

Significance
Human CMV (HCMV) is a major cause of birth defects, an opportunistic infection in untreated HIV/AIDS, and a life-threatening complication in immunosuppressed transplant patients. HCMV infects multiple different cell types, so understanding mechanisms supporting the entry of the virus into different cells is crucial to deciphering its pathogenesis. Two viral glycoprotein complexes (trimeric and pentameric complexes) are known to control HCMV tropism in different cell types. Here we have refined our understanding of the mechanism by which the cell-surface receptor tyrosine kinase, PDGF receptor-α, supports the entry of HCMV into fibroblasts.
Keywords: herpesvirus, tropism, cellular receptor
Abstract
Human CMV (HCMV) exhibits a broad cell tropism that depends on two virion glycoprotein complexes: a trimeric complex (gH/gL/gO) that facilitates viral infection primarily in fibroblasts and a pentameric complex (gH/gL/pUL128-pUL130-pUL131A) that mediates infection in epithelial and endothelial cells. We performed genome-wide CRISPR screens in which the PDGF receptor-α (PDGFRα) was identified as the most significant cellular gene product essential for infection by HCMV virions containing only trimeric complex (trimer-only virus). Trimer-only virus did not enter PDGFRα knockout fibroblasts. By using knockout fibroblasts, the extracellular domain of PDGFRα required for virus entry was mapped, and the intracellular tyrosine kinase domain was shown to be nonessential. In addition, direct cell-to-cell spread of virus from knockout cells transfected with trimer-only viral DNA was blocked, despite the production of infectious virus in the transfected cells. In contrast to trimer-only virus, wild-type HCMV virions containing both trimeric and pentameric complexes entered PDGFRα knockout cells, reinforcing the view that fibroblasts contain a second, independent receptor for the pentameric complex. Importantly, however, wild-type virus entered the knockout fibroblasts at reduced efficiency compared with parental fibroblasts, arguing that the cellular receptor for the virion pentameric complex is limiting or that virions are produced containing different relative amounts of the two glycoprotein complexes. Finally, ectopic expression of PDGFRα in ARPE-19 epithelial cells and THP-1 monocytic cells, which have little to no endogenous PDGFRα expression, markedly enhanced their susceptibility to trimer-only virions. In sum, our data clarify several key determinants of HCMV tropism.
Human CMV (HCMV) is the prototypical β-herpesvirus. The HCMV virion envelope surrounds a tegument layer and capsid, which contains a 235-kb double-stranded DNA genome (1). HCMV exhibits broad tissue tropism and can be found in epithelial, endothelial, fibroblast, muscle, myeloid, and neuronal cells in viremic patients (2, 3). The viral and cellular determinants mediating entry into different cell types have been only partially defined. Two virus-encoded glycoprotein complexes on the virion envelope, a pentameric complex (gH/gL/pUL128-pUL130-pUL131A) and a trimeric complex (gH/gL/gO), are thought to control tropism (4, 5). We refer to these complexes as “pentamer” and “trimer” and to viruses containing the trimer but lacking the pentameric complex as “trimer-only” virus in this paper. The pentamer complex is crucial for HCMV entry into epithelial and endothelial cells in vitro (6–8). Viral genes encoding components of the pentamer complex are preserved in clinical strains, such as TB40/E (9), Merlin (10), and FIX (6), while laboratory strains, such as AD169, contain a mutation in an ORF encoding one of the pentamer components, UL128, UL130, and UL131A, disabling pentamer formation and function (11). Restoration of the pentamer in AD169 restores its capacity to infect epithelial cells (8), while mutations within constituents of the pentamer in the clinical isolate FIX disable its ability to infect epithelial cells (6).
The mechanism by which the pentamer interacts with cell-surface proteins to facilitate viral entry into epithelial cells is only partially understood. A recent study has shown that knockdown of either ErbB1/EGFR or ErbB2/HER2 reduces entry of a clinical isolate (VR1814) into ARPE-19 epithelial cells (12). However, several studies using EGFR-blocking antibodies have concluded that EGFR is dispensable for entry into ARPE-19 cells (13, 14). In contrast, HCMV entry into fibroblasts has been well studied, with multiple cellular determinants reported to be involved, including heparan sulfate (15), EGFR (13, 16), integrins (17, 18), PDGF receptor-α (PDGFRα) (12, 14, 19–21), and THY-1 (22). Considerable evidence points to PDGFRα as a trimer-specific receptor: (i) knockdown of PDGFRα reduced HCMV entry into fibroblasts (12, 19, 21); (ii) biochemical and structural experiments have shown direct binding of PDGFRα to trimer (12, 21); (iii) a soluble derivative of the PDGFRα extracellular domain inhibits trimer-only HCMV entry (20, 21); and (iv) PDGFRα is not required for the entry of HCMV lacking trimer into fibroblasts (20, 21).
Here we used an unbiased loss-of-function screen to identify host factors essential for HCMV infection. The screen identified PDGFRα as the most significant cellular factor required for trimer-only virus replication in human foreskin fibroblasts (HFFs). We generated clonal PDGFRα knockout (PDGFRα-KO) HFFs, and they could not be infected with trimer-only HCMV, although they remained susceptible to virus containing both trimer and pentamer complexes. By ectopically expressing different fragments of PDGFRα in the PDGFRα-KO cells, we showed that the extracellular IgG-like domain 3 of PDGFRα is required for entry of trimer-only viruses. Ectopic expression of PDGFRα in PDGFRα-KO HFFs, ARPE-19 epithelial cells, or THP-1 monocytes, which express little or no endogenous PDGFRα, was sufficient to restore susceptibility to infection with trimer-only virus. Efficient infection of unmodified PDGFRα-low cell types, such as peripheral blood monocytes, could be achieved by growth of pentamer-containing HCMV strains in ARPE-19 epithelial cells, which maintained robust virion pentamer expression. These studies support a model whereby PDGFRα is needed for entry of virions containing the trimer but not pentamer complex, point to an unidentified cell-surface receptor for the pentamer complex, and reveal a striking difference in the pentamer protein levels of virions produced in epithelial cells versus fibroblasts—a difference that influences the range of cell types that the virus can enter in subsequent rounds of infection.
Results
A Genome-Wide CRISPR Screen Confirms an Essential Role for PDGFRα in Trimer-Only HCMV Infection.
To identify host factors required for HCMV infection, we performed genome-wide CRISPR screens in HFFs using the HCMV trimer-only strain AD169, which contains a frame-shift insertion in the UL131A pentamer component (7, 8), and trimer-only strain Merlin (pAL1111), which has a nonsense mutation in the UL128 pentamer component (10). HFFs modified with a high-coverage CRISPR library (23, 24) were infected with HCMV, and surviving, infection-resistant cells were collected and subjected to sequence analysis. A modified sequencing strategy was implemented to streamline analysis and reduce cost (Materials and Methods); 98.4% of the reads recovered from surviving cells after AD169 infection were from five CRISPR guide RNAs (gRNAs) targeting PDGFRα, as were 33.2% of the reads from surviving cells from Merlin (pAL1111) infection targeted PDGFRα (Table 1 and Dataset S1). In both screens, MAGeCK analysis (25) clearly identified PDGFRα as the most significant cellular gene required for trimer-only HCMV infection (Fig. 1).
Table 1.
Next-generation sequencing read counts of gRNA sequences present in the surviving HFFs after AD169 or Merlin (pAL1111) infection
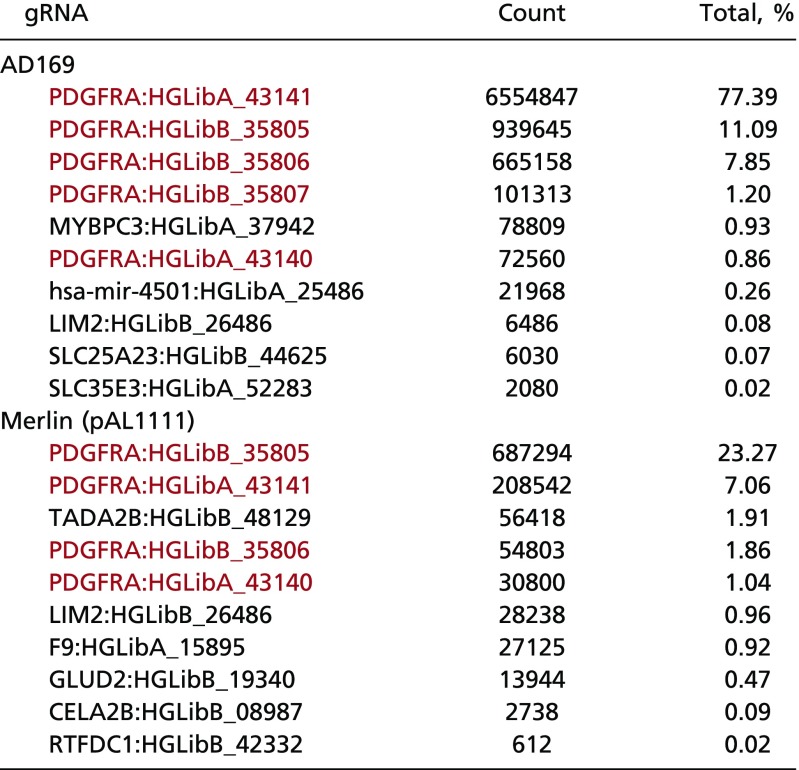 |
The top 10 gRNAs in terms of read counts are listed, with gRNAs against PDGFRα present in the surviving cells in red.
Fig. 1.

HFFs expressing Cas9 and gRNAs targeting PDGFRα survived trimer-only HCMV infection in a genome-wide CRISPR screen. Statistical analysis identified PDGFRα as the most significant hit from the screens of AD169 (A) and Merlin (B), using the MAGeCK algorithm.
To validate the screen results, we used CRISPR treatment followed by FACS sorting to generate a mixed population of PDGFRα-low HFF cells (SI Appendix, Fig. S1 A and B) and challenged them with GFP-expressing versions of HCMV strains AD169 and Merlin. GFP expression was monitored as a surrogate for viral entry. Cells transduced with nontargeting or PDGFRβ gRNAs, served as controls. Infection of the PDGFRα-low cells by AD169-GFP or Merlin-GFP (pAL1158) showed that the majority of cells did not express GFP by 1 d postinfection (dpi), while the PDGFRβ-low cells, or cells expressing control gRNAs, were >70% GFP-positive (SI Appendix, Fig. S1C). The difference in GFP expression was reflected in virus yields; PDGFRα-low cells produced 100- to 1,000-fold less infectious progeny compared with the control cells (SI Appendix, Fig. S1D). These data show that suppression of PDGFRα reduces the entry of trimer-only virions into fibroblasts.
The low percentage of residual infected cells observed after infection of mixed PDGFRα-low cells (SI Appendix, Fig. S1C) could be the result of a secondary cellular receptor for trimer-only virus, or it could be the result of read-through CRISPR modifications allowing partial PDGFRα expression. To differentiate between these possibilities, we generated clonal PDGFRα-KO HFFs using CRISPR. Three clones of PDGFRα-KO HFF cells were generated using three different gRNAs. PDGFRα cell-surface expression was virtually eliminated in all clones (Fig. 2A). Parental HFF cells were also cloned to obtain a population of similarly aged control cells (CN-9). Clonal PDGFRα-KO cells were completely resistant to infection with trimer-only virions, showing no indication of cytopathic effect (Fig. 2B) and no significant cell-free virus production (Fig. 2C). We conclude that PDGFRα is the sole cellular receptor for trimer-only HCMV in HFFs.
Fig. 2.

Trimer-only HCMV cannot initiate infection in PDGFRα-KO HFF cells. (A) Single-cell cloning of HFFs transduced with lentivirus expressing Cas9 and gRNAs against PDGFRα generated three PDGFRα-KO clones from three independent gRNAs. HFFs (CN-9) that underwent the same single-cell cloning process served as an aged control. Surface staining of PDGFRα and PDGFRβ was performed to confirm the PDGFRα-KO phenotype. (B) Cells in A were infected with Merlin-GFP (pAL1158) at a multiplicity of 3 FFU per cell, and cell morphology was monitored at 5 dpi. (C) Cell-free virus production at 5 dpi was measured by plaque assay. Mean ± SD is presented from three biological replicates, and the red line indicates the limit of detection.
PDGFRα Ig-Like Domain 3, but Not the Kinase Domain, Is Required for Trimer-Mediated HCMV Entry.
Given the low background provided by the PDGFRα-KO cells, we performed a mapping experiment to identify the extracellular domain of the receptor required for trimer-only virion entry. PDGFRα-KO cells (clone 1-10) expressing different truncated versions of PDGFRα were assayed for HCMV entry by measuring IE1 expression after infection with Merlin-GFP (pAL1158). PDGFRα constructs lacking Ig-like domains 1 and 2 supported virus entry, whereas those lacking domains 1, 2, and 3 completely abrogated entry (Fig. 3 A and B). This analysis does not rule out the possibility that domains 4 and 5 also contribute but clearly demonstrates an essential role for domain 3 in entry. We also measured viral genome replication in PDGFRα-KO cells (clone 1-10) producing a PDGFRα derivative lacking its C-terminal kinase domain (Fig. 3C, ΔC). The loss of the kinase domain did not cause a significant reduction in viral genome copy number (Fig. 3D).
Fig. 3.

IgG-like domain 3 of PDGFRα is required for trimer-only HCMV entry to fibroblasts. (A) PDGFRα-KO cells (clone 1-10) were transduced with lentiviruses expressing V5-tagged, full-length, or deleted variants of PDGFRα. Transduced cells were infected with Merlin-GFP (pAL1158) at a multiplicity of 3 FFU per cell, and IE1-positive cells were quantified at 24 hpi by flow cytometry. (B and C) Expression of V5-tagged full-length and deleted PDGFRα variants was confirmed by Western blot. (D) HFF or PDGFRα-KO cells (clone 1-10) expressing full-length or C-terminus-deleted PDGFRα in C was infected by AD169, TB40/E, or Merlin (pAL1111) at multiplicity of infection of 3 FFU per cell. Viral genome replication in infected cells was measured by qPCR at 4 dpi. D, IgG-like domain; SP, signal peptide; TM, transmembrane domain.
PDGFRα Is Required for Cell-to-Cell Spread of Trimer-Only Virus, but Not Virus Containing the Pentamer.
In addition to extracellular transmission by cell-free virus particles, HCMV can spread directly from cell to cell, which likely serves to avoid the humoral immune response (26, 27). Accordingly, we tested whether HCMV could spread from cell to cell in the absence of PDGFRα. Since trimer-only HCMV does not infect PDGFRα-KO cells (Fig. 2), we electroporated infectious HCMV DNA contained in a BAC into control and PDGFRα-KO cells and cultured them with or without anti-CMV immune globulin (CytoGam), which neutralizes extracellular HCMV virions. These experiments were performed with BAC DNA encoding trimer-only AD169-GFP, trimer-only Merlin-GFP (pAL1158; UL128−), and Merlin-GFP (pAL1160; UL128+). pAL1160 has a repaired UL128 locus and produces pentamer-containing virions (10). Whereas pAL1160 spread and produced plaques in PDGFRα-KO cells in the presence or absence of neutralizing antibody, virus did not spread from the initial cells transfected by the BAC DNAs containing trimer-only HCMV genomes (Fig. 4A). Even though they did not spread to neighboring cells, the trimer-only viruses generated infectious progeny in the BAC-transfected PDGFRα-KO cells, which was detected by transferring supernatants of transfected KO cells to HFF cells (Fig. 4B).
Fig. 4.

PDGFRα is important for cell-to-cell spread of trimer-only virus, but not for spread of virus containing the pentameric complex. (A) AD169-GFP, Merlin-GFP (pAL1158), or Merlin-GFP (pAL1160) BAC DNA was electroporated into CN-9 and PDGFRα-KO cells. The culture medium was supplemented with 6% CytoGam or PBS control and medium was changed daily. At 16 d after electroporation, immunofluorescence analysis of IE1-positive cells was performed to assay the spread of virus. White arrows point to the single cells producing IE1 from HCMV BACs. (B) Supernatants from CN-9 and 1-10 cells of the PBS group in A between day 8 and day 12 after electroporation were added to HFF cells to assay the presence of progeny viruses. Images (GFP or bright field) were recorded at 17 dpi. (C) CN-9 and 1-10 cells were electroporated with AD169-GFP BAC DNA, and GFP-positive cells were sorted at 6 d after electroporation. The same number (140) of GFP-positive CN-9 or 1-10 cells was added to cultures of HFF or 1-10 cells. GFP expression was recorded by fluorescent microscopy after 14 d of coculture. White arrows point to the single cells producing GFP from HCMV BACs.
We also sorted GFP-positive cells after electroporation of AD169-GFP BAC DNA into PDGFRα-positive (CN-9) or PDGFRα-KO (1-10) cells and plated equal numbers (140) of the sorted cells with HFF or 1-10 cells. Irrespective of the electroporated cell population’s PDGFRα status, infectious progeny spread throughout the monolayer of HFFs after 14 d of coculture (Fig. 4C, Top). In contrast, only single infected cells were observed when 1-10 cells were cocultured with GFP-positive 1-10 or CN-9 cells (Fig. 4C, Bottom). In sum, infectious progeny is produced following electroporation of trimer-only virus DNA into fibroblasts lacking PDGFRα, but direct cell-to-cell spread of the progeny is blocked in the absence of the receptor.
Pentamer-Containing HCMV Virions Enter PDGFRα-KO HFFs Through an EGFR/HER2-Independent Mechanism.
Given that pentamer-containing virions spread from cell to cell in the absence of PDGFRα, we tested the competency of pentamer-containing virions to directly enter PDGFRα-KO fibroblasts. As pentamer components rapidly mutate during passage in fibroblasts (28, 29), we propagated UL131A-repaired AD169 (AD169rUL131A-GFP) or UL128-repaired Merlin-GFP (pAL1160) in ARPE-19 epithelial cells, which maintains epithelial tropism (29). Trimer-only (pentamer-deficient) strains were prepared in fibroblasts. We first tested the entry efficiencies of matched trimer-only and pentamer-containing AD169 and Merlin strains, assaying virus-directed GFP or IE1 expression, respectively (Fig. 5). As anticipated, fibroblast-propagated trimer-only strains were not able to enter PDGFRα-KO cells. However, epithelial cell-propagated, pentamer-containing viruses retained the ability to enter PDFGRα-KO cells, albeit at 30–50% reduced efficiency (Fig. 5). These results argue that laboratory stocks of HCMV rapidly distribute into mixed populations of trimer- and pentamer-dominant virions after replication in epithelial cells. To test this hypothesis, we propagated virus from a BAC clone of pentamer-containing TB40/E in either fibroblasts or ARPE-19 epithelial cells and quantified PDGFRα-dependent entry of the resulting preparations. After two passages in epithelial cells, TB40/E-epi virions could infect ∼65% of PDGFRα-KO cells, whereas after two passages in fibroblasts TB40/E-fibro virions could only infect 15% of PDGFRα-KO cells at a multiplicity of 3 focus-forming units (FFU) per cell (Fig. 6A). Analysis of virion glycoproteins in virions produced from fibroblasts infected by TB40/E-fibro or TB40/E-epi confirmed markedly altered ratios of trimer and pentamer constituents in TB40/E-fibro and TB40/E-epi (Fig. 6B). These data show that PDGFRα is dispensable for entry of pentamer-dominant virions and reveal that the host cell type can dramatically alter the levels of trimer and pentamer complexes in HCMV virion populations.
Fig. 5.

Virus containing pentamer enters PDGFRα-KO HFF cells using an alternative route(s). HFFs, PDGFRα-KO cell clones (1-10 and 10-4), and CN-9 control cells were infected with viruses produced from fibroblasts or epithelial cells at a multiplicity of 3 FFU per cell. At 2 dpi, GFP expression from viral genomes was measured by flow cytometry to assay the percentage of infected cells, except for Merlin-GFP-epi and Merlin-GFP-fibro, where IE1 expression was measured to assay percentage of infected cells due to the dim GFP signal. The suffixes -epi and -fibro designate virus stocks produced in ARPE-19 epithelial and HFF fibroblast cells, respectively.
Fig. 6.

Pentamer-containing TB40/E raised from ARPE-19 epithelial cells enters PDGFRα-KO HFFs more efficiently than TB40/E grown from fibroblasts. (A) HFFs, PDGFRα-KO cell clones (1-10 and 10-4), and CN-9 control cells were infected with TB40/E-GFP produced from fibroblasts or epithelial cells at a multiplicity of 3 FFU per cell. At 2 dpi, GFP expression from viral genomes was measured by flow cytometry to assay the percentage of infected cells. The suffixes -epi and -fibro designate virus stocks produced in ARPE-19 epithelial and HFF fibroblast cells, respectively. (B) Western blot analysis of virions produced from MRC-5 cells infected by TB40/E-fibro or TB40/E-epi for gH/gL complexes (nonreducing conditions) using anti-gH antibody and for viral major capsid protein (MCP; reducing conditions) serving as loading control for virion proteins.
It has been proposed that EGFR and HER2 can mediate HCMV entry into epithelial cells (12). We tested whether EGFR or HER2 was necessary for entry of pentamer-containing virus into PDGFRα-KO or ARPE-19 cells. Knockdown of EGFR, HER2, or both EGFR and HER2 was efficient (SI Appendix, Fig. S2, Top) and had no significant inhibitory effect on entry of AD169rUL131A-GFP-epi into PDGFRα-KO fibroblasts. However, entry into ARPE-19 cells was reduced from 68% in cells receiving a nontargeting shRNA to 53% and 54%, respectively, in cells treated with shHER2 or shEGFR + shHER2 (SI Appendix, Fig. S2B, Bottom). To further explore a possible modest effect of double knockdowns in ARPE-19 cells, the experiment was repeated using the original shRNAs plus a second set of shRNAs, together with a second virus, TB40/E-epi, and little to no inhibition of entry was observed (SI Appendix, Fig. S3). These results suggest that an unidentified cellular receptor is required for entry of pentamer-containing virions into cells lacking sufficient PDGFRα expression.
Ectopic Expression of PDGFRα Enables Trimer-Only HCMV Entry into PDGFRα-Low Epithelial and Monocytic Cells.
ARPE-19 cells have no detectable cell-surface expression of PDGFRα (Fig. 7A, Top). If PDGFRα is the receptor for trimer-only virus, then its ectopic expression should restore susceptibility of ARPE-19 cells to trimer-only HCMV strains. To test this idea, we generated ARPE-19 epithelial cells stably expressing PDGFRα (Fig. 7A, Top) and then assayed the entry of trimer-only viruses. Addition of PDGFRα greatly increased their susceptibility to trimer-only HCMV viruses (Fig. 7A, Bottom). For example, the receptor increased the portion of AD169-infected ARPE-19 cells from 2.1 to 95%, and for Merlin (pAL1111) the increase was from 1.4 to 89%. We also tested the effect of exogenously expressed PDGFRα on the monocytic cell line THP-1, a commonly utilized, but notoriously difficult to infect, cell type for HCMV studies. As expected, these cells display little to no surface PDGFRα (Fig. 7B, Top). Expression of PDGFRα in these cells greatly increased their susceptibility to trimer-only HCMV viruses (Fig. 7B, Bottom). Furthermore, ectopic expression of PDGFRα enhanced the entry of TB40/E-fibro from 28 to 91% for ARPE-19 cells and from 1.4 to 58% for THP-1 cells (Fig. 7). The enhanced entry of fibroblast-passaged, pentamer-containing clinical strains, like TB40/E, into ARPE-19 cells expressing PDGFRα supports our conclusion that trimer-dominant virions are the major population within TB40/E stocks prepared in fibroblasts (Fig. 6B). We also confirmed the requirement of IgG-like domain 3 of PDGFRα for trimer-only virus (AD169-GFP) using ARPE-19 cells expressing different truncated versions of PDGFRα (SI Appendix, Fig. S4). Furthermore, ARPE-19 cells expressing full-length PDGFRα versus cells producing a PDGFRα derivative lacking its C-terminal kinase domain showed no differences in expression of GFP (SI Appendix, Fig. S4A) and IE1 (SI Appendix, Fig. S4C) or viral DNA accumulation (SI Appendix, Fig. S4D) following infection with trimer-only viruses [AD169 and Merlin (pAL1111)]. This observation confirms the result in fibroblasts (Fig. 3), again arguing that the PDGFRα signaling pathway is not required for the entry of trimer-only viruses.
Fig. 7.

Ectopically expressed PDGFRα renders ARPE-19 and THP-1 cells susceptible to trimer-only virus entry. Cell-surface staining of PDGFRα on ARPE-19 (A) or THP-1 (B) cells following no treatment (control), infection with a lentivirus lacking an insert (vector), or infection with a lentivirus expressing PDGFRα. (Top) Cells were subjected to flow cytometry using the indicated antibodies. (Bottom) Cells were infected with AD169-fibro, Merlin-fibro (pAL1111), or TB40/E-fibro at a multiplicity of 3 FFU per cell, and IE1 expression was measured at 24 hpi (ARPE-19) or 18 hpi (THP-1) to assay the percentage of infected cells.
Trimer-only HCMV virions enter fibroblasts by plasma membrane fusion (30) but enter epithelial cells by endocytosis (31, 32). Therefore, we tested which entry route trimer-only virions utilized in PDGFRα-expressing ARPE-19 cells. Consistent with previous reports (31, 32), treatment of epithelial cells with bafilomycin A1 (BFA) or NH4Cl, which neutralize endosomes and inhibit endocytosis, reduced TB40/E-fibro entry by ∼50% in control ARPE-19 cells (SI Appendix, Fig. S5). However, entry of trimer-only viruses into PDGFRα-expressing ARPE-19 cells was insensitive to either compound, suggesting that expression of PDGFRα shifted the entry mechanism to direct cell fusion in epithelial cells.
Pentamer-Containing Virus Produced from Epithelial Cells Efficiently Enters Monocytes.
PDGFRα is not expressed on peripheral blood monocytes (33), which are frequently used to as a model for nonpermissive HCMV infection and latency. Given that propagation of clinical HCMV strains in epithelial cells was able to maintain epithelial tropism and low ratios of trimer to pentamer virion complexes (Fig. 6), we compared entry of fibroblast and epithelial propagated stocks into freshly isolated primary monocytes. Purified CD14+ monocytes (Fig. 8A) were isolated from three healthy donors and did not express detectable cell-surface PDGFRα (Fig. 8B). The monocytes were infected with pentamer-containing Merlin-epi (pAL1120; UL128+) or trimer-only Merlin-fibro (pAL1111; UL128−) at a multiplicity of 3 FFU per cell and analyzed for intracellular IE1 expression at 16 h postinfection (hpi). Merlin-epi initiated IE1 expression in ∼40% of the infected monocytes, while Merlin-fibro was unable to initiate detectable IE1 expression in the monocytes, even at a multiplicity of 10 FFU per cell (Fig. 8C).
Fig. 8.

Virus containing the pentameric complex produced from epithelial cells efficiently enters primary human monocytes. CD14+ monocytes were purified from three donors and cell-surface staining of CD14 (A) or PDGFRα and PDGFRβ (B) were analyzed before culture in medium containing autologous serum. Purified CD14+ monocytes were infected with Merlin-epi (3 FFU per cell) or Merlin-fi (10 and 3 FFU/cell) for 16 h and intracellular staining of IE1 was performed to assay the percentage of infected cells (C).
We conclude that the lack of PDGFRα expression in monocytes severely restricts their infection by virus preparations deficient in the pentamer complex. Pentamer-containing viruses enter these cells via a PDGFRα-independent pathway.
Discussion
By executing a genome-wide CRISPR screen (Fig. 1 and Table 1) and by generating PDGFRα-KO fibroblasts (Fig. 2), we have confirmed and extended the prevailing view that PDGFRα is essential for trimer-only HCMV virions to enter fibroblasts. A role for PDGFRα in HCMV entry was first reported by Cobbs and coworkers (19), who showed that knockdown of PDGFRα in human fibroblasts inhibited the entry of both trimer-only viruses (Towne, AD169) and a pentamer-containing virus (strain TR). Since all of the viruses were produced in fibroblasts, our results (Fig. 6B) predict that the TR virions likely contained modest levels of pentamer. Cobbs and coworkers (19) also demonstrated that HCMV induces phosphorylation of PDGFRα and activates the downstream PI3K signaling pathway. Subsequent in vitro experiments have shown that PDGFRα binds to a recombinant trimer (gH/gL/gO), and structural analysis of the gH/gL/gO/PDGFRα complex has demonstrated that PDGFRα directly interacts with gO (12). This conclusion was reinforced by neutralization of HCMV infectivity in fibroblasts by a recombinant PDGFRα ectodomain (12, 20, 21) and via coimmunoprecipitation experiments showing that an antibody to gH could precipitate PDGFRα from fibroblasts infected with trimer-only, but not gO-deficient, virions (21). The latter study also demonstrated that entry of TB40/E into HEK293 cells was not dependent on the kinase domain of PDGFRα, suggesting that the PDGFRα ectodomain, but not receptor signaling, was required for HCMV entry (21). Our work confirms these observations by showing that trimer-only viruses cannot enter human fibroblasts in which PDGFRα has been genetically ablated (Fig. 2) and the receptor signaling is not required for HCMV infectivity (Fig. 3D and SI Appendix, Fig. S4). In addition, as Merlin (strain pAL1111) contains the complete ULb′ region while AD169 does not, our results demonstrate that entry of trimer-only viruses via PDGFRα is independent of the viral ULb′ region.
Our study also provides insights into the role of the PDGFRα in HCMV infection. We used PDGFRα-KO fibroblasts as recipients for ectopically expressed derivatives of PDGFRα to show that deletion of the N-terminal extracellular domain (amino acids 24–201) of the receptor does not influence entry of a trimer-only virus into fibroblasts, but extension of the deletion to include the D3 domain (amino acids 202–318) blocks entry (Fig. 3). The D3 domain is required for PDGF-AA homodimers to bind PDGFRα (34), so it is possible that the gO binding site partially overlaps the PDGF binding site on the receptor. Earlier work has shown that an N-terminal fragment (amino acids 91–130) of PDGFRα blocks HCMV infection of fibroblasts (20), but this region was not required in our study. This polypeptide, which spans the junction of Ig domains D1 and D2, has a stretch of amino acid homology to D3 (SI Appendix, Fig. S6), which may explain its inhibitory effect.
We also used PDGFRα-KO fibroblasts to demonstrate that cell-to-cell spread of trimer-only virus requires PDGFRα (Fig. 4), in agreement with a recent study showing that siRNA-mediated knockdown of PDGFRα blocked spread of trimer-only virus (21). Our PDGFRα-KO cells completely eliminated cell-to-cell spread (Fig. 4A), and the fact that PDGFRα-KO cells transfected with infectious HCMV DNA were able to produce infectious progeny (Fig. 4 B and C) eliminates the possibility that a block before the generation of competent virions was responsible for the failure of virus to spread. The mechanism by which PDGFRα mediates cell-to-cell spread of trimeric virus is not clear. It is possible that surface presentation of gO or gH/gL/gO on the infected cells interacts with PDGFRα on neighboring cells to permit cell fusion, which in turn facilitates virus transit between cells. Alternatively, virus might spread between cells by exiting from an infected cell and immediately binding to an adjacent cell expressing the receptor. We favor the former possibility, since we have shown that a mutant HCMV, lacking the virion pUL99 protein, fails to assemble enveloped virions but nevertheless spreads directly from cell to cell with normal efficiency (35).
Our present study further reveals that PDGFRα expressed in fibroblasts not only allows entry of trimer-only virus but it also improves the efficiency with which pentamer-containing virus produced in epithelial cells enters fibroblasts (Figs. 5 and 6). This is evident in the 30–50% reduction in entry into PDGFRα-KO fibroblasts versus control fibroblasts at the same passage level. The pentamer must direct entry through a PDGFRα-independent pathway, but this pathway is either limiting or virus produced in epithelial cells is not fully competent to utilize this pathway. The latter possibility could occur by the production of particles with differing relative amounts of pentamer in epithelial cells, perhaps due to natural variation in the assembly process. Propagation in epithelial cells would then provide strong selective pressure to maintain a large proportion of pentamer-dominant virions, while propagation in fibroblasts would select for trimer-dominant virions. This model is consistent with our entry data (Figs. 5, 6A, and 7A, Bottom), as well as our analysis of trimer and pentamer levels on fibroblast and epithelial propagated HCMV viruses (Fig. 6B). Thus, we favor a model in which only a portion of virus particles in pentamer-containing virus stocks are competent to enter fibroblasts independently of PDGFRα. It is difficult to know if this is an artifact of in vitro cell culture or more generally descriptive of virus grown in epithelial cells within an infected host. Conceivably, virions with different host-cell affinities are produced following infection of epithelial cells. A stochastic process that generates virus particles with different receptor specificities could facilitate movement of progeny virus to a distant cell without being captured by receptor on the surface of the cell that produced it.
Our observation that a substantial proportion of pentamer-containing viruses enter PDGFRα-KO cells provides very strong evidence for the existence of an unidentified pentamer receptor on PDGFRα-low cells, such as ARPE-19. Curiously, although we have been able to efficiently infect a variety of primary and transformed epithelial cells with epithelial-propagated HCMV stocks (8, 36), THP-1 cells were recalcitrant to infection with epithelial-prepared stocks, suggesting that some myeloid cells may lack both the trimer and pentamer receptors. An important, outstanding question is how HCMV enters into myeloid progenitor cells, such as CD34+ hematopoietic stem cells, which are thought to be the main reservoir for latent HCMV in vivo.
A practical implication of our study relates to the efficient infection of monocytes by pentamer-containing clinical strains of HCMV. The use of clinical strains has greatly facilitated studies of HCMV in epithelial, endothelial, CD34+ hematopoietic stem cells, and monocytes, all of which are important cell types for productive or latent infection by HCMV in vivo (11, 37). The competence of clinical strains to infect these cell types has been credited to the preservation of the pUL128, pUL130, and pUL131A subunits of the pentameric complex (6–8, 37). Whereas the Merlin clinical strain grown in fibroblasts (Merlin-fibro) did not detectably infect human peripheral blood monocytes, Merlin-epi was capable of infecting 36, 39, and 45% of monocytes from three independent donors (Fig. 8). These data highlight the importance of using epithelial cells to propagate clinical strains for study of HCMV in at least some clinically important cell types.
CRISPR KO screens are very powerful tools for investigation of host–pathogen interactions, but there are, of course, several limitations to the screen we have performed. The approach excludes cellular genes that are essential for cell viability, so our screen tested only nonessential cellular genes for a role in HCMV infection. In addition, the high multiplicity of infection employed (20 FFU per cell) put a stringent selection pressure on the assay. Cellular genes like PDGFRα, which are absolutely required for trimer-only virus cell entry scored in the assay, but disruption of cellular genes that augment infection would likely not protect cells from being killed by the virus. Furthermore, the cytomegalovirus IE1 and IE2 proteins, which are expressed at the start of infection, are promiscuous transcriptional regulatory proteins that exert profound effects on cellular physiology (38). As a consequence, loss of a cell function leading to a block at a later stage of the viral replication cycle might fail to score in our CRISPR screen due to IE1/2-mediated cell death. Despite its limitations, our screen identified additional cellular genes that might be important for HCMV replication (Dataset S1), including the deubiquitinating enzyme USP17L20. HCMV encodes its own deubiquitinating enzyme, pUL48, which facilitates its growth at a low multiplicity of infection (39) and implies the importance of deubiquitinating activity during HCMV infection. In addition, Lim2, a cell junctional calmodulin-binding protein, may be required for HCMV infection given the important role of calmodulin-dependent kinase kinase (CaMKK) for HCMV-mediated glycolytic activation and viral replication (40).
Materials and Methods
Cells, Viruses, and Plasmids.
Primary HFFs (41) and diploid human embryonic lung fibroblasts (MRC-5; ATCC CCL-171) were cultured in DMEM (D5796; Sigma) supplemented with 10% FBS, 1× MEM Non-Essential Amino Acids Solution, 1 mM sodium pyruvate, 1× GlutaMAX, 10 mM Hepes, pH 7.4, 100 units/mL penicillin, and 95 µg/mL streptomycin. ARPE-19 cells (ATCC CRL-2302) were cultured in a 1:1 mix of DMEM and Ham’s Nutrient Mixture F12 (SH30026; HyClone) with the same supplements as HFF and MRC-5. THP-1 cells (ATCC TIB-202) were cultured in RPMI-1640 medium supplemented with 10% FBS, 100 units/mL penicillin, and 95 µg/mL streptomycin. The 293FT cells were maintained in DMEM supplemented with 10% newborn calf serum, 100 units/mL penicillin, and 95 µg/mL streptomycin.
To generate PDGFRα/β-low cell populations, HFFs were transduced with lentiviruses expressing Cas9 and nontargeting gRNAs, or gRNAs against PDGFRα or PDGFRβ. Fourteen days after transduction, HFF cells transduced with gRNAs against PDGFRα and PDGFRβ were sorted by flow cytometry after cell-surface staining of PDGFRα and PDGFRβ, respectively. Cell-surface staining of PDGFRα and PDGFRβ was also measured before and after sorting.
To generate clonal PDGFRα-KO cells, lentiviral vector encoding Cas9 and gRNA (lentiCRISPRv2-gRNA) were transfected into 293FT cells, along with psPAX2 and pMD2.G, to produce lentivirus. HFF cells were subsequently transduced by the lentivirus and selected with 1.5 µg/mL puromycin (P8833; Sigma) and cloned via single-cell plating in 96-well plates. PDGFRα-KO candidates were screened by cell-surface staining using flow cytometry. To generate the control cells that had undergone a similar number of cell doublings, the parental HFF cells without lentivirus transduction were processed through the same single-cell cloning process.
To generate ARPE-19 cells expressing PDGFRα, lentiviruses generated from pLenti6-UBC-PDGFRα-PGK-blast and empty vector , a gift from J. Munger, University of Rochester, Rochester, NY (42), were used to transduce ARPE-19 cells in the presence of 6 µg/mL polybrene for 18 h, and then cells were selected with 10 µg/mL blasticidin for 3 d, beginning at 2 d after transduction. To generate THP-1 cells expressing PDGFRα, lentiviruses generated from pLV-EF1a-PDGFRα-FL-no-V5-IRES-puro and empty vector were used to transduce THP-1 cells and selected with 2 µg/mL puromycin for 4 d, beginning at 2 d after transduction.
CD14+ monocytes were purified from peripheral blood samples of three healthy donors (New York Blood Center) using CD14 microbeads (130-050-201; Miltenyi Biotec). Before culture in Iscove’s modified Dulbecco’s medium supplemented with 10% autologous human serum, monocytes were stained with CD14 or PDGFRα/β antibodies to confirm purity and surface expression as described below.
BAC infectious clones of AD169 (pAD/Cre) (43), AD169-GFP (pAD-GFP) (44), Merlin (pAL1111; UL128−) (10), Merlin-GFP (pAL1158; UL128−; IE2-IRES-GFP) (10), Merlin-GFP (pAL2159; UL128−; IE2-P2A-GFP), a gift from R. Stanton, Cardiff University, Cardiff, UK, TB40/E (TB40-BAC4) (9), and TB40/E-GFP (TB40/Ewt-GFP) (45) were electroporated into MRC-5 cells and subsequently passaged once more in MRC-5 cells to generate virus stocks. BAC infectious clones of AD169rUL131A-GFP (pADrUL131-C4) (8), Merlin (pAL1120; UL128+) (10), Merlin-GFP (pAL1160; UL128+; IE2-IRES-GFP) (10), TB40/E (TB40-BAC4) (9), and TB40/E-GFP (TB40/Ewt-GFP) (45) were electroporated into ARPE-19 cells and subsequently propagated once more in ARPE-19 cells to generate virus stocks. For electroporated cells, when cytopathic effect reached ∼100%, either supernatant (AD169) or cells (clinical strains) were passaged to corresponding cell type to amplify the virus, where only supernatant was collected and concentrated by ultracentrifugation with 20% sorbitol solution as cushion.
For titration of virus stocks, virus stocks (-fibro) grown from MRC-5 cells were titrated on HFF cells and virus stocks (-epi) grown from ARPE-19 cells were titrated on ARPE-19 cells using fluorescent focus assay. Briefly, cells were infected by serial dilutions of virus stocks for 1 h at 37 °C and fixed at 24 hpi for indirect immunofluorescence assay using a mouse monoclonal antibody against IE1 (1B12) (46). To measure virus production from infected cells, end-point dilution assay (TCID50) (47) or plaque assay were performed, the latter of which has a better limit of detection to measure the virus production from PDGFRα-KO cells.
Plasmids were obtained or generated as follows. gRNAs from the GeCKOv2 library or designed using DESKGEN Cloud software (Desktop Genetics) were cloned into lentiCRISPRv2 (52961; Addgene) following the vendor’s oligo cloning protocol. These CRISPR clones include lentiCRIPSRv2-PDGFRα-gRNA-#1/#8/#10, lentiCRISPRv2-PDGFRβ-gRNA-#11, and lentiCRISPRv2-non-targeting-gRNA-#1/#2, using primers listed in SI Appendix, Table S1.
For domain mapping of PDGFRα, full-length and a series of truncated PDGFRα derivatives were made in the backbone of pLV-EF1a-IRES-puro (85132; Addgene) or pLV-EF1a-IRES-hygro (85134; Addgene). The template for PDGFRα was pLenti6-UBC-PDGFRα-PGK-blast, a gift from J. Munger (42), where PDGFRα was amplified from BJ fibroblast cDNA and contains five silent mutations (T303C, A1692G, A1701G, A2094G, and T3222C) compared with PDGFRα isoform 1 (NM_006206.5; CCDS3495.1). Full-length PDGFRα was amplified using primers Ra-FL-V5_Fwd and Ra-FL_Rev and cloned into pLV-EF1a-IRES-puro, designated pLV-EF1a-PDGFRα-FL-no-V5-IRES-puro. To complement PDGFRα in PDGFRα-KO clone 1-10, a PDGFRα-gRNA-#1–resistant PDGFRα-expressing plasmid was made from pLenti6-UBC-PDGFRα-PGK-blast by site-directed mutagenesis using primers PDGFRα_gRNA1_Q5_Fwd and PDGFRα_gRNA1_Q5_Rev. Based on this gRNA-#1–resistant PDGFRα clone, the full-length and various truncated clones with C-terminal V5 tag were amplified by PCR using primers listed in SI Appendix, Table S1 and cloned into pLV-EF1a-IRES-hygro.
To produce EGFR and HER2-deficient cells, nonmammalian shRNA (SHC002; Sigma) and shRNAs targeting EGFR and HER2 (SI Appendix, Table S1) were cloned into pLKO.1-puro (8453; Addgene), pLKO.1 hygro (24150; Addgene), or pLKO.1-blast (26655; Addgene). PDGFRα-KO clone 1-10 cells or ARPE-19 cells were transduced with lentiviruses expressing shRNAs with 5 µg/mL polybrene overnight and selected with 200 µg/mL hygromycin, 10 µg/mL blasticidin, or both for 3 d.
Genome-Scale CRISPR KO (GeCKO) Screen.
Libraries A and B of human GeCKO lentiviral gRNA library v2 (lentiCRISPRv2; 1000000048; Addgene) (23, 24) were transformed and amplified in Endura electrocompetent cells (60242; Lucigen) following the protocol associated with the library. A 1:1 mix of libraries A and B was transfected into 293FT cells using polyethylenimine (23966-2; Polysciences), along with psPAX2 and pMD2.G to generate a lentivirus stock of the complete GeCKOv2 library. 9 × 107 HFF cells were transduced with the library at multiplicity of ∼0.5 IU per cell for 18 h in the presence of 6 μg/mL polybrene (H9268; Sigma) followed by feeding with fresh medium. Two days after transduction, a final concentration of 1.5 μg/mL puromycin was added to the cells for 2 d and surviving cells were subsequently amplified. Twelve days posttransduction, genomic DNA was extracted from 6 × 107 cells using the Qiagen blood and cell culture DNA midi kit (13343; Qiagen) to serve as a baseline control or infected with AD169 or Merlin (pAL1111) at multiplicity of 20 FFU per cell for 1 h. At 26 dpi, DNA was extracted from the surviving cells on the infected plates using a QIAamp DNA mini or micro kit (51304 and 56304; Qiagen).
To amplify the gRNA locus from control or surviving cells, genomic DNA samples (5 μg) were used as template for the first round of PCR in 50-μL reaction volume using Herculase II Fusion DNA Polymerase (600679; Agilent), v2Adaptor_F and v2Adaptor_R primer pairs (SI Appendix, Table S1), and cycling conditions of 1× 2 min at 95 °C; 18× (20 s at 95 °C; 20 s at 60 °C; 30 s at 72 °C), and 1× 3 min at 72 °C. For control samples, 48 reactions of first-round PCR were performed (the number of reactions was determined to achieve 300× coverage). The size of GeCKOv2 library is 1.2 × 105 gRNAs; for 300× coverage, at least 1.2 × 105 × 300 = 3.6 × 107 Cas9/gRNA-carrying cells were needed. Given 6.6 μg gDNA per 1 × 106 cells, 3.6 × 107 cells correspond to 240 μg gDNA. Since 5 μg gDNA was used as template per 50-μL reaction, 48 reactions were needed to maintain 300× coverage. For surviving cells, all of the gDNA samples were used for PCR amplification.
To add the Illumina flow cell attachment sequence, sequencing primer, and barcode to the PCR product, 5 μL of pooled first PCR product was used as template with a unique forward primer with barcode for each sample and a universal reverse primer (SI Appendix, Table S1) for the second round of PCR (16–20 cycles) using the same polymerase and cycling conditions mentioned above for the first round. To avoid PCR overamplification, which could lead to a gRNA representation bias, small aliquots of reactions were taken at various times and assayed by using the Qubit dsDNA HS Assay Kit (Q32854; Invitrogen). Aliquots of the product were loaded onto 2% E-Gel EX Agarose Gels (G401002; Thermo Fisher) to separate the desired 317-bp DNA from second PCR from the 288-bp first PCR product. The 317-bp bands were subsequently excised and purified using QIAquick Gel Extraction Kit (28704; Qiagen) following the vendor’s protocol, except that the gel slice was dissolved with 3× QG buffer at 37 °C. The libraries were examined on Agilent Bioanalyzer DNA High Sensitivity chips for size distribution and quantified by Qubit fluorometer (Invitrogen). Based on the desired sequence read depth, appropriate amounts of second PCR products were mixed and purified using the MinElute PCR purification kit (28004; Qiagen). The library pools were sequenced using an Illumina HiSeq 2500 System as single-end 75-nt reads, with a 10% control phiX library spike in to mitigate the monotemplate issue for the sequencing system. Raw sequencing reads were filtered by Illumina HiSeq Control Software and only the pass-filter reads, which were around 2.5–3.0 × 108 per sequencing run, were demultiplexed by barcode splitting (no mismatch allowed) and then analyzed by using the MAGeCK (version 0.5.1) algorithm (25). Briefly, the MAGeCK “count” command was used to generate gRNA read count tables from raw fastq files. After count table generation, the MAGeCK RRA algorithm (with default parameters) was used to compare surviving cells following infection with AD169 or Merlin (pAL1111) with control cells, and the positively selected RRA score is used to rank genes in Fig. 1.
Protein Analyses.
For flow cytometry, cells were trypsinized, resuspended in FACS buffer (0.2% BSA in PBS), and incubated with fluorophore-conjugated antibodies for 20 min at 4 °C. The following antibodies were used: PE-PDGFRα (clone αR1; 556002; BD), PE-PDGFRβ (clone 28D4; 558821; BD), PE-isotype control (clone G155-178; 555574; BD), PE-EGFR (clone EGFR.1; 555997; BD), PE-HER2 (clone Neu 24.7; 340552; BD), PE-Cy7-CD14 (clone M5E2; 557742; BD), and PE-Cy7-isotype control (clone G155-178; 557907; BD). The samples were subsequently analyzed using a BD LSRII flow cytometer or sorted using a Bio-Rad S3e cell sorter.
For intracellular staining of the viral IE1 protein for analysis by flow cytometry, the FIX and PERM kit (GAS003; Invitrogen) was used with modifications to its protocol. Briefly, cells were trypsinized and stained with live/dead fixable blue dead cell stain (L23105; Invitrogen). Stained cells were then incubated with reagent A for 3 min at room temperature before adding cold methanol to permeabilize the nuclear membrane. They were then incubated with IE1 antibody (20 µL; 1B12) (46) in reagent B for 30 min at room temperature and subsequently with Alexa Flour 647 conjugated goat anti-mouse IgG (A-21235; Invitrogen) in staining buffer for 30 min at room temperature before data acquisition using a BD LSRII flow cytometer. Flow data were analyzed using FlowJo vX software, where cells were gated on FSC vs. SSC to eliminate debris, FSC-A vs. FSC-H to eliminate out aggregates, and FSC vs. DAPI or fixable blue dead cell stain to eliminate dead cells from subsequent plots.
For indirect immunofluorescence assays, cells were fixed in cold methanol for 10 min and incubated with anti-IE1 (1B12; 1:50 dilution) in 10% normal goat serum at 4 °C overnight. Alexa Flour 488 conjugated goat anti-mouse IgG (A-11001; Invitrogen) was used for visualization of infected cells using a Nikon TE2000 widefield platform fitted with an Andor Zyla sCMOS camera.
For Western blot assays, total cell protein (15–20 µg) was separated in 7.5% SDS/PAGE gels, transferred to nitrocellulose membrane, and incubated with mouse anti-V5 antibody (V8012, 1:2,000; Sigma) and rabbit anti–β-actin antibody (926-42210, 1:2,000; LI-COR) or rabbit anti-PDGFRα (5241, 1:1,000; CST) and mouse anti–α-tubulin (sc-32293, 1:1,000; Santa Cruz) overnight at 4 °C. Goat anti-mouse 800CW (926-32210, 1:5,000; LI-COR) and goat anti-rabbit 680RD (926-68071, 1:5,000; LI-COR) were then added at room temperature for 1 h, before scanning using an Odyssey CLx infrared imager. A previously described procedure (48) was followed to analyze trimer and pentamer of HCMV virions using anti-gH (AP86; undiluted culture supernatant), a gift from W. Britt, University of Alabama at Birmingham, Birmingham, AL, and anti-major capsid protein (MCP, 28-4; ascites fluid), a gift from W. Britt.
PDGFRα Domain Mapping.
Lentiviruses expressing full-length or truncated PDGFRα were made from 293FT cells as described above. PDGFRα-KO clone 1-10 cells and ARPE-19 were transduced with lentiviruses in the presence of 5 µg/mL polybrene overnight, and 200 µg/mL hygromycin (10687010; Thermo Fisher) was added at 2 d after transduction for 3 d. Selected clones were briefly expanded and seeded for infection with AD169-GFP-fibro or Merlin-GFP-fibro (pAL2159) at 3 multiplicities of infection or harvested for protein analysis in RIPA buffer. At 1 dpi, cells infected by AD169-GFP-fibro were directly analyzed for GFP expression by flow cytometry, and cells infected by Merlin-GFP-fibro (pAL2159) were processed for intracellular IE1 staining as described above.
Virus Entry and Spread Assays.
To assay viral entry in the presence of endosome acidification inhibitors, ARPE-19 cells and ARPE-19 cells transduced with lentivirus expressing PDGFRα or an empty vector were pretreated with 40 nM BFA or 40 µM NH4Cl for 1 h before infection with TB40/E-fibro, Merlin-fibro, or AD169-fibro at a multiplicity of 3 FFU per cell. Infected cells were then cultured with inhibitor and intracellular staining of viral IE1 protein was performed to measure the percentage of infected cells at 20 hpi.
To analyze viral genome replication, infected HFF cells were harvested for genomic DNA extraction using QIAamp DNA Mini Kit (51304; Qiagen). The samples were analyzed using primer sets against UL44 or genomic GAPDH locus (49) (SI Appendix, Table S1) and Power SYBR Green Master Mix (4367660; Thermo Scientific) by QuantStudio 6 real-time PCR system.
To assay direct cell-to-cell virus spread, AD169-GFP, Merlin-GFP (pAL1158), or Merlin-GFP (pAL1160) BAC DNA was electroporated into CN-9 or PDGFRα-KO cells. The cultures contained 6% Cytogam or PBS control and medium was changed daily. Sixteen days after electroporation, immunofluorescence analysis of IE1-positive cells was performed to assay the spread of virus as described above.
Supplementary Material
Acknowledgments
We thank N. Sanjana (Broad Institute/New York University), W. Li (George Washington University), and Q. Ding (Princeton University) for help on the GeCKO screen; L. Parsons and M. Cahn (Princeton University) for computing assistance; H. Yin (Massachusetts Institute of Technology) for advice on the PDGFRα-low sorting strategy; J. Munger (University of Rochester) for PDGFRα constructs; E. Murphy (Forge Life Science) for TB40/E-GFP BAC; R. Stanton and G. Wilkinson (Cardiff University) for Merlin BACs; W. Britt (University of Alabama at Birmingham) for gH and MCP antibodies; C. DeCoste, J. Grady, and K. Rittenback (Princeton University) for assistance with flow cytometry and sorting; and G. Laevsky (Princeton University) for assistance with confocal imaging. This work was supported by NIH Grant AI1112951 (to T.S.), New Jersey Commission on Cancer Research Postdoctoral Fellowship DFHS15PPC027 (to K.W.), and Ruth L. Kirschstein National Research Service Award AI106175 (to A.O.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1806305115/-/DCSupplemental.
References
- 1.Shenk TE, Stinski MF, editors. Human Cytomegalovirus. Springer; Berlin: 2008. [PubMed] [Google Scholar]
- 2.Sinzger C, Digel M, Jahn G. Human Cytomegalovirus, Current Topics in Microbiology and Immunology. Springer; Berlin: 2008. Cytomegalovirus cell tropism; pp. 63–83. [DOI] [PubMed] [Google Scholar]
- 3.Britt W. Human Cytomegalovirus, Current Topics in Microbiology and Immunology. Springer; Berlin: 2008. Manifestations of human cytomegalovirus infection: Proposed mechanisms of acute and chronic disease; pp. 417–470. [DOI] [PubMed] [Google Scholar]
- 4.Gerna G, Revello MG, Baldanti F, Percivalle E, Lilleri D. The pentameric complex of human cytomegalovirus: Cell tropism, virus dissemination, immune response and vaccine development. J Gen Virol. 2017;98:2215–2234. doi: 10.1099/jgv.0.000882. [DOI] [PubMed] [Google Scholar]
- 5.Podlech J, Reddehase MJ, Adler B, Lemmermann NAW. Principles for studying in vivo attenuation of virus mutants: Defining the role of the cytomegalovirus gH/gL/gO complex as a paradigm. Med Microbiol Immunol. 2015;204:295–305. doi: 10.1007/s00430-015-0405-2. [DOI] [PubMed] [Google Scholar]
- 6.Hahn G, et al. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J Virol. 2004;78:10023–10033. doi: 10.1128/JVI.78.18.10023-10033.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang D, Shenk T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc Natl Acad Sci USA. 2005;102:18153–18158. doi: 10.1073/pnas.0509201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang D, Shenk T. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J Virol. 2005;79:10330–10338. doi: 10.1128/JVI.79.16.10330-10338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sinzger C, et al. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J Gen Virol. 2008;89:359–368. doi: 10.1099/vir.0.83286-0. [DOI] [PubMed] [Google Scholar]
- 10.Stanton RJ, et al. Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J Clin Invest. 2010;120:3191–3208. doi: 10.1172/JCI42955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilkinson GWG, et al. Human cytomegalovirus: Taking the strain. Med Microbiol Immunol. 2015;204:273–284. doi: 10.1007/s00430-015-0411-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kabanova A, et al. Platelet-derived growth factor-α receptor is the cellular receptor for human cytomegalovirus gHgLgO trimer. Nat Microbiol. 2016;1:16082. doi: 10.1038/nmicrobiol.2016.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isaacson MK, Feire AL, Compton T. Epidermal growth factor receptor is not required for human cytomegalovirus entry or signaling. J Virol. 2007;81:6241–6247. doi: 10.1128/JVI.00169-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vanarsdall AL, Wisner TW, Lei H, Kazlauskas A, Johnson DC. PDGF receptor-α does not promote HCMV entry into epithelial and endothelial cells but increased quantities stimulate entry by an abnormal pathway. PLoS Pathog. 2012;8:e1002905. doi: 10.1371/journal.ppat.1002905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Compton T, Nowlin DM, Cooper NR. Initiation of human cytomegalovirus infection requires initial interaction with cell surface heparan sulfate. Virology. 1993;193:834–841. doi: 10.1006/viro.1993.1192. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Huong S-M, Chiu ML, Raab-Traub N, Huang E-S. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature. 2003;424:456–461. doi: 10.1038/nature01818. [DOI] [PubMed] [Google Scholar]
- 17.Feire AL, Koss H, Compton T. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc Natl Acad Sci USA. 2004;101:15470–15475. doi: 10.1073/pnas.0406821101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Huang DY, Huong S-M, Huang E-S. Integrin alphavbeta3 is a coreceptor for human cytomegalovirus. Nat Med. 2005;11:515–521. doi: 10.1038/nm1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soroceanu L, Akhavan A, Cobbs CS. Platelet-derived growth factor-α receptor activation is required for human cytomegalovirus infection. Nature. 2008;455:391–395. doi: 10.1038/nature07209. [DOI] [PubMed] [Google Scholar]
- 20.Stegmann C, et al. A derivative of platelet-derived growth factor receptor alpha binds to the trimer of human cytomegalovirus and inhibits entry into fibroblasts and endothelial cells. PLoS Pathog. 2017;13:e1006273. doi: 10.1371/journal.ppat.1006273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Y, et al. Human cytomegalovirus glycoprotein complex gH/gL/gO uses PDGFR-α as a key for entry. PLoS Pathog. 2017;13:e1006281. doi: 10.1371/journal.ppat.1006281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Q, Fischer E, Cohen JI. Cell surface THY-1 contributes to human cytomegalovirus entry via a macropinocytosis-like process. J Virol. 2016;90:9766–9781. doi: 10.1128/JVI.01092-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shalem O, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li W, et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. doi: 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murrell I, et al. The pentameric complex drives immunologically covert cell-cell transmission of wild-type human cytomegalovirus. Proc Natl Acad Sci USA. 2017;114:6104–6109. doi: 10.1073/pnas.1704809114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sattentau Q. Avoiding the void: Cell-to-cell spread of human viruses. Nat Rev Microbiol. 2008;6:815–826. doi: 10.1038/nrmicro1972. [DOI] [PubMed] [Google Scholar]
- 28.Dargan DJ, et al. Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J Gen Virol. 2010;91:1535–1546. doi: 10.1099/vir.0.018994-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murrell I, et al. Genetic stability of bacterial artificial chromosome-derived human cytomegalovirus during culture in vitro. J Virol. 2016;90:3929–3943. doi: 10.1128/JVI.02858-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Compton T, Nepomuceno RR, Nowlin DM. Human cytomegalovirus penetrates host cells by pH-independent fusion at the cell surface. Virology. 1992;191:387–395. doi: 10.1016/0042-6822(92)90200-9. [DOI] [PubMed] [Google Scholar]
- 31.Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J Virol. 2006;80:710–722. doi: 10.1128/JVI.80.2.710-722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang D, Yu Q-C, Schröer J, Murphy E, Shenk T. Human cytomegalovirus uses two distinct pathways to enter retinal pigmented epithelial cells. Proc Natl Acad Sci USA. 2007;104:20037–20042. doi: 10.1073/pnas.0709704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan G, Nogalski MT, Yurochko AD. Activation of EGFR on monocytes is required for human cytomegalovirus entry and mediates cellular motility. Proc Natl Acad Sci USA. 2009;106:22369–22374. doi: 10.1073/pnas.0908787106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lokker NA, et al. Functional importance of platelet-derived growth factor (PDGF) receptor extracellular immunoglobulin-like domains. Identification of PDGF binding site and neutralizing monoclonal antibodies. J Biol Chem. 1997;272:33037–33044. doi: 10.1074/jbc.272.52.33037. [DOI] [PubMed] [Google Scholar]
- 35.Silva MC, Schröer J, Shenk T. Human cytomegalovirus cell-to-cell spread in the absence of an essential assembly protein. Proc Natl Acad Sci USA. 2005;102:2081–2086. doi: 10.1073/pnas.0409597102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oberstein A, Shenk T. Cellular responses to human cytomegalovirus infection: Induction of a mesenchymal-to-epithelial transition (MET) phenotype. Proc Natl Acad Sci USA. 2017;114:E8244–E8253. doi: 10.1073/pnas.1710799114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goodrum F, Reeves M, Sinclair J, High K, Shenk T. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood. 2007;110:937–945. doi: 10.1182/blood-2007-01-070078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paulus C, Nevels M. The human cytomegalovirus major immediate-early proteins as antagonists of intrinsic and innate antiviral host responses. Viruses. 2009;1:760–779. doi: 10.3390/v1030760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim ET, Oh SE, Lee Y-O, Gibson W, Ahn J-H. Cleavage specificity of the UL48 deubiquitinating protease activity of human cytomegalovirus and the growth of an active-site mutant virus in cultured cells. J Virol. 2009;83:12046–12056. doi: 10.1128/JVI.00411-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McArdle J, Schafer XL, Munger J. Inhibition of calmodulin-dependent kinase kinase blocks human cytomegalovirus-induced glycolytic activation and severely attenuates production of viral progeny. J Virol. 2011;85:705–714. doi: 10.1128/JVI.01557-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bresnahan WA, Hultman GE, Shenk T. Replication of wild-type and mutant human cytomegalovirus in life-extended human diploid fibroblasts. J Virol. 2000;74:10816–10818. doi: 10.1128/jvi.74.22.10816-10818.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu S, Schafer X, Munger J. Expression of oncogenic alleles induces multiple blocks to human cytomegalovirus infection. J Virol. 2016;90:4346–4356. doi: 10.1128/JVI.00179-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu D, Smith GA, Enquist LW, Shenk T. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J Virol. 2002;76:2316–2328. doi: 10.1128/jvi.76.5.2316-2328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Terhune S, et al. Human cytomegalovirus UL38 protein blocks apoptosis. J Virol. 2007;81:3109–3123. doi: 10.1128/JVI.02124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Connor CM, Murphy EA. A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation, resulting in infectious progeny. J Virol. 2012;86:9854–9865. doi: 10.1128/JVI.01278-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu H, Shen Y, Shenk T. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J Virol. 1995;69:7960–7970. doi: 10.1128/jvi.69.12.7960-7970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reed LJ, Muench H. A simple method of estimating fifty per cent endpoints. Am J Epidemiol. 1938;27:493–497. [Google Scholar]
- 48.Zhou M, Lanchy J-M, Ryckman BJ. Human cytomegalovirus gH/gL/gO promotes the fusion step of entry into all cell types whereas gH/gL/UL128-131 broadens virus tropism through a distinct mechanism. J Virol. 2015;89:8999–9009. doi: 10.1128/JVI.01325-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu K, Orozco D, Hearing P. The adenovirus L4-22K protein is multifunctional and is an integral component of crucial aspects of infection. J Virol. 2012;86:10474–10483. doi: 10.1128/JVI.01463-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.