inflammation in endothelial cells (Vion, et al., 2017). Using genetically-engineered mice of TFEB, a master gene for autophagy and lysosome biogenesis, Fan and colleagues revealed that TFEB governs angiogenesis through activation of AMPKα and autophagy (Y. Fan, et al, 2018). More evidence has linked autophagy to a wider cascade of vascular processes including calcification of vessel wall, atherosclerosis and pulmonary hypertension (Nussenzweig, et al., 2015; Vion, et al., 2017). Furthermore, suppression of autophagy has also been observed pressure overload-induced cardiac dysfunction (Atsuko Nakai, et al., 2007). It is believed that a transitory suppression of autophagic activity may serve as an early contribution to the adaptive increase in myocardial workload. A recent genome-wide association (GWAS) study involving 5205 hypertensive patients and 5320 controls revealed a common variant in the damage-regulated autophagy modulator locus associated with hypertension (Larsson, et al., 2013). In general, autophagy is deemed to confer resistance against arterial or pulmonary hypertension (Lee, et al, 2011). Paradoxically, increased autophagy possibly via oxidative stress was believed to be responsible for pulmonary arterial hypertension-induced ventricular hypertrophy and diastolic heart failure (Rawat, et al., 2014). Currently, direct evidence for autophagy-regulated blood pressure is still lacking while a consensus seems to suggest a paracrine regulation of vasoactive substances in endothelium by autophagy. Further study is needed to discern the role for autophagy in vascular biology and blood pressure regulation, and the emerging strategies to target autophagy process for therapeutic benefit.
g. Autophagy and renal injury
Over nutrition and the development of obesity, insulin resistance with the compensatory hyperinsulinemia, elevations in systolic blood pressure with hypertension and dyslipidemia are metabolic disorders that also affect kidney function (Jia & Sowers, 2015; Kimura, Isaka, & Yoshimori, 2017; Sorop, et al, 2017; Whaley-Connell & Sowers, 2017). The mechanisms are complex but generally involve increases in intraglomerular pressure, hyperfiltration, and alterations in tubuloglomerular feedback, which are hemodynamic in origin, but also hypertrophic, proliferative, and inflammatory and oxidative pathways that govern extracellular matrix deposition and fibrosis formation. These cellular metabolic responses govern the onset and development of kidney disease. In this context, much of the work done to investigate the metabolic origins of kidney disease have been done in experimental models of obese, insulin resistant, or diabetic rodents. Several studies in diabetic models have demonstrated that activation of mTOR as well as inaction of AMPK downregulate autophagy in the kidney and specifically in the proximal tubule which governs to a great extent the absorptive, lysosomal capacity of the kidney (Serra, et al., 2010). In this context, a recent group used a proximal tubule specific autophagy deficient mouse line with Atg5 gene deletion to reverse the effects of a high fat diet on proteinuria and kidney dysfunction (Yamahara, et al., 2013). Within this study, autophagy mediated the extent of injury through mTOR. In a similar autophagy deficient line, the loss of Atg5 mediated hypertrophy of the proximal tubule and fibrosis with accumulation of cytosolic amorphous material (Kimura, et al., 2011), these findings were augmented in response to ischemia-reperfusion injury and supported that autophagy regulates to a great extent proximal tubule function. Another line of evidence suggests that autophagy mediates to a great extent immune function in the kidney. In autophagy deficient cell lines, the calcineurin inhibitor cyclosporine mediates mitochondrial function, ROS formation and TGF-ß production (Kimura, et al., 2013; Serkova, Klawitter, & Niemann, 2003).
h. Autophagy, ischemia heart disease and cardiorenal metabolic syndrome-associated cardiac anomalies
Autophagy plays an essential role in the maintenance of cardiac geometry and function (Delbridge, Mellor, Taylor, & Gottlieb, 2017). Evidence of autophagy in human heart disease was first reported in patients with dilated cardiomyopathy (Shimomura, et al., 2001). A role for dysregulated autophagy was later confirmed in various heart diseases including ischemic and cardiorenal metabolic syndrome-related heart anomalies (Delbridge, et al., 2017). Analysis of autophagy gene fingerprint in human ischemia-reperfusion injury revealed elevated autophagic activity (increased LC3-I levels and LC3-II/LC3-I ratios) (K. K. Singh, et al., 2014). On the other hand, autophagy activity is reduced and lysosomal accumulation is increased in peripheral leukocytes of myocardial infarction patients (G. Wu, et al, 2011 ), denoting the complexity and tissue specificity in autophagy following ischemia-reperfusion injury. More studies revealed a double-edged sword for autophagy in myocardial ischemia-reperfusion injury. Data from our lab and others depicted a myocardial protective role for autophagy in ischemia phase. However, during reperfusion, autophagy is deemed detrimental and promotes excessive myocardial cell death (H. Ma, et al., 2011; Sciarretta, Maejima, Zablocki, & Sadoshima, 2018). Further data from our group depicted disparate roles of AMPK and Akt in the regulation of autophagy during ischemia and reperfusion phases, respectively (H. Ma, et al, 2011). These data suggested the therapeutic potential of autophagy in the management of ischemic heart diseases.
Onset and development of cardiac remodeling and contractile dysfunction are common in cardiorenal metabolic syndrome with data supporting a role for autophagy in the pathology of cardiac anomalies. Using an atherogenic diet-induced Ossabaw swine model of cardiorenal metabolic syndrome (obesity, dyslipidemia, and insulin resistance), increased cardiac output and lowered myocardial perfusion were noted with increased protein levels of Atg5 but decreased ULK1, Beclin1 and the LC3BII/I ratio (Z. L. Li, et al., 2012). In another study using high fat, high cholesterol diet-fed hypercholesterolemic swine, activated upstream and downstream mTOR signaling, suppressed autophagy, and overt cardiac hypertrophy were noted, confirming a role of mTOR in cardiac autophagy dysrégulation (Glazer, Osipov, Clements, Sellke, & Bianchi, 2009). Using a 12-week 60% high fat diet feeding regimen, mice developed cardiorenal metabolic syndrome including obesity, hyperglycemia, hyperinsulinemia and dyslipidemia. Cardiac function was suppressed although autophagy was unaffected. Interestingly, a carbon monoxidereleasing molecule (CORM-3) promoted cardiac autophagy and improved cardiac function without affecting metabolic syndrome (Lancel, et al., 2012). Along the same line, caloric restriction improve myocardial autophagy and metabolic syndrome in mice fed 58% high fat diet for ten weeks, which prompted obesity insulin resistance and dyslipidemia (Cui, Yu, Wang, Gao, & Li, 2013). Data from our own group suggested that a 45% high fat diet intake for 20 weeks led to obesity, glucose intolerance, dyslipidemia, and insulin resistance. Heart function was compromised in these high fat diet-fed mice including reduced cardiac output and fractional shortening, cardiomyocyte contractile function and intracellular Ca2+ handling (X. Xu, Hua, et al., 2013). Our data further revealed apparently normal autophagy initiation along with suppressed autophagolysosome degradation (autophagy flux) in hearts from these fat diet-fed mice. In general, it is believed that abrogating autophagy defects by modulating autophagy signaling pathways or direct autophagy induction may help to rescue cardiac dysfunction in cardiorenal metabolic syndrome (F. Wang Jia, & Rodrigues, 2017). At this point, the precise role of autophagy is still controversial as different levels or even same levels of autophagy may exert distinct effects on the myocardium at a given disease stage. To this end, determining the precise role of autophagy in ischemia and cardiorenal metabolic disease-associated heart dysfunction should help to further understand disease pathogenesis and identify novel strategies for the optimal management of heart disease.
i. Autophagy and diabetic cardiomyopathy
Diabetic cardiomyopathy refers to a heart muscle disease independent of macro- and micro-vascular pathology, and contributes to the high risk of heart failure and high mortality in diabetic patients (Jia, Hill, & Sowers, 2018). Recent evidence has indicated altered autophagy in both type 1 and type 2 diabetic hearts. In particular, cardiac autophagy and autophagic flux were suppressed in type 1 diabetes mellitus such as streptozotocin and OVE26 mice, a genetic model of type 1 diabetes (Z. Xie, et al., 2011; X. Xu, Kobayashi, et al., 2013). Reduced levels of Atg12, the Atg12-Atg5 complex and AMPK activity may contribute to decreased autophagosome formation and autophagy in these typet diabetic models. Paradoxically, type 1 diabetes-induced cardiac injury was overtly attenuated in Beclin 1- and Atg16-deficient mice, and was exacerbated by beclin 1 overexpression in the same model of type 1 diabetes, favoring the notion of an adaptive response for decreased autophagy in type 1 diabetes (X. Xu, Kobayashi, et at., 2013). Given that autophagy favors this autoimmune process (Fierabracci, 2014), decreased autophagic activity in the pancreas might protect ß cells, to limit diabetes progression and cardiac damage.
Genetic ablation of Atg7 in pancreatic ß cells promoted islet degeneration, impaired glucose tolerance and insulin secretion (Ebato, et al., 2008; Fujitani, Ebato, Uchida, Kawamori, & Watada, 2009), denoting a role for autophagy in preserving normal cellular architecture and function. However, myocardial autophagic flux was reported to be increased in fructose diet-induced type 2 diabetes, leading to pathologic remodeling of the heart (Mellor, Bell, Young, Ritchie, & Delbridge, 2011). This is consistent with the upregulation of autophagy in human type 2 diabetic pancreatic ß cells (Masini, et al., 2009). Munasinghe and colleagues recently showed that excessive autophagy may trigger loss of myocardial cells and compromised cardiac function (Munasinghe, et al., 2016). Nonetheless, cardiomyocytes isolated from db/db type 2 diabetic mice and high fat diet-induced obese mice displayed decreased autophagic activity (Sciarretta, et al., 2012). This is in line with the observation of decreased cardiac Atg5 albeit no change in LC3B) in a type 2 diabetic patient cohort (Montaigne, et al., 2014).
V. Intervention Targeting Autophagy for the Management of Cardiorenal Metabolic Diseases
a. Pharmacological intervention
The clinical guidelines for management of the cardiorenal metabolic syndrome target individual components of the syndrome to achieve cardiovascular benefits (Rask Larsen, et al., 2018). The main objective of cardiorenal metabolic therapy includes management of dyslipidemia, hypertension, glucose metabolism and obesity. Currently, the first-line pharmacological therapeutics for the cardiorenal metabolic syndrome are statins for dyslipidemia, renin-angiotensin-aldosterone system (RAAS) inhibitors for hypertension, metformin or sodium/glucose cotransporter 2 (SGLT) inhibitors or glucagon-like peptide 1 receptor agonists (GLP-1RAs) for glucose intolerance, and the GLP-1RA liraglutide for weight control (Rask Larsen, et al., 2018). Ample preclinical evidence has depicted the promises of autophagy modulators such as mTOR inhibitors, imatinib and trehalose in the management of various metabolic and cardiovascular diseases (Castillo, et al., 2013; Lim, et al., 2014; Q. Xie, et al., 2017; Y. Zhang, et al., 2018). Natural products from plants may also exert beneficial effect in metabolic tissues through enhancing autophagy (Fan, et al., 2017). As a key catabolic process for digestion and removal of cytoplasmic components, modulating autophagic activity, through targeting regulatory players in the core autophagy machinery, may affect disease processes (Y. Zhang et al., 2018). However, most of approved usage of autophagy modulators is for cancer therapy (Marinkovic, Sprung Buljubasic, & Novak, 2018). Targeting autophagy for the FDA approved drugs for cardiorenal metabolic syndrome remains merely associative at this stage. Several double-blind, placebo-controlled randomized trials mandated by the US Food and Drug Administration (FDA) examined cardiovascular safety of anti-diabetic and other metabolic agents in patients with type 2 diabetes. A role for autophagy induction has been indicated for the FDA-approved insulin sensitizer metformin in the management of pre-diabetes and diabetes (Jiang et at, 2014; Kalender, et at, 2010). Another class of insulin sensitizers, the PPAR-γ agonist thiazolidinediones, has also been shown to either stimulate (S. Yan, Yang Chen, Xi, & Jiang 2014) or suppress (H. Li, Zhang Yang & Wang 2017; Yao, Zheng & Zhang 2015) autophagy although the permissive role of autophagy regulation has not been validated for thiazolidinediones in metabolic diseases. More autophagy inducers such as rapamycin, trehalose and resveratrol may also benefit insulin resistance through improved insulin sensitivity, ß-cell fonction, and diabetic phenotypes, as indicated in Table 2 (Lim, et al., 2014; Rocchi & He, 2015).
Table 2:
FDA approved drugs with a known indication on metabolism that may taget autophagy
| Drug Name | Primary Mechanisms of Action | Indication | Reference |
|---|---|---|---|
| Carbamazepine | Reduces inositol and Ins(l,4,5)P3 levels | Liver disease, dwarfism | (Hidvegi, et al., 2010; C. W. Lin, et al., 2013; Mullan, et al, 2017) |
| Chloroquine | Blocks lysosome-autophagosome fusion and lysosomal protein degradation |
Rheumatoid arthritis and autoimmune diseases, cancer? | (Morgan, ét al., 2014; Parkhitko, et al, 2011) |
| Empagliflozin | SGLT2 inhibitor, AMPK- dependent inhibition of mitochondrial fission |
Diabetes | (Zhou, étal., 2018) |
| Erlotinib hydrochloride |
Inhibits the PI3K-AKT-mTOR signaling pathway |
Non-small cell lung Cancer | (Gorzalczany, et al., 2011; W. Han, et al., 2011) |
| Everolimus | Inhibits mTOR signaling | Atherosclerosis, myocardial information and cardiac remodeling | (Martinet, ét al., 2014) |
| Ezetimibe | Cholesterol absorption inhibitor | Steatohepatitis | (S. H. Kim, ét al., 2017) |
| Ghrelin | Inhibits mTOR signaling | Diabetes, liver disease | (Chung, Choi, & Park, 2018; Ezquerro, Fruhbeck, & Rodriguez, 2017) |
| Liraglutide | Inhibits mTOR signaling | Diabetes, liver disease | (Q. He, et al., 2016) |
| Metformin | Upregulates AMPK | Type 2 diabetes | (Jiang, ét al., 2014); (Kalender, et al , 2010) |
| Pioglitazone | Inhibits mTOR signaling | Type 2 diabetes, obesity, insulin resistance | (Matsuura, et al., 2015) |
| Rapamycin (and Rapalogue, CCI-779) |
Inhibits mTORCl | Cancer, Metabolic disease | (Majumder, et al., 2004; Morgan-Bathke, ét al., 2014) |
| Resveratrol | Activates sirtuin 1 and inhibits ribosomal protein S6 kinase |
Cancer, Metabolic disease | (Armour, ét al., 2009; Opipari, et al, 2004) |
| Rilmenidine | Reduces cAMP levels | Huntington’s disease | (F. Lin & Qin, 2013; Rose, ét al., 2010) |
| Trehalose | Increase cardiac | Myocardial Infarction | (Sciarretta, Yee, et al., 2018) |
| LC3-II levels, induces autophagy | |||
| Sodium valproate | Reduces inositol and Ins(1,4,5)P3 levels | Neurodegenerative diseases | (Sarkar, et al., 2005) |
| Sorafenib | Inhibits mTOR signaling | Cancers | (Prieto-Dominguez, et al., 2016) |
| Statins | Inhibits geranylgeranyl biosynthesis and activates AMPK | Prostate cancer, squamous cell carcinomas | (L. Ma, Niknejad, Gorn-Hondermann, Dayekh, & Dimitroulakos, 2012) |
| Trastuzumab | Anti-HER2 antibody (inhibition of HER2, mTOR | Breast cancer, cardiotoxic agent | (Mohan, Shen, Endo, ElZarrad, & Wu, 2016) |
| Troglitazone | Induces autophagy, apoptosis and necroptosis | Bladder cancer | (S. Yan, et al., 2014) |
| Rosiglitazone | Reduces autophagy | Traumatic spinal cord injury, traumatic brain injury | (H. Li, et al., 2017; Yao, et al., 2015) |
| Sitagliptin | Augments or restores autophagy | Diabetic cardiovascular complications | (Dai, et al., 2018; Murase, et al., 2015) |
| MK-626 | Enhances autophagy and restores insulin secretion | High fat diet-induced obesity | (L. Liu, et al., 2016) |
| GLP-1 analogue | Induces autophagy; inhibits excessive autophagy | Hepatic lipid accumulation; obesity-related glomerulopathy | (H. Guo, et al., 2018; Q. He, et al., 2016) |
Incretin-based therapies exhibit some promises in the treatment of cardiorenal metabolic diseases. Dipeptidyl peptidase-4 (DPP-4) inhibitors suppress the breakdown of GLP-1 and improve glucose and metabolic control in diabetic and/or obese patients (Scheen, 2018). Recent evidence suggested that DPP-4 inhibitors such as sitagliptin may exert its therapeutic effect against diabetic cardiovascular complications through augmenting autophagy in an AMPK-ULK1 dependent manner (Dai, et al., 2018; Murase, et at, 2015). DPP-4 inhibitor MK-626 was reported to restore insulin secretion via autophagy induction in high fat diet-induced mice (L. Liu, Liu, & Yu, 2016). It is noteworthy that the cardiovascular effects of DPP-4 inhibitors remain controversial with inconsistent findings in clinical trials (Fadini, et al., 2017; Verma, et at, 2017). DPP-4 inhibitors such as saxagliptin exhibit a poor cardiovascular profile with high prevalence of hospitalization for heart failure. Sitagliptin displayed little effect on the risk of heart failure although a recent randomized controlled trial Sitagliptin Preventive Study of Intima-Media Thickness Evaluation (SPIKE) suggested that sitagliptin retarded the progression of carotid intima-media thickness in diabetic patients compared with conventional treatment (Fadini, et al., 2017). More evidence has indicated a role for autophagy regulation in GLP-1-offered cardiorenal metabolic benefits. GLP-1 may exert beneficial effect on glucose and lipid homeostasis through autophagy-dependent mechanism in pancreatic ß-cells, hépatocytes and other cell types (Arden, 2018; Q. He, Sha, Sun, Zhang, & Dong, 2016). Interestingly, GLP-1 treatment was also reported to improve diabetic retinopathy through alleviation of autophagy in a GLP-1 receptor-ERK1/2-HDAC-dependent manner. Likewise, GLP-1 analogue was found to prevent obesity-associated glomerulopathy by inhibiting excessive autophagy in podocytes (H. Guo, et al., 2018). It should be noted the heterogeneity of GLP-l-offered cardiovascular benefit. For example, Liraglutide and semaglutide seem to lower the risk of composite cardiovascular outcomes whereas lixisenatide displays no reduction or increase in cardiovascular risk (Scheen, 2018). The SGLT2 inhibitor agents, are a new class of agents that reduces blood pressure, weight, visceral adiposity, hyperinsulinemia, albuminuria, and oxidative stress in patients with type 2 diabetes and metabolic diseases (Secrest, Udell, & Filion, 2017). In particular, SGLT2 inhibitors reduce body weight from 2.9 kg for monotherapy to 4.7 kg when used in combination with metformin. In the completed clinical trials to assess SGLT2 inhibitors, empagliflozin and canagliflozin displayed reduced risk of composite cardiovascular endpoints (Ford, et al., 2007; Secrest, et al., 2017). Recent findings from our own lab suggested that empagliflozin rescued myocardial microvascular injury in diabetes via AMPK-mediated inhibition of mitochondrial fission (Zhou, et al., 2018) although direct impact on mitophagy by SGLT-2 inhibitors remains to be explored.
Other therapies:
Lipid lowering drugs, ß-adrenergic blockers and RAAS inhibitors have all been reported to offer beneficial effect against oxidative stress, inflammation, insulin resistance, and progression of cardiorenal metabolic outcomes. Statins, aspirins, ß-blockers, ACE inhibitors (ACEI), and angiotensin II receptor blockers (ARB) have all exhibited benefits on reducing the cardiovascular mortality due to proper risk management including myocardial infarction, stroke and hypertension (Rask Larsen, et al., 2018). It was estimated that 44% of the decrease in cardiovascular mortality between 1980 and 2000 was due to risk factor modification with 24% attributable to reduction in cholesterol and 20% attributable to reduction in blood pressure (Ford, et al., 2007). Although autophagy regulation is not considered the main mechanism of action for any of these agents, the ability to modulate autophagy may have contributed to the beneficial action of these pharmacological agents in the maintenance of cellular homeostasis and cardiorenal metabolic function. For example, oral or stent-based delivery of the autophagy inducers such as rapamycin, rapalogs or everolimus possess pleiotropic anti-atherosclerotic effects through control or retardation of atherosclerotic growth and destabilization of atherosclerotic plaques (Martinet, De Loof, & De Meyer, 2014). It was also suggested that ACEI or ARB rescued impaired autophagy in high glucose-induced endothelial apoptosis (F. Chen, et al., 2014). It is of note that regulation on autophagy may only provide add-on benefit for these agents in the management of cardiorenal metabolic anomalies beyond their well-characterized mechanism of action. Most recent studies reported a role for micro RNAs in cardiorenal metabolic diseases. For example, miR199a-5p was reported to suppress hepatic insulin sensitivity via inhibition of Atgl4-mediated autophagy (B. Li, et al., 2018). Dubinsky and colleagues identified let-7 miRNAas an amino acid sensor to prevent mTORCl activation and trigger autophagy (Dubinsky, et al., 2014). More evidence also suggested increased levels of miRNA-9 and miRNA-124a in obese subjects and overexpression of these micro RNAs confers lipid accumulation in adipose tissues (Mentzel et al., 2015). Thus, identification of the roles of the different miRNAs may provide novel therapeutic targets for prevention of metabolic diseases in future.
b. Life style modification, exercise, cessation of smoking.
Although available information is still controversial with regards to the role of autophagy in the pathogenesis of metabolic diseases, autophagy regulation is increasingly appreciated for the control of metabolic and cardiovascular homeostasis (R. Guo, et al., 2013; Maixner, et al., 2016). However, inconsistent data were using various animal species and strain with regards to the role of autophagy in cardiorenal metabolic syndrome. In particular, effective and specific pharmacological regulators for autophagy still remain somewhat limited, making it pertinent to search for potent non-pharmacological measures to regulate autophagy. Indeed, the changes in autophagy in cardiorenal metabolic syndrome were found to be heavily influenced by many factors, including age, gender, and life style factors. Ample evidence has suggested that caloric restriction, weight loss, regular physical exercise and smoking cessation can positively modify metabolic abnormalities, improve systemic and tissue insulin sensitivity, and lessen the progression of CVD (Ryter, Koo, & Choi, 2014). The Diabetes Prevention Program regimen showed that therapeutic lifestyle changes (TLCs) such as dietary modifications and physical activity had a far better impact than pharmacological options. For example, exercise as part of lifestyle intervention can prevent impairment of cardiac function and increase LV ejection fraction by 2% to 5% in patients with obesity and type 2 diabetes (Jia, Jia, & Sowers, 2016). In a recent clinical trial involving 100 obese older patients with stable heart failure and preserved ejection fraction, caloric restriction diet and aerobic exercise training, both known facilitators of autophagy, were reported to enhance peak oxygen consumption, and the effects may be additive (Kitzman, et al., 2016). Physical exercise is deemed a physiological stimulus for metabolic adaptations to benefit human health. Although the beneficial responses of exercise were traditionally credited to the biosynthesis of metabolic proteins and organelles, recycling of cellular components by autophagy has recently emerged as a unique mechanism in the adaptive responses to exercise (Halling & Pilegaard, 2017). Exercise may turn on BCL2-mediated autophagy, thus contributing to the beneficial metabolic effects of exercise against high fat diet intake (C. He, et al., 2012). It is perceived that a biphasic autophagy response may be mobilized during exercise, resulting in both an acute induction and a long-term potentiation of autophagy. A number of transcriptional paradigm have been indicated in exercise-facilitated autophagy responses including Forkhead box O (Foxo), TFEB, p53 and PGC-1α, to fuel sustained autophagic activity (Vainshtein & Hood, 2016). More recent study indicated that aerobic exercise training improves the quality of life, cardiorespiratory fitness, and cardiorenal metabolic profile in overweight/obese women with polycystic ovary syndrome (PCOS) (Brennan, et al., 2017; Costa, et al., 2018). PCOS is associated with excess weight gain and multiple pregnancy complications, including preeclampsia, gestational diabetes, and large babies, due to hypothalamic-pituitary-ovarian (HPO) defect-induced endocrine abnormalities (e.g., hyperandrogenism and hyperinsulinemia) (Bani Mohammad & Majdi Seghinsara, 2017). Both elevated (D. Li, et al., 2018) and suppressed (Su, et al., 2017) autophagy were noted in PCOS depending on models or timing of PCOS development. Although direct evidence for autophagy in lifestyle modification (control of weight gain)-offered first-line treatment for PCOS (Brennan, et al., 2017) is still lacking, exercise-associated cellular machineries such as activation of AMPK and autophagy have been confirmed to rescue against disrupted implantation process in PCOS due to uterine defects (Y. Zhang, et al., 2017). These findings suggest potential therapeutic usefulness of metformin and autophagy in PCOS patients with uterine defect. More in depth work is needed to consolidate the precise interplay between physical exercise and autophagy in metabolic disorders such as PCOS. More evidence also depicted a role for autophagy in cigarette smoking-induced chronic anomalies (G. Wang, et al., 2017), denoting the therapeutic promises of targeting autophagy in smoking-induced chronic diseases such as pulmonary diseases although further study is needed for metabolic disorders. Perhaps more intriguingly, physical activity, but not dietary intake, alleviates the FTO polymorphism-induced metabolic anomalies in humans (Petkeviciene, et al., 2016), suggesting a gene-environment interaction. Thus, a better understanding of the role for autophagy in life style modification-induced benefit against metabolic disorders should shed some lights towards a better management of the cardiorenal metabolic syndrome. It is noteworthy that bariatric surgery is also regarded as one of the most effective strategies to control obesity and CVD since bariatric surgery can reduce body fat and offer beneficial effects on maintaining normal adipocyte and adipose tissue function lessening dyslipidemia, insulin resistance, dysglycemic hypertension, and associated CVD (Jia, et al., 2016).
c. Epigenetic modulation
Autophagy is primarily a cytoplasmic event to remove damaged and long-lived materials in stressed conditions. However, recent evidence has depicted occurrence of transcriptional and epigenetic regulation of autophagy within the nucleus (Baek & Kim, 2017; Fullgrabe, Klionsky, & Joseph, 2014). Epigenetic DNA modifications [chromatin remodeling, DNA méthylation (at the 5’-position of cytosine residues within CpG dinucleotides), histone modifications (acetylation, méthylation, phosphorylation, ubiquitination, sumoylation) and small noncoding RNA] develop in response to environmental factors to promote phenotypic changes of metabolic traits (Kunes, et al., 2015). These gene-environment interplays converge at the metabolic-epigenome-genome axis to determine the metabolic traits. Although the overall risk of metabolic diseases can be largely attributed to poor-/over-nutrition, unhealthy life style, drinking and smoking in adult life, evidence has suggested that predisposition of metabolic anomalies start early in life, such as in fetal and perinatal periods through modifiable epigenetic changes (Smith & Ryckman, 2015). These metabolic changes (e.g., the “thrifty phenotype”) are visible not only in individuals exposed to environmental factors but also in subsequent progeny for multiple generations, a phenomenon termed as transgenerational inheritance. In particular, maternal undernutrition or overnutrition affects covalent modifications of fetal genome and its associated histones that are carried forward to subsequent generations for risk of cardiorenal metabolic syndrome. To this end, a better understanding of the epigenetic cues for various metabolic derangements should be critical for phenotypic change in adulthood and prevention of cardiorenal metabolic syndrome (Kunes, et al., 2015). Several tightly controlled transcriptional factors (e.g., TFEB and ZKSCAN3), and histone modifications [CARM1 H3R17 methyltransferase, acetylated Lys16 of histone 4 (H4K16ac), SIRT1 H4K16 deacetylase, hMOF H4K16 acetyltransferase, EZH2 H3K27 methyltransferase and dimethylated H3K9 (H3K9me2)] have been associated with autophagy and may provide short-term transcriptional and potential long-term regulation on autophagy and cell fate (Fullgrabe, et al., 2014). Moreover, amino acids (e.g., glycine, histidine, and methionine) and vitamins (B6, B12 and folate), with a known regulatory role on autophagy, provide methyl donors for DNA and protein méthylation. Intervention targeting epigenetically disturbed metabolic pathways via dietary supplementation with nutrients (functional amino acids and vitamins) has been shown to affect one-carbon-unit metabolism, gene expression, as well as protein méthylation and acetylation, resulting in improved health and well-being of affected offspring (Jia, et at, 2016). Nonetheless, whether autophagy regulation contributes to the beneficial effect of dietary nutrients remains elusive at this time. Food nutrients and metabolic supply-demand dynamics constitute environmental factors that interact with our genome influencing health and disease states.
While acute and rapid response of autophagy typically happens in the cytoplasm, prolonged starvation leads to transcriptional activation and epigenetic changes such as histone modification in the control of autophagy and cell fate. Recent evidence has depicted a tie between histone H3R17 dimethylation and autophagy (Shin, Kim, Kim, & Baek, 2016). Glucose starvation increases H3R17 dimethylation resulting from coactivator-associated arginine methyltransferase 1 (CARM1) induction in the nucleus. It is perceived that CARM1 serves as a coactivator of TFEB for activation of autophagy and lysosomal genes. Linder glucose-rich state, nuclear C ARM1 is degraded by the ubiquitin-dependent SKP2-SCF E3 ubiquitin ligase. When glucose starvation persists, AMPK gets accumulated in the nucleus to promote downregulation of SKP2 in the nucleus thus allowing CARM1 to escape from SCF E3 ubiquitin ligase, resulting in the transcription of various autophagy-related genes (H. J. Shin, et al., 2016). The role for H3R17 dimethylation in autophagy received consolidation from the autophagy suppressing property in compounds (e.g., ellagic acid) that block H3R17 méthylation (H. R. Shin, et al., 2016). These kinds of histone modifications in the nucleus seem to be a prototype of protein stabilization in certain cellular compartments during starvation-induced autophagy (Baek & Kim, 2017). Acetylation (such as in H4K16) has also emerged as an integral regulatory part for autophagy. The genome-wide investigation suggested that rapamycin promotes H4K16 deacetylation (coinciding with downregulation of hMOF without changing Sirt1 levels or activity) and associated transcriptional activation of autophagy-related genes (Baek & Kim, 2017; Fullgrabe, et al., 2014). The importance of acetylation in autophagy regulation also received support by the role of acetyl-CoA (as the sole acetyl-group donor for protein acetylation) in epigenomics and metabolism. In the setting of metabolic disorder, the mitochondrial network is essential to the metabolic-epigenome-genome regulation of various metabolites (NAD+, acetyl CoA, ATP) serving as substrates/cofactors for acetyl transferases and deacetylases (e.g. sirtuins) (Aon, Cortassa, Juhaszova, & Sollott, 2016). The nucleo-cytosolic acetyl-CoA functions as a phylogenetically conserved inhibitor of starvation-induced and aging-associated autophagy, and therefore serves as a metabolic rheostat, connecting metabolic state to autophagy regulation through protein acetylation. This notion receives support from the inverse relationship between acetylation of histones/cytosolic proteins and autophagy (Schroeder, et al., 2014). More evidence suggests that acetyl-CoA-suppressed autophagy involves regulation of histone acetylation and changes in autophagy-relevant transcriptome (Eisenberg, et al., 2014). Understanding of DNA méthylation and histone modification landscape in autophagy-related metabolic diseases should be a promising area for study of disease pathogenesis and drug development for metabolic control.
VI. Future Perspectives and Challenge in Autophagy Drug Development
A tremendous effort has been engaged towards the management of the cardiorenal metabolic syndrome although the advance in new drugs and procedures still face many problems and challenges. Perhaps genetics, epigenetics and precision medicine will unlock some of the mysteries of cardiorenal metabolic syndrome and the metabolic anomalies that underlie the cardiovascular sequelae, but this will take time and more immediate solutions are needed. Recent intensive attempts in the pharmacological modulation of autophagy have offered new avenues in the management of cardiorenal metabolic syndrome and metabolic derangement (Cheng, et al., 2013 ; Maixner, et al., 2016). A common denominator in the onset and development of cardiorenal metabolic syndrome and associated metabolic derangement is the maintenance of metabolism, adiposity, proteostasis, and organelle function (Cheng, et al., 2013; Maixner, et al., 2016). Ample evidence denotes a pivotal role for autophagy as a major mechanism to maintain metabolic homeostasis (Maixner, et al., 2016). As such, a number of autophagy-targeting compounds have been developed and approved for use in humans (Table 2), although some of these agents are still suboptimal for clinical practice. Currently, induction of autophagy and autophagic flux is predominantly dependent on nutrient sensors such as mTORand AMPK, and the insulin-IGF1 cascade. Autophagy modulators may be classified into mTOR-dependent and -independent. mTOR and AMPK regulate autophagy via direct phosphorylation of their downstream proteins. It is believed that hyperactivation of mTOR may serve as a main contributor for metabolic derangement by suppressing autophagy. For example, activation of skeletal muscle MTORC1 may result in autophagy inhibition and myopathy (Castets, et al., 2013). Thus nutrient signaling via mTOR provides a central mechanism and a possible drug target for intervention of autophagy- and lysosomal-related anomalies including adiposity (Settembre, Fraldi, Medina, & Ballabio, 2013). Recent evidence has depicted a pivotal role for mTOR phosphorylation of TFEB at the lysosomal membrane for autophagy flux regulation (Martina, Chen, Gucek, & Puertollano, 2012). More evidence has suggested the contribution to sustained transcriptional regulation of autophagy (Fleming Noda, Yoshimori, & Rubinsztein, 2011).
One other added benefit in targeting autophagy for pharmacotherapy may be related to the development of cell resistance to antineoplastic therapies. In these situations, interference with autophagic response (in combination of chemotherapy) should represent a good therapeutic strategy to enhance the effects of chemotherapy and improve the clinical outcomes, given that autophagy inhibits chemotherapy-induced apoptosis (Aveic & Tonini, 2016).
VII. Summary and Conclusion
In conclusion, cardiorenal metabolic syndrome is of growing relevance for public health, not only in general population but also in subpopulations that are more prone to the syndrome (Hemkens & Bucher, 2014). Accumulating evidence has emphasized a major role for autophagy in adiposity and metabolic regulation. Both transcriptional and epigenetic regulatory pathways can offer key information in the disease pathogenesis and provide attractive entry points for drug development in an effort to manipulate autophagy activity. Alternatively, non-pharmacological approaches such as life style modification (e.g., exercise and diet control) may also contribute to autophagy induction and benefit organismal health (Lapierre, et al., 2015). Nonetheless, successful development of therapeutic autophagy agents requires a much thorough understanding of metabolic profiling in the pathogenesis but also the regulatory networks of transcriptional factors, epigene tic modulations, their potential interactions among each other or with other factors (Cheng et al., 2013).
VIII. Acknowledgements
This review is dedicated to the late Prof Gerald M. Reaven, MD, who coined the concept of “Syndrome X”. The authors received support in part from the American Diabetes Association (7–13-BS-142-BR), NSFC81522004, NSFC81570225, NSFC81770261, RO1 HL73101–01A and RO1 HL107910–01 and the US veterans Affairs Merit System (0018). The authors wish to greatly appreciate the skillful graphical assistance of Ms. Sai Ma from the University of Wyoming College of Health Sciences, Laramie, WY 82071 USA. We wish to express our sincere apology for those authors whose important work was unable to be cited due to space limitation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain
DISCLOSURE STATEMENT: The authors have nothing to disclose.
REFERENCES
- Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, Fruchart JC, James WP, Loria CM, & Smith SC Jr. (2009). Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation, 120, 1640–1645. [DOI] [PubMed] [Google Scholar]
- Alberti KG, Zimmet P, & Shaw J (2005). The metabolic syndrome--a new worldwide definition. Lancet, 366, 1059–1062. [DOI] [PubMed] [Google Scholar]
- Altman BJ, & Rathmell JC (2012). Metabolic stress in autophagy and cell death pathways. Cold Spring Harb Perspect Biol, 4, a008763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres AM, Kooren JA, Parker SJ, Tucker KC, Ravindran N, Ito BR, Huang C, Venkatraman V, Van Eyk JE, Gottlieb RA, & Mentzer RM Jr. (2016). Discordant signaling and autophagy response to fasting in hearts of obese mice: Implications for ischemia tolerance. Am J Physiol Heart Circ Physiol, 311, H219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, Juhaszova M, & Sollott SJ (2016). Mitochondrial health, the epigenome and healthspan. Clin Sci (Lond), 130, 1285–1305.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arden C (2018). A role for Glucagon-Like Peptide-1 in the regulation of beta-cell autophagy. Peptides, 100, 85–93. [DOI] [PubMed] [Google Scholar]
- Armour SM, Baur JA, Hsieh SN, Land-Bracha A, Thomas SM, & Sinclair DA (2009). Inhibition of mammalian S6 kinase by resveratrol suppresses autophagy. Aging (Albany NY), 1, 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwell M, Gunn P, & Gibson S (2012). Waist-to-height ratio is a better screening tool than waist circumference and BMI for adult cardiometabolic risk factors: systematic review and meta-analysis. Obes Rev, 13, 275–286. [DOI] [PubMed] [Google Scholar]
- Aune D, Sen A, Norat T, Janszky I, Romundstad P, Tonstad S, & Vatten LJ (2016). Body Mass Index, Abdominal Fatness, and Heart Failure Incidence and Mortality: A Systematic Review and Dose-Response Meta-Analysis of Prospective Studies. Circulation, 133, 639–649.. [DOI] [PubMed] [Google Scholar]
- Aveic S, & Tonini GP (2016). Resistance to receptor tyrosine kinase inhibitors in solid tumors: can we improve the cancer fighting strategy by blocking autophagy? Cancer Cell Int, 16, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek SH, & Kim KI (2017). Epigenetic Control of Autophagy: Nuclear Events Gain More Attention. Mol Cell, 65, 781–785. [DOI] [PubMed] [Google Scholar]
- Bani Mohammad M, & Majdi Seghinsara A (2017). Polycystic Ovary Syndrome (PCOS), Diagnostic Criteria, and AMH. Asian Pac J Cancer Prev, 18, 17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow AD, & Thomas DC (2015). Autophagy in diabetes: beta-cell dysfunction, insulin resistance, and complications. DNA Cell Biol, 34, 252–260. [DOI] [PubMed] [Google Scholar]
- Bartlett JJ, Trivedi PC, & Pulinilkunnil T (2017). Autophagic dysregulation in doxorubicin cardiomyopathy. J Mol Cell Cardiol, 104, 1–8. [DOI] [PubMed] [Google Scholar]
- Bassuk SS, & Manson JE (2005). Epidemiological evidence for the role of physical activity in reducing risk of type 2 diabetes and cardiovascular disease. J Appl Physiol (1985), 99, 1193–1204. [DOI] [PubMed] [Google Scholar]
- Beltran-Sanchez H, Harhay MO, Harhay MM, & McElligott S (2013). Prevalence and trends of metabolic syndrome in the adult U.S. population, 1999–2010. J Am Coll Cardiol, 62, 697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatti JS, Bhatti GK, & Reddy PH (2016). Mitochondrial dysfunction and oxidative stress in metabolic disorders - A step towards mitochondria based therapeutic strategies. Biochim Biophys Acta. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilanges B, Alliouachene S, Pearce W, Morelli D, Szabadkai G, Chung YL, Chicanne G, Valet C, Hill JM, Voshol PJ, Collinson L, Peddie C, Ali K, Ghazaly E, Rajeeve V, Trichas G, Srinivas S, Chaussade C, Salamon RS, Backer JM, Scudamore CL, Whitehead MA, Keaney EP, Murphy LO, Semple RK, Payrastre B, Tooze SA, & Vanhaesebroeck B (2017). Vps34 PI 3-kinase inactivation enhances insulin sensitivity through reprogramming of mitochondrial metabolism. Nat Commun, 8, 1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botoseneanu A, Ambrosius WT, Beavers DP, de Rekeneire N, Anton S, Church T, Folta SC, Goodpaster BH, King AC, Nicklas BJ, Spring B, Wang X, & Gill TM (2015). Prevalence of metabolic syndrome and its association with physical capacity, disability, and self-rated health in Lifestyle Interventions and Independence for Elders Study participants. J Am Geriatr Soc, 63, 222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan L, Teede H, Skouteris H, Linardon J, Hill B, & Moran L (2017). Lifestyle and Behavioral Management of Polycystic Ovary Syndrome. J Womens Health (Larchmt), 26, 836–848. [DOI] [PubMed] [Google Scholar]
- Burguete-Garcia AI, Martinez-Nava GA, Valladares-Salgado A, Bermudez Morales VH, Estrada-Velasco B, Wacher N, Peralta-Romero J, Garcia-Mena J, Parra E, & Cruz M (2014). Association of beta1 and beta3 adrenergic receptors gene polymorphisms with insulin resistance and high lipid profiles related to type 2 diabetes and metabolic syndrome. Nutr Hosp, 29, 1327–1334. [DOI] [PubMed] [Google Scholar]
- Burkewitz K, Zhang Y, & Mair WB (2014). AMPK at the nexus of energetics and aging. Cell Metab, 20, 10–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona-Gutierrez D, Hughes AL, Madeo F, & Ruckenstuhl C (2016). The crucial impact of lysosomes in aging and longevity. Ageing Res Rev, 32, 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castets P, Lin S, Rion N, Di Fulvio S, Romanino K, Guridi M, Frank S, Tintignac LA, Sinnreich M, & Ruegg MA (2013). Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab, 17, 731–744. [DOI] [PubMed] [Google Scholar]
- Castillo K, Nassif M, Valenzuela V, Rojas F, Matus S, Mercado G, Court FA, van Zundert B, & Hetz C (2013). Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy, 9, 1308–1320. [DOI] [PubMed] [Google Scholar]
- Castrejon-Tellez V, Rodriguez-Perez JM, Perez-Torres I, Perez-Hernandez N, Cruz-Lagunas A, Guarner-Lans V, Vargas-Alarcon G, & Rubio-Ruiz ME (2016). The Effect of Resveratrol and Quercetin Treatment on PPAR Mediated Uncoupling Protein (UCP-) 1, 2, and 3 Expression in Visceral White Adipose Tissue from Metabolic Syndrome Rats. Int J Mol Sci, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceylan-Isik AF, Guo KK, Carlson EC, Privratsky JR, Liao SJ, Cai L, Chen AF, & Ren J (2009). Metallothionein abrogates GTP cyclohydrolase I inhibition-induced cardiac contractile and morphological defects: role of mitochondrial biogenesis. Hypertension, 53, 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang E, Kim L, Park SE, Rhee EJ, Lee WY, Oh KW, Park SW, & Park CY (2015). Ezetimibe improves hepatic steatosis in relation to autophagy in obese and diabetic rats. World J Gastroenterol, 21, 7754–7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HH, Moro A, Takakura K, Su HY, Mo A, Nakanishi M, Waldron RT, French SW, Dawson DW, Hines OJ, Li G, Go VLW, Sinnett-Smith J, Pandol SJ, Lugea A, Gukovskaya AS, Duff MO, Rosenberg DW, Rozengurt E, & Eibl G (2017). Incidence of pancreatic cancer is dramatically increased by a high fat, high calorie diet in KrasG12D mice. PLoS One, 12, e0184455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Chen B, Xiao FQ, Wu YT, Wang RH, Sun ZW, Fu GS, Mou Y, Tao W, Hu XS, & Hu SJ (2014). Autophagy protects against senescence and apoptosis via the RAS-mitochondria in high-glucose-induced endothelial cells. Cell Physiol Biochem, 33, 1058–1074. [DOI] [PubMed] [Google Scholar]
- Chen Y, Xu C, Yan T, Yu C, & Li Y (2015). omega-3 Fatty acids reverse lipotoxity through induction of autophagy in nonalcoholic fatty liver disease. Nutrition, 31, 1423–1429 e1422. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Ren X, Hait WN, & Yang JM (2013). Therapeutic targeting of autophagy in disease: biology and pharmacology. Pharmacol Rev, 65, 1162–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi AM, Ryter SW, & Levine B (2013). Autophagy in human health and disease. N Engl J Med, 368, 1845–1846. [DOI] [PubMed] [Google Scholar]
- Chung H, Choi J, & Park S (2018). Ghrelin protects adult rat hippocampal neural stem cells from excessive autophagy during oxygen-glucose deprivation. Endocr J, 65, 63–73. [DOI] [PubMed] [Google Scholar]
- Costa EC, de Sa JCF, Stepto NK, Costa IBB, Farias-Junior LF, da Nobrega Tomaz Moreira S, Soares EMM, Lemos T, Browne RAV, & Azevedo GD (2018). Aerobic Training Improves Quality of Life in Women with Polycystic Ovary Syndrome. Med Sci Sports Exerc. [DOI] [PubMed] [Google Scholar]
- Cui M, Yu H, Wang J, Gao J, & Li J (2013). Chronic caloric restriction and exercise improve metabolic conditions of dietary-induced obese mice in autophagy correlated manner without involving AMPK. J Diabetes Res, 2013, 852754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva IV, Rodrigues JS, Rebelo I, Miranda JPG, & Soveral G (2018). Revisiting the metabolic syndrome: the emerging role of aquaglyceroporins. Cell Mol Life Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Zeng J, Yan X, Lin Q, Wang K, Chen J, Shen F, Gu X, Wang Y, Chen J, Pan K, Cai L, Wintergerst KA, & Tan Y (2018). Sitagliptin-mediated preservation of endothelial progenitor cell function via augmenting autophagy enhances ischaemic angiogenesis in diabetes. J Cell Mol Med, 22, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meyer GR, Grootaert MO, Michiels CF, Kurdi A, Schrijvers DM, & Martinet W (2015). Autophagy in vascular disease. Circ Res, 116, 468–479. [DOI] [PubMed] [Google Scholar]
- Delbridge LMD, Mellor KM, Taylor DJ, & Gottlieb RA (2017). Myocardial stress and autophagy: mechanisms and potential therapies. Nat Rev Cardiol, 14, 412–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lullo L, Bellasi A, Russo D, Cozzolino M, & Ronco C (2017). Cardiorenal acute kidney injury: Epidemiology, presentation, causes, pathophysiology and treatment. Int J Cardiol, 227, 143–150. [DOI] [PubMed] [Google Scholar]
- Dommermuth R, & Ewing K (2018). Metabolic Syndrome: Systems Thinking in Heart Disease. Prim Care, 45, 109–129. [DOI] [PubMed] [Google Scholar]
- Dong F, Li Q, Sreejayan N, Nunn JM, & Ren J (2007). Metallothionein prevents high-fat diet induced cardiac contractile dysfunction: role of peroxisome proliferator activated receptor gamma coactivator 1alpha and mitochondrial biogenesis. Diabetes, 56, 2201–2212. [DOI] [PubMed] [Google Scholar]
- Dong Q, Xing W, Su F, Liang X, Tian F, Gao F, Wang S, & Zhang H (2017). Tetrahydroxystilbene Glycoside Improves Microvascular Endothelial Dysfunction and Ameliorates Obesity-Associated Hypertension in Obese ZDF Rats Via Inhibition of Endothelial Autophagy. Cell Physiol Biochem, 43, 293–307. [DOI] [PubMed] [Google Scholar]
- Dorn GW 2nd, (2011). Nix Nought Nothing: fairy tale or real deal. J Mol Cell Cardiol, 51, 497–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du D, Hu L, Wu J, Wu Q, Cheng W, Guo Y, Guan R, Wang Y, Chen X, Yan X, Zhu D, Wang J, Zhang S, Guo Y, & Xia C (2017). Neuroinflammation contributes to autophagy flux blockage in the neurons of rostral ventrolateral medulla in stress-induced hypertension rats. J Neuroinflammation, 14, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubinsky AN, Dastidar SG, Hsu CL, Zahra R, Djakovic SN, Duarte S, Esau CC, Spencer B, Ashe TD, Fischer KM, MacKenna DA, Sopher BL, Masliah E, Gaasterland T, Chau BN, Pereira de Almeida L, Morrison BE, & La Spada AR (2014). Let-7 coordinately suppresses components of the amino acid sensing pathway to repress mTORC1 and induce autophagy. Cell Metab, 20, 626–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont N, Chauhan S, Arko-Mensah J, Castillo EF, Masedunskas A, Weigert R, Robenek H, Proikas-Cezanne T, & Deretic V (2014). Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr Biol, 24, 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran RV, MacKenzie ED, Boulahbel H, Frezza C, Heiserich L, Tardito S, Bussolati O, Rocha S, Hall MN, & Gottlieb E (2013). HIF-independent role of prolyl hydroxylases in the cellular response to amino acids. Oncogene, 32, 4549–4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, Azuma K, Hirose T, Tanaka K, Kominami E, Kawamori R, Fujitani Y, & Watada H (2008). Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab, 8, 325–332. [DOI] [PubMed] [Google Scholar]
- Egom EE, Pharithi RB, Shiwani HA, Khan B, Kruzliak P, El-Hiani Y, & Maher V (2017). Time to redefine body mass index categories in chronic diseases? Spotlight on obesity paradox. Int J Food Sci Nutr, 1–11. [DOI] [PubMed] [Google Scholar]
- Eisenberg T, Schroeder S, Buttner S, Carmona-Gutierrez D, Pendl T, Andryushkova A, Marino G, Pietrocola F, Harger A, Zimmermann A, Magnes C, Sinner F, Sedej S, Pieber TR, Dengjel J, Sigrist S, Kroemer G, & Madeo F (2014). A histone point mutation that switches on autophagy. Autophagy, 10, 1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elouej S, Belfki-Benali H, Nagara M, Lasram K, Attaoua R, Sallem OK, Kamoun I, Chargui M, Romdhane L, Jamoussi H, Turki Z, Abid A, Ben Slama C, Bahri S, Abdelhak S, Grigorescu F, Ben Romdhane H, & Kefi R (2016). Association of rs9939609 Polymorphism with Metabolic Parameters and FTO Risk Haplotype Among Tunisian Metabolic Syndrome. Metab Syndr Relat Disord, 14, 121–128. [DOI] [PubMed] [Google Scholar]
- Evans TD, Jeong SJ, Zhang X, & Razani B (2018). TFEB and trehalose drive the macrophage autophagy-lysosome system to protect against atherosclerosis. Autophagy, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans TD, Sergin I, Zhang X, & Razani B (2017). Target acquired: Selective autophagy in cardiometabolic disease. Sci Signal, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezquerro S, Fruhbeck G, & Rodriguez A (2017). Ghrelin and autophagy. Curr Opin Clin Nutr Metab Care, 20, 402–408. [DOI] [PubMed] [Google Scholar]
- Fadini GP, Saragoni S, Russo P, Degli Esposti L, Vigili de Kreutzenberg S, Melazzini M, & Avogaro A (2017). Intraclass differences in the risk of hospitalisazion for heart failure among type 2 diabetic patients initiating a dipeptydil peptidase-4 inhibitor or a sulphonylurea. Results from the OsMed Health-DB registry. Diabetes Obes Metab. [DOI] [PubMed] [Google Scholar]
- Fan M, Jiang H, Zhang Y, Ma Y, Li L, & Wu J (2018). Liraglutide Enhances Autophagy and Promotes Pancreatic beta Cell Proliferation to Ameliorate Type 2 Diabetes in High-Fat-Fed and Streptozotocin-Treated Mice. Med Sci Monit, 24, 2310–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Lu H, Liang W, Garcia-Barrio MT, Guo Y, Zhang J, Zhu T, Hao Y, Zhang J, & Chen YE (2018). Endothelial TFEB Positively Regulates Post-Ischemic Angiogenesis. Circ Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Wang N, Rocchi A, Zhang W, Vassar R, Zhou Y, & He C (2017). Identification of natural products with neuronal and metabolic benefits through autophagy induction. Autophagy, 13, 41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez AF, Barcena C, Martinez-Garcia GG, Tamargo-Gomez I, Suarez MF, Pietrocola F, Castoldi F, Esteban L, Sierra-Filardi E, Boya P, Lopez-Otin C, Kroemer G, & Marino G (2017). Autophagy couteracts weight gain, lipotoxicity and pancreatic beta-cell death upon hypercaloric pro-diabetic regimens. Cell Death Dis, 8, e2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierabracci A (2014). The putative role of proteolytic pathways in the pathogenesis of Type 1 diabetes mellitus: the ‘autophagy’ hypothesis. Med Hypotheses, 82, 553–557. [DOI] [PubMed] [Google Scholar]
- Fleming A, Noda T, Yoshimori T, & Rubinsztein DC (2011). Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol, 7, 9–17. [DOI] [PubMed] [Google Scholar]
- Ford ES, Ajani UA, Croft JB, Critchley JA, Labarthe DR, Kottke TE, Giles WH, & Capewell S (2007). Explaining the decrease in U.S. deaths from coronary disease, 1980–2000. N Engl J Med, 356, 2388–2398. [DOI] [PubMed] [Google Scholar]
- Fu Q, Wang Q, & Xiang YK (2017). Insulin and beta Adrenergic Receptor Signaling: Crosstalk in Heart. Trends Endocrinol Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujitani Y, Ebato C, Uchida T, Kawamori R, & Watada H (2009). beta-cell autophagy: A novel mechanism regulating beta-cell function and mass: Lessons from beta-cell-specific Atg7-deficient mice. Islets, 1, 151–153. [DOI] [PubMed] [Google Scholar]
- Fujitani Y, Ueno T, & Watada H (2010). Autophagy in health and disease. 4. The role of pancreatic beta-cell autophagy in health and diabetes. Am J Physiol Cell Physiol, 299, C1–6. [DOI] [PubMed] [Google Scholar]
- Fullgrabe J, Klionsky DJ, & Joseph B (2014). The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol, 15, 65–74. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Pietrocola F, Levine B, & Kroemer G (2014). Metabolic control of autophagy. Cell, 159, 1263–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbossa SG, & Folli F (2017). Vitamin D, sub-inflammation and insulin resistance. A window on a potential role for the interaction between bone and glucose metabolism. Rev Endocr Metab Disord, 18, 243–258. [DOI] [PubMed] [Google Scholar]
- Gatica D, Lahiri V, & Klionsky DJ (2018). Cargo recognition and degradation by selective autophagy. Nat Cell Biol, 20, 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giricz Z, Koncsos G, Rajtik T, Varga ZV, Baranyai T, Csonka C, Szobi A, Adameova A, Gottlieb RA, & Ferdinandy P (2017). Hypercholesterolemia downregulates autophagy in the rat heart. Lipids Health Dis, 16, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazer HP, Osipov RM, Clements RT, Sellke FW, & Bianchi C (2009). Hypercholesterolemia is associated with hyperactive cardiac mTORC1 and mTORC2 signaling. Cell Cycle, 8, 1738–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go AS, Bauman MA, King SMC, Fonarow GC, Lawrence W, Williams KA, & Sanchez E (2014). An effective approach to high blood pressure control: a science advisory from the American Heart Association, the American College of Cardiology, and the Centers for Disease Control and Prevention. Journal of the American College of Cardiology, 63, 1230–1238. [DOI] [PubMed] [Google Scholar]
- Gorzalczany Y, Gilad Y, Amihai D, Hammel I, Sagi-Eisenberg R, & Merimsky O (2011). Combining an EGFR directed tyrosine kinase inhibitor with autophagy-inducing drugs: a beneficial strategy to combat non-small cell lung cancer. Cancer Lett, 310, 207–215. [DOI] [PubMed] [Google Scholar]
- Gosadi IM (2016). Assessment of the environmental and genetic factors influencing prevalence of metabolic syndrome in Saudi Arabia. Saudi Med J, 37, 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan G, Whaley-Connell A, Mugo M, Stump C, & Sowers JR (2005). The cardiometabolic syndrome as a cardiovascular risk factor. Am J Med Sci, 330, 311–318. [DOI] [PubMed] [Google Scholar]
- Grootaert MOJ, Moulis M, Roth L, Martinet W, Vindis C, Bennett MR, & De Meyer GRY (2018). Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc Res. [DOI] [PubMed] [Google Scholar]
- Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, Gordon DJ, Krauss RM, Savage PJ, Smith SC Jr., Spertus JA, & Costa F (2006). Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute scientific statement. Curr Opin Cardiol, 21, 1–6. [DOI] [PubMed] [Google Scholar]
- Guo H, Wang B, Li H, Ling L, Niu J, & Gu Y (2018). Glucagon-like peptide-1 analog prevents obesity-related glomerulopathy by inhibiting excessive autophagy in podocytes. Am J Physiol Renal Physiol, 314, F181–F189. [DOI] [PubMed] [Google Scholar]
- Guo L, Huang JX, Liu Y, Li X, Zhou SR, Qian SW, Liu Y, Zhu H, Huang HY, Dang YJ, & Tang QQ (2013). Transactivation of Atg4b by C/EBPbeta promotes autophagy to facilitate adipogenesis. Mol Cell Biol, 33, 3180–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R, Nair S, Zhang Y, & Ren J (2017). Adiponectin deficiency rescues high-fat diet-induced hepatic injury, apoptosis and autophagy loss despite persistent steatosis. Int JObes (Lond), 41, 1403–1412. [DOI] [PubMed] [Google Scholar]
- Guo R, Zhang Y, Turdi S, & Ren J (2013). Adiponectin knockout accentuates high fat diet-induced obesity and cardiac dysfunction: role of autophagy. Biochim Biophys Acta, 1832, 1136–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haim Y, Bluher M, Slutsky N, Goldstein N, Kloting N, Harman-Boehm I, Kirshtein B, Ginsberg D, Gericke M, Guiu Jurado E, Kovsan J, Tarnovscki T, Kachko L, Bashan N, Gepner Y, Shai I, & Rudich A (2015). Elevated autophagy gene expression in adipose tissue of obese humans: A potential non-cell-cycle-dependent fonction of E2F1. Autophagy, 11, 2074–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halling JF, & Pilegaard H (2017). Autophagy-Dependent Beneficial Effects of Exercise. Cold Spring Harb Perspect Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han CY (2016). Roles of reactive oxygen species on insulin resistance in adipose tissue. Diabetes Metab J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Pan H, Chen Y, Sun J, Wang Y, Li J, Ge W, Feng L, Lin X, Wang X, & Jin H (2011). EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS One, 6, e18691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder LM, Bunkenborg J, & Andersen JS (2014). Inducing autophagy: a comparative phosphoproteomic study of the cellular response to ammonia and rapamycin. Autophagy, 10, 339–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG (2014). AMPK--sensing energy while talking to other signaling pathways. Cell Metab, 20, 939–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatami M, Saidijam M, Yadegarzari R, Borzuei S, Soltanian A, Arian MS, & Goodarzi MT (2016). Peroxisome Proliferator-Activated Receptor-gammaGene Expression and Its Association with Oxidative Stress in Patients with Metabolic Syndrome. Chonnam Med J, 52, 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, Korsmeyer S, Packer M, May HI, Hill JA, Virgin HW, Gilpin C, Xiao G, Bassel-Duby R, Scherer PE, & Levine B (2012). Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature, 481, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, & Klionsky DJ (2009). Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet, 43, 67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Wei Y, Sun K, Li B, Dong X, Zou Z, Liu Y, Kinch LN, Khan S, Sinha S, Xavier RJ, Grishin NV, Xiao G, Eskelinen EL, Scherer PE, Whistler JL, & Levine B (2013). Beclin 2 functions in autophagy, degradation of G protein-coupled receptors, and metabolism. Cell, 154, 1085–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Sha S, Sun L, Zhang J, & Dong M (2016). GLP-1 analogue improves hepatic lipid accumulation by inducing autophagy via AMPK/mTOR pathway. Biochem Biophys Res Commun, 476, 196–203. [DOI] [PubMed] [Google Scholar]
- He S, & Liang C (2015). Frameshift mutation of UVRAG: Switching a tumor suppressor to an oncogene in colorectal cancer. Autophagy, 11, 1939–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemkens LG, & Bucher HC (2014). HIV infection and cardiovascular disease. Eur Heart J, 35, 1373–1381. [DOI] [PubMed] [Google Scholar]
- Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, Maurice N, Mukherjee A, Goldbach C, Watkins S, Michalopoulos G, & Perlmutter DH (2010). An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science, 329, 229–232. [DOI] [PubMed] [Google Scholar]
- Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, Figueroa ME, & Passegue E (2017). Autophagy maintains the metabolism and fonction of young and old stem cells. Nature, 543, 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohn A, Konig J, & Jung T (2016). Metabolic Syndrome, Redox State, and the Proteasomal System. Antioxid Redox Signal, 25, 902–917. [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, & Auwerx J (2012). Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol, 13, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu HC, Liu CH, Tsai YC, Li SJ, Chen CY, Chu CH, & Chen MF (2016). Time-dependent cellular response in the liver and heart in a dietary-induced obese mouse model: the potential role of ER stress and autophagy. Eur J Nutr, 55, 2031–2043. [DOI] [PubMed] [Google Scholar]
- Hua Y, & Nair S (2015). Proteases in cardiometabolic diseases: Pathophysiology, molecular mechanisms and clinical applications. Biochim Biophys Acta, 1852, 195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura Y, Kominami E, Tanaka K, & Komatsu M (2008). Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy, 4, 1063–1066.. [DOI] [PubMed] [Google Scholar]
- Inami Y, Yamashina S, Izumi K, Ueno T, Tanida I, Ikejima K, & Watanabe S (2011). Hepatic steatosis inhibits autophagic proteolysis via impairment of autophagosomal acidification and cathepsin expression. Biochem Biophys Res Commun, 412, 618–625. [DOI] [PubMed] [Google Scholar]
- Invitti C, Gilardini L, & Viberti G (2004). Obesity and the metabolic syndrome in children and adolescents. N Engl J Med, 351, 1146–1148; author reply 1146–1148. [PubMed] [Google Scholar]
- Jaber N, Dou Z, Chen JS, Catanzaro J, Jiang YP, Ballou LM, Selinger E, Ouyang X, Lin RZ, Zhang J, & Zong WX (2012). Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci US A, 109, 2003–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob JA, Salmani JM, Jiang Z, Feng L, Song J, Jia X, & Chen B (2017). Autophagy: An overview and its roles in cancer and obesity. Clin Chim Acta, 468, 85–89. [DOI] [PubMed] [Google Scholar]
- James HA, O’Neill BT, & Nair KS (2017). Insulin Regulation of Proteostasis and Clinical Implications. Cell Metab, 26, 310–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen HJ, van Essen P, Koenen T, Joosten LA, Netea MG, Tack CJ, & Stienstra R (2012). Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology, 153, 5866–5874. [DOI] [PubMed] [Google Scholar]
- Ji Y, Wu Z, Dai Z, Sun K, Wang J, & Wu G (2016). Nutritional epigenetics with a focus on amino acids: implications for the development and treatment of metabolic syndrome. J Nutr Biochem, 27, 1–8. [DOI] [PubMed] [Google Scholar]
- Jia G, Hill MA, & Sowers JR (2018). Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ Res, 122, 624–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Jia Y, & Sowers JR (2016). Contribution of Maladaptive Adipose Tissue Expansion to Development of Cardiovascular Disease. Compr Physiol, 7, 253–262. [DOI] [PubMed] [Google Scholar]
- Jia G, & Sowers JR (2015). Autophagy: a housekeeper in cardiorenal metabolic health and disease. Biochim Biophys Acta, 1852, 219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Whaley-Connell A, & Sowers JR (2018). Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia, 61, 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jialal I, & Devaraj S (2018). Subcutaneous adipose tissue biology in metabolic syndrome. Horm Mol Biol Clin Investig. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Huang W, Wang J, Xu Z, He J, Lin X, Zhou Z, & Zhang J (2014). Metformin plays a dual role in MIN6 pancreatic beta cell function through AMPK-dependent autophagy. Int J Biol Sci, 10, 268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Ro SH, Cao J, Otto NM, & Kim DH (2010). mTOR regulation of autophagy. FEBS Lett, 584, 1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, Nguyen YH, Kang TM, Yoon KH, Kim JW, Jeong YT, Han MS, Lee MK, Kim KW, Shin J, & Lee MS (2008). Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab, 8, 318–324. [DOI] [PubMed] [Google Scholar]
- Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, Kemp BE, Bardeesy N, Dennis P, Schlager JJ, Marette A, Kozma SC, & Thomas G (2010). Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab, 11, 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapahi P, Kaeberlein M, & Hansen M (2016). Dietary restriction and lifespan: Lessons from invertebrate models. Ageing Res Rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Rodriguez-Navarro JA, Arias E, Kiffin R, Sahu S, Schwartz GJ, Cuervo AM, & Singh R (2011). Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab, 14, 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ (2010). The genetics of ageing. Nature, 464, 504–512. [DOI] [PubMed] [Google Scholar]
- Keramati AR, Fathzadeh M, Go GW, Singh R, Choi M, Faramarzi S, Mane S, Kasaei M, Sarajzadeh-Fard K, Hwa J, Kidd KK, Babaee Bigi MA, Malekzadeh R, Hosseinian A, Babaei M, Lifton RP, & Mani A (2014). A form of the metabolic syndrome associated with mutations in DYRK1B. N Engl J Med, 370, 1909–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaldoun SA, Emond-Boisjoly MA, Chateau D, Carriere V, Lacasa M, Rousset M, Demignot S, & Morel E (2014). Autophagosomes contribute to intracellular lipid distribution in enterocytes. Mol Biol Cell, 25, 118–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lim YM, & Lee MS (2018). The Role of Autophagy in Systemic Metabolism and Human-Type Diabetes. Mol Cells, 41, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JA, Wei Y, & Sowers JR (2008). Role of mitochondrial dysfunction in insulin resistance. Circ Res, 102, 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim DH, Hur KY, Kim HK, Ko T, Han J, Kim HL, Kim J, Back SH, Komatsu M, Chen H, Chan DC, Konishi M, Itoh N, Choi CS, & Lee MS (2013). Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med, 19, 83–92. [DOI] [PubMed] [Google Scholar]
- Kim KH, & Lee MS (2014). Autophagy--a key player in cellular and body metabolism. Nat Rev Endocrinol, 10, 322–337. [DOI] [PubMed] [Google Scholar]
- Kim SH, Kim G, Han DH, Lee M, Kim I, Kim B, Kim KH, Song YM, Yoo JE, Wang HJ, Bae SH, Lee YH, Lee BW, Kang ES, Cha BS, & Lee MS (2017). Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy, 13, 1767–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmelman AC, & White E (2017). Autophagy and Tumor Metabolism. Cell Metab, 25, 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Isaka Y, & Yoshimori T (2017). Autophagy and kidney inflammation. Autophagy, 13, 997–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, Rakugi H, & Isaka Y (2011). Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol, 22, 902–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Takahashi A, Takabatake Y, Namba T, Yamamoto T, Kaimori JY, Matsui I, Kitamura H, Niimura F, Matsusaka T, Soga T, Rakugi H, & Isaka Y (2013). Autophagy protects kidney proximal tubule epithelial cells from mitochondrial metabolic stress. Autophagy, 9, 1876–1886. [DOI] [PubMed] [Google Scholar]
- Kitzman DW, Brubaker P, Morgan T, Haykowsky M, Hundley G, Kraus WE, Eggebeen J, & Nicklas BJ (2016). Effect of Caloric Restriction or Aerobic Exercise Training on Peak Oxygen Consumption and Quality of Life in Obese Older Patients With Heart Fa ilure With Preserved Ejection Fraction: A Randomized Clinical Trial. JAMA, 315, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloting N, & Bluher M (2014). Adipocyte dysfunction, inflammation and metabolic syndrome. Rev Endocr Metab Disord, 15, 277–287. [DOI] [PubMed] [Google Scholar]
- Kosacka J, Nowicki M, Paeschke S, Baum P, Bluher M, & Kloting N (2018). Up-regulated autophagy: as a protective factor in adipose tissue of WOKW rats with metabolic syndrome. Diabetol Metab Syndr, 10, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovsan J, Bluher M, Tarnovscki T, Kloting N, Kirshtein B, Madar L, Shai I, Golan R, Harman-Boehm I, Schon MR, Greenberg AS, Elazar Z, Bashan N, & Rudich A (2011). Altered autophagy in human adipose tissues in obesity. J Clin Endocrinol Metab, 96, E268–277. [DOI] [PubMed] [Google Scholar]
- Kunes J, Vaneckova I, Mikulaskova B, Behuliak M, Maletinska L, & Zicha J (2015). Epigenetics and a new look on metabolic syndrome. Physiol Res, 64, 611–620. [DOI] [PubMed] [Google Scholar]
- Lamb CA, Dooley HC, & Tooze SA (2013). Endocytosis and autophagy: Shared machinery for degradation. Bioessays, 35, 34–45. [DOI] [PubMed] [Google Scholar]
- Lampert MA, & Gustafsson AB (2018). Balancing Autophagy for a Healthy Heart. Curr Opin Physiol, 1, 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancel S, Montaigne D, Marechal X, Marciniak C, Hassoun SM, Decoster B, Ballot C, Blazejewski C, Corseaux D, Lescure B, Motterlini R, & Neviere R (2012). Carbon monoxide improves cardiac function and mitochondrial population quality in a mouse model of metabolic syndrome. PLoS One, 7, e41836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapierre LR, Kumsta C, Sandri M, Ballabio A, & Hansen M (2015). Transcriptional and Epigenetic Regulation of Autophagy in Aging. Autophagy, 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson E, Wahlstrand B, Hedblad B, Hedner T, Kjeldsen SE, Melander O, & Lindahl P (2013). Hypertension and genetic variation in endothelial-specific genes. PLoS One, 8, e62035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavie CJ, McAuley PA, Church TS, Milani RV, & Blair SN (2014). Obesity and cardiovascular diseases: implications regarding fitness, fatness, and severity in the obesity paradox. J Am Coll Cardiol, 63, 1345–1354. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Smith A, Guo L, Alastalo TP, Li M, Sawada H, Liu X, Chen ZH, Ifedigbo E, Jin Y, Feghali-Bostwick C, Ryter SW, Kim HP, Rabinovitch M, & Choi AM (2011). Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med, 183, 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu SY, Tsai YC, Chen WC, Hsu CH, Lee YM, & Cheng PY (2018). Raspberry ketone induces brown-like adipocyte formation through suppression of autophagy in adipocytes and adipose tissue. J Nutr Biochem, 56, 116–125. [DOI] [PubMed] [Google Scholar]
- Levine B, & Klionsky DJ (2004). Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell, 6, 463–477. [DOI] [PubMed] [Google Scholar]
- Levine B, & Klionsky DJ (2017). Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker’s yeast fuel advances in biomedical research. Proc Natl Acad Sci U S A, 114, 201–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Packer M, & Codogno P (2015). Development of autophagy inducers in clinical medicine. J Clin Invest, 125, 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Wu X, Chen H, Zhuang C, Zhang Z, Yao S, Cai D, Ning G, & Su Q (2018). miR199a-5p inhibits hepatic insulin sensitivity via suppression of ATG14-mediated autophagy. Cell Death Dis, 9, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, & Lumey LH (2016). Exposure to the Chinese famine of 1959–61 in early life and current health conditions: a systematic review and meta-analysis. Lancet, 388 Suppl 1, S63. [DOI] [PubMed] [Google Scholar]
- Li D, You Y, Bi FF, Zhang TN, Jiao J, Wang TR, Zhou YM, Shen ZQ, Wang XX, & Yang Q (2018). Autophagy is activated in the ovarian tissue of polycystic ovary syndrome. Reproduction, 155, 85–92. [DOI] [PubMed] [Google Scholar]
- Li H, Zhang Q, Yang X, & Wang L (2017). PPAR-gamma agonist rosiglitazone reduces autophagy and promotes functional recovery in experimental traumaticspinal cord injury. Neurosci Lett, 650, 89–96. [DOI] [PubMed] [Google Scholar]
- Li J, & Cai Y (2015). The dual effects of autophagy in myocardial hypertrophy. Acta Cardiol, 70, 493–498. [DOI] [PubMed] [Google Scholar]
- Li J, & Pfeffer SR (2016). Lysosomal membrane glycoproteins bind cholesterol and contribute to lysosomal cholesterol export. Elife, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Dou X, Ning H, Song Q, Wei W, Zhang X, Shen C, Li J, Sun C, & Song Z (2017). SIRT3 Acts As a Negative Regulator of Autophagy Dictating Hepatocyte Susceptibility to Lipotoxicity. Hepatology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Li H, Yang D, Yu X, Irwin DM, Niu G, & Tan H (2017). Excessive Autophagy Activation and Increased Apoptosis Are Associated with Palmitic Acid-Induced Cardiomyocyte Insulin Resistance. J Diabetes Res, 2017, 2376893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Gong H, Yang S, Yang L, Fan Y, & Zhou Y (2017). Pectic Bee Pollen Polysaccharide from Rosa rugosa Alleviates Diet-Induced Hepatic Steatosis and Insulin Resistance via Induction of AMPK/mTOR-Mediated Autophagy. Molecules, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZL, Woollard JR, Ebrahimi B, Crane JA, Jordan KL, Lerman A, Wang SM, & Lerman LO (2012). Transition from obesity to metabolic syndrome is associated with altered myocardial autophagy and apoptosis. Arterioscler Thromb Vasc Biol, 32, 1132–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YM, Lim H, Hur KY, Quan W, Lee HY, Cheon H, Ryu D, Koo SH, Kim HL, Kim J, Komatsu M, & Lee MS (2014). Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nat Commun, 5, 4934. [DOI] [PubMed] [Google Scholar]
- Lin CW, Zhang H, Li M, Xiong X, Chen X, Dong XC, & Yin XM (2013). Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol, 58, 993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F, & Qin ZH (2013). Degradation of misfolded proteins by autophagy: is it a strategy for Huntington’s disease treatment? J Huntingtons Dis, 2, 149–157. [DOI] [PubMed] [Google Scholar]
- Liu H, Javaheri A, Godar RJ, Murphy J, Ma X, Rohatgi N, Mahadevan J, Hyrc K, Saftig P, Marshall C, McDaniel ML, Remedi MS, Razani B, Urano F, & Diwan A (2017). Intermittent fasting preserves beta-cell mass in obesity-induced diabetes via the autophagy-lysosome pathway. Autophagy, 13, 1952–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, & Debnath J (2016). The Evolving, Multifaceted Roles of Autophagy in Cancer. Adv Cancer Res, 130, 1–53. [DOI] [PubMed] [Google Scholar]
- Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, Tanaka KE, & Czaja MJ (2015). Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy, 11, 271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Liao JZ, He XX, & Li PY (2017). The role of autophagy in hepatocellular carcinoma: friend or foe. Oncotarget. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Liu J, & Yu X (2016). Dipeptidyl peptidase-4 inhibitor MK-626 restores insulin secretion through enhancing autophagy in high fat diet-induced mice. Biochem Biophys Res Commun, 470, 516–520. [DOI] [PubMed] [Google Scholar]
- Liu Y, Takahashi Y, Desai N, Zhang J, Serfass JM, Shi YG, Lynch CJ, & Wang HG (2016). Bif-1 deficiency impairs lipid homeostasis and causes obesity accompanied by insulin resistance. Sci Rep, 6, 20453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long L, Yang X, Southwood M, Lu J, Marciniak SJ, Dunmore BJ, & Morrell NW (2013). Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ Res, 112, 1159–1170. [DOI] [PubMed] [Google Scholar]
- Ludwig DS (2016). Lifespan Weighed Down by Diet. JAMA, 315, 2269–2270. [DOI] [PubMed] [Google Scholar]
- Ma D, Molusky MM, Song J, Hu CR, Fang F, Rui C, Mathew AV, Pennathur S, Liu F, Cheng JX, Guan JL, & Lin JD (2013). Autophagy deficiency by hepatic FIP200 deletion uncouples steatosis from liver injury in NAFLD. Mol Endocrinol, 27, 1643–1654.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Guo R, Yu L, Zhang Y, & Ren J (2011). Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur Heart J, 32, 1025–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Niknejad N, Gorn-Hondermann I, Dayekh K, & Dimitroulakos J (2012). Lovastatin induces multiple stress pathways including LKB1/AMPK activation that regulate its cytotoxic effects in squamous cell carcinoma cells. PLoS One, 7, e46055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrigal-Matute J, & Cuervo AM (2016). Regulation of Liver Metabolism by Autophagy. Gastroenterology, 150, 328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahu I, & Domingos AI (2017). The sympathetic neuro-adipose connection and the control of body weight. Exp Cell Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiano C, Hue O, Morin AJ, & Moullec G (2016). Prevalence of overweight and obesity among children and adolescents with intellectual disabilities: a systematic review and meta-analysis. Obes Rev. [DOI] [PubMed] [Google Scholar]
- Maixner N, Bechor S, Vershinin Z, Pecht T, Goldstein N, Haim Y, & Rudich A (2016). Transcriptional Dysregulation of Adipose Tissue Autophagy in Obesity. Physiology (Bethesda), 31, 270–282. [DOI] [PubMed] [Google Scholar]
- Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, Loda M, Lane HA, & Sellers WR (2004). mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med, 10, 594–601. [DOI] [PubMed] [Google Scholar]
- Mandviwala T, Khalid U, & Deswal A (2016). Obesity and Cardiovascular Disease: a Risk Factor or a Risk Marker? Curr Atheroscler Rep, 18, 21. [DOI] [PubMed] [Google Scholar]
- Mao Y, Yu F, Wang J, Guo C, & Fan X (2016). Autophagy: a new target for nonalcoholic fatty liver disease therapy. Hepat Med, 8, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marasco MR, & Linnemann AK (2018). beta-Cell Autophagy in Diabetes Pathogenesis. Endocrinology, 159, 2127–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinkovic M, Sprung M, Buljubasic M, & Novak I (2018). Autophagy Modulation in Cancer: Current Knowledge on Action a nd Therapy. Oxid Med Cell Longev, 2018, 8023821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, Schroeder S, Pendl T, Harger A, Niso-Santano M, Zamzami N, Scoazec M, Durand S, Enot DP, Fernandez AF, Martins I, Kepp O, Senovilla L, Bauvy C, Morselli E, Vacchelli E, Bennetzen M, Magnes C, Sinner F, Pieber T, Lopez-Otin C, Maiuri MC, Codogno P, Andersen JS, Hill JA, Madeo F, & Kroemer G (2014). Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell, 53, 710–725. [DOI] [PubMed] [Google Scholar]
- Marsh BJ, Soden C, Alarcon C, Wicksteed BL, Yaekura K, Costin AJ, Morgan GP, & Rhodes CJ (2007). Regulated autophagy controls hormone content in secretory-deficient pancreatic endocrine beta-cells. Mol Endocrinol, 21, 2255–2269. [DOI] [PubMed] [Google Scholar]
- Martina JA, Chen Y, Gucek M, & Puertollano R (2012). MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy, 8, 903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinet W, De Loof H, & De Meyer GR (2014). mTOR inhibition: a promising strategy for stabilization of atherosclerotic plaques. Atherosclerosis, 233, 601–607. [DOI] [PubMed] [Google Scholar]
- Martinez MC, & Andriantsitohaina R (2017). Extracellular Vesicles in Metabolic Syndrome. Circ Res, 120, 1674–1686. [DOI] [PubMed] [Google Scholar]
- Masini M, Bugliani M, Lupi R, del Guerra S, Boggi U, Filipponi F, Marselli L, Masiello P, & Marchetti P (2009). Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia, 52, 1083–1086. [DOI] [PubMed] [Google Scholar]
- Matsuura N, Asano C, Nagasawa K, Ito S, Sano Y, Minagawa Y, Yamada Y, Hattori T, Watanabe S, Murohara T, & Nagata K (2015). Effects of pioglitazone on cardiac and adipose tissue pathology in rats with metabolic syndrome. Int J Cardiol, 179, 360–369. [DOI] [PubMed] [Google Scholar]
- Medvedev R, Hildt E, & Ploen D (2017). Look who’s talking-the crosstalk between oxidative stress and autophagy supports exosomal-dependent release of HCV particles. Cell Biol Toxicol, 33, 211–231. [DOI] [PubMed] [Google Scholar]
- Mellor KM, Bell JR, Young MJ, Ritchie RH, & Delbridge LM (2011). Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J Mol Cell Cardiol, 50, 1035–1043. [DOI] [PubMed] [Google Scholar]
- Meng Q, & Cai D (2011). Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. J Biol Chem, 286, 32324–32332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentzel CM, Anthon C, Jacobsen MJ, Karlskov-Mortensen P, Bruun CS, Jorgensen CB, Gorodkin J, Cirera S, & Fredholm M (2015). Gender and Obesity Specific MicroRNA Expression in Adipose Tissue from Lean and Obese Pigs. PLoS One, 10, e0131650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Fullgrabe J, Jackson A, Jimenez Sanchez M, Karabiyik C, Licitra F, Lopez Ramirez A, Pavel M, Puri C, Renna M, Ricketts T, Schlotawa L, Vicinanza M, Won H, Zhu Y, Skidmore J, & Rubinsztein DC (2017). Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron, 93, 1015–1034. [DOI] [PubMed] [Google Scholar]
- Mizushima N (2018). A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol, 20, 521–527. [DOI] [PubMed] [Google Scholar]
- Mohan N, Shen Y, Endo Y, ElZarrad MK, & Wu WJ (2016). Trastuzumab, but Not Pertuzumab, Dysregulates HER2 Signaling to Mediate Inhibition of Autophagy and Increase in Reactive Oxygen Species Production in Human Cardiomyocytes. Mol Cancer Ther, 15, 1321–1331. [DOI] [PubMed] [Google Scholar]
- Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, Potelle C, El Arid JM, Mouton S, Sebti Y, Duez H, Preau S, Remy-Jouet I, Zerimech F, Koussa M, Richard V, Neviere R, Edme JL, Lefebvre P, & Staels B (2014). Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation, 130, 554–564. [DOI] [PubMed] [Google Scholar]
- Moreira MC, Pinto IS, Mourao AA, Fajemiroye JO, Colombari E, Reis AA, Freiria-Oliveira AH, Ferreira-Neto ML, & Pedrino GR (2015). Does the sympathetic nervous system contribute to the pathophysiology of metabolic syndrome? Front Physiol, 6, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan-Bathke M, Harris ZI, Arnett DG, Klein RR, Burd R, Ann DK, & Limesand KH (2014). The Rapalogue, CCI-779, improves salivary gland function following radiation. PLoS One, 9, e113183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MJ, Gamez G, Menke C, Hernandez A, Thorburn J, Gidan F, Staskiewicz L, Morgan S, Cummings C, Maycotte P, & Thorburn A (2014). Regulation of autophagy and chloroquine sensitivity by oncogenic RAS in vitro is context-dependent. Autophagy, 10, 1814–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullan LA, Mularczyk EJ, Kung LH, Forouhan M, Wragg JM, Goodacre R, Bateman JF, Swanton E, Briggs MD, & BootHandford RP (2017). Increased intracellular proteolysis reduces disease severity in an ER stress-associated dwarfism. J Clin Invest, 127, 3861–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munasinghe PE, Riu F, Dixit P, Edamatsu M, Saxena P, Hamer NS, Galvin IF, Bunton RW, Lequeux S, Jones G, Lamberts RR, Emanueli C, Madeddu P, & Katare R (2016). Type-2 diabetes increases autophagy in the human heart through promotion of Beclin-1 mediated pathway. Int J Cardiol, 202, 13–20. [DOI] [PubMed] [Google Scholar]
- Murase H, Kuno A, Miki T, Tanno M, Yano T, Kouzu H, Ishikawa S, Tobisawa T, Ogasawara M, Nishizawa K, & Miura T (2015). Inhibition of DPP-4 reduces acute mortality after myocardial infarction with restoration of autophagic response in type 2 diabetic rats. Cardiovasc Diabetol, 14, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscogiuri G, De Martino MC, Negri MR, Pivonello C, Simeoli C, Orio F, Pivonello R, & Colao A (2017). Adr enal mass: insight into pathogenesis and a common link with insulin resistance. Endocrinology, 158, 1527–1532. [DOI] [PubMed] [Google Scholar]
- Naidu S, Ponnampalvanar S, Kamaruzzaman SB, & Kamarulzaman A (2017). Prevalence of Metabolic Syndrome Among People Living with HIV in Developing Countries: A Systematic Review. AIDS Patient Care STDS, 31, 1–13. [DOI] [PubMed] [Google Scholar]
- Nair S, & Ren J (2012). Autophagy and cardiovascular aging: lesson learned from rapamycin. Cell Cycle, 11, 2092–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, & Asahi M (2007). The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nature medicine, 13, 619–624. [DOI] [PubMed] [Google Scholar]
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, & Otsu K (2007). The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med, 13, 619–624. [DOI] [PubMed] [Google Scholar]
- Nakatogawa H, & Ohsumi Y (2014). Autophagy: close contact keeps out the uninvited. Curr Biol, 24, R560–562. [DOI] [PubMed] [Google Scholar]
- Neeland IJ, Poirier P, & Despres JP (2018). Cardiovascular and Metabolic Heterogeneity of Obesity: Clinical Challenges and Implications for Management. Circulation, 137, 1391–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri T, Tarantino E, Orsenigo M, Reid JF, Gariboldi M, Zambetti M, Pierotti MA, & Pilotti S (2010). Chromosome band 17q21 in breast cancer: significant association between beclin 1 loss and HER2/NEU amplification. Genes Chromosomes Cancer, 49, 901–909. [DOI] [PubMed] [Google Scholar]
- Nielsen J (2017). Systems Biology of Metabolism: A Driver for Developing Personalized and Precision Medicine. Cell Metab, 25, 572–579. [DOI] [PubMed] [Google Scholar]
- Nishida K, & Otsu K (2016). Autophagy during cardiac remodeling. J Mol Cell Cardiol, 95, 11–18. [DOI] [PubMed] [Google Scholar]
- Nussenzweig SC, Verma S, & Finkel T (2015). The role of autophagy in vascular biology. Circ Res, 116, 480–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwankwo T, Yoon SS, Burt V, & Gu Q (2013). Hypertension among adults in the United States: National Health and Nutr ition Examination Survey, 2011–2012. NCHS data brief, 1–8. [PubMed] [Google Scholar]
- Oga EA, & Eseyin OR (2016). The Obesity Paradox and Heart Failure: A Systematic Review of a Decade of Evidence. J Obes, 2016, 9040248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara Y, Itakura E, Kono N, Mizushima N, Arai H, Nara A, Mizukami T, & Yamamoto A (2014). Stearoyl-CoA desaturase 1 activity is required for autophagosome formation. J Biol Chem, 289, 23938–23950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opipari AW Jr., Tan L, Boitano AE, Sorenson DR, Aurora A, & Liu JR (2004). Resveratrol-induced autophagocytosis in ovarian cancer cells. Cancer Res, 64, 696–703. [DOI] [PubMed] [Google Scholar]
- Ouimet M, Franklin V, Mak E, Liao X, Tabas I, & Marcel YL (2011). Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab, 13, 655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiardi C, Mirzoyan Z, Zola S, Parisi F, Vingiani A, Pasini ME, & Bellosta P (2017). The Stearoyl-CoA Desaturase-1 (Desat1) in Drosophila cooperated with Myc to Induce Autophagy and Growth, a Potential New Link to Tumor Survival. Genes (Basel), 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palizban A, Rezaei M, Khanahmad H, & Fazilati M (2017). Transcription factor 7-like 2 polymorphism and context-specific risk of metabolic syndrome, type 2 diabetes, and dyslipidemia. J Res Med Sci, 22, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paniagua JA (2016). Nutrition, insulin resistance and dysfunctional adipose tissue determine the different components of metabolic syndrome. World J Diabetes, 7, 483–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Kim SH, Lee MS, & Kim MS (2017). Epigenetic modification by dietary factors: Implications in metabolic syndrome. Mol Aspects Med, 54, 58–70. [DOI] [PubMed] [Google Scholar]
- Parkhitko A, Myachina F, Morrison TA, Hindi KM, Auricchio N, Karbowniczek M, Wu JJ, Finkel T, Kwiatkowski DJ, Yu JJ, & Henske EP (2011). Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proc Natl Acad Sci U S A, 108, 12455–12460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry TL, & Willis MS (2016). Cardiac ubiquitin ligases: Their role in cardiac metabolism, autophagy, cardioprotectio n and therapeutic potential. Biochim Biophys Acta, 1862, 2259–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Perez A, Vilarino-Garcia T, Fernandez-Riejos P, Martin-Gonzalez J, Segura-Egea JJ, & Sanchez-Margalet V (2017). Role of leptin as a link between metabolism and the immune system. Cytokine Growth Factor Rev. [DOI] [PubMed] [Google Scholar]
- Perrone-Filardi P, Paolillo S, Costanzo P, Savarese G, Trimarco B, & Bonow RO (2015). The role of metabolic syndrome in heart failure. Eur Heart J, 36, 2630–2634. [DOI] [PubMed] [Google Scholar]
- Petkeviciene J, Smalinskiene A, Klumbiene J, Petkevicius V, Kriaucioniene V, & Lesauskaite V (2016). Physical activity, but not dietary intake, attenuates the effect of the FTO rs9939609 polymorphism on obesity and metabolic syndrome in Lithuanian adult population. Public Health, 135, 23–29. [DOI] [PubMed] [Google Scholar]
- Pfeifer U, Föhr J, Wilhelm W, & Dämmrich J (1987). Short-term inhibition of cardiac cellular autophagy by isoproterenol. Journal of molecular and cellular cardiology, 19, 1179–1184. [DOI] [PubMed] [Google Scholar]
- Pickles S, Vigie P, & Youle RJ (2018). Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr Biol, 28, R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, & Kroemer G (2015). Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab, 21, 805–821. [DOI] [PubMed] [Google Scholar]
- Pradeepa R, Surendar J, Indulekha K, Chella S, Anjana RM, & Mohan V (2016). Prevalence of Metabolic Syndrome and its Association with Coronary Artery Disease Among an Urban Elderly South Indian Population (CURES- 145). J Assoc Physicians India, 64, 20–25. [PubMed] [Google Scholar]
- Prieto-Dominguez N, Ordonez R, Fernandez A, Garcia-Palomo A, Muntane J, Gonzalez-Gallego J, & Mauriz JL (2016). Modulation of Autophagy by Sorafenib: Effects on Treatment Response. Front Pharmacol, 7, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Q, Zhang Z, Orwig A, Chen S, Ding WX, Xu Y, Kunz RC, Lind NRL, Stamler JS, & Yang L (2018). S- Nitrosoglutathione Reductase Dysfunction Contributes to Obesity-Associated Hepatic Insulin Resistance via Regulating Autophagy. Diabetes, 67, 193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan W, Hur KY, Lim Y, Oh SH, Lee JC, Kim KH, Kim GH, Kim SW, Kim HL, Lee MK, Kim KW, Kim J, Komatsu M, & Lee MS (2012). Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia, 55, 392–403. [DOI] [PubMed] [Google Scholar]
- Rambold AS, Cohen S, & Lippincott-Schwartz J (2015). Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell, 32, 678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rask Larsen J, Dima L, Correll CU, & Manu P (2018). The pharmacological management of metabolic syndrome. Expert Rev Clin Pharmacol, 11, 397–410. [DOI] [PubMed] [Google Scholar]
- Rawat DK, Alzoubi A, Gupte R, Chettimada S, Watanabe M, Kahn AG, Okada T, McMurtry IF, & Gupte SA (2014). Increased reactive oxygen species, metabolic maladaptation, and autophagy contribute to pulmonary arterial hypertension-induced ventricular hypertrophy and diastolic heart failure. Hypertension, 64, 1266–1274. [DOI] [PubMed] [Google Scholar]
- Reaven GM (1988). Banting lecture 1988. Role of insulin resistance in human disease. Diabetes, 37, 1595–1607. [DOI] [PubMed] [Google Scholar]
- Reaven GM (1993). Role of insulin resistance in human disease (syndrome X): an expanded definition. Annu Rev Med, 44, 121–131. [DOI] [PubMed] [Google Scholar]
- Ren J, & Anversa P (2015). The insulin-like growth factor I system: physiological and pathophysiological implication in cardiovascular diseases associated with metabolic syndrome. Biochem Pharmacol, 93, 409–417. [DOI] [PubMed] [Google Scholar]
- Ren J, Pulakat L, Whaley-Connell A, & Sowers JR (2010). Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J Mol Med (Berl), 88, 993–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, & Taegtmeyer H (2015). Too much or not enough of a good thing - The Janus faces of autophagy in cardiac fuel and protein homeostasis. J Mol Cell Cardiol. [DOI] [PubMed] [Google Scholar]
- Rocchi A, & He C (2015). Emerging roles of autophagy in metabolism and metabolic disorders. Front Biol (Beijing), 10, 154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Duran A, Selloum M, Champy MF, Diez-Guerra FJ, Flores JM, Serrano M, Auwerx J, Diaz-Meco MT, & Moscat J (2006). Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab, 3, 211–222. [DOI] [PubMed] [Google Scholar]
- Rose C, Menzies FM, Renna M, Acevedo-Arozena A, Corrochano S, Sadiq O, Brown SD, & Rubinsztein DC (2010). Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum Mol Genet, 19, 2144–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotter I, Skonieczna-Zydecka K, Kosik-Bogacka D, Adler G, Ryl A, & Laszczynska M (2016). Relationships between FTO rs9939609, MC4R rs17782313, and PPARgamma rs1801282 polymorphisms and the occurrence of selected metabolic and hormonal disorders in middle-aged and elderly men - a preliminary study. Clin Interv Aging, 11, 1723–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland TJ, Sweet ME, Mestroni L, & Taylor MR (2016). Danon disease - dysregulation of autophagy in a multisystem disorder with cardiomyopathy. J Cell Sci, 129, 2135–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Marino G, & Kroemer G (2011). Autophagy and aging. Cell, 146, 682–695. [DOI] [PubMed] [Google Scholar]
- Ruiz-Ramirez A, Lopez-Acosta O, Barrios-Maya MA, & El-Hafidi M (2016). Cell Death and Heart Failure in Obesity: Role of Uncoupling Proteins. Oxid Med Cell Longev, 2016, 9340654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo M, & Russo GL (2018). Autophagy inducers in cancer. Biochem Pharmacol. [DOI] [PubMed] [Google Scholar]
- Rybstein MD, Bravo-San Pedro JM, Kroemer G, & Galluzzi L (2018). The autophagic network and cancer. Nat Cell Biol, 20, 243–251. [DOI] [PubMed] [Google Scholar]
- Ryter SW, Koo JK, & Choi AM (2014). Molecular regulation of autophagy and its implications for metabolic diseases. Curr Opin Clin Nutr Metab Care, 17, 329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, & Sadoshima J (2015). Molecular Mechanisms of Mitochondrial Autophagy/Mitophagy in the Heart. Circ Res, 116, 1477–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltiel AR, & Olefsky JM (2017). Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest, 127, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, & Rubinsztein DC (2005). Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol, 170, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, & Sabatini DM (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell, 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheen AJ (2018). Cardiovascular Effects of New Oral Glucose-Lowering Agents: DPP-4 and SGLT-2 Inhibitors. Circ Res, 122, 1439–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder S, Pendl T, Zimmermann A, Eisenberg T, Carmona-Gutierrez D, Ruckenstuhl C, Marino G, Pietrocola F, Harger A, Magnes C, Sinner F, Pieber TR, Dengjel J, Sigrist SJ, Kroemer G, & Madeo F (2014). Acetyl-coenzyme A: a metabolic master regulator of autophagy and longevity. Autophagy, 10, 1335–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciarretta S, Forte M, Frati G, & Sadoshima J (2018). New Insights Into the Role of mTOR Signaling in the Cardiovascular System. Circ Res, 122, 489–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciarretta S, Maejima Y, Zablocki D, & Sadoshima J (2018). The Role of Autophagy in the Heart. Annu Rev Physiol, 80, 1–26. [DOI] [PubMed] [Google Scholar]
- Sciarretta S, Yee D, Nagarajan N, Bianchi F, Saito T, Valenti V, Tong M, Del Re DP, Vecchione C, Schiro ne L, Forte M, Rubattu S, Shirakabe A, Boppana VS, Volpe M, Frati G, Zhai P, & Sadoshima J (2018). Trehalose-Induced Activation of Autophagy Improves Cardiac Remodeling After Myocardial Infarction. J Am Coll Cardiol, 71, 1999–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, Condorelli G, & Sadoshima J (2012). Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation, 125, 1134–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secrest MH, Udell JA, & Filion KB (2017). The cardiovascular safety trials of DPP-4 inhibitors, GLP-1 agonists, and SGLT2 inhibitors. Trends Cardiovasc Med, 27, 194–202. [DOI] [PubMed] [Google Scholar]
- Seravalle G, & Grassi G (2016). Sympathetic Nervous System, Hypertension, Obesity and Metabolic Syndrome. High Blood Press Cardiovasc Prev, 23, 175–179. [DOI] [PubMed] [Google Scholar]
- Serkova N, Klawitter J, & Niemann CU (2003). Organ-specific response to inhibition of mitochondrial metabolism by cyclosporine in the rat. Transpl Int, 16, 748–755. [DOI] [PubMed] [Google Scholar]
- Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, Rentsch KM, Spanaus KS, Senn O, Kristanto P, Scheffel H, Weishaupt D, & Wuthrich RP (2010). Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med, 363, 820–829. [DOI] [PubMed] [Google Scholar]
- Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, Wollenberg AC, Di Bernardo D, Chan L, Irazoqui JE, & Ballabio A (2013). TFEB controls cellular lipid metabolism through a starvation- induced autoregulatory loop. Nat Cell Biol, 15, 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Medina DL, & Ballabio A (2013). Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol, 14, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Niso-Santano M, Adjemian S, Takehara T, Malik SA, Minoux H, Souquere S, Marino G, Lachkar S, Senovilla L, Galluzzi L, Kepp O, Pierron G, Maiuri MC, Hikita H, Kroemer R, & Kroemer G (2012). Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol Cell, 48, 667–680. [DOI] [PubMed] [Google Scholar]
- Shi R, Guberman M, & Kirshenbaum LA (2017). Mitochondrial quality control: The role of mitophagy in aging. Trends Cardiovasc Med. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yoshimura K, Furuya N, Koike M, Ueno T, Komatsu M, Arai H, Tanaka K, Kominami E, & Uchiyama Y (2009). The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem Biophys Res Commun, 382, 419–423. [DOI] [PubMed] [Google Scholar]
- Shimano H, & Sato R (2017). SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol, 13, 710–730. [DOI] [PubMed] [Google Scholar]
- Shimomura H, Terasaki F, Hayashi T, Kitaura Y, Isomura T, & Suma H (2001). Autophagic degeneration as a possible mechanism of myocardial cell death in dilated cardiomyopathy. Jpn Circ J, 65, 965–968. [DOI] [PubMed] [Google Scholar]
- Shin HJ, Kim H, Oh S, Lee JG, Kee M, Ko HJ, Kweon MN, Won KJ, & Baek SH (2016). AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature, 534, 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HR, Kim H, Kim KI, & Baek SH (2016). Epigenetic and transcriptional regulation of autophagy. Autophagy, 12, 2248–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmulewitz D, Heath SC, Blundell ML, Han Z, Sharma R, Salit J, Auerbach SB, Signorini S, Breslow JL, Stoffel M, & Friedman JM (2006). Linkage analysis of quantitative traits for obesity, diabetes, hypertension, and dyslipidemia on the island of Kosrae, Federated States of Micronesia. Proc Natl Acad Sci U S A, 103, 3502–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman AI, & Mangelsdorf DJ (2005). Retinoid x receptor heterodimers in the metabolic syndrome. N Engl J Med, 353, 604–615. [DOI] [PubMed] [Google Scholar]
- Singh KK, Yanagawa B, Quan A, Wang R, Garg A, Khan R, Pan Y, Wheatcroft MD, Lovren F, Teoh H, & Verma S (2014). Autophagy gene fingerprint in human ischemia and reperfusion. J Thorac Cardiovasc Surg, 147, 1065–1072 e1061. [DOI] [PubMed] [Google Scholar]
- Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, & Czaja MJ (2009). Autophagy regulates adipose mass and differentiation in mice. J Clin Invest, 119, 3329–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha-Hikim AP, Sinha-Hikim I, & Friedman TC (2017). Connection of Nicotine to Diet-Induced Obesity and Non-Alcoholic Fatty Liver Disease: Cellular and Mechanistic Insights. Front Endocrinol (Lausanne), 8, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha RA, Singh BK, & Yen PM (2017). Reciprocal Crosstalk Between Autophagic and Endocrine Signaling in Metabolic Homeostasis. Endocr Rev, 38, 69–102. [DOI] [PubMed] [Google Scholar]
- Sinha RA, Singh BK, & Yen PM (2018). Direct effects of thyroid hormones on hepatic lipid metabolism. Nat Rev Endocrinol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slutsky N, Vatarescu M, Haim Y, Goldstein N, Kirshtein B, Harman-Boehm I, Gepner Y, Shai I, Bashan N, Bluher M, & Rudich A (2016). Decreased adiponectin links elevated adipose tissue autophagy with adipocyte endocrine dysfunction in obesity. Int J Obes (Lond), 40, 912–920. [DOI] [PubMed] [Google Scholar]
- Smith CJ, & Ryckman KK (2015). Epigenetic and developmental influences on the risk of obesity, diabetes, and metabolic syndrome. Diabetes Metab Syndr Obes, 8, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Tan J, Miao Y, Li M, & Zhang Q (2017). Crosstalk of autophagy and apoptosis: Involvement of the dual role o f autophagy under ER stress. J Cell Physiol. [DOI] [PubMed] [Google Scholar]
- Song X, Shen Q, Fan L, Yu Q, Jia X, Sun Y, Bai W, & Kang J (2018). Dehydroepiandrosterone-induced activation of mTORC1 and inhibition of autophagy contribute to skeletal muscle insulin resistance in a mouse model of polycystic ovary syndrome. Oncotarget, 9, 11905–11921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorop O, Olver TD, van de Wouw J, Heinonen I, van Duin RW, Duncker DJ, & Merkus D (2017). The microcirculation: a key player in obesity-associated cardiovascular disease. Cardiovasc Res, 113, 1035–1045. [DOI] [PubMed] [Google Scholar]
- Stolz A, Ernst A, & Dikic I (2014). Cargo recognition and trafficking in selective autophagy. Nat Cell Biol, 16, 495–501. [DOI] [PubMed] [Google Scholar]
- Su Y, Wu J, He J, Liu X, Chen X, Ding Y, Zhang C, Chen W, Wang Y, & Gao R (2017). High insulin impaired ovarian function in early pregnant mice and the role of autophagy in this process. Endocr J. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, Yamachika S, He F, & Karin M (2016). p62/SQSTM1-Dr. Jekyll and Mr. Hyde that prevents oxidative stress but promotes liver cancer. FEBS Lett, 590, 2375–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeman NM, Skovso S, Page MM, Lim GE, & Johnson JD (2017). A causal role for hyperinsulinemia in obesity. J Endocrinol, 232, R173–R183. [DOI] [PubMed] [Google Scholar]
- Tuomikoski P, & Savolainen-Peltonen H (2017). Vasomotor symptoms and metabolic syndrome. Maturitas, 97, 61–65. [DOI] [PubMed] [Google Scholar]
- Vainshtein A, & Hood DA (2016). The regulation of autophagy during exercise in skeletal muscle. J Appl Physiol (1985), 120, 664–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Greevenbroek MM, Schalkwijk CG, & Stehouwer CD (2016). Dysfunctional adipose tissue and low-grade inflammation in the management of the metabolic syndrome: current practices and future advances. F1000Res, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan KL, & Mattison JA (2016). Obesity and Aging in Humans and Nonhuman Primates: A Mini-Review. Gerontology, 62, 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S, Goldenberg RM, Bhatt DL, Farkouh ME, Quan A, Teoh H, Connelly KA, Leiter LA, & Friedrich JO (2017). Dipeptidyl peptidase-4 inhibitors and the risk of heart failure: a systematic review and meta-analysis. CMAJ Open, 5, E152–E177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vion AC, Kheloufi M, Hammoutene A, Poisson J, Lasselin J, Devue C, Pic I, Dupont N, Busse J, Stark K, Lafaurie-Janvore J, Barakat AI, Loyer X, Souyri M, Viollet B, Julia P, Tedgui A, Codogno P, Boulanger CM, & Rautou PE (2017). Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc Natl Acad Sci U S A, 114, E8675–E8684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Jia J, & Rodrigues B (2017). Autophagy, Metabolic Disease, and Pathogenesis of Heart Dysfunction. Can J Cardiol, 33, 850–859. [DOI] [PubMed] [Google Scholar]
- Wang G, Zhou H, Strulovici-Barel Y, Al-Hijji M, Ou X, Salit J, Walters MS, Staudt MR, Kaner RJ, & Crystal RG (2017). Role of OSGIN1 in mediating smoking-induced autophagy in the human airway epithelium. Autophagy, 13, 1205–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, & Ren J (2018). Role of autophagy and regulatory mechanisms in alcoholic cardiomyopathy. Biochim Biophys Acta, 1864, 2003–2009. [DOI] [PubMed] [Google Scholar]
- Webster BR, Scott I, Traba J, Han K, & Sack MN (2014). Regulation of autophagy and mitophagy by nutrient availability and acetylation. Biochim Biophys Acta, 1841, 525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, An XR, Jin SJ, Li XX, & Xu M (2018). Inhibition of insulin resistance by PGE1 via autophagy-dependent FGF21 pathway in diabetic nephropathy. Sci Rep, 8, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whaley-Connell A, & Sowers JR (2014). Basic science: Pathophysiology: the cardiorenal metabolic syndrome. J Am Soc Hypertens, 8, 604–606.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whaley-Connell A, & Sowers JR (2017). Obesity and kidney disease: from population to basic science and the search for new therapeutic targets. Kidney Int, 92, 313–323. [DOI] [PubMed] [Google Scholar]
- Wu G, Liu L, Huang J, Pang S, Wei G, Cui Y, & Yan B (2011). Alterations of autophagic-lysosomal system in the peripheral leukocytes of patients with myocardial infarction. Clin Chim Acta, 412, 1567–1571. [DOI] [PubMed] [Google Scholar]
- Wu JJ, Quijano C, Chen E, Liu H, Cao L, Fergusson MM, Rovira II, Gutkind S, Daniels MP, Komatsu M, & Finkel T (2009). Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging (Albany NY), 1, 425–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Wu QQ, Duan MX, Liu C, Yuan Y, Yang Z, Liao HH, Fan D, & Tang QZ (2018). TAX1BP1 overexpression attenuates cardiac dysfunction and remodeling in STZ-induced diabetic cardiomyopathy in mice by regulating autophagy. Biochim Biophys Acta, 1864, 1728–1743. [DOI] [PubMed] [Google Scholar]
- Xie Q, Lin Q, Li D, & Chen J (2017). Imatinib induces autophagy via upregulating XIAP in GIST882 cells. Biochem Biophys Res Commun, 488, 584–589. [DOI] [PubMed] [Google Scholar]
- Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D, & Zou MH (2011). Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes, 60, 1770–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu A, & Sweeney G (2015). Emerging role of autophagy in mediating widespread actions of ADIPOQ/adiponectin. Autophagy, 11, 723–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Bucala R, & Ren J (2013). Macrophage migration inhibitory factor deficiency augments doxorubicin-induced cardiomyopathy. J Am Heart Assoc, 2, e000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, & Ferrante AW Jr. (2013). Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab, 18, 816–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Hua Y, Nair S, Zhang Y, & Ren J (2013). Akt2 knockout preserves cardiac function in high-fat diet-induced obesity by rescuing cardiac autophagosome maturation. J Mol Cell Biol, 5, 61–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, Gulick J, Yue Z, Robbins J, Epstein PN, & Liang Q (2013). Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem, 288, 18077–18092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Pacheco BD, Leng L, Bucala R, & Ren J (2013). Macrophage migration inhibitory factor plays a permissive role in the maintenance of cardiac contractile function under starvation through regulation of autophagy. Cardiovasc Res, 99, 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, & Ren J (2015). Macrophage migration inhibitory factor (MIF) knockout preserves cardiac homeostasis through alleviating Aktmediated myocardial autophagy suppression in high-fat diet-induced obesity. Int J Obes (Lond), 39, 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamahara K, Kume S, Koya D, Tanaka Y, Morita Y, Chin-Kanasaki M, Araki H, Isshiki K, Araki S, Haneda M, Matsusaka T, Kashiwagi A, Maegawa H, & Uzu T (2013). Obesity-mediated autophagy insufficiency exacerbates proteinuria-induced tubulointerstitial lesions. J Am Soc Nephrol, 24, 1769–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S, Huda N, Khambu B, & Yin XM (2017). Relevance of autophagy to fatty liver diseases and potential therapeutic applications. Amino Acids. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S, Yang X, Chen T, Xi Z, & Jiang X (2014). The PPARgamma agonist Troglitazone induces autophagy, apoptosis and necroptosis in bladder cancer cells. Cancer Gene Ther, 21, 188–193. [DOI] [PubMed] [Google Scholar]
- Yan WJ, Dong HL, & Xiong LZ (2013). The protective roles of autophagy in ischemic preconditioning. Acta Pharmacol Sin, 34, 636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li P, Fu S, Calay ES, & Hotamisligil GS (2010). Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab, 11, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Zhang Y, & Ren J (2018). Autophagic Regulation of Lipid Homeostasis in Cardiometabolic Syndrome. Front Cardiovasc Med, 5, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Zheng K, & Zhang X (2015). Rosiglitazone exerts neuroprotective effects via the suppression of neuronal autophagy and apoptosis in the cortex following traumatic brain injury. Mol Med Rep, 12, 6591–6597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D’Agostino D, Planavsky N, Lupfer C, Kanneganti TD, Kang S, Horvath TL, Fahmy TM, Crawford PA, Biragyn A, Alnemri E, & Dixit VD (2015). The ketone metabolite beta- hydroxybutyrate blocks NLRP3 infammasome-mediated inflammatory disease. Nat Med, 21, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, & Heintz N (2003). Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A, 100, 15077–15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, & Pan D (2003). Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol, 5, 578–581. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Hu M, Meng F, Sun X, Xu H, Zhang J, Cui P, Morina N, Li X, Li W, Wu XK, Brannstrom M, Shao R, & Billig H (2017). Metformin Ameliorates Uterine Defects in a Rat Model of Polycystic Ovary Syndrome. EBioMedicine, 18, 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, & Ren J (2016). Epigenetics and obesity cardiomyopathy: From pathophysiology to prevention and management. Pharmacol Ther, 161, 52–66. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Sowers JR, & Ren J (2018). Targeting autophagy in obesity: from pathophysiology to management. Nat Rev Endocrinol, 14, 356–376. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Hao CJ, Li CG, Zang DJ, Zhao J, Li XN, Wei AH, Wei ZB, Yang L, He X, Zhen XC, Gao X, Speakman JR, & Li W (2014). Mutation of SLC35D3 causes metabolic syndrome by impairing dopamine signaling in striatal D1 neurons. PLoS Genet, 10, e1004124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Wang S, Zhu P, Hu S, Chen Y, & Ren J (2018). Empagliflozin rescues diabetic myocardial microvascular injury via AMPK- mediated inhibition of mitochondrial fission. Redox Biol, 15, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylke JW, & Bauchner H (2016). The Unrelenting Challenge of Obesity. JAMA, 315, 2277–2278. [DOI] [PubMed] [Google Scholar]