

Abstract
Background
Reconstructing individual transmission events in an infectious disease outbreak can provide valuable information and help inform infection control policy. Recent years have seen considerable progress in the development of methodologies for reconstructing transmission chains using both epidemiological and genetic data. However, only a few of these methods have been implemented in software packages, and with little consideration for customisability and interoperability. Users are therefore limited to a small number of alternatives, incompatible tools with fixed functionality, or forced to develop their own algorithms at considerable personal effort.
Results
Here we present outbreaker2, a flexible framework for outbreak reconstruction. This R package re-implements and extends the original model introduced with outbreaker, but most importantly also provides a modular platform allowing users to specify custom models within an optimised inferential framework. As a proof of concept, we implement the within-host evolutionary model introduced with TransPhylo, which is very distinct from the original genetic model in outbreaker, and demonstrate how even complex model results can be successfully included with minimal effort.
Conclusions
outbreaker2 provides a valuable starting point for future outbreak reconstruction tools, and represents a unifying platform that promotes customisability and interoperability. Implemented in the R software, outbreaker2 joins a growing body of tools for outbreak analysis.
Keywords: Transmission, Epidemics, Chain, Tree, Genomics, Software, MCMC, Bayesian, Likelihood
Background
Determining ‘who infected whom’ during an outbreak can yield precious insights into transmission dynamics of an infectious disease and subsequently inform infection control policies. Transmission tree reconstruction has been used to specify the contribution of individual cases and locations to overall transmission [1], characterise heterogeneous infectiousness within outbreaks [2, 3], evaluate the impact of control measures on transmission intensity [4, 5] and identify transmission routes [6]. Consequently there exists significant interest in designing methodologies for the inference of transmission trees from outbreak data, including temporal data (e.g. date of symptom onset), contact data, pathogen whole genome sequences (WGS) and geographic locations.
A large number of studies have addressed this problem in recent years (Table 1) [6–15]. These approaches differ in multiple ways, including in their underlying epidemiological models (e.g. SIR [8, 11], SEIR [9, 12] or branching process models [7, 14, 15]) and genetic models (e.g. non-phylogenetic [7, 10, 12, 16] or phylogenetic models [11, 13–15]), as well as their ability to account for unobserved cases and multiple infectious introductions. This methodological diversity is beneficial, providing various theoretical frameworks for outbreak reconstruction in different epidemic scenarios.
Table 1.
Studies on outbreak reconstruction and their availability as software
| Study | Available as software | Study | Available as software |
|---|---|---|---|
| Cottam et al. [23] | ✗ | Numinnen et al. [24] | ✗ |
| Aldrin et al. [25] | ✗ | Hall et al. [11] | ✓ |
| Jombart et al. [26] | ✓ | Worby et al. [10] | ✓ |
| Ypma et al. [27] | ✗ | Lau et al. [12] | ✗ |
| Morelli et al. [28] | ✗ | Soubeyrand [29] | ✗ |
| Ypma et al. [6] | ✗ | De Maio et al. [13] | ✓ |
| Stadler et al. [30] | ✓ | Kenah et al. [31] | ✗ |
| Jombart et al. [7] | ✓ | Klinkenberg et al. [14] | ✓ |
| Didelot et al. [8] | ✓ | Worby et al. [32] | ✗ |
| Mollentze et al. [9] | ✗ | Didelot et al. [15] | ✓ |
Unfortunately, the implementation of these methodologies in a user-friendly computational framework to encourage their use by the wider scientific community has so far remained limited. Primarily, a large proportion of the methods described in the literature is not available as readily useable software (Table 1), requiring the user either to directly modify the original code if it is available, or implement the algorithm themselves if not. Moreover, the existing software tools were developed in parallel with little consideration for interoperability, accepting similar types of data or outputting similar results in different formats. This results in unnecessary time spent preparing and formatting the data, and hinders effective comparison of results produced under different models. Finally, the current software are generally inflexible, with few options to specify algorithm behaviour without modifying the often complex source code (e.g. [7]). This ranges from simple implementation issues, such as being limited to specific distributions for priors [14, 15], to more fundamental restrictions on the inferential process itself, in that the underlying epidemiological and genetic models are hardcoded and not customisable by the user.
To address these issues, we have developed outbreaker2, a flexible software tool for outbreak reconstruction. outbreaker2 exploits the fact that most transmission tree inference methods, though based on very different models, are generally implemented in a similar manner. Most consider the same data, namely WGS and some form of temporal data (i.e. dates of symptom onset and assumptions on the distribution of incubation and infectious periods [7, 9, 12, 14, 15], or explicitly defined exposure intervals [10, 13]). The majority are also implemented in a Bayesian framework, and therefore describe prior distributions on parameters and likelihood functions that evaluate the plausibility of a given parameter sets under specific transmission and evolutionary models. Unobserved data, including the transmission tree itself, times of infection and unobserved cases, are generally modelled using augmented data [17]. Finally most methods use a Markov Chain Monte-Carlo (MCMC) algorithm to derive samples from the posterior distributions, using often complex proposal functions to explore alternative transmission scenarios.
outbreaker2 generalises this procedure and allows the user to implement their own models by specifying custom prior distributions, likelihood functions and movement functions, which are then employed within a wider inference framework. This enables sophisticated customisation of the algorithm with minimal effort by the user, allowing a greater focus on methodological developments rather than their implementation. Importantly, it also permits for different modules to be developed and easily combined, so that outbreak reconstruction approaches can be tailored to specific diseases and epidemiological contexts. outbreaker2 is implemented as a package for the R software [18], as part of a larger toolkit for epidemics analysis developed under the R Epidemics Consortium (www.repidemicsconsortium.org). In the following, we explain the rationale of this implementation and illustrate its modularity using a simple case study.
Implementation
outbreaker2 is written in R and C++, making extensive use of Rcpp [19] to facilitate the integration of C++ into R. The original method for outbreak reconstruction introduced with outbreaker [7] has been entirely re-implemented using a modular and highly customisable approach (Fig. 1). This was achieved by distinguishing the architecture which underpins the inferential process from model-specific regions of code (i.e. code that varies between model implementations), and treating these components as independent modules (Fig. 1). Non-specific components of the implementation include data and overall configuration infrastructure, as well as all post-processing of outputs including summaries and graphics. The three central, model-specific components of our Bayesian inference framework are the prior distributions, likelihood functions and MCMC functions defining movements of the parameters. By abstracting these components into algorithmic functions with predefined input and output structures, we designed simple procedures allowing users to customise most aspects of the outbreak reconstruction, including the model itself and the MCMC used to explore the parameter space (Fig. 1). The following sections describes the structure of the core components of outbreaker2, and the mechanisms by which they can be changed. Extensive documentation, including a full description of the API and example of customised models, are available from the outbreaker2 website (http://www.repidemicsconsortium.org/outbreaker2/).
Fig. 1.
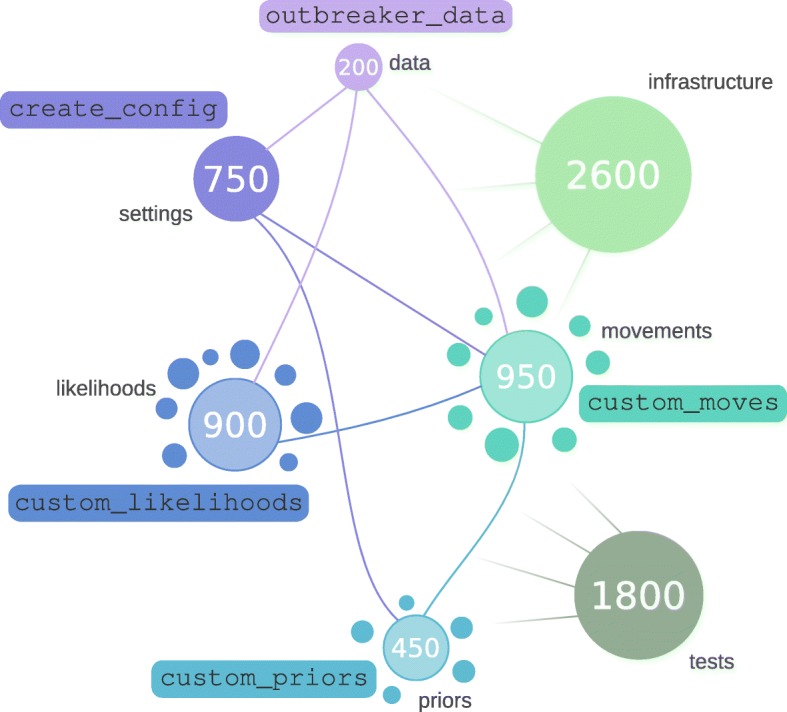
Schematic representation of the code design of outbreaker2. Each disk represents a different component of the code. Disk size matches the size of the corresponding component, indicated by numbers (in lines of code, rounded to 50). Separate disks for likelihoods, priors and movements indicate independent C++ modules. Links represent flows of information between components, colored according to the input. Infrastructure and tests are globally connected to all components. Functions indicated within rectangles are entry points into the code, indicating possible customisation by the user
Object classes
outbreaker2 defines several S3 object classes used to transfer information across modules. The outbreaker_data class stores the data which remains unchanged throughout the inference process. Users pass temporal data (e.g. sampling times) as a vector of dates, and genetic data as either DNA sequences (DNAbin objects [20]) or phylogenetic trees (phylo objects [20]). Generation time and incubation period distributions are also specified by the user. Extensive data validation is achieved by the constructor of this class to prevent often intractable errors at a later stage. The outbreaker_config class stores the global properties of the algorithm and can be optionally specified by the user. Importantly, this allows the user to declare which parameters and augmented data should be moved and inferred during the MCMC procedure. Again, the constructor of this class ensures validation of the inputs. The outbreaker_param class is used internally for storing a single state in the MCMC chain, and describes parameters and augmented data. Objects of this class are proposed, accepted or rejected, and sampled during the MCMC procedure. The advantage of this fixed internal structure is that it greatly simplifies writing new, customised movement functions. Finally, results of the reconstruction are output as outbreaker_chains objects, for which various methods (e.g. plot, print, summary) have been defined to summarise and visualise results, or carry on further secondary analyses.
Custom prior distributions
A prior distribution describes the probability of observing a parameter given our previous knowledge of the infectious disease under observation. In outbreaker2, custom priors are specified as functions with a single argument of class outbreaker_param, which return a log-probability of a given parameter value (all probabilities are treated on a log scale). Custom priors can take any shape as long as this structure is satisfied. Priors can be specified for each parameter in the model by passing a named list of functions to the priors argument of the outbreaker function.
Custom likelihood functions
Likelihood functions define the probability of a set of parameters (outbreaker_param object) given some observed data (outbreaker_data object) under a specific model. In outbreaker2, the overall likelihood is decomposed in separate likelihood components, which can be evaluated independently during the MCMC and therefore boost computer efficiency. Customised likelihood functions can be specified by the user for each of these components, to use alternative epidemiological or evolutionary models. Note that additional likelihood components can also be added, in which case the overall likelihood function will also need to be re-defined. Likelihood components can have any form, as long as they take an outbreaker_data and an outbreaker_param objects as arguments, and return a log-probability. As a result, users can combine different epidemiological and evolutionary models to fit specific needs.
Custom movements functions
In an MCMC algorithm, movement functions are used to update the set of parameters and augmented data, from one MCMC iteration to the next. For example, a commonly used strategy is to use a Metropolis-Hastings move, where an update is first proposed and then accepted or rejected depending on its likelihood. Well designed movement functions are necessary to achieve efficient chain convergence and ensure rapid and representative sampling from the posterior distribution. In outbreaker2, the MCMC is decomposed as a list of movement functions, each of which is evaluated at each step of the chain.
Given the size and complexity of the parameter space when inferring temporally resolved transmission trees with unobserved cases, efficient movement functions are difficult and time-intensive to develop. outbreaker2 allows users to access the optimised, default movement functions for various parameters and augmented data (including, crucially, the transmission tree) while using custom prior distributions and likelihood functions. Default movements, likelihood and prior functions can all be accessed through the function get_cpp_api, so that these components can be used when designing new MCMC procedures.
Unlike priors and likelihood functions which always take the same arguments, movement functions may have varying arguments including the data, general settings, custom priors and likelihoods. To simplify the specification of custom movements by the user, outbreaker2 only requires that new movement functions have an outbreaker_param object for first argument; further arguments such as data and custom likelihood components are automatically detected, and internally replaced by the corresponding components of the code. In other words, the whole machinery of the code is added seamlessly to custom movement functions where it is needed. Importantly, as the acceptance-rejection step is specified within movement functions, users are not restricted to Metropolis-Hastings methods [21], and could use alternative MCMCs such as a Gibbs sampler [22].
Results and discussion
Implementing a custom model
The main asset of outbreaker2 is its ability to define new models easily. We tested this flexibility by implementing the genetic model developed by Didelot et al. [8, 15] in the TransPhylo package. In contrast to the model of evolution used by outbreaker2 [7], which treats mutations between all transmission pairs as independent events, TransPhylo uses a phylogenetic tree to account for patterns of common evolution amongst the sampled isolates. Briefly, TransPhylo takes a time-stamped phylogenetic tree as input, and explores ways of “coloring” this tree with one color for each infected host, thus revealing the evolution that occurred within this host. Transmission events are therefore also represented as the points of transition from one color to another, or in other words from one host to another. This approach is completely different from the one implemented in the default setting of outbreaker2, making it a good case study for the flexibility of implementation of custom models in the outbreaker2 framework.
The genetic likelihood of TransPhylo was already implemented within the original package, and was therefore easily passed on to outbreaker2 as a custom likelihood. To account for restrictions on the topology of the transmission tree in the TransPhylo model, a custom movement function on ancestries and infection times was also developed. This work was implemented in the R package o2mod.TransPhylo (standing for ‘outbreaker2 module: TransPhylo’), which infers transmission trees using the TransPhylo genetic model while benefiting from the epidemiological model exploiting data on the incubation period and generation time distributions [7], extending the original Wallinga & Teunis model [5]. The total effort required to implement this model was minimal: only 185 lines of code (LOC) were necessary to design o2mod.TransPhylo, which is negligible compared to the 1434 LOC in the original TransPhylo package, or the 7633 LOC of outbreaker2.
We compared the performance of o2mod.TransPhylo and TransPhylo by reconstructing simulated outbreaks, using the simulator in the phybreak package described described by Klinkenberg et al. [14]. We used epidemiological and evolutionary parameters of Ebola virus as a plausible use case (Table 2), and assumed a linearly growing within-host pathogen population size. A total of 100 outbreaks each with 20 cases were simulated, and reconstructed using o2mod.TransPhylo, TransPhylo, and the default outbreaker2 algorithm.
Table 2.
Epidemiological and evolutionary parameters for Ebola virus. When several studies are cited, the mean value weighted by the sample size of the study was used
Algorithm performance
MCMC chains of o2mod.TransPhylo converged rapidly, and mixed more efficiently than those of TransPhylo as demonstrated both by visual chain inspection (Fig. 2) and lower autocorrelation between log-likelihood values (0.31 and 0.72 at a lag of 50, respectively). This resulted in a significantly higher effective sample size per iteration (95.2 and 27.2 across a 5000 iteration window, respectively). However, individual iterations were significantly slower, taking on average 555.2 s per 1000 iterations, compared to 4.6 s for TransPhylo and 4.5 s for the basic model of outbreaker2. The vast majority of computational time by o2mod.TransPhylo was spent in the custom functions, which could be re-written in C++ for a performance boost. However, running times of o2mod.TransPhylo were acceptable for a complex Bayesian model: final results (well-mixed chain with 10,000 iterations) could be obtained in 1.5 h on a standard desktop computer. Users can therefore implement their models entirely in R and expect reasonable runtimes.
Fig. 2.

MCMC traces of posterior likelihood for o2mod.TransPhylo and the original TransPhylo package
Model outputs
Visual inspection of ancestry assignments for a single outbreak suggests that outbreaker2 successfully explored the posterior distribution, and demonstrates that the inference framework is general enough to accommodate new models (Fig. 3). Encouragingly, o2mod.TransPhylo and TransPhylo appear to describe highly similar posterior distributions of ancestries, and agree on many assignments even if these have a very low posterior frequency.
Fig. 3.

Posterior distribution of ancestry assignments using o2mod.TransPhylo and the original TransPhylo package. The size of each circle indicates the frequency of a given individual (“infector”) in the posterior distribution of infectors for a given case (“infectee”). An infector of 0 (bottom row) indicates that the individual is the index case. Black crosses represent the true simulated ancestries
To better compare the ancestry assignments made by o2mod.TransPhylo and TransPhylo, we used a consensus tree, defined as the tree with the highest posterior infector probability for each case, as a summary statistic. Across 100 outbreaks, on average 76.5% of ancestry assignments were equivalent. This represents a significant increase over the baseline similarity between the default outbreaker2 model and TransPhylo, which agree on only 41.3% of ancestries on average, as confirmed by individual comparisons of consensus trees in reconstructed outbreaks (Fig. 4). It is important to note that 100% agreement between o2mod.TransPhylo and TransPhylo is not expected, as the epidemiological model of the latter parametrizes an offspring distribution and incorporates additional prior knowledge on its shape. However, the significant convergence in results upon using a custom likelihood acts as a proof of concept that outbreaker2 can accurately recreate high level behaviour of largely different inference frameworks, and therefore represents a promising starting point for the implementation of future models.
Fig. 4.

Similarity of consensus trees inferred by o2mod.TransPhylo and outbreaker2 compared to TransPhylo. The consensus tree of a reconstructed outbreak is defined as the tree with the ancestor of the highest posterior probability for each case. The similarity between consensus trees is calculated as the proportion of identically assigned ancestries. The x-axis indicates individual simulated outbreaks. Each outbreak was reconstructed once using the default outbreaker2 model (white dots), and once using o2mod.TransPhylo (black dots). The colour of the line represents the change in similarity to the consensus tree returned by the original TransPhylo package
Future developments
outbreaker2 is introduced as a flexible platform for outbreak reconstruction. We believe most future developments will occur through the creation of new modules by the community, distributed as separate R packages. Further adjustments may be made to accommodate additional epidemiological and evolutionary data and parameters currently not implemented in outbreaker2, and which may limit the scope for additional modules. Such changes will however be merely incremental, and should not represent any substantial development challenges.
Conclusion
outbreaker2 is a highly flexible outbreak reconstruction tool that can implement complex epidemiological and genetic models within an optimised and robust transmission tree inference framework. It allows users to focus on model development rather than software implementation, and provides a unifying platform for outbreak reconstruction tools that promotes interoperability and ease of use. We encourage the development of extensions to outbreaker2 by the wider scientific community, with the goal of accumulating an extensive and sophisticated repertoire of methods for outbreak reconstruction within the R software.
Availability and requirements
Project name: outbreaker2
Project home page: http://www.repidemicsconsortium.org/outbreaker2/
Project development page: https://github.com/reconhub/outbreaker2
Operating system(s): Platform independent
Programming language: R, C++
Other requirements: C++ 11
License: MIT
Any restrictions to use by non-academics: None
Acknowledgements
We are thankful to the R community for sustaining the development of free, open-source statistical software, to github (http://www.github.com) for providing code hosting facilities, and to travis (https://travis-ci.org/), appveyor (https://www.appveyor.com/), and codecov (https://codecov.io/) for providing free resources for unit testing.
Funding
FC is funded by the Wellcome Trust.
XD is funded by the UK Medical Research Council.
RF is funded by UK Medical Research Council Centre for Outbreak Analysis and Modelling.
NF is funded by UK Medical Research Council; UK National Institute for Health Research under the Health Protection Research Unit initiative; National Institute of General Medical Sciences under the Models of Infectious Disease Agent Study initiative; Bill and Melinda Gates Foundation.
AC is funded by the Medical Research Council Centre for Outbreak Analysis and Modelling.
TJ is funded by the National Institute for Health Research - Health Protection Research Unit for Modelling Methodology, and by the Medical Research Council Centre for Outbreak Analysis and Modelling.
The funders had no role in the design of the study, the collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
The datasets generated and analysed during the current study are available in the github repository https://github.com/finlaycampbell/BMC_outbreaker2.
About this supplement
This article has been published as part of BMC Bioinformatics Volume 19 Supplement 11, 2018: Proceedings from the 6th Workshop on Computational Advances in Molecular Epidemiology (CAME 2017). The full contents of the supplement are available online at https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-19-supplement-11.
Abbreviations
- LOC
Lines of code
- MCMC
Markov Chain Monte Carlo
Authors’ contributions
FC wrote the majority of the manuscript, run the analyses, and contributed to model development and software implementation. XD developed the o2mod.TransPhylo module for outbreaker2, and edited the manuscript. RF provided advice on code design. AC and NF provided advice on the outbreaker2 model. TJ wrote the outbreaker2 package, supervised new developments, and contributed to writing the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Finlay Campbell, Email: f.campbell15@imperial.ac.uk.
Xavier Didelot, Email: x.didelot@imperial.ac.uk.
Rich Fitzjohn, Email: r.fitzjohn@imperial.ac.uk.
Neil Ferguson, Email: neil.ferguson@imperial.ac.uk.
Anne Cori, Email: a.cori@imperial.ac.uk.
Thibaut Jombart, Phone: 0044 (0)20 7594 3658, Email: thibautjombart@gmail.com.
References
- 1.Faye O, Boëlle P-Y, Heleze E, Faye O, Loucoubar C, Magassouba N, et al. Chains of transmission and control of Ebola virus disease in Conakry, Guinea, in 2014: an observational study. Lancet Infect Dis. 2015;15:320–326. doi: 10.1016/S1473-3099(14)71075-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lloyd-Smith JO, Schreiber SJ, Kopp PE, Getz WM. Superspreading and the effect of individual variation on disease emergence. Nature. 2005;438:355–359. doi: 10.1038/nature04153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Althaus CL. Ebola superspreading. Lancet Infect Dis. 2015;15:507–508. doi: 10.1016/S1473-3099(15)70135-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferguson NM, Donnelly CA, Anderson RM. Transmission intensity and impact of control policies on the foot and mouth epidemic in Great Britain. Nature. 2001;413:542–548. doi: 10.1038/35097116. [DOI] [PubMed] [Google Scholar]
- 5.Wallinga J, Teunis P. Different epidemic curves for severe acute respiratory syndrome reveal similar impacts of control measures. Am J Epidemiol. 2004;160:509–516. doi: 10.1093/aje/kwh255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ypma RJF, van Ballegooijen WM, Wallinga J. Relating phylogenetic trees to transmission trees of infectious disease outbreaks. Genetics. 2013;195:1055–1062. doi: 10.1534/genetics.113.154856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jombart T, Cori A, Didelot X, Cauchemez S, Fraser C, Ferguson N. Bayesian reconstruction of disease outbreaks by combining epidemiologic and genomic data. PLoS Comput Biol. 2014;10:e1003457. doi: 10.1371/journal.pcbi.1003457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Didelot X, Gardy J, Colijn C. Bayesian inference of infectious disease transmission from whole-genome sequence data. Mol Biol Evol. 2014;31:1869–1879. doi: 10.1093/molbev/msu121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mollentze N, Nel LH, Townsend S, le Roux K, Hampson K, Haydon DT, et al. A Bayesian approach for inferring the dynamics of partially observed endemic infectious diseases from space-time-genetic data. Proc Biol Sci. 2014;281:20133251. doi: 10.1098/rspb.2013.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Worby CJ, O’Neill PD, Kypraios T, Robotham JV, De Angelis D, Cartwright EJP, et al. Reconstructing transmission trees for communicable diseases using densely sampled genetic data. Ann Appl Stat. 2016;10:395–417. doi: 10.1214/15-AOAS898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall M, Woolhouse M, Rambaut A. Epidemic Reconstruction in a Phylogenetics Framework: Transmission Trees as Partitions of the Node Set. PLoS Comput Biol. 2015;11:e1004613. doi: 10.1371/journal.pcbi.1004613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lau MSY, Marion G, Streftaris G, Gibson G. A Systematic Bayesian Integration of Epidemiological and Genetic Data. PLoS Comput Biol. 2015;11:e1004633. doi: 10.1371/journal.pcbi.1004633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Maio N, Wu C-H, Wilson DJ. SCOTTI: Efficient Reconstruction of Transmission within Outbreaks with the Structured Coalescent. PLoS Comput Biol. 2016;12:e1005130. doi: 10.1371/journal.pcbi.1005130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klinkenberg D, Backer JA, Didelot X, Colijn C, Wallinga J. Simultaneous inference of phylogenetic and transmission trees in infectious disease outbreaks. PLoS Comput Biol. 2017;13:e1005495. doi: 10.1371/journal.pcbi.1005495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Didelot X, Fraser C, Gardy J, Colijn C. Genomic infectious disease epidemiology in partially sampled and ongoing outbreaks. Mol Biol Evol. 2017; 10.1093/molbev/msw275. [DOI] [PMC free article] [PubMed]
- 16.Teunis P, Heijne JCM, Sukhrie F, van Eijkeren J, Koopmans M, Kretzschmar M. Infectious disease transmission as a forensic problem: who infected whom? J R Soc Interface. 2013;10:20120955. doi: 10.1098/rsif.2012.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanner MA, Wong WH. The Calculation of Posterior Distributions by Data Augmentation. J Am Stat Assoc. 1987;82:528. doi: 10.1080/01621459.1987.10478458. [DOI] [Google Scholar]
- 18.R Development Core Team R . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. 2011. [Google Scholar]
- 19.Eddelbuettel D, Francois R. Rcpp: Seamless R and C++ Integration. J Stat Softw. 2011;40:1–18. [Google Scholar]
- 20.Popescu A-A, Huber KT, Paradis E. ape 3.0: New tools for distance-based phylogenetics and evolutionary analysis in R. Bioinformatics. 2012;28:1536–1537. doi: 10.1093/bioinformatics/bts184. [DOI] [PubMed] [Google Scholar]
- 21.Hastings WK. Monte Carlo sampling methods using Markov chains and their applications. Biometrika. 1970;57:97–109. doi: 10.1093/biomet/57.1.97. [DOI] [Google Scholar]
- 22.Geman S, Geman D. Stochastic relaxation, gibbs distributions, and the bayesian restoration of images. IEEE Trans Pattern Anal Mach Intell. 1984;6:721–741. doi: 10.1109/TPAMI.1984.4767596. [DOI] [PubMed] [Google Scholar]
- 23.Cottam EM, Thébaud G, Wadsworth J, Gloster J, Mansley L, Paton DJ, et al. Integrating genetic and epidemiological data to determine transmission pathways of foot-and-mouth disease virus. Proc Biol Sci. 2008;275:887–895. doi: 10.1098/rspb.2007.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Numminen E, Chewapreecha C, Sirén J, Turner C, Turner P, Bentley SD, et al. Two-phase importance sampling for inference about transmission trees. Proc Biol Sci. 2014;281:20141324. doi: 10.1098/rspb.2014.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aldrin M, Lyngstad TM, Kristoffersen AB, Storvik B, Borgan Ø, Jansen PA. Modelling the spread of infectious salmon anaemia among salmon farms based on seaway distances between farms and genetic relationships between infectious salmon anaemia virus isolates. J R Soc Interface. 2011;8:1346–1356. doi: 10.1098/rsif.2010.0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jombart T, Eggo RM, Dodd PJ, Balloux F. Reconstructing disease outbreaks from genetic data: a graph approach. Heredity. 2011;106:383–390. doi: 10.1038/hdy.2010.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ypma RJF, Bataille AMA, Stegeman A, Koch G, Wallinga J, van Ballegooijen WM. Unravelling transmission trees of infectious diseases by combining genetic and epidemiological data. Proc Biol Sci. 2012;279:444–450. doi: 10.1098/rspb.2011.0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morelli MJ, Thébaud G, Chadœuf J, King DP, Haydon DT, Soubeyrand S. A Bayesian inference framework to reconstruct transmission trees using epidemiological and genetic data. PLoS Comput Biol. 2012;8:e1002768. doi: 10.1371/journal.pcbi.1002768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soubeyrand S. Construction of semi-Markov genetic-space-time SEIR models and inference. Journal de la Société Française de Statistique. 2016;157:129–152. [Google Scholar]
- 30.Stadler T, Bonhoeffer S. Uncovering epidemiological dynamics in heterogeneous host populations using phylogenetic methods. Philos Trans R Soc Lond Ser B Biol Sci. 2013;368:20120198. doi: 10.1098/rstb.2012.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kenah E, Britton T, Halloran ME, Longini IM., Jr Molecular Infectious Disease Epidemiology: Survival Analysis and Algorithms Linking Phylogenies to Transmission Trees. PLoS Comput Biol. 2016;12:e1004869. doi: 10.1371/journal.pcbi.1004869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Worby Colin J, Lipsitch Marc, Hanage William P. Shared Genomic Variants: Identification of Transmission Routes Using Pathogen Deep-Sequence Data. American Journal of Epidemiology. 2017;186(10):1209–1216. doi: 10.1093/aje/kwx182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.WHO Ebola Response Team Ebola Virus Disease in West Africa — The First 9 Months of the Epidemic and Forward Projections. N Engl J Med. 2014;371:1481–1495. doi: 10.1056/NEJMoa1411100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.WHO Ebola Response Team West African Ebola Epidemic after One Year — Slowing but Not Yet under Control. N Engl J Med. 2015;372:584–587. doi: 10.1056/NEJMc1414992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoenen T, Groseth A, Feldmann F, Marzi A, Ebihara H, Kobinger G, et al. Complete Genome Sequences of Three Ebola Virus Isolates from the 2014 Outbreak in West Africa. Genome Announc. 2014;2:647–648. doi: 10.1128/genomeA.01331-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gire SK, Goba A, Andersen KG, Sealfon RSG, Park DJ, Kanneh L, et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science. 2014;345:1369–1372. doi: 10.1126/science.1259657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tong Yi-Gang, Shi Wei-Feng, Liu Di, Qian Jun, Liang Long, Bo Xiao-Chen, Liu Jun, Ren Hong-Guang, Fan Hang, Ni Ming, Sun Yang, Jin Yuan, Teng Yue, Li Zhen, Kargbo David, Dafae Foday, Kanu Alex, Chen Cheng-Chao, Lan Zhi-Heng, Jiang Hui, Luo Yang, Lu Hui-Jun, Zhang Xiao-Guang, Yang Fan, Hu Yi, Cao Yu-Xi, Deng Yong-Qiang, Su Hao-Xiang, Sun Yu, Liu Wen-Sen, Wang Zhuang, Wang Cheng-Yu, Bu Zhao-Yang, Guo Zhen-Dong, Zhang Liu-Bo, Nie Wei-Min, Bai Chang-Qing, Sun Chun-Hua, An Xiao-Ping, Xu Pei-Song, Zhang Xiang-Li-Lan, Huang Yong, Mi Zhi-Qiang, Yu Dong, Yao Hong-Wu, Feng Yong, Xia Zhi-Ping, Zheng Xue-Xing, Yang Song-Tao, Lu Bing, Jiang Jia-Fu, Kargbo Brima, He Fu-Chu, Gao George F., Cao Wu-Chun. Genetic diversity and evolutionary dynamics of Ebola virus in Sierra Leone. Nature. 2015;524(7563):93–96. doi: 10.1038/nature14490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baize Sylvain, Pannetier Delphine, Oestereich Lisa, Rieger Toni, Koivogui Lamine, Magassouba N'Faly, Soropogui Barrè, Sow Mamadou Saliou, Keïta Sakoba, De Clerck Hilde, Tiffany Amanda, Dominguez Gemma, Loua Mathieu, Traoré Alexis, Kolié Moussa, Malano Emmanuel Roland, Heleze Emmanuel, Bocquin Anne, Mély Stephane, Raoul Hervé, Caro Valérie, Cadar Dániel, Gabriel Martin, Pahlmann Meike, Tappe Dennis, Schmidt-Chanasit Jonas, Impouma Benido, Diallo Abdoul Karim, Formenty Pierre, Van Herp Michel, Günther Stephan. Emergence of Zaire Ebola Virus Disease in Guinea. New England Journal of Medicine. 2014;371(15):1418–1425. doi: 10.1056/NEJMoa1404505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and analysed during the current study are available in the github repository https://github.com/finlaycampbell/BMC_outbreaker2.